

Abstract
Atherosclerosis, a chronic inflammatory disease of vascular wall, is a progressive pathophysiological process with lipids oxidation/depositing initiation and innate/adaptive immune responses. The coordination of multi systems covering oxidative stress, dysfunctional endothelium, diseased lipid uptake, cell apoptosis, thrombotic and pro-inflammatory responding as well as switched SMCs contributes to plaque growth. In this circumstance, inevitably, targeting these processes is considered to be effective for treating atherosclerosis. Arriving, retention and working of payload candidates mediated by targets in lesion direct ultimate therapeutic outcomes. Accumulating a series of scientific studies and clinical practice in the past decades, lesion homing delivery strategies including stent/balloon/nanoparticle-based transportation worked as the potent promotor to ensure a therapeutic effect. The objective of this review is to achieve a very brief summary about the effective therapeutic methods cooperating specifical targets and positioning-delivery strategies in atherosclerosis for better outcomes.
Keywords: Atherosclerosis, targets, site-homing delivery, stent, balloon, nanoparticle
Introduction
Cardiovascular diseases (CVDs) are increasing prevalence in the worldwide with an estimated 17.9 million deaths representing 32% of all world’s deaths in 2019 (data from World Health Organization), and atherosclerosis is the manifest contributor to CVDs, marked by these major clinical entities including ischemic heart disease (IHD), ischemic stroke, and peripheral arterial disease (PAD). According to the Global Burden of Disease, of the total deaths, 16.2% and 11.6% were due to IHD and stroke respectively. Under this circumstance, vast researches have got started to resolve atherosclerotic conditions.
Atherosclerosis, chronic inflammation of the vessel wall, is a progressive pathophysiological process, characterized by lipids depositing initiation and innate/adaptive immune responses. Responding to disturbed flow, phenotypic transformation of endothelial cells from rest phenotype into proatherogenic phenotype, which commonly is described as the starting point of atherosclerosis and introduces excessive oxidative system activation, prothrombotic effects, foam cells formation, inflammatory releasing and sensitized SMCs, as shown in Figure 1. Extensive studies have proved that targeting these pro-atherosclerotic processes can effectively postpone the growth of plaque and even make it regression. In the following description, therapeutic targets in the progression of atherosclerosis will be introduced.
Figure 1.

The main progresses of atheromatous plaques.
After initiating atherogenesis by disturbed flow, most arterial wall cells (covering ECs, SMCs, and macrophages expressing NOX) participate in producing oxidants, like ROS. Lipoproteins highly accumulate in tunica intima in two main pathways, including increased permeability for lipoproteins and rising lipoprotein affinity receptors, 1 such as LDL receptors for LDL uptake and LOX-1 corresponding to the passing of oxLDL. Meanwhile, dysfunctional endothelium up-regulates adhesion molecules (P-selectin, ICAM-1, and ICAM-1) and chemokines MCP-1(CCL2), 2 CCL5 and CX3CL1 for highly attracting circulating monocytes into the vascular media. 3 Besides MCP-1, oxidized LDL itself is a direct chemoattractant for monocytes. 4 Subsequently, infiltrating monocytes differentiate into inflammatory macrophages (M1 macrophages) and MCSF released by injured EC drives this transformation. 4 M1 macrophages display increased scavenge receptors (SR-A1, CD36, and LOX-1) on cytomembrane for taking in oxLDL, unregulated enzymes in the cell (ACAT1 turning oxLDL into cholesterol esters, hydrolase, and lipase for disassemble cholesterol esters to fatty acids and free cholesterol), downregulated inversus cholesterol transporter (ABCA1, ABCG, and SR-BI), which makes a dent in cholesterol efflux, enhancement of lipids afflux and accumulation of oxLDL, cholesterol and cholesterol esters, giving rise to foam cells formation. Furthermore, proinflammatory macrophages and foam cells secrete signaling molecules including inflammatory factors, ROS as well as growth factors (PDGF), stimulating SMCs migration and proliferation, accelerating the growth of plaque in arterial walls. SMCs imbibe oxLDL by elevated LOX-1 on the surface and change into lipid-laden foam cells. 5 Platelets answering the imperfect endothelium and succedent thrombosis are associated with the all stages of atherosclerotic process. Thrombosis-promoting molecules exposed from ECM in damaged endothelial system interplay with platelets, and trigger them. Accordingly, activated platelets generate active signals covering CD40L 6 and CCL5, CXCL4(PF-4), CXCL12(SDF-1) and CXCL8(IL-8), etc. to prompt atheroma development by exacerbating inflammatory reaction. 7 Importantly, attracting other platelets and immune/inflammation cells (monocytes, macrophages, and B cells) aggravates inflammatory processes and thrombi by expressing signaling molecules CD40L.6,8 And frequently, CD40L also triggers endothelial inflammation via CD40 on ECs. 8
Generally, atherosclerotic disease has a blocked vessel lumen. As known, stenosis can be divided into several grades, namely less than 50%, 50%, 70%, and exceeding 70%. 9 In angiography-guided therapies, the patients with more than 70% diameter luminal narrowing would accept interventional treatment. While, vessel lumen with 50%–70% narrowing would be treated with intervention or only medical therapy, which decided by operator’s assessment. 10 According to the collection of scientific studies and clinical practices/trials, systemic administrations evoked by nanoparticles and interventional strategies induced by stent and balloon have positive impacts on treating atherosclerotic plaque. Worked as vehicles for drugs, nanoparticles encapsulating therapeutic agents with improved blood half-life selectively get to lesional sites and release agents for treating, and tethering of locating moieties to particles can give this system better performance of active targeting. 11 Stent opens blocked blood vessel and maintains its luminal structure, but results in restenosis. Seeing that balloon also makes blood vessel unobstructed, balloon, especially drug eluting balloon, is mainly applied to solve in-stent restenosis and small artery disease with de nove lesion.12,13 The safety and effectiveness of balloon angioplasty and stenting procedures have been approved beyond doubt, although there are postoperative complications and limitations, seen in the later parts. Noteworthily, some efforts to ameliorate these minus factors, primarily adding payloads, proved to be of avail. In this review, nanoparticles, stents, and balloons were described as site-specific drug delivery platforms, shown as Figure 2.
Figure 2.

The stenosis severity-guided delivery strategies.
Taken together both pathological circumstance and positioning-delivery strategies, the objective of this review is to achieve a very brief summary about the tried and true therapeutic methods connecting specifical targets and positioning-delivery strategies in atherosclerosis, mainly aiming at treatment targets in program of inflammation, lipid metabolism, coagulation, apoptosis as well as lesion-populating cells, and local-fixed transportation formulations including stent, balloon, and nanoparticles.
Focusing on several pivotal cells
Inflamed endothelial cells
Considering activated endothelium in atheromatous plaque, available active targets may be centered on expressed adhesion molecules, receptors as well as impaired function, such as P selectins, ICAM 1, VCAM 1, αVβ3 integrin, and LOX-1 major receptors for LDL uptake by ECs, 14 biosynthesis of NO, MCP-1 secretion. Overexpressed P and E selectins, ICAM 1, VCAM 1 induced by inflammatory stimuli, mediating leukocytes adhering to lesional region, are the hallmark of atherosclerosis. Sterilizing these adhesive molecules plays a positive role in prevention of plaque progression. Sager et al. 15 proposed that small interfering RNA (siRNA) targeting P and E selectins, ICAM1, ICAM2, and VCAM1 would combat leukocyte recruitment into plaque and lesional inflammation. Alicaforsen, an antisense phosphorothioate oligonucleotide, another antagonist of ICAM-1, selectively inactivates ICAM-1 mRNA coinciding with lessened membrane-bound protein ICAM-1. 16 Meanwhile, active atherosclerotic plaques excessively expressed P-selectin embodying bioactivity of plaques and getting prominent during thrombus formation. 17 Powerful blocking-up in P-selectin actions inhibited the inflammatory and thrombotic events, which incidentally decreased neointimal hyperplasia after balloon injury. 18 Seeing that high affinity and specificity of fucoidan for P selectins exceeding PSGL-1, stronger fucoidan-P selectins interaction blocking P selectins activities may abolish selectin-dependent recruitment of leukocytes.19,20 The main ligands of P-selectin on sensitized ECs and platelets incorporate innate PSGL-1 on the membrane of leukocytes and other recognition effector Sialyl Lewis X, synthetic Sialyl Lewis × mimics, inclacumab, sulfated oligosaccharides as well as polysaccharides (such as fucoidan, heparin and dextran sulfate), reacting with P-selectin.19,20 Beyond anticoagulation, heparin is also seen as a direct modulator of adhesion mediated by P-selectin. Sevuparin, a heparin-derived polysaccharide, keeps potent anti-P-selectin activities (anti-adhesive feature) without the anticoagulation properties. 21 CX3CL1/CX3CR1 process also exerts a possible therapeutic target, since CX3CL1 on inflamed ECs mediates monocytes recruitment onto the pathological vessel wall via CX3CR1 of monocytes and this interaction of CX3CL1 and its receptor also stimulates SMC migration, platelet activation and neo-angiogenesis. 22 Disruption of CX3CL1/CX3CR1 interaction with CX3CL1-Fc prevented monocyte-endothelial cell reacting and reduced atherosclerosis formation. 23 More than targeted therapies, growing proof validated that adhesion molecules could serve as location targets. Peptide ligands decoration for binding to ICAM-1, such as fibrinogen-derived peptide (NNQKIVNLKEKVAQLEA) and the sequence VHPKQHR, yielded a specific and high-affinity system directing to inflamed endothelial surface in atherosclerotic lesions.24,25 For orientating atheroma, superfluous VCAM-1 can straight tether ligand-modified objects to lesional site, and these ligands included specific antibody or some peptides, like anti-VCAM-1 antibody, anti-VCAM-1 nanobody, and VHSPNKK.26–28 The ligand with VHSPNKK sequence also blocked leukocyte-endothelium interactions. 28 Activated or growing cells under pathological conditions (such as differentiated macrophages and angiogenic endothelial cells) reveals high density αVβ3 integrin, but the counterpart is minimal in quiescent cells of normal vascular tissues, 29 and RGD or RGD derivatives could achieve ligation to αVβ3-positive cells in atherosclerosis. 30 Beyond expressing adhesion molecules for capturing monocytes, TGF-β stimulation through TGFβR1/2 in endothelium drives inflammatory phenotype of EC, which enhances atherogenesis. 31 Inhibition of endothelial TGF-β-TGFβRs signaling might be effective in arresting progression of building plaque by reducing vascular inflammation. 32
Regulating phenotype-switched SMCs
In atherosclerosis, SMC proliferation is held responsible for plaque growth. And, in the past decades, the phenotypic heterogeneity theory of SMCs, also called as phenotypic switching from a contractile/quiescent phenotype supporting the arterial structure toward a synthetic one with increased migration, replication, and protein synthesis activities, has been expounded partly. Conventionally, this process is believed to have a bearing on atherosclerosis. Dapperly, VSMC plasticity also contains the transformed phenotypes of resembling foam cells, macrophage-like cells, and MSC-like cells. 33
Theoretically, reversing or inhibiting those pro-transformation programs (as seen in Figure 3) would hold back phenotypic changes and put switched SMCs into reverse. In one example, Vengrenyuk and his team 34 validated that maintaining the producing of myocardin could conduce to contractile phenotypic SMCs after cholesterol loading. As a key enhancer in regulating phenotypic transition of SMCs, KLF4 bears on atherosclerotic plaque pathogenesis. Knocking down KLF4 in SMCs specifically cuts down lesion size, while heightens plaque stability. 35 Furthermore, alternative targets miR-143 and miR-145 accelerate myocardin expression for keeping SMCs in a contractile state and also affect a network of transcription factors relating to Klf4, myocardin, and Elk-1, which generate repressed proliferation of SMCs.34,36,37 And, the inhibiting effect of miR-145 for SMC modulating is also due to regulating the L-type calcium channel expression partly. 38 Intriguingly, normal SMCs with a quiescent, differentiated state express calcium ion channels. In parallel with cell proliferation, L-type calcium channel (LTCC) will die away and the recurring of LTCCα1C (a LTCC subunit) is followed by reappearance of contractile phenotype markers in an earlier investigation. 39 In this study, the authors stated that activated RhoA, ERK1/2, and p38 MAPK pathways inspired inhibited LTCCα1C production under PDGF stimulation and fluvastatin upregulated LTCCα1C expression via inactivating those pathways for retaining a more differentiated VSMC phenotype. Indeed, it had been corroborated that increased miR-133 could coax modulated SMCs back to quiescence for regulating VSMC growth via suppressing the expression of transcription factor Sp-1. 40 Furthermore, KLF5 also takes an important part in regulating SMC phenotype and inhibition of this factor (e.g. by synthetic retinoid Am80) could achieve suppressed smooth muscle phenotypic modulation. 41 Dramatically, STAT3 (activator of transcription 3) protein also contributes to SMC phenotypic switch by interacting with myocardin. 42 Naturally, PERK-STAT3-MRTFA signaling axis could serve as a target. 43 A PERK inhibitor GSK2606414 obstructed STAT3 while triggering SRF by dampening down PERK activity in smooth muscle cells, which suppressed SMC’s phenotypic change. 43 In another case, crocin effectively prevented VSMCs proliferation and phenotypic switch induced by PDGF-BB through STAT3 pathway. 44 In SMC phenotype switching, abrogating of glycolytic enzyme PKM2 (pyruvate kinase muscle 2) obtained inhibiting effects of SMC proliferation, migration, phenotypic switching and neointimal hyperplasia, accompanied with decreased ERK (extracellular signal-regulated kinase), mTOR (mammalian target of rapamycin), and STAT3 signaling. 45 To recapitulate briefly, any signal molecule for promoted activation of contractile differentiation and inhibited synthetic/dedifferentiated pathway, including SRF, myocardin and myocardin related transcription factor (MRTFs), or SRF/CArG-box complex and KLF4, might work as a mediator in modulated SMCs. 46
Figure 3.

The main external stimulus-mediated phenotypic modulation of SMCs.
Diverse circumstance stimuli (e.g. platelet-derived growth factor-BB/DD, oxidized phospholipids, cholesterol, and inflammatory factors-TNFα and IL-1β) could evoke transition of SMCs phenotype toward synthetic type with loss of contractile markers involving SMα-actin, SM22 α, SMMHC, and others, mainly via KLF4 and MEK-ERK1/2-Elk-1 pathway.34,47–51 For details, in MEK-ERK1/2-Elk-1 pathway, phosphorylated Elk-1 replaced myocardin from SRF, which reduced differentiation marker genes. 48 And, the mechanisms of KLF4 mediated SMC phenotypic modulation have been described as several types, including KLF4 direct integrating to SMC marker gene promoters; blocking SRF/CArG-box binding; lessoning myocardin production and HDACs recruiting. 49
Apoptotic cells and efferocytosis of MAs in atherosclerosis
Regulating apoptosis procedures of plaque-residing cells
Rupture-prone plaque is typically actuated by cell death (may be a marker of plaque instability), fundamentally macrophages (in necrotic core) and smooth muscle cells (in fibrous cap) apoptosis germinating necrotic core change in size and fibrous cap thinning respectively. 52 Apoptosis accompanies the whole process of atherosclerosis. As a momentous feature of atherosclerosis, decided by cell type and plaque stage, apoptosis of cell exerts profitable and deleterious effects. Importantly, foam cell apoptosis contributes to the formation of the acellular lipid core and endothelial apoptosis is directly seen at post-stenotic area with low shear stress. 53 EC apoptosis is a contributor of initiating plaque development, involving in EC dysfunction, increased endothelium permeability, thrombosis and instable plaque. 54 Depending on cell types, the stimulus for apoptosis (different dead pathway) as well as cell at different pathological stages, apoptosis regulation needs to be considered in detail. The brief apoptotic pathways of several pivotal cells are shown in Figure 4. In the early stages of atherosclerosis, increased macrophage apoptosis diminished lesion cellularity and decreased lesion progression, 55 but in advanced plaque, decreased macrophage apoptosis reduced necrotic core formation and lesion size, promoted plaque stability. 56 Generally, induction of VSMC apoptosis could be beneficial for lessening cellular accumulation and following stenosis. However, after vessel injury including after angioplasty and stenting, protection against apoptosis aids to reduce neointima formation. 57 In the whole, conceived therapeutic strategies may selectively facilitate apoptosis of macrophages and SMCs in early lesional vessel and preclude death of macrophages at late stage, SMC after injury and EC in all expanding of plaque.
Figure 4.

Apoptosis and efferocytosis of vascular wall cells (endothelial cells, smooth muscle cells and macrophages) in atherosclerosis.
Centering on MicroRNAs (miRNAs) has found that miRNAs affect apoptosis of vascular cells conducing to the pathogenesis of atherosclerosis. Impediment of EC apoptosis has attracted for designing novel means against atherosclerosis, and the following all may be potential therapeutic targets for atherosclerosis: miR-210 upregulation repressed PDK1 favoring endothelial apoptosis 58 ; miR-26a with anti-apoptotic effect downregulating TRPC3 or TRPC6 overexpression alleviating the development of atherosclerosis59,60; MicroRNA-122 promoting endothelial cell apoptosis by targeted XIAP inhibition 61 ; MiR-365 potentiating ox-LDL-induced ECs apoptosis paralleling damaged Bcl-2 expression 62 ; MiR-429-mediated down regulation of Bcl-2 giving impetus to atherosclerosis-associated endothelial cell apoptosis 63 ; MicroRNA-142-3p also monitoring endothelial cell apoptosis. 64 Absorbed in regulatory roles of micro RNA in apoptosis, Chen et al. 65 proposed that inciting expression of miR-26a by tanshinol could attenuate the endothelial cells apoptosis for endothelial protection and dwindling formation of atherosclerosis. Additionally, Liang et al. 66 unfolded that direct inhibition of p38 via MiR-124 overexpression restrained macrophage apoptosis accompanied by climbing anti-inflammatory cytokines and dropping pro-inflammatory factors. Pointing toward another example, Tian et al. 67 also drew a conclusion that inhibiting Fas/FasL pathway by D4F (apolipoprotein A-I mimetic peptide) prevented macrophages from ox-LDL-induced apoptosis. For breaking TNFα-TNFR-1 signal transmission, Cho’s group 68 subdued TNFR-1 expression by trafficking small interfering RNA to win anti-apoptotic effects of EC with higher ratio of anti-apoptotic factor (Bcl-xL) to pro-apoptotic factor (Bax) as well as apparently stronger HUVEC proliferation and capillary formation caused by angiogenic factors (KDR/Flk-1 and eNOS). Another critical mitogen-activated protein kinase (MAPK) signaling pathway provides pro-apoptotic signals through JNK or p38 activation or pro-survival signals through ERK1/2, ERK5 activation.69–71 ERK5 engaging in PKB/Akt survival pathway protects cell from apoptosis. 72 Raising ERK5 in macrophages (such as by statins) upregulated macrophage efferocytosis halting plaque formation. 73 All in all, given regulating apoptosis, besides suppression of death signal (by anti-oxidants 74 ) and signal transmission, elevating Bcl-2/Bax ratio for decreased cytochrome c excretion, apoptosome formation, sensitizing PI3K-Akt signals with diminished Bax/Bad and caspase 3, or raising IAPs expression also have been regard as beneficial adjustments.
Apoptosis mainly embraces mitochondria dependent apoptosis (Bcl protein family, anti-apoptosis Bcl-2/Bcl-XL/A1, and pro-apoptosis protein Bax/Bad/trBid) and receptor-mediated apoptosis. TNF receptors family (TNFR1, Fas) binding death ligands (TNFα, Fas ligand) sensitize caspase 8 and caspase 3 sequentially, and produced caspase 3 is the onset of apoptosis and sensitive caspase 8 turns Bid into trBid localizing to mitochondria for enhancing mitochondrial dependent apoptosis. Once mitochondria responds to death signals, pro-apoptosis members (Bax/Bad/trBid) are conducive to mitochondrial cytochrome c release, forming apoptosome with APAF-1 and pro-caspase 9 to activate caspase 9 and caspase 3 in order. Bcl-2/Bcl-XL/A1, IAPs (inhibitors of apoptosis proteins) including XIAP (X-chromosome linked IAP), cIAP1, cIAP2, NAIP (neuronal apoptosis inhibitor protein), active PI3K/Akt signal, and some survival factors help cell live. Apoptotic cells display pro-phagocytic signals, especially phosphatidylserine (PS), for clearance of phagocytes. Adequate efferocytosis is imperative to defend against atherosclerosis, producing inflammation-counteracted TGFβ and IL-10. But, impaired efferocytosis (increased cell death, sufficient numbers of phagocytes, and damaged engulfment mechanisms) in atheroma, including overloaded macrophages forming foam cells, oxidized LDL and phospholipids insensitizing phagocytic receptors (SR-BI) or bridging molecules (MFGI8), hampered devouring molecules expression (SR-BI or LRP1), HMGB1 binding αvβ3 and PS, shedding of MERTK and LRP1 as well as TNFα/TNFR1 pathway evoking CD47 (a do not eat me signaling, repulsing phagocytes) boosting, causes defective clearance and conversion of apoptotic into necrotic cells, emerging proatherogenic factors (IL-1β, IL-6, TNFα, HMGB1) and secondary necrosis55,71,75–77.
Enhancing effective efferocytosis
Efferocytosis, referring to scavenging/engulfment of apoptotic cells by phagocytic cells like macrophages, activates anti-inflammatory, and proresolving signaling pathways that are crucial for the resolution of inflammation and effective efferocytosis of apoptotic can suppress inflammation and growth of necrotic core. 78 Efficient efferocytosis by macrophages takes a pivotal part in limiting the progression of atherosclerotic plaque. Three prerequisites depicted in Figure 4, apoptotic cell ligands (mainly PS), efferocytotic receptors (MERTK, LRP1, TG2, SRBI and integrin αvβ5, αvβ3), and bridging molecules (MFGE8, Protein S, Gas6, and complement C1q), collectively determine the clearance of apoptotic cell in vascular lesion.55,71,75 Increased cell death, sufficient number of phagocytes and blocked digestion pathway induced by oxLDL competition inhibition, shielded PS, occupied linker molecules, decreased efferocytotic receptors, along with augmented CD 47-rejection phagocytosis via discerning SIRPα engender impaired efferocytosis 79 in atherosclerosis, playing a major role in extending lesion. Strengthening efficient efferocytosis is a promising and pivotal way to limit the progression and vulnerability of atherosclerotic plaque. During atherogenesis, progressively upregulated CD47 co-localizes to necrotic core, and special blocking-up anti-phagocytic CD47 signaling could restore eliminating of diseased and apoptotic SMCs or macrophages to prevent atherosclerosis.80,81 Targeting CD47-SIRPα axis by miR-378a that depletes SIRPα level could hoist the phagocytic activity of oxLDL-stimulated macrophages. 82 SHP-1, a downstream effector molecule of CD47-SIRPα signaling, suppressed phagocytic function, and PEG-functionalized single-walled carbon nanotubes (SWNTs) with monocyte/macrophage-selectivity loaded by SHP-1 inhibitors (NSC-87877) accumulated within lesional macrophages and renewed lesional phagocytosis, linked to lowered the plaque burden and inflammatory gene levels. 83 TPI-1 (tyrosine phosphatase inhibitor 1, toward SHP-1 inhibition)-carrying SWNTs also could reactivate macrophage efferocytosis. 84 Huang et al. 85 also gave a description that increased SHP-1 expression during carotid atherosclerotic plaque progression possessed a position by macrophage polarization-mediated efferocytosis and deleting SHP-1 function could shift macrophages toward an anti-inflammatory phenotype preferring to promote efferocytosis. Attentionally, CD47 linking ligand TSP1 reinforces ROS release and abates eNOS activation and NO production. Consequently, inhibiting CD47 may ameliorate atheroma, as a consequence of elevated NO levels and decreased oxidative stress. 86 Another instrumental thing may be an increase of “eat me” signals of dying cells and PS functions as a therapeutic target to heighten phagocytosis of apoptotic cells. Schutters et al. 87 employed RGD-introduced annexin A5 interacting with αvβ3/5 on the phagocytes to target expressed phosphatidylserine (PS) for enhanced engulfment of apoptotic cells and IL-10 secretion.
Aiming at critical pathological proceeding
Targeting of lipid metabolism process
Targets in forward direction
The phagocytosis of oxidized low density lipoprotein is implicated in endothelial dysfunction, inflammation, formation of foam cells, migration and proliferation of smooth muscle cells, platelets activation, cell apoptosis, and atherosclerotic plaque instability. 88 Oxidized low density lipoprotein transits through endothelium in connect with LOX-1, and under the pathologic pathway of atherogenesis, undue presence of scavenger receptors on induced-atherogenesis cells (mainly LOX-1, CD36, SR-A1 on macrophages and LOX-1 on SMCs) is a significant cause of foam cell production. Resting platelets constitutively express CD36, mediating OxLDL binding to platelets, and activated platelets exhibit LOX-1. 89 Thus, scavenger receptors appear to be an available target of curing and arriving in plaque sites.
LOX-1, a cell-surface receptor for oxidized LDL (Ox-LDL), is dramatically associated with atherosclerosis. Its expression of human carotid arteries in advanced atherosclerotic plaques has been observed in intimal smooth muscle cells, as well as macrophages, endothelial cells, and active platelets. 90 LOX-1 is also colocalized with the apoptotic cells highly in lesional site. 91 Ishino et al. 92 deemed LOX-1 was expressed in the macrophage-rich lipid core area. And they also took advantage of anti-LOX-1 antibody to direct LOX-1 receptor for imaging of vulnerable plaque, 93 indicating the effectiveness of targeting LOX-1 for plaque homing. Analogously, targeting LOX-1 can realize controlled delivery of therapeutic agents into arterial plaques. Saito et al. 94 found that LOX1-targeted liposome loading fasudil notably prevented intimal hypertrophy and matrix metalloproteinase-9 expression. And anti-LOX1 antibody bound liposomes reached carotid artery lesions with effect.
As a therapeutic target, given that remarkable increase of LOX-1 in the neointima after balloon injury has been reported, the loss-function of LOX-1 has obvious inhibitory effects on intimal hyperplasia, oxidative stress, leukocyte infiltration by anti-LOX-1 antibody administration. 95 Gene silencer also has been recognized as a feasible means to LOX-1 deficiency, and PIP (pyrrole-imidazole polyamide) is an optional molecule for inhibiting the expression of LOX-1, monocyte chemoattractant protein-1, intercellular adhesion molecule-1, matrix metalloproteinase-9, and thickening neointimal. 96 Kaimin et al. 97 designed PIP targeting LOX-1 eluting stents and unmasked that the stent could dwindle the area of neointima and in-stent restenosis (ISR) without impairing re-endothelialization. Fan et al. 98 revealed that casein kinase 2-interacting protein-1 (CKIP-1) minified LOX-1 transcription on account of interplaying with proteasome activator REGγ for promoted degradation of transcriptional factor Oct-1. CKIP-1 harbors a protective role during foam cell formation and atherosclerosis. 99 Attractively, use of scavenger receptor inhibitors (for example, CD36 inhibitors AP5055/AP5258 100 and micellar nanolipoblockers (NLBs) functionalized with anionic carboxylate group for SR-A 101 ) or molecules competing with oxLDLs for binding to SRs (such as long-chain fatty acids 102 ) can prevent scavenger receptors-dependent oxLDL uptake, which is a promising avenue of the treatment and prevention of atherosclerotic development. Considering Ox-LDL metabolic process, well-directedly suppressed ACAT1 expression or promoted transfer-out proteins for cutting down cholesterol gathering in macrophages could limit further foam cell formation. Liraglutide could down-regulate ACAT1 with suppressed macrophage foam cell formation, which interdicts the development of atherosclerotic plaque. 103
Modulating reversing procedure
Possible mechanisms of rapid regression of atheroma plaque relate to efflux of cholesterol from phagocyte, emigration of foam cells out of plaque, influx of healthy phagocytes for remove necrotic debris, and other components of the plaque.104,105 This part would focus on transfer-out of cholesterol. Backward cholesterol transport occupies an important position against atherosclerosis development, and cholesterol out from foam cells has been identified as one powerful therapeutic strategy for meliorating lesion. ABCA 1 is the key reverse cholesterol transporter mediating cholesterol exporting from cells. Chen et al. 106 reported that carvedilol could boost cholesterol out and ABCA1 expression for halting atherosclerosis possibly through NF-κB. A great deal of targets can pose as a primer mover for contributory ABCA1 expression.
Enabled liver X receptor (LXRs, LXRα and LXRβ forms, ligand-activated transcription factors), upon ligands binding, augment target genes expression involved in reverse cholesterol transport (considerable ABCA1 and ABCG1 prompting cholesterol excretion) and mitigate proinflammatory gene expression (TNFα, IL-1β, and CCL2.107,108 Some endogenous (non-enzymatically generated oxysterols-weak or no agonistic activity, and other cholesterol derivatives-24(S)-hydroxycholesterol, FF-MAS, desmosterol) and exogenous (T0901317, GW3965) agonists are widely accepted as LXR ligands.107,108 LXRs signaling can be seen as a potent signal for cholesterol efflux from foam cells, some reports had elaborated this property. Quercetin (one of flavonoids) motivates increasing cholesterol efflux from foam cells derived from ox-LDL-induced macrophages through PPARγ-LXRα (more ABCA1 expression) pathway. 109 PPARγ (peroxisome proliferator-activated receptor-γ) motivating significantly enhances foam/macrophages to expel cholesterol through the expression of ABCA1 and LXRα, and functional ABCA1 expression is necessary for PPARγ-induced cholesterol efflux from macrophages. Ren et al. 110 uncovered that mangiferin promoted cholesterol efflux of acetylated LDL-loaded macrophage via elevated ABCA1 and ABCG1 mediated by the activated PPARγ-LXRα pathway and ameliorated atherogenesis (decreased plaque size). Libby and Plutzky 111 summarized that the activation of PPARγ also minified biomarkers of inflammation (TNF-α, IL-1β), inflammatory associated adhesion molecules, oxLDL-internalized receptors (LOX-1, scavenger receptor A) as well as MMPs expression, which suppressed inflammation and atherosclerosis. Zimmer et al. 112 also authenticated CD (cyclodextrin)-mediated LXR agonism exhibited the anti-atherosclerotic and anti-inflammatory effects with augmented removal of cholesterol and regression of plaque. For permitting cholesterol out from foam cells or macrophages and amending atherosclerosis, Han et al. 113 availed Urolithin A (UA) to promote cholesterol efflux from formed foam cells and attenuate cholesterol accumulation via modulating related microRNA-33a (decreased miR-33a but increased expression ABCA1 and ABCG1) and ERK/AMPK/SREBP1ignaling pathways. More specifically, miR-33a strongly repressed the levels of ABCA1 to dampen cellular cholesterol efflux, inhibition of endogenous miR-33a downwards adjusted ABCA1 expression. 114 Additionally, inhibiting macrophage miR-34a to up-modulate ABCA1 and ABCG1 gives impetus to cholesterol efflux or reverse transport, which gooses atherosclerosis regression. miR-34a inhibition also alters M1 into M2 macrophage polarization via liver X receptor, together with shrinking inflammation (reduced TNF-α, IL-6, and MCP-1). 115 PCSK9 downregulates ABCA1 gene and protein expression for weak cholesterol efflux and PCSK9 could also serve as a powerful therapeutic target. 116 As a regulator of LDLR (LDL receptor) and LDL-C (LDL cholesterol), the peptide-based anti-PCSK9 vaccines could obtain long-term therapeutic effect against atherosclerosis. 117
Inflammation pathway
Classical targets: NLRP3/IL-1β pathway and downstream mediators
Atherosclerosis development is not merely caused by accumulating lipid within the arterial wall, also a chronic inflammatory disease responsible for vascular injury. As a consequence, targeting inflammation itself has stimulated novel approaches to reduce cardiovascular events and risks induced by atherosclerotic walls. IL-1β is a primary form of circulating IL-1 that induces various secondary inflammatory cytokines (including IL-6, TNFα) synthetizing when answering vascular injury and is manufactured in the setting of NLRP3 inflammasome activation. 118 The activation of the NLRP3 inflammasome (more likely driven by cholesterol crystal) drives the initiation and progression of atherosclerosis fundamentally by predominant IL-1β effect originating from macrophages. 119 NLRP3 inflammasome activation is discovered mainly in macrophages/monocytes and foam cells, sporadically in SMCs, ECs, and T cells in plaque site. And NLRP3, ASC and caspase 1 as well as IL-1β and IL-18 increase in arterial wall. 120 A growing elaboration sharpens that, owing to PRRs signaling, ox-LDL, ROS and cholesterol/calcium phosphate crystals phagocytosis (proatherogenic mediators), NLRP3 pathway gets activated, which enables the next IL-1β and IL-18 release, causing vascular inflammation in the progression of atherosclerosis,121–123 as shown in Figure 5. IL-1β acts on cells (abundant SMCs, ECs, and macrophages) in the diseased vessel system, then alters cell functions including inflammatory transform releasing IL-6 and TNF-α, autocrine factors, and self-active state, resulting in endothelial dysfunction, infiltrative monocytes, MAs inflamed state and SMC proliferation (a catastrophic road of atherogenesis).124–126 Taking these together, pointing at NLRP3 inflammasome, IL-1β and downstream inflammatory mediators (IL-6, TNF-α) may obtain positive outcome for impeding atherosclerotic extension. In these regards, there are three directions for working: smothering of IL-1β emerging by inhibiting active NLRP3 inflammasome; incapacitating IL-1β through blocking IL-1 receptors binding or nullifying IL-1β itself; making generative secondary inflammatory factors (IL-6, TNF-α) ineffective for abrogating cells inflammatory response.
Figure 5.

The key pro-atherogenic role of NLRP3 inflammasome and IL-1β in initiation and development in atheroma plaque.
Combating NLRP3 inflammasome activation by thioredoxin-1 significantly mitigates ROS-stimulating NLRP3 generation, IL-1β secretion and gets atherosclerosis stunted, unfolding atheroprotective functions. 127 Some other tactics of indirect or direct depressing NLRP3 inflammasome have been demonstrated to block inflammasome activation for diminished IL-1β with selectivity and advantage, embodying small-molecule inhibitors (MCC950, 128 β-hydroxybutyrate, arglabin) and microRNA(microRNA-223, suppressing NLRP3 protein expression), 129 and more inhibitors also had been summarized by Zahid and their colleagues. 130 The new therapeutic strategies inhibiting NLRP3 inflammasome activation are burgeoning. Peng’s group 131 unveiled 13-methylberberine opposed NLRP3 inflammasome activation for inhibited cell injury induced by H2O2. Melatonin inhibits NLRP3 inflammasome and pyroptosis with diminished caspase1, IL-1βand IL 18 production and suppressive NF-κB activation. 132 Pyroptosis (inflammatory form of cell death), observed in monocytes, macrophages, dendritic cells, VSMCs, vascular endothelial cells, is dependent on caspase-1 and triggered by activated inflammasomes. 133 As a pyroptosis promoter, active enzyme caspase-1 also plays a forceful part in aiming inflammation. Caspase-1 retardant, VX-765, remarkably reduces VSMCs pyroptosis and IL-1β processing in OxLDL circumstance and impedes the growth of atherosclerosis. 134 As a pivotal intermediator in inflammatory responses, with regulating a myriad of pro-inflammatory genes, the transcription factor NF-κB also partakes inflammasome regulation. 135 Growing evidence indicates that interfering with NF-κB signaling could mediate vascular inflammation, and some nuclear factor kappa B inhibitors have been investigated in different cell models, like resveratrol for endothelial cells, lactucopicrin for macrophages and NLS (NF-kB nuclear localization sequence) peptide for SMCs and MAs.136–139
For taking aim at primary and secondary inflammatory products, antagonists or therapeutic agents have got groundbreaking findings for anti-inflammation or atheroprotection by directly targeting TNF-α with etanercept, adalimumab, TNF-specific antibody CDP571, TNF receptor–Fc fusion protein, IL-1β with canakinumab, gevokizumab, LY2189102, IL-6 with tocilizumab, IL-1R with anakinra126,140–142 or indirectly inhibiting the production of these factors, like silencing tumor necrosis factor alpha converting enzyme (TACE, cleaving precursor of TNF-α) expression. 143 IRAK4, recognized as a result of danger signal acting on TLR or IL-1 receptors, is an essential signal transducer downstream of TLR and IL-1 receptors. 144 IRAK4 of macrophages as a target, is directly reacted by FC-99 (benzenediamine derivate), and, as a result, this treatment attenuates proinflammatory mediators (TNF-a, IL-6, MCP-1) production. 145 Extensive inhibitors of IRAK4 have been reviewed and used, such as N-Acyl-2-aminobenzimidazole inhibitors, a diarylamide and an unrelated imidazo[1,2-a] pyridine series of IRAK4 inhibitors and quinazoline based inhibitors.144,146,147 However, targeting any inflammatory mediators cannot completely block all the inflammatory pathways in atherosclerosis.
The cytokine interleukin-1β (IL-1β) rooting in macrophages is a major driver in pathogenesis of atherosclerosis. Priming and activating signals trigger active NLRP3 inflammasome drawing forth IL-1β and IL-18. ROS, disturbed flow, phagocytic calcium phosphate crystals, and cholesterol crystal (CC) or CC from untaken oxLDL position themselves as enable signals of NLRP3 inflammasome. And danger signals recognizing receptors (IL-1βwith IL-1R, NETs/fibrinogen/ CC with TLRs, oxLDL-dependent CD36/TLR4/TLR6 heterotrimer, as well as TNFαengaged TNFR) sever as priming signal to trigger NLRP3 inflammasome activation via NF-κB pathway. Activated NLRP3 inflammasome elicits capase-1 responsible for cleaving pro-IL-1 β and pro-IL-18 for IL-18 and IL-1β generation. For one thing, IL-1β ligating to IL-1R as a priming signal irritates added IL-1β production. For another, IL-1β acts on cells in plaques (including ECs, SMCs, MAs) via IL-1 receptor family, contributing to cells sensitization for enhanced pro-inflammatory factors production (IL-6, IL-1, and TNFα). Graver ECs dysfunction, increased vascular permeability and anabatic expression of MCP-1, TF, and adhesion molecules cause pro-coagulation effects, leukocyte infiltration. Autocrine factor PDGF of SMCs works on SMC proliferation. Of note, these cells biosynthesize ascending matrix metalloproteinase, MMPs 2/9 of ECs for erosion, MMP 3 and MMPs 2/9 of SMCs for remodeling and migration respectively, and MMP 1/8/13 of macrophages for plaque rupture (collagenase).119,148,149 For the enzyme caspase-1 (a promoter of apoptosis) mentioned earlier, it can trigger pyroptosis (the inflammatory cell death), precipitating the development of atherosclerosis. 124
Some momentous regulators for inflammation
PCSK9
PCSK9 (the proprotein convertase subtilisin/kexin type 9), an enzyme, possibly wields its pro-atherogenic power through inherent pro-inflammatory effects, beyond regulation of cholesterol homeostasis.150,151 Uplifted PCSK9 secreted by ECs, SMCs as well as macrophages in plaque regions is provoked by immoderate oxLDL gather, inflammatory milieu, 152 a mass of ROS generation. Synthetic PCSK9 has pleiotropic effects on atherogenesis, covering modulation of inflammatory, engulfing oxLDL, apoptosis/autophagy, and cholesterol efflux, specifically seen in Figure 6. More importantly, PCSK9 directly increases inflammation in atherosclerotic lesion. 153 The deficiency of PCSK9 could reduce atherosclerosis markedly. 154 Targeted treatments against PCSK9 overtly lessens inflammation, endothelial dysfunction, and plaque size.154–156 As an emerging target for treating atherosclerosis, the therapeutic capacity of PCSK9 has been confirmed. Monoclonal antibodies, gene silencing, and mimetic peptide (inspired by EGF-A binding domain of the LDLR, which interacts with PCSK9) are several methods of restraining PCSK9 for exhibiting anti-atherosclerosis effects.156–158 Such as, Tang et al. found that, in terms of PCSK9 quantity, PCSK9 was more clearly observed in the atherosclerotic plaques than normal aortic tissues. Gene interference, specifically blocking PCSK9 expression, weakened inflammatory factors secretion (TNF-α, IL-1β) and plaque area. 155 What’s more, other researchers also exposed inhibition of PCSK9 could inhibit HUVEC apoptosis induced by ox-LDL via Bcl/Bax–caspase9–caspase3 pathway. 159
Figure 6.

The effect of PCSK9 on atherosclerotic progression.116,160,161 Covered by inflammatory conditions, oxLDL and ROS at the plaque site, response cells (ECs, SMCs, and MAs) synthesize PCSK9, elevating SR (LOX-1, SRA, CD36) expression for more oxLDL intake, lowering ABCA1 and ABCG1 with decrease cholesterol outflow. Under this modulation, lipid-laden cells emerge as foam cells. Concomitantly, PCSK9 upregulates adhesion molecules VCAM-1 expression (more monocytes trafficking), and provokes macrophages to liberate inflammatory molecules (TNFα and IL-1β) based on TLR4/NF-κB pathway activation.
Protease-activated receptors (PARs)
Veritably, inflammation and coagulation systems converge at injury and plaque tissue, which can be delineated as proinflammatory factors bring about active coagulation pathway, in return, coagulation considerably contributes to inflammation. 162 Tissue factor (TF, on the membrane or in the plasma) is deemed as a cross-talked linker between inflammation and coagulation system, and in response to inflammatory mediators (TNFα, IL-1β), ROS, LPS or other injurious stimuli, heightened TF expression is witnessed.163,164 TF activates FVII, forming TF/FVIIa complex which signals to generate FXa and thrombin, which fix to PARs for inflammation through PAR-2 and PAR-1, respectively. 164 Thrombin interacts with PAR-1, -3, and -4 by the great affinity (PAR-1 is the major receptor of thrombin), FXa is usually sensitive to PAR-1, -2, and -4 (PAR-2 is the most prime for FXa and not activatable by thrombin, may function as a FVIIa/FXa complex receptor). Veritably, PAR1 and PAR2 appeared at MA, EC, SMC and monocytes.165–170 Thrombin and FXa activate a variety of cell types including MA, SMC, and EC for actuating cellular inflammatory response, which works in arterial injury and in neointima of human atherosclerotic lesions. Based on the precondition, PAR1 and PAR2 are the charming targets. Hara et al. 171 found that FXa-PAR-2 signaling activates macrophages and promotes vascular inflammation, increasing atherosclerosis involving MCP-1, IL-6 and TNF-α production, activation of NF-κB (upregulated inflammation through NF-κB pathway). It is trust worthy that PAR-2 is a underlying remedial target. Contributing PAR route on endothelial cells causes vWF, TF, adhesion molecules expression, and P-selectin relocation onto the cell surface. Vorapaxar treatment for competitive binding to PAR-1 (inhibiting thrombin-induced PAR-1 activation) realized lower coagulation activation, inflammatory response and endothelial activation, containing descending level of TNF-α, IL-6, vWF, and soluble E-selectin. 172
GLP-1R/GLP-1 signal
At present, the localization of GLP-1R (glucagon-like peptide-1 receptor) in blood vessels is on vascular smooth muscle, ECs, platelets, and monocytes/macrophages. And native GLP-1 (glucagon-like peptide-1, thought as anti-atherogenic and anti-inflammatory actions) or GLP-1 analogs (liraglutide, semaglutide, exendin-4) as GLP-1R agonists heighten GLP-1R expression for modifying cells or tissue function. Overall, GLP-1R signaling in multiple pathways impacts abnormal vascular tissue possibly relating to enhanced plaque stability, endothelial function and subdued smooth muscle proliferation, platelet aggregation, oxidative stress, and inflammation with attenuating atherosclerotic development.173–175 Some investigators considered that GLP-1Rs deadened plaque tissue pullulation and affected atherosclerosis through an anti-inflammatory mechanism.176,177 GLP-1R activation in endothelial cells blunted eNOS uncoupling and vascular inflammation, prevented vascular oxidative stress, and elevated NO bioavailability. 175 Furthermore, GLP-1 and its analogs prevented the development of aortic atherosclerotic lesions by reducing the monocyte/macrophage infiltration and macrophage foam cell formation. 103 Shrinkage of GLP-1 inactivation and degradation has shown as anti-inflammatory effects. Dipeptidyl peptidase IV (DPP-IV), CD26, a cell-surface, and secreted peptidase with increased dipeptidyl peptidase-4 activities in atherosclerosis, is in charge of degrading GLP-1.178–182 Dipeptidyl peptidase-IV (DPP-IV) inhibition has been widely appreciated as a possible therapeutic target for atherosclerosis, and its anti-atherogenesis could be explained by increased GLP-1 biological activity for direct vascular protective effects with better EC function, impaired monocytes/macrophage inflammation.183,184 Matsubara et al. 184 concluded that des-fluoro-sitagliptin (DFS), a DPP-IV inhibitor, augmented GLP-1 activity to reduce the releasing of proinflammatory mediators and ROS in macrophages, as well as attenuate EC dysfunction, exhibiting antiatherogenic effects.
The coagulation system
The rupture of an atherosclerotic plaque primarily triggers arterial thrombosis formation. As plaque ruptures, exposed collagen and von Willebrand factors(vWF), acting as specific platelet cell-surface receptors, capture circulating platelets to the site via bonding integrin α2β1, GPVI, and GPIb on platelets, respectively. And GPIbα mediates platelets to adhere to injured and inflamed endothelium. After adhering, major platelet integrin αIIbβ3 and GPIbα binds other platelets, then leading to rapid growth of the thrombus. As a more important role, high concentration of tissue factor exiting in lesions generated by atherogenic cells, referring to active platelets, monocytes/macrophages, inflamed ECs, SMCs, and foam cells, notably initiates the extrinsic coagulation pathway. Ultimately, combining faulty platelets, stable thrombus rich in fibrin, platelets and red cells take shape, which further enlarges ongoing plaque size. More concerned, stent implantation for treating narrowing vessel tissue would trigger clotting formation, encompassing acute (0–24 h), subacute(24 h to 30 days), late (30 days to 1 year), and very late (beyond 1 year-likely associated with hypersensitivity reaction, excessive fibrin deposition, or neoatherosclerosis) stent thrombosis, sharing similar mechanisms with thrombus formation.185,186 As a consequence, concentrating on thrombosis formation and platelets is a charming way to AS (atherosclerosis) targeting, therapy, and suppressing plaques growing.
Platelets
Platelets occupy an important position in developing thrombus and plaques, looking like Figure 7. And targeting it for preventing thrombosis mainly covers specifically inhibiting the receptors themselves, inactivating the promoters of platelets and hindering these promoters to yield. Hypothetically, blocking-up of platelets adhesion to bare collagen (via GPVI, α2β1), vWF (via GPIb) after plaque fracture and injured/inflamed endothelium (such as via GPIbα binding to P-selectin and vWF) could decrease vascular occlusion with effect. Currently, anti-GPVI treatment could be a powerful strategy to specifically passivate the collagen-GPVI pathway in platelets, reviewed by Nieswandt et al. 188 Meanwhile, other payloads have been employed as efficacious antithrombotic agents, containing assorted receptors inhibitors: glycoprotein IIb/IIIa inhibitors-abciximab, eptifibatide, tirofiban, and lamifiban 189 ; ADP receptor antagonists-Y2P1 inhibitor A2P5P and P2Y12 inhibitors (AR-C69931MX, clopidogrel, ticagrelor, and prasugrel190,191); incapacitating thromboxane A2 receptor by thromboxane receptor antagonists, such as ifetroban, domitroban, variprost, and others, more details seen in Kontogiorgis’s review. 192 Peptide (SP-14 with sequence SHIHGDYSSPSGAP) is also used to inhibit the binding of TXA2 to TP receptor for reducing platelet aggregation. 193 More than blocking receptors, extinguishing platelet autocrine factors (ADP, TXA2) production is also a potent strategy for opposing thrombogenesis, and in some cases, TXA2 biosynthesis could be blocked powerfully by aspirin targeting COX-1 194 or thromboxane synthase inhibitors(e.g. dazoxiben, dazmagrel, pirmagrel, isbogrel, and ozagrel). 192 TXA2 was produced via cyclooxygenase (COX)-thromboxane synthase pathway. 192 Later, bifunctional regulators with TP receptors blocking and thromboxane synthase inhibition were also exploited, including ribogrel, terbogrel, picotamide and BM-351/573, etc. 192 Not merely incurring platelet aggregation and activation, TXA2 may lead to plaque evolution and thrombus in human atherosclerosis, and infiltrated monocytes and macrophages in lesion make TXA2, which constitutes a significant source of TXA2. 195 Worthily mentioning, synergistic effect of blocking TP receptor and selectively suppression of TXA2 may have a positive impact on reducing plaque tissue. 196 Thrombin is a central part of forming fibrinous thrombus and platelet acting, and PAR-1 is the main thrombin receptor on platelets. PAR-1 antagonists have properties of antiplatelet and anti-arterial thrombosis, and vorapaxar is a novel antiplatelet agent that selectively inhibits the cellular actions of thrombin through antagonism of PAR-1, 197 some other PAR-1 antagonists had been summarized by Chackalamannil. 165 A newly-found receptor, GLP-1R, may serve as an antiplatelet-target. For instance, GLP-1 and other GLP-1 receptor agonists (liraglutide, exenatide) treatment could hinder platelets aggregation and thrombosis induced by collagen and thrombin very likely through triggering of GLP-1R signaling for increased cAMP level.198,199 Once platelets are in active state, PS flips to the outer side of the plasma membrane, exerting pro-coagulation activity with initiation and propagation of coagulation via formed TF/FVIIa complex and assembly of intrinsic tenase (FVIIIa/FIXa), prothrombinase (FVa/FXa) on PS-exposed platelets. 200 Based on this feature, outside PS owns both targeting properties and therapeutic capacities for platelets-containing thrombi. Competitive ligation of PS-binding ligands (such as lactadherin and annexin V) to PS inhibits platelet prothrombinase and factor Xase activity, effectively restrains FXa, and thrombin generation, which leads to delayed thrombosis formation. 201 According to the avidity of PS and annexin V, annexin V acted as a guiding molecules for selective targeting of platelet-containing thrombi. 202 Annexin V fusing curative stuffs, such as Kunitz protease inhibitor (KPI), 203 hirudin, 204 are shown to have thrombosis inhibitory activity with specifical PS affinity. Jing et al. 202 constructed disintegrin protein echistatin-annexin V system to obtain an antithrombotic effect via competitively binding to αIIbβ3 integrin resulting in reduced fibrinogen linking and platelet aggregation, and to PS molecules for dwindling prothrombinase complex and thrombin formation.
Figure 7.

The major portion of platelets in growing atherosclerotic plaques and prothrombotic events. There are abundant membrane-anchored receptors for adhering to injured/inflamed endothelial layer (GPIbα for P-selectin and vWF, PSGL-1 binding to P-selectin, CD40L-CD40 interaction, CX3CR1 for CX3CL1), ruptured lesion (GPVIα and α2β1 for collagen, GPIb for vWF), monocytes/macrophages (P-selectin for PSGL-1, CX3CR1 for CX3CL1), and active platelets (GPIbα and αIIbβ3 grasp additional platelets). PAR1/PAR4 coupling thrombin and P2Y1/P2Y12 binding ADP quietly potentiate platelet activation. Thrombin binding PAR promotes TXA2 and ADP producing, in addition to this, more ADP generating has a lot to do with TXA2-bound TP receptors. 187
Coagulation factor as targets
Figure 8 represents the extrinsic and intrinsic clotting pathways. Antithrombotic modulations come in all shapes and sizes, mainly centering on neutralizing thrombogenic molecules through various proteins, antibodies, peptides, aptamers, oligonucleotide, and other small molecules. Notably, targeting pro-coagulation factors gets attractive, including TF, thrombin, FXIIa, FXII, FXa, FXIa, kallikrein, and so on. Designedly making these coagulation factors invalid has been confirmed potently antithrombotic capacities. Concretely, targets and their corresponding inhibitors are showed, mainly delineated as: thrombin is a prime target, directly/indirectly inhibited by antithrombin activators(heparin), warfarin, hirudin and its derivatives (lepirudin, desirudin, bivalirudin), dabigatran, argatroban, 205 thrombin-specific aptamers (HD1 and NU172), 206 thrombalexin (TLN), 207 and avathrin 208 ; aptamers (RNA11F7t and RNABA4), rivaroxaban, apixaban, edoxaban, ACH-11, are effective for FXa blocking.206,209,210 The factor Xa can excite platelets by PAR-1 pathway, so tempering FXa with rivaroxaban would be antiplatelet effect more than reduced atherothrombotic events through PAR-1 derived platelet activation. 211 Sterilizing intrinsic factors FXIIa, FXII, and FXIa are also prevention of thrombosis formation by: RNA aptamer R4cXII-1 targeting FXIIa and FXII 206 ; Infestin-4 with FXIIa inhibitory effects 212 ; BF9, 213 Boophilin, 214 and DEF (an mAb to FXIa) 215 for inhibiting FXIa as well as Ir-CPI (Ixodes ricinus contact phase inhibitor) binding both FXIIa and FXIa. 216 For preventing undesirable thrombotic events, some natural anticoagulants have also been investigated, including antithrombin, activated protein C (APC), tissue factor pathway inhibitor (TFPI), and protein S. In some examples, protein S, a regulator in intrinsic coagulation pathway, subdues FIXa and Xase complex (FIXa–FVIIIa) through tethering to FIXa heparin-binding exosite, thereby in keeping with lower FX activation. 217 And, protein S also activates protein C for suppressing FV into FVa and FVIII into FVIIIa. 206 TFPI, found in endothelial cells and platelets, plays an anticoagulant part in early stages of clotting cascade and regulates tissue factor (TF)-induced coagulation via inactivating FXa and binding to FVIIa of TF/factor VIIa complex for inhibiting further FX activation. 218 As a cell-signaling receptor, TF also keeps a direct proinflammatory role for macrophages/SMC and promotes migratory and mutagenic effects of SMCs, so that TFPI has protective effects against MA/SMC inflammation and SMC migration and proliferation in addition to abating thrombus formation.219–221
Figure 8.

The extrinsic and intrinsic clotting pathway. 226 Ample TF combining coagulation factor FVIIa transforms FIX into FIXa, and FX into FXa, is closely linked to extrinsic and intrinsic coagulation pathway, finally meeting at bio-synthesizing thrombin and stable fibrin clot formation imbued with platelets and erythrocytes. More than TF-FVIIa pathway, FXII interacts with negatively charged surface or molecules, followed by forming FIXa and FXIa resulting from activated FXII (FXIIa), which means intrinsic pathway starts.
Coagulation holds a vital position in the onset of atherosclerosis, on good grounds, depriving effects of coagulation factors, beyond preventing sequential thrombotic events and abolishing subsequent occlusive arterial thrombus formation, may also be remarkably effective in impeding atherosclerotic progression or postoperative restenosis. Nationally, targeting coagulation factor Xa or thrombin has been elucidated as a promising treatment for holding back plaque starting and propagation or injury-induced neointima formation, and promoting lesion stability and plaque regression.210,222,223 Posthuma et al. 210 corroborated and extended that, except for inhibiting newly-formed plaque and increased stability of brittle plaque, FXa inhibition by rivaroxaban also facilitated regression of pre-existing atherosclerotic lesion with reduced macrophages, enhanced collagen deposition, diminished necrotic core, lower expression of PARs, thrombin, FXa, and MMPs. Some other researchers had also made it clear that direct thrombin inhibition by anticoagulant dabigatran improved endothelial function, reduced atherosclerotic lesion size, collagen content, and oxidative stress (ROS production), 224 retarded the initiation and progression of lesion and macrophage accumulation in Apolipoprotein E-deficient mice. 225
Important ROS generation system
Reactive oxygen species (ROS) participates in the modulation of cell functions and biological processes for promoted atherosclerotic progression, shown as oxidative modification of lipoproteins, inflammatory response, EC dysfunction and SMC proliferation and phenotypic switching, promoted cell death, etc. 227 Scavenging of generated ROS and arresting of its emergence have been considered potential for counteracting atherosclerosis. ROS-scavenging polysaccharide β-cyclodextrin (TPCD) nanoparticles with Tempol (a free radical scavenger) and phenylboronic acid pinacol ester (PBAP, for eliminating hydrogen peroxide effectively) evidently have inhibitory effects for atherosclerosis development by elimination of overproduced ROS, which led to diminished ROS-induced inflammation and apoptosis in macrophages, and inhibited foam cell formation. 228 And, NADPH oxidases (NOX) and renin-angiotensin systems are important sources of ROS in the cardiovascular system, shown in Figure 9. NADPH oxidases (NOX) system exists in nearly all plaque cell types including EC, SMC, MA, monocytes, and platelets.229,230 NADPH oxidase (nicotinamide adenine dinucleotide phosphate oxidase) may serve as a significant target. Neutralizing agents targeting NADPH oxidase, such as antibody, peptide or siRNA have been demonstrated to attenuate neointimal formation after arterial injury. 231 Nanoparticles loaded by siRNA targeting NOX2 were transferred into balloon injured artery in an atherosclerotic rat model, which would prevent neointimal area and lumen loss. 231 An available Nox inhibitor VAS2870 could inhibit ROS liberation induced by PDGF in SMCs and abolish PDGF-guided SMC migration effectively. 232 The importance of Ang II in ROS formation in renin-angiotensin system had been stressed over recent years. Blocking Ang II-induced ROS generation involves two parts: abolishing of Ang II biosynthesis (Ang II biosynthesis means that angiotensinogen is cleaved by renin to form angiotensin I, and subsequently generated angiotensin I is converted into Ang II under angiotensin-converting enzyme (ACE) 233 ); blocking the interaction between Ang II and AT1R. For combating oxidative stress, the synergism of ACE inhibitor and AT1 receptor blocker had been observed. 234
Figure 9.
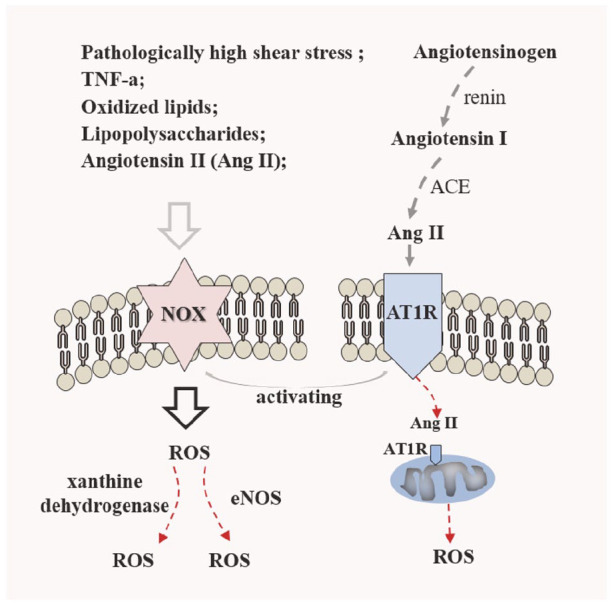
The NOX-derived ROS pathway and important renin-angiotensin system. The mediators of NOX activity and expression are involved in excessive proatherogenic factors, such as pathological shear stress, TNFα, PDGF, oxLDL, and important angiotensin II (Ang II). 237 By renin-angiotensin pathway, generated Ang II linking to angiotensin type 1 receptor (AT1R) activates membrane-bound NADPH oxidase in ECs, SMCs, and macrophages, 227 and it also traffics to AT1R on outer mitochondria membranes, inducing mitochondria-derived ROS. 236 Importantly, NOX-derived ROS aggravates ROS overproduction by acting on eNOS and xanthine dehydrogenase. 230
More than inducing ROS generation for inflammatory expression, synthetic SMC, EC dysfunction as well as cell apoptosis, Ang II-AT1R interaction guides apoptosis by Fas pathway, synthetic response of SMC by JAK2-STAT or JAK2-ERK1/2 pathway, 227 and stimulates angiogenic action for enhanced plaque neovessel formation, 235 which all resulted in aggravated atherosclerosis. Wu et al. 235 considered that renin inhibitor aliskiren, suppressing angiotensin II (Ang II) biosynthesis, reduced the atherosclerotic plaque area and plaque neovessel density, even to the extent of decreasing the vascular inflammatory action. An absorbing point in Ang II is that activation of Ang II is involved in all phases of atherosclerosis, presented as stimulating TF expression, resultant endothelial dysfunction, promoting to form foam cells via regulating LOX-1 expression (SMCs) and contributory oxLDL absorption, inducing apoptosis of ECs and SMCs. 236 Thus, blocking-up in Ang II-AT1AR (Angiotensin II Type 1A Receptor) could be attainable tactics for disposing of atherosclerosis through antagonizing AT1AR or lessoning Ang II.
Strategies of lesion-localizing delivery for treating atherosclerosis
Site-specific treatments of atherosclerotic plaque, relating to inhibition of restenosis and thrombus formation or modifying plaque with less prone to rupture/grow and promoting plaque regression, demand therapeutic agents to collect to the target lesion and into the vessel wall and cells. The section mainly states the tactics of effective loading enriched in atherosclerotic lesion, like Figure 10, including balloon catheter-driven local delivery, stent-based, and nanoparticle models.
Figure 10.
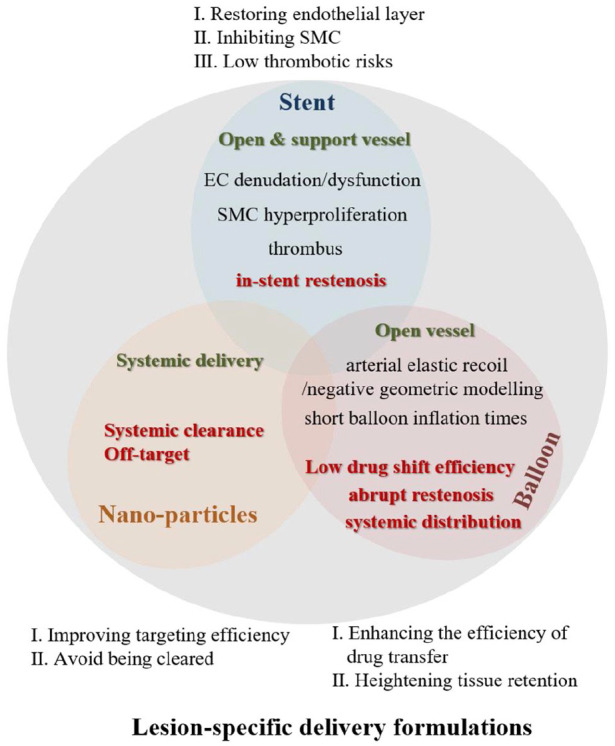
The schematic representation of the main lesion-specific delivery formulations.
Systematic delivery mode by nanoparticles
Bio-recognition based on natural ligand-receptor interaction
Biomarkers including changed cells (proinflammatory MA, inflamed EC, and switched SMC), biological factors and ECM components (collagen, vWF, fibrin) as well as deposited clots in plaque have been applied for the location of nanoparticles with therapeutic benefit in atherosclerosis. Bio-recognition based on natural ligand-receptor interaction may work as a potent formulation and resident cells in lesion contribute to this procedure. Currently, all kinds of ligands for targeting treatments embrace antibodies, peptides, aptamers including RNA- or DNA-based ligands. Growing evidence demonstrates that scavenger receptors highly express in atherosclerosis and targeting these molecules with affinitive ligands (like decadeoxyguanine 238 and DNA oligonucleotides 239 for SRA and anti-LOX1 antibody for LOX-1) could make therapeutic drugs get to lesion site. 94 Fasudil, a rho-kinase inhibitor, is capsulized into liposomes embellished with anti-LOX1 antibody and successfully reaches the arterial plaque with inhibited intimal hypertrophy. 94 Based on leukocytes binding to endothelial ICAM-1 by LFA-1 (lymphocyte function-associated antigen-1) integrin, inflamed leukocyte-mimetic nanoparticles with LFA-1 I domain preferentially reach the site of inflammation. 240 Antibodies 241 and peptides targeting ICAM-1 (cyclo(1,12)PenITDGEATDSGC peptide 242 ) and VCAM-1 (VHPKQHR 243 and 18F-4V peptide 244 ) also have emerged. More than targeting to inflamed endothelium, a specific sequence with VHSPNKK motif also blocks the interaction between leukocyte and endothelial layer. 28 Furthermore, targeting the underlying basement membrane had become another important point, such as KLWVLPK peptide binding to collagen. 245 Nanoparticles coupling with KLWVLPK peptide for paclitaxel delivery exhibits greater vascular retention in vivo. 245 And this particles system also transports IL-10 to atherosclerotic plaques for deactivating macrophages and T cells and resolving acute inflammation. 246 Modery’s group 247 had designed RGD and EWVDV peptides-modified liposomes to target active αIIbβ3 and P-selectin respectively, and manifested these particles had higher selectivity as well as retention to activated platelets. The CREKA peptide can mediate nanogels loaded by recombinant hirudin to get to fibrous clots, winning anticoagulant therapy by binding to fibrin and fibronectin. 248 Based on high affinity of CREKA for fibrin as well as abundance of fibrin and H2O2 in thrombi, Kang et al. 249 developed a fibrin-targeted nano-platform composed of tirofiban (a glycoprotein IIb/IIIa receptor inhibitor), FBAP for H2O2-scavenging, BAP with H2O2-response, and proved its antioxidant and antithrombotic activity. p32(gClqR), also named as p33, p32, C1qBP, HABP-1, is biosynthesized on the surface, in cells and as a secreted protein. Sharply inflammatory region and atherosclerotic plaque, exist in violently activated cells including active platelet (or collagen-induced aggregation), inflamed endothelial cells as well as inflammatory answering cells (macrophages/monocytes), foam cells and smooth muscle cells, highly expressing p32, in return, p32 activation irritates these cells to be active.250,251 Sufficient grounds have been available as the affinity interaction of LyP-1 (a 9 residues peptide, CGNKRTRGC) and p32. LyP-1-carrying nanoparticles exposed enhanced affinity to macrophage in vitro and converged at carotid lesions in vivo. 252 LyP-1-coated nanoparticles could penetrate the plaque to advantage, substantially accumulate in the plaque interior not at the surface of the plaque. 253 LyP-1 liposomes loaded by GW3965 also locate to atherosclerotic plaques and cut down the number of macrophages. 254
Biomimetic targeting strategies
Membrane cloaking derived from platelet and macrophage
Platelets participate in inflammatory response and clot formation in atheroma through reacting with activated endothelial layer, subendothelial layer (collagen and vWF), or leukocyte.7,255 Considering this intrinsic affinity of platelet to plaques, mimicking of platelet morphology, size, flexibility, and surface biology has been followed with interest. 256 In one example, Anselmo’s group took nanoparticles and PAH/BSA as template and flexible shell separately to imitate the morphology and mechanical flexibility of platelets, followed by bearing the collagen-binding peptide (CBP; [GPO]7), vWF binding peptide (VBP, TRYLRIHPQSQVHQI) and integrin GPIIb-IIIa linked FMP peptide (GRGDS, linear fibrinogen-mimetic peptide) for limited biological functions, and validated that this mimetic particles coupled to activated natural platelets and injured endothelial sites. 256 As a novel interfacing approach, the application of cell membrane has many natural advantages based on its multiple biological properties and functions, such as immunosuppression and selective recognition. 257 On treatment and checking, platelet membrane cloaking maintains inherent platelet properties with immune evasion (originating from CD47), suppressed complement system activation (due to CD55, CD59), localizing to clotting wall of atherosclerosis or angioplasty-induced denuded/injured vessels (on account of GPIb-IX-V, GPIa-IIa, and GPVI).258,259 Homing efficacy of platelet membrane coating to plaque may give the credit to their membrane proteins, such as GPIb linking to vWF, GPVI and integrin α2β1 (GPIa/IIa) tying to collagen, GPIIb/IIIa, and GPVI fixing to fibrin. 260 At present, except for detection of the condition, 261 growing evidence confirmed that platelet membrane modification significantly elevates therapeutic effects for atheromatous plaque, ascribing to more effective homing capacity to the focus. 262 Encapsulation rapamycin with platelet membrane-coated nanoparticles effectively homes to plaques in atherosclerotic mice modes, and evidently weakens the progression of atherosclerosis. 262 In another example, GSK2606414 (PERK inhibitor)-loaded nanoclusters were coated by platelet membrane, which thwarted phenotypic modulation of SMC and EC dysfunction, mitigated restenosis, and thrombosis in the rat model of carotid artery balloon angioplasty. 43 Absorbingly, platelet membrane fused by other counterpart of selective cell would carry more perfect properties combining various function of both source cells. 263
Termed macrophage naturally homing to the inflammatory plaque, such as macrophage membrane with high expression of α4 integrin reacting with the vascular cell adhesion molecule-1 (VCAM-1), 264 macrophage membrane embellishment has been applied to target and solve atherosclerosis. 265 Given that atherosclerosis is characterized by inflammation and ROS overproduction, Gao et al. 266 prepared macrophage membrane coated ROS-responsive nanoparticles (NPs) for improved therapeutic efficacy in atherosclerosis with reduced inflammation and plaque burden, owing to specific targeting of inflammatory site as well as tied and sequestered multiped pro-inflammatory substances by membrane antigens (TNFR2, CD36, and CCR2).
Lipoprotein modalities (mimicking HDL)
Lipoproteins, innate plasma particles transporting lipid, are commonly classified as five groups according to their density and size: chylomicrons, high-density (HDL), low-density (LDL), intermediate-density (IDL), and very low-density lipoprotein (VLDL). 267 HDL exerts to remove excess cholesterol from cells (e.g. foam cells/macrophages in atherosclerotic plaques), reduce inflammation, and improve endothelial function. HDL has certain key features, including: nano-diameter with 7- to 13-nm range; a hydrophobic core of predominately cholesteryl esters covered by phospholipids monolayer with embedded apolipoproteins, especially apolipoprotein (apo) A-I 268 ; bio-functions involving cholesterol carriage, anti-inflammatory effects, antimicrobial activity, anticoagulation, and specifically targeting several cell surface receptors (such as scavenger receptor B1, ATP-binding cassette A1, and ABCG1 for removing the excess cholesterol from cells).269–272 Considering HDL’s nature with inherent plaque affinity, forming “HDL-like” nanoparticles by simulating its structure and elements is now contributing to targeting treatments.
Rather than achieving the characteristics of HDL itself, recombined HDL with or without decoration is used for a novel potent transit system for agents directly entering into plaque. HDL is naturally marked by binding to and interacting with macrophage cells and hepatocytes during reverse cholesterol transportation, which has an important protective effect in atherosclerosis by reverse efflux of cholesterol from plaque macrophages. 273 In Cormode’s study, inorganic nanocrystal replacing the hydrophobic core of HDL incorporated phospholipid to come into being micelles. Whereafter, for producing HDL-like nanoparticles, apo A-I, the primary protein constituent of HDL and providing targeting abilities, had been used as a working component. Their research results validated that these artificially analogous particles to natural HDL possessed the capacity of being specifically ingested by macrophages with high affinity. 274 As a proof-of-principle study, these nanocrystal core HDL had lodged in the atherosclerotic wall in mice pathological model. In addition, the targeting abilities of imitated HDL particles to inflammatory artery had been verified in a wide of investigation, and these particles could be used as a carrier for therapeutic drugs or imaging agents. Marrache and Dhar 275 developed the biodegradable synthetic HDL mimic containing hydrophobic core, and a phospholipid bilayer coat decorated with triphenylphosphonium (TPP) and apolipoprotein (apo) A-I mimetic 4F peptide as a vulnerable plaques targeting agent to lodge in atherosclerotic areas, markedly in mitochondria. Lameijer et al. 276 used HDL biomimetic particles to successfully transport small molecule 68770028 (a CD40-TRAF6 inhibitor) to plaque, gaining immunotherapy with impaired monocyte migration and recruitment and reduced plaque macrophage content. 4F peptide-modified nano-system also was applied to successfully home GW3965 to plaque, which reduced plaque burden without increased liver toxicity. 277 Artificial HDL particles have limited cholesterol efflux ability, tailoring artificial HDL properties by HA recognizing overexpressing CD44 receptors in injured endothelial, would exhibit anti-atherogenic effects with a greater cholesterol efflux capacity and better targeting efficiency. 278 Luthi et al. 279 unveiled that orienting size and surface composition of high-density lipoprotein (HDL) biomimics altered the biomimic-mediated the cholesterol binding capacities and efflux cholesterol from macrophage cells.
Apoptotic mimicry
“Eat me” signals of apoptotic cell are composed of phosphatidylserine (PS), intercellular adhesion molecule-3 (ICAM-3), carbohydrates, and calreticulin, selectively recognized by phagocytes via phagocytic receptors. 280 On account of phagocytic ligand-induced effective recognition and engulfment, apoptotic bionics equipped with “eat me” signal molecules could target to the inflammatory macrophages in atherosclerosis and PS equipment has been widely concerned. 281 Naturally occurring membrane molecular alterations is ubiquitous in the process of apoptosis. Significantly, phosphatidylserine (PS) abandons membrane asymmetry and flips to the surface of apoptotic cells, and externalized PS is the best representative “eat me” signal for macrophages recognition to remove dead cells. 282 Conversely, PS can pass for a “tethering” ligand for macrophages. Zhao et al. 283 prepared PS-modified microbubble and proved its targeting capability to activated macrophages. Wu et al. 284 designed the biomimic liposome embellished with cRGDfK and PS to delivery pioglitazone (PIO, a PPARγ agonist ) into atherosclerotic macrophages, which kept macrophages weakened the release of IL-1β and TNFα, and strengthened the secretion of anti-inflammatory substances (such as 1L-4 and IL-10) with attenuated progression of atherosclerosis. The cRGDfK modification facilitated liposomes to fasten onto activated ECs in plaque and penetrate into plaque, followed that intra-plaque particles were intendedly discerned and absorbed by macrophages owing to PS signal. Sometimes, PS also articulating other phagocytic ligand, such as oxidized cholesterol ester derivative cholesterol-9-carboxynonanoate, co-mediated inflammatory macrophages targeting. 285 Significantly, Hosseini et al. 286 gave proof that phosphatidylserine liposomes (PSLs) simulating apoptotic cells attenuated atherosclerosis by targeting B1a cell activation. Another hot spot, the growing evidence has been certificated that specifically targeting PS could also act as delivery system for imaging and treating agents, and several frequently-used PS affine ligands have been used in this policy, including annexin V, 87 lactadherin or its PS-binding C domains-fused protein, 287 and some PS-recognizing peptides PSP1 288 and LIKKPF. 289
Interventional device-based delivery approaches to lesion
Stent as coping style of local delivery
As an intervention program of occlusive atherosclerotic arteries, stent opens vessel via balloon expansion and maintains its original tubular shape for remodeling blood flow. From the earliest bare metal stents (BMS) to current drug-eluting stents (DES) and bioabsorbable stents (BRS), the development of stents has been in line with clinical outcomes. 290 After stenting, vascular injury response (endothelial disruption, overgrowth of SMCs) often favors re-narrowing of treated arteries. Surmounting in-stent restenosis is a long-term goal in the field of stent technology. Compared with permanent one, novel bioabsorbable stents degrade naturally with vascular healing, which requires the stent degradation rate to match the mechanical properties for supporting artery and velocity of mending. Considering this, for BRS, regulating degradation rate should be paid close attention. In this part, eternal stent would be uniquely reviewed. For stent-guided local transport, coatings on stents clearly occupy an important position at performance. From the earliest bare metal stent to the current coated stent, investigators have indicated that coated technologies endued stent more exceptional service properties, such as gold-coated NIR stents. 291 In efforts to coat metal stents, drug-eluting designs, bionic tactics with virous functional factors have been available.
Drug-eluting designs
As a drug carrier platform, stent systems are expected to achieve high local drug concentration with low systemic toxicity and long actuation duration. Therapeutic agents and their loading systems are two important research directions. A large number of clinical drugs or ones under ongoing trails had been reviewed, involving in anti-inflammatory, antiproliferative, and immunosuppressive drugs, including sirolimus and its analogs (e.g. everolimus, zotarolimus, biolimus A9), paclitaxel, dexamethasone, batimastat, 17-beta estradiol, actinomycin D, or tacrolimus.292,293 Antiplatelet drugs also have been being under investigation. Ticagrelor, a P2Y12 receptor inhibitor, is coated onto stent. More than affecting platelet, the ticagrelor eluting stents preserves endothelial recovery and favors suppressive smooth muscle proliferation. 294 The drug release profile largely determines the overall performance of stents in a variety of evaluation models. 291 Polymers, synthetic or biological polymers, acting as versatile basal layers demanding to provide good biocompatibility (blood or tissue), generally are utilized to incorporate pharmacologic agents for mediating drug release behavior, covering poly(ethylene-co-vinylacetate), poly(n-butylmethacrylate), poly(styrene-b-isobutylene-b-styrene) fluoropolymer, phosphorylcholine, poly(lactic acid) (PLA), and poly(lactic-co-glycolic acid) (PLGA). 295 Additionally, biological molecules, hyaluronan (HA) and chitosan (CH) polyelectrolyte multilayers 296 or phosphorylcholine, 297 also may be potential candidate polymers for local drug delivery. Adequate coating techniques are essential to acquire excellent drug loading and releasing. Dipping, spray, electrospunning nanofiber 298 as well as hydrogel299,300 coating have been developed. Nevertheless, increasing researchers held the view that polymer coating could answer for SMC proliferation, endothelial incompetence, proinflammatory responses, and thrombotic events. 301 In this regard, polymer-free drug-coated stents have been seen. Considering drug-binding matrix without polymer, some stent developers had exploited nitrogen-doped titanium oxide (N-TiO2) coatings for drug elution, and found that this polymer-free everolimus (EVL)-eluting stent prevented the platelet adhesion and restenosis, compared with bare metal stent. 302 Some novel smooth muscle-sensitive drugs are essential, as drugs against SMC proliferation often inhibit EC proliferation and migration without distinction, which leads to a detrimental effect on successful vascular repair. CTP synthase1 inhibitors and PERK inhibitors exhibit cell-specific inhibitory effects on VSMC not ECs.43,303 Tang et al. 304 demonstrated that cyclopentenyl cytosine (CPEC), a CTP synthase inhibitor, promoted contractile SMC showing pro-angiogenic property, and lessened neointima formation after injury, while accelerating re-endothelialization. Of note, CPEC-induced SMCs energized proliferative and migratory effects of EC via a pro-angiogenic paracrine effect.
Enhancing or restoring the normal function of cells populating lesion
The health and continuous endothelium occupy an important position in regulating local hemostasis, thrombogenesis as well as VSMC proliferation, particularly at the injured locus post-stenting. Meddling endothelial cell selectively and specifically for promoting ECs repopulation has been presumed to be a rational choice to boost vascular repair and cripple neointima formation after stent placement. 305 Generally, coating stent with biological factors, such as proteins (antibodies, ECM proteins), cytokines and chemokines, gas signal molecule (nitric oxide, NO), or adapters (DNA, RNA, and peptides) for joining stent surface with the endothelial wall, has been utilized to make an increment in attachment, migration, vitality, and function of endothelial cells. As the native microenvironment provided by ECM, multifarious ECM-derived proteins, active peptide sequence, or short fragment of protein have been surfaced onto stent for improving endothelial cell, covering fibronectin (FN) 306 or FN+ phosphatidylcholine, 307 fibrinogen, 308 laminin, 309 collagen, 310 vitronectin, 311 and their derived peptide: fibronectin fragments RGD, REDV, GRGDSP, and PHSRN; YIGSR, PDSGR, LRE, IKLLI, and IKVAV enriched in laminin; and DGEA and CAG from collagen type I 312 and collagen IV 313 respectively. Furthermore, other types of peptides also have emerged. SVVYGLR, synthetic peptide with binding integrins such as α4β1 and α9β1 and mimicking VEGF, enhances EC adhesion, migration, and maintains ECs proliferation.314,315 Immobilizing P8RI (a soluble synthetic peptide, like a CD31 agonist) onto stent could favor vascular endothelium repair, prevent in-stent stenosis and thrombosis. 316 Other than those enhancements, biological factors can provide reinforced cues for positive effects in inducing ECs repopulating and function. Chemotactic/growth factors including CXCL1, 317 SDF-1, VEGF, 318 chemokine (CC motif) ligand 2 (CCL2) and insulin-like growth factor-1 (IGF-1), 319 NGF, 320 hepatocyte growth factor (HGF), 321 sphingosine 1-phosphate (S1P), 322 erythropoietin or recombinant erythropoietin 323 have been applied to thrust complete endothelial covering via augmenting attachment, proliferation, migration, or nitrogen monoxide (NO) production. Noteworthily, NO releasing from ECs naturally plays an important role in anti-thrombosis and anti-restenosis through inhibiting excessive proliferation of smooth muscle cells, propagating wall endothelium, and deterring adhesion/aggregation of platelets. Justifiably, covering materials with NO-producing layers has caught hold of attention of researchers. At present, NO transportation platform mainly includes NO-releasing materials (involving NO donors like NONOates, SNAP and RSNOs, and NO-releasing peptide like the sequence GTAGLIGQ linked with polylysine KKKKK 324 ) and NO-generating strategies (covering loading of catalyzer selenocystamine, CuII and NOS gene, eNOS and iNOS).325–327 Fixation of antibody can work as another potent candidate, as antibody fragments scFv, single chain fragment variable binding to VEGFR2 mediating VEGFR2-positive ECs to adhere and reproduce on the stent surface. 328 Meanwhile, burgeoning gene therapies also serve as the encouragement of migratory and proliferative effects of vascular endothelial cells, such as ZNF580 gene, 310 DNA encoding for human VEGF 329 and CD39-encoding mRNA. 330
Responding to mechanical injury (e.g. stenting, balloon injury) and pathological stimulus (e.g. growth factor PDGF, inflammatory cytokines TNFα), activated SMCs evinces augmented proliferative and migratory activities associating with their de-differentiation, which induced the narrowing of vascular lumen or neointima formation in stent. 331 For SMCs in abnormal state, some efforts have been done. In some cases, as inhibiting SMC activity potently and stimulating EC proliferation modestly, perlecan, and a perlecan-inducing compound (e.g. RUS3108, inducing perlecan synthesis in SMC) may be used for stents to prevent ISR (in-stent restenosis). 332 And, through regulating SMC phenotypic modulation, diminishing in restenosis after stenting is possible. In light of synthetic smooth muscle phenotype featured with active RhoA expression, Huang et al. 333 built RhoA inhibitor-eluting stent with rhosin loading, and subsequently demonstrated that this RhoA inhibitor down-regulated YAP (Yes-associated protein) expression in SMC phenotypic modulation, which attenuated neointimal formation. Furtherly, selective Sp-1 inhibition with Mithramycin A also abrogates partly restenosis via disturbing YAP-mediated SMC phenotypic modulation. 334 And, sorafenib couples to myocardin by competing with YAP for increased SRF-myocardin interaction, which modulates SMC phenotypic switching and attenuates in-stent restenosis. 335 Blocking signal transduction of PDGF-PDGFR pathway works as another candidate. PDGF receptor (PDGFR) tyrosine kinase inhibitor, Sunitinib, targets SMC proliferation, necrosis of SMCs and migration, mediating reduced neointimal formation after stenting. 336 Stent-based delivery of D-65495, a bis(1H-2-indolyl)methanone, another PDGF-receptor inhibitor, could cause diminished neointima formation. 337 Through jamming the G0/G1 cell cycle and PDGF receptor β-Akt pathway, statins prevent SMCs from the reception of PDGF-BB signal for inhibited the pathological proliferation and migration of SMCs. Rosuvastatin inhibits the smooth-muscle-cell phenotypic modulation in PDGF-BB-induced synthetic SMC model and stents covered by heparin and rosuvastatin can reduce the incidence of stenosis and late thrombosis. 338
As answering environmental stimuli (i.e. ox-LDL), macrophages in plaque could convert their polarized forms to M1 macrophage, which would release pro-inflammatory factors and reactive oxygen or nitrogen species. Inevitably, a corresponding M2 macrophage also have been deemed as immunosuppression and eliminating inflammation of lesion. 339 Given phenotypic tunability of macrophages, a novel formulation transforming inflammatory state of plaque macrophages to anti-inflammatory M2 macrophages might be used to treat atherosclerosis. To name a few, Zhao et al. 340 constructed nanospindles with TiO2 and Ti4Ni2O on NiTi alloy, which manipulated macrophages to produce a favorable immune microenvironment with TGF-β, BMP-2, and VEGF for enhancing the functionality of the ECs. Similarly, Xu and co-workers 341 considered that induced M2 macrophage polarization could secrete VEGF for promoted endothelialization through ERK1/2 and PI3K/AKT pathways.
Capturing new cells or guiding neighboring ECs to injury site
Trapping circulating cells (covering ECs, EPCs, and SMCs) from blood flow is an alternative option of winning self-endothelialization. Endothelial progenitor cells (EPCs) are generally defined as a group of cells featuring differentiation into mature endothelial cells, 342 which have great application potential to expedite reendothelialization and limit intimal thickening in stent usage. Coupling of the stuff possessing excellent ligand accessibility to receptors on EPCs to stent has succeeded in self-endothelialized implants, including antibodies and peptides. In one example, linked CGRGDS peptide onto materials could harvest ECFCs and HBOECs under shear stress, which has the ability to grow into successive worked endothelial layer in vivo. 343 Recently, antibody-immobilized stent has been designed to catch EPCs (particularly late stage of EPCs) efficiently and specifically. Park et al. 344 unwrapped that stents integrating anti-CD146 antibody could capture more late-EPCs as well as mesenchymal stem cells (MSCs) in vitro. More biomarkers of EPCs have emerged in this research field, touching upon CD34, 345 CD133, 346 vascular endothelial-cadherin (VE-cadherin), 347 CD146, 344 endoglin, 348 vascular endothelial growth factor type 2 receptor (VEGFR2), 328 CD144, and CD309. 349 Fixation of antibody for bounding these biomarkers onto materials could potently attract these cells to pathological sites. Antibody fragments scFv, single chain fragment variable binding to VEGFR2 can be utilized to guide attachment and multiplying of VEGFR2-positive cells covering circulating ECs and EPCs. 328 What’s more, specific peptide ligands also have been defined, involving integrin-combined peptide ligand, such as αvβ3-integrin-binding peptide cyclic Arg-Gly-Asp peptide 350 and LXW7 (an octamer disulfide cyclic peptide (cGRGDdvc)). 351 In the example, LXW7 potently and specifically immobilized to αvβ3-integrin of EPC/EC, but presented weaker binding to platelets and no binding to THP-1 monocytes. A 12-mer peptide, termed as TPS (TPSLEQRTVYAK), an EPC-specific ligand, also unfolds high affinity and specific binding ability for human endothelial progenitor cells (EPCs). 352 WKYMVm (Trp-Lys-Tyr-Met-Val-D-Met), a hexapeptide, according to Li’s group, 353 possessed a fixed characteristics showing up remarkable EPC-induced recruitment for vasculogenesis guided by activating the formyl peptide receptor. Newly, HGGVRLY, hemocompatible peptide-1 (HCP-1), α4β1 integrin ligand, a recently defined stem cell homing peptide, could combine with EPCs and boost them spreading, which confirmed by Hsu et al. 354 Going further, HGGVRLY also fastened a high proportion of BMSC (bone marrow stromal cells) having VCAM-1. In other investigations, HCP-1-immobilized surface was very productive in gaining mitigated thrombus by lowing platelet adhesion and enhanced endothelial cell adhesion.355,356 Another α4β1 integrin ligand, REDV displaying peptide sequences Arg-Glu-Asp-Val, has been recognized as optionally and effectively catching endothelial cells (ECs) in fluid shear stress. 357 Markedly, for mesenchymal stem cells, another promoter cell, incremental evidence had argued that MSCs possessed inhibitory potential to restenosis via boosting re-endothelialization 358 and anti-inflammatory effects of SMC promoted by paracrine factors from MSCs. 359 Additionally, DNA aptamers 360 also could act as potent cell-bonded molecules, especially for harvesting self-seeding of EPCs onto vascular stent. More than promoted migration and proliferation for ECs, ZNF580 or VEGF gene also served as an attractor of EPCs in circulating blood. 310 Another type of candidate, introduction of growth (NGF, nerve growth factor) 320 /chemotactic CXCL1 (CXCL1 binding his receptor CXCR2) 317 factors on stent had been investigated in homing circulating EPCs.
Balloon catheter-based systems
Balloon catheter-driven local delivery comes to the foreground for lesional trafficking in recent years. Based on existing circumstances, compared with old plain balloon, apart from opening artery and improving blood flow, coated balloons could carrier a sufficient dose of an effective payload, for example paclitaxel, to the lesional tissue. Upon the balloons inflated, the drugs, adhering onto balloon membrane or hidden underneath the folds wrapped around the shaft, are pushed into the wall of arterial blood vessel and surrounding tissue. 361 The loss and transfer to the wall can be balanced by designing physical surface features or coating formulation of the balloons. For coating balloon, some elements would be appropriately added, such as medication for treatment, an inactive excipient with aiding in drug transfer. More often, balloon surface would be also designed, such as micro-porous. Furthermore, designing balloon catheter itself has emerged as a novel approach of local liquid delivery to achieve uniform agents into vessel wall, such as occlusion perfusion catheter. Occlusion perfusion catheter (OPC), a catheter containing three balloons, comprises two compliant occlusion balloons defining the treatment chamber at both ends and a center space occupying balloon (treatment chamber) through which the therapeutic agent would be infused. Perfusion catheter opens a direct manner of delivering payloads into medial artery for handling restenosis. 362 Bunch et al. exploited the occlusion perfusion catheter as an actual drug delivery catheter to locally deliver liquid paclitaxel to the stenotic lesions and assessed the feasibility, safety, and efficacy of this tactic for disposing of restenosis. Ultimately, they demonstrated that liquid paclitaxel treatment using occlusion perfusion catheter could be safely and effectively prevent restenosis. 363 In this part, therapeutic ingredients coated balloons will be introduced mainly.
Effective payloads
Lesion-homing delivery via active principle (like antiproliferative drug, gene, antithrombotic agents as well as cells) laden balloon may occupy the crude preponderance with leaving no foreign bodies in the blood vessel in management of in-stent restenosis and de novo lesion in small-vessel. For remedying atherosclerotic arterial disease, drug-coating balloon has emerged as an available alternative. The balloons carrying medicine (such as cytotoxic agent paclitaxel and sirolimus) coat the inner side of atherosclerotic vessels, which arrests or puts off adverse complications and inhibits pathological pathway of atherosclerosis and neointimal hyperplasia.364–366 Paclitaxel, a highly lipophilic antineoplastic drug showing sustained inhibition of the cell proliferation, produces a rapid inhibitory effects after the exposure of cells or tissues to paclitaxel for just a few seconds to a few minutes. 367 Based on paclitaxel eluting, drug-coated balloons have recently displayed enhanced efficacy in treating recurrence of stenosis.368,369 During treatment of coronary in-stent restenosis, early studies suggested that paclitaxel-coated balloon catheter with 3 μg/mm2 of balloon surface area apparently weakened the secondary occurrence of stenosis, and second stent implantation and sustained drug release at the site of injury may not be necessary for the inhibition of restenosis. 370 Meanwhile, Bonaventura et al. generalized that, differing from drug-eluting stents, handling with drug-coated balloon carrying paclitaxel received lower late lumen loss and similar restenosis and revascularization rates to DES for treating small-vessel de novo lesions. 371 Chowdhury 372 represented that diminished inflammation and plaque burden in nonobstructive lesions had been observed in paclitaxel drug-coated balloon treatment and proved that paclitaxel drug-coated balloon with suppressed capacity for atherosclerotic proceedings could safely and potentially serve as regional anti-atherosclerosis therapy. Another candidate sirolimus is also a coping style of reducing intimal growth. In one investigation, various sirolimus coating (crystalline coating and amorphous one) had been produced, and their inhibitory capability of neointimal proliferation had been confirmed, although they showed significantly unlike residence time in the coronary artery wall. 373 Of particularly note is that characteristics of drug molecules themselves embodying lipophilicity, molecular size and surface charge, would affect drug penetration and cell uptake into specified target, which had been narrated by Tesfamariam in detail. 374
For local administration with angioplasty balloon catheter, inhibition of thrombosis is also of great concern. Back in 1994, Nunes et al. 375 transported a synthetic antithrombin to the arterial wall for suppressive platelet-dependent thrombosis via a hydrogel-coated angioplasty balloon catheter, concurrently without altered bleeding parameters. Other antithrombotic drugs (heparin, argatroban) had been sent to injured arteries via porous balloon catheter to prevent thrombosis or partial intimal hyperplasia without obstructing system coagulability.376,377 Furthermore, over the past decades, balloon catheter-based systems have been described as useful mode of therapeutical gene shipping for treating cardiovascular conditions, incorporating micro-needle, double-balloon, channel balloon, and porous balloon catheters, while, for efficiently, selectively and rapidly mediating gene transfer into given cell types of artery wall, appropriate system should be opted.378,379 For the efficient delivery and long-term expression of therapeutic genes targeting the medial SMCs, Pankajakshan et al. applied irrigating balloon catheter to transfer gene (SM22α promoter) wrapped in a AAV2/9 vector into intended site after inflating balloon catheter with minute pores to injure the coronary artery SMCs. In the light of their exploration, object gene was successfully expressed in the middle layer with lasting for at least 2 months. 380 On these foundations, suppress intimal hyperplasia via balloon catheter-based gene delivery is a promising concept. More than the foregoing payloads, endothelial cells also had been conveyed to balloon-dilated rabbit arteries through a local delivery catheter (dispatch), and adhered onto scathed arteries with modest cellular retention. 381
Modifying of the balloon surface
Considering more effective local drug delivery for facilitated drug retention on balloon and shift into tissue while weakened drug loss into blood, modifying balloon surface is considered to be applicable and potential, including adding excipients (iopromide, dimethyl sulfate, dextrane, urea, polyethylene glycol, and so on) or carriers/coatings (hydrogel, nanoparticle, and polyethylene oxide coating) and microstructure (perforating, arrays).365,382 The immobilization of ionic monomers (acrylic acid and 2(dimethylamino) ethyl methacrylate) onto polyethylene balloon catheters meets an instantaneous local high concentration of argatroban (an available anti-thrombin agent) within dilated artery, though encountering a decrease after restoring blood flow. 383 Given surface design could manifest discrepant kinetic characteristics for a certain kind, different in rate of release, binding capacity to treated sites, systemic drug concentration, tissue penetration depth, and retention capacity. Obviously, adding controlled release coating, such as hydrogel or others, could obtain better pharmacokinetics. In one example, Dick et al. employed a porous balloon catheter and a gel-coated one as model of local drug delivery system and compared the differences of drug behavior between them. Evidently, it could be concluded that, compared to the hydrogel-coated balloon, the porous one obtained more payloads within treated segment at the cost of higher system concentration, which rendered low transport efficiency. 384 Tomaru et al. 376 used a novel porous balloon catheter consisting of dumbbell-shaped balloon with 12 small holes as the delivery system and demonstrated that this new porous balloon catheter created less mechanical vascular trauma comparing with the conventional ones. Microstructure of surface determines the drug transfer efficiency and retention efficiency in treated vessel. Developing microneedle array as drug supporter also enables to eliminate low drug delivery efficiency of standard DEB, which implies the superior therapeutic efficacy for atherosclerosis. And microneedle drug eluting balloon is proven to be suitable solution for intraluminal delivery led by microneedles embedding tissue and going deep into the tunica media of the vascular tissue. 385 Still further, Tzafriri and his partners 386 devised various micro-features (amorphous, flaky, and microneedle) on balloon surface to move paclitaxel, observed that microneedle system reaped larger coverage, higher tissue concentration than amorphous/flaky surface. Based on nanomotors with the property of penetrating into target cell, Huang et al. 387 developed a novel balloon-coating with a paclitaxel packaged into MJAMS/aV (platelet membrane coated Janus mesoporous silica nanomotor modified with anti-VCAM-1 antibody) for the long-term anti-proliferative effects of paclitaxel. Specifically, nanomotor could penetrate into the atherosclerotic plaque deeply, which promoted by near-infrared (NIR) light, wrapping of nanomotor with platelet membrane minimized drug loss before reaching the plaque and anti-VCAM-1 antibody linked to the excessive VCAM-1of the plaque site. Concurrently, solvents also pose as a controller of healing potion transfer. Take for example, when methanol, water, or acetone were used as solvent for paclitaxel-contained balloon, high drug loss into blood entailed low drug transfer rates to the applied site. But used ethyl acetate-solutions, smaller drug loss and more un-transferred PTX onto balloon had been observed. 388
Comprehensive analysis of these delivery strategies
As a systematic transportation strategy, more than therapeutic agents, the transport mode on the basis of nanoparticles encounters their challenges: how to colocalize and how to work. Its final therapeutic effects are derived from drug molecule, efficiency of targeted transport as well as degree of plaque damage caused by particle fixation. In this regard, the specificity of molecules (including therapeutic molecules and targeting moieties) embellished onto/into nanoparticles is required for positioning, retention and cure. More than payloads, the shape and size of particles may induce the discrepancies in targeting ability and distribution. Zhang et al. 389 elaborated that the order of targeting capacity is platelet (the highest), cylinder, blade, sphere and brick (the lowest), and enlarged size mediated the increased targeting efficiency. Moreover, the level of plaque injury is closely related to the changes in hydrodynamic characteristics caused by particles. Virtually, the onset, growth and rupture of atherosclerosis are very sensitive to fluid dynamics. Hemodynamics plays a central part in the development of atherosclerosis, and shear stress, the friction force exerted by blood flow on the blood vessel wall, is a key driving force. 390 According to the principle of hydrodynamics in atherogenesis, the regions of predisposition for plaque forming possess some clear attributes: curvature, branching, bifurcation, tapering, or external attachment (like plaque) inducing disturbed flow. 391 In these susceptible zones, weaker shear force acts on endothelial cells, comparing with protected zones. 392 And, in these areas of vascular stenosis, the cap of the plaque presents the maximum wall shear stress (WSS) and the regions with low WSS exist at the rear of plaques like shoulder. 393 High shear stress tends to make plaque rupture, while the sites with low shear stress are more likely to obtain lipid deposition. Additionally, aggravated stenosis is parallel to the rising WSS. 393 Given these conditions, the effect of transport vehicle induced hemodynamic changes on plaque progression should be considered. And seeing the principle of fluid dynamic in stenosed vascular lumen, nanoparticles themselves can be used as drug agents for regulating the hemodynamics of stenotic artery toward atheroprotective flow environment, like minimizing the stresses on the wall and resistance to blood flow. 394 Injected nanoparticles with different size, shape, and dose (volume fraction) have distinct effects upon mechanical forces in plaque, which induces plaque injury of different severity. Generally, nanoparticles with platelet shape show the highest WSS, meaning the most serious damage to plaque, compared with cylinder-, brick-, and sphere-shaped nanoparticles. 395 Enlarged dose and particle size would trigger appearance of high shear stress, which may lead to the rupture of plaque.389,395 Together, more than optimum presentation of therapeutic or targeted molecules, it is necessary to optimize the particle body design.
As a major interventional formulation, the long-term effectiveness of stent implantation in the treatment of atherosclerosis is approved. But its adverse consequences have also attracted attention. Fast recovery of blood flow and in situ supply of therapeutic molecules provide favorable factors. Thrombotic and inflammatory properties of the material itself accelerate negative effects. And acute injury (endothelium denudation/dysfunction) caused by stent expansion was once dubbed the important cause in the formation of stenosis. For dispelling ultimate unfavorable performance, multifarious drugs are loaded onto the surface of stent for restoring endothelial layer, SMC-selective prevention, or lowering the thrombosis/inflammation of materials. Ulteriorly, in addition to drug dependent wall response, it has been deliberated that the local hemodynamic forces induced by rigid protruding and malapposed struts after stent graft are a significant inducement of in-stent restenosis and thrombosis. Insufficient stent design (strut thickness and shape) and deployment (i.e. underexpansion inducing larger distance of the struts from the vessel wall) may impede healing response post-stent. 396 Concretely, in stented segments, the zones between the struts have a strong disposition to growing neointima considering low WSS in these regions while high WSS on the surface of struts linked to platelet activation. And, the large struts engender heavy protruding and hindrance for blood flow while less embedding into vascular lumen, accompanied by increased shear rate. Regions with high shear rate reside in the top and bottom of malapposed struts, forming a thrombotic surface. As a consequence of incomplete stent expansion, residual stenosis further deteriorates the mechanical environment of dilated artery (i.e. decreasing WSS, more disturbed), thus accelerates the stent failure with restenosis, stent thrombosis. 397 And, it is obvious that severe residual stenosis heralds more disturbance. Compared with non-streamlined struts (rectangular), struts with appropriate streamlining (circular/ovoid) could abate the magnitude of shear rate at the top of strut and the area of recirculation regions around the struts. 398 Overall, the final clinical outcome of stent implantation is the comprehensive result of drugs, stent design and implantation technology.
As another alternative mode of drug transportation based on interventional device, more than opening blocked blood vessels quickly, balloon transport also has advantages in uniform drug distribution on arterial wall, immediate drug release, a shorter duration of antiplatelet therapy, and no long-term residue of foreign material. Now, balloon dilatation is widely used to deal with in-stent restenosis implicated in EDS and bare metal stent, neo-plaques at small vessels and bifurcated vessels. 12 But, the expansion of balloon causes vascular injury of the host artery, featured by endothelial dissection and damage, which expedites ensuing acute platelet deposition, SMC hyperplasia, and restenosis. The acute and subsequent results of balloon angioplasty are the most susceptible to inflation pressure, balloon size (balloon to vessel ratio), and time (inflating time). High inflation pressure obtains high acute lumen acquisition, but results in severe vascular trauma with significantly higher incidence of dissection, thrombus and restenosis. And, unrelated expansion pressure, excessive balloon size leads to large vascular wall trauma and restenosis. 399 As reported Li et al., 400 excessively rapid balloon dilatation also works as a considerable cause of arterial dissection (referring to a tear along the inner wall of an artery). Commonly, it is recommended that inflated time of 30–60 s at nominal pressure (about 7–8 atm) and matched balloon-to-vessel ratio (about 0.8–1.0) are appropriate to avert vascular dissection. 12 But, for drug delivery, expansion time should be considered more carefully. Notably, due to elastic recoil of blood vessels after deflating balloon or insufficient dilatation, the existence of residual stenosis limits the therapeutic effect of balloon. Based on hemodynamic analysis, less loss of blood vessel gain helps to maintain a normal hydrodynamic environment, as large residual stenosis implies large vessel wall shear stress, increasing the risk of restenosis. 401 Significantly, selecting the optimal balloon size and expansion pressure, small residual stenosis could be obtained. Optimizing balloon design is also a way to achieve small residual stenosis. By the combination of incision (cutting the lumen of the thickened vascular wall and possibly breaking the elastic force) and dilatation of the plaque, cutting balloon angioplasty minimizes arterial wall trauma and decreases residual stenosis for lessened neoproliferative response, and subsequent restenosis.402,403 For inhibiting the response of excessive SMC proliferation and eventual restenosis induced by trauma, the selected drug is loaded on the balloon surface. Low drug transport efficiency to target tissue and the use of nonspecific drugs are still the main limitations. In order to enhance drug transport efficiency, coating technologies (like hydrogel), balloon surface design as well as the employment of drug carriers have been carefully recommended. Ultimately, integrating these factors, the optimization of therapeutic agent selection, balloon design (surface and construct), and surgical procedure improves the targeting delivery of treatment drugs into plaque and lessens the trauma of target artery for accelerated positive vascular repair response.
Conclusions and perspectives
Inevitably, the pathological progression of atherosclerosis is the ramification of cross- and cascade-response of multi-pathways, covering macrophage inflammatory phenotype and damaged phagocytosis, inflamed endothelium, phenotype-modulated SMC, abnormal lipid metabolism, production of reactive oxygen species as well as deposited thrombus, etc. Based on this circumstance, it is a legitimate basis for treating atherosclerotic plaque to eliminate/inhibit pathogenic factors and revive native cell functions. Focusing on how the therapeutic agents reach the lesion, stent-, balloon-, or nanoparticles-based delivery modes are under processive investigation. Naturally, since the idea delivery strategies require maximal bioavailability of target tissue/cells in plaque for therapeutic molecules to restore functions of health vascular, delivery modes mediated by stent, balloon, or nanoparticles counter their drawbacks. Post-stenting restenosis induced by vascular injury response (e.g. endothelial discontinuity and growth of SMC), late thrombus and nonspecific therapeutic drugs (like antiproliferative agents) limits stent-based treatment. Additionally, coagulation and inflammation induced by stent itself should be solved, like ticagrelor 294 for resolving coagulation and recombinant CD47 404 for reduced inflammatory cell attachment. For this formulation guided by balloon,374,385 mostly relying on EC-toxic drugs, arterial elastic recoil, and negative geometric modeling may cause an abrupt restenosis and short balloon inflation times (30–60 s) would be not conducive to drug shift efficiency from balloon surface to the luminal lesion. Moreover, cytotoxic levels associated with high drug concentrations locally and systemic distribution also should be considered. Therefore, stent/balloon-free strategies meet the clinical needs. The handicaps of systemic transportation induced by nanoparticles are concentrated on how to arrive in plaque tissue/cells successively rather than being cleared. And this systemic trafficking scheme is required to achieve lessened plaque progression, potentiated regression and stability.
From the perspective of therapeutic molecules, the effects should be directionality and uniqueness with annihilating harmful factors while protecting or promoting favorable events one or more. In the case of RGD presenting in fibronectin, fibrinogen, von Willebrand factor, vitronectin, and thrombospondin, 189 this peptide captures platelets via binding to αIIbβ3 (GPIIb/IIIa), αvβ3, and α5β3, while blocks fibrinogen binding to platelets guided by competitive effect for αIIbβ3. RGD on surface would capture platelets. More importantly, integrins αvβ3 and α5β3 are also expressed on endothelial cells. 405 So, RGD can link ECs and platelets. Therefore, selected payloads should be specific for treated targets. General drug-eluting stents have impeded neointimal smooth muscle cell hyperplasia, but aggravating dysfunction of endothelial cell, which lead to late thrombogenic events and post-angioplasty restenosis. For supporting stent applications, new specific agents should alleviate disease phenotypes of SMCs but facilitate health one of ECs and nonthrombogenic effects. The novel PERK inhibition (like GSK2606414) mitigated phenotypic switching of smooth muscle cells by activating SRF and inhibiting STAT3, while rescued endothelial cells from impaired growth and releasing of prothrombogenic tissue factor. 43
Taken together, given reliving more normal functions of narrowing vessels, election of appropriate drugs and local delivery modes is required for: treating inflammation and restoring effective efferocytosis; targeting thrombi; preventing monocyte recruiting and subsequent macrophage accumulation; altering lipid metabolism; holding back SMC phenotypic modulation, reducing ROS production, and protecting endothelium. And, based on different shift strategies, the future directions should be considered separately as depiction of Figure 11.
Figure 11.

The promising treatment of atherosclerosis in development and the trends.
Footnotes
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the financial support of National Natural Science Foundation of China (No. 31,870,955#) and National Key Research and Development of China (2020YFC1107303#).
ORCID iD: Li Li  https://orcid.org/0000-0001-8834-9837
https://orcid.org/0000-0001-8834-9837
References
- 1. Liu Y, Chen BPC, Lu M, et al. Shear stress activation of SREBP1 in endothelial cells is mediated by integrins. Arterioscler Thromb Vasc Biol 2002; 22: 76–81. [DOI] [PubMed] [Google Scholar]
- 2. Gu L, Okada Y, Clinton SK, et al. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor–deficient mice. Mol Cell 1998; 2: 275–281. [DOI] [PubMed] [Google Scholar]
- 3. Mestas J, Ley K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc Med 2008; 18: 228–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Steinberg D. Low density lipoprotein oxidation and its pathobiological significance. J Biol Chem 1997; 272: 20963–20966. [DOI] [PubMed] [Google Scholar]
- 5. Hossain E, Ota A, Karnan S, et al. Lipopolysaccharide augments the uptake of oxidized LDL by up-regulating lectin-like oxidized LDL receptor-1 in macrophages. Mol Cell Biochem 2015; 400: 29–40. [DOI] [PubMed] [Google Scholar]
- 6. Lievens D, Zernecke A, Seijkens T, et al. Platelet CD40L mediates thrombotic and inflammatory processes in atherosclerosis. Blood 2010; 116: 4317–4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nording H, Baron L, Langer HF. Platelets as therapeutic targets to prevent atherosclerosis. Atherosclerosis 2020; 307: 97–108. [DOI] [PubMed] [Google Scholar]
- 8. Henn V, Slupsky JR, Gräfe M, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 1998; 391: 591–594. [DOI] [PubMed] [Google Scholar]
- 9. Min JK, Chandrashekhar Y. Atherosclerosis, stenosis, and ischemia: one primary, one secondary, and one tertiary. JACC Cardiovasc Imaging 2018; 11: 531–533. [DOI] [PubMed] [Google Scholar]
- 10. Ahmadi A, Stone GW, Leipsic J, et al. Association of coronary stenosis and plaque morphology with fractional flow reserve and outcomes. JAMA Cardiol 2016; 1: 350–357. [DOI] [PubMed] [Google Scholar]
- 11. Bejarano J, Navarro-Marquez M, Morales-Zavala F, et al. Nanoparticles for diagnosis and therapy of atherosclerosis and myocardial infarction: evolution toward prospective theranostic approaches. Theranostics 2018; 8: 4710–4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Her A-Y, Shin E-S, Bang LH, et al. Drug-coated balloon treatment in coronary artery disease: Recommendations from an Asia-Pacific Consensus Group. Cardiol J 2021; 28: 136–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kleber F, Mathey D, Rittger H, et al. How to use the drug-eluting balloon: recommendations by the German consensus group. EuroIntervention 2011; 7: K125–K128. [DOI] [PubMed] [Google Scholar]
- 14. Schnitzler JG, Dzobo KE, Nurmohamed NS, et al. Surmounting the endothelial barrier for delivery of drugs and imaging tracers. Atherosclerosis 2020; 315: 93–101. [DOI] [PubMed] [Google Scholar]
- 15. Sager HB, Dutta P, Dahlman JE, et al. RNAi targeting multiple cell adhesion molecules reduces immune cell recruitment and vascular inflammation after myocardial infarction. Sci Transl Med 2016; 8: 342ra80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reinisch W, Hung K, Hassan-Zahraee M, et al. Targeting endothelial ligands: ICAM-1/alicaforsen, MAdCAM-1. J Crohns Colitis 2018; 12: S669–S677. [DOI] [PubMed] [Google Scholar]
- 17. Perkins LA, Anderson CJ, Novelli EM. Targeting P-selectin adhesion molecule in molecular imaging: P-selectin expression as a valuable imaging biomarker of inflammation in cardiovascular disease. J Nucl Med 2019; 60: 1691–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang K, Zhou Z, Zhou X, et al. Prevention of intimal hyperplasia with recombinant soluble P-selectin glycoprotein ligand-immunoglobulin in the porcine coronary artery balloon injury model. J Am Coll Cardiol 2001; 38: 577–582. [DOI] [PubMed] [Google Scholar]
- 19. Bachelet L, Bertholon I, Lavigne D, et al. Affinity of low molecular weight fucoidan for P-selectin triggers its binding to activated human platelets. Biochim Biophys Acta Gen Subjects 2009; 1790: 141–146. [DOI] [PubMed] [Google Scholar]
- 20. Alfonso F, Angiolillo DJ. Targeting P-selectin during coronary interventions: the elusive link between inflammation and platelets to prevent myocardial damage. J Am Coll Cardiol 2013; 61: 2056–2059. [DOI] [PubMed] [Google Scholar]
- 21. Kanter J, Lanzkron S. Innovations in targeted anti-adhesion treatment for sickle cell disease. Clin Pharmacol Ther 2020; 107: 140–146. [DOI] [PubMed] [Google Scholar]
- 22. Apostolakis S, Spandidos D. Chemokines and atherosclerosis: focus on the CX3CL1/CX3CR1 pathway. Acta Pharmacol Sin 2013; 34: 1251–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Riopel M, Vassallo M, Ehinger E, et al. CX3CL1-Fc treatment prevents atherosclerosis in Ldlr KO mice. Mol Metab 2019; 20: 89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Garnacho C, Muro S. ICAM-1 targeting, intracellular trafficking, and functional activity of polymer nanocarriers coated with a fibrinogen-derived peptide for lysosomal enzyme replacement. J Drug Target 2017; 25: 786–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kheirolomoom A, Kim CW, Seo JW, et al. Multifunctional nanoparticles facilitate molecular targeting and miRNA delivery to inhibit atherosclerosis in ApoE-/- mice. ACS Nano 2015; 9: 8885–8897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bala G, Blykers A, Xavier C, et al. Targeting of vascular cell adhesion molecule-1 by 18F-labelled nanobodies for PET/CT imaging of inflamed atherosclerotic plaques. Eur Heart J Cardiovasc Imaging 2016; 17: 1001–1008. [DOI] [PubMed] [Google Scholar]
- 27. Rouleau L, Berti R, Ng VWK, et al. VCAM-1-targeting gold nanoshell probe for photoacoustic imaging of atherosclerotic plaque in mice. Contrast Media Mol Imaging 2013; 8: 27–39. [DOI] [PubMed] [Google Scholar]
- 28. Kelly KA, Allport JR, Tsourkas A, et al. Detection of vascular adhesion molecule-1 expression using a novel multimodal nanoparticle. Circ Res 2005; 96: 327–336. [DOI] [PubMed] [Google Scholar]
- 29. Antonov AS, Antonova GN, Munn DH, et al. αVβ3 integrin regulates macrophage inflammatory responses via PI3 kinase/Akt-dependent NF-κB activation. J Cell Physiol 2011; 226: 469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yoo JS, Lee J, Jung JH, et al. SPECT/CT imaging of high-risk atherosclerotic plaques using integrin-binding RGD dimer peptides. Sci Rep 2015; 5: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Howe KL, Fish JE. Transforming endothelial cells in atherosclerosis. Nat Metab 2019; 1: 856–857. [DOI] [PubMed] [Google Scholar]
- 32. Chen P-Y, Qin L, Li G, et al. Endothelial TGF-β signalling drives vascular inflammation and atherosclerosis. Nat Metab 2019; 1: 912–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res 2016; 118: 692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vengrenyuk Y, Nishi H, Long X, et al. Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler Thromb Vasc Biol 2015; 35: 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shankman LS, Gomez D, Cherepanova OA, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med 2015; 21: 628–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cordes KR, Sheehy NT, White MP, et al. MiR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 2009; 460: 705–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Santulli G. MicroRNAs distinctively regulate vascular smooth muscle and endothelial cells: functional implications in angiogenesis, atherosclerosis, and in-stent restenosis. Adv Exp Med Biol 2015; 887: 53–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Turczyńska KM, Hellstrand P, Swärd K, et al. Regulation of vascular smooth muscle mechanotransduction by micrornas and L-type calcium channels. Commun Integr Biol 2013; 6: e22278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ouyang QF, Han Y, Lin ZH, et al. Fluvastatin upregulates the α 1C subunit of CaV1.2 channel expression in vascular smooth muscle cells via RhoA and ERK/p38 MAPK pathways. Dis Markers 2014; 2014: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Torella D, Iaconetti C, Catalucci D, et al. MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ Res 2011; 109: 880–893. [DOI] [PubMed] [Google Scholar]
- 41. Fujiu K, Manabe I, Ishihara A, et al. Synthetic retinoid Am80 suppresses smooth muscle phenotypic modulation and in-stent neointima formation by inhibiting KLF5. Circ Res 2005; 97: 1132–1141. [DOI] [PubMed] [Google Scholar]
- 42. Liao XH, Wang N, Zhao DW, et al. STAT3 protein regulates vascular smooth muscle cell phenotypic switch by interaction with myocardin. J Biol Chem 2015; 290: 19641–19652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang B, Zhang M, Urabe G, et al. PERK inhibition mitigates restenosis and thrombosis: a potential low-thrombogenic antirestenotic paradigm. JACC Basic Transl Sci 2020; 5: 245–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tong L, Qi G. Crocin prevents platelet-derived growth factor BB-induced vascular smooth muscle cells proliferation and phenotypic switch. Mol Med Rep 2018; 17: 7595–7602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jain M, Dhanesha N, Doddapattar P, et al. Smooth muscle cell-specific PKM2 (pyruvate kinase muscle 2) promotes smooth muscle cell phenotypic switching and neointimal hyperplasia. Arterioscler Thromb Vasc Biol 2021; 41: 1724–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mack CP. Signaling mechanisms that regulate smooth muscle cell differentiation. Arterioscler Thromb Vasc Biol 2011; 31: 1495–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thomas JA, Deaton RA, Hastings NE, et al. PDGF-DD, a novel mediator of smooth muscle cell phenotypic modulation, is upregulated in endothelial cells exposed to atherosclerosis-prone flow patterns. Am J Physiol Heart Circ Physiol 2009; 296: H442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yoshida T, Gan Q, Owens GK. Krüppel-like factor 4, Elk-1, and histone deacetylases cooperatively suppress smooth muscle cell differentiation markers in response to oxidized phospholipids. Am J Physiol Cell Physiol 2008; 295: C1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Deaton RA, Gan Q, Owens GK. Sp1-dependent activation of KLF4 is required for PDGF-BB-induced phenotypic modulation of smooth muscle. Am J Physiol Hear Circ Physiol 2009; 296: H1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Basatemur GL, Jørgensen HF, Clarke MCH, et al. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol 2019; 16: 727–744. [DOI] [PubMed] [Google Scholar]
- 51. Clément N, Gueguen M, Glorian M, et al. Notch3 and IL-1β exert opposing effects on a vascular smooth muscle cell inflammatory pathway in which NF-κB drives crosstalk. J Cell Sci 2007; 120: 3352–3361. [DOI] [PubMed] [Google Scholar]
- 52. Kolodgie FD, Petrov A, Virmani R, et al. Targeting of apoptotic macrophages and experimental atheroma with radiolabeled annexin V: a technique with potential for noninvasive imaging of vulnerable plaque. Circulation 2003; 108: 3134–3139. [DOI] [PubMed] [Google Scholar]
- 53. Mallat Z, Tedgui A. Apoptosis in the vasculature: mechanisms and functional importance. Br J Pharmacol 2000; 130: 947–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Choy JC, Granville DJ, Hunt DWC, et al. Endothelial cell apoptosis: biochemical characteristics and potential implications for atherosclerosis. J Mol Cell Cardiol 2001; 33: 1673–1690. [DOI] [PubMed] [Google Scholar]
- 55. Linton MF, Babaev VR, Huang J, et al. Macrophage apoptosis and efferocytosis in the pathogenesis of atherosclerosis. Circ J 2016; 80: 2259–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Seimon T, Tabas I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. J Lipid Res 2009; 50: S382–S387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stoneman V, Bennett M. Role of apoptosis in atherosclerosis and its therapeutic implications. Clin Sci 2004; 107: 343–354. [DOI] [PubMed] [Google Scholar]
- 58. Li Y, Yang C, Zhang L, et al. MicroRNA-210 induces endothelial cell apoptosis by directly targeting PDK1 in the setting of atherosclerosis. Cell Mol Biol Lett 2017; 22: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Feng M, Xu D, Wang L. MiR-26a inhibits atherosclerosis progression by targeting TRPC3. Cell Biosci 2018; 8: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang Y, Qin W, Zhang L, et al. MicroRNA-26a prevents endothelial cell apoptosis by directly targeting TRPC6 in the setting of atherosclerosis. Sci Rep 2015; 5(1): 940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li Y, Yang N, Dong B, et al. MicroRNA-122 promotes endothelial cell apoptosis by targeting XIAP: therapeutic implication for atherosclerosis. Life Sci 2019; 232: 116590. [DOI] [PubMed] [Google Scholar]
- 62. Qin B, Xiao B, Liang D, et al. MicroRNAs expression in ox-LDL treated HUVECs: mir-365 modulates apoptosis and Bcl-2 expression. Biochem Biophys Res Commun 2011; 410: 127–133. [DOI] [PubMed] [Google Scholar]
- 63. Zhang T, Tian F, Wang J, et al. Atherosclerosis-associated endothelial cell apoptosis by MiR-429-mediated down regulation of Bcl-2. Cell Physiol Biochem 2015; 37: 1421–1430. [DOI] [PubMed] [Google Scholar]
- 64. Qin B, Shu Y, Long L, et al. MicroRNA-142-3p induces atherosclerosis-associated endothelial cell apoptosis by directly targeting rictor. Cell Physiol Biochem 2018; 47: 1589–1603. [DOI] [PubMed] [Google Scholar]
- 65. Chen C, Cheng G, Yang X, et al. Tanshinol suppresses endothelial cells apoptosis in mice with atherosclerosis via lncRNA TUG1 up-regulating the expression of miR-26a. Am J Transl Res 2016; 8: 2981–2991. [PMC free article] [PubMed] [Google Scholar]
- 66. Liang X, Wang L, Wang M, et al. MicroRNA-124 inhibits macrophage cell apoptosis via targeting p38/MAPK signaling pathway in atherosclerosis development. Aging 2020; 12: 13005–13022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tian H, Yao ST, Yang NN, et al. D4F alleviates macrophage-derived foam cell apoptosis by inhibiting the NF-κB-dependent Fas/FasL pathway. Sci Rep 2017; 7: 7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cho SW, Hartle L, Son SM, et al. Delivery of small interfering RNA for inhibition of endothelial cell apoptosis by hypoxia and serum deprivation. Biochem Biophys Res Commun 2008; 376: 158–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Drew BA, Burow ME, Beckman BS. MEK5/ERK5 pathway: the first fifteen years. Biochim Biophys Acta 2012; 1825: 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nithianandarajah-Jones GN, Wilm B, Goldring CEP, et al. ERK5: structure, regulation and function. Cell Signal 2012; 24: 2187–2196. [DOI] [PubMed] [Google Scholar]
