

Abstract
Rare neurogenetic disorders are collectively common, affecting 3% of the population, and often manifest with complex multiorgan comorbidity. With advances in genetic, -omics, and computational analysis, more children can be diagnosed and at an earlier age. Innovations in translational research facilitate the identification of treatment targets and development of disease-modifying drugs such as gene therapy, nutraceuticals, and drug repurposing. This increasingly allows targeted therapy to prevent the often devastating manifestations of rare neurogenetic disorders. In this perspective, successes in diagnosis, prevention, and treatment are discussed with a focus on inherited disorders of metabolism. Barriers for the identification, development, and implementation of rare disease-specific therapies are discussed. New methodologies, care networks, and collaborative frameworks are proposed to optimize the potential of personalized genomic medicine to decrease morbidity and improve lives of these vulnerable patients.
INTRODUCTION
With advances in genetic and -omics technologies, more children with rare neurogenetic disorders—that is, neurodevelopmental disorders (NDDs) and inherited metabolic disorders (IMDs)—can be diagnosed at an earlier age (Blau et al. 2022). Early genetic diagnosis is paramount for the movement toward disease-modifying and other personalized treatments that have great potential for improving health and quality of life (Sahin and Sur 2015; Vissers et al. 2016). These approaches may help many individuals, because NDDs are collectively common, affecting 3% of the population directly and another 5% indirectly—that is, as family members coping with care and potential recurrence risks. Patients are often affected by severe and complex somatic and neuropsychiatric comorbidity, including intellectual disability (ID), epilepsy, cognitive and behavioral disturbances, sensory deficits, and other organ dysfunction. Because of their lifelong care needs on multiple life domains, these patient populations present a challenging task for health-care providers and systems to provide optimal personalized care, taking both the characteristics of the disorder as well as the individual care needs into account.
The opportunities for personalized therapeutic interventions, both symptomatic and disease-modifying, are drastically increasing for NDDs, and even more for IMDs. These vary from diets and vitamins, (repurposed) medications, organ and stem cell transplants, to RNA and gene therapy (Sun et al. 2017). However, implementation via therapeutic trials for these often-small patient populations face specific challenges in methodology and outcome measures. Additionally, implementation of personalized medicine is hampered by financial, organizational, and regulatory barriers. Translation of novel treatments to patient care with guaranteed access and reimbursement is cumbersome and often unsuccessful at both the local and international levels. This results in an unacceptable situation: Novel treatments do not reach the patient, and there is lack of evidence-based care.
To address the current state of personalized medicine for IMDs, we review several success stories to show opportunities for improvement in trial design, outcome measures, and collaborative efforts. We put forward how this knowledge can be extended to other NDDs and propose acceleration of personalized medicine via our flywheel model as depicted in Figure 1.
Figure 1.
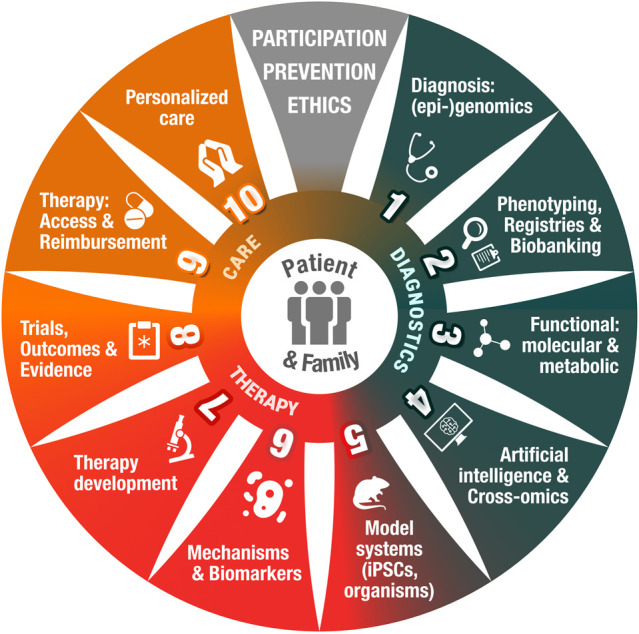
Catalyzing personalized medicine for all rare disease patients. This flywheel depicts the patient and family at the center of the rare diseases community's translational research and care activities, aimed at providing accurate diagnosis and counseling, effective and accessible therapies, and ultimately tailored care and prevention. Patients are partners in the process, strengthening the transdisciplinary team of basic and laboratory scientists, paramedical clinicians, and ethicists who unite expertise and efforts to catalyze this flywheel to leave no rare disease patient behind. We apply this model in the Emma Center for Personalized Medicine at Amsterdam UMC. (Copyright: Health2Media.)
ADVANCES IN DIAGNOSTICS
Numerous studies have shown advances in diagnosing presymptomatic individuals, acutely ill children, and children and adults with developmental delay, epilepsy, neurocognitive deterioration, or other illnesses. Rapid diagnosis prevents diagnostic odysseys, helps caregivers with acceptance and connecting with patient organizations for peer support, and allows accurate counseling regarding prognosis and recurrence risk. Increasingly, a genetic diagnosis allows for personalized medicine—that is, the implementation of etiology-driven health monitoring and treatments, with the potential of drastically changing the course of a disorder.
Population-based genetic screening has provided enormous opportunities to diagnose rare diseases before clinical disease onset. Strategies and timing vary from preconception and preimplantation carrier screening in parents to prenatal and neonatal screening programs in offspring; historically, genetic testing was considered too slow, yet next-generation sequencing (NGS) analysis has been shown to yield diagnoses and treatment potential in 73% of families with acutely ill children (Soden et al. 2014). NGS has also revolutionized the discovery of variants in new genes that are associated with rare Mendelian diseases (Tarailo-Graovac et al. 2016; Boycott and Ardigó 2018; Quaio et al. 2020). With exome sequencing, the diagnostic yield can be as high as 41% for rare pediatric conditions (Quaio et al. 2020) and 39% for early-onset epilepsies (Demos et al. 2019), providing a direct potential for the redirection of care in 15.6% and 39%, respectively. Studies in adults with epilepsy, autism spectrum disorder, and ID have shown similar results (Snoeijen-Schouwenaars et al. 2019; Satterstrom et al. 2020). The combination of deep clinical and biochemical phenotyping with whole-exome sequencing (WES) provided a diagnosis in 68% of children with developmental delay and a metabolic phenotype with a change in treatment in 44% (Tarailo-Graovac et al. 2016).
However, despite the diagnostic success of WES, at least one-third of patients with a rare disease phenotype still remain undiagnosed. Without a molecular diagnosis, targeted treatment is not possible. The reasons for missing heritability are diverse and include technical limitations (e.g., missed coding variants by WES, structural abnormalities, intronic variants), complex mechanisms (e.g., tissue-specific somatic mosaicism, allelic expression imbalance [Falkenberg et al. 2017]), short tandem repeat expansions (van Kuilenburg et al. 2019), polygenic inheritance (as recently described in as many as 4%–9% of patients with rare diseases), and lack of evidence for or against the causality of a particular candidate variant (Posey et al. 2017).
Whole-genome sequencing (WGS) has a higher sensitivity than WES for certain coding variants, insertions/deletions, copy-number variants (CNVs), chromosomal rearrangements, inversions, transposons, or causal variants in regulatory regions. WGS may therefore be an effective technique to identify the genetic basis of unresolved IMD especially if combined with RNA sequencing (Kremer et al. 2017). RNA therapy is increasingly available for splice-site variants and related complex molecular mechanisms (Kim et al. 2019). Interpretation of gene variants of unknown significance, discovery of incidental findings and carriership for genetic disorders (Petrikin et al. 2015), and societal stigma need careful evaluation. Despite these challenges, parents show high acceptance and few regrets when being apprised of results (Cakici et al. 2020).
Beyond exome and genome sequencing, other “-omics” are changing the diagnostic landscape (van Karnebeek et al. 2018). Genome-wide methylation analysis (methylomics) can also identify biological signals that support the causality of disease genes. For example, the methylation profiling of several rare diseases associated with gene defects suspected of having an effect on methylation status identified diagnostic signatures for more than 48 neurological disorders (Schenkel et al. 2017; Sadikovic et al. 2021).
Advanced technologies such as metabolomics, lipidomics, and glycomics techniques enable simultaneous profiling of thousands of metabolites, lipids, and glycans in body fluids, tissues, and cells. Together with model organisms, these approaches offer a possible pathway to study the phenotypes that remain unsolved after advanced WES or WGS (Vaz et al. 2019). In such “systems biology” approaches, powerful bioinformatics and close communication with molecular genetics, IMD clinicians, laboratory specialists, and scientists are essential to determine the phenotypic, genetic, biochemical, and functional data. Using methods such as gene expression, CRISPR–Cas technologies, and genetic engineering of cell models and model organisms (“functional genetics”), it is possible to unravel disease mechanisms, identify biomarkers for diagnosis and disease activity, and develop therapeutic strategies in a so-called translational metabolism model (van Karnebeek et al. 2018; Wanders et al. 2019; Boycott et al. 2020). This principle is illustrated by a study in which 90% of the 41 families with an unexplained neurometabolic phenotype could still be diagnosed by applying a combination of phenomics and multiomics, and in which 11 new disease genes and more than 20 new phenotypes were identified (Tarailo-Graovac et al. 2016). In addition to an increased diagnostic yield, the above study also showed that the chosen multiomics approach in combination with deep phenotyping can lead to new pathophysiological insights, enabling (new) optimized treatment in >40% of patients. The added value of untargeted metabolomics to WES and phenomics has been illustrated with the discovery of a new IMD: NANS-congenital disorder of glycosylation (CDG). This condition was identified in nine patients with ID and bone abnormalities as a result of this novel sialic acid synthesis defect (van Karnebeek et al. 2016). N-acetyl-mannosamine was subsequently identified as a biomarker in several Dutch patients. Recently, a deep phenotyping study of NANS-CDG showed that this progressive neurometabolic disease has typical magnetic resonance imaging (MRI) abnormalities with a genotype–phenotype correlation (den Hollander et al. 2021). Zebrafish studies showed that intervention with sialic acid corrected the phenotype, leading to a human clinical trial now under way, as well as iPSC studies for additional treatments.
Predictive Medicine: Phenotypic Modifiers and Prognosis
Knowledge of factors that determine and influence the course of the disorders is necessary to optimize prognosis and correct timing of interventions. This is exemplified by X-linked adrenoleukodystrophy for which there is no possibility to predict the course at diagnosis. Timely hematopoietic stem cell transplantation (HSCT) can, however, prevent very serious, ultimately fatal, degeneration of the central nervous system (Kemp et al. 2016). This disease manifestation only occurs in a limited number of affected boys for whom presently very frequent MRI screening of the brain is the only available prevention to identify first significant changes in the brain. Phenotypic discordance in individuals with the same genotype, including siblings and monozygotic twins, suggests that additional (epi-)genetic modifiers are needed to activate the demyelination process. A recent proof-of-principle study applying a combination of various -omics technologies identified the differences or phenotypic modifiers in six brothers discordant for early and late disease onset (Richmond et al. 2020). Further research to generate these insights has become even more essential now that screening for X-linked adrenoleukodystrophy (ALD) is included in the newborn screening program (Barendsen et al. 2020). Gene therapy for this debilitating disorder is also becoming available (Eichler et al. 2017). In addition, knowledge about modifiers may also be used to develop preventive measures and develop new therapies targeting protective modifiers enhancing resilience.
FROM DIAGNOSIS TOWARD TREATMENT
Ideally, early diagnosis allows disease-modifying and other therapies to exert their effect in the crucial “neurodevelopmental time window,” potentially preventing (progression of) epileptic encephalopathies, ID, and autism (Kotulska et al. 2021; van Karnebeek and Jaggumantri 2015) as well as somatic complications (Krueger et al. 2010). Treatment targets can be identified at the genomic, epigenomic-transcriptomic, and metabolomic level using model systems, such as (differentiated) induced pluripotent stem cells (iPSCs), organoids, and organisms. Deep phenotyping of these models allows for biomarker identification. Additionally, potentially treatable manifestations can be identified via radiological, electrophysiological, hematological, somatic, neuropsychiatric, and contextual characterization.
Figure 1 illustrates the many steps needed for treatment success represented by the different blades in our flywheel. Such a formidable task in our experience can only be achieved by uniting expertise and efforts in a dedicated personalized medicine center. Detailed evaluation of the effect of existing and future interventions (diet, nutritional supplements, repurposed/medication, transplantation, cellular, and RNA/gene therapy) on relevant outcome measures in vitro, using various -omics technologies and cell models (Wagstaff et al. 2021), and in vivo in trials that are methodology designed for rare diseases, are an important part of this center (Stockler-Ipsiroglu et al. 2021).
INHERITED METABOLIC DISORDERS AS A MODEL
Over the past century IMDs elegantly illustrate how knowledge of the precise defect in genetic or biochemical pathways has led to therapies that target pathophysiological features at the molecular or cellular level. Although individually rare, the collective frequency of IMDs is significant and estimated at 1 in 800 newborns. These monogenic diseases are often caused by the deficiency of an enzyme or transporter. The resulting deficiencies cause a blockage in the metabolic process, resulting in an accumulation of substrate and a shortage of one or more important building materials that cause progressive damage to organs such as the brain, heart, liver, kidneys, or eyes. IMDs are now the largest group of monogenic disorders (n > 1600) for which targeted treatments are available or under development, ranging from an adapted diet, nutritional supplements, enzyme therapy, bone marrow transplantation, and medication to cellular and gene/RNA therapy (Ferreira et al. 2021; Hoytema van Konijnenburg 2021).
Increasingly, in certain groups of patients with symptoms such as ID, previously considered nontreatable, IMDs are recognized as a substantial etiological group. For example, a recent study of 518 patients with ID in which a systematic screening algorithm using directed mass spectrometry as a first-line screening test, found IMD as a cause in 8% of the subjects examined, the majority of whom appeared to be treatable (van Karnebeek 2014).
Only good understanding of the pathophysiology enables development and evaluation of targeted treatments for IMDs. The best-known example concerns the therapy for phenylketonuria, based on research by Følling in 1934. Examination of two children with ID and a distinctive odor from the urine due to excessive amounts of phenylpyruvic acid indicated a causal deficiency of the enzyme phenylalanine hydroxylase (Centerwall and Centerwall 2000). Based on this pathophysiological insight, Dr. H. Bickel was the first to successfully treat PKU patients with a phenylalanine restricted diet in 1954. This finding paved the way for the development of a neonatal screening method (heel prick) for early detection of PKU patients, which Guthrie first described in 1962. Introduction of the heel prick screening program for PKU in newborns has led to the prevention of severe cognitive impairment in individuals around the world. However, the resulting lifelong diet is a burden and challenges compliance. Therefore, new treatment options have emerged such as BH4 as cofactor supplementation to catalyze residual enzyme activity in a personalized fashion and, more recently, subcutaneous PAH enzyme replacement, and gene replacement therapy, currently in the trial phase. These successes and ongoing developments are the result of a close collaboration between researchers, specialists, and the PKU patient association. The PKU story is a fine example of “preventive, predictive, personalized, and participatory” medicine (P4 medicine; Fig. 2), a model increasingly applied over the past five decades to provide optimal care for IMD patients (Hood et al. 2012).
Figure 2.

P4 medicine model.
Another illustrative example, to which our group has contributed intensively, is pyridoxine-dependent epilepsy (PDE). In 2005, this severe encephalopathic epilepsy, known since 1951 as treatment-resistant neonatal seizures that only respond to vitamin B6 (pyridoxine), was unraveled simultaneously in the Netherlands and the United Kingdom as a neurometabolic disease resulting from a lysine breakdown disorder leading to accumulation of potentially toxic metabolites (including alpha-amino-adipic acid semialdehyde, α-AASA) (Mills et al. 2006). This explains the ID that occurs in 75% of patients, despite adequate epilepsy control with pyridoxine (Bok et al. 2012). In search of a better treatment, we started the International PDE Consortium to develop, in close collaboration with patients and families (P4: participatory), an adjuvant treatment to reduce α-AASA production. This resulted in the so-called lysine reduction therapy (LRT), a dietary treatment in combination with arginine supplementation, leading to an improvement in epilepsy control and psychomotor development in treated patients (P4: personalized) (Coughlin et al. 2015). The evidence for these improved outcomes with LRT as adjunctive treatment was reinforced (level 2b) by the International PDE Registry study. Based on this, the PDE care pathway has been developed and the PDE international consensus guidelines have been published (Coughlin et al. 2021).
The next hurdle, the identification of a reliable biomarker (2-OPP) for PDE that can be detected by means of neonatal screening bloodspot cards in order to be able to start treatment in time (P4: preventive), has been overcome (Engelke et al. 2021). In the meantime, further research into better treatment options is ongoing (e.g., via upstream enzyme inhibition). The success of diagnosis, treatment, early detection, and prevention in PDE is in stark contrast to that in the vast majority of IMDs, characterized by physical and mental disabilities, which are often progressive and sometimes fatal. The diagnosis is often made relatively late, and treatment is often only supportive and palliative for at least 1250 IMDs. In addition, it is plausible that there are still many tens or even hundreds of IMDs that are not yet recognized as such, and for which there are therefore no treatment developments as yet (Ferreira et al. 2019; Warmerdam et al. 2020).
Other recent success stories include specific amino acid supplementations for the acyl-tRNA synthetase 1 deficiencies and nicotinamide riboside for the redox defects (Kok et al. 2021; Zapata-Pérez et al. 2021). Furthermore, for conditions considered more NDDs than IMD, progress is being made as well, exemplified by L-serine supplementation to rescue loss of function of the N-methyl-D-Aspartate (NMDR) receptor for Grin2B-NDD (Krey et al. 2022). This treatment improves psychomotor development and seizures.
Recent advances for non-nutraceutical IMD interventions include Asfotase alpha as tissue-nonspecific alkaline phosphatase (TNSALP) enzyme replacement therapy for hypophosphatemia, which successfully treats the debilitating bone phenotype as well as the epilepsy in affected patients (Bianchi and Vai 2019). Recently, lumasiran, an RNAi therapeutic for primary hyperoxaluria type 1, was reported to reduce urinary oxalate excretion, the cause of progressive kidney failure in PH1 (Garrelfs et al. 2021). Another impressive example of personalized medicine is the development of milasen (a splice-site modulating antisense oligonucleotide) tailored to Mila suffering CLN7 and its administration in an N-of-1 study within 1 year of diagnosis (Kim et al. 2019). Initially seizures improved but ultimately the disease was fatal. Excitingly, gene therapies are becoming a reality as well. Recently EMA approved Libmeldy, an ex vivo gene therapy with lentivirus vector-transduced autologous CD34+ stem cells, for treatment of metachromatic leukodystrophy (Fumagalli et al. 2022).
NEUROGENETIC DISORDER TRIALS AND TRIBULATIONS
Personalized Trial Design
Heterogeneity and small numbers pose a challenge to generate evidence for rare diseases such as IMDs (Stocker et al. 2020). Somatic and neuropsychiatric manifestations typically show great inter- and intra- individual variability of the disorder over time, hampering conventional trial designs. In addition, randomized controlled trials (RCTs) have often shown great differences in treatment response resulting in negative results on a group level (Erickson et al. 2017; Overwater et al. 2019), potentially leading to the missing out on important therapies for individual patients. This variability in manifestations, treatment targets, and treatment response calls for a new framework for personalized medicine trial methodology.
Properly executed single-case experimental designs (SCEDs) such as the N-of-1 (A-B-A) design or the multiple baseline design (Tate and Perdices 2019) may provide a powerful alternative to larger randomized controlled trials in rare disorders and a much needed bridge between practice and science (Müller et al. 2021). N-of-1 studies are randomized, controlled, multiple crossover trials in a single patient and closely follow indications of causality. Whereas traditionally RCTs generally assess an average treatment effect on a group level, N-of-1 series identify individual characteristics that may modify response to the intervention, addressing this important question of variability in treatment response. Series of SCEDs will allow cross-disorder comparisons and investigation of generalizability to the whole population affected with these disorders and/or comorbidity.
It has been proposed that targeted disorders or comorbidity should be stable over time to be eligible for conducting an N-of-1 study. IMDs are, however, often (neuro)degenerative disorders, resulting in an unstable and often variable natural course across patients. As the natural course of NDDs unfolds, this variable course increasingly applies to many other NDDs (Antonarakis et al. 2020; Rosso et al. 2020). However, even for unstable manifestations, effects may be observed by tracing the overall enduring effect on the personal course, including (multiple) baseline, placebo, and follow-up measurements. In this way, disease-modifying treatment options can be investigated, theoretically expecting a more enduring effect on the individual's natural course for disease-modifying drugs versus a temporary effect for symptomatic drug treatments.
In all cases, a personalized baseline is warranted to observe manifestations without any intervention. Follow-ups will add internal validity and information about the effectiveness and tolerance of an intervention, calling for longitudinal monitoring and phenotyping in patient registries for optimal retro- and prospective collection of evidence. The deep phenotyping mentioned above can help to identify (surrogate) biomarkers for monitoring of the disease course and response to treatments. For optimal generalizability to patients with the same disorder, sample size calculations will provide information on the number of inclusions needed (Senn 2019). For analyzing the results, both mixed-effects models and Bayesian models can properly address the inter- and intrapatient variability in series (Senn 2016).
Personalized Outcome Measures
Because IMDs comprise a vulnerable patient group often affected by severe comorbidity and complex environmental factors, there is a great need for personalized and disorder-specific outcome measures. These measures should strive for optimal patient relevance as well as pathophysiological insights, with minimally invasive procedures such as digital apps, including objective and biological outcomes, validated symptom checklists, neuropsychological assessments, and personalized outcomes. The International Classification of Functioning and Disability (ICF) of the World Health Organization (Kostanjsek 2011) provides a framework to select a combination of outcome measures that capture all components for better understanding of the impact of a diagnosis on all life domains, providing relevant interventions for optimal quality of life.
Instruments such as patient-reported outcome measures (Slade et al. 2018), Goal Attainment Scaling (Gaasterland et al. 2019), or experience-sampling methods (van Os et al. 2017) may be considered, enabling quantitative expression of meaningful subjective patient experiences while translating these into evidence. As patients with ID can often not report their clinical condition, this places a demand on parents and caregivers, and proxy-friendly assessment tools are required to ensure trial compliance. This personalized approach has the potential of maximizing treatment and trial adherence that is both patient-centered and evidence-based. To further foster this adherence, patient involvement in the intervention, design, and outcome measures will greatly contribute to the experienced relevancy and enthusiasm of participants (Gaasterland et al. 2018).
Unfortunately, outcome measures are often not available or validated for these rare NDD/IDM cohorts, and even in validation studies the property responsiveness to change is often not assessed. Extensive investigation of responsiveness to therapy is essential in measuring the effectiveness of interventions, as lack of responsiveness may have resulted in the often-disappointing trial results in NDDs in the past (Budimirovic et al. 2017).
FROM GENES, TO TRIALS, TO CARE
Because opportunities to treat IMDs/NDDs are emerging at a steady rate, we need to adapt and seize the opportunity. The patients deserve and demand a new paradigm in translational research as well as care. What is needed to adjust our health-care system to these new possibilities?
Into the Future with P4 Medicine
Our care for individuals with IMDs/NDDs will have to adapt to a “per patient approach,” using multidisciplinary collaborations, ensuring optimal relevance and alignment with patient needs. P4 (Hood 2006) medicine can be used as a model to empower clinicians to implement local and national care paths, optimally integrated with research, education, and guidelines (Fig. 2). P4 medicine is preferably provided in a full facility academic clinic, where all involved research and clinical disciplines can interact to provide holistic care to the patient and family. An example is our Emma Center for Personalized Medicine in Amsterdam, which is organized in three different patient friendly portals as illustrated by the green, red, and orange colors in Figure 1. We aim to provide timely diagnosis (blades 1–4), innovate therapy (blades 5–8), and tailored care (blades 9–10) to rare diseases patients, ultimately implementing this via local and international care networks and guidelines.
After genetic diagnosis, care, and treatment targets, recommendations should be implemented in counseling on all life domains using the ICF framework, preferably simultaneously establishing and educating a local care network. Increasingly, expert centers for specific disorders and patient populations are established on national levels, and in Europe international collaborations are encouraged by European Reference Centers such as MetabERN and ERN ITHACA. In this way, patient-friendly transmural care networks can be established, preferably supported by shared electronic patient records. Accessible information on diagnosis and therapies will lower the threshold for health-care providers and patients alike. This is enabled by freely accessible apps that recommend IMD differential diagnoses and treatments such as IEMBase (www.iembase.org; Lee et al. 2018) and Treatable ID (www.treatable-id.org; Hoytema van Konijnenburg et al. 2021).
Registries, preferably patient-owned, are necessary at local and (inter)national levels to monitor features on all life domains as well as longitudinal patient-reported outcomes. Hence, patients will have information on the course of their individual functioning and can evaluate severity of comorbidity or effectiveness of interventions retro- and prospectively. Such lifelong monitoring including digital technologies can prevent that the patient with ID will be “known well by no one” in adulthood (Camfield and Camfield 2011).
There are ethical considerations to consider—for example, to ensure that individuals without a diagnosis receive the same expert care that is increasingly available for specific disorders (Mueller et al. 2016). Additionally, genetic diagnostics and personalized care should become accessible for lower-income countries, but also for adults with ID. As life expectancy of individuals with NDDs has increased (Coppus 2013; Stepien et al. 2021), now the largest NDD population comprises adults, many of whom are not diagnosed and thus missing out on personalized medicine.
New Collaborative Frameworks
Ideally, care and trials would be provided in a “transmural” fashion, in academic multidisciplinary expert centers when necessary and by a local care team when possible, to minimize burden on the individual patient and caregivers.
When a treatment is identified at individual or group level, speedy translation into trials and care is necessary for optimal prognosis of the individual. This calls for close collaboration of patients and their clinicians with fundamental researchers, methodologists, statisticians, outcome measure specialists, patient organizations, legal, ethical, legislators, and reimbursement experts. These experts should be involved from the start, as previous personalized trial approaches have failed because of their limited involvement or support. After achieving consensus on relevance, design, statistics, outcome measures, and ethical and legal considerations, acceptance and reimbursement should follow quickly per individual or at a group level.
These collaborations should be in place at a local as well as (inter)national levels, supported by knowledge platforms providing information and education on trial design, outcome measures, and regulatory issues in IMDs and NDDs. Training programs including patient-educators will help students and local and academic clinicians to understand the manifestations and impact of IMDs and NDDs on all ICF domains of the patient and their families. Allowing students to attend multidisciplinary clinical and research meetings will help in understanding the full scope of diagnostic, treatment, and implementation endeavors needed to improve our patients’ lives. International exchange programs including fellowships for clinicians will assist in understanding the care needed and decrease health disparities.
But how will we pay for this? Patient advocacy groups and health-care providers will continue to drive national and international regulatory and funding agencies to address this public health issue of rare diseases. Consensus on care needs—for instance, access to orphan medicinal products (Annemans et al. 2017)—will further drive personalized care and can be supported by validation studies (Rode et al. 2005). New platforms such as the National Centers of Excellence Programs for Rare Disorders in the United States, European Reference Networks, and Medicine for Society in the Netherlands provide a way forward, creating new frameworks involving academia, pharma, government, patients, and other stakeholders to ensure affordability and equity for rare disease therapies. Patient preferences and priorities should be considered every step of the way, and ethical, legal, and social considerations (ELSI) must be weighed carefully. Not everything that can be done should be done, certainly not without rigorous study and multistakeholder evaluation (Adhikari et al. 2020). This pertains to every blade in the flywheel, including preventive measures and population-based screening programs. The latter now can use genomic technologies to identify conditions undetectable by mass spectrometry; studies are ongoing to evaluate risks and benefits. This is much needed to achieve the second goal of the International Rare Diseases Research Consortium: namely, to have 1000 new therapies for rare diseases approved by 2027.
CONCLUSION: TOWARD PERSONALIZED, RATIONAL CARE
Personalized “genes first” medicine has great potential to decrease morbidity and improve lives of patients with IMDs and NDDs. For optimal integration of novel diagnostic and treatment options, new technologies, methodologies, and collaborative frameworks are necessary. Care models such as P4 medicine may empower collaborations to provide care on all life domains and integrate care with research, supported by technical innovations. Together, scientists, patients, ELSI experts, and health-care professionals can unite forces and accelerate personalized medicine, ultimately to leave no rare disease patient behind.
Competing Interest Statement
The authors have declared no competing interest.
Acknowledgments
We are grateful to Mr. Roderick Houben (Health2Media) for the design of the figures and to Ms. Joyce de Bree (Amsterdam UMC) for administrative support. C.D.v.K. is a member of Metab-ERN. N.I.W. is a member of the European Reference Network for Rare Neurological Diseases (ERN-RND), project ID 739510. A.M.v.E. is a member of ERN ITHACA.
REFERENCES
- Adhikari AN, Gallagher RC, Wang Y, Currier RJ, Amatuni G, Bassaganyas L, Chen F, Kundu K, Kvale M, Mooney SD, et al. 2020. The role of exome sequencing in newborn screening for inborn errors of metabolism. Nat Med 26: 1392–1397. 10.1038/s41591-020-0966-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annemans L, Aymé S, Le Cam Y, Facey K, Gunther P, Nicod E, Reni M, Roux JL, Schlander M, Taylor D, et al. 2017. Recommendations from the European working group for value assessment and funding processes in rare diseases (ORPH-VAL). Orphanet J Rare Dis 12: 50. 10.1186/s13023-017-0601-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis SE, Skotko BG, Rafii MS, Strydom A, Pape SE, Bianchi DW, Sherman SL, Reeves RH. 2020. Down syndrome. Nat Rev Dis Primers 6: 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barendsen RW, Dijkstra IME, Visser WF, Alders M, Bliek J, Boelen A, Bouva MJ, van der Crabben SN, Elsinghorst E, van Gorp AGM, et al. 2020. Adrenoleukodystrophy newborn screening in the Netherlands (SCAN Study): the X-factor. Front Cell Dev Biol 8: 499. 10.3389/fcell.2020.00499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi ML, Vai S. 2019. Alkaline phosphatase replacement therapy. Adv Exp Med Biol 1148: 201–232. 10.1007/978-981-13-7709-9_10 [DOI] [PubMed] [Google Scholar]
- Blau N, Dionisi Vici C, Ferreira CR, Vianey-Saban C, van Karnebeek CDM. 2022. Physician's guide to the diagnosis, treatment, and follow-up of inherited metabolic diseases, 2nd ed. Springer Nature; Switzerland, Cham. [Google Scholar]
- Bok LA, Halbertsma FJ, Houterman S, Wevers RA, Vreeswijk C, Jakobs C, Struys E, Van Der Hoeven JH, Sival DA, Willemsen MA. 2012. Long-term outcome in pyridoxine-dependent epilepsy. Dev Med Child Neurol 54: 849–854. 10.1111/j.1469-8749.2012.04347.x [DOI] [PubMed] [Google Scholar]
- Boycott KM, Ardigó D. 2018. Addressing challenges in the diagnosis and treatment of rare genetic diseases. Nat Rev Drug Discov 17: 151–152. 10.1038/nrd.2017.246 [DOI] [PubMed] [Google Scholar]
- Boycott KM, Campeau PM, Howley HE, Pavlidis P, Rogic S, Oriel C, Berman JN, Hamilton RM, Hicks GG, Lipshitz HD, et al. 2020. The Canadian Rare Diseases Models and Mechanisms (RDMM) network: connecting understudied genes to model organisms. Am J Hum Genet 106: 143–152. 10.1016/j.ajhg.2020.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budimirovic DB, Berry-Kravis E, Erickson CA, Hall SS, Hessl D, Reiss AL, King MK, Abbeduto L, Kaufmann WE. 2017. Updated report on tools to measure outcomes of clinical trials in fragile X syndrome. J Neurodev Disord 9: 14. 10.1186/s11689-017-9193-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cakici JA, Dimmock DP, Caylor SA, Gaughran M, Clarke C, Triplett C, Clark MM, Kingsmore SF, Bloss CS. 2020. A prospective study of parental perceptions of rapid whole-genome and -exome sequencing among seriously ill infants. Am J Hum Genet 107: 953–962. 10.1016/j.ajhg.2020.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camfield P, Camfield C. 2011. Transition to adult care for children with chronic neurological disorders. Ann Neurol 69: 437–444. 10.1002/ana.22393 [DOI] [PubMed] [Google Scholar]
- Centerwall SA, Centerwall WR. 2000. The discovery of phenylketonuria: the story of a young couple, two affected children, and a scientist. Pediatrics 105: 89–103. 10.1542/peds.105.1.89 [DOI] [PubMed] [Google Scholar]
- Coppus AMW. 2013. People with intellectual disability: what do we know about adulthood and life expectancy? Dev Disabil Res Rev 18: 6–16. 10.1002/ddrr.1123 [DOI] [PubMed] [Google Scholar]
- Coughlin CR, van Karnebeek CDM, Al-Hertani W, Shuen AY, Jaggumantri S, Jack RM, Gaughan S, Burns C, Mirsky DM, Gallagher RC, et al. 2015. Triple therapy with pyridoxine, arginine supplementation and dietary lysine restriction in pyridoxine-dependent epilepsy: neurodevelopmental outcome. Mol Genet Metab 116: 35–43. 10.1016/j.ymgme.2015.05.011 [DOI] [PubMed] [Google Scholar]
- Coughlin CR, Tseng LA, Abdenur JE, Ashmore C, Boemer F, Bok LA, Boyer M, Buhas D, Clayton PT, Das A, et al. 2021. Consensus guidelines for the diagnosis and management of pyridoxine-dependent epilepsy due to α-aminoadipic semialdehyde dehydrogenase deficiency. J Inherit Metab Dis 44: 178–192. 10.1002/jimd.12332 [DOI] [PubMed] [Google Scholar]
- Demos M, Guella I, DeGuzman C, McKenzie MB, Buerki SE, Evans DM, Toyota EB, Boelman C, Huh LL, Datta A, et al. 2019. Diagnostic yield and treatment impact of targeted exome sequencing in early-onset epilepsy. Front Neurol 10: 434. 10.3389/fneur.2019.00434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hollander B, Rasing A, Post MA, Klein WM, Oud MM, Brands MM, de Boer L, Engelke UFH, van Essen P, Fuchs SA, et al. 2021. NANS-CDG: delineation of the genetic, biochemical, and clinical spectrum. Front Neurol 12: 668640. 10.3389/fneur.2021.668640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler F, Duncan C, Musolino PL, Orchard PJ, De Oliveira S, Thrasher AJ, Armant M, Dansereau C, Lund TC, Miller WP, et al. 2017. Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy. N Engl J Med 377: 1630–1638. 10.1056/nejmoa1700554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelke UFH, van Outersterp RE, Merx J, van Geenen FAMG, van Rooij A, Berden G, Huigen MCDG, Kluijtmans LAJ, Peters TMA, Al-Shekaili HH, et al. 2021. Untargeted metabolomics and infrared ion spectroscopy identify biomarkers for pyridoxine-dependent epilepsy. J Clin Invest 131: e148272. 10.1172/JCI148272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson CA, Davenport MH, Schaefer TL, Wink LK, Pedapati EV, Sweeney JA, Fitzpatrick SE, Brown WT, Budimirovic D, Hagerman RJ, et al. 2017. Fragile X targeted pharmacotherapy: lessons learned and future directions. J Neurodev Disord 9: 7. 10.1186/s11689-017-9186-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenberg KD, Braverman NE, Moser AB, Steinberg SJ, Klouwer FCC, Schlüter A, Ruiz M, Pujol A, Engvall M, Naess K, et al. 2017. Allelic expression imbalance promoting a mutant PEX6 allele causes Zellweger spectrum disorder. Am J Hum Genet 101: 965–976. 10.1016/j.ajhg.2017.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira CR, van Karnebeek CDM, Vockley J, Blau N. 2019. A proposed nosology of inborn errors of metabolism. Genet Med 21: 102–106. 10.1038/s41436-018-0022-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira CR, Rahman S, Keller M, Zschocke J. 2021. An international classification of inherited metabolic disorders (ICIMD). J Inherit Metab Dis 44: 164–177. 10.1002/jimd.12348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli F, Calbi V, Natali Sora MG, Sessa M, Baldoli C, Rancoita PMV, Ciotti F, Sarzana M, Fraschini M, Zambon AA, et al. 2022. Lentiviral haematopoietic stem-cell gene therapy for early-onset metachromatic leukodystrophy: long-term results from a non-randomised, open-label, phase 1/2 trial and expanded access. Lancet 399: 372–383. 10.1016/S0140-6736(21)02017-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaasterland CMW, Jansen-van der Weide MC, Vroom E, Leeson-Beevers K, Kaatee M, Kaczmarek R, Bartels B, van der Pol WL, Roes KCB, van der Lee JH. 2018. The POWER-tool: recommendations for involving patient representatives in choosing relevant outcome measures during rare disease clinical trial design. Health Policy (New York) 122: 1287–1294. 10.1016/j.healthpol.2018.09.011 [DOI] [PubMed] [Google Scholar]
- Gaasterland CMW, van der Weide MCJ, Roes KCB, van der Lee JH. 2019. Goal attainment scaling as an outcome measure in rare disease trials: a conceptual proposal for validation. BMC Med Res Methodol 19: 227. 10.1186/s12874-019-0866-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrelfs SF, Frishberg Y, Hulton SA, Koren MJ, O'Riordan WD, Cochat P, Deschênes G, Shasha-Lavsky H, Saland JM, van't Hoff WG, et al. 2021. Lumasiran, an RNAi therapeutic for primary hyperoxaluria type 1. N Engl J Med 384: 1216–1226. 10.1056/nejmoa2021712 [DOI] [PubMed] [Google Scholar]
- Hood L. 2006. Health care of the 21st century: predictive, preventive, personalized and participatory (P4) medicine. J Oral Maxillofacial Surg 64. 10.1016/j.joms.2006.06.302 [DOI] [Google Scholar]
- Hood L, Balling R, Auffray C. 2012. Revolutionizing medicine in the 21st century through systems approaches. Biotechnol J 7: 992–1001. 10.1002/biot.201100306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoytema van Konijnenburg EMM, Wortmann SB, Koelewijn MJ, Tseng LA, Houben R, