

Abstract
The ‘apple-shaped’ anatomical pattern that accompanies visceral adiposity increases risk for multiple chronic diseases, including conditions that impact the brain, such as diabetes and hypertension. However, distinguishing between the consequences of visceral obesity, as opposed to visceral adiposity-associated metabolic and cardiovascular pathologies, presents certain challenges. This review summarizes current literature on relationships between adipose tissue distribution and cognition in preclinical models and highlights unanswered questions surrounding the potential role of tissue- and cell type-specific insulin resistance in these effects. While gaps in knowledge persist related to insulin insensitivity and cognitive impairment in obesity, several recent studies suggest that cells of the neurovascular unit contribute to hippocampal synaptic dysfunction, and this review interprets those findings in the context of progressive metabolic dysfunction in the CNS. Signalling between cerebrovascular endothelial cells, astrocytes, microglia, and neurons has been linked with memory deficits in visceral obesity, and this article describes the cellular changes in each of these populations with respect to their role in amplification or diminution of peripheral signals. The picture emerging from these studies, while incomplete, implicates pro-inflammatory cytokines, insulin resistance, and hyperglycemia in various stages of obesity-induced hippocampal dysfunction. As in the parable of the five blind wanderers holding different parts of an elephant, considerable work remains in order to assemble a model for the underlying mechanisms linking visceral adiposity with age-related cognitive decline.
Keywords: Diabetes, insulin, sex difference, astrocyte, microglia, blood-brain barrier, memory, tumor necrosis factor alpha, interleukin-1beta, leptin, adiponectin
1. Introduction and evolutionary perspectives
The central nervous system is a unique metabolic environment characterized by high cellular metabolism and limited local energy storage (Kety and Schmidt, 1948; Sokoloff et al, 1955). These characteristics evolved in tandem with intricate coordination between local cerebral blood flow and neuronal activity, or ‘neurovascular coupling,’ as revealed by comparative anatomical measures, physiological proxies, and empirically based computational models of different species (Pontzer et al, 2016; Armstrong, 1983; Hayward and Baker, 1969; Karbowski, 2007). Historically, the expansion of human brain size and complexity has occurred in the context of food scarcity (Bellisari, 2008). Cellular specialization in nutrient sensing at the circumventricular organs, which lack a fully formed blood-brain barrier (BBB), predates mammalian evolution (Kiecker et al, 2017; Tsuneki, 1986). Neurons in each of the circumventricular organs have been implicated in sensing of glucose, ketones, and hormones such as insulin and leptin (Faouzi et al, 2007; Simpson et al, 2007; Kang et al, 2004; El Messari et al, 1998; Djogo et al, 2016). The conservation of ‘sensor neurons’ and their integration into energy balance circuits in different species implicates homeostatic control as one of the earliest drivers of brain evolution.
Unlike the neurons in the peripheral nervous system, the mammalian CNS exhibits limited regenerative capacity (Pleasure et al, 1999). Even under intact conditions, synaptic transmission is accompanied by extracellular ion and nucleotide gradients that are similar to damage-associated molecular patterns (DAMPs) in peripheral tissues. Specifically, neuronal firing is accompanied by localized bursts of extracellular ATP known to promote chemotaxis and polarization of lymphocytes outside of the CNS (Di Virgilio et al, 2020; Kitajima et al, 2020). As a consequence, increases in brain size likely emerged after the development of regulatory mechanisms for control of interactions between the immune system and the brain. Consistent with this idea, a functional BBB with endothelial tight junctions and limited paracellular permeability is present in species with high regenerative capacity in the adult CNS, such as the zebrafish Danio rerio (Kishimoto et al, 2012; Jeong et al, 2008) and the axolotl Ambystoma mexicanum (Amomoto et al, 2016; Lazzari et al, 2008), and in species with limited CNS regeneration in adulthood, such as the frog Xenopus laevis (Lee-Liu et al, 2017; De Jesús Andino et al, 2016) and most reptiles, birds, and mammals (Sánchez Alvarado and Tsonis, 2006; O’Brown et al, 2018).
While segregation between the peripheral immune system, comprised of hematopoetic lineage cells (and their derived signaling factors), and the CNS was originally framed as absolute, recent years have seen increasing recognition of dynamic interactions with cells in the brain parenchyma. Many of these interactions occur at the blood-brain and blood-cerebrospinal fluid interfaces, where vascular mural cells and choroid plexus epithelial cells coordinate the entry and exit of solutes, cytokines, peptide hormones, and protein aggregates (Abbott et al, 2006; Lun et al, 2015). The intercellular networks at each of the above interfaces are complex; taking the BBB as an example, arteries, arterioles, capillaries, and veins exhibit variable degrees of coverage and regulation by endothelial cells, pericytes, and vascular smooth muscle cells (VSMCs) on the luminal (blood) side (Abbott et al, 2006; Zhao et al, 2015). On the brain side, the above-listed populations of vascular mural cells interact with astroglial endfeet and are innervated by interneurons (Abbott et al, 2006), although this innervation remains incompletely characterized with respect to its function and structure. Sandwiched in between the glia limitans on the brain side and the basal lamina of the cerebral vasculature are the perivascular macrophages (pvMΦ) (Lapenna et al, 2018; Kierdorf et al, 2019). pvMΦ have been implicated in various neuroimmune interactions, including diapedesis and infiltration of bone marrow-derived immune cell populations from the vasculature and cytokine signaling between the brain and periphery (Serrats et al, 2010; Lapenna et al, 2018). However, these functions overlap with those of vascular mural cells, and the degree of anatomical specification in different vascular beds in various brain regions remains incompletely understood.
Given the cellular diversity at the blood-brain and blood-CSF interfaces, cerebral responses to perturbation of energy metabolism are likely to be complex and dynamic over time. The temporal kinetics of these reponses are likely to differ between classical metabolic circuits, such as the brainstem and hypothalamus; regions implicated in hedonic control of feeding, such as the prefrontal cortex and ventral tegmental area; and regions that mediate environment-context associations, such as the hippocampus and amygdala. This review considers neuroimmune responses to visceral adiposity in the hippocampus, with reference to the many unanswered questions regarding initation and propogation of synaptic dysfunction in these conditions. Insulin resistance develops hetereogeneously in different cell types and tissues, but a complete picture of CNS feedback has yet to emerge. This review covers the metabolic characteristics of mice with tissue-specific insulin insensitivity and identifies candidate signaling factors for feedback regulation of brain function. The physiological consequences of cell type-specific insulin insensitivity in the brain are reviewed with respect to their direct and indirect consequences for neurons. Dysfunctional interactions at the blood-brain and blood-cerebrospinal fluid barriers are considered, with emphasis on the consequences of subsequent neuronal and glial exposure to peripheral signals in obesity. Theoretical models for visceral adiposity-driven neuroinflammation are proposed, with reference to local and long-range effects on cells located within or adjacent to the brain parenchyma. The mechanistic and correlative interactions described in this review were identified based on publications listed in PubMed, Scopus, and Google Scholar. The overarching theme emerging from these studies is that feedback regulation of the hippocampus and other brain regions not directly implicated in homeostatic control likely occurs in discrete stages with chronic obesity. Delineating the peripheral metabolic and immunological signatures of progressive cognitive dysfunction in obesity could uncover critical windows for therapeutic intervention.
1.1. Visceral adiposity, cognition, and heterogeneous insulin resistance across cell types and tissues
Individuals with visceral obesity exhibit earlier and more frequent cognitive impairment with aging in longitudinal and twin studies (Jagust et al, 2005; Elias et al, 2005; Whitmer et al, 2008; Debette et al, 2011; Xu et al, 2011). While statistical covariation-based approaches suggest that this relationship occurs independently of other metabolic risk factors, such as insulin resistance (Whitmer et al, 2008, Xu et al, 2011), understanding the underlying mechanisms for visceral obesity-associated cognitive risk is fraught with caveats. Visceral obesity is also a major risk factor for the development of insulin resistance, which is a complex phenomenon involving progressive loss of sensitivity to the glucose-lowering effects of insulin in different tissues, including muscle, liver, and adipose tissue. Even in nondiabetic young adults, whole-body insulin sensitivity was correlated with cognitive performance (Bove et al, 2013; Bove et al, 2016), reflecting the critical role of energy homeostasis as a determinant of brain function. Preclinical research on obesity and cognition is gradually progressing from associational studies linking high-fat diet and memory impairment (Greenwood and Winocur, 1996; Molteni et al, 2002), to designs that incorporate static and/or dynamic measures of whole-body glycemic control (Stranahan et al, 2008; McNay et al, 2010; Morrison et al, 2010; Porter et al, 2011; Lavin et al, 2011; Jeon et al, 2012; Pancani et al, 2013; Grayson et al, 2014; Hao et al, 2016; Spencer et al, 2017; Wang et al, 2018; Petrault et al, 2019; Yamamoto et al, 2019; Guo et al, 2020). However, relationships between tissue-specific insulin resistance and cognition remain underexplored.
Given the diverse (and growing) list of feedback signals from adipose tissue, muscle, and liver that regulate cognition (Fig.1), tissue-specific insulin resistance has the potential to impact synaptic function independently of (or in coordination with) dysfunctional insulin signaling in the brain. Tissue-specific insulin resistance emerges earlier than whole-body deficits in glycemic control (reviewed by Czech, 2017), and can be assessed clinically and preclinically using glucose clamps, opening the potential for earlier identification of cognitive risk. Section 1.1.1 provides an overview of preclinical studies on tissue-specific insulin resistance, with reference to potential feedback regulation of circuits in the medial temporal lobe, and section 1.1.2 highlights the direct and indirect effects of insulin receptor deletion in cells located within or adjacent to the CNS.
Figure 1. Multisystem impact of tissue-specific insulin receptor deletion.
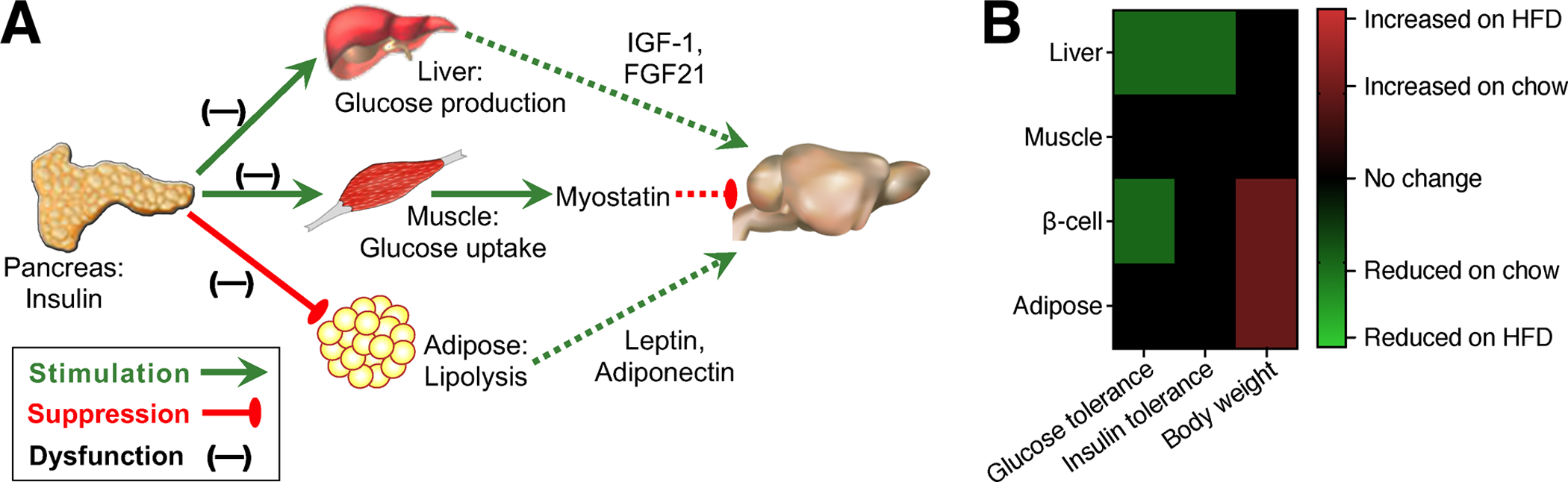
A) Tissue-specific effects of constitutive deletion of insulin receptor (IR) in pancreatic beta-cells, muscle, liver, or white adipose tissue. Dashed lines represent circulating factors that regulate cognition and are disrupted in tissue-specific IR knockouts. B) Organism-level metabolic outcomes following selective deletion of IR in peripheral metabolic organs. Abbreviations: HFD, high-fat diet.
1.1.1. Tissue-specific insulin resistance and feedback regulation of the medial temporal lobe
In chronic overnutrition, the demand placed upon the insulin-producing pancreatic beta-cells leads to cellular stress and eventual exhaustion, converting insulin resistant diabetes to insulin deficiency. Elegant cell type-specific inactivation of insulin receptor (IR) in pancreatic beta-cells, hepatocytes, skeletal muscle, adipocytes, and neurons indicates that compartmentalized responses to insulin govern different aspects of obesity-associated pathophysiology (Fig.1A–B). Ablation of IR expression in beta-cells results in defective glucose-stimulated insulin secretion, while constitutive deletion in hepatocytes interferes with insulin suppression of hepatic glucose output and increases fat mass (Kulkarni et al, 1999; Michael et al, 2000; Fig.1B). With respect to potential feedback regulation of cognition, LIRKO mice exhibit reductions in circulating insulin-like growth factor-1 (IGF-1) (Ling et al, 2018), a hepatokine with known positive effects on hippocampal synaptic plasticity and cognition (Fig.1A; for review, see Fernandez and Torres-Aleman, 2012). Fibroblast growth factor-21 (FGF21) is another liver-derived signaling factor previously shown to enhance insulin sensitivity, increase energy expenditure, and attenuate obesity (Fig.1A; reviewed by Gasser et al, 2017). Administration of exogenous FGF21 also rescues cognition, attenuates obesity, and reduces local inflammation in the hippocampus with dietary obesity (Wang et al, 2018). However, restoration of hepatic insulin sensitivity is unlikely to underlie these effects, as LIRKO are still responsive to the insulin-sensitizing and anti-obesity effects of exogenous FGF21 (Emanuelli et al, 2014). Although this remains an active area of research, the effects of FGF21 on body weight likely involve neurons in the ventromedial hypothalamus, while effects on insulin sensitivity are at least partially mediated by actions on adipose tissue (Jensen-Cody et al, 2020; BonDurant et al, 2017). The restorative effects of FGF21 could be mediated by direct actions on hippocampal neurons, as reported for the ventromedial hypothalamus (Wang et al, 2018; Jensen-Cody et al, 2020); alternatively, FGF21 could rescue hippocampal function via indirect effects on adipose tissue (BonDurant et al, 2017).
Constitutive IR ablation in skeletal muscle increases fat mass and impairs insulin sensitivity, as determined by hyperinsulinemic-euglycemic clamp (Fig.1A–B; Kim et al 2000; Bruning et al, 1998). More recently, muscle-specific insulin receptor overexpression was linked with a distinct and opposing phenotype characterized by increased lean mass and age-dependent improvements in glucose tolerance (Wang et al, 2020). While considerably less is known about the CNS impact of secreted factors from skeletal muscle (‘myokines’), emerging evidence supports potential interactions with brain regions outside of canonical metabolic circuits. Increased levels of the myokine myostatin, which inhibits muscle growth and promotes atrophy (reviewed by Pedersen and Febbraio, 2012), were recently linked with cognitive impairment in Alzheimer’s disease (AD) model mice (Lin et al, 2019). This relationship likely occurs independently of skeletal muscle insulin receptors, as MIRKO mice exhibit local and systemic reductions in myostatin (Fig.1A; Li et al, 2019). The trophic myokine irisin, generated following cleavage of transmembrane protein fibronectin type III domain-containing protein 5 (FNDC5), also regulates cognition in AD mice (Lourenco et al, 2019), but changes in irisin/FNDC5 have yet to be examined following selective manipulation of skeletal muscle insulin signaling.
The metabolic phenotype of muscle-specific IRKO mice (MIRKO) differs from that of white fat-specific IRKO mice (FIRKO), which exhibit reduced fat mass (Fig.1A–B; Bluher et al, 2002). The degree of reduction in fat mass varies between different adipose cre drivers, resulting in dramatically different phenotypes. Under the aP2 promoter, which generated moderate reductions in fat mass and serum leptin levels, FIRKO extended lifespan and were protected against age-related reductions in glycemic control (Bluher et al, 2003; Bluher et al, 2002). Under the adiponectin promoter, fat mass was severely reduced, causing ectopic lipid deposition, hypoleptinemia, and fasting hyperglycemia on standard chow (Softic et al, 2016). The aP2 and Adiponectin cre drivers differ in recombination efficacy and specificity (reviewed by Kang et al, 2014), and in this case, the serendipitous dose-response underscored the nonlinear relationship between fat mass and metabolic phenotype. Although CNS endpoints have yet to examined in either line of FIRKO mice, the phenotype of the adiponectin-cre driver is reminiscent of Seipin knockouts, which model congenital generalized lipodystrophy (Cui et al, 2011). Seipin knockouts and Adiponectin-cre/IRKO mice both exhibit severe hypoleptinemia and reduced serum adiponectin levels (Cui et al, 2011; Softic et al, 2016). Constitutive ablation of Seipin in the nervous system impairs memory and induces widespread glial reactivity (Qian et al, 2016), and leptin and adiponectin have independently been shown to support hippocampal synaptic function and memory (Fig.1A; for leptin, see Irving and Harvey, 2021; for adiponectin, Rizzo et al, 2010). While the potential synergistic effects of combined deprivation remain unexplored, those data will likely emerge from future studies. Published work in this area carries certain caveats associated with the developmental effects of IR knockdown; however, any differences between prior work in constitutive cre lines and future studies in inducible knockouts could lead to fascinating new questions related to critical periods for the observed phenotypes. Moreover, the underlying logic used to dissect the functional impact of insulin receptor ablation in various tissues is an excellent model for inquiry into other complex diseases.
1.1.2. Consequences of dysfunctional insulin signaling in vascular mural cells, astrocytes, and neurons
Endothelial insulin receptors are engaged during signal transduction and during influx of insulin into brown fat, muscle, and brain (Konishi et al, 2017; Banks et al, 1997). However, current knowledge surrounding the progression of cellular insulin insensitivity and dysfunctional transport in different tissues remains incomplete, likely due to the scarcity of experimental strategies for tissue-specific manipulation of endothelial cells. Despite this limitation, recent studies implicate impaired insulin transport into the CNS as a mechanism for disordered glucoregulation and perturbation of body weight homeostasis (Fig.2A–B; Garcia-Caceres et al, 2016; Konishi et al, 2017). Inducible ablation of IR in astrocytes impaired glycemic control in adult mice on normal chow, and these effects accompanied by cerebral hypometabolism, determined by PET imaging (Fig.2A; Garcia-Caceres et al, 2016). In a similar vein, constitutive ablation of IR among VE-cadherin-expressing endothelial cells introduces a temporal delay in insulin receptor phosphorylation across multiple brain regions, including the hippocampus, and in peripheral tissues such as skeletal muscle and brown adipose tissue (Konishi et al, 2017). In lean mice, disruption of endothelial insulin receptor expression was associated increased cerebrovascular permeability to 3kDa fluorescent dextran and reductions in hypothalamic tight junction protein expression (Konishi et al, 2017). With high-fat diet-induced obesity, constitutive deletion of endothelial IR increased susceptibility to insulin resistance and impaired glycemic control, as determined by IPITT and OGTT (Fig.2A–B; Konishi et al, 2017). Successful disruption of endothelial insulin receptor expression in VE-cadherinCRE/IRfl/fl mice was determined by western blotting for insulin receptor targets in whole aorta and binding of FITC-conjugated insulin in lectin-labeled cortical vascular profiles (Konishi et al, 2017). These approaches revealed a partial knockdown, which is consistent with subsequent findings in this model using radiolabeled insulin (Rhea et al, 2018). It should be noted that endothelial IR were knocked down throughout development (Konishi et al, 2017), and astroglial IR were ablated in adulthood (Garcia-Caceres et al, 2016). While both manipulations impaired glycemic control, the lack of endothelial IR during development may have contributed to the reductions in BBB integrity reported in VE-cadherinCRE/IRfl/fl mice (Konishi et al, 2017). The results of both studies are broadly consistent with prior work in rats with dietary obesity, which revealed insensitivity to the anorexic effects of intra-3rd ventricular insulin (Begg et al, 2013), and resistance to the procognitive effects of intrahippocampal insulin delivery using microdialysis (McNay et al, 2010). However, fundamental questions have yet to be answered regarding the onset and progression of cellular insulin resistance among diverse cell populations throughout the brain.
Figure 2. Direct and indirect effects of insulin receptor deletion in specific populations that reside within or adjacent to the CNS.
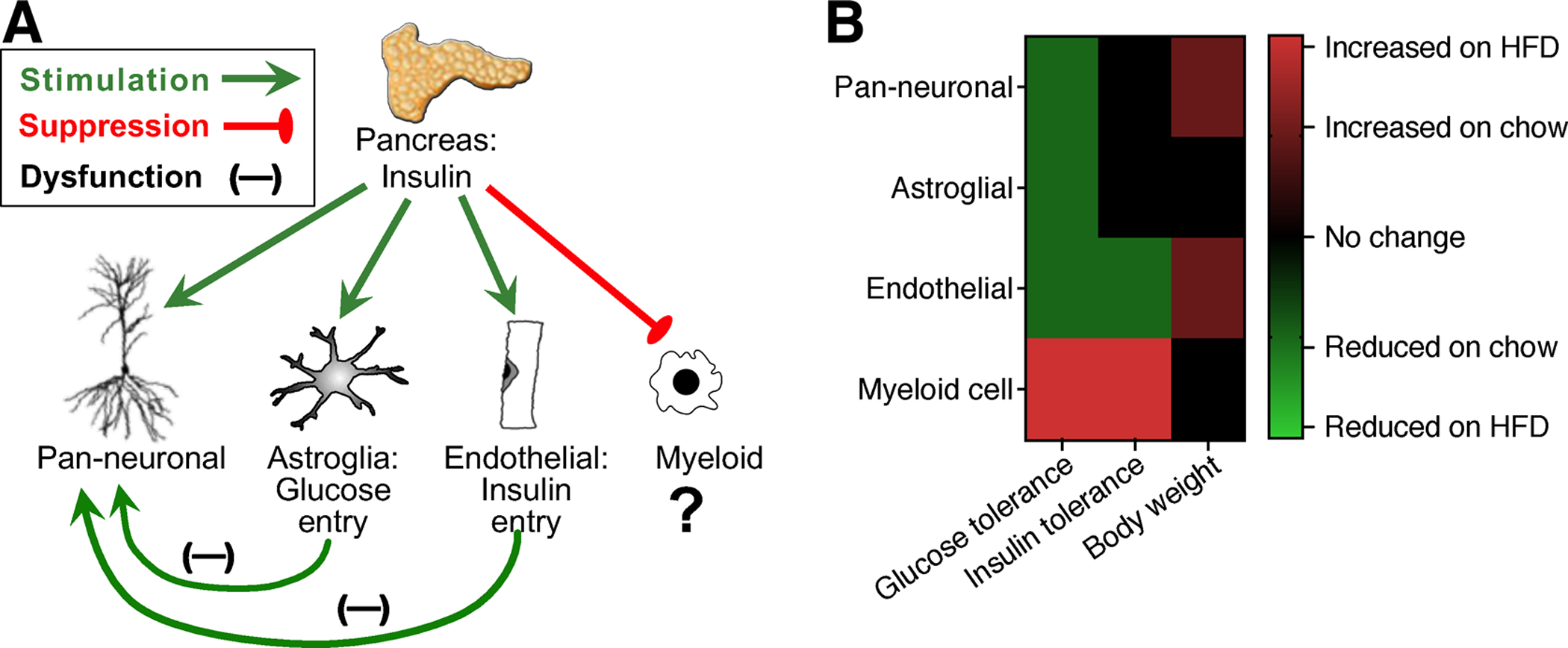
A) Cell type-specific and paracrine effects of IR ablation in neurons, astrocytes, endothelial cells, and myeloid cells. B) Impact of cell type-specific IR deletion on glucoregulation and body weight homeostasis. Abbreviations: HFD, high-fat diet.
Insulin enters the CNS via saturable transporters, likely via receptor-mediated transcytosis (Abbott et al, 2006). The same insulin receptor involved in cellular signaling is thought to mediate blood-to-brain transport, based on minimal influx of radiolabeled insulin following heat-inactivation (Banks et al, 1997). However, given the importance of insulin signaling for metabolic homeostasis, reproduction, and lifespan (Tower, 2017), it would not be surprising if redundant mechanisms governed CNS influx. This conclusion was indirectly supported by persistent entry of blood-borne radiolabeled insulin into the brains of VE-cadherinCRE/IRfl/fl mice and in the brains of Wt mice treated with S961, a peptide antagonist of IR (Rhea et al, 2018). While one interpretation involves IR-independent entry of circulating insulin into the brain, insulin will bind to IGF-1 receptors (and vice versa), albeit with much lower affinity (Schumacher et al, 1991). In the absence of serum binding proteins, insulin also competes with IGF-1 for receptor-mediated transport across the BBB (Yu et al 2006). While the initial report characterizing VE-cadherinCRE/IRfl/fl mice validated deletion and loss of IR signaling in the aorta, published RNA sequencing data support the possibility of tissue- and cell type-specific differences in IR and IGF1R expression. Specifically, capillary endothelial cells in the brain express higher levels of both IR and IGF1R than capillary endothelial cells of the lung (Vanlandewijck et al, 2018). Higher relative expression of IGF1R among capillary endothelial cells in the brain may therefore translate into greater potential for transport of circulating insulin by IGF1R after constitutive ablation of IR in VE-cadherin-expressing endothelial cells. Within the CNS, data from early postnatal (Zhang et al, 2014) and adult brain (Vanlandewijck et al, 2018) indicates that both IR and IGF1R are expressed at higher levels in endothelial cells, relative to microglia, astrocytes, and neurons.
In comparison with the multisystem impact of IR ablation in peripheral tissues, the effects of deletion in specific neuronal populations were more subtle. Selective IRKO in AgRP or POMC neurons did not alter food intake, body weights, or glucose homeostasis, but AgRP-specific IRKO disrupted insulin suppression of hepatic glucose production (Konner et al, 2007). By contrast, pan-neuronal IR deletion under the Nestin promoter increased body weight, fat mass, and fasting glucose levels (Fig.2A–B; Bruning et al, 2000; Fisher et al, 2005). Cell type-specific ablation of IR under the LysM promoter, which targets peripheral myeloid cells and microglia (reviewed by Blank and Prinz, 2016), reduced adipose tissue inflammation and improved glycemic control in mice with dietary obesity (Fig.2A–B; Mauer et al, 2010). However, the potential impact of microglia-specific IR deletion remains to be seen.
1.2. Disruption of CNS barriers and immune cell infiltration with visceral obesity
Cerebrovascular dysfunction can be initiated by inflammatory responses in the brain parenchyma, or may occur following exposure to damage-associated molecular patterns in circulation (Zhao et al, 2015). Neurodegenerative proteopathies such as AD and Parkinson’s disease are examples of intrinsic CNS disorders that disrupt cerebrovascular homeostasis, while HIV dementia and autoimmune encephalitis involve degradation by blood-borne factors. There has been limited investigation into whether cerebrovascular dysfunction is a cause or a consequence of inflammation in the brain parenchyma in obesity, but evidence exists for either scenario. On the brain side, neuronal firing is accompanied by transient increases in both local cerebral blood flow and vascular permeability (Vazana et al, 2016; for review, see Iadecola, 2017). Dietary obesity disrupts glutamatergic synaptic plasticity in the hippocampus (Stranahan et al, 2008; Pancani et al, 2013; Hao et al, 2016; Yamamoto et al, 2019; Guo et al, 2020), and it is possible that obesity-induced cerebrovascular deficits could be secondary to dysfunctional neurovascular coupling. On the blood side, C-reactive protein (CRP) is perhaps the most well-characterized circulating biomarker of inflammation in obesity (Kahn et al, 2006). Cerebrovascular exposure to CRP promotes BBB breakdown and elicits maladaptive glial reactivity following entry into the brain parenchyma (Kuhlmann et al, 2009; Hsuchou et al, 2012). This is just one hypothetical pathway in which peripheral inflammation might drive obesity-induced cerebrovascular dysfunction. In reality, the progression of cerebrovascular dysfunction in obesity likely involves a diverse set of interactions between cells of the CNS and vascular mural cells, which includes endothelial cells, pericytes, and vascular smooth muscle cells (VSMCs) surrounding larger arteries descending from the pial surface.
Until recently, the degree of transcriptional and structural heterogeneity among vascular mural cells in different brain regions has been a topic for speculation. However, a powerful new RNA-sequencing study recently elucidated certain fundamental principles governing endothelial cell heterogeneity in arterioles, capillaries, and venuoles (Vanlandewijck et al, 2018). Unlike the phenotypic clustering of gene expression observed between pericytes and arteriole-associated VSMCs, cerebrovascular endothelial cells in arteries, capillaries, and veins exhibited gradual, overlapping genetic signatures (Vanlandewijck et al, 2018). Consistent with anatomical gradients in gene expression, structural and functional decrements in cerebrovascular homeostasis are evident across multiple brain regions in obesity. Increases in BBB permeability have been reported in the hippocampus, arcuate nucleus of the hypothalamus, and prefrontal cortex in rodent models (Kanoski et al, 2010; Yi et al, 2012; Jais et al, 2015; Yamamoto et al, 2019). Anatomically, both the hippocampus and the arcuate nucleus lie adjacent to the cerebral ventricles, and the arcuate nucleus has a uniquely permissive cerebrovascular interface that enables local signaling between peripheral myeloid cells and microglia (Valdearcos et al, 2017). The hippocampus is also situated posterior to the subfornical organ (SFO), which lacks a BBB, but bundled myelinated axons of the fimbria separate the rostral pole of the hippocampus from the SFO. The ventral hippocampus is bordered by the lateral ventricles, but here again, myelinated axons of the fimbria demarcate the boundary and may therefore constrain paracrine interactions by limiting volume transmission. These anatomical constraints remain hypothetical at present, as heterogeneous patterns of obesity-induced synaptic dysfunction in different brain areas have not yet been causally linked with cell-autonomous or environmental features.
Anatomically, there are fewer constraints on inter-cellular signaling between hippocampal neurons and ependymal cells lining the dorsal third ventricle. Ventricular capillaries are highly fenestrated and lack tight junctions, rendering them more permeable to water-soluble signaling factors (reviewed by Lun et al, 2015). Adjacent epithelial cells form polarized inter-cellular adhesions, with tight junctions predominating at the apical surface facing the ventricles, and less-stringent adherens junctions linking cells at the basal surface facing the stroma (for review, see Neman and Chen, 2016). With aging, choroid plexus epithelial cells become compressed, potentially leading to disruption of cell-cell contacts and increased permeability of the BCSFB (Serot et al, 2001). Bone marrow-derived lymphocytes, including macrophages and T-cells, are present in cerebrospinal fluid and indirect evidence suggests that local production of cytokines influences hippocampal neuroplasticity and cognition (reviewed by Filiano et al 2017). Disruption of the blood-cerebrospinal fluid (CSF) barrier has yet to be examined in obesity, but given the greater basal permeability of the BCSF interface, interactions are likely and may represent an early event during the progression of obesity.
Circulating immune cells patrol the cerebral vasculature but do not penetrate the intact BBB (reviewed by Daneman and Engelhardt et al, 2017). Confinement of circulating immune cells is attributable to cellular and structural features of the luminal surface, with tight junction proteins essentially sealing the gaps between adjacent endothelial cells. Increased cerebrovascular permeability is accompanied by loss of tight junction protein expression in rodent models of obesity and diabetes (Kanoski et al, 2010; Ouyang et al, 2014; Stranahan et al, 2016), and we recently provide comprehensive evidence for disruption of tight junction ultrastructure (Yamamoto et al, 2019). This disruption could be intrinsically mediated by endothelial cells responding to the circulating milieu in obesity, or could be mediated by local interactions between endothelial cells and immune cell populations that patrol the cerebral vasculature. Conditional ablation of myeloid lineage cells protects against insulin resistance with high-fat feeding, and also blunted the induction of MMP-9 (Mauer et al, 2010). It is therefore possible that myeloid cell-specific insulin resistance might erode the BBB by increasing MMP-9 in circulating macrophages, but this scenario has yet to be addressed experimentally. Macrophage infiltration into the CNS has been reported in both genetic and dietary obesity (Buckman et al, 2014; Stranahan et al, 2016; Guo et al, 2020), and understanding the mechanism for this process could distinguish between signals that degrade the cerebrovascular barrier and chemokines that attract or repel circulating lymphocytes.
1.3. Visceral adiposity, interleukin-1beta, and neuroimmune feedback on the brain
During the early stages of obesity, adipose tissue expansion is accompanied by angiogenesis, which maintains tissue oxygenation and a relatively benign immunological environment (reviewed comprehensively by Crewe et al, 2017 and by Graupera and Claret, 2018). Over time, obesity-induced adipose tissue expansion outpaces angiogenesis, placing individual adipocytes further away from blood vessels, which exacerbates lipid overload-associated cellular stress by promoting hypoxia (Crewe et al 2017). Concurrent hypoxia and lipid overload eventually cause adipocyte cell death, which continues the cycle of macrophage infiltration and pro-inflammatory cytokine production (Graupera and Claret, 2018). These processes involve the pro-inflammatory cytokines interleukin-1beta (IL1b) and tumor necrosis factor alpha (TNFa), with the latter produced following lipid overload, and the former produced mainly by macrophages following infiltration and exposure to stressed adipocytes in obese VAT (Nagareddy et al, 2014; Hotamisligil et al, 1993). We recently used VAT transplantation to investigate the underlying mechanisms for visceral obesity-induced cognitive impairment (Guo et al, 2020). Cumulative exposure to local inflammation is accompanied by pre-assembly of cytosolic ‘inflammasome’ complexes comprised of a Nod-like receptor protein (NLRP), a caspase-1 precursor, and an adaptor protein (ASC; Wen et al, 2012). Formation of inflammasome complexes around the cytoplasmic receptor Nlrp3 contributes to inflammation in VAT with genetic or dietary obesity (Nagareddy et al, 2014) and promotes the development of obesity-induced insulin resistance (Vandanmagsar et al, 2011). Whole-body NLRP3 knockout mice exhibit protection against age-related cognitive decline, reduced hippocampal astrogliosis, improved glycemic control, and attenuation of trabecular bone loss (Youm et al, 2013). When crossed with an APP/PS1 mouse model of Alzheimer’s disease, the resultant APP/PS1/Nlrp3−/− mice exhibited reduced amyloid pathology, improved hippocampus-dependent memory and synaptic plasticity, and enhanced microglial internalization of amyloid-beta (Heneka et al, 2013).
In our recent publication, whole-body Nlrp3−/− mice were protected against obesity-induced cognitive deficits, and exhibited intact hippocampal long-term potentiation, despite comparable severity of obesity (Guo et al, 2020). Protection against obesity-induced hippocampal dysfunction in Nlrp3−/− mice was accompanied by reduced levels of IL1b in VAT, serum, and hippocampus. However, it was not possible to draw causal inferences from correlations in different tissues. We therefore transplanted VAT from obese Nlrp3+/+ and Nlrp3−/− donors into lean Wt recipients. In previous work, we determined that VAT transplants from genetically obese and diabetic db/db mice impaired memory and activated microglia in lean Wt recipients (Erion et al, 2014). Visceral fat transplants from Wt mice with high-fat diet-induced obesity also impaired hippocampal function, with associated activation of microglia based on cellular and morphological measures (Guo et al, 2020). Transplants from comparably obese Nlrp3−/− donors had no effect on memory, synaptic physiology, or microglial reactivity, and recipients were also protected against increases in circulating and hippocampal IL1b (Guo et al, 2020).
After observing correlations between Nlrp3 induction in VAT, hippocampal IL1b accumulation, and cognitive dysfunction, we investigated whether microglial IL1 receptor 1 (IL1R1) activation underlies these effects. Inducible ablation of IL1R1 in CX3CR1creERT2 mice protected against microglial activation without influencing the severity of dietary obesity (Guo et al, 2020). CX3CR1creERT2/IL1R1fl/fl mice were also protected against obesity-induced hippocampal dysfunction (Guo et al, 2020). To examine whether similar protection might occur following VAT transplantation, lean CX3CR1creERT2/IL1R1fl/fl mice and nontransgenic littermates received VAT transplants from Wt donor mice with dietary obesity. Transgenic mice were protected against hippocampal dysfunction and microglial activation following VAT transplantation, and ex vivo stimulation experiments revealed a potential role for microglial IL1R1 signaling in local amplification of IL1b (Guo et al, 2020). Taken together, these studies suggest that NLRP3 induction in VAT leads to local IL1b production, increased penetration of IL1b into the CNS, and cognitive deficits following IL1R1 activation on CX3CR1+ cells in the nervous system. We interpreted these effects as reflecting the actions of IL1R1 activation among microglia because microglia represent the predominant CX3CR1-expressing population in the CNS, but it should be noted that CX3CR1 is also expressed by long-lived CNS border-associated macrophages (BAMs) in the meninges and perivascular space (reviewed in Kierdorf et al, 2019). We cannot rule out a potential role for BAMs in transport or amplification of IL1, or in any other aspect of CNS inflammation with obesity or VAT transplantation. However, future work using the microglia-specific TMEM119creERT2 line (Kaiser and Feng, 2019) will eventually shed light on this issue.
1.4. Tumor necrosis factor alpha-driven inflammation and cognitive impairment in obesity
We focused on IL1b signaling as a link between visceral obesity and cognitive dysfunction due to the well-characterized suppression of learning and synaptic plasticity that occurs following exposure to high levels of IL1b (Hein et al, 2012; Erion et al, 2014; Lynch, 2015) and previously reported relationships with visceral adiposity (Nagareddy et al, 2014; Vandanmagsar et al, 2011). However, IL1b is by no means the only cytokine generated by VAT in obesity with known links to cognitive dysfunction. Adipose tissue contains adipocytes, endothelial cells, lymphatic vessels, and immune cells including macrophages, T-cells, innate lymphoid cells, and eosinophils. Even in lean individuals, visceral fat contains more immune cells than subcutaneous fat, likely due to the evolutionary advantages of having a pool of immune cells in close proximity to the gut, which would facilitate resolution of inflammation in response to ingested pathogens or irritants (Harman-Boehm et al, 2007; West-Eberhard 2020). Adipocytes expand and proliferate during overnutrition, and shrink with lipolysis during subsequent periods of caloric deficit. Eventually, as the proliferative capacity of adipocyte precursors is depleted, continued demands for lipid storage cause cellular stress, leading to production of tumor necrosis-alpha (TNFa) by adipocytes (reviewed by Hotamisligil, 2017). This section considers the role of TNFa in peripheral metabolic dysfunction (1.4.1); neuroimmune regulation of brain and behavior following TNFa inhibition (1.4.2), and the role of TNFa in obesity-induced synaptic deficits (1.4.3).
1.4.1. Tumor necrosis factor signaling and metabolic dysfunction in obesity
The tumor necrosis factor superfamily includes Tnfa, Tnfb (aka lymphotoxin, LT), Nerve Growth factor (Ngf), vascular endothelial cell-growth inhibitor (VEGFI), and receptor activator of nuclear factor-kB (NFKb) ligand (RANKL), among others (for review, see Aggarwal, 2003). The functional significance of the TNF superfamily was initially identified based on tumor regression after bacterial infection, well before sequencing and characterization of the two archetypal TNFs (Aggarwal, 2003). Adipocyte-derived TNFa acts as a chemoattractant for circulating macrophages and monocyte precursors, which accumulate in VAT and skew toward a pro-inflammatory state in obesity (reviewed by Hotamisligil, 2017). Recruited macrophages amplify local production of TNFa and other pro-inflammatory cytokines, such as interleukin-6 (IL6), monocyte chemoattractant protein 1 (MCP1, aka CCL2), and interferon-gamma in VAT (reviewed comprehensively by Crewe et al 2017). Local induction of pro-inflammatory cytokines is accompanied by increases in circulating TNFa (Mauer et al, 2010), although the degree to which circulating concentrations track local synthesis in adipose tissue has yet to be fully adequately resolved (Zhou et al, 2011).
TNFa is expressed as a membrane-bound monomer and as a soluble trimer with paracrine and autocrine functions (see reviews by Aggarwal 2003 and by Croft et al, 2009). Soluble TNFa (sTNFa) binds to TNFR1 (aka Tnfrsf1a, p55) with relatively greater affinity than TNFR2 (aka Tnfrsf1b, p75; Lang et al, 2016) in cells that express both receptors. Outside of immune and vascular mural cell populations, Tnfr2 expression is rare, while Tnfr1 is essentially ubiquitous (Zhang et al, 2014; Vanlandewijck et al, 2018; Lein et al, 2007). Ligand binding elicits rapid clustering and trimerization of TNF receptors, which have similar ligand binding regions and variable intracellular signaling domains (Aggarwal 2003; Croft et al, 2009). The intracellular signaling domain of Tnfr1 contains a death domain and recruits caspase-dependent apoptotic and caspase-independent necrotic cascades (Aggarwal 2003). Tnfr1 activation also recruits mitogen-activated protein kinase (MAPK) signaling and activates NFKb by promoting proteosomal degradation of IkBa (Aggarwal 2003). Interestingly, the intracellular region of Tnfr1 contains a neutral sphingomyelinase activation domain, linking activation of Tnfr1 with ceramide accumulation. By contrast, the intracellular domain of Tnfr2 contains two TNFR-associated factor (TRAF) sequences (TRAF1, TRAF2) and lacks a death domain. Depending on cell type and prior history of activation, TRAFs exert pro- or anti-inflammatory effects by forming complexes with inhibitor of NFkB alpha (IkBa), IkB kinase-beta (IKKb), or NFkB-inducing kinase (Croft et al, 2009; Aggarwal, 2003). Activation of Tnfr2/TRAF signaling also regulates cell survival by recruiting Pi3K/Akt (Croft et al, 2009). Consistent with dichotomous pro- and anti-inflammatory effects of TNFR signaling, TNFa is required for physiologically beneficial adipogenesis within 72hr of HFD exposure, but also promotes adipocyte hyperplasia and inflammation with long-term HFD consumption (Wernstedt Asterholm et al, 2014; Hotamisligil et al, 1993). While TNF signaling exerts heterogeneous effects on adipose tissue over time, there is consensus that TNF-driven inflammation drives the onset of insulin resistance in chronic obesity (Hotamisligil, 2017).
1.4.2. Impact of TNFa inhibition on metabolic pathology and behavior in preclinical models
Receptors for TNFa are ‘shed’ into the extracellular space, allowing them to compete with endogenous TNFs for ligand binding, resulting in net suppression of receptor activation (Aggarwal, 2003). Negative regulation of TNFa signaling by soluble receptors and the importance of TNFa signaling in chronic inflammatory conditions led to the development and approval of TNF-scavenging antibodies for clinical use in 1998. Infliximab is a monoclonal human:mouse chimeric antibody against TNFa. It neutralizes both membrane-bound and soluble TNFa, including sTNFa bound to TNF receptors, without affecting TNFb. Infliximab exerts variable degrees of cytotoxicity on TNFa-producing cells (Scott and Kingsley, 2006), and systemic administration attenuated hypothalamic mitochondrial dysfunction in mice with short-term dietary obesity (Carraro et al, 2018). Adalimumab is another monoclonal antibody raised against human TNFa that sequesters endogenous TNFa (both soluble and membrane-bound) without influencing TNFb and also elicits cytotoxicity among TNFa-producing cells (Bain and Brazil, 2003). While not used frequently in rodents, there is published evidence that systemic treatment with adalimumab attenuates fasting hyperglycemia and reduces circulating levels of TNFa and IL-6 (Shuwa et al, 2018).
Unlike the monoclonal antibodies, Etanercept is a recombinant fusion protein comprised of the extracellular region of human TNFR2 and the intracellular tail of a human immunoglobulin Fc receptor. Etanercept blocks both TNFa and TNFb signaling without cytotoxicity towards TNF-producing cell populations (Mitoma et al, 2008). While etanercept is used experimentally in rodents, the ligand binding domain exhibits approximately 70% homology, and there is limited data on how variability in the amino acid sequence might impact CNS bioavailability following peripheral administration. However, direct intracerebroventricular infusions of etanercept reduced behavioral anxiety in leptin receptor mutant (db/db) mice with genetic obesity, mimicking the behavioral effects of reducing endogenous hippocampal TNFa with caloric restriction in this model (Fourrier et al, 2019). More recently, systemic treatment with the brain-penetrant soluble TNFa inhibitor XPro1595 was shown to reduce hippocampal pro-inflammatory cytokine gene expression and attenuate behavioral anxiety in mice with dietary obesity (De Sousa Rodrigues et al, 2019). These effects were accompanied by attenuation of fasting hyperinsulinemia (De Sousa Rodrigues et al, 2019), and were in line with previously reported reductions in neuroinflammation in AD model mice treated with XPro1595 (Cavanagh et al, 2016; MacPherson et al, 2017). XPro1595 is currently under evaluation in Phase I clinical trials for the treatment of Alzheimer’s disease. Given that other TNF inhibitors have been shown to reduce risk of diabetes among at-risk populations (reviewed by Hotamisligil, 2017), such as individuals with visceral obesity, repurposing these drugs could have combined therapeutic benefits.
1.4.3. Central actions of TNFa in obesity
Peripheral TNFa enters the nervous system via transporters that saturate in the picomolar range (Gutierrez et al, 1993). Data from whole-body Tnf receptor knockout mice and Wt mice treated with scavenging antibodies suggests that receptor-mediated transport is carried out by the same receptors involved in signaling (Pan and Kastin, 2002). In the CNS, TNFR1 and TNFR2 play redundant roles in blood-to-brain influx of TNFa, based on nearly complete elimination of transport in TNFR1/TNFR2 double knockout mice and minimal impact on transport kinetics in TNFR1 or TNFR2 single-knockout mice (Pan and Kastin, 2002). This pattern differs from that of the spinal cord, where TNFR1 and TNFR2 make independent and additive contributions to influx of circulating TNFa (Pan and Kastin, 2002). At the blood-brain and blood-CSF interfaces, TNFR1 is robustly expressed by cerebrovascular endothelial cells, vascular smooth muscle cells, pericytes, and choroid plexus epithelial cells (Vanlandewijck et al, 2018, Lun et al, 2015), while TNFR2 is expressed by endothelial cells, albeit at lower levels than in microglia and brain-resident macrophages (Vanlandewijck et al, 2018).
Parenchymal expression of TNFa mRNA is restricted to microglia, with nonexistent or minimal gene expression by astrocytes, neurons, and cells at the blood-brain and blood-CSF interfaces (Zhang et al, 2014; Vanlandewijck et al, 2018; Lun et al, 2015). Two independent single-cell RNA sequencing studies indicate that Tnfr1 receptors are ubiquitously expressed by neurons, astrocytes, and microglia, abeit with nearly 10-fold higher enrichment among microglia, relative to other cell populations (Zhang et al, 2014). TNFR2 expression is low or undetectable in neurons and astrocytes, but is expressed by microglia (Zhang et al, 2014; Vanlandewijck et al, 2018). While the cellular signaling interactions have yet to be fully elucidated, there is considerable evidence that TNFa exerts dose-dependent, developmentally regulated effects on synaptic function. In the CA1 subfield of hippocampal slice preparations from neonatal rodents, exposure to low nanomolar concentrations of TNFa regulates both activity-dependent and homeostatic plasticity by promoting AMPA receptor insertion (reviewed by Stellwagen et al, 2006). These effects were likely limited to early postnatal development, as activity-dependent long-term potentiation in hippocampal area CA1 in acute slices from adult animals is suppressed by similar doses of TNFa (Tancredi et al, 1992). Microglia were indirectly implicated as a potential source for TNFa-mediated synaptic deficits in adult mice, as diptheria toxin-mediated conditional ablation of microglia maintained NMDA/AMPA current ratios and prevented synaptic loss in hippocampal area CA1 after sciatic nerve injury (Liu et al, 2017).
Hippocampal dentate gyrus granule neurons differ from CA1 pyramidal neurons in their morphology, resting membrane potential, and firing rates (for review, see Spruston and McBain, 2007). The dentate gyrus subgranular zone exhibits ongoing proliferation and neurogenesis in adult rodents, but the ontogeny of developmentally generated dentate granule neurons also differs from area CA1. In rodents, the majority of CA1 neurons are generated during prenatal development, between E10-E18 (for review, see Frotscher and Seress, 2007). This timeframe differs from the ontogeny of most granule neurons, which are generated between E16-P10 in rodents (Hastings and Gould, 2002). Subfield-specific variability in developmental neurogenesis is not unique to the rodent hippocampus, as hippocampal area CA1 develops during the first trimester in human fetuses and is mostly complete by week 16 of gestation (Frotscher and Seress, 2007). Gestational neurogenesis in the dentate gyrus starts during the first trimester, but is not complete until approximately 30wk later, well into the third trimester (Frotscher and Seress, 2007).
Considering the different developmental ontogenies of dentate granule neurons, versus CA1 pyramidal cells, it is challenging to align the reported window for TNFa-mediated homeostatic plasticity in CA1 with a similar period of maturation in the dentate. In fact, given the relatively greater developmental heterogeneity among dentate granule neurons, relative to CA1 neurons, direct and accurate comparisons of the two subfields may not be feasible. Synaptic physiological regulation by TNFa at medial perforant path synapses in the dentate gyrus clearly differs from responses in CA1, but these differences could arise due to variability in either the dose-response relationship or the developmental window for TNFa-mediated synaptic scaling. Specifically, early postnatal exposure to low nanomolar concentrations of TNFa elicits homeostatic increases in CA1 neuron excitability via coordinated AMPA receptor insertion and removal of GABAA receptors (reviewed by Pribiag and Stellwagen, 2014). In contrast to the postsynaptic response observed at Schaffer collateral synapses in area CA1, early developmental exposure to picomolar concentrations of TNFa enhances presynaptic glutamate release, creating stronger excitatory drive onto dentate granule neurons (Habbas et al, 2015). Exposure to nanomolar concentrations of TNFa suppresses LTP in the dentate gyrus of the adult brain without altering basal synaptic transmission (Butler et al, 2004; Curran et al, 2003), but developmental regulation of homeostatic and activity-dependent responses to TNFa remain incompletely understood in this subfield. Given the distinct contributions of dentate granule neurons and CA1 neurons to pattern separation and spatial memory (reviewed in Wosiski-Kuhn and Stranahan, 2013), it would be worthwhile to examine subfield-specific responses to TNFa in greater detail. Obesity-induced synaptic dysfunction has been reported in the dentate gyrus and CA1 subfields (Stranahan et al, 2008; Hao et al, 2016; Pancani et al, 2013, Guo et al, 2020), but trans-synaptic mechanisms for impaired plasticity remain unexplored. Addressing gaps in knowledge surrounding TNFa and malfunction at different relays within hippocampal circuitry could generate additional support for repurposing existing TNFa-suppressing therapies.
1.5. Multiple models of interaction between visceral fat and the brain
The literature on metabolic and immune responses to visceral obesity is very broad, as is the ever-growing literature on neuroimmune regulation of brain function. However, there are some systems-level interactions that are initiated primarily in VAT, with known consequences for neurons and glia. Under the ‘adipo-centric’ model, cytokines and hormones from stressed adipocytes are released into circulation, and subsequently enter the brain via transporters or at ‘gaps’ in the BBB (Fig.3A). TNFa is one example of a VAT-derived signal that accumulates in the brain with obesity (Hotamisligil et al, 1993; Jeon et al, 2012; Dey et al, 2014). Although transport mechanisms exist for blood-to-brain influx of TNFa (Gutierrez et al, 1993), the origins of increased central TNFa have yet to be determined in dietary obesity, and are likely to differ between circumventricular areas and regions that lie beyond the BBB. Other adipose-derived hormones, including adiponectin and leptin, are associated with distinct functional consequences for inflammation in the brain and peripheral tissues (reviewed by Tilg and Moschen, 2006). In the case of adiponectin, these consequences are anti-inflammatory (Spranger et al, 2006; Chabry et al, 2015; Li et al, 2021), while leptin evokes pro-inflammatory responses in microglia and peripheral myeloid cells (Fourrier et al, 2019; Dey et al, 2014; Erion et al, 2014). Leptin is also preferentially expressed in subcutaneous fat, which is less prone to inflammation than the visceral compartment, while adiponectin is expressed by white adipocytes in the subcutaneous and visceral depots (Tilg and Moschen, 2006). Although downregulation of adiponectin in obesity could recruit the ‘adipo-centric’ CNS model by removing an opposing anti-inflammatory signal, brain vascular mural cells express both AdipoR1 and AdipoR2 (Spranger et al, 2006; Vanlandewijck et al, 2018). Exposure to physiological concentrations of adiponectin suppresses lipopolysaccharide (LPS) induced expression of the pro-inflammatory cytokine IL6 in cerebral microvessels, leaving open the possibility of indirect effects on cells of the brain parenchyma (Spranger et al, 2006). Peripherally administered adiponectin accumulates in CSF, without evidence of systemic efflux, and ICV delivery exerts clear physiological effects on energy expenditure (Qi et al, 2004; Kubota et al, 2007). However, specific adiponectin transporters (or sites of penetration) have not yet been identified, and the potential role of cellular ‘gatekeepers’ in the circumventricular organs and at the BCSF/BBB interfaces has not yet been resolved.
Figure 3. Mechanistic interactions between visceral adipose tissue and the CNS.
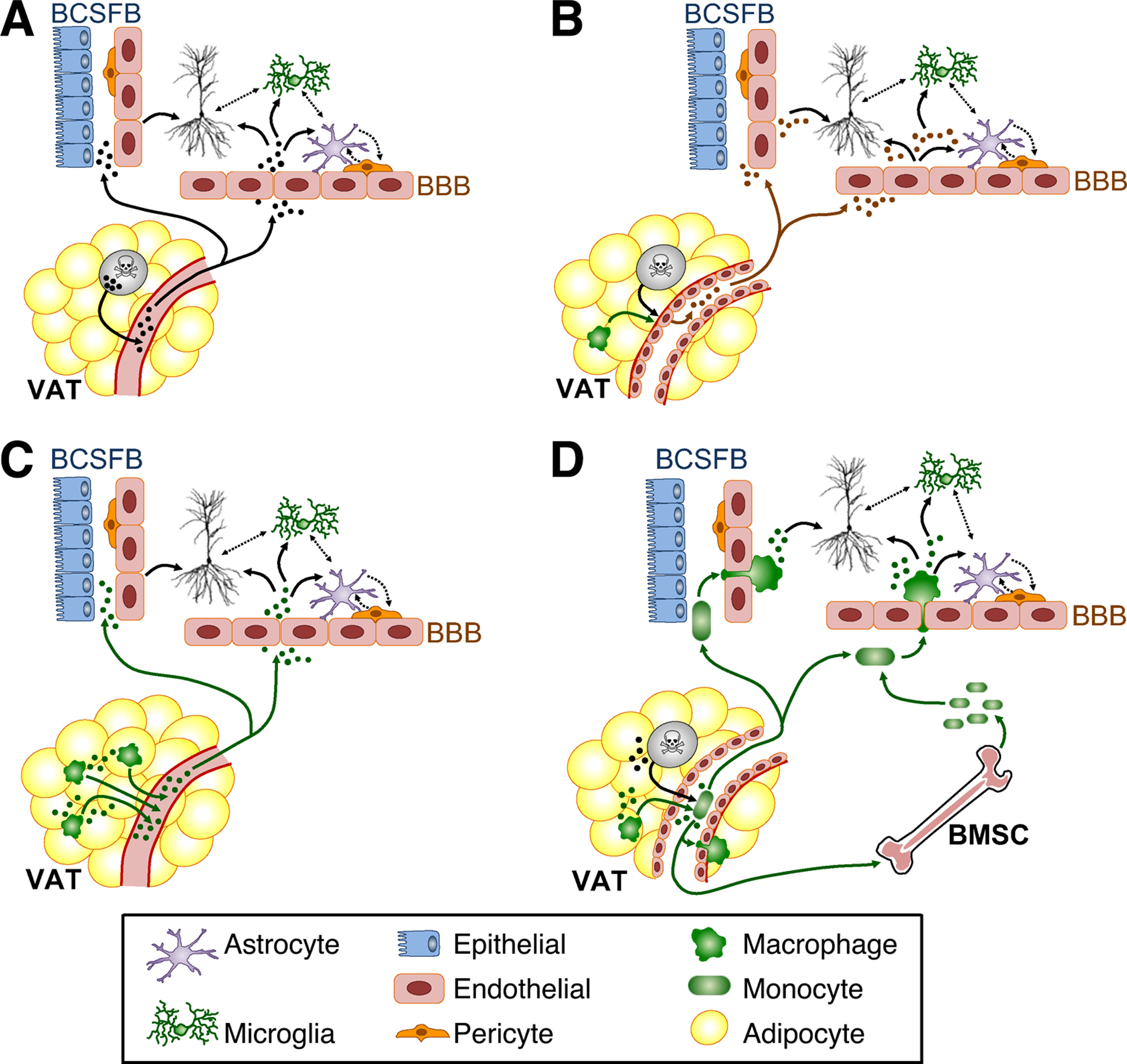
A) The ‘adipo-centric’ model for neuroimmune regulation of hippocampal function by stressed adipocytes (gray) in visceral adipose tissue (VAT). B) The ‘vasculo-centric’ model for immune regulation of hippocampal function in visceral obesity. C) Long-range model for immunological signaling between VAT and hippocampus. D) Trafficking model for immune signaling between VAT and hippocampus. For all panels, solid arrows represent known routes of entry and cellular targets for adipocyte-derived signals. Dashed arrows represent potential inter-cellular interactions. Abbreviations: BMSCs, bone marrow stem cells (BMSCs); BBB, blood-brain barrier; BCSFB, blood-cerebrospinal fluid barrier.
A complementary ‘vasculo-centric’ model for neuroinflammation in visceral obesity incorporates local inter-cellular interactions in VAT, and potential cellular interactions at interfaces between the brain and periphery (Fig.3B). In VAT, bidirectional communication between signaling between adipocytes, adipose tissue macrophages, and vascular endothelial cells governs angiogenesis, adipocyte precursor cell differentiation, and insulin resistance in adipose tissue (comprehensively reviewed by Crewe et al, 2017). Under the ‘vasculo-centric’ model, adipocyte-derived signals, alone or in concert with cytokines from adipose tissue macrophages, act on tissue-resident vascular mural cells, leading to release of endothelium-derived factors into circulation (Fig.3B). For example, reductions in adiponectin synthesis with dietary obesity removes a tonic anti-inflammatory signal that suppresses vascular inflammation, smooth muscle cell proliferation, and platelet aggregation (Matsuda et al, 2002; Ouichi et al, 1999; Kato et al, 2006). The functional impact on vascular mural cells could be direct, as endothelial cells in VAT express both AdipoR1 and AdipoR2, and incubation with adiponectin suppressed high glucose-induced generation of ROS in vitro (Chen et al, 2021; Ouedraogo et al, 2006). Alternatively, adiponectin reduces induction of pro-inflammatory cytokines, including TNFa, in LPS-stimulated macrophages (Yokota et al, 2000), opening the possibility for indirect regulation of endothelial reactivity via adiponectin-mediated reductions in TNFa production by ATMs (Fig.3B). The two scenarios are not mutually exclusive, as stimulation with adiponectin also suppresses activation of IKB kinase (IKKb), a biological readout for NFkB, following in vitro exposure to TNFa in endothelial cells (Wu et al, 2007). In the in vivo setting, surprisingly few circulating factors have been identified with known origins in VAT endothelium, but this gap in knowledge likely reflects the scarcity of experimental strategies for tissue-specific manipulation of endothelial cells. Here again, TNFa represents a prime example of a signal originating in VAT, with potential amplification by VAT endothelial cells prior to increases in circulation and established routes into the CNS (Hotamisligil et al, 1993; Maeda et al, 2002; Gutierrez et al, 1993; Pan and Kastin, 2002).
Under the long-range cytokine model, adipose tissue macrophages release pro-inflammatory cytokines into circulation, with direct and indirect consequences for neurons and glia. Upon entry into the CNS, cytokine signals from VAT would then bind directly to their receptors on neurons, astrocytes, and microglia; alternatively, low-level cytokine exposure could initiate an auto-amplification loop in microglia and/or pvMΦ. As an example, we recently demonstrated that obesity exposes the brain to peripheral IL1b, which is sensed and amplified by IL1R1 activation on CX3CR1-expressing microglia and other CNS myeloid cells (Guo et al, 2020). VAT transplantation from an obese Wt, but not a similarly obese NLRP3−/− donor recapitulates this outcome, indicating that NLRP3 induction in VAT signals to the brain via IL1b (Fig.3C). In addition to the long-range cytokine signaling model, there are several possible scenarios in which immune cell trafficking could mediate interactions between visceral adiposity and the CNS. For example, signals from stressed adipocytes and/or adipose tissue macrophages could activate and polarize circulating lymphocytes that subsequently traffic into the cerebral vasculature and/or CSF compartments, allowing for signaling interactions with cells of the brain parenchyma (Fig.3D). These signaling interactions could be direct, via local production of cytokines transported across the BBB or BCSFB, or via diapedesis and infiltration into the CNS proper. Alternatively, VAT-activated lymphocytes that traffic to the BBB/BCSFB could regulate glia and neurons indirectly via interactions with cerebrovascular mural cells and/or epithelial cells lining the cerebral ventricles (Fig.3D).
Another variation on the trafficking model involves amplification of pro-inflammatory signals from VAT following an increase in the pool of circulating lymphocytes, similar to that previously reported in obese mice and humans (Nagareddy et al, 2014). Under this scenario, signals from VAT promote the proliferation of bone marrow stem cells (BMSCs) and/or circulating precursors (Fig.3D). Lymphocytosis could then amplify pro-inflammatory signals locally in adipose tissue after infiltration and differentiation into adipose tissue macrophages; alternatively or in addition, the expanded pool of circulating lymphocytes could traffic into the cerebral vasculature or CSF, as previously reported in other models of chronic inflammation (for review, see Filiano et al, 2017 and Lun et al, 2015). Trafficking into either compartment would enable direct interaction between peripheral lymphocytes and cells of the brain parenchyma after infiltration, or indirect signaling through interactions with vascular mural cells. The potential relevance of this scenario is supported by the following lines of evidence: first, obesity is accompanied by expansion of circulating lymphocytes in mice and humans (Nagareddy et al, 2014; Breznik et al, 2018); second, dietary obesity promotes the entry of peripheral myeloid cells into the CNS (Buckman et al, 2014; Guo et al, 2020); and third, interactions between pvMΦ and brain vascular endothelial cells regulate neuroinflammation and circuit recruitment under pro-inflammatory conditions (Serrats et al, 2010; Lapenna et al, 2018; Kierdorf et al, 2019).
Obesity-induced lymphocytosis occurs independently of systemic hyperglycemia and insulin resistance, but requires induction of TNF (Nagareddy et al, 2014; Breznik et al, 2018). However, trafficking is involved in both pro-inflammatory and pro-resolving effects on the CNS, as local production of anti-inflammatory cytokines by CSF lymphocytes supports learning and memory (reviewed by Filiano et al, 2017). We recently explored this model while investigating the protective effects of subcutaneous fat on the brain (Guo et al, 2021). Unlike VAT, subcutaneous fat is home to a heterogeneous population of white and ‘beige’ adipocytes that expend energy, similar to brown fat (reviewed comprehensively by Rosen and Spiegelman, 2014). The transcription factor PRDM16 is required for ‘beiging,’ and type 2 (anti-inflammatory) cytokine signaling also regulates beige fat formation (Qiu et al, 2014). To examine relationships between beige fat and cognition, Wt mice with dietary obesity received SAT transplants from a lean Wt donor, or an AdipoqCRE/PRDM16fl/fl mouse lacking beige adipocytes. After observing beige adipose-dependent reinstatement of microglial quiescence and memory (Guo et al, 2021), we considered the potential role of lymphocyte trafficking between resident and transplanted SAT and the brain in these effects. Subcutaneous fat transplants from a lean donor expressing a fluorescent reporter in all tissues revealed no evidence of immune cell trafficking into the brain, but subsequent transplantation experiments in scid mice lacking most lymphocyte subsets revealed that recipient-derived immune cells were required for the procognitive and anti-inflammatory effects of SAT transplantation (Guo et al, 2021). While these studies indirectly implicate host lymphocytes in beige adipose-dependent reinstatement of hippocampal function and immunoquiescence, the relative contributions of long-range signaling and lymphocyte trafficking (Fig.3C–D) remain to be determined.
1.6. Understanding immunometabolic regulation of brain function in obese females
Developmental exposure to sex steroids determines adipose tissue distribution, with females exhibiting greater subcutaneous adiposity than males under normal conditions (Krotkieski et al, 1983; reviewed comprehensively by Palmer and Clegg, 2015). There is also substantial evidence for differences in basal insulin sensitivity, with females exhibiting significantly greater physiological responses to insulin (Macotela et al, 2009). These factors likely contribute to the relatively greater prevalence of type 2 diabetes in men (Kuhl et al, 2005), and the protection against obesity-induced insulin resistance reported in female rodents (Medrikova et al, 2012). Although sex differences in adipose tissue expansion after obesogenic diet consumption are well-established (Palmer and Clegg, 2015), surgical removal of SAT does not influence glucose tolerance in female HFD mice (Shi et al, 2007), or in genetically obese and diabetic Zucker rats (Liszka et al, 1998), suggesting that sexually dimorphic vulnerability to metabolic pathology is unlikely to occur as a simple function of subcutaneous fat mass.
Innate and adaptive immunity are also sexually dimorphic, and sex differences vary over the reproductive lifespan (for review, see Giefing-Kröll et al, 2015). Postpubertal females exhibit stronger innate immune responses, retain higher concentrations of antibodies, and exhibit higher rates of T cell proliferation upon re-exposure (Giefing-Kröll et al, 2015). Adult females are also more vulnerable to autoimmune disorders, relative to males (Giefing-Kröll et al, 2015). These disparities are diminished in postmenopausal women and ovariectomized rodents, suggesting that gonadal steroids underlie prior sexual dimorphisms (Giefing-Kröll et al, 2015). This relationship has been explored, and largely upheld, in female rodents on regular chow (Rettew et al, 2009; Loram et al, 2012). In rodent models of obesity, elegant bone marrow chimera studies uncovered cell-autonomous sex differences in myelopoesis (Singer et al, 2015). Before the onset of HFD, irradiated males and females received a 1:1 mixture of bone marrow stem cells from congenic male and female donors expressing distinct CD45 variants to enable tracking (Singer et al, 2015). After recovery, recipient mice were maintained on HFD for varying durations before analysis of male donor-derived and female donor-derived macrophages in perigonadal VAT. This beautifully designed and executed study revealed that male donor-derived ATMs preferentially accumulate in VAT, irrespective of signals from the environment (Singer et al, 2015). When interpreted in the context of extensively replicated reports of macrophage infiltration and male-typical hypertrophy in VAT from ovariectomized rodents (Rettew et al, 2009), the study by Singer et al (2015) demonstrates that the organizational effects of exposure to male or female sex steroids on bone marrow stem cells are a stronger determinant of myelopoesis than environmental signals or acute exposure to a completely different hormonal milieu.
Resident microglia express estrogen receptors (alpha and beta; Ishihara et al, 2015) and respond differently to stimulation in cycling females, relative to males and acyclic females. Specifically, ovariectomized young females and aged females in reproductive senescence exhibit microglial hypersensitivity to immune challenge in vivo and in vitro (Loram et al, 2012, Saijo et al, 2011). Estrus cycle-dependent variability has received less attention, but endogenous fluctuations in estrogen regulate neuroinflammation and functional outcomes in ischemia (Xiong et al, 2015). There is also evidence that microglia regulate sexual dimorphisms in the developing brain. The medial preoptic area (MPOA) of the hypothalamus is significantly larger in males, and the developmental effects of estradiol (derived from testosterone) underlie structural differences (reviewed by Crews, 2005). Microglia in the developing MPOA of females are more quiescent, whereas microglia in the developing MPOA of males express higher levels of inflammatory markers, including prostaglandins and cyclo-oxygenases (Lenz et al, 2013). Developmental microglial activation in males promotes synaptogenesis and increased MPOA volume, and these features are recapitulated by early postnatal estradiol administration in females (Lenz et al, 2013; Amateau et al, 2004). Sex differences in microglial activation and synaptogenesis in the MPOA could be independent processes, or they could be mediated by microglial interactions with neurons and other CNS cell populations. Although sex steroids pass freely through the blood-brain barrier, brain vascular endothelial cells (BVECs) are also functionally regulated by estrogen, which protects against BBB breakdown after systemic treatment with bacterial lipopolysaccharides (Maggioli et al, 2015). Taken together, these lines of evidence indicate that CNS responses to inflammation are subject to multilevel regulation by reproductive hormones. Moving forward, it will be fascinating to see how cellular and tissue-specific trajectories of inflammation and insulin resistance vary as a function of biological sex.
2. How did we get here? and Future Directions
Relationships between peripheral signals and brain function have fascinated neuroscientists for over a hundred years, beginning with functional characterization of the blood-brain barrier by Edwin E. Goldmann in 1913 using dyes developed by immunologist Paul Ehrlich (for comprehensive historical review, see Bentivoglio and Kristensson, 2014). However, the concept of an impenetrable ‘barrier’ is gradually giving way to the idea of a dynamic gate, with entry and efflux determined by signaling from between cells at the luminal and abluminal interfaces. Refinement of canonical doctrines surrounding CNS immune privilege took place in parallel with, but mostly separate from, research on the impact of drastic fluctuations in diet composition in the brain. Highly palatable, calorically dense foods once reserved for the wealthy were suddenly, in the evolutionary equivalent of a millisecond, available for at-will consumption.
As obesity was reaching pandemic status, studies of insulin and leptin revealed a critical role for neuronal sensing of hormones and cytokines in CNS control of metabolic homeostasis. These findings, together with the systematic functional characterization of cerebrovascular transport mechanisms by Banks and colleagues (Erickson and Banks, 2018), instigated the current, more dynamic reinterpretation of the cerebrovascular interface. Disruption of cerebrovascular permeability in obesity was first reported in humans, based on correlations between cerebrospinal fluid IgG levels and body mass index (Gustafson et al, 2007). This observation emerged approximately 10 years after initial reports of correlations between obesity and cognitive impairment in population-based studies (Kilander et al, 1997). While subsequent studies of obesity and cognitive impairment in humans reported variable outcomes (Sturman et al, 2008; van den Berg et al, 2007), studies that incorporated measures of adipose tissue distribution, as opposed to body mass index, revealed more consistent relationships between visceral obesity and age-related cognitive impairment (Jagust et al, 2005; Elias et al, 2005; Whitmer et al, 2008; Debette et al, 2011; Xu et al, 2011; Metzler-Baddeley et al, 2019).
The translational goal of studying relationships between adipose tissue distribution and cognition is twofold: first, determine whether visceral adiposity is a predictive biomarker for cognitive decline; and second, understand how visceral adiposity drives risk of age-related cognitive impairment and develop strategies to prevent or attenuate risk. In rodents, most (but not all; see Mielke et al, 2006) data supports the underlying premise that dietary obesity impairs cognition (Molteni et al, 2002; Granholm et al, 2008; McNay et al, 2010; Morrison et al, 2010; Grayson et al, 2014; Hao et al, 2016; Spencer et al, 2017), and generalizing across studies suggests that preferential accumulation of visceral fat is correlated with obesity-induced cognitive dysfunction (Erion et al, 2014; Petrault et al, 2019; Guo et al, 2020; Guo et al, 2021). Moving forward, it will be intriguing to carry out high-fat diet studies in transgenic mice with preferential accumulation of visceral fat, such as the 11beta-hydroxysteroid dehydrogenase-1 (11bHSD1) overexpression model (Masuzaki et al, 2001), and in mice that acquire visceral-like features in subcutaneous fat, as reported in adipose-specific G-protein suppressor 2 knockouts (GPS2; Drareni et al, 2018). Animal models with depot-specific adipocyte dysfunction will generate additional insight into relationships between adipose distribution and cognition, and combined with targeted disruption of signaling between adipocytes and peripheral immune cells, will advance understanding of signaling between fat and the hippocampus.
The links between impaired glycemic control and obesity-induced cognitive dysfunction present certain conceptual challenges for use of adipose tissue distribution as a risk factor or criterion for targeted intervention. The onset and progression of tissue- and cell type-specific insulin resistance is heterogeneous, even among commonly used inbred mouse strains (Ayala et al, 2010; Goren et al, 2004), and individual differences are likely to be magnified in species with greater genetic diversity, such as humans. The recent focus on identifying and characterizing individuals with ‘metabolically healthy’ obesity (Stefan et al, 2013), and parallel characterization of normal-weight individuals with impaired glycemic control (Stefan et al, 2017), is consistent with expected patterns in a genetically heterogeneous population. It is interesting to note that the textbook curve representing progression from insulin resistance to beta-cell exhaustion and insulin dependence closely resembles trajectories of cognitive aging over the lifespan. While kinetic similarity does not demonstrate mechanistic overlap, joint investigation of cognitive and metabolic aging using longitudinal approaches in outbred rodents will facilitate the identification of ‘critical windows’ for obesity-associated cognitive risk, similar to that reported in humans (Whitmer et al, 2008; Debette et al, 2011). Expanding beyond early adulthood into the developing and aged brain will also allow for more precise delineation of ‘critical periods,’ which may differ as a function of cell type and circuit. The impact of maternal HFD on stress responses was initially characterized in rats over 10 years ago (Tamashiro et al, 2009); this pioneering study revealed increased vulnerability to obesity and insulin resistance in the offspring of dams maintained on HFD, with or without concurrent exposure to chronic mild stress during gestation. Since that report, there have been >900 publications on maternal HFD in models ranging from mice to nonhuman primates, covering a range of behaviorally relevant circuits (Sullivan et al, 2010; Dias et al 2020). Further investigation into these effects could shed light on the long-term consequences of developmental high-fat diets for cognition over the lifespan.
Given evolutionary predispositions and the widespread availability of palatable, calorically dense foods, rates of obesity are unlikely to be reduced in the near future. Several international societies including the American Diabetes Association, International Diabetes Foundation, and the Japanese Diabetes Society have recently issued guidelines for routine evaluation of cognitive status among diabetic patients (Biessels and Whitmer, 2020). If the goal is to use obesity and its comorbidities as predictive risk factors for neurodevelopmental and neurodegenerative disease, it is reasonable to model experimental criteria for impaired glycemic control on clinical definitions, while taking into account physiological differences between rodents and humans. Investigating the underlying mechanisms for obesity-induced cognitive risk should be driven by the goal of attenuating risk without exacerbating pre-existing societal prejudices against overweight and obese individuals.
Acknowledgements:
These studies were supported by a grant from the National Institutes of Health (R01DK110586).
Footnotes
Declaration of interest: None.
References
- Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006. Jan;7(1):41–53. [DOI] [PubMed] [Google Scholar]
- Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003. Sep;3(9):745–56. [DOI] [PubMed] [Google Scholar]
- Amamoto R, Huerta VG, Takahashi E, Dai G, Grant AK, Fu Z, Arlotta P. Adult axolotls can regenerate original neuronal diversity in response to brain injury. Elife. 2016. May 9;5:e13998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amateau SK, McCarthy MM (2004) Induction of PGE2 by estradiol mediates developmental masculinization of sex behavior. Nat Neurosci 7:643–650. [DOI] [PubMed] [Google Scholar]
- Armstrong E Relative brain size and metabolism in mammals. Science. 1983. Jun 17;220(4603):1302–4. [DOI] [PubMed] [Google Scholar]
- Ayala JE, Samuel VT, Morton GJ, Obici S, Croniger CM, Shulman GI, Wasserman DH, McGuinness OP; NIH Mouse Metabolic Phenotyping Center Consortium. Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis Model Mech. 2010. Sep-Oct;3(9–10):525–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain B, Brazil M. Adalimumab. Nat Rev Drug Discov. 2003. Sep;2(9):693–94. [DOI] [PubMed] [Google Scholar]
- Banks WA, Jaspan JB, Huang W, Kastin AJ. Transport of insulin across the blood-brain barrier: saturability at euglycemic doses of insulin. Peptides. 1997;18(9):1423–9. [DOI] [PubMed] [Google Scholar]
- Begg DP, Mul JD, Liu M, Reedy BM, D’Alessio DA, Seeley RJ, Woods SC. Reversal of diet-induced obesity increases insulin transport into cerebrospinal fluid and restores sensitivity to the anorexic action of central insulin in male rats. Endocrinology. 2013. Mar;154(3):1047–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellisari A Evolutionary origins of obesity. Obes Rev. 2008. Mar;9(2):165–80. [DOI] [PubMed] [Google Scholar]
- Bentivoglio M, Kristensson K. Tryps and trips: cell trafficking across the 100-year-old blood-brain barrier. Trends Neurosci. 2014. Jun;37(6):325–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biessels GJ, Whitmer RA. Cognitive dysfunction in diabetes: how to implement emerging guidelines. Diabetologia. 2020. Jan;63(1):3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank T, Prinz M. CatacLysMic specificity when targeting myeloid cells? Eur J Immunol. 2016. Jun;46(6):1340–2. [DOI] [PubMed] [Google Scholar]
- Blüher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003. Jan 24;299(5606):572–4. [DOI] [PubMed] [Google Scholar]
- Blüher M, Michael MD, Peroni OD, Ueki K, Carter N, Kahn BB, Kahn CR. Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev Cell. 2002. Jul;3(1):25–38. [DOI] [PubMed] [Google Scholar]
- BonDurant LD, Ameka M, Naber MC, Markan KR, Idiga SO, Acevedo MR, Walsh SA, Ornitz DM, Potthoff MJ. FGF21 Regulates Metabolism Through Adipose-Dependent and -Independent Mechanisms. Cell Metab. 2017. Apr 4;25(4):935–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bove RM, Brick DJ, Healy BC, Mancuso SM, Gerweck AV, Bredella MA, Sherman JC, Miller KK. Metabolic and endocrine correlates of cognitive function in healthy young women. Obesity (Silver Spring). 2013. Jul;21(7):1343–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bove RM, Gerweck AV, Mancuso SM, Bredella MA, Sherman JC, Miller KK. Association between adiposity and cognitive function in young men: Hormonal mechanisms. Obesity (Silver Spring). 2016. Apr;24(4):954–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breznik JA, Naidoo A, Foley KP, Schulz C, Lau TC, Loukov D, Sloboda DM, Bowdish DME, Schertzer JD. TNF, but not hyperinsulinemia or hyperglycemia, is a key driver of obesity-induced monocytosis revealing that inflammatory monocytes correlate with insulin in obese male mice. Physiol Rep. 2018. Dec;6(23):e13937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, Klein R, Krone W, Müller-Wieland D, Kahn CR. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000. Sep 22;289(5487):2122–5. [DOI] [PubMed] [Google Scholar]
- Brüning JC, Michael MD, Winnay JN, Hayashi T, Hörsch D, Accili D, Goodyear LJ, Kahn CR. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell. 1998. Nov;2(5):559–69. [DOI] [PubMed] [Google Scholar]
- Buckman LB, Hasty AH, Flaherty DK, Buckman CT, Thompson MM, Matlock BK, Weller K, Ellacott KL. Obesity induced by a high-fat diet is associated with increased immune cell entry into the central nervous system. Brain Behav Immun. 2014. Jan;35:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MP, O’Connor JJ, Moynagh PN. Dissection of tumor-necrosis factor-alpha inhibition of long-term potentiation (LTP) reveals a p38 mitogen-activated protein kinase-dependent mechanism which maps to early-but not late-phase LTP. Neuroscience. 2004;124(2):319–26. [DOI] [PubMed] [Google Scholar]
- Carraro RS, Souza GF, Solon C, Razolli DS, Chausse B, Barbizan R, Victorio SC, Velloso LA. Hypothalamic mitochondrial abnormalities occur downstream of inflammation in diet-induced obesity. Mol Cell Endocrinol. 2018. Jan 15;460:238–245. [DOI] [PubMed] [Google Scholar]
- Cavanagh C, Tse YC, Nguyen HB, Krantic S, Breitner JC, Quirion R, Wong TP. Inhibiting tumor necrosis factor-alpha before amyloidosis prevents synaptic deficits in an Alzheimer’s disease model. Neurobiol Aging. 2016. Nov;47:41–49. [DOI] [PubMed] [Google Scholar]
- Chabry J, Nicolas S, Cazareth J, Murris E, Guyon A, Glaichenhaus N, Heurteaux C, Petit-Paitel A. Enriched environment decreases microglia and brain macrophages inflammatory phenotypes through adiponectin-dependent mechanisms: Relevance to depressive-like behavior. Brain Behav Immun. 2015. Nov;50:275–287. [DOI] [PubMed] [Google Scholar]
- Chen Q, Lai SM, Xu S, Tan Y, Leong K, Liu D, Tan JC, Naik RR, Barron AM, Adav SS, Chen J, Chong SZ, Ng LG, Ruedl C. Resident macrophages restrain pathological adipose tissue remodeling and protect vascular integrity in obese mice. EMBO Rep. 2021. Aug 4;22(8):e52835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Levy JD, Zhang Y, Frontini A, Kolodin DP, Svensson KJ, Lo JC, Zeng X, Ye L, Khandekar MJ, Wu J, Gunawardana SC, Banks AS, Camporez JP, Jurczak MJ, Kajimura S, Piston DW, Mathis D, Cinti S, Shulman GI, Seale P, Spiegelman BM. Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell. 2014. Jan 16;156(1–2):304–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crewe C, An YA, Scherer PE. The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis. J Clin Invest. 2017. Jan 3;127(1):74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews D Evolution of neuroendocrine mechanisms that regulate sexual behavior. Trends Endocrinol Metab. 2005. Oct;16(8):354–61. [DOI] [PubMed] [Google Scholar]
- Croft M The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. 2009. Apr;9(4):271–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X, Wang Y, Tang Y, Liu Y, Zhao L, Deng J, Xu G, Peng X, Ju S, Liu G, Yang H. Seipin ablation in mice results in severe generalized lipodystrophy. Hum Mol Genet. 2011. Aug 1;20(15):3022–30. [DOI] [PubMed] [Google Scholar]
- Curran BP, Murray HJ, O’Connor JJ. A role for c-Jun N-terminal kinase in the inhibition of long-term potentiation by interleukin-1beta and long-term depression in the rat dentate gyrus in vitro. Neuroscience. 2003;118(2):347–57 [DOI] [PubMed] [Google Scholar]
- Czech MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med. 2017. Jul 11;23(7):804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Engelhardt B. Brain barriers in health and disease. Neurobiol Dis. 2017. Nov;107:1–3. [DOI] [PubMed] [Google Scholar]
- De Jesús Andino F, Jones L, Maggirwar SB, Robert J. Frog Virus 3 dissemination in the brain of tadpoles, but not in adult Xenopus, involves blood brain barrier dysfunction. Sci Rep. 2016. Mar 2;6:22508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sousa Rodrigues ME, Houser MC, Walker DI, Jones DP, Chang J, Barnum CJ, Tansey MG. Targeting soluble tumor necrosis factor as a potential intervention to lower risk for late-onset Alzheimer’s disease associated with obesity, metabolic syndrome, and type 2 diabetes. Alzheimers Res Ther. 2019. Dec 31;12(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debette S, Seshadri S, Beiser A, Au R, Himali JJ, Palumbo C, Wolf PA, DeCarli C. Midlife vascular risk factor exposure accelerates structural brain aging and cognitive decline. Neurology. 2011. Aug 2;77(5):461–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey A, Hao S, Erion JR, Wosiski-Kuhn M, Stranahan AM. Glucocorticoid sensitization of microglia in a genetic mouse model of obesity and diabetes. J Neuroimmunol. 2014. Apr 15;269(1–2):20–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Virgilio F, Sarti AC, Coutinho-Silva R. Purinergic signaling, DAMPs, and inflammation. Am J Physiol Cell Physiol. 2020. May 1;318(5):C832–C835. [DOI] [PubMed] [Google Scholar]
- Dias CT, Curi HT, Payolla TB, Lemes SF, Betim Pavan IC, Torsoni MA, Simabuco FM, Lambertucci RH, Mendes da Silva C. Maternal high-fat diet stimulates the proinflammatory pathway and increases the expression of Tryptophan Hydroxylase 2 (TPH2) and brain-derived neurotrophic factor (BDNF) in adolescent mice hippocampus. Neurochem Int. 2020. Oct;139:104781. [DOI] [PubMed] [Google Scholar]
- Djogo T, Robins SC, Schneider S, Kryzskaya D, Liu X, Mingay A, Gillon CJ, Kim JH, Storch KF, Boehm U, Bourque CW, Stroh T, Dimou L, Kokoeva MV. Adult NG2-Glia Are Required for Median Eminence-Mediated Leptin Sensing and Body Weight Control. Cell Metab. 2016. May 10;23(5):797–810. [DOI] [PubMed] [Google Scholar]
- Drareni K, Ballaire R, Barilla S, Mathew MJ, Toubal A, Fan R, Liang N, Chollet C, Huang Z, Kondili M, Foufelle F, Soprani A, Roussel R, Gautier JF, Alzaid F, Treuter E, Venteclef N. GPS2 Deficiency Triggers Maladaptive White Adipose Tissue Expansion in Obesity via HIF1A Activation. Cell Rep. 2018. Sep 11;24(11):2957–2971.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Messari S, Leloup C, Quignon M, Brisorgueil MJ, Penicaud L, Arluison M. Immunocytochemical localization of the insulin-responsive glucose transporter 4 (Glut4) in the rat central nervous system. J Comp Neurol. 1998. Oct 5;399(4):492–512. [DOI] [PubMed] [Google Scholar]
- Elias MF, Elias PK, Sullivan LM, Wolf PA, D’Agostino RB. Obesity, diabetes and cognitive deficit: The Framingham Heart Study. Neurobiol Aging. 2005. Dec;26 Suppl 1:11–6. [DOI] [PubMed] [Google Scholar]
- Emanuelli B, Vienberg SG, Smyth G, Cheng C, Stanford KI, Arumugam M, Michael MD, Adams AC, Kharitonenkov A, Kahn CR. Interplay between FGF21 and insulin action in the liver regulates metabolism. J Clin Invest. 2014. Feb;124(2):515–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson MA, Banks WA. Neuroimmune Axes of the Blood-Brain Barriers and Blood-Brain Interfaces: Bases for Physiological Regulation, Disease States, and Pharmacological Interventions. Pharmacol Rev. 2018. Apr;70(2):278–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erion JR, Wosiski-Kuhn M, Dey A, Hao S, Davis CL, Pollock NK, Stranahan AM. Obesity elicits interleukin 1-mediated deficits in hippocampal synaptic plasticity. Journal of Neuroscience, 34(7):2618–2631 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faouzi M, Leshan R, Björnholm M, Hennessey T, Jones J, Münzberg H. Differential accessibility of circulating leptin to individual hypothalamic sites. Endocrinology. 2007. Nov;148(11):5414–23. [DOI] [PubMed] [Google Scholar]
- Fernandez AM, Torres-Alemán I. The many faces of insulin-like peptide signalling in the brain. Nat Rev Neurosci. 2012. Mar 20;13(4):225–39. [DOI] [PubMed] [Google Scholar]
- Filiano AJ, Gadani SP, Kipnis J. How and why do T cells and their derived cytokines affect the injured and healthy brain? Nat Rev Neurosci. 2017. Jun;18(6):375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher SJ, Brüning JC, Lannon S, Kahn CR. Insulin signaling in the central nervous system is critical for the normal sympathoadrenal response to hypoglycemia. Diabetes. 2005. May;54(5):1447–51. [DOI] [PubMed] [Google Scholar]
- Fourrier C, Bosch-Bouju C, Boursereau R, Sauvant J, Aubert A, Capuron L, Ferreira G, Layé S, Castanon N. Brain tumor necrosis factor-alpha mediates anxiety-like behavior in a mouse model of severe obesity. Brain Behav Immun. 2019. Mar;77:25–36. [DOI] [PubMed] [Google Scholar]
- Frotscher M, Seress L. Morphological development of the hippocampus. Chapter in: Andersen P, Morris R, Amaral D, Bliss TV, O’Keefe J (Eds). The Hippocampus Book. 2007. Oxford University Press, p. 115–128. [Google Scholar]
- García-Cáceres C, Quarta C, Varela L, Gao Y, Gruber T, Legutko B, Jastroch M, Johansson P, Ninkovic J, Yi CX, Le Thuc O, Szigeti-Buck K, Cai W, Meyer CW, Pfluger PT, Fernandez AM, Luquet S, Woods SC, Torres-Alemán I, Kahn CR, Götz M, Horvath TL, Tschöp MH. Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell. 2016. Aug 11;166(4):867–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser E, Moutos CP, Downes M, Evans RM. FGF1 - a new weapon to control type 2 diabetes mellitus. Nat Rev Endocrinol. 2017. Oct;13(10):599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giefing-Kröll C, Berger P, Lepperdinger G, Grubeck-Loebenstein B. How sex and age affect immune responses, susceptibility to infections, and response to vaccination. Aging Cell. 2015. Jun;14(3):309–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goren HJ, Kulkarni RN, Kahn CR. Glucose homeostasis and tissue transcript content of insulin signaling intermediates in four inbred strains of mice: C57BL/6, C57BLKS/6, DBA/2, and 129X1. Endocrinology. 2004. Jul;145(7):3307–23. [DOI] [PubMed] [Google Scholar]
- Granholm AC, Bimonte-Nelson HA, Moore AB, Nelson ME, Freeman LR, Sambamurti K. Effects of a saturated fat and high cholesterol diet on memory and hippocampal morphology in the middle-aged rat. J Alzheimers Dis. 2008. Jun;14(2):133–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graupera M, Claret M. Endothelial Cells: New Players in Obesity and Related Metabolic Disorders. Trends Endocrinol Metab. 2018. Nov;29(11):781–794. [DOI] [PubMed] [Google Scholar]
- Grayson BE, Fitzgerald MF, Hakala-Finch AP, Ferris VM, Begg DP, Tong J, Woods SC, Seeley RJ, Davidson TL, Benoit SC. Improvements in hippocampal-dependent memory and microglial infiltration with calorie restriction and gastric bypass surgery, but not with vertical sleeve gastrectomy. Int J Obes (Lond). 2014. Mar;38(3):349–56. [DOI] [PubMed] [Google Scholar]
- Greenwood CE, Winocur G. Cognitive impairment in rats fed high-fat diets: a specific effect of saturated fatty-acid intake. Behav Neurosci. 1996. Jun;110(3):451–9. [DOI] [PubMed] [Google Scholar]
- Guo DH, Yamamoto M, Hernandez CM, Khodadadi H, Baban B, Stranahan AM. Beige adipocytes mediate the neuroprotective and anti-inflammatory effects of subcutaneous fat in obese mice. Nat Commun. 2021. Jul 30;12(1):4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo DH, Yamamoto M, Hernandez CM, Khodadadi H, Baban B, Stranahan AM. Visceral adipose NLRP3 impairs cognition in obesity via IL-1R1 on CX3CR1+ cells. J Clin Invest. 2020. Apr 1;130(4):1961–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson DR, Karlsson C, Skoog I, Rosengren L, Lissner L, Blennow K. Mid-life adiposity factors relate to blood-brain barrier integrity in late life. J Intern Med. 2007. Dec;262(6):643–50. [DOI] [PubMed] [Google Scholar]
- Gutierrez EG, Banks WA, Kastin AJ. Murine tumor necrosis factor alpha is transported from blood to brain in the mouse. J Neuroimmunol. 1993. Sep;47(2):169–76. [DOI] [PubMed] [Google Scholar]
- Habbas S, Santello M, Becker D, Stubbe H, Zappia G, Liaudet N, Klaus FR, Kollias G, Fontana A, Pryce CR, Suter T, Volterra A. Neuroinflammatory TNFα Impairs Memory via Astrocyte Signaling. Cell. 2015. Dec 17;163(7):1730–41. [DOI] [PubMed] [Google Scholar]
- Hao S, Dey A, Yu X, Stranahan AM. Dietary obesity reversibly induces synaptic stripping by microglia and impairs hippocampal plasticity. Brain Behav Immun. 2016. Jan;51:230–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman-Boehm I, Blüher M, Redel H, Sion-Vardy N, Ovadia S, Avinoach E, Shai I, Klöting N, Stumvoll M, Bashan N, Rudich A. Macrophage infiltration into omental versus subcutaneous fat across different populations: effect of regional adiposity and the comorbidities of obesity. J Clin Endocrinol Metab. 2007. Jun;92(6):2240–7. [DOI] [PubMed] [Google Scholar]
- Hastings NB, Gould E. Rapid extension of axons into the CA3 region by adult-generated granule cells. J Comp Neurol. 1999. Oct 11;413(1):146–54. [DOI] [PubMed] [Google Scholar]
- Hayward JN, Baker MA. A comparative study of the role of the cerebral arterial blood in the regulation of brain temperature in five mammals. Brain Res. 1969. Dec;16(2):417–40. [DOI] [PubMed] [Google Scholar]
- Hein AM, Zarcone TJ, Parfitt DB, Matousek SB, Carbonari DM, Olschowka JA, O’Banion MK. Behavioral, structural and molecular changes following long-term hippocampal IL-1β overexpression in transgenic mice. J Neuroimmune Pharmacol. 2012. Mar;7(1):145–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013. Jan 31;493(7434):674–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993. Jan 1;259(5091):87–91. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. 2017. Feb 8;542(7640):177–185. [DOI] [PubMed] [Google Scholar]
- Hsuchou H, Kastin AJ, Pan W. Blood-borne metabolic factors in obesity exacerbate injury-induced gliosis. J Mol Neurosci. 2012. Jun;47(2):267–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron. 2017. Sep 27;96(1):17–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving A, Harvey J. Regulation of hippocampal synaptic function by the metabolic hormone leptin: Implications for health and disease. Prog Lipid Res. 2021. Apr;82:101098. [DOI] [PubMed] [Google Scholar]
- Ishihara Y, Itoh K, Ishida A, Yamazaki T. Selective estrogen-receptor modulators suppress microglial activation and neuronal cell death via an estrogen receptor-dependent pathway. J Steroid Biochem Mol Biol. 2015. Jan;145:85–93. [DOI] [PubMed] [Google Scholar]
- Jagust W, Harvey D, Mungas D, Haan M. Central obesity and the aging brain. Arch Neurol. 2005. Oct;62(10):1545–8. [DOI] [PubMed] [Google Scholar]
- Jais A, Solas M, Backes H, Chaurasia B, Kleinridders A, Theurich S, Mauer J, Steculorum SM, Hampel B, Goldau J, Alber J, Förster CY, Eming SA, Schwaninger M, Ferrara N, Karsenty G, Brüning JC. Myeloid-Cell-Derived VEGF Maintains Brain Glucose Uptake and Limits Cognitive Impairment in Obesity. Cell. 2016. May 5;165(4):882–95. [DOI] [PubMed] [Google Scholar]
- Jensen-Cody SO, Flippo KH, Claflin KE, Yavuz Y, Sapouckey SA, Walters GC, Usachev YM, Atasoy D, Gillum MP, Potthoff MJ. FGF21 Signals to Glutamatergic Neurons in the Ventromedial Hypothalamus to Suppress Carbohydrate Intake. Cell Metab. 2020. Aug 4;32(2):273–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon BT, Jeong EA, Shin HJ, Lee Y, Lee DH, Kim HJ, Kang SS, Cho GJ, Choi WS, Roh GS. Resveratrol attenuates obesity-associated peripheral and central inflammation and improves memory deficit in mice fed a high-fat diet. Diabetes. 2012. Jun;61(6):1444–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong JY, Kwon HB, Ahn JC, Kang D, Kwon SH, Park JA, Kim KW. Functional and developmental analysis of the blood-brain barrier in zebrafish. Brain Res Bull. 2008. Mar 28;75(5):619–28. [DOI] [PubMed] [Google Scholar]
- Kahn SE, Zinman B, Haffner SM, O’Neill MC, Kravitz BG, Yu D, Freed MI, Herman WH, Holman RR, Jones NP, Lachin JM, Viberti GC; ADOPT Study Group. Obesity is a major determinant of the association of C-reactive protein levels and the metabolic syndrome in type 2 diabetes. Diabetes. 2006. Aug;55(8):2357–64. [DOI] [PubMed] [Google Scholar]
- Kaiser T, Feng G. Tmem119-EGFP and Tmem119-CreERT2 Transgenic Mice for Labeling and Manipulating Microglia. eNeuro. 2019. Aug 26;6(4):ENEURO.0448–18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang L, Routh VH, Kuzhikandathil EV, Gaspers LD, Levin BE. Physiological and molecular characteristics of rat hypothalamic ventromedial nucleus glucosensing neurons. Diabetes. 2004. Mar;53(3):549–59. [DOI] [PubMed] [Google Scholar]
- Kang S, Kong X, Rosen ED. Adipocyte-specific transgenic and knockout models. Methods Enzymol. 2014;537:1–16. [DOI] [PubMed] [Google Scholar]
- Kanoski SE, Zhang Y, Zheng W, Davidson TL. The effects of a high-energy diet on hippocampal function and blood-brain barrier integrity in the rat. J Alzheimers Dis. 2010;21(1):207–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski J. Global and regional brain metabolic scaling and its functional consequences. BMC Biol. 2007. May 9;5:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Kashiwagi H, Shiraga M, Tadokoro S, Kamae T, Ujiie H, Honda S, Miyata S, Ijiri Y, Yamamoto J, Maeda N, Funahashi T, Kurata Y, Shimomura I, Tomiyama Y, Kanakura Y. Adiponectin acts as an endogenous antithrombotic factor. Arterioscler Thromb Vasc Biol. 2006. Jan;26(1):224–30. [DOI] [PubMed] [Google Scholar]
- Kety SS, Schmidt CF. THE NITROUS OXIDE METHOD FOR THE QUANTITATIVE DETERMINATION OF CEREBRAL BLOOD FLOW IN MAN: THEORY, PROCEDURE AND NORMAL VALUES. J Clin Invest. 1948. Jul;27(4):476–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiecker C The origins of the circumventricular organs. J Anat. 2018. Apr;232(4):540–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierdorf K, Masuda T, Jordão MJC, Prinz M. Macrophages at CNS interfaces: ontogeny and function in health and disease. Nat Rev Neurosci. 2019. Sep;20(9):547–562. [DOI] [PubMed] [Google Scholar]
- Kilander L, Nyman H, Boberg M, Lithell H. Cognitive function, vascular risk factors and education. A cross-sectional study based on a cohort of 70-year-old men. J Intern Med. 1997. Oct;242(4):313–21. [DOI] [PubMed] [Google Scholar]
- Kim JK, Michael MD, Previs SF, Peroni OD, Mauvais-Jarvis F, Neschen S, Kahn BB, Kahn CR, Shulman GI. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J Clin Invest. 2000. Jun;105(12):1791–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto N, Shimizu K, Sawamoto K. Neuronal regeneration in a zebrafish model of adult brain injury. Dis Model Mech. 2012;5:200–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitajima N, Takikawa K, Sekiya H, Satoh K, Asanuma D, Sakamoto H, Takahashi S, Hanaoka K, Urano Y, Namiki S, Iino M, Hirose K. Real-time in vivo imaging of extracellular ATP in the brain with a hybrid-type fluorescent sensor. Elife. 2020. Jul 10;9:e57544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi M, Sakaguchi M, Lockhart SM, Cai W, Li ME, Homan EP, Rask-Madsen C, Kahn CR. Endothelial insulin receptors differentially control insulin signaling kinetics in peripheral tissues and brain of mice. Proc Natl Acad Sci U S A. 2017. Oct 3;114(40):E8478–E8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Könner AC, Janoschek R, Plum L, Jordan SD, Rother E, Ma X, Xu C, Enriori P, Hampel B, Barsh GS, Kahn CR, Cowley MA, Ashcroft FM, Brüning JC. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 2007. Jun;5(6):438–49. [DOI] [PubMed] [Google Scholar]
- Kovats S Estrogen receptors regulate innate immune cells and signaling pathways. Cell Immunol. 2015. Apr;294(2):63–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krotkiewski M, Björntorp P, Sjöström L, Smith U. Impact of obesity on metabolism in men and women. Importance of regional adipose tissue distribution. J Clin Invest. 1983. Sep;72(3):1150–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota N, Yano W, Kubota T, Yamauchi T, Itoh S, Kumagai H, Kozono H, Takamoto I, Okamoto S, Shiuchi T, Suzuki R, Satoh H, Tsuchida A, Moroi M, Sugi K, Noda T, Ebinuma H, Ueta Y, Kondo T, Araki E, Ezaki O, Nagai R, Tobe K, Terauchi Y, Ueki K, Minokoshi Y, Kadowaki T. Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab. 2007. Jul;6(1):55–68. [DOI] [PubMed] [Google Scholar]
- Kuhl J, Hilding A, Ostenson CG, Grill V, Efendic S, Båvenholm P. Characterisation of subjects with early abnormalities of glucose tolerance in the Stockholm Diabetes Prevention Programme: the impact of sex and type 2 diabetes heredity. Diabetologia. 2005. Jan;48(1):35–40. [DOI] [PubMed] [Google Scholar]
- Kuhlmann CR, Librizzi L, Closhen D, Pflanzner T, Lessmann V, Pietrzik CU, de Curtis M, Luhmann HJ. Mechanisms of C-reactive protein-induced blood-brain barrier disruption. Stroke. 2009. Apr;40(4):1458–66. [DOI] [PubMed] [Google Scholar]
- Kulkarni RN, Brüning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell. 1999. Feb 5;96(3):329–39. [DOI] [PubMed] [Google Scholar]
- Lang I, Füllsack S, Wyzgol A, Fick A, Trebing J, Arana JA, Schäfer V, Weisenberger D, Wajant H. Binding Studies of TNF Receptor Superfamily (TNFRSF) Receptors on Intact Cells. J Biol Chem. 2016. Mar 4;291(10):5022–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapenna A, De Palma M, Lewis CE. Perivascular macrophages in health and disease. Nat Rev Immunol. 2018. Nov;18(11):689–702. [DOI] [PubMed] [Google Scholar]
- Lavin DN, Joesting JJ, Chiu GS, Moon ML, Meng J, Dilger RN, Freund GG. Fasting induces an anti-inflammatory effect on the neuroimmune system which a high-fat diet prevents. Obesity (Silver Spring). 2011. Aug;19(8):1586–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzari M, Bettini S, Ciani F, Franceschini V. Glucose transporter distribution in the vessels of the central nervous system of the axolotl Ambystoma mexicanum (Urodela: Ambystomatidae). Anat Rec (Hoboken). 2008. Oct;291(10):1293–300. [DOI] [PubMed] [Google Scholar]
- Lee-Liu D, Méndez-Olivos EE, Muñoz R, Larraín J. The African clawed frog Xenopus laevis: A model organism to study regeneration of the central nervous system. Neurosci Lett. 2017. Jun 23;652:82–93. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007. Jan 11;445(7124):168–76. [DOI] [PubMed] [Google Scholar]
- Lenz KM, Nugent BM, Haliyur R, McCarthy MM. Microglia are essential to masculinization of brain and behavior. J Neurosci. 2013. Feb 13;33(7):2761–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ME, Lauritzen HPMM, O’Neill BT, Wang CH, Cai W, Brandao BB, Sakaguchi M, Tao R, Hirshman MF, Softic S, Kahn CR. Role of p110a subunit of PI3-kinase in skeletal muscle mitochondrial homeostasis and metabolism. Nat Commun. 2019. Jul 30;10(1):3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Zhao S, Zhang Z, Zhu Y, Gliniak CM, Vishvanath L, An YA, Wang MY, Deng Y, Zhu Q, Shan B, Sherwood A, Onodera T, Oz OK, Gordillo R, Gupta RK, Liu M, Horvath TL, Dixit VD, Scherer PE. Adiponectin preserves metabolic fitness during aging. Elife. 2021. Apr 27;10:e65108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YS, Lin FY, Hsiao YH. Myostatin Is Associated With Cognitive Decline in an Animal Model of Alzheimer’s Disease. Mol Neurobiol. 2019. Mar;56(3):1984–1991. [DOI] [PubMed] [Google Scholar]
- Ling AV, Gearing ME, Semova I, Shin DJ, Clements R, Lai ZW, Biddinger SB. FoxO1 Is Required for Most of the Metabolic and Hormonal Perturbations Produced by Hepatic Insulin Receptor Deletion in Male Mice. Endocrinology. 2018. Mar 1;159(3):1253–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liszka TG, Dellon AL, Im M, Angel MF, Plotnick L. Effect of lipectomy on growth and development of hyperinsulinemia and hyperlipidemia in the Zucker rat. Plast Reconstr Surg. 1998. Sep;102(4):1122–7. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhou LJ, Wang J, Li D, Ren WJ, Peng J, Wei X, Xu T, Xin WJ, Pang RP, Li YY, Qin ZH, Murugan M, Mattson MP, Wu LJ, Liu XG. TNF-α Differentially Regulates Synaptic Plasticity in the Hippocampus and Spinal Cord by Microglia-Dependent Mechanisms after Peripheral Nerve Injury. J Neurosci. 2017. Jan 25;37(4):871–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loram LC, Sholar PW, Taylor FR, Wiesler JL, Babb JA, Strand KA, Berkelhammer D, Day HE, Maier SF, Watkins LR. Sex and estradiol influence glial pro-inflammatory responses to lipopolysaccharide in rats. Psychoneuroendocrinology. 2012. Oct;37(10):1688–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourenco MV, Frozza RL, de Freitas GB, Zhang H, Kincheski GC, Ribeiro FC, Gonçalves RA, Clarke JR, Beckman D, Staniszewski A, Berman H, Guerra LA, Forny-Germano L, Meier S, Wilcock DM, de Souza JM, Alves-Leon S, Prado VF, Prado MAM, Abisambra JF, Tovar-Moll F, Mattos P, Arancio O, Ferreira ST, De Felice FG. Exercise-linked FNDC5/irisin rescues synaptic plasticity and memory defects in Alzheimer’s models. Nat Med. 2019. Jan;25(1):165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun MP, Monuki ES, Lehtinen MK. Development and functions of the choroid plexus-cerebrospinal fluid system. Nat Rev Neurosci. 2015. Aug;16(8):445–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MA. Neuroinflammatory changes negatively impact on LTP: A focus on IL-1β. Brain Res. 2015. Sep 24;1621:197–204. [DOI] [PubMed] [Google Scholar]
- Macotela Y, Boucher J, Tran TT, Kahn CR. Sex and depot differences in adipocyte insulin sensitivity and glucose metabolism. Diabetes. 2009. Apr;58(4):803–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson KP, Sompol P, Kannarkat GT, Chang J, Sniffen L, Wildner ME, Norris CM, Tansey MG. Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol Dis. 2017. Jun;102:81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, Furuyama N, Kondo H, Takahashi M, Arita Y, Komuro R, Ouchi N, Kihara S, Tochino Y, Okutomi K, Horie M, Takeda S, Aoyama T, Funahashi T, Matsuzawa Y. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med. 2002. Jul;8(7):731–7. [DOI] [PubMed] [Google Scholar]
- Maggioli E, McArthur S, Mauro C, Kieswich J, Kusters DH, Reutelingsperger CP, Yaqoob M, Solito E. Estrogen protects the blood-brain barrier from inflammation-induced disruption and increased lymphocyte trafficking. Brain Behav Immun. 2016. Jan;51:212–22. [DOI] [PubMed] [Google Scholar]
- Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, Flier JS. A transgenic model of visceral obesity and the metabolic syndrome. Science. 2001. Dec 7;294(5549):2166–70. [DOI] [PubMed] [Google Scholar]
- Matsuda M, Shimomura I, Sata M, Arita Y, Nishida M, Maeda N, Kumada M, Okamoto Y, Nagaretani H, Nishizawa H, Kishida K, Komuro R, Ouchi N, Kihara S, Nagai R, Funahashi T, Matsuzawa Y. Role of adiponectin in preventing vascular stenosis. The missing link of adipo-vascular axis. J Biol Chem. 2002. Oct 4;277(40):37487–91. [DOI] [PubMed] [Google Scholar]
- Mauer J, Chaurasia B, Plum L, Quast T, Hampel B, Blüher M, Kolanus W, Kahn CR, Brüning JC. Myeloid cell-restricted insulin receptor deficiency protects against obesity-induced inflammation and systemic insulin resistance. PLoS Genet. 2010. May 6;6(5):e1000938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNay EC, Ong CT, McCrimmon RJ, Cresswell J, Bogan JS, Sherwin RS. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol Learn Mem. 2010. May;93(4):546–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medrikova D, Jilkova ZM, Bardova K, Janovska P, Rossmeisl M, Kopecky J. Sex differences during the course of diet-induced obesity in mice: adipose tissue expandability and glycemic control. Int J Obes (Lond). 2012. Feb;36(2):262–72. [DOI] [PubMed] [Google Scholar]
- Metzler-Baddeley C, Mole JP, Leonaviciute E, Sims R, Kidd EJ, Ertefai B, Kelso-Mitchell A, Gidney F, Fasano F, Evans J, Jones DK, Baddeley RJ. Sex-specific effects of central adiposity and inflammatory markers on limbic microstructure. Neuroimage. 2019. Apr 1;189:793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael MD, Kulkarni RN, Postic C, Previs SF, Shulman GI, Magnuson MA, Kahn CR. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell. 2000. Jul;6(1):87–97. [PubMed] [Google Scholar]
- Mielke JG, Nicolitch K, Avellaneda V, Earlam K, Ahuja T, Mealing G, Messier C. Longitudinal study of the effects of a high-fat diet on glucose regulation, hippocampal function, and cerebral insulin sensitivity in C57BL/6 mice. Behav Brain Res. 2006. Dec 15;175(2):374–82. [DOI] [PubMed] [Google Scholar]
- Mitoma H, Horiuchi T, Tsukamoto H, Tamimoto Y, Kimoto Y, Uchino A, To K, Harashima S, Hatta N, Harada M. Mechanisms for cytotoxic effects of anti-tumor necrosis factor agents on transmembrane tumor necrosis factor alpha-expressing cells: comparison among infliximab, etanercept, and adalimumab. Arthritis Rheum. 2008. May;58(5):1248–57. [DOI] [PubMed] [Google Scholar]
- Molteni R, Barnard RJ, Ying Z, Roberts CK, Gómez-Pinilla F. A high-fat, refined sugar diet reduces hippocampal brain-derived neurotrophic factor, neuronal plasticity, and learning. Neuroscience. 2002;112(4):803–14. [DOI] [PubMed] [Google Scholar]
- Morrison CD, Pistell PJ, Ingram DK, Johnson WD, Liu Y, Fernandez-Kim SO, White CL, Purpera MN, Uranga RM, Bruce-Keller AJ, Keller JN. High fat diet increases hippocampal oxidative stress and cognitive impairment in aged mice: implications for decreased Nrf2 signaling. J Neurochem. 2010. Sep;114(6):1581–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagareddy PR, Kraakman M, Masters SL, Stirzaker RA, Gorman DJ, Grant RW, Dragoljevic D, Hong ES, Abdel-Latif A, Smyth SS, Choi SH, Korner J, Bornfeldt KE, Fisher EA, Dixit VD, Tall AR, Goldberg IJ, Murphy AJ. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab. 2014. May 6;19(5):821–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neman J, Chen TC. The Choroid Plexus and Cerebrospinal Fluid: Emerging Roles in CNS Development, Maintenance, and Disease Progression. Academic Press, 2016. [Google Scholar]
- O’Brown NM, Pfau SJ, Gu C. Bridging barriers: a comparative look at the blood-brain barrier across organisms. Genes Dev. 2018. Apr 1;32(7–8):466–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouchi N, Kihara S, Arita Y, Maeda K, Kuriyama H, Okamoto Y, Hotta K, Nishida M, Takahashi M, Nakamura T, Yamashita S, Funahashi T, Matsuzawa Y. Novel modulator for endothelial adhesion molecules: adipocyte-derived plasma protein adiponectin. Circulation. 1999. Dec 21–28;100(25):2473–6. [DOI] [PubMed] [Google Scholar]
- Ouedraogo R, Wu X, Xu SQ, Fuchsel L, Motoshima H, Mahadev K, Hough K, Scalia R, Goldstein BJ. Adiponectin suppression of high-glucose-induced reactive oxygen species in vascular endothelial cells: evidence for involvement of a cAMP signaling pathway. Diabetes. 2006. Jun;55(6):1840–6. [DOI] [PubMed] [Google Scholar]
- Ouyang S, Hsuchou H, Kastin AJ, Wang Y, Yu C, Pan W. Diet-induced obesity suppresses expression of many proteins at the blood-brain barrier. J Cereb Blood Flow Metab. 2014. Jan;34(1):43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer BF, Clegg DJ. The sexual dimorphism of obesity. Mol Cell Endocrinol. 2015. Feb 15;402:113–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan W, Kastin AJ. TNFalpha transport across the blood-brain barrier is abolished in receptor knockout mice. Exp Neurol. 2002. Apr;174(2):193–200. [DOI] [PubMed] [Google Scholar]
- Pancani T, Anderson KL, Brewer LD, Kadish I, DeMoll C, Landfield PW, Blalock EM, Porter NM, Thibault O. Effect of high-fat diet on metabolic indices, cognition, and neuronal physiology in aging F344 rats. Neurobiol Aging. 2013. Aug;34(8):1977–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen BK, Febbraio MA. Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol. 2012. Apr 3;8(8):457–65. [DOI] [PubMed] [Google Scholar]
- Pétrault O, Pétrault M, Ouk T, Bordet R, Bérézowski V, Bastide M. Visceral adiposity links cerebrovascular dysfunction to cognitive impairment in middle-aged mice. Neurobiol Dis. 2019. Oct;130:104536. [DOI] [PubMed] [Google Scholar]
- Pleasure DE. Regeneration in the Central and Peripheral Nervous Systems. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects. 6th edition. Siegel GJ, Agranoff BW, Albers RW, et al. , editors. Philadelphia: Lippincott-Raven; 1999. [Google Scholar]
- Pontzer H, Brown MH, Raichlen DA, Dunsworth H, Hare B, Walker K, Luke A, Dugas LR, Durazo-Arvizu R, Schoeller D, Plange-Rhule J, Bovet P, Forrester TE, Lambert EV, Thompson ME, Shumaker RW, Ross SR. Metabolic acceleration and the evolution of human brain size and life history. Nature. 2016. May 19;533(7603):390–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DW, Irwin N, Flatt PR, Hölscher C, Gault VA. Prolonged GIP receptor activation improves cognitive function, hippocampal synaptic plasticity and glucose homeostasis in high-fat fed mice. Eur J Pharmacol. 2011. Jan 15;650(2–3):688–93. [DOI] [PubMed] [Google Scholar]
- Pribiag H, Stellwagen D. Neuroimmune regulation of homeostatic synaptic plasticity. Neuropharmacology. 2014. Mar;78:13–22. [DOI] [PubMed] [Google Scholar]
- Qi Y, Takahashi N, Hileman SM, Patel HR, Berg AH, Pajvani UB, Scherer PE, Ahima RS. Adiponectin acts in the brain to decrease body weight. Nat Med. 2004. May;10(5):524–9. [DOI] [PubMed] [Google Scholar]
- Qian Y, Yin J, Hong J, Li G, Zhang B, Liu G, Wan Q, Chen L. Neuronal seipin knockout facilitates Abeta-induced neuroinflammation and neurotoxicity via reduction of PPARgamma in hippocampus of mouse. J Neuroinflammation. 2016. Jun 10;13(1):145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y, Nguyen KD, Odegaard JI, Cui X, Tian X, Locksley RM, Palmiter RD, Chawla A. Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell. 2014. Jun 5;157(6):1292–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettew JA, Huet YM, Marriott I. Estrogens augment cell surface TLR4 expression on murine macrophages and regulate sepsis susceptibility in vivo. Endocrinology. 2009. Aug;150(8):3877–84. [DOI] [PubMed] [Google Scholar]
- Rhea EM, Rask-Madsen C, Banks WA. Insulin transport across the blood-brain barrier can occur independently of the insulin receptor. J Physiol. 2018. Oct;596(19):4753–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo MR, Fasano R, Paolisso G. Adiponectin and Cognitive Decline. Int J Mol Sci. 2020. Mar 16;21(6):2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell. 2014. Jan 16;156(1–2):20–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo K, Collier JG, Li AC, Katzenellenbogen JA, Glass CK. An ADIOL-ERβ-CtBP transrepression pathway negatively regulates microglia-mediated inflammation. Cell. 2011. May 13;145(4):584–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez Alvarado A, Tsonis PA. Bridging the regeneration gap: genetic insights from diverse animal models. Nat Rev Genet. 2006. Nov;7(11):873–84. [DOI] [PubMed] [Google Scholar]
- Schumacher R, Mosthaf L, Schlessinger J, Brandenburg D, Ullrich A. Insulin and insulin-like growth factor-1 binding specificity is determined by distinct regions of their cognate receptors. J Biol Chem. 1991. Oct 15;266(29):19288–95. [PubMed] [Google Scholar]
- Scott DL, Kingsley GH. Tumor necrosis factor inhibitors for rheumatoid arthritis. N Engl J Med. 2006. Aug 17;355(7):704–12. [DOI] [PubMed] [Google Scholar]
- Serot JM, Foliguet B, Béné MC, Faure GC. Choroid plexus and ageing in rats: a morphometric and ultrastructural study. Eur J Neurosci. 2001. Sep;14(5):794–8. [DOI] [PubMed] [Google Scholar]
- Serrats J, Schiltz JC, García-Bueno B, van Rooijen N, Reyes TM, Sawchenko PE. Dual roles for perivascular macrophages in immune-to-brain signaling. Neuron. 2010. Jan 14;65(1):94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Strader AD, Woods SC, Seeley RJ. The effect of fat removal on glucose tolerance is depot specific in male and female mice. Am J Physiol Endocrinol Metab. 2007. Oct;293(4):E1012–20. [DOI] [PubMed] [Google Scholar]
- Shuwa HA, Dallatu MK, Yeldu MH, Ahmed HM, Nasir IA. Effects of Adalimumab, an Anti-tumour Necrosis Factor-Alpha (TNF-α) Antibody, on Obese Diabetic Rats. Malays J Med Sci. 2018. Jul;25(4):51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson IA, Carruthers A, Vannucci SJ. Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J Cereb Blood Flow Metab. 2007. Nov;27(11):1766–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer K, Maley N, Mergian T, DelProposto J, Cho KW, Zamarron BF, Martinez-Santibanez G, Geletka L, Muir L, Wachowiak P, Demirjian C, Lumeng CN. Differences in Hematopoietic Stem Cells Contribute to Sexually Dimorphic Inflammatory Responses to High Fat Diet-induced Obesity. J Biol Chem. 2015. May 22;290(21):13250–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Softic S, Boucher J, Solheim MH, Fujisaka S, Haering MF, Homan EP, Winnay J, Perez-Atayde AR, Kahn CR. Lipodystrophy Due to Adipose Tissue-Specific Insulin Receptor Knockout Results in Progressive NAFLD. Diabetes. 2016. Aug;65(8):2187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokoloff L, Mangol R, Wechsler RL, Kenney C, Kety SS. The effect of mental arithmetic on cerebral circulation and metabolism. J Clin Invest. 1955. Jul;34(7, Part 1):1101–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer SJ, D’Angelo H, Soch A, Watkins LR, Maier SF, Barrientos RM. High-fat diet and aging interact to produce neuroinflammation and impair hippocampal- and amygdalar-dependent memory. Neurobiol Aging. 2017. Oct;58:88–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger J, Verma S, Göhring I, Bobbert T, Seifert J, Sindler AL, Pfeiffer A, Hileman SM, Tschöp M, Banks WA. Adiponectin does not cross the blood-brain barrier but modifies cytokine expression of brain endothelial cells. Diabetes. 2006. Jan;55(1):141–7. [PubMed] [Google Scholar]
- Spruston N, McBain C. Structural and functional properties of hippocampal neurons. Chapter in: Andersen P, Morris R, Amaral D, Bliss TV, O’Keefe J (Eds). The Hippocampus Book. 2007. Oxford University Press, p. 133–166. [Google Scholar]
- Stefan N, Häring HU, Hu FB, Schulze MB. Metabolically healthy obesity: epidemiology, mechanisms, and clinical implications. Lancet Diabetes Endocrinol. 2013. Oct;1(2):152–62. [DOI] [PubMed] [Google Scholar]
- Stefan N, Schick F, Häring HU. Causes, Characteristics, and Consequences of Metabolically Unhealthy Normal Weight in Humans. Cell Metab. 2017. Aug 1;26(2):292–300. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci. 2005. Mar 23;25(12):3219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, Hao S, Dey A, Yu X, Baban B. Blood-brain barrier breakdown promotes macrophage infiltration and cognitive impairment in leptin receptor-deficient mice. J Cereb Blood Flow Metab. 2016. Dec;36(12):2108–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, Norman ED, Lee K, Cutler RG, Telljohann RS, Egan JM, Mattson MP. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus. 2008;18(11):1085–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturman MT, de Leon CF, Bienias JL, Morris MC, Wilson RS, Evans DA. Body mass index and cognitive decline in a biracial community population. Neurology. 2008. Jan 29;70(5):360–7. [DOI] [PubMed] [Google Scholar]
- Sullivan EL, Grayson B, Takahashi D, Robertson N, Maier A, Bethea CL, Smith MS, Coleman K, Grove KL. Chronic consumption of a high-fat diet during pregnancy causes perturbations in the serotonergic system and increased anxiety-like behavior in nonhuman primate offspring. J Neurosci. 2010. Mar 10;30(10):3826–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamashiro KL, Terrillion CE, Hyun J, Koenig JI, Moran TH. Prenatal stress or high-fat diet increases susceptibility to diet-induced obesity in rat offspring. Diabetes. 2009. May;58(5):1116–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tancredi V, D’Arcangelo G, Grassi F, Tarroni P, Palmieri G, Santoni A, Eusebi F. Tumor necrosis factor alters synaptic transmission in rat hippocampal slices. Neurosci Lett. 1992. Nov 9;146(2):176–8. [DOI] [PubMed] [Google Scholar]
- Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006. Oct;6(10):772–83. [DOI] [PubMed] [Google Scholar]
- Tower J Sex-Specific Gene Expression and Life Span Regulation. Trends Endocrinol Metab. 2017. Oct;28(10):735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuneki K A survey of occurrence of about seventeen circumventricular organs in brains of various vertebrates with special reference to lower groups. J Hirnforsch. 1986;27(4):441–70. [PubMed] [Google Scholar]
- Valdearcos M, Douglass JD, Robblee MM, Dorfman MD, Stifler DR, Bennett ML, Gerritse I, Fasnacht R, Barres BA, Thaler JP, Koliwad SK. Microglial Inflammatory Signaling Orchestrates the Hypothalamic Immune Response to Dietary Excess and Mediates Obesity Susceptibility. Cell Metab. 2017. Jul 5;26(1):185–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg E, Biessels GJ, de Craen AJ, Gussekloo J, Westendorp RG. The metabolic syndrome is associated with decelerated cognitive decline in the oldest old. Neurology. 2007. Sep 4;69(10):979–85. [DOI] [PubMed] [Google Scholar]
- Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011. Feb;17(2):179–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanlandewijck M, He L, Mäe MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Laviña B, Gouveia L, Sun Y, Raschperger E, Räsänen M, Zarb Y, Mochizuki N, Keller A, Lendahl U, Betsholtz C. A molecular atlas of cell types and zonation in the brain vasculature. Nature. 2018. Feb 22;554(7693):475–480. [DOI] [PubMed] [Google Scholar]
- Vazana U, Veksler R, Pell GS, Prager O, Fassler M, Chassidim Y, Roth Y, Shahar H, Zangen A, Raccah R, Onesti E, Ceccanti M, Colonnese C, Santoro A, Salvati M, D’Elia A, Nucciarelli V, Inghilleri M, Friedman A. Glutamate-Mediated Blood-Brain Barrier Opening: Implications for Neuroprotection and Drug Delivery. J Neurosci. 2016. Jul 20;36(29):7727–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskuhl R Sex differences in autoimmune diseases. Biol Sex Differ. 2011. Jan 4;2(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Yu Y, Cai W, Batista TM, Suk S, Noh HL, Hirshman M, Nigro P, Li ME, Softic S, Goodyear L, Kim JK, Kahn CR. Muscle-Specific Insulin Receptor Overexpression Protects Mice From Diet-Induced Glucose Intolerance but Leads to Postreceptor Insulin Resistance. Diabetes. 2020. Nov;69(11):2294–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Yuan J, Yu Z, Lin L, Jiang Y, Cao Z, Zhuang P, Whalen MJ, Song B, Wang XJ, Li X, Lo EH, Xu Y, Wang X. FGF21 Attenuates High-Fat Diet-Induced Cognitive Impairment via Metabolic Regulation and Anti-inflammation of Obese Mice. Mol Neurobiol. 2018. Jun;55(6):4702–4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Ting JP, O’Neill LA. A role for the NLRP3 inflammasome in metabolic diseases--did Warburg miss inflammation? Nat Immunol. 2012. Mar 19;13(4):352–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernstedt Asterholm I, Tao C, Morley TS, Wang QA, Delgado-Lopez F, Wang ZV, Scherer PE. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 2014. Jul 1;20(1):103–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West-Eberhard MJ. Nutrition, the visceral immune system, and the evolutionary origins of pathogenic obesity. Proc Natl Acad Sci U S A. 2019. Jan 15;116(3):723–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmer RA, Gustafson DR, Barrett-Connor E, Haan MN, Gunderson EP, Yaffe K. Central obesity and increased risk of dementia more than three decades later. Neurology. 2008. Sep 30;71(14):1057–64. [DOI] [PubMed] [Google Scholar]
- Wosiski-Kuhn M, Stranahan AM. From pattern separation to mood regulation: multiple roles for developmental signals in the adult dentate gyrus. Front Cell Neurosci. 2013. Jun 26;7:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Mahadev K, Fuchsel L, Ouedraogo R, Xu SQ, Goldstein BJ. Adiponectin suppresses IkappaB kinase activation induced by tumor necrosis factor-alpha or high glucose in endothelial cells: role of cAMP and AMP kinase signaling. Am J Physiol Endocrinol Metab. 2007. Dec;293(6):E1836–44. [DOI] [PubMed] [Google Scholar]
- Xiong X, Xu L, Wei L, White RE, Ouyang YB, Giffard RG. IL-4 Is Required for Sex Differences in Vulnerability to Focal Ischemia in Mice. Stroke. 2015. Aug;46(8):2271–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu WL, Atti AR, Gatz M, Pedersen NL, Johansson B, Fratiglioni L. Midlife overweight and obesity increase late-life dementia risk: a population-based twin study. Neurology. 2011. May 3;76(18):1568–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Guo DH, Hernandez CM, Stranahan AM. Endothelial Adora2a Activation Promotes Blood-Brain Barrier Breakdown and Cognitive Impairment in Mice with Diet-Induced Insulin Resistance. J Neurosci. 2019. May 22;39(21):4179–4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi CX, Tschöp MH, Woods SC, Hofmann SM. High-fat-diet exposure induces IgG accumulation in hypothalamic microglia. Dis Model Mech. 2012. Sep;5(5):686–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota T, Oritani K, Takahashi I, Ishikawa J, Matsuyama A, Ouchi N, Kihara S, Funahashi T, Tenner AJ, Tomiyama Y, Matsuzawa Y. Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood. 2000. Sep 1;96(5):1723–32. [PubMed] [Google Scholar]
- Youm YH, Grant RW, McCabe LR, Albarado DC, Nguyen KY, Ravussin A, Pistell P, Newman S, Carter R, Laque A, Münzberg H, Rosen CJ, Ingram DK, Salbaum JM, Dixit VD. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013. Oct 1;18(4):519–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Kastin AJ, Pan W. Reciprocal interactions of insulin and insulin-like growth factor I in receptor-mediated transport across the blood-brain barrier. Endocrinology. 2006. Jun;147(6):2611–5. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014. Sep 3;34(36):11929–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and Dysfunction of the Blood-Brain Barrier. Cell. 2015. Nov 19;163(5):1064–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Leeman SE, Amar S. Signaling mechanisms in the restoration of impaired immune function due to diet-induced obesity. Proc Natl Acad Sci U S A. 2011. Feb 15;108(7):2867–72. [DOI] [PMC free article] [PubMed] [Google Scholar]