- 71. Martinet W, Coornaert I, Puylaert P, et al. Macrophage death as a pharmacological target in atherosclerosis. Front Pharmacol 2019; 10: 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nithianandarajah-Jones G, Wilm B, Goldring CP, et al. The role of ERK5 in endothelial cell function. Biochem Soc Trans 2014; 42: 1584–1589. [DOI] [PubMed] [Google Scholar]
- 73. Heo KS, Cushman HJ, Akaike M, et al. ERK5 activation in macrophages promotes efferocytosis and inhibits atherosclerosis. Circulation 2014; 130: 180–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zeisel SH. Antioxidants suppress apoptosis. J Nutr 2004; 134: 3179S–3180S. [DOI] [PubMed] [Google Scholar]
- 75. Van Vré EA, Ait-Oufella H, Tedgui A, et al. Apoptotic cell death and efferocytosis in atherosclerosis. Arterioscler Thromb Vasc Biol 2012; 32: 887–893. [DOI] [PubMed] [Google Scholar]
- 76. Boada-Romero E, Martinez J, Heckmann BL, et al. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol 2020; 21: 398–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Blanc-Brude OP, Teissier E, Castier Y, et al. IAP survivin regulates atherosclerotic macrophage survival. Arterioscler Thromb Vasc Biol 2007; 27: 901–907. [DOI] [PubMed] [Google Scholar]
- 78. Zhang Y, Wang Y, Zhou D, et al. Angiotensin II deteriorates advanced atherosclerosis by promoting MerTK cleavage and impairing efferocytosis through the AT1R/ROS/p38 MAPK/ADAM17 pathway. Am J Physiol Cell Physiol 2019; 317: C776–C787. [DOI] [PubMed] [Google Scholar]
- 79. Morrissey MA, Kern N, Vale RD. CD47 ligation repositions the inhibitory receptor SIRPA to suppress integrin activation and phagocytosis. Immunity 2020; 53: 290–302.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ryan JJ. CD47-Blocking antibodies and atherosclerosis. JACC Basic Transl Sci 2016; 1: 413–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kojima Y, Volkmer JP, McKenna K, et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 2016; 536: 86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chen W, Li X, Wang J, et al. MiR-378a modulates macrophage phagocytosis and differentiation through targeting CD47-SIRPα axis in atherosclerosis. Scand J Immunol 2019; 90: e12766. [DOI] [PubMed] [Google Scholar]
- 83. Flores AM, Hosseini-Nassab N, Jarr KU, et al. Pro-efferocytic nanoparticles are specifically taken up by lesional macrophages and prevent atherosclerosis. Nat Nanotechnol 2020; 15: 154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhang Y, Ye J, Hosseini-Nassab N, et al. Macrophage-targeted single walled carbon nanotubes stimulate phagocytosis via pH-dependent drug release. Nano Res 2021; 14: 762–769. [Google Scholar]
- 85. Huang Y, Zhu L, Tan J, et al. Correlation between SHP-1 and carotid plaque vulnerability in humans. Cardiovasc Pathol 2020; 49: 107258. [DOI] [PubMed] [Google Scholar]
- 86. Cheng Q, Gu J, Adhikari BK, et al. Is CD47 a potentially promising therapeutic target in cardiovascular diseases?: role of CD47 in cardiovascular diseases. Life Sci 2020; 247: 117426. [DOI] [PubMed] [Google Scholar]
- 87. Schutters K, Kusters DHM, Chatrou MLL, et al. Cell surface-expressed phosphatidylserine as therapeutic target to enhance phagocytosis of apoptotic cells. Cell Death Differ 2013; 20: 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Pirillo A, Norata GD, Catapano AL. LOX-1, OxLDL, and atherosclerosis. Mediators Inflamm 2013; 2013: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Chen M, Kakutani M, Naruko T, et al. Activation-dependent surface expression of LOX-1 in human platelets. Biochem Biophys Res Commun 2001; 282: 153–158. [DOI] [PubMed] [Google Scholar]
- 90. Kataoka H, Kume N, Miyamoto S, et al. Expression of lectinlike oxidized low-density lipoprotein receptor-1 in human atherosclerotic lesions. Circulation 1999; 99: 3110–3117. [DOI] [PubMed] [Google Scholar]
- 91. Li DY, Chen HJ, Staples ED, et al. Oxidized low-density lipoprotein receptor LOX-1 and apoptosis in human atherosclerotic lesions. J Cardiovasc Pharmacol Ther 2002; 7: 147–153. [DOI] [PubMed] [Google Scholar]
- 92. Ishino S, Mukai T, Kume N, et al. Lectin-like oxidized LDL receptor-1 (LOX-1) expression is associated with atherosclerotic plaque instability—analysis in hypercholesterolemic rabbits. Atherosclerosis 2007; 195: 48–56. [DOI] [PubMed] [Google Scholar]
- 93. Ishino S, Mukai T, Kuge Y, et al. Targeting of lectinlike oxidized low-density lipoprotein receptor 1 (LOX-1) with 99mTc-labeled Anti–LOX-1 antibody: potential agent for imaging of vulnerable plaque. J Nucl Med 2008; 49: 1677–1685. [DOI] [PubMed] [Google Scholar]
- 94. Saito A, Shimizu H, Doi Y, et al. Immunoliposomal drug-delivery system targeting lectin-like oxidized low-density lipoprotein receptor-1 for carotid plaque lesions in rats: laboratory investigation. J Neurosurg 2011; 115: 720–727. [DOI] [PubMed] [Google Scholar]
- 95. Hinagata J, Kakutani M, Fujii T, et al. Oxidized LDL receptor LOX-1 is involved in neointimal hyperplasia after balloon arterial injury in a rat model. Cardiovasc Res 2006; 69: 263–271. [DOI] [PubMed] [Google Scholar]
- 96. Yao EH, Fukuda N, Ueno T, et al. Novel gene silencer pyrrole-imidazole polyamide targeting lectin-like oxidized low-density lipoprotein receptor-1 attenuates restenosis of the artery after injury. Hypertension 2008; 52: 86–92. [DOI] [PubMed] [Google Scholar]
- 97. Kaimin L, Ziwen Z, Lianglong C. Effects of PIP targeting LOX-1 eluting stents on in-stent restenosis and re-endothelialisation in rat abdominal aorta stenting models. Heart 2011; 97: A66–A67. [Google Scholar]
- 98. Fan J, Liu L, Liu Q, et al. CKIP-1 limits foam cell formation and inhibits atherosclerosis by promoting degradation of Oct-1 by REGγ. Nat Commun 2019; 10: 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Fu L, Zhang L. Physiological functions of CKIP-1: from molecular mechanisms to therapy implications. Ageing Res Rev 2019; 53: 100908. [DOI] [PubMed] [Google Scholar]
- 100. Geloen A, Helin L, Geeraert B, et al. CD36 inhibitors reduce postprandial hypertriglyceridemia and protect against diabetic dyslipidemia and atherosclerosis. PLoS One 2012; 7: e37633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Plourde NM, Kortagere S, Welsh W, et al. Structure−activity relations of nanolipoblockers with the atherogenic domain of human macrophage scavenger receptor A. Biomacromolecules 2009; 10: 1381–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Takai M, Kozai Y, Tsuzuki S, et al. Unsaturated long-chain fatty acids inhibit the binding of oxidized low-density lipoproteins to a model CD36. Biosci Biotechnol Biochem 2014; 78(2): 238–244. [DOI] [PubMed] [Google Scholar]
- 103. Tashiro Y, Sato K, Watanabe T, et al. A glucagon-like peptide-1 analog liraglutide suppresses macrophage foam cell formation and atherosclerosis. Peptides 2014; 54: 19–26. [DOI] [PubMed] [Google Scholar]
- 104. Williams KJ, Feig JE, Fisher EA. Rapid regression of atherosclerosis: insights from the clinical and experimental literature. Nat Clin Pract Cardiovasc Med 2008; 5: 91–102. [DOI] [PubMed] [Google Scholar]
- 105. Spady DK. Reverse cholesterol transport and atherosclerosis regression. Circulation 1999; 100: 576–578. [DOI] [PubMed] [Google Scholar]
- 106. Chen SJ, Tsui PF, Chuang YP, et al. Carvedilol ameliorates experimental atherosclerosis by regulating cholesterol efflux and exosome functions. Int J Mol Sci 2019; 20: 5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Wójcicka G, Jamroz-Wiśniewska A, Horoszewicz K, et al. Liver X receptors (LXRs). Part I: Structure, function, regulation of activity, and role in lipid metabolism. Postpy Hig Med Dośw 2007; 61: 736–759. [PubMed] [Google Scholar]
- 108. Sodhi RK, Singh N. Liver X receptors: emerging therapeutic targets for Alzheimer’s disease. Pharmacol Res 2013; 72: 45–51. [DOI] [PubMed] [Google Scholar]
- 109. Sun L, Li E, Wang F, et al. Quercetin increases macrophage cholesterol efflux to inhibit foam cell formation through activating PPARγ-ABCA1 pathway. Int J Clin Exp Pathol 2015; 8: 10854–10860. [PMC free article] [PubMed] [Google Scholar]
- 110. Ren K, Li H, Zhou HF, et al. Mangiferin promotes macrophage cholesterol efflux and protects against atherosclerosis by augmenting the expression of ABCA1 and ABCG1. Aging 2019; 11: 10992–11009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Libby P, Plutzky J. Inflammation in diabetes mellitus: role of peroxisome proliferator-activated receptor-α and peroxisome proliferator-activated receptor-γ agonists. Am J Cardiol 2007; 99: 27–40. [DOI] [PubMed] [Google Scholar]
- 112. Zimmer S, Grebe A, Bakke SS, et al. Cyclodextrin promotes atherosclerosis regression via macrophage reprogramming. Sci Transl Med 2016; 8: 333ra50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Han QA, Su D, Shi C, et al. Urolithin A attenuated ox-LDL-induced cholesterol accumulation in macrophages partly through regulating miR-33a and ERK/AMPK/SREBP1 signaling pathways. Food Funct 2020; 11: 3432–3440. [DOI] [PubMed] [Google Scholar]
- 114. Kostopoulou F, Malizos KN, Papathanasiou I, et al. MicroRNA-33a regulates cholesterol synthesis and cholesterol efflux-related genes in osteoarthritic chondrocytes. Arthritis Res Ther 2015; 17: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Xu Y, Xu Y, Zhu Y, et al. Macrophage miR-34a is a key regulator of cholesterol efflux and atherosclerosis. Mol Ther 2020; 28: 202–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Adorni MP, Cipollari E, Favari E, et al. Inhibitory effect of PCSK9 on abca1 protein expression and cholesterol efflux in macrophages. Atherosclerosis 2017; 256: 1–6. [DOI] [PubMed] [Google Scholar]
- 117. Momtazi-Borojeni AA, Jaafari MR, Badiee A, et al. Therapeutic effect of nanoliposomal PCSK9 vaccine in a mouse model of atherosclerosis. BMC Med 2019; 17: 223. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 118. Moriya J. Critical roles of inflammation in atherosclerosis. J Cardiol 2019; 73: 22–27. [DOI] [PubMed] [Google Scholar]
- 119. Warnatsch A, Ioannou M, Wang Q, et al. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015; 349: 316–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Baldrighi M, Mallat Z, Li X. NLRP3 inflammasome pathways in atherosclerosis. Atherosclerosis 2017; 267: 127–138. [DOI] [PubMed] [Google Scholar]
- 121. Karasawa T, Takahashi M. Role of NLRP3 inflammasomes in atherosclerosis. J Atheroscler Thromb 2017; 24: 443–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Grebe A, Hoss F, Latz E. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ Res 2018; 122: 1722–1740. [DOI] [PubMed] [Google Scholar]
- 123. Minutoli L, Puzzolo D, Rinaldi M, et al. ROS-mediated NLRP3 inflammasome activation in brain, heart, kidney, and testis ischemia/reperfusion injury. Oxid Med Cell Longev 2016; 2016: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Jin Y, Fu J. Novel insights into the NLRP3 inflammasome in atherosclerosis. J Am Heart Assoc 2019; 8: e012219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Libby P. Interleukin-1 beta as a target for atherosclerosis therapy: biological basis of CANTOS and beyond. J Am Coll Cardiol 2017; 70: 2278–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Donath MY. Targeting inflammation in the treatment of type 2 diabetes: time to start. Nat Rev Drug Discov 2014; 13: 465–476. [DOI] [PubMed] [Google Scholar]
- 127. Wang Y, Ji N, Gong X, et al. Thioredoxin-1 attenuates atherosclerosis development through inhibiting NLRP3 inflammasome. Endocrine 2020; 70: 65–70. [DOI] [PubMed] [Google Scholar]
- 128. Coll RC, Robertson AAB, Chae JJ, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 2015; 21: 248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Shao BZ, Xu ZQ, Han BZ, et al. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol 2015; 6: 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Zahid A, Li B, Kombe AJK, et al. Pharmacological inhibitors of the nlrp3 inflammasome. Front Immunol 2019; 10: 2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Peng Z, Zhan H, Shao Y, et al. 13-Methylberberine improves endothelial dysfunction by inhibiting NLRP3 inflammasome activation via autophagy induction in human umbilical vein endothelial cells. Chin Med 2020; 15: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Zhang Y, Liu X, Bai X, et al. Melatonin prevents endothelial cell pyroptosis via regulation of long noncoding RNA MEG3/miR-223/NLRP3 axis. J Pineal Res 2018; 64: e12449. [DOI] [PubMed] [Google Scholar]
- 133. Zhaolin Z, Guohua L, Shiyuan W, et al. Role of pyroptosis in cardiovascular disease. Cell Prolif 2019; 52: e12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Li Y, Niu X, Xu H, et al. VX-765 attenuates atherosclerosis in ApoE deficient mice by modulating VSMCs pyroptosis. Exp Cell Res 2020; 389: 111847. [DOI] [PubMed] [Google Scholar]
- 135. Liu T, Zhang L, Joo D, et al. NF-κB signaling in inflammation. Signal Transduct Target Ther 2017; 2(1): 7023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Song D, Fang G, Mao SZ, et al. Selective inhibition of endothelial NF-κB signaling attenuates chronic intermittent hypoxia-induced atherosclerosis in mice. Atherosclerosis 2018; 270: 68–75. [DOI] [PubMed] [Google Scholar]
- 137. He L, Weng H, Li Q, et al. Lactucopicrin inhibits cytoplasmic dynein-mediated NF-κB activation in inflammated macrophages and alleviates atherogenesis in apolipoprotein E-deficient mice. Mol Nutr Food Res 2021; 65: e2000989. [DOI] [PubMed] [Google Scholar]
- 138. Mallavia B, Recio C, Oguiza A, et al. Peptide inhibitor of NF-κB translocation ameliorates experimental atherosclerosis. Am J Pathol 2013; 182: 1910–1921. [DOI] [PubMed] [Google Scholar]
- 139. Csiszar A, Smith K, Labinskyy N, et al. Resveratrol attenuates TNF-α-induced activation of coronary arterial endothelial cells: role of NF-κB inhibition. Am J Physiol Hear Circ Physiol 2006; 291: 1694–1699. [DOI] [PubMed] [Google Scholar]
- 140. Tousoulis D, Oikonomou E, Economou EK, et al. Inflammatory cytokines in atherosclerosis: current therapeutic approaches. Eur Heart J 2016; 37: 1723–1732. [DOI] [PubMed] [Google Scholar]
- 141. Ridker PM. Targeting inflammatory pathways for the treatment of cardiovascular disease. Eur Heart J 2014; 35: 540–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Sager HB, Heidt T, Hulsmans M, et al. Targeting interleukin-1β reduces leukocyte production after acute myocardial infarction. Circulation 2015; 132: 1880–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Zhao X, Kong J, Zhao Y, et al. Gene silencing of TACE enhances plaque stability and improves vascular remodeling in a rabbit model of atherosclerosis. Sci Rep 2015; 5: 17939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Wang Z, Wesche H, Stevens T, et al. IRAK-4 inhibitors for inflammation. Curr Top Med Chem 2009; 9: 724–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Dou H, Song Y, Liu X, et al. A novel benzenediamine derivate rescued mice from experimental sepsis by attenuating proinflammatory mediators via IRAK4. Am J Respir Cell Mol Biol 2014; 51: 191–200. [DOI] [PubMed] [Google Scholar]
- 146. Chaudhary D, Robinson S, Romero DL. Recent advances in the discovery of small molecule inhibitors of interleukin-1 receptor-associated kinase 4 (IRAK4) as a therapeutic target for inflammation and oncology disorders. J Med Chem 2015; 58: 96–110. [DOI] [PubMed] [Google Scholar]
- 147. Smith GF, Altman MD, Andresen B, et al. Identification of quinazoline based inhibitors of IRAK4 for the treatment of inflammation. Bioorg Med Chem Lett 2017; 27: 2721–2726. [DOI] [PubMed] [Google Scholar]
- 148. Boraschi D, Tagliabue A. The interleukin-1 receptor family. Semin Immunol 2013; 25: 394–407. [DOI] [PubMed] [Google Scholar]
- 149. Puhlmann M, Weinreich DM, Farma JM, et al. Interleukin-1β induced vascular permeability is dependent on induction of endothelial tissue factor (TF) activity. J Transl Med 2005; 3: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Ding Z, Pothineni NVK, Goel A, et al. PCSK9 and inflammation: role of shear stress, pro-inflammatory cytokines, and LOX-1. Cardiovasc Res 2020; 116: 908–915. [DOI] [PubMed] [Google Scholar]
- 151. Li S, Zhang Y, Xu RX, et al. Proprotein convertase subtilisin-kexin type 9 as a biomarker for the severity of coronary artery disease. Ann Med 2015; 47: 386–393. [DOI] [PubMed] [Google Scholar]
- 152. Feingold KR, Moser AH, Shigenaga JK, et al. Inflammation stimulates the expression of PCSK9. Biochem Biophys Res Commun 2008; 374: 341–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Lin XL, Xiao LL, Tang ZH, et al. Role of PCSK9 in lipid metabolism and atherosclerosis. Biomed Pharmacother 2018; 104: 36–44. [DOI] [PubMed] [Google Scholar]
- 154. Sun H, Krauss RM, Chang JT, et al. PCSK9 deficiency reduces atherosclerosis, apolipoprotein b secretion, and endothelial dysfunction. J Lipid Res 2018; 59: 207–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Tang ZH, Peng J, Ren Z, et al. New role of PCSK9 in atherosclerotic inflammation promotion involving the TLR4/NF-κB pathway. Atherosclerosis 2017; 262: 113–122. [DOI] [PubMed] [Google Scholar]
- 156. Kühnast S, van der Hoorn JWA, Pieterman EJ, et al. Alirocumab inhibits atherosclerosis, improves the plaque morphology, and enhances the effects of a statin. J Lipid Res 2014; 55: 2103–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Denis M, Marcinkiewicz J, Zaid A, et al. Gene inactivation of proprotein convertase subtilisin/kexin type 9 reduces atherosclerosis in mice. Circulation 2012; 125: 894–901. [DOI] [PubMed] [Google Scholar]
- 158. Urban D, Pöss J, Böhm M, et al. Targeting the proprotein convertase subtilisin/kexin type 9 for the treatment of dyslipidemia and atherosclerosis. J Am Coll Cardiol 2013; 62: 1401–1408. [DOI] [PubMed] [Google Scholar]
- 159. Wu CY, Tang ZH, Jiang L, et al. PCSK9 siRNA inhibits HUVEC apoptosis induced by ox-LDL via Bcl/Bax–caspase9–caspase3 pathway. Mol Cell Biochem 2012; 359: 347–358. [DOI] [PubMed] [Google Scholar]
- 160. Ding Z, Liu S, Wang X, et al. Cross-talk between LOX-1 and PCSK9 in vascular tissues. Cardiovasc Res 2015; 107: 556–567. [DOI] [PubMed] [Google Scholar]
- 161. Ding Z, Liu S, Wang X, et al. Hemodynamic shear stress via ROS modulates PCSK9 expression in human vascular endothelial and smooth muscle cells and along the mouse aorta. Antioxid Redox Signal 2015; 22: 760–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Levi M, van der Poll T. Inflammation and coagulation. Crit Care Med 2010; 38: S26–S34. [DOI] [PubMed] [Google Scholar]
- 163. Foley JH, Conway EM. Cross talk pathways between coagulation and inflammation. Circ Res 2016; 118: 1392–1408. [DOI] [PubMed] [Google Scholar]
- 164. Posma JJ, Grover SP, Hisada Y, et al. Roles of coagulation proteases and PARs (protease-activated receptors) in mouse models of inflammatory diseases. Arterioscler Thromb Vasc Biol 2019; 39: 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Chackalamannil S. Thrombin receptor (protease activated receptor-1) antagonists as potent antithrombotic agents with strong antiplatelet effects. J Med Chem 2006; 49: 5389–5403. [DOI] [PubMed] [Google Scholar]
- 166. O'Brien PJ, Prevost N, Molino M, et al. Thrombin responses in human endothelial cells. J Biol Chem 2000; 275: 13502–13509. [DOI] [PubMed] [Google Scholar]
- 167. Daubie V, Cauwenberghs S, Senden NHM, et al. Factor Xa and thrombin evoke additive calcium and proinflammatory responses in endothelial cells subjected to coagulation. Biochim Biophys Acta Mol Cell Res 2006; 1763: 860–869. [DOI] [PubMed] [Google Scholar]
- 168. Koo BH, Chung KH, Hwang KC, et al. Factor xa induces mitogenesis of coronary artery smooth muscle cell via activation of PAR-2. FEBS Lett 2002; 523: 85–89. [DOI] [PubMed] [Google Scholar]
- 169. Zuo P, Zuo Z, Wang X, et al. Factor Xa induces pro-inflammatory cytokine expression in RAW 264.7 macrophages via protease-activated receptor-2 activation. Am J Transl Res 2015; 7: 2326–2334. [PMC free article] [PubMed] [Google Scholar]
- 170. Loeffen R, Spronk HMH, ten Cate H. The impact of blood coagulability on atherosclerosis and cardiovascular disease. J Thromb Haemost 2012; 10(7): 1207–1216. [DOI] [PubMed] [Google Scholar]
- 171. Hara T, Phuong PT, Fukuda D, et al. Protease-activated receptor-2 plays a critical role in vascular inflammation and atherosclerosis in apolipoprotein E-deficient mice. Circulation 2018; 138: 1706–1719. [DOI] [PubMed] [Google Scholar]
- 172. Schoergenhofer C, Schwameis M, Gelbenegger G, et al. Inhibition of protease-activated receptor (PAR1) reduces activation of the endothelium, coagulation, fibrinolysis and inflammation during human endotoxemia. Thromb Haemost 2018; 118: 1176–1184. [DOI] [PubMed] [Google Scholar]
- 173. Drucker D. The cardiovascular biology of glucagon-like peptide-1. Cell Metab 2016; 24: 15–30. [DOI] [PubMed] [Google Scholar]
- 174. Song X, Jia H, Jiang Y, et al. Anti-atherosclerotic effects of the glucagon-like peptide-1 (GLP-1) based therapies in patients with type 2 diabetes mellitus: a meta-analysis. Sci Rep 2015; 5: 10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Helmstädter J, Frenis K, Filippou K, et al. Endothelial GLP-1 (glucagon-like peptide-1) receptor mediates cardiovascular protection by liraglutide in mice with experimental arterial hypertension. Arterioscler Thromb Vasc Biol 2020; 40: 145–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Rakipovski G, Rolin B, Nøhr J, et al. The GLP-1 analogs liraglutide and semaglutide reduce atherosclerosis in ApoE−/− and LDLr−/− mice by a mechanism that includes inflammatory pathways. JACC Basic Transl Sci 2018; 3: 844–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Wu YC, Wang WT, Lee SS, et al. Glucagon-like peptide-1 receptor agonist attenuates autophagy to ameliorate pulmonary arterial hypertension through Drp1/NOX and Atg-5/Atg-7/Beclin-1/LC3β pathways. Int J Mol Sci 2019; 20: 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Yang G, Li Y, Cui L, et al. Increased plasma dipeptidyl peptidase-4 activities in patients with coronary artery disease. PLoS One 2016; 11: e0163027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Hong O, Lee S, Yoo SJ, et al. Gemigliptin inhibits interleukin-1β-induced endothelial- mesenchymal transition via canonical-bone morphogenetic protein pathway. Endocrinol Metabol 2020; 35: 384–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Matheeussen V, Waumans Y, Martinet W, et al. Dipeptidyl peptidases in atherosclerosis: expression and role in macrophage differentiation, activation and apoptosis. Basic Res Cardiol 2013; 108: 350. [DOI] [PubMed] [Google Scholar]
- 181. Zhong J, Rao X, Rajagopalan S. An emerging role of dipeptidyl peptidase 4 (DPP4) beyond glucose control: Potential implications in cardiovascular disease. Atherosclerosis 2013; 226: 305–314. [DOI] [PubMed] [Google Scholar]
- 182. Choi SH, Park S, Oh CJ, et al. Dipeptidyl peptidase-4 inhibition by gemigliptin prevents abnormal vascular remodeling via NF-E2-related factor 2 activation. Vasc Pharmacol 2015; 73: 11–19. [DOI] [PubMed] [Google Scholar]
- 183. Hirano T, Mori Y. Anti-atherogenic and anti-inflammatory properties of glucagon-like peptide-1, glucose-dependent insulinotropic polypepide, and dipeptidyl peptidase-4 inhibitors in experimental animals. J Diabetes Invest 2016; 7: 80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Matsubara J, Sugiyama S, Sugamura K, et al. A dipeptidyl peptidase-4 inhibitor, des-fluoro-sitagliptin, improves endothelial function and reduces atherosclerotic lesion formation in apolipoprotein E-deficient mice. J Am Coll Cardiol 2012; 59: 265–276. [DOI] [PubMed] [Google Scholar]
- 185. Reejhsinghani R, Lotfi AS. Prevention of stent thrombosis: challenges and solutions. Vasc Health Risk Manag 2015; 11: 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Chen JP, Hou D, Pendyala L, et al. Drug-eluting stent thrombosis: the Kounis hypersensitivity-associated acute coronary syndrome revisited. JACC Cardiovasc Interv 2009; 2: 583–593. [DOI] [PubMed] [Google Scholar]
- 187. Woulfe D, Yang J, Brass L. ADP and platelets: the end of the beginning. J Clin Investig 2001; 107: 1503–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Nieswandt B, Pleines I, Bender M. Platelet adhesion and activation mechanisms in arterial thrombosis and ischaemic stroke. J Thromb Haemost 2011; 9: 92–104. [DOI] [PubMed] [Google Scholar]
- 189. Bhatt DL, Topol EJ. Current role of platelet glycoprotein IIb/IIIa inhibitors in acute coronary syndromes. J Am Med Assoc 2000; 284: 1549–1558. [DOI] [PubMed] [Google Scholar]
- 190. Storey RF, Sanderson HM, White AE, et al. The central role of the P 2t receptor in amplification of human platelet activation, aggregation, secretion and procoagulant activity. Br J Haematol 2000; 110: 925–934. [DOI] [PubMed] [Google Scholar]
- 191. Dornbos D, Katz JS, Youssef P, et al. Glycoprotein IIb/IIIa inhibitors in prevention and rescue treatment of thromboembolic complications during endovascular embolization of intracranial aneurysms. Clin Neurosurg 2018; 82: 268–277. [DOI] [PubMed] [Google Scholar]
- 192. Kontogiorgis C, Hadjipavlou-Litina D. Thromboxane synthase inhibitors and thromboxane A2 receptor antagonists: a quantitative structure activity relationships (QSARs) analysis. Curr Med Chem 2010; 17: 3162–3214. [DOI] [PubMed] [Google Scholar]
- 193. Ye X, Chen M, Chen Y, et al. Isolation and characterization of a novel antithrombotic peptide from enzymatic hydrolysate of agkistrodon acutus venom. Int J Pept Res Ther 2015; 21: 343–351. [Google Scholar]
- 194. Pascale S, Petrucci G, Dragani A, et al. Aspirin-insensitive thromboxane biosynthesis in essential thrombocythemia is explained by accelerated renewal of the drug target. Blood 2012; 119: 3595–3603. [DOI] [PubMed] [Google Scholar]
- 195. Gabrielsen A, Qiu H, Bäck M, et al. Thromboxane synthase expression and thromboxane A2 production in the atherosclerotic lesion. J Mol Med 2010; 88: 795–806. [DOI] [PubMed] [Google Scholar]
- 196. Petri MH, Tellier C, Michiels C, et al. Effects of the dual TP receptor antagonist and thromboxane synthase inhibitor EV-077 on human endothelial and vascular smooth muscle cells. Biochem Biophys Res Commun 2013; 441: 393–398. [DOI] [PubMed] [Google Scholar]
- 197. Morrow DA, Braunwald E, Bonaca MP, et al. Vorapaxar in the secondary prevention of atherothrombotic events. New Engl J Med 2012; 366: 1404–1413. [DOI] [PubMed] [Google Scholar]
- 198. Jia G, Aroor AR, Sowers JR. Glucagon-like peptide 1 receptor activation and platelet function: beyond glycemic control. Diabetes 2016; 65: 1487–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Steven S, Jurk K, Kopp M, et al. Glucagon-like peptide-1 receptor signalling reduces microvascular thrombosis, nitro-oxidative stress and platelet activation in endotoxaemic mice. Br J Pharmacol 2017; 174: 1620–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Chen HH, Vicente CP, He L, et al. Fusion proteins comprising annexin V and Kunitz protease inhibitors are highly potent thrombogenic site-directed anticoagulants. Blood 2005; 105: 3902–3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Shi J, Pipe SW, Rasmussen JT, et al. Lactadherin blocks thrombosis and hemostasis in vivo: correlation with platelet phosphatidylserine exposure. J Thromb Haemost 2008; 6: 1167–1174. [DOI] [PubMed] [Google Scholar]
- 202. Jing J, Sun Y. An αIIbβ3- and phosphatidylserine (PS)-binding recombinant fusion protein promotes PS-dependent anticoagulation and integrin-dependent antithrombosis. J Biol Chem 2019; 294: 6670–6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Yeh CH, Chen TP, Wang YC, et al. Potent cardioprotection from ischemia-reperfusion injury by a two-domain fusion protein comprising annexin V and Kunitz protease inhibitor. J Thromb Haemost 2013; 11: 1454–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Yuan H, Yang X, Hua ZC. Optimization of expression of an Annexin V-Hirudin chimeric protein in Escherichia coli. Microbiol Res 2004; 159: 147–156. [DOI] [PubMed] [Google Scholar]
- 205. Aliter KF, Al-Horani RA. Thrombin inhibition by argatroban: potential therapeutic benefits in COVID-19. Cardiovasc Drugs Ther 2021; 35: 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Liu M, Zaman K, Fortenberry YM. Overview of the therapeutic potential of aptamers targeting coagulation factors. Int J Mol Sci 2021; 22: 3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Hamaoui K, Gowers S, Boutelle M, et al. Organ pretreatment with cytotopic endothelial localizing peptides to ameliorate microvascular thrombosis and perfusion deficits in ex vivo renal hemoreperfusion models. Transplantation 2016; 100: e128–e139. [DOI] [PubMed] [Google Scholar]
- 208. Iyer JK, Koh CY, Kazimirova M, et al. Avathrin: a novel thrombin inhibitor derived from a multicopy precursor in the salivary glands of the ixodid tick, Amblyomma variegatum. FASEB J 2017; 31: 2981–2995. [DOI] [PubMed] [Google Scholar]
- 209. Chen M, Ye X, Ming X, et al. A novel direct factor Xa inhibitory peptide with anti-platelet aggregation activity from Agkistrodon acutus venom hydrolysates. Sci Rep 2015; 5: 10846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Posthuma JJ, Posma JJN, van Oerle R, et al. Targeting coagulation factor Xa promotes regression of advanced atherosclerosis in apolipoprotein-E deficient mice. Sci Rep 2019; 9: 3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Petzold T, Thienel M, Dannenberg L, et al. Rivaroxaban reduces arterial thrombosis by inhibition of FXA-driven platelet activation via protease activated receptor-1. Circ Res 2020; 126: 486–500. [DOI] [PubMed] [Google Scholar]
- 212. Krupka J, May F, Weimer T, et al. The coagulation factor XIIa inhibitor rhainfestin-4 improves outcome after cerebral ischemia/reperfusion injury in rats. PLoS One 2016; 11: e0146783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Ding L, Hao J, Luo X, et al. The Kv1.3 channel-inhibitory toxin BF9 also displays anticoagulant activity via inhibition of factor XIa. Toxicon 2018; 152: 9–15. [DOI] [PubMed] [Google Scholar]
- 214. Assumpção TC, Ma D, Mizurini DM, et al. In vitro mode of action and anti-thrombotic activity of boophilin, a multifunctional Kunitz protease inhibitor from the midgut of a tick vector of babesiosis, rhipicephalus microplus. PLoS Negl Trop Dis 2016; 10: e0004298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Ely LK, Lolicato M, David T, et al. Structural basis for activity and specificity of an anticoagulant anti-FXIa monoclonal antibody and a reversal agent. Structure 2018; 26: 187–198.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Pireaux V, Tassignon J, Demoulin S, et al. Anticoagulation with an inhibitor of factors XIa and XIIa during cardiopulmonary bypass. J Am Coll Cardiol 2019; 74: 2178–2189. [DOI] [PubMed] [Google Scholar]
- 217. Plautz WE, Sekhar Pilli VS, Cooley BC, et al. Anticoagulant protein S targets the factor IXa heparin-binding exosite to prevent thrombosis. Arterioscler Thromb Vasc Biol 2018; 38: 816–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Chowdary P. Inhibition of tissue factor pathway inhibitor (TFPI) as a treatment for haemophilia: rationale with focus on concizumab. Drugs 2018; 78: 881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Winckers K, ten Cate H, Hackeng TM. The role of tissue factor pathway inhibitor in atherosclerosis and arterial thrombosis. Blood Rev 2013; 27: 119–132. [DOI] [PubMed] [Google Scholar]
- 220. Espada S, Stavik B, Holm S, et al. Tissue factor pathway inhibitor attenuates ER stress-induced inflammation in human m2-polarized macrophages. Biochem Biophys Res Commun 2017; 491: 442–448. [DOI] [PubMed] [Google Scholar]
- 221. Cunningham MA, Romas P, Hutchinson P, et al. Tissue factor and factor VIIa receptor/ligand interactions induce proinflammatory effects in macrophages. Blood 1999; 94: 3413–3420. [PubMed] [Google Scholar]
- 222. Xue M, Ren S, Welch S, et al. Hirulog-like peptide reduces balloon catheter injury induced neointima formation in rat carotid artery without increase in bleeding tendency. J Vasc Res 2001; 38: 144–152. [DOI] [PubMed] [Google Scholar]
- 223. Kadoglou NPE, Moustardas P, Katsimpoulas M, et al. The beneficial effects of a direct thrombin inhibitor, dabigatran etexilate, on the development and stability of atherosclerotic lesions in apolipoprotein E-deficient mice. Cardiovasc Drugs Ther 2012; 26: 367–374. [DOI] [PubMed] [Google Scholar]
- 224. Lee IO, Kratz MT, Schirmer SH, et al. The effects of direct thrombin inhibition with dabigatran on plaque formation and endothelial function in apolipoprotein E-deficient mice. J Pharmacol Exp Ther 2012; 343: 253–257. [DOI] [PubMed] [Google Scholar]
- 225. Preusch MR, Ieronimakis N, Wijelath ES, et al. Dabigatran etexilate retards the initiation and progression of atherosclerotic lesions and inhibits the expression of oncostatin M in apolipoprotein E-deficient mice. Drug Des Dev Ther 2015; 9: 5203–5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Kalathottukaren MT, Kizhakkedathu JN. Mechanisms of blood coagulation in response to biomaterials: extrinsic factors. In: Siedlecki C. (ed.) Hemocompatibility of biomaterials for clinical applications: blood-biomaterials interactions. Cambridge: Woodhead Publishing, 2018, pp.29–49. [Google Scholar]
- 227. Schmidt-Ott KM, Kagiyama S, Phillips MI. The multiple actions of angiotensin II in atherosclerosis. Regul Pept 2000; 93: 65–77. [DOI] [PubMed] [Google Scholar]
- 228. Wang Y, Li L, Zhao W, et al. Targeted therapy of atherosclerosis by a broad-spectrum reactive oxygen species scavenging nanoparticle with intrinsic anti-inflammatory activity. ACS Nano 2018; 12: 8943–8960. [DOI] [PubMed] [Google Scholar]
- 229. Kattoor AJ, Pothineni NVK, Palagiri D, et al. Oxidative stress in atherosclerosis. Curr Atheroscler Rep 2017; 19: 42. [DOI] [PubMed] [Google Scholar]
- 230. Drummond GR, Sobey CG. Endothelial NADPH oxidases: which NOX to target in vascular disease? Trends Endocrinol Metab 2014; 25: 452–463. [DOI] [PubMed] [Google Scholar]
- 231. Li JM, Newburger PE, Gounis MJ, et al. Local arterial nanoparticle delivery of siRNA for NOX2 knockdown to prevent restenosis in an atherosclerotic rat model. Gene Ther 2010; 17: 1279–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Freyhaus H, Huntgeburth M, Wingler K, et al. Novel Nox inhibitor VAS2870 attenuates PDGF-dependent smooth muscle cell chemotaxis, but not proliferation. Cardiovasc Res 2006; 71: 331–341. [DOI] [PubMed] [Google Scholar]
- 233. Weiss D, Sorescu D, Taylor WR. Angiotensin II and atherosclerosis. Am J Cardiol 2001; 87: 25–32. [DOI] [PubMed] [Google Scholar]
- 234. Panth N, Paudel KR, Parajuli K. Reactive oxygen species: a key hallmark of cardiovascular disease. Adv Med 2016; 2016: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Wu H, Cheng XW, Hu L, et al. Renin inhibition reduces atherosclerotic plaque neovessel formation and regresses advanced atherosclerotic plaques. Atherosclerosis 2014; 237: 739–747. [DOI] [PubMed] [Google Scholar]
- 236. Kolakovic A, Zivkovic M, Stankovic A. Involvement of renin-angiotensin system in atherosclerosis. In: Tolekova A. (ed.) Renin-angiotensin system: past, present and Future. London: IntechOpen, 2017, pp.15–38. [Google Scholar]
- 237. Manea A, Simionescu M. Nox enzymes and oxidative stress in atherosclerosis. Front Biosci 2012; 4: 651–670. [DOI] [PubMed] [Google Scholar]
- 238. Rensen P, Gras JCE, Lindfors E, et al. Selective targeting of liposomes to macrophages using a ligand with high affinity for the macrophage scavenger receptor class A. Curr Drug Discov Technol 2006; 3: 135–144. [DOI] [PubMed] [Google Scholar]
- 239. Zhang L, Tian XY, Chan CKW, et al. Promoting the delivery of nanoparticles to atherosclerotic plaques by DNA coating. ACS Appl Mater Interfaces 2019; 11: 13888–13904. [DOI] [PubMed] [Google Scholar]
- 240. Chen X, Wong R, Khalidov I, et al. Inflamed leukocyte-mimetic nanoparticles for molecular imaging of inflammation. Biomaterials 2011; 32: 7651–7661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Zern BJ, Chacko AM, Liu J, et al. Reduction of nanoparticle avidity enhances the selectivity of vascular targeting and PET detection of pulmonary inflammation. ACS Nano 2013; 7: 2461–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Zhang N, Chittasupho C, Duangrat C, et al. PLGA nanoparticle−peptide conjugate effectively targets intercellular cell-adhesion molecule-1. Bioconjug Chem 2008; 19: 145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Nahrendorf M, Jaffer FA, Kelly KA, et al. Noninvasive vascular cell adhesion molecule-1 imaging identifies inflammatory activation of cells in atherosclerosis. Circulation 2006; 114: 1504–1511. [DOI] [PubMed] [Google Scholar]
- 244. Nahrendorf M, Keliher E, Panizzi P, et al. 18F-4V for PET–CT imaging of VCAM-1 expression in atherosclerosis. JACC Cardiovasc Imaging 2009; 2: 1213–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Chan JM, Zhang L, Tong R, et al. Spatiotemporal controlled delivery of nanoparticles to injured vasculature. Proc Natl Acad Sci 2010; 107: 2213–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Kamaly N, Fredman G, Fojas JJR, et al. Targeted interleukin-10 nanotherapeutics developed with a microfluidic chip enhance resolution of inflammation in advanced atherosclerosis. ACS Nano 2016; 10: 5280–5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Modery CL, Ravikumar M, Wong TL, et al. Heteromultivalent liposomal nanoconstructs for enhanced targeting and shear-stable binding to active platelets for site-selective vascular drug delivery. Biomaterials 2011; 32: 9504–9514. [DOI] [PubMed] [Google Scholar]
- 248. Xu X, Huang X, Zhang Y, et al. Self-regulated hirudin delivery for anticoagulant therapy. Sci Adv 2020; 6: eabc0382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Kang C, Gwon S, Song C, et al. Fibrin-targeted and H2O2-responsive nanoparticles as a theranostics for thrombosed vessels. ACS Nano 2017; 11: 6194–6203. [DOI] [PubMed] [Google Scholar]
- 250. Ghebrehiwet B, Geisbrecht BV, Xu X, et al. The C1q receptors: focus on gC1qR/p33 (C1qBP, p32, HABP-1). Semin Immunol 2019; 45: 101338. [DOI] [PubMed] [Google Scholar]
- 251. Peerschke E, Minta J, Zhou S, et al. Expression of gC1q-R/p33 and its major ligands in human atherosclerotic lesions. Mol Immunol 2004; 41: 759–766. [DOI] [PubMed] [Google Scholar]
- 252. Uchida M, Kosuge H, Terashima M, et al. Protein cage nanoparticles bearing the lyp-1 peptide for enhanced imaging of macrophage-rich vascular lesions. ACS Nano 2011; 5: 2493–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Hamzah J, Kotamraju VR, Seo JW, et al. Specific penetration and accumulation of a homing peptide within atherosclerotic plaques of apolipoprotein E-deficient mice. Proc Natl Acad Sci 2011; 108: 7154–7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Benne N, Martins Cardoso R, Boyle AL, et al. Complement receptor targeted liposomes encapsulating the liver X receptor agonist GW3965 accumulate in and stabilize atherosclerotic plaques. Adv Healthc Mater 2020; 9: 2000043. [DOI] [PubMed] [Google Scholar]
- 255. Chen J, López JA. Interactions of platelets with subendothelium and endothelium. Microcirculation 2005; 12: 235–246. [DOI] [PubMed] [Google Scholar]
- 256. Anselmo AC, Modery-Pawlowski CL, Menegatti S, et al. Platelet-like nanoparticles: mimicking shape, flexibility, and surface biology of platelets to target vascular injuries. ACS Nano 2014; 8: 11243–11253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Xuan M, Shao J, Li J. Cell membrane-covered nanoparticles as biomaterials. Natl Sci Rev 2019; 6: 551–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Banskota S, Yousefpour P, Chilkoti A. Cell-based biohybrid drug delivery systems: the best of the synthetic and natural worlds. Macromol Biosci 2017; 17: 1600361. [DOI] [PubMed] [Google Scholar]
- 259. Hu CMJ, Fang RH, Wang KC, et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature 2015; 526: 118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Yang H, Song Y, Chen J, et al. Platelet membrane-coated nanoparticles target sclerotic aortic valves in ApoE−/− mice by multiple binding mechanisms under pathological shear stress. Int J Nanomed 2020; 15: 901–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Wei X, Ying M, Dehaini D, et al. Nanoparticle functionalization with platelet membrane enables multifactored biological targeting and detection of atherosclerosis. ACS Nano 2018; 12: 109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Song Y, Huang Z, Liu X, et al. Platelet membrane-coated nanoparticle-mediated targeting delivery of rapamycin blocks atherosclerotic plaque development and stabilizes plaque in apolipoprotein E-deficient (ApoE−/−) mice. Nanomed Nanotechnol Biol Med 2019; 15: 13–24. [DOI] [PubMed] [Google Scholar]
- 263. Dehaini D, Wei X, Fang RH, et al. Erythrocyte-platelet hybrid membrane coating for enhanced nanoparticle functionalization. Adv Mater 2017; 29(16): 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Cao H, Dan Z, He X, et al. Liposomes coated with isolated macrophage membrane can target lung metastasis of breast cancer. ACS Nano 2016; 10: 7738–7748. [DOI] [PubMed] [Google Scholar]
- 265. Peng R, Ji H, Jin L, et al. Macrophage-based therapies for atherosclerosis management. J Immunol Res 2020; 2020: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Gao C, Huang Q, Liu C, et al. Treatment of atherosclerosis by macrophage-biomimetic nanoparticles via targeted pharmacotherapy and sequestration of proinflammatory cytokines. Nat Commun 2020; 11: 2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Wu M, Chen C, Wang Z, et al. Separating extracellular vesicles and lipoproteins via acoustofluidics. Lab Chip 2019; 19: 1174–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Cormode DP, Fisher EA, Stroes ESG, et al. High-density lipoprotein is a nanoparticle, but not all nanoparticles are high-density lipoprotein. Proc Natl Acad Sci 2013; 110: 3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Haghikia A, Landmesser U. High-density lipoproteins: effects on vascular function and role in the immune response. Cardiol Clin 2018; 36: 317–327. [DOI] [PubMed] [Google Scholar]
- 270. Meilhac O, Tanaka S, Couret D. High-density lipoproteins are bug scavengers. Biomolecules 2020; 10: 598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Navab M, Reddy ST, Van Lenten BJ, et al. HDL and cardiovascular disease: Atherogenic and atheroprotective mechanisms. Nat Rev Cardiol 2011; 8: 222–232. [DOI] [PubMed] [Google Scholar]
- 272. Chen J, Zhang X, Millican R, et al. High density lipoprotein mimicking nanoparticles for atherosclerosis. Nano Converg 2020; 7: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Skajaa T, Cormode DP, Jarzyna PA, et al. The biological properties of iron oxide core high-density lipoprotein in experimental atherosclerosis. Biomaterials 2011; 32: 206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Cormode DP, Skajaa T, van Schooneveld MM, et al. Nanocrystal core high-density lipoproteins: A multimodality contrast agent platform. Nano Lett 2008; 8: 3715–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Marrache S, Dhar S. Biodegradable synthetic high-density lipoprotein nanoparticles for atherosclerosis. Proc Natl Acad Sci 2013; 110: 9445–9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Lameijer M, Binderup T, van Leent MMT, et al. Efficacy and safety assessment of a TRAF6-targeted nanoimmunotherapy in atherosclerotic mice and non-human primates. Nat Biomed Eng 2018; 2: 279–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Mansukhani NA, Peters EB, So MM, et al. Peptide amphiphile supramolecular nanostructures as a targeted therapy for atherosclerosis. Macromol Biosci 2019; 19: e1900066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Zhang M, He J, Jiang C, et al. Plaque-hyaluronidase-responsive high-density-lipoprotein-mimetic nanoparticles for multistage intimal-macrophage-targeted drug delivery and enhanced anti-atherosclerotic therapy. Int J Nanomedicine 2017; 12: 533–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Luthi AJ, Zhang H, Kim D, et al. Tailoring of biomimetic high-density lipoprotein nanostructures changes cholesterol binding and efflux. ACS Nano 2012; 6: 276–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Naeini MB, Bianconi V, Pirro M, et al. The role of phosphatidylserine recognition receptors in multiple biological functions. Cell Mol Biol Lett 2020; 25: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Maiseyeu A, Bagalkot V. In vitro uptake of apoptotic body mimicking phosphatidylserine-quantum dot micelles by monocytic cell line. Nanoscale Res Lett 2014; 9(1): 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Harre U, Keppeler H, Ipseiz N, et al. Moonlighting osteoclasts as undertakers of apoptotic cells. Autoimmun 2012; 45: 612–619. [DOI] [PubMed] [Google Scholar]
- 283. Zhao R, Jiang J, Li H, et al. Phosphatidylserine-microbubble targeting-activated microglia/macrophage in inflammation combined with ultrasound for breaking through the blood–brain barrier. J Neuroinflammation 2018; 15: 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Wu Y, Zhang Y, Dai L, et al. An apoptotic body-biomimic liposome in situ upregulates anti-inflammatory macrophages for stabilization of atherosclerotic plaques. J Control Release 2019; 316: 236–249. [DOI] [PubMed] [Google Scholar]
- 285. Bagalkot V, Badgeley MA, Kampfrath T, et al. Hybrid nanoparticles improve targeting to inflammatory macrophages through phagocytic signals. J Control Release 2015; 217: 243–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Hosseini H, Li Y, Kanellakis P, et al. Phosphatidylserine liposomes mimic apoptotic cells to attenuate atherosclerosis by expanding polyreactive IgM producing b1a lymphocytes. Cardiovasc Res 2015; 106: 443–452. [DOI] [PubMed] [Google Scholar]
- 287. Kim S, Bae SM, Seo J, et al. Advantages of the phosphatidylserine-recognizing peptide PSP1 for molecular imaging of tumor apoptosis compared with annexin V. PLoS One 2015; 10: e0121171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Kooijmans SAA, Gitz-Francois JJJM, Schiffelers RM, et al. Recombinant phosphatidylserine-binding nanobodies for targeting of extracellular vesicles to tumor cells: A plug-and-play approach. Nanoscale 2018; 10: 2413–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Khoshbakht S, Beiki D, Geramifar P, et al. 18FDG-labeled LIKKPF: a PET tracer for apoptosis imaging. J Radioanal Nucl Chem 2016; 310: 413–421. [Google Scholar]
- 290. Hoare D, Bussooa A, Neale S, et al. The future of cardiovascular stents: bioresorbable and integrated biosensor technology. Adv Sci 2019; 6: 1900856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. McKittrick CM, Kennedy S, Oldroyd KG, et al. Modelling the impact of atherosclerosis on drug release and distribution from coronary stents. Ann Biomed Eng 2016; 44: 477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Duda SH, Poerner TC, Wiesinger B, et al. Drug-eluting stents: Potential applications for peripheral arterial occlusive disease. J Vasc Interv Radiol 2003; 14: 291–301. [DOI] [PubMed] [Google Scholar]
- 293. Yin RX, Yang DZ, Wu JZ. Nanoparticle drug- and gene-eluting stents for the prevention and treatment of coronary restenosis. Theranostics 2014; 4: 175–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Lee CH, Hsieh MJ, Liu KS, et al. Promoting vascular healing using nanofibrous ticagrelor-eluting stents. Int J Nanomedicine 2018; 13: 6039–6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Ma X, Wu T, Robich MP. Drug-eluting stent coatings. Interv Cardiol 2012; 4: 73–83. [Google Scholar]
- 296. Thierry B, Winnik FM, Merhi Y, et al. Bioactive coatings of endovascular stents based on polyelectrolyte multilayers. Biomacromolecules 2003; 4: 1564–1571. [DOI] [PubMed] [Google Scholar]
- 297. Whelan DM, Van Der Giessen WJ, Krabbendam SC, et al. Biocompatibility of phosphorylcholine coated stents in normal porcine coronary arteries. Heart 2000; 83: 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Aditya G, Parmeshwar D, Mukty S. Nanofiberous coating for bare metal stents: A comparative study of coaxial and monoaxial modes. Mater Today Proc 2019; 18: 1108–1115. [Google Scholar]
- 299. Choi J, Konno T, Takai M, et al. Regulation of cell proliferation by multi-layered phospholipid polymer hydrogel coatings through controlled release of paclitaxel. Biomaterials 2012; 33: 954–961. [DOI] [PubMed] [Google Scholar]
- 300. Liu J, Qu S, Suo Z, et al. Functional hydrogel coatings. Natl Sci Rev 2021; 8: nwaa254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Strohbach A, Busch R. Polymers for cardiovascular stent coatings. Int J Polym Sci 2015; 2015: 1–11. [Google Scholar]
- 302. Park DS, Bae IH, Jeong MH, et al. In vitro and in vivo evaluation of a novel polymer-free everolimus-eluting stent by nitrogen-doped titanium dioxide film deposition. Mater Sci Eng C 2018; 91: 615–623. [DOI] [PubMed] [Google Scholar]
- 303. Tang R, Cui XB, Wang JN, et al. CTP synthase 1, a smooth muscle–sensitive therapeutic target for effective vascular repair. Arterioscler Thromb Vasc Biol 2013; 33: 2336–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Tang R, Zhang G, Chen SY. Smooth muscle cell proangiogenic phenotype induced by cyclopentenyl cytosine promotes endothelial cell proliferation and migration. J Biol Chem 2016; 291: 26913–26921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Douglas G, Van Kampen E, Hale AB, et al. Endothelial cell repopulation after stenting determines in-stent neointima formation: effects of bare-metal vs. Drug-eluting stents and genetic endothelial cell modification. Eur Heart J 2013; 34: 3378–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Jin Z, Yan X, Liu G, et al. Fibronectin modified TiO2 nanotubes modulate endothelial cell behavior. J Biomater Appl 2018; 33: 44–51. [DOI] [PubMed] [Google Scholar]
- 307. Montaño-Machado V, Noël C, Chevallier P, et al. Interaction of phosphorylcholine with fibronectin coatings: surface characterization and biological performances. Appl Surf Sci 2017; 396: 1613–1622. [Google Scholar]
- 308. Tersteeg C, Roest M, Mak-Nienhuis EM, et al. A fibronectin-fibrinogen-tropoelastin coating reduces smooth muscle cell growth but improves endothelial cell function. J Cell Mol Med 2012; 16: 2117–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Liu T, Hu Y, Tan J, et al. Surface biomimetic modification with laminin-loaded heparin/poly-L-lysine nanoparticles for improving the biocompatibility. Mater Sci Eng C 2017; 71: 929–936. [DOI] [PubMed] [Google Scholar]
- 310. Jana S. Endothelialization of cardiovascular devices. Acta Biomater 2019; 99: 53–71. [DOI] [PubMed] [Google Scholar]
- 311. Hazawa M, Yasuda T, Noshiro K, et al. Vitronectin improves cell survival after radiation injury in human umbilical vein endothelial cells. FEBS Open Bio 2012; 2: 334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Motwani MS, Rafiei Y, Tzifa A, et al. In situ endothelialization of intravascular stents from progenitor stem cells coated with nanocomposite and functionalized biomolecules. Biotechnol Appl Biochem 2011; 58: 2–13. [DOI] [PubMed] [Google Scholar]
- 313. Khan M, Yang J, Shi C, et al. Surface tailoring for selective endothelialization and platelet inhibition via a combination of SI-ATRP and click chemistry using Cys–Ala–Gly-peptide. Acta Biomater 2015; 20: 69–81. [DOI] [PubMed] [Google Scholar]
- 314. Lei Y, Rémy M, Labrugère C, et al. Peptide immobilization on polyethylene terephthalate surfaces to study specific endothelial cell adhesion, spreading and migration. J Mater Sci Mater Med 2012; 23: 2761–2772. [DOI] [PubMed] [Google Scholar]
- 315. Le Saux G, Plawinski L, Parrot C, et al. Surface bound VEGF mimicking peptide maintains endothelial cell proliferation in the absence of soluble VEGF in vitro. J Biomed Mater Res A 2016; 104: 1425–1436. [DOI] [PubMed] [Google Scholar]
- 316. Diaz-Rodriguez S, Rasser C, Mesnier J, et al. Coronary stent CD31-mimetic coating favours endothelialization and reduces local inflammation and neointimal development in vivo. Eur Heart J 2021; 42: 1760–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Simsekyilmaz S, Liehn EA, Weinandy S, et al. Targeting in-stent-stenosis with RGD- and CXCL1-coated mini-stents in mice. PLoS One 2016; 11: e0155829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Hu T, Lin S, Du R, et al. Design, preparation and performance of a novel drug-eluting stent with multiple layer coatings. Biomater Sci 2017; 5: 1845–1857. [DOI] [PubMed] [Google Scholar]
- 319. Viana IMMN, de Almeida MES, Lins MP, et al. Combined effect of insulin-like growth factor-1 and CC chemokine ligand 2 on angiogenic events in endothelial cells. PLoS One 2015; 10: e0121249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Song H, Wu T, Yang X, et al. Surface modification with NGF-loaded chitosan/heparin nanoparticles for improving biocompatibility of cardiovascular stent. Stem Cells Int 2021; 2021: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Choi DH, Kang SN, Kim SM, et al. Growth factors-loaded stents modified with hyaluronic acid and heparin for induction of rapid and tight re-endothelialization. Colloids Surf B Biointerfaces 2016; 141: 602–610. [DOI] [PubMed] [Google Scholar]
- 322. Levine YC, Li GK, Michel T. Agonist-modulated regulation of AMP-activated protein kinase (AMPK) in endothelial cells: evidence for an AMPK → Rac1 → Akt → endothelial nitric-oxide synthase pathway. J Biol Chem 2007; 282: 20351–20364. [DOI] [PubMed] [Google Scholar]
- 323. Koutsoumparis A, Vassili A, Bakopoulou A, et al. Erythropoietin (rhEPOa) promotes endothelial transdifferentiation of stem cells of the apical papilla (SCAP). Arch Oral Biol 2018; 96: 96–103. [DOI] [PubMed] [Google Scholar]
- 324. Alexander GC, Hwang PT, Chen J, et al. Nanomatrix coated stent enhances endothelialization but reduces platelet, smooth muscle cell, and monocyte adhesion under physiologic conditions. ACS Biomater Sci Eng 2018; 4: 107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Rao J, Pan Bei H, Yang Y, et al. Nitric oxide-producing cardiovascular stent coatings for prevention of thrombosis and restenosis. Front Bioeng Biotechnol 2020; 8: 578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Fishbein I, Guerrero DT, Alferiev IS, et al. Stent-based delivery of adeno-associated viral vectors with sustained vascular transduction and iNOS-mediated inhibition of in-stent restenosis. Gene Ther 2017; 24: 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Sharif F, Hynes SO, Cooney R, et al. Gene-eluting stents: adenovirus-mediated delivery of eNOS to the blood vessel wall accelerates re-endothelialization and inhibits restenosis. Mol Ther 2008; 16: 1674–1680. [DOI] [PubMed] [Google Scholar]
- 328. Wawrzyńska M, Kraskiewicz H, Paprocka M, et al. Functionalization with a VEGFR2-binding antibody fragment leads to enhanced endothelialization of a cardiovascular stent in vitro and in vivo. J Biomed Mater Res B Appl Biomater 2020; 108: 213–224. [DOI] [PubMed] [Google Scholar]
- 329. Walter DH, Cejna M, Diaz-Sandoval L, et al. Local gene transfer of phVEGF-2 plasmid by gene-eluting stents: an alternative strategy for inhibition of restenosis. Circulation 2004; 110: 36–45. [DOI] [PubMed] [Google Scholar]
- 330. Abraham MK, Nolte A, Reus R, et al. In vitro study of a novel stent coating using modified CD39 messenger RNA to potentially reduce stent angioplasty-associated complications. PLoS One 2015; 10: e0138375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Koga JI, Aikawa M. Crosstalk between macrophages and smooth muscle cells in atherosclerotic vascular diseases. Vasc Pharmacol 2012; 57: 24–28. [DOI] [PubMed] [Google Scholar]
- 332. Segev A, Nili N, Osherov A, et al. A perlecan-inducing compound significantly inhibits smooth muscle cell function and in-stent intimal hyperplasia: novel insights into the diverse biological effects of perlecan. EuroIntervention 2010; 6: 134–140. [PubMed] [Google Scholar]
- 333. Huang C, Zhou M, Zheng X. RhoA inhibitor-eluting stent attenuates restenosis by inhibiting YAP signaling. J Vasc Surg 2019; 69: 1581–1589.e1. [DOI] [PubMed] [Google Scholar]
- 334. Huang C, Zhao J, Zhu Y. Drug-eluting stent targeting Sp-1-attenuated restenosis by engaging YAP-mediated vascular smooth muscle cell phenotypic modulation. J Am Heart Assoc 2020; 9: e014103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. Huang C, Zhang W, Zhu Y. Drug-eluting stent specifically designed to target vascular smooth muscle cell phenotypic modulation attenuated restenosis through the YAP pathway. Am J Physiol Heart Circ Physiol 2019; 317: H541–H551. [DOI] [PubMed] [Google Scholar]
- 336. Huang C, Mei H, Zhou M, et al. A novel PDGF receptor inhibitor-eluting stent attenuates in-stent neointima formation in a rabbit carotid model. Mol Med Rep 2017; 15: 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337. Jandt E, Mutschke O, Mahboobi S, et al. Stent-based release of a selective PDGF-receptor blocker from the bis-indolylmethanon class inhibits restenosis in the rabbit animal model. Vasc Pharmacol 2010; 52: 55–62. [DOI] [PubMed] [Google Scholar]
- 338. Liu Y, Liu P, Song Y, et al. A heparin–rosuvastatin-loaded P(LLA-CL) nanofiber-covered stent inhibits inflammatory smooth-muscle cell viability to reduce in-stent stenosis and thrombosis. J Nanobiotechnol 2021; 19: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Oh B, Lee CH. Functionalized cardiovascular stents: cardiovascular stents incorporated with stem cells. In: Wall G, Podbielska H, Wawrzynska M. (eds) Functionalised Cardiovascular Stents. Cambridge: Woodhead Publishing, 2018, pp.251–290. [Google Scholar]
- 340. Zhao Y, Wang Z, Bai L, et al. Regulation of endothelial functionality through direct and immunomodulatory effects by Ni-Ti-O nanospindles on NiTi alloy. Mater Sci Eng C 2021; 123: 112007. [DOI] [PubMed] [Google Scholar]
- 341. Xu WC, Dong X, Ding JL, et al. Nanotubular TiO2 regulates macrophage M2 polarization and increases macrophage secretion of VEGF to accelerate endothelialization via the ERK1/2 and PI3K/AKT pathways. Int J Nanomedicine 2019; 14: 441–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Reed MJ, Karres N, Eyman D, et al. Endothelial precursor cells. Stem Cell Rev 2007; 3: 218–225. [DOI] [PubMed] [Google Scholar]
- 343. Wang X, Cooper S. Adhesion of endothelial cells and endothelial progenitor cells on peptide-linked polymers in shear flow. Tissue Eng Part A 2013; 19: 1113–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Park KS, Kang SN, Kim DH, et al. Late endothelial progenitor cell-capture stents with CD146 antibody and nanostructure reduce in-stent restenosis and thrombosis. Acta Biomater 2020; 111: 91–101. [DOI] [PubMed] [Google Scholar]
- 345. Sethi R, Lee CH. Endothelial progenitor cell capture stent: Safety and effectiveness. J Interv Cardiol 2012; 25: 493–500. [DOI] [PubMed] [Google Scholar]
- 346. Duan Y, Yu S, Xu P, et al. Co-immobilization of CD133 antibodies, vascular endothelial growth factors, and REDV peptide promotes capture, proliferation, and differentiation of endothelial progenitor cells. Acta Biomater 2019; 96: 137–148. [DOI] [PubMed] [Google Scholar]
- 347. Lim WH, Seo WW, Choe W, et al. Stent coated with antibody against vascular endothelial-cadherin captures endothelial progenitor cells, accelerates re-endothelialization, and reduces neointimal formation. Arterioscler Thromb Vasc Biol 2011; 31: 2798–2805. [DOI] [PubMed] [Google Scholar]
- 348. Rossi E, Bernabeu C, Smadja DM. Endoglin as an adhesion molecule in mature and progenitor endothelial cells: a function beyond TGF-β. Front Med 2019; 6: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349. Bashth OS, Elkhodiry MA, Laroche G, et al. Surface grafting of Fc-binding peptides as a simple platform to immobilize and identify antibodies that selectively capture circulating endothelial progenitor cells. Biomater Sci 2020; 8: 5465–5475. [DOI] [PubMed] [Google Scholar]
- 350. Tesfamariam B. Drug release kinetics from stent device-based delivery systems. J Cardiovasc Pharmacol 2008; 51: 118–125. [DOI] [PubMed] [Google Scholar]
- 351. Hao D, Xiao W, Liu R, et al. Discovery and characterization of a potent and specific peptide ligand targeting endothelial progenitor cells and endothelial cells for tissue regeneration. ACS Chem Biol 2017; 12: 1075–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Li Q, Wang Z, Zhang S, et al. Functionalization of the surface of electrospun poly(epsilon-caprolactone) mats using zwitterionic poly(carboxybetaine methacrylate) and cell-specific peptide for endothelial progenitor cells capture. Mater Sci Eng C 2013; 33: 1646–1653. [DOI] [PubMed] [Google Scholar]
- 353. Li L, Liu W, Zhao Y, et al. Dual-peptide-functionalized nanofibrous scaffolds recruit host endothelial progenitor cells for vasculogenesis to repair calvarial defects. ACS Appl Mater Interfaces 2020; 12: 3474–3493. [DOI] [PubMed] [Google Scholar]
- 354. Hsu YI, Mahara A, Yamaoka T. Identification of circulating cells interacted with integrin α4β1 ligand peptides REDV or HGGVRLY. Peptides 2021; 136: 170470. [DOI] [PubMed] [Google Scholar]
- 355. Liu Y, Mahara A, Kambe Y, et al. Endothelial cell adhesion and blood response to hemocompatible peptide 1 (HCP-1), REDV, and RGD peptide sequences with free N-terminal amino groups immobilized on a biomedical expanded polytetrafluorethylene surface. Biomater Sci 2021; 9: 1034–1043. [DOI] [PubMed] [Google Scholar]
- 356. Munisso MC, Yamaoka T. Peptide with endothelial cell affinity and antiplatelet adhesion property to improve hemocompatibility of blood-contacting biomaterials. Pept Sci 2019; 111: e24114. [Google Scholar]
- 357. Plouffe BD, Njoka DN, Harris J, et al. Peptide-mediated selective adhesion of smooth muscle and endothelial cells in microfluidic shear flow. Langmuir 2007; 23: 5050–5055. [DOI] [PubMed] [Google Scholar]
- 358. Kim AK, Kim MH, Kim DH, et al. Inhibitory effects of mesenchymal stem cells in intimal hyperplasia after balloon angioplasty. J Vasc Surg 2016; 63: 510–517. [DOI] [PubMed] [Google Scholar]
- 359. Iso Y, Usui S, Toyoda M, et al. Bone marrow-derived mesenchymal stem cells inhibit vascular smooth muscle cell proliferation and neointimal hyperplasia after arterial injury in rats. Biochem Biophys Rep 2018; 16: 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Hoffmann J, Paul A, Harwardt M, et al. Immobilized DNA aptamers used as potent attractors for porcine endothelial precursor cells. J Biomed Mater Res A 2008; 84: 614–621. [DOI] [PubMed] [Google Scholar]
- 361. Speck U, Stolzenburg N, Peters D, et al. How does a drug-coated balloon work? Overview of coating techniques and their impact. J Cardiovasc Surg 2016; 57: 3–11. [PubMed] [Google Scholar]
- 362. Atigh MK, Turner E, Christians U, et al. The use of an occlusion perfusion catheter to deliver paclitaxel to the arterial wall. Cardiovasc Ther 2017; 35: e12269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. Bunch F, Nair P, Aggarwala G, et al. A universal drug delivery catheter for the treatment of infrapopliteal arterial disease using liquid therapy. Catheter Cardiovasc Interv 2020; 96: 393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364. Kayssi A, Al-Atassi T, Oreopoulos G, et al. Drug-eluting balloon angioplasty versus uncoated balloon angioplasty for peripheral arterial disease of the lower limbs. Cochrane Database Syst Rev 2016; 2016: CD011319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Li M, Guo C, Lv YH, et al. Drug-coated balloon versus drug-eluting stent in de novo small coronary vessel disease: a systematic review and meta-analysis. Medicine 2019; 98: e15622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Jeger RV, Eccleshall S, Wan Ahmad WA, et al. Drug-coated balloons for coronary artery disease: third report of the international DCB consensus group. JACC Cardiovasc Interv 2020; 13: 1391–1402. [DOI] [PubMed] [Google Scholar]
- 367. Moneta GL. Local delivery of paclitaxel to inhibit restenosis during angioplasty of the leg. Yearb Vasc Surg 2009; 2009: 189–190. [Google Scholar]
- 368. Yuan Y, Cheng W, Lu H. Drug-eluting balloon versus plain balloon angioplasty for the treatment of failing hemodialysis access: a systematic review and meta-analysis. Ann Vasc Surg 2020; 64: 389–396. [DOI] [PubMed] [Google Scholar]
- 369. Scheinert D, Duda S, Zeller T, et al. The LEVANT i (lutonix paclitaxel-coated balloon for the prevention of femoropopliteal restenosis) trial for femoropopliteal revascularization: first-in-human randomized trial of low-dose drug-coated balloon versus uncoated balloon angioplasty. JACC Cardiovasc Interv 2014; 7: 10–19. [DOI] [PubMed] [Google Scholar]
- 370. Unverdorben M, Vallbracht C, Cremers B, et al. Paclitaxel-coated balloon catheter versus paclitaxel-coated stent for the treatment of coronary in-stent restenosis. Circulation 2009; 119: 2986–2994. [DOI] [PubMed] [Google Scholar]
- 371. Bonaventura K, Schwefer M, Yusof AKM, et al. Systematic scoring balloon lesion preparation for drug-coated balloon angioplasty in clinical routine: results of the PASSWORD observational study. Adv Ther 2020; 37: 2210–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372. Chowdhury MM, Singh K, Albaghdadi MS, et al. Paclitaxel drug-coated balloon angioplasty suppresses progression and inflammation of experimental atherosclerosis in rabbits. JACC Basic Transl Sci 2020; 5: 685–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373. Clever YP, Peters D, Calisse J, et al. Novel sirolimus-coated balloon catheter. Circ Cardiovasc Interv 2016; 9: e003543. [DOI] [PubMed] [Google Scholar]
- 374. Tesfamariam B. Local arterial wall drug delivery using balloon catheter system. J Control Release 2016; 238: 149–156. [DOI] [PubMed] [Google Scholar]
- 375. Nunes GL, Hanson SR, King SB, et al. Local delivery of a synthetic antithrombin with a hydrogel-coated angioplasty balloon catheter inhibits platelet-dependent thrombosis. J Am Coll Cardiol 1994; 23: 1578–1583. [DOI] [PubMed] [Google Scholar]
- 376. Tomaru T, Kobayakawa N, Onitake A, et al. Local antithrombotic therapy using a novel porous balloon catheter. Jpn Heart J 2000; 41: 87–95. [DOI] [PubMed] [Google Scholar]
- 377. Tomaru T, Yoshimura A, Aoki N, et al. Local delivery of antithrombotic drug inhibits neointimal hyperplasia following arterial injury. J Interv Cardiol 1997; 10: 51–60. [Google Scholar]
- 378. Marshall DJ, Palasis M, Lepore JJ, et al. Biocompatibility of cardiovascular gene delivery catheters with adenovirus vectors: an important determinant of the efficiency of cardiovascular gene transfer. Mol Ther 2000; 1: 423–429. [DOI] [PubMed] [Google Scholar]
- 379. Varenne O, Gerard RD, Sinnaeve P, et al. Percutaneous adenoviral gene transfer into porcine coronary arteries: is catheter-based gene delivery adapted to coronary circulation? Hum Gene Ther 1999; 10: 1105–1115. [DOI] [PubMed] [Google Scholar]
- 380. Pankajakshan D, Makinde TO, Gaurav R, et al. Successful transfection of genes using AAV-2/9 vector in swine coronary and peripheral arteries. J Surg Res 2012; 175: 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381. Dillavou E, Cupp P, Consigny PM. Delivery of endothelial cells to balloon-dilated rabbit arteries with use of a local delivery catheter. J Vasc Interv Radiol 2001; 12: 601–605. [DOI] [PubMed] [Google Scholar]
- 382. Turner EA, Atigh MK, Erwin MM, et al. Coating and pharmacokinetic evaluation of air spray coated drug coated balloons. Cardiovasc Eng Technol 2018; 9: 240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383. Richey T, Iwata H, Oowaki H, et al. Surface modification of polyethylene balloon catheters for local drug delivery. Biomaterials 2000; 21: 1057–1065. [DOI] [PubMed] [Google Scholar]
- 384. Dick A, Kromen W, Jüngling E, et al. Quantification of horseradish peroxidase delivery into the arterial wall in vivo as a model of local drug treatment: comparison between a porous and a gel-coated balloon catheter. Cardiovasc Intervent Radiol 1999; 22: 389–393. [DOI] [PubMed] [Google Scholar]
- 385. Lee K, Lee J, Lee SG, et al. Microneedle drug eluting balloon for enhanced drug delivery to vascular tissue. J Control Release 2020; 321: 174–183. [DOI] [PubMed] [Google Scholar]
- 386. Tzafriri AR, Muraj B, Garcia-Polite F, et al. Balloon-based drug coating delivery to the artery wall is dictated by coating micro-morphology and angioplasty pressure gradients. Biomaterials 2020; 260: 120337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Huang Y, Li T, Gao W, et al. Platelet-derived nanomotor coated balloon for atherosclerosis combination therapy. J Mater Chem B 2020; 8: 5765–5775. [DOI] [PubMed] [Google Scholar]
- 388. Kempin W, Kaule S, Reske T, et al. In vitro evaluation of paclitaxel coatings for delivery via drug-coated balloons. Eur J Pharm Biopharm 2015; 96: 322–328. [DOI] [PubMed] [Google Scholar]
- 389. Zhang X, Zheng L, Luo M, et al. Evaluation of particle shape, size and magnetic field intensity for targeted delivery efficiency and plaque injury in treating atherosclerosis. Powder Technol 2020; 366: 63–72. [Google Scholar]
- 390. Mehta V, Tzima E. A turbulent path to plaque formation. Nature 2016; 540: 531–532. [DOI] [PubMed] [Google Scholar]
- 391. Texon M. Atherosclerosis: its hemodynamic basis and implications. Med Clin North Am 1974; 58: 257–268. [DOI] [PubMed] [Google Scholar]
- 392. Malek AM, Alper SL. Hemodynamic shear stress and its role in atherosclerosis. JAMA 1999; 282: 2035–2042. [DOI] [PubMed] [Google Scholar]
- 393. Gholipour A, Ghayesh MH, Zander A. Nonlinear biomechanics of bifurcated atherosclerotic coronary arteries. Int J Eng Sci 2018; 133: 60–83. [Google Scholar]
- 394. Ijaz S, Nadeem S. Biomedical theoretical investigation of blood mediated nanoparticles (Ag-Al2O3/blood) impact on hemodynamics of overlapped stenotic artery. J Mol Liq 2017; 248: 809–821. [Google Scholar]
- 395. Zhang X, Wang E, Ma L, et al. Analysis of hemodynamics and heat transfer of nanoparticle-injected atherosclerotic patient: considering the drag force and slip between phases of different particle shapes and volume fractions. Int J Therm Sci 2021; 159: 106637. [Google Scholar]
- 396. Ng J, Bourantas CV, Torii R, et al. Local hemodynamic forces after stenting: implications on restenosis and thrombosis. Arterioscler Thromb Vasc Biol 2017; 37: 2231–2242. [DOI] [PubMed] [Google Scholar]
- 397. Fan Z, Dong L, Liu X, et al. Effects of residual stenosis on carotid artery after stent implantation: a numerical study. Med Novel Technol Devices 2022; 13: 100105. [Google Scholar]
- 398. Jiménez JM, Prasad V, Yu MD, et al. Macro- and microscale variables regulate stent haemodynamics, fibrin deposition and thrombomodulin expression. J R Soc Interface 2014; 11: 20131079–20131079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399. Sarembock IJ, LaVeau PJ, Sigal SL, et al. Influence of inflation pressure and balloon size on the development of intimal hyperplasia after balloon angioplasty: A study in the atherosclerotic rabbit. Circulation 1989; 80: 1029–1040. [DOI] [PubMed] [Google Scholar]
- 400. Li G, Qiao H, Lin H, et al. Application of drug-coated balloons for intracranial atherosclerosis disease: a systematic review. Clin Neurol Neurosurg 2022; 213: 107065. [DOI] [PubMed] [Google Scholar]
- 401. Decorato I, Salsac AV, Legallais C, et al. Influence of an arterial stenosis on the hemodynamics within an arteriovenous fistula (AVF): comparison before and after balloon-angioplasty. Cardiovasc Eng Technol 2014; 5: 233–243. [Google Scholar]
- 402. Martí V, Salas E, Aymat RM, et al. Influence of residual stenosis in determining restenosis after cutting balloon angioplasty. Catheter Cardiovasc Interv 2000; 49: 410–414. [DOI] [PubMed] [Google Scholar]
- 403. Kariya S, Tanigawa N, Kojima H, et al. Percutaneous transluminal cutting-balloon angioplasty for hemodialysis access stenoses resistant to conventional balloon angioplasty. Acta Radiol 2006; 47: 1017–1021. [DOI] [PubMed] [Google Scholar]
- 404. Slee JB, Alferiev IS, Nagaswami C, et al. Enhanced biocompatibility of CD47-functionalized vascular stents. Biomaterials 2016; 87: 82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405. Adamson K, Spain E, Prendergast U, et al. Fibrinogen motif discriminates platelet and cell capture in peptide-modified gold micropore arrays. Langmuir 2018; 34: 715–725. [DOI] [PubMed] [Google Scholar]