

Abstract
Case summary
An 18-month-old castrated male domestic shorthair cat was presented with a 2-month history of collapse and severe weakness, particularly affecting the pelvic limbs. A biceps femoris muscle biopsy revealed excessive variability in myofibre size, mild necrosis, minimal centronucleation and scattered 10 μm intracytoplasmic oval inclusions. The inclusions appeared amphophilic with haematoxylin and eosin, blue with Gomori trichrome and unstained with nicotinamide adenine dinucleotide dehydrogenase tetrazolium reductase staining. ATPase staining revealed a normal mosaic pattern and atrophy of both type 1 and 2 myofibres. The pathological diagnosis was a myopathy with inclusions. In contrast to previous feline myofibre inclusions previously reported in the literature, inclusions were not identified after immunohistochemistry using anti-desmin, tubulin, spectrin, laminin, LAMP and LC3 antibodies. After supportive care and corticosteroid treatment, clinical improvement was noted and the cat was discharged 10 days after initial presentation. Clinical and neurological re-examinations were performed at 1, 3, 6 and 9 months after discharge. Owner contact at both 10 and 30 months post-discharge confirmed that persistent muscular weakness was present.
Relevance and novel information
This case report describes a novel and slowly progressive feline myopathy associated with oval amphophilic inclusions unreactive to immunostaining, which have not been previously reported in feline myopathies.
Keywords: Myopathy, inclusion, muscle, immunostaining
Introduction
Feline muscular weakness is reported in multiple acquired conditions such as myasthenia gravis, myositis and polyneuropathies.1–3 In other cases, feline muscular weakness can be associated with hereditary musculoskeletal disorders. In humans, biomolecular and genetic mutation tests have enabled precise classification of hereditary muscular disorders. In the cat, a diagnosis is often based on histological changes observed on a muscle biopsy and associated supportive signs.4–13
This case report describes the clinical and histopathological findings of a cat that was initially presented with muscular weakness. A muscle biopsy revealed sarcoplasmic oval inclusions associated with fibre atrophy, centronucleation and mild inflammation. Immunostaining did not show any similarities with the other feline congenital muscular disorders previously described in the literature. Symptomatic treatment led to clinical improvement, but mild clinical signs persisted after discharge. Even if the definitive biomolecular abnormality ultimately remained unknown, this case report describes atypical inclusions in myofibres that have not previously been described in the cat.
Case description
An 18-month-old male castrated and vaccinated domestic shorthair cat was presented with a 2-month history of prolonged periods of recumbency. Litter mates were reported to be healthy at the time of diagnosis. Muscle weakness, particularly affecting the pelvic limbs, an abnormal gait with episodes of collapse, difficulties in swallowing and a progressive onset were reported. Two weeks prior to referral the cat deteriorated acutely and aphonia was reported. The cat was stabilised with fluid therapy and analgesics prior to referral.
On physical examination, severe paresis was present; the cat was unable to stand even with assistance and presented in lateral recumbency. Generalised pain, hypothermia (36.8 ºC) and bradycardia (80 beats per min) were observed. Mild pain was identified on palpation of all muscles. Normal proprioception and hypotonia were observed on the four limbs, pelvic limb reflexes were diminished and cranial nerve examination revealed a bilaterally decreased palpebral reflex. Differential diagnoses included muscular disorders (including myopathies and myositis), neuromuscular junction disorders (myasthenia gravis, botulism, polyradiculoneuritis) and polyneuropathies (congenital, infectious, ischaemic neuromyopathy or paraneoplastic disorders). Polyarthritis and meningitis, although considered less likely were included in the differential list.
Haematology and electrolytes were within the reference intervals (RIs). Most biochemical parameters were within the RIs except for serum creatine kinase and lactate dehydrogenase, which were both slightly increased (605 UI/l [RI 0–340] and 2136 UI/l [RI 0–180], respectively). Arthrocentesis of both carpi and tarsi revealed grossly normal synovial fluid (volume, colour and viscosity) and, as such, cytological evaluation was not performed. A lumbar cerebrospinal tap was performed and cerebrospinal fluid proteins (0.1 g/l) and cell count (0 cell/µl) were within the RIs. A neostigmine test (neostigmine 0.04 mg/kg IM [Prostigmine; Mylan]) did not result in any clinical improvement in the 6 h following the injection. The neostigmine test is not 100% sensitive for myasthenia gravis, so an acetylcholine receptor antibody test was recommended but refused by the owners on financial grounds. Serological tests for feline leukaemia antigen, feline immunodeficiency virus antibody and toxoplasma antibodies were negative. No abnormalities were detected on thoracic and spinal radiographs.
Fluid therapy (lactated Ringer’s 3 ml/kg/h IV) and analgesia (buprenorphine 20 µg/kg SC q8h [Vetergesic NDV, Ceva]) were administered. Prednisolone (0.5 mg/kg PO q12h [Prednicortone; Dechra]) therapy was initiated as an inflammatory myopathy or myositis were considered possible. After 10 days of treatment, the cat was pain-free and ambulatory. Limb proprioception and palpebral reflexes were normal. Muscular weakness was still present with frequent episodes of recumbency after any exercise.
An underlying myopathy was suspected. Once the cat was considered stable for anaesthesia, a surgical muscle biopsy was obtained. Under anaesthesia (diazepam 0.25 mg/kg IV [Diazepam; TVM]; alfaxalone 4 mg/kg IV [Alfaxan]; morphine 0.1 mg/kg IV [Cooper]; isoflurane [Isovet]), a biopsy from the left biceps femoris muscle was obtained and sent immediately for histological evaluation at 4 ºC. On arrival at the laboratory, the muscle biopsy was snap frozen in isopentane cooled with liquid nitrogen, prior to transverse serial sectioning (8 µm thick). Routine staining techniques were utilised: haematoxylin, eosin and saffron (HES), Gomori trichrome, periodic acid–Schiff and histoenzymological procedures (nicotinamide adenine dinucleotide dehydrogenase tetrazolium reductase [NADH-TR]) and ATPase following preincubation at pH 4.2, 4.63 and 10.4 for fibre typing. 14 Histopathological analysis was performed by a board-certified veterinary pathologist.
Histopathological analysis revealed generalised changes in the muscle, characterised by excessive variability in myofibre size with occasional hypertrophic fibres (up to 80 µm) and numerous atrophic fibres (<20 μm), some of which had fragmented or clear sarcoplasm associated with ongoing necrosis (Figure 1). Ten percent of fibres had central myonuclei, consistent with mild regenerative response. Centronucleation was not observed in any of the C-type fibres. Inflammation was mild and mainly comprised mononuclear cells surrounding necrotic fibres. Fibrosis was absent. Approximatively 1–2% of fibres showed a single 10 μm intrasarcoplasmic, centrally located oval-shaped inclusion. Fibres containing more than one inclusion were rarely observed. All inclusions were similar in appearance, amphophilic with HES staining, staining navy blue with Gomori trichrome and unstained with both NADH-TR reagent and periodic acid–Schiff (Figures 1 and 2). ATPase typing did not show any alteration in either normal or atrophic fibre type distribution. Inclusions were present both in type 1 and 2 fibres. Fibre atrophy, centronucleation and mild necrosis without any fibre-type grouping was consistent with a primary muscle pathology. Histopathological diagnosis was consistent with a myopathy with intrasarcoplasmic inclusions.
Figure 1.

Muscular histopathological changes. Transverse sections of a fresh-frozen muscle biopsy from the left biceps muscle stained with (a–c) haematoxylin, eosin and saffron (HES) and (d–e) Gomori trichrome navy blue staining (GT). (a,d) × 100; (b,e) × 200; (c,f) × 400. Atrophic fibres were observed (yellow and white arrows) and most contained inclusions, which appeared amphophilic on HES (white arrowheads) and blue on GT (yellow arrowheads)
Figure 2.

Morphology of the inclusions. Transverse section of a fresh-frozen muscle biopsy from the left biceps muscle (× 400). (a) Haematoxylin, eosin and saffron staining: a 10 µm intrasarcoplasmic oval shape amphophilic inclusion is located at the centre of a myofibre (arrowhead). (b) Nicotinamide adenine dinucleotide dehydrogenase tetrazolium reductase reaction staining: two atrophic myofibres exhibit similar oval-shaped inclusions in the form of unstained halos (arrowheads)
Additional muscle sample sections were used for immunohistochemistry. Antibody markers used in the study are summarised in Table 1. Negative and positive controls were obtained, with an additional muscle section, with omission of the primary antibody, and with a section from a healthy feline biceps femoris muscle. Tissue distribution and cellular localisation of these proteins were unchanged. The inclusions were not stained by any of the immunohistochemical markers. Rare intrasarcoplasmic figures of macrophages identified by a LC3-positive signal was consistent with phagocytosis induced by necrosis (Figure 3).
Table 1.
Proteins and corresponding antibodies used for immunostaining
| Protein | Antibody reference | Dilution | Laboratory |
|---|---|---|---|
| Laminin | Rabbit polyclonal antibody, L9393 | 1:100 | Sigma-Aldrich |
| Mouse monoclonal antibody, D5 | 1:50 | Developmental Studies Hybridoma Bank | |
| Alpha tubulin | Mouse monoclonal antibody GTX628802, clone GT114 | 1:200 | Gentex Corporation |
| Beta tubulin | Mouse monoclonal antibody MMS-435P, clone TU51 | 1:500 | BioLegend |
| Desmin | Mouse monoclonal antibody M0760, clone D33 | 1:200 | Dako |
| Spectrin | Mouse monoclonal antibody NCL-SPEC1 | 1:100 | Novocastra Laboratories |
| LAMP | Mouse monoclonal antibody ab25082 | 1:400 | Abcam |
| LC3 | Rabbit polyclonal antibody L7543 | 1:100 | Sigma-Aldrich |
Figure 3.
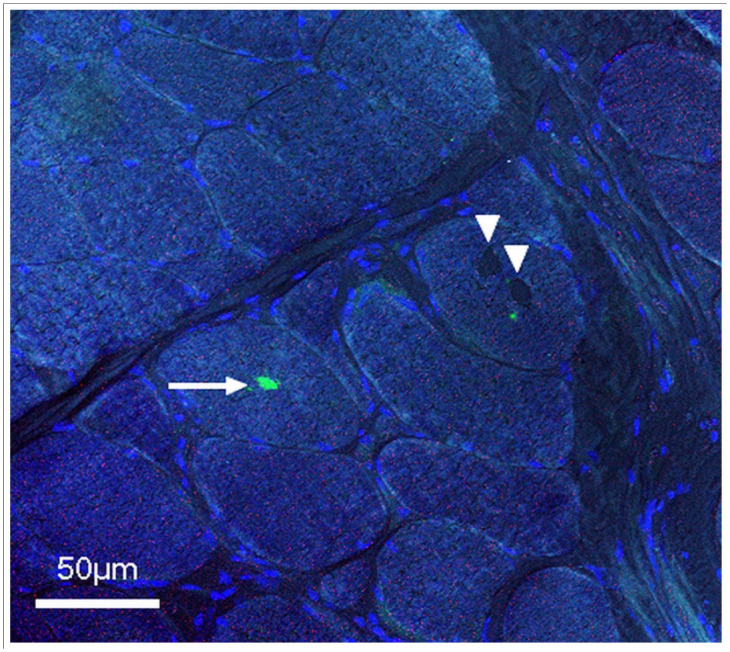
LC3 immunostainings of a transverse section of a fresh-frozen muscle biopsy from the left biceps muscle (× 400). Inclusions (arrowheads) are distinguishable but are not marked by LC3 antibodies. A macrophage located inside the fibre (arrow) is marked by LC3, suggesting macrophage phagocytosis secondary to fibre necrosis. Nuclei counterstained in blue and phase contrast to depict fibre limits
Two days postoperatively, the cat was discharged on prednisolone (0.5 mg/kg PO q12h for 21 days and 50% tapering doses every 7 days). Corticosteroid therapy was continued owing to a perceived improvement in clinical signs associated with treatment. Re-examinations were performed at 1, 3, 6, 9 and 30 months postoperatively. The owners reported some improvement in clinical signs over this time period; however, intermittent periods of collapse, exercise intolerance, polypnoea and prolonged recovery from exercise were still present. During all re-examinations between 1 and 30 months, the cat was exercise intolerant but was no longer recumbent. The creatine kinase blood level was 113 U/l and 688 U/l at 3 and 6 months, respectively. At the time of writing (>2 years after initial diagnosis), the cat had stable disease without any ongoing treatment.
Discussion
In this case, muscular weakness prompted a muscle biopsy, which subsequently revealed a myopathy with inclusions. Muscular weakness remained after discharge, but the cat had stable disease for at least 4 years post-diagnosis.
Slowly progressive to static evolution is commonly reported in human congenital myopathies, 11 and has been described in feline nemaline rod congenital myopathy. 6 Comparatively, the life expectancy of cats with feline dystrophies is generally less than 3 years.4,12,13 Clinical signs were widely dominated by muscular weakness. Muscular weakness is the most commonly observed clinical sign in both human and feline muscular disorders.3,5,7,9,11–13,15,16 In this study, we also observed abnormal gait, episodes of collapse, dysphonia/aphonia and difficulty in swallowing, which have been reported in other feline myopathies.6,7,9,15 However, we did not observe stridor, tremor or cervical ventroflexion, which have also been reported.6,9,15 Some signs more suggestive of congenital human myopathies were also observed in our study, such as hyporeflexia, hypotonia and absence of mental alteration. 11 Creatine kinase was only mildly increased in our study. This parameter is generally increased in other feline muscular disorders, such as muscular dystrophy, 4 toxoplasmosis, 1 necrotising myopathy 5 or nemaline rods myopathy. 6 Clinical evaluation is currently the best way to assess the severity of the disease as there are no blood parameters that accurately reflect the severity of the muscular lesions.5,13,15,17,18 Except for hypokalaemic myopathy, 19 no specific treatment is available for myopathic cats, and only supportive care can be given. Corticosteroids can improve some inflammatory or necrotising myopathies in both cats and in humans.5,20
A fresh biceps femoris biopsy was obtained, which is a good option for feline muscle examination. 21 Histopathological changes were dominated by atrophy of both type 1 and 2 fibres and mild necrosis. These findings excluded feline dystrophic disorders, which are dominated by replacement of muscle fibres by adipose and connective tissue.8,10,12,13 Significant centronucleation (>3%) was observed in our study, but it was less severe than would usually be identified in human or canine centronuclear myopathies;11,22,23 these changes suggested regeneration secondary to mild necrosis. The most specific morphological alterations observed were 10 μm intra-sarcoplasmic oval-shaped inclusions, which were amphophilic in HES and navy blue with Gomori trichrome stain. The other muscular disorders that can show fibre inclusion in humans were not consistent with the epidemioclinical findings of our study.24–26 Infectious causes and neoplastic changes were not identified. Inclusions had a different morphology than feline nemaline rods6,9 or alpha glucosidase deficiency, 7 and affected both fibre types, contrary to that seen in tubulin-inclusion myopathy, 15 which has not previously been reported in either humans or cats (Table 2).
Table 2.
Comparative pathology of different feline myopathies reporting sarcoplasmic inclusions
| Myopathy | Nemaline rods myopathy | Glycogen storage IV | Tubulin reactive inclusions | Oval amphophilic inclusion bodies |
|---|---|---|---|---|
| Authors’ description | Cooper et al, 6 Kube et al 9 | Fyfe 7 | Shelton et al 15 | The present study |
| Underlying effects | Possible surplus of Z-band material | Glycogen branching enzyme deficiency | Unknown | Unknown |
| Inheritance | Possibly autosomal recessive | Autosomal recessive | Unknown | Unknown |
| Epidemiology | DSH (USA, Belgium) | Norwegian Forest Cat (USA, Europe) | Devon Rex, DSH (USA) | DSH (France) |
| Onset | 6–18 months | <5 months | <8 months | 16 months |
| Age of death | <2 years | 9–13.5 months | >4 years | >20 months |
| Clinical signs | ||||
| Abnormal gait | Wobbly hypermetric | Bunny hopping | Loss of balance and collapse | Loss of balance and collapse |
| Muscular weakness | Exercise intolerance Recumbency after walking (Cooper et al6) |
Prolonged decubitus | Mild cervical ventroflexion Prolonged decubitus |
Muscular weakness Prolonged decubitus |
| Muscle atrophy | Progressive | Progressive | Mild to moderate | – |
| Difficulties in feeding | + (Kube et al9) | + | + | + |
| Tremor | + | + | – | – |
| Myotatic reflexes | Decreased | Decreased | Normal | Decreased to normal |
| Comorbidities | Weak voice (Cooper et al6) | Stillbirth Cardiomyopathy Persistent hyperthermia |
Upper respiratory infection | Intermittent aphonia |
| Histological lesions | ||||
| Atrophy | + | + | – | + |
| Necrosis | + (Cooper et al6) | + | – | + |
| Fibrosis | + (Cooper et al6) | + (myocardium) | – | – |
| Elevated centronucleation | + (Cooper et al6) | + | – | + |
| Fibre type mostly affected | 2A | Unspecified | 2B | Both equal |
| Inclusion morphology | Rod-like: very few to filling the whole fibre H&E: not obvious Gomori trichrome: red (Cooper et al6) to blue (Kube et al9) |
Numerous clusters of round shapes H&E: pale blue PS: red purple |
Oval: mostly 1 to a few in a fibre H&E: eosinophilic Gomori trichrome: bright red PAS: positive around the inclusions NADH/SDH: dark blue rim around the inclusions |
Oval: 1 to a few in a fibre H&E: amphophilic Gomori trichrome: purple NADH: unstained halo around inclusions |
| Immunofluorescences abnormalities | Sarcoplasmic accumulation of dystrophin, desmin and spectrin Diffuse alpha-actinin distribution |
Not realised | Tubulin reactive inclusion Normal distribution of desmin, amyloid, ubiquitin and MLF1 |
Normal distribution of desmin, tubulin, spectrin, laminin, LAMP and LC3 Positive LC3 activated macrophages |
DSH = domestic shorthair; H&E = haematoxylin and eosin; PAS = periodic acid–Schiff; NADH = nicotinamide adenine dinucleotide dehydrogenase; SDH = succinic deshydrogenase; MLF1 = succinic deshydrogenase; + = present; – = absent
In humans, biomolecular and genetic testing provides a more precise classification of hereditary muscular disorders, such as dystrophies, congenital myopathies, myotonic dystrophies, myasthenic syndrome and metabolic myopathies. 11
In humans, sarcoplasmic inclusions associated with congenital myopathies (such as rods or cores) are differentiated from other inclusions using immunostaining. 11 Several antibodies used in the diagnosis of human myopathies are available and have been found to cross-react with feline muscle proteins. 27 In this study, antibodies previously tested in feline dystrophies or myopathies, such as desmin, tubulin, spectrin and laminin were used.9,12,13,15 Sarcoplasmic desmin accumulation and diffuse alpha-actinin distribution were not observed in our case, whereas they are associated with feline and human nemaline rod myopathy.9,11 In feline tubulin and human core myopathies, inclusions are highlighted by tubulin and desmin, respectively.11,15 In our study, distribution of these proteins was similar to the control. LAMP and LC3, two autophagy-related proteins that have been highlighted in some human myopathies, were also used.28,29 In our study, macrophages were positive for LC3, but no inclusions were highlighted by this antibody.
Genetic diagnosis is available for three feline muscular hereditary disorders: COLQ for Devon Rex and Sphynx congenital myasthenic syndrome; CLCN1 for myotonia; and WNK4 for periodic hypokalaemic polymyopathy.19,30,31 Feline myopathy classification is currently lacking, owing to a lack of tests to identify mutations.
Limitations of this case report include the absence of identification of the biochemical nature of the inclusion. Electron microscopy has been previously used to characterise nemaline rods and glycogen storage myopathies,6,7,9 and would have been useful here to determine the nature of the inclusions. The current lack of specific immunostaining described in the literature to characterise feline muscular disorders has also limited the exploration of this myopathy.9,12,13,15 An additional limitation was the absence of electromyography, nerve biopsy or MRI, which may have allowed us to exclude a neuropathic disorder with more certainty.
Conclusions
This study describes a slowly evolving muscular disorder in the cat, characterised histopathologically by atrophy and sarcoplasmic inclusions. Given the ‘one medicine’ approach, better characterisation of feline muscular disorders using molecular biology and mutation identification must be encouraged in the future.
Footnotes
Accepted: 2 February 2022
Conflict of interest: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical approval: The work described in this manuscript involved the use of non-experimental (owned or unowned) animals. Established internationally recognised high standards (‘best practice’) of veterinary clinical care for the individual patient were always followed and/or this work involved the use of cadavers. Ethical approval from a committee was therefore not specifically required for publication in JFMS Open Reports. Although not required, where ethical approval was still obtained, it is stated in the manuscript.
Informed consent: Informed consent (verbal or written) was obtained from the owner or legal custodian of all animal(s) described in this work (experimental or non-experimental animals, including cadavers) for all procedure(s) undertaken (prospective or retrospective studies). For any animals or people individually identifiable within this publication, informed verbal consent for their use in the publication was obtained from the people involved.
ORCID iD: Eliot Gougeon  https://orcid.org/0000-0003-3876-4818
https://orcid.org/0000-0003-3876-4818
References
- 1. Butts DR, Langley-Hobbs SJ. Lameness, generalised myopathy and myalgia in an adult cat with toxoplasmosis. JFMS Open Rep 2020; 6. DOI: 10.1177/2055116920909668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cavana P, Sammartano F, Capucchio MT, et al. Peripheral neuropathy in a cat with renal lymphoma. J Feline Med Surg 2009; 11: 869–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mignan T, Targett M, Lowrie M. Classification of myasthenia gravis and congenital myasthenic syndromes in dogs and cats. J Vet Intern Med 2020; 34: 1707–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carpenter JL, Hoffman EP, Romanul FC, et al. Feline muscular dystrophy with dystrophin deficiency. Am J Pathol 1989; 135: 909–919. [PMC free article] [PubMed] [Google Scholar]
- 5. Chow J, Lam A, Shelton D. Progressive increase in creatine kinase activity in an anorexic cat with necrotizing myopathy. JFMS Open Rep 2021; 7. DOI: 10.1177/20551169211031790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cooper BJ, De Lahunta A, Gallagher EA, et al. Nemaline myopathy of cats. Muscle Nerve 1986; 9: 618–625. [DOI] [PubMed] [Google Scholar]
- 7. Fyfe JC. Glycogen storage disease in cats. J Am Vet Med Assoc 1995; 206: 286. [PubMed] [Google Scholar]
- 8. Gaschen F, Burgunder JM. Changes of skeletal muscle in young dystrophin-deficient cats: a morphological and morphometric study. Acta Neuropathol 2001; 101: 591–600. [DOI] [PubMed] [Google Scholar]
- 9. Kube SA, Vernau KM, LeCouteur RA, et al. CM with abundant nemaline rods in a cat. Neuromuscul Disord 2006; 16: 188–191. [DOI] [PubMed] [Google Scholar]
- 10. Martin PT, Shelton GD, Dickinson PJ, et al. Muscular dystrophy associated with alpha-dystroglycan deficiency in Sphynx and Devon Rex cats. Neuromuscul Disord 2008; 18: 942–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. North KN, Wang CH, Clarke N, et al. Approach to the diagnosis of congenital myopathies. Neuromuscul Disord 2014; 24: 97–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. O’Brien DP, Johnson GC, Liu LA, et al. Laminin alpha 2 (merosin)-deficient muscular dystrophy and demyelinating neuropathy in two cats. J Neurol Sci 2001; 189: 37–43. [DOI] [PubMed] [Google Scholar]
- 13. Poncelet L, Résibois A, Engvall E, et al. Laminin alpha2 deficiency-associated muscular dystrophy in a Maine Coon cat. J Small Anim Pract 2003; 44: 550–552. [DOI] [PubMed] [Google Scholar]
- 14. Dubowitz V, Sewry CA. Muscle biopsy: a practical approach. 3rd ed. Philadelphia, PA: Saunders, 2006. [Google Scholar]
- 15. Shelton GD, Sturges BK, Lyons LA, et al. Myopathy with tubulin-reactive inclusions in two cats. Acta Neuropathol 2007; 114: 537–542. [DOI] [PubMed] [Google Scholar]
- 16. Toll J, Cooper B, Altschul M. Congenital myotonia in 2 domestic cats. J Vet Intern Med 1998; 12: 116–119. [DOI] [PubMed] [Google Scholar]
- 17. Aroch I, Keidar I, Himelstein A, et al. Diagnostic and prognostic value of serum creatine-kinase activity in ill cats: a retrospective study of 601 cases. J Feline Med Surg 2010; 12: 466–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Valentine BA, Kornegay JN, Cooper BJ. Clinical electromyographic studies of canine X-linked muscular dystrophy. Am J Vet Res 1989; 50: 2145–2147. [PubMed] [Google Scholar]
- 19. Malik R, Musca FJ, Gunew MN, et al. Periodic hypokalaemicpolymyopathy in Burmese and closely related cats: a review including the latest genetic data. J Feline Med Surg 2015; 17: 417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pipitone N, Salvarani C. Treatment of inflammatory myopathies. Expert Rev Clin Immunol 2018; 14: 607–621. [DOI] [PubMed] [Google Scholar]
- 21. Braund KG. Skeletal muscle biopsy. Semin Vet Med Surg Small Anim 1989; 4: 108–115. [PubMed] [Google Scholar]
- 22. Maurer M, Mary J, Guillaud L, et al. Centronuclear myopathy in Labrador Retrievers: a recent founder mutation in the PTPLA gene has rapidly disseminated worldwide. PLoS One 2012; 7: e46408. DOI: 10.1371/journal.pone.0046408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maxie M, Jubb K, Kennedy P, et al. Jubb, Kennedy, and Palmer’s pathology of domestic animals. 5th ed, vol 1. Edinburgh: Elsevier Saunders, 2007. [Google Scholar]
- 24. Kovarsky J, Schochet SS, Jr, McCormick WF. The significance of target fibers: a clinicopathologic review of 100 patients with neurogenic atrophy. Am J Clin Pathol 1973; 59: 790–797. [DOI] [PubMed] [Google Scholar]
- 25. Lambrianides S, Kinnis E, Cleanthous M, et al. A novel case of inclusion body myositis and myasthenia gravis. Neuromuscul Disord 2019; 29: 771–775. [DOI] [PubMed] [Google Scholar]
- 26. Yoshimura K, Morihata H, Takeda K, et al. A case of hyperkalemic periodic paralysis presenting progressive myopathy with tubular aggregates [in Japanese]. Rinsho Shinkeigaku 2018; 58: 663–667. [DOI] [PubMed] [Google Scholar]
- 27. Gaschen F, Jaggy A, Jones B. Congenital diseases of feline muscle and neuromuscular junction. J Feline Med Surg 2004; 6: 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hiniker A, Daniels BH, Lee HS, et al. Comparative utility of LC3, p62 and TDP-43 immunohistochemistry in differentiation of inclusion body myositis from polymyositis and related inflammatory myopathies. Acta Neuropathol Commun 2013; 1: 29. DOI: 10.1186/2051-5960-1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Manso AM, Hashem SI, Nelson BC, et al. Systemic AAV9. LAMP2B injection reverses metabolic and physiologic multiorgan dysfunction in a murine model of Danon disease. Sci Transl Med 2020; 12. DOI: 10.1126/scitranslmed. aax1744. [DOI] [PubMed] [Google Scholar]
- 30. Gandolfi B, Grahn RA, Creighton EK, et al. COLQ variant associated with Devon Rex and Sphynx feline hereditary myopathy. Anim Genet 2015; 46: 711–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gandolfi B, Daniel RJ, O’Brien DP, et al. A novel mutation in CLCN1 associated with feline myotonia congenita. PLoS One 2014; 9: e109926. DOI: 10.1371/journal.pone.0109926. [DOI] [PMC free article] [PubMed] [Google Scholar]