

Abstract
Ethylmalonic encephalopathy (MIM #602473) is a rare autosomal recessive metabolic condition caused by biallelic variants in ETHE1 (MIM #608451), characterized by global developmental delay, infantile hypotonia, seizures, and microvascular damage. The microvascular changes result in a pattern of relapsing spontaneous diffuse petechiae and purpura, positional acrocyanosis, and pedal edema, hemorrhagic suffusions of mucous membranes, and chronic diarrhea. Here, we describe an instructive case in which ethylmalonic encephalopathy masqueraded as meningococcal septicemia and shock. Ultrarapid whole-genome testing (time to result 60 h) and prompt biochemical analysis facilitated accurate diagnosis and counseling with rapid implementation of precision treatment for the metabolic crisis related to this condition. This case provides a timely reminder to consider rare genetic diagnoses when atypical features of more common conditions are present, with an early referral to ensure prompt biochemical and genomic diagnosis.
Keywords: acrocyanosis, ankle clonus, central hypotonia, cerebral ischemia, delayed fine motor development, delayed gross motor development, ethylmalonic aciduria, petechiae, progressive encephalopathy, recurrent cerebral hemorrhage
INTRODUCTION
Undifferentiated presentations in acute pediatrics form the backbone of clinical practice with evidenced-based care pathways used to improve outcomes and support timely patient care. Meningococcemia and its timely management are sentinel cases for a health-care system and the quality of care provided. This severe infection typically presents with septic shock and a characteristic purpuric rash that is often considered pathognomonic for the condition. Rapid administration of antimicrobial and supportive therapies is best practice; however, cases with atypical features or that fail to respond to standard treatment require consideration of alternate diagnoses.
Although individually uncommon, rare genetic diseases collectively account for 15.9% of pediatric hospital admissions (Gjorgioski et al. 2020), and early recognition may allow the introduction of targeted therapies. The introduction of ultrarapid genomic sequencing in the diagnosis of seriously ill neonates and children is revolutionizing the early detection of rare genetic and metabolic diseases, resulting in some cases in the early institution of life-saving interventions (Lunke et al. 2020; Dimmock et al. 2021; Owen et al. 2021; Stark and Ellard 2022). We present a case of a rare metabolic condition, ethylmalonic encephalopathy (MIM #602473), masquerading as meningococcemia with atypical features. This case provides a timely reminder to consider rare genetic diagnoses when atypical features of more common conditions are present, with an early referral for a prompt genomic and biochemical diagnosis being key to management.
RESULTS
Clinical Presentation and Family History
The patient, IV.III, is a 9-mo-old female who presented with an acute deterioration, involving depressed conscious state, hypotonia, reduced peripheral perfusion with only central pulses palpable, and a rapidly evolving widespread purpuric rash (Fig. 1). She was initially assessed at home by paramedics who considered the rash to be pathognomonic for meningococcal septicemia. The patient's spontaneous respiratory effort was supported with bag-valve-mask and oxygen, intra-osseous access was placed, ceftriaxone and hydrocortisone administered, and a fluid bolus commenced during time-critical transport to the pediatric emergency department. Upon arrival to the pediatric emergency department, the patient was hypothermic (33.7°C), minimally responsive, and poorly perfused, with a marked metabolic acidosis (lactate 6.6 mmol/L, normal range 0.5–2.0). Peripheral inotropes were commenced, and the patient was intubated and transferred to Pediatric Intensive Care for stabilization. There were no prolonged periods of hypoxia during hospital admission. Investigations on admission are shown in Table 1.
Figure 1.

Presenting features: cutaneous manifestations. Clinical photographs taken on day 4 of admission demonstrating widespread petechiae and purpura to the face, upper and lower limbs, and torso (not shown).
Table 1.
Presenting features: initial investigation results
| Normal range | Day 0 | Day 1 | |
|---|---|---|---|
| Blood gas | |||
| pH | 7.32–7.43V | 7.31V | 7.33A |
| 7.35–7.45A | |||
| pCO2 | 38–61V mmHg | 23V | 44A |
| 32–45A mmHg | |||
| HCO3 | 22–32V,A mmol/L | 12V | 23A |
| Base excess | −3–+3V,A mmol/L | −15V | -3A |
| Lactate | 0.5–2.0V,A mmol/L | 6.6V | 1.4A |
| Glucose | 3–7.7V,A mmol/L | 21.6V,a | 5.3A |
| Full blood picture | |||
| Hb | 100–140 g/L | 126 | 86 |
| WCC | 5–18 × 109/L | 22.5a | 11.6 |
| Neutrophils | 1.5–9 × 109/L | 9.72a | 8.10 |
| Platelets | 150–450 × 109/L | 618a | 441 |
| Markers of muscle breakdown | |||
| CK | 30–150 U/L | 68 | - |
| Troponin I (HS) | 0–40 ng/L | 137 | - |
| Coagulation profile | |||
| INR | 0.8–1.2 | 1.2 | 1.2 |
| APTT | 22–32 sec | 26 sec | 25 |
| Fibrinogen | 1.5–4.0 g/L | 1.5 | 2.9 |
| Inflammatory markers | |||
| CRP | 0–5 mg/L | 0.5 | 13 |
| Procalcitonin | 0.00–0.07 mcg/L | 0.32 | - |
| Ferritin | 20–310 mcg/L | 29 | 29 |
| CSF | |||
| RCC | × 106/L | 1240 | 760 |
| Polymorphonuclear cells | × 106/L | 4 | 2 |
| Protein | 0.15–0.45 g/L | 0.32 | 0.33 |
| Glucose | 2.5–5.0 mmol/L | 8 | 3.9 |
| Lactate | 1.2–2.8 mmol/L | 5.5 | 2.6 |
| Molecular microbiology | |||
| Meningococcal PCR serum | Not detected | Negative | Negative |
| Meningococcal PCR CSF | Not detected | Negative | Negative |
| Enterovirus PCR CSF | Not detected | Negative | Negative |
| SARS-CoV-2 PCR | Not detected | Negative | Negative |
(Hb) Hemoglobin, (WCC) white cell count, (CK) creatine kinase, (INR) international normalized ratio, (APTT) activated partial thromboplastin time, (CRP) C-reactive protein, (RCC) red cell count, (CSF) cerebrospinal fluid, (PCR) polymerase chain reaction, (V) venous, (A) arterial.
aPost-hydrocortisone
Supportive management in the pediatric intensive care unit (PICU) over the next 48 h included inotropic support, fluid resuscitation, and ventilation together with empiric antimicrobial agents. In contrast to what was expected, markers of inflammation (C-reactive protein [CRP] and procalcitonin) remained low and the neutrophil and platelet count remained high with no evidence of disseminated intravascular coagulopathy, and needle aspiration of the purpuric rash was negative for bacteria on Gram stain. Following a normal head computed tomography (CT), a lumbar puncture was performed 20 h after admission and repeated on day 2, both with cerebrospinal fluid (CSF) samples demonstrating low cell counts (Table 1). Supportive care resulted in improvement in the metabolic acidosis and correction of lactic acidosis. An echocardiogram demonstrated a structurally normal heart with dilated left ventricle with mildly depressed systolic function, as commonly seen in critically unwell patients in the setting of an acidosis. Echocardiographic findings normalized within 4 d. Inotropes were ceased, and the patient was extubated 48 h postpresentation. No organism was isolated on blood, urine, or CSF cultures. Autoimmune markers including antinuclear antibody (ANA), antineutrophil antibody (ANCA), double-stranded DNA (dsDNA), and rheumatoid factor (RF) antibodies were normal.
Magnetic resonance imaging (MRI) of the brain performed on day 2 demonstrated an unusual pattern of acute-on-chronic changes (Fig. 2). Reduced size of the left cerebral hemisphere, bilateral T2 hyperintense, and T1 hypointense periventricular lesions without restricted diffusion were consistent with chronic, potentially in utero, diffuse ischemic damage to the left cerebral hemisphere white matter and more focal chronic infarcts in the periventricular white matter, respectively, similar to the appearances of typical preterm ischemic white matter injury. In addition, however, we identified bilateral punctate T2 hyperintense lesions demonstrating true restricted diffusion involving the basal ganglia and thalami, with some lesions demonstrating susceptibility-related signal loss consistent with hemorrhagic infarcts. This biphasic appearance of acute and established ischemic pathology and hemorrhagic infarcts was not consistent with meningococcemia but rather a noninfective, potentially genetic, vasculitis, with provisional diagnoses of ADA2- or STING1-related vasculitis proposed based on the MRI abnormalities, especially given the cutaneous rash and systemic clinical presentation. With increasing suspicion of a noninfective cause to explain the patient's condition, referrals to neurology, clinical genetics, and metabolic medicine were made on day 4/5 of admission. Electroencephalogram (EEG) on day 6 demonstrated encephalopathy without overt seizure activity.
Figure 2.
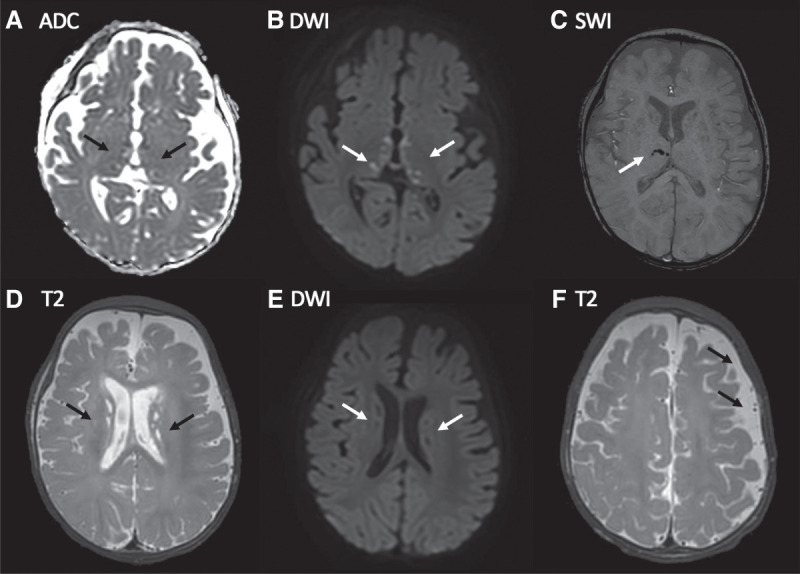
Presenting features: magnetic resonance imaging (MRI) brain findings. MRI day 2 postadmission: (A) Apparent diffusion coefficient (ADC) map and (B) corresponding diffusion-weighted image (DWI) show multiple punctate lesions in the thalami and globi pallidi that are associated with true diffusion restriction consistent with small acute (likely <7 d old) infarcts (arrows). (C) Susceptibility-weighted image (SWI) showing signal loss in a few of the right thalamic lesions consistent with microhemorrhage (arrow). (D) T2-weighted image demonstrating numerous T2 hyperintense periventricular punctate lesions that have no corresponding restricted diffusion (E) consistent with nonacute infarcts. (F) T2-weighted axial image demonstrating subtle widening of the left frontoparietal subarachnoid space (arrows) likely due to reduced left hemispheric volume consequent upon microscopic remote ischemic white matter/subplate injury postnatally or prenatally.
Further information was sought at this time about the family history. The patient's parents were consanguineous (first cousins) of Afghani ethnicity (see pedigree, Fig. 3). The mother had two previous pregnancies. Her first pregnancy ended in a termination of pregnancy at 26 wk gestation for an antenatal diagnosis of arthrogryposis. Genetic testing at this time identified a previously reported (Sandaradura et al. 2018) homozygous TNNT3 (MIM #600692) variant, NM_006757.3(TNNT3):c.681 + 1G > A, which explained the fetus’ clinical presentation with a diagnosis of distal arthrogryposis (MIM #618435). Subsequent testing of the parents confirmed that both were unaffected carriers for this variant. Her second pregnancy ended in the live birth of a male infant, who is now 3-yr-old and remains well, with reported normal neurodevelopment. The mother's third pregnancy ended in the live birth of the current patient. She had genetic testing for the TNNT3 variant antenatally revealing unaffected status. Her neurodevelopment was delayed in comparison to her brother, with hypotonia, and delayed gross and fine motor on review of family videos. In the previous months, there had been instances of profound positional acrocyanosis (Fig. 4). On the day of presentation, her parents noticed sustained clonus (Supplemental Video 1). There was no other family history of abnormal neurodevelopment or other neurological conditions.
Figure 3.

Pedigree.
Figure 4.
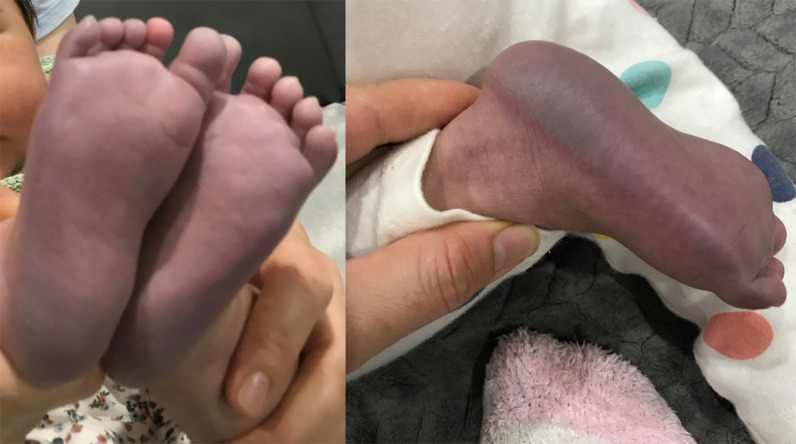
Presenting features: positional acrocyanosis. Photographs taken by the family in the weeks prior to presentation showing positional acrocyanosis.
Given the history of hypotonia, symmetry of MRI findings, encephalopathy, purpuric and petechial rash, and recurrence of lactatemia in the absence of acidosis or perfusion concerns on day 3, a genetic or metabolic cause for the patient's presentation was considered most likely, and she was approved for inclusion in a rapid genomic diagnosis program, the Australian Genomics Acute Care study (Lunke et al. 2020), on day 4 of admission. Following genetic counseling, the parents provided written consent for trio whole-genome sequencing (WGS) and samples collected on day 5 of admission. Urine metabolic investigations were requested and performed in tandem with ultrarapid WGS sequencing.
Genomic Analyses
WGS Results
Results of ultrarapid WGS were reported on day 6, 60 h after receipt of samples, and identified a homozygous frameshift variant in exon 2 of ETHE1 (MIM #608451), c.131_132del; p.(Glu44Valfs*62) (Table 2). This variant was classified according to modified ACMG/AMP criteria (Richards et al. 2015). The variant c.131_132del results in a frameshift and is predicted to cause nonsense mediated decay (NMD) and loss of protein with a premature termination codon (PVS1). Loss of function is an established mechanism of disease with multiple other splice site, frameshift, nonsense and missense variants classified as likely pathogenic and pathogenic in ClinVar. This variant has been previously reported in homozygous and compound heterozygous state in individuals affected with ethylmalonic encephalopathy (Tiranti et al. 2009; Dionisi-Vici et al. 2016; Boyer et al. 2018) (PM3_strong), with cosegregation demonstrated in one family (Tiranti et al. 2009). The variant is present in a large population database at an allele frequency of <0.01 for a recessive condition (3 heterozygotes, 0 homozygotes, highest population specific allele frequency 0.000054, East Asian 1/18380) (gnomAD v2.1.1 https://gnomad.broadinstitute.org/ (accessed 2-Oct-2021) (PM2_supporting). The variant was confirmed by trio analysis to be maternally and paternally inherited (biallelic). Based on modified American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) criteria (Richards et al. 2015), this variant was classified as pathogenic. This result is consistent with a diagnosis of ethylmalonic encephalopathy (MIM #602473) and is concordant with the results of the urine metabolic screen and organic acids analysis (Table 3; see below).
Table 2.
Whole-genome sequencing results: variant table
| Gene | Chromosome | HGVS DNA | HGVS protein | Variant type | Predicted effect | gnomAD | Zygosity | Inheritance | dbSNP ID | ClinVar ID | Classifi cation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ETHE1 | Chr 19 | NM_014297.5: c.131_132delAG | p.(Glu44Valfs*62) | Frameshift | Premature truncation codon | 3/251040 | Homo-zygous | Biallelic | rs761827730 | 504493 | Class 5; pathogenic |
Table 3.
Biochemical analyses
| Result | |
|---|---|
| Urine metabolic screen | |
| C4 carnitine | ++ |
| C4 glycine | +++ |
| C6 glycine | ++ |
| C5 dicarboxylic | ++ |
| C5 carnitine | + |
| C5 dicarboxyl carnitine | + |
| C5 glycine | + |
| Urine organic acid and acylglycine screen | |
| Adipic acid | + |
| Ethylmalonic acid | >+++ |
| Hexanoyl glycine | >+++ |
| Isobutyryl glycine | >+++ |
| Isovaleryl glycine | ++ |
| 2-Methylbutyryl glycine | + |
| Methyl succinic acid | ++ |
| Urine metabolites | |
| Thiosulfate | High |
Phenotypic Analyses
Urine Metabolic Screen
The urine metabolic screen and organic acids analysis (Table 3) demonstrated a pattern consistent with either multiple acyl-CoA dehydrogenase deficiency (MADD) or ethylmalonic encephalopathy (C4–C6 acylcarnitines/acylglycines, ethylmalonic acid, isovalerylglycine, isobutyrylglycine, 2 methylbutyrylglycine, and methylsuccinic). Urine thiosulfate levels were subsequently analyzed and found to be elevated. Urine thiosulfate is a marker of abnormal hydrogen sulfide metabolism, which is more specific for ethylmalonic encephalopathy and not expected to be increased in MADD.
Newborn Screening Results (Retrospective)
Following receipt of the results from the urine metabolic screen and WGS, the patient's newborn blood spot screening analysis was retrospectively reviewed (Fig. 5). This showed increased C4 and C5 carnitine, relative to controls, consistent with ethylmalonic encephalopathy. However, the values were below the decision limits for a sample recollection, and the sample was therefore not flagged for further action at the time.
Figure 5.
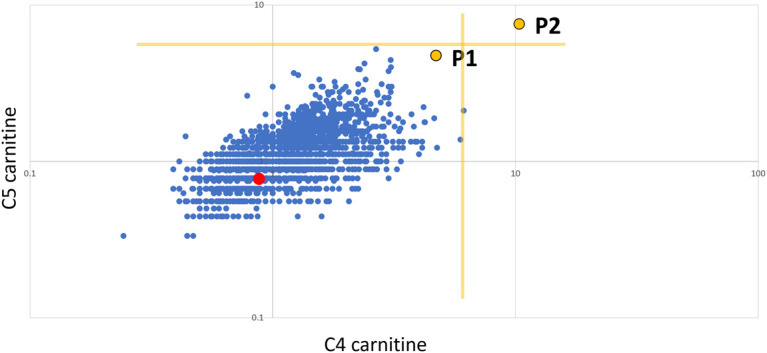
Retrospective reanalysis of newborn blood spot screening (NBS). C5 carnitine vs C4 carnitine expressed as multiples of median (log:log scale). P1 is the proband, P2 is an unrelated neonate with a confirmed ETHE1 defect, the red dot is the unaffected sibling of P1, the blue dots represent control neonates (n = 3215), and the yellow lines indicate decision limits for a repeat sample (∼99.95%ile).
Patient Follow-Up and Clinical Outcomes
The initial results of the urine metabolic studies showed increases in several metabolites (ethylmalonic, hexanoyl glycine, and glutaryl carnitine) that initially suggested the possibility of multiple acyl CoA dehydrogenase deficiency. The patient was started on riboflavin, coenzyme Q10, and carnitine enterally on day 5, with intravenous N-acetylcysteine (NAC), metronidazole, and a low protein intake added to reduce the enteric hydrogen sulfide (H2S) production and levels on day 6. Increased ethylmalonic acid excretion is not specific for ethylmalonic encephalopathy, as it is also increased in short chain acyl-CoA dehydrogenase deficiency (MIM #201470) and several genetic disorders causing multiple acyl-CoA dehydrogenase deficiency. Increased thiosulfate appears to be a reliable means of distinguishing ethylmalonic encephalopathy from these other disorders with increased ethylmalonic excretion (Blau et al. 2014). Targeted analysis of thiosulfate (a metabolite of hydrogen sulfide) in the urine was performed with results received after genomic sequencing.
Despite the targeted medical management, the patient's lactate levels rose from normal levels on day 3 with a subsequent decline in conscious state requiring reintubation on day 4. Between day 3 and day 5 a persistent lactemia of between 6 and 11.5 mmol/L (normal 1.2–2.8 mmol/L) reflected ongoing metabolic crisis. On day 8 of admission, she was noted to have fixed dilated pupils with progressive loss of brainstem function over the following 24 h. Head CT demonstrated diffuse supratentorial hypoxic ischemic injury with supratentorial edema and transtentorial herniation. A nuclear medicine scan on day 9 confirmed brain death. A specific trigger for the metabolic decompensation was not identified.
The family will have further follow-up and both grief and genetic counseling. The family has already indicated that they intend on having further children; as such these results will have major utility for informed counseling and decision-making for the family.
DISCUSSION
We present a case of a rare metabolic condition, ethylmalonic encephalopathy, masquerading as meningococcal septicemia, a common and life-threatening pediatric condition. This case provides a timely reminder to consider rare genetic diagnoses when atypical features of more common conditions are present—such as the patient's normal inflammatory markers, negative cultures, ongoing lactic acidemia without metabolic acidosis, and fluctuating conscious state. Shared and unique features of meningococcal septicemia and ethylmalonic encephalopathy are summarized in Table 4. Chronic symmetric MRI brain changes and preceding developmental and neurological concerns, together with a history of consanguinity, were suggestive of an underlying metabolic disorder and increased the probability of a genomic diagnosis. The specific findings on the urine metabolic and organic acid screens focused the genomic analysis, with ultrarapid genomic sequencing confirming the diagnosis of ethylmalonic encephalopathy for this family.
Table 4.
Comparison of meningococcal septicemia and ethylmalonic encephalopathy
| Meningococcal septicemia | Ethylmalonic encephalopathy | |
|---|---|---|
| Shock | + | +/− |
| Petechiae | + | + |
| Purpura | + | + |
| Peripheral perfusion | Poor | Poor |
| Positional acrocyanosis | - | + |
| Lactate/metabolic acidosis | High, normalizes | High, persistent |
| DIC | +/− | − |
| Altered conscious state | + | + |
| Seizures | +/− | +/− |
| Altered respiratory status | + | + |
| Diarrhea | +/− | +/− |
| Inflammatory markers | High | Normal/low |
| White cell count | High | Normal |
| Response to antimicrobials | Rapid | No |
| Abnormal CSF cell count/biochemistry | Yes | No |
| Meningococcal PCR or culture positive in blood or CSF | +/− | No |
| MRI brain changes | Acute | Acute on chronic/chronic |
| Abnormal neurodevelopmental history | Unlikely | Likely |
| Abnormal neurological signs | Follows rash | Precedes rash |
| Abnormal urine metabolic screen | Unlikely | Likely |
(DIC) Disseminated intravascular coagulation, (CSF) cerebrospinal fluid, (PCR) polymerase chain reaction, (MRI) magnetic resonance imaging, (+) feature is present in most cases, (−) feature is not present in most cases, (+/−) feature may or may not be present.
Ethylmalonic encephalopathy (MIM #602473) is a rare autosomal recessive metabolic condition, caused by biallelic variants in ETHE1 (MIM #608451). It is associated with global developmental delay, generalized infantile hypotonia, chronic diarrhea, seizures, and microvascular damage. The microvascular changes result in a pattern of diffuse and spontaneous relapsing petechiae and purpura, orthostatic acrocyanosis, and pedal edema, together with hemorrhagic suffusions of mucosal surfaces. It is characterized by a buildup of hydrogen sulfide and thiosulfate toxic metabolites. Neurologic deterioration often accelerates following illnesses and metabolic crises, with children usually dying in the first decade. Natural history and prognosis are altered by the timing of initiation of therapies such as supportive therapy, NAC, metronidazole, and tailored diets. Early initiation of NAC, metronidazole, and modified diet has been shown to alter neurodevelopmental trajectory as well as reduce the frequency of crises (Viscomi et al. 2010; Boyer et al. 2018). There is preliminary evidence for continuous renal replacement therapy (CRRT) for thiosulfate clearance and to assist in a crisis (Cardelo Autero et al. 2021). Although it does not reverse prior cerebral damage, liver transplantation stabilizes, or even improves, the disease course in ethylmalonic encephalopathy, reducing the frequency and severity of crises and representing an opportunity for intervention after early detection before irreversible neurological damage occurs (Tam et al. 2019; Zhou et al. 2020; Olivieri et al. 2021).
The diagnosis is suggested by clinical findings and substantiated by biochemical testing showing increased serum lactate, C4–C5 acylcarnitine esters, plasma, and urine thiosulfate and urinary ethylmalonic acid. Documented mild cases, late-onset cases, and those with only intermittent biochemical evidence are reported (Di Rocco et al. 2006; Kitzler et al. 2019; Ersoy et al. 2020). Brain MRI often demonstrates symmetric patchy T2-weighted signals in the basal ganglia, periventricular white matter and dentate nuclei, brain stem, and cerebellar white matter (Di Meo et al. 1993; Tiranti et al. 2004). In some instances, cortical atrophy and diffuse leukoencephalopathy are present (Di Meo et al. 1993; Tiranti et al. 2004).
Genetic conditions result in prolonged length of stay, increased case complexity, and increased readmissions (Gjorgioski et al. 2020). As the proportion of pediatric patients admitted to hospitals with underlying genetic causes increases and case complexity and total hospitalization costs increase, ultrarapid genomic testing will become an increasingly important tool to achieve diagnoses in a time- and cost-efficient manner. Considering rare genetic conditions, especially in patients whose clinical presentation is atypical, will enable earlier test initiation and avoidance of unnecessary investigations and treatments. Although atypical presentations of common conditions are often more likely than rare genetic conditions, the clinical impact of a diagnosis can be significant, and early genomic diagnosis may be lifesaving, particularly for metabolic conditions. These often present with crises and critical illness on the background of mild chronic concerns and may occur with minor environmental triggers. Metabolic disorders often have specific emergency management plans, and as such diagnosis is critical to timely and targeted therapy.
Despite the extremely rapid biochemical and genomic diagnosis and appropriate treatment, the outcome for this patient and family was still poor, prompting us to consider the utility of screening approaches in the reproductive and newborn settings. Considering the large proportion of consanguineous relationships in some communities it is important to remember that there is a residual risk of ∼3% for additional recessive diseases after an initial genetic diagnosis, such as in this family (AlAbdi et al. 2021). This further raises the question about improved access for families to reproductive carrier screening both before and after the birth of a child with an autosomal recessive disease, especially in the context of consanguinity. ETHE1 is included in most comprehensive reproductive carrier screening platforms and could have been avoided if it was more widely available to couples, especially those at higher risk. In contrast TNNT3 is not included on most platforms. A low-risk reproductive carrier screening result does not exclude a rare genetic condition, and as such clinicians should remain vigilant.
Ethylmalonic encephalopathy is potentially identifiable by newborn blood spot screening (NBS) using C4/C5 carnitine levels. This is illustrated by an unrelated child with ethylmalonic encephalopathy and C4/C5 carnitine levels above NBS decision limits (P2 in Fig. 5). P2 presented clinically in the first few days of life, suggesting that later-onset patients, such as the one presented here, may be more difficult to detect on NBS because of lower metabolite levels. Ethylmalonic encephalopathy is not currently included on NBS in many countries, influenced by its low prevalence and lack of a definitive treatment. However, evidence for the impact on morbidity including improved neurodevelopment and reduced metabolic crises suggests that this may soon change (Boyer et al. 2018). Early administration of NAC, metronidazole, and specific diets results in improved clinical outcomes and biochemical markers (Boyer et al. 2018). In the case of the proband, IV.III, the NBS C4/C5 levels were increased but did not reach the decision limits for recollection. This reinforces the need to remain vigilant for metabolic conditions in older infants and children despite a normal NBS result. Data on the sensitivity of NBS for ethylmalonic encephalopathy are lacking; cases such as ours suggest that at least some cases will be missed on NBS. This is in keeping with other metabolic conditions—in particular, those that require stress or a trigger to cause abnormalities in metabolic analytes (Elmonem and van den Heuvel 2021). Despite the limitations of NBS to detect ethylmalonic encephalopathy, retrospective analysis of NBS results from high-risk cases, such as this one, may provide useful diagnostic or confirmatory evidence. NBS for several inborn errors of metabolism is currently based on tandem mass spectrometry, but research and debate are currently ongoing on the potential transition to genomic sequencing for NBS as an opportunity for population health screening initiatives (Woerner et al. 2021), which may result in earlier diagnosis and treatment of rare inborn errors of metabolism such as ethylmalonic encephalopathy that are not routinely included on current NBS panels.
In summary, this case provides a timely reminder to consider rare genetic diagnoses when atypical features of more common conditions are present, with an early referral for a prompt genomic diagnosis. It also raises questions regarding the timing and utility of genomic sequencing as part of newborn and reproductive carrier screening programs.
METHODS
Genomic Sequencing
Genomic sequencing was performed by a clinically accredited laboratory at the Victorian Clinical Genetics Services, using a WGS technique. WGS was performed using massively parallel sequencing (MPS) (Nextera DNA Flex Library Prep kit (Illumina Sequencers) with a mean target coverage of 30×, and a minimum of 90% of bases sequenced to at least 10×. Sequencing data was generated using Illumina NovoSeq 6000 instruments using S2 flow cells and a 1 × 150-bp paired-end run configuration. Data were processed, including read alignment to the reference genome (GRCh38) and variant calling, using Cpipe (Sadedin et al. 2015) or a functionally equivalent analysis with the Illumina DRAGEN System (Illumina). Variant analysis and interpretation within the selected target region (RefSeq genes ±1 kb) was performed using Alissa Interpret (Agilent). Variants were annotated against all RefSeq gene transcripts and reported following Human Genome Variation Society (HGVS) nomenclature (den Dunnen et al. 2016). Copy-number variants (CNVs) were screened for using an internal CNV detection tool (Sadedin et al. 2018). Curation of variants was phenotype-driven with a precurated custom gene list for variant prioritization. Variant classification was based on modified ACMG/AMP guidelines in line with the ClinGen Sequence Variant Interpretation working group (Richards et al. 2015; Harrison et al. 2019). Consistent with our clinical accreditation, variants that meet minimum quality criteria for clinical reporting are not in general validated using orthogonal methods.
Urine Metabolic Screen
Urine organic acids were determined by standard gas chromatography-mass spectrometry methodology. Comprehensive targeted urine metabolite screening was performed by flow injection tandem mass spectrometry as previously described (Pitt et al. 2002). Urine thiosulfate was determined by the addition of a −113>−80 m/z transition to the above platform.
Newborn Screening
NBS for amino acids and acyl carnitines was performed by standard flow injection tandem mass spectrometry after butylation.
ADDITIONAL INFORMATION
Data Deposition and Access
The variant was submitted to ClinVar (https://www.ncbi.nih.nlm.gov/clinvar/) and can be found under accession number SCV002016238.1. Raw sequencing data could not be deposited because of lack of patient consent.
Ethics Statement
The patient's family provided written consent for publication of deidentified patient information, results, pedigree, clinical photography, and videography.
Acknowledgments
Trio WGS of the patient and parents was performed as part of the Australian Genomics Acute Care study with Human Research Ethics Committee approval HREC/16/MH251 and with funding from the Medical Research Futures Fund (GHFM76747).
Author Contribution
All authors have contributed to patient care and publication and approved the current version of the manuscript and its submission to CSH Molecular Case Studies.
Funding
The research conducted at the Murdoch Children's Research Institute was supported by the Victorian Government's Operational Infrastructure Support Program. The Chair in Genomic Medicine awarded to J.C. is generously supported by The Royal Children's Hospital Foundation.
Competing Interest Statement
The authors have declared no competing interest.
Referees
Joshi Stephen
Anonymous
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- AlAbdi L, Alrashseed S, Alsulaiman A, Helaby R, Imtiaz F, Alhamed M, Alkuraya FS. 2021. Residual risk for additional recessive diseases in consanguineous couples. Genet Med 23: 2448–2454. 10.1038/s41436-021-01289-5 [DOI] [PubMed] [Google Scholar]
- Blau N, Duran M, Gibson KM, Dionisi-Vici C. 2014. Physician's guide to the diagnosis, treatment, and follow-up of inherited metabolic diseases. Springer-Verlag, Berlin. [Google Scholar]
- Boyer M, Sowa M, Di Meo I, Eftekharian S, Steenari MR, Tiranti V, Abdenur JE. 2018. Response to medical and a novel dietary treatment in newborn screen identified patients with ethylmalonic encephalopathy. Mol Genet Metab 124: 57–63. 10.1016/j.ymgme.2018.02.008 [DOI] [PubMed] [Google Scholar]
- Cardelo Autero N, Cordón Martínez AM, Ramos-Fernández JM. 2021. Ethylmalonic encephalopathy: phenotype-genotype description and review of its management. Neurologia (Engl Ed) 36: 729–731. 10.1016/j.nrl.2021.01.004 [DOI] [PubMed] [Google Scholar]
- den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, Roux AF, Smith T, Antonarakis SE, Taschner PE. 2016. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat 37: 564–569. 10.1002/humu.22981 [DOI] [PubMed] [Google Scholar]
- Di Meo I, Lamperti C, Tiranti V. 1993. Ethylmalonic encephalopathy. In GeneReviews® (ed. Adam MP, Ardinger HH, Pagon RA, et al. ). University of Washington, Seattle, WA. [PubMed] [Google Scholar]
- Dimmock D, Caylor S, Waldman B, Benson W, Ashburner C, Carmichael JL, Carroll J, Cham E, Chowdhury S, Cleary J, et al. 2021. Project Baby Bear: rapid precision care incorporating rWGS in 5 California children's hospitals demonstrates improved clinical outcomes and reduced costs of care. Am J Hum Genet 108: 1231–1238. 10.1016/j.ajhg.2021.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dionisi-Vici C, Diodato D, Torre G, Picca S, Pariante R, Giuseppe Picardo S, Di Meo I, Rizzo C, Tiranti V, Zeviani M, et al. 2016. Liver transplant in ethylmalonic encephalopathy: a new treatment for an otherwise fatal disease. Brain 139: 1045–1051. 10.1093/brain/aww013 [DOI] [PubMed] [Google Scholar]
- Di Rocco M, Caruso U, Briem E, Rossi A, Allegri AE, Buzzi D, Tiranti V. 2006. A case of ethylmalonic encephalopathy with atypical clinical and biochemical presentation. Mol Genet Metab 89: 395–397. 10.1016/j.ymgme.2006.05.010 [DOI] [PubMed] [Google Scholar]
- Elmonem MA, van den Heuvel LP. 2021. Editorial: Newborn screening for inborn errors of metabolism: is it time for a globalized perspective based on genetic screening? Front Genet 12: 758142. 10.3389/fgene.2021.758142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ersoy M, Tiranti V, Zeviani M. 2020. Ethylmalonic encephalopathy: clinical course and therapy response in an uncommon mild case with a severe ETHE1 mutation. Mol Genet Metab Rep 25: 100641. 10.1016/j.ymgmr.2020.100641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjorgioski S, Halliday J, Riley M, Amor DJ, Delatycki MB, Bankier A. 2020. Genetics and pediatric hospital admissions, 1985 to 2017. Genet Med 22: 1777–1785. 10.1038/s41436-020-0871-9 [DOI] [PubMed] [Google Scholar]
- Harrison SM, Biesecker LG, Rehm HL. 2019. Overview of Specifications to the ACMG/AMP Variant Interpretation Guidelines. Curr Protoc Hum Genet 103: e93. 10.1002/cphg.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitzler TM, Gupta IR, Osterman B, Poulin C, Trakadis Y, Waters PJ, Buhas DC. 2019. Acute and chronic management in an atypical case of ethylmalonic encephalopathy. JIMD Rep 45: 57–63. 10.1007/8904_2018_136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunke S, Eggers S, Wilson M, Patel C, Barnett CP, Pinner J, Sandaradura SA, Buckley MF, Krzesinski EI, de Silva MG, et al. 2020. Feasibility of ultra-rapid exome sequencing in critically ill infants and children with suspected monogenic conditions in the Australian public health care system. J Am Med Assoc 323: 2503–2511. 10.1001/jama.2020.7671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivieri G, Martinelli D, Longo D, Grimaldi C, Liccardo D, Di Meo I, Pietrobattista A, Sidorina A, Semeraro M, Dionisi-Vici C. 2021. Ethylmalonic encephalopathy and liver transplantation: long-term outcome of the first treated patient. Orphanet J Rare Dis 16: 229. 10.1186/s13023-021-01867-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen MJ, Niemi AK, Dimmock DP, Speziale M, Nespeca M, Chau KK, Van Der Kraan L, Wright MS, Hansen C, Veeraraghavan N, et al. 2021. Rapid sequencing-based diagnosis of thiamine metabolism dysfunction syndrome. N Engl J Med 384: 2159–2161. 10.1056/NEJMc2100365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt JJ, Eggington M, Kahler SG. 2002. Comprehensive screening of urine samples for inborn errors of metabolism by electrospray tandem mass spectrometry. Clin Chem 48: 1970–1980. 10.1093/clinchem/48.11.1970 [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadedin SP, Dashnow H, James PA, Bahlo M, Bauer DC, Lonie A, Lunke S, Macciocca I, Ross JP, Siemering KR, et al. 2015. Cpipe: a shared variant detection pipeline designed for diagnostic settings. Genome Med 7: 68. 10.1186/s13073-015-0191-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadedin SP, Ellis JA, Masters SL, Oshlack A. 2018. Ximmer: a system for improving accuracy and consistency of CNV calling from exome data. Gigascience 7: giy112. 10.1093/gigascience/giy112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandaradura SA, Bournazos A, Mallawaarachchi A, Cummings BB, Waddell LB, Jones KJ, Troedson C, Sudarsanam A, Nash BM, Peters GB, et al. 2018. Nemaline myopathy and distal arthrogryposis associated with an autosomal recessive TNNT3 splice variant. Hum Mutat 39: 383–388. 10.1002/humu.23385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark Z, Ellard S. 2022. Rapid genomic testing for critically ill children: time to become standard of care? Eur J Hum Genet 30: 142–149. 10.1038/s41431-021-00990-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam A, AlDhaheri NS, Mysore K, Tessier ME, Goss J, Fernandez LA, D'Alessandro AM, Schwoerer JS, Rice GM, Elsea SH, et al. 2019. Improved clinical outcome following liver transplant in patients with ethylmalonic encephalopathy. Am J Med Genet A 179: 1015–1019. 10.1002/ajmg.a.61104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiranti V, D'Adamo P, Briem E, Ferrari G, Mineri R, Lamantea E, Mandel H, Balestri P, Garcia-Silva MT, Vollmer B, et al. 2004. Ethylmalonic encephalopathy is caused by mutations in ETHE1, a gene encoding a mitochondrial matrix protein. Am J Hum Genet 74: 239–252. 10.1086/381653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiranti V, Viscomi C, Hildebrandt T, Di Meo I, Mineri R, Tiveron C, Levitt MD, Prelle A, Fagiolari G, Rimoldi M, et al. 2009. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat Med 15: 200–205. 10.1038/nm.1907 [DOI] [PubMed] [Google Scholar]
- Viscomi C, Burlina AB, Dweikat I, Savoiardo M, Lamperti C, Hildebrandt T, Tiranti V, Zeviani M. 2010. Combined treatment with oral metronidazole and N-acetylcysteine is effective in ethylmalonic encephalopathy. Nat Med 16: 869–871. 10.1038/nm.2188 [DOI] [PubMed] [Google Scholar]
- Woerner AC, Gallagher RC, Vockley J, Adhikari AN. 2021. The use of whole genome and exome sequencing for newborn screening: challenges and opportunities for population health. Front Pediatrics 9: 663752. 10.3389/fped.2021.663752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou GP, Qu W, Zhu ZJ, Sun LY, Wei L, Zeng ZG, Liu Y. 2020. Compromised therapeutic value of pediatric liver transplantation in ethylmalonic encephalopathy: a case report. World J Gastroenterol 26: 6295–6303. 10.3748/wjg.v26.i40.6295 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The variant was submitted to ClinVar (https://www.ncbi.nih.nlm.gov/clinvar/) and can be found under accession number SCV002016238.1. Raw sequencing data could not be deposited because of lack of patient consent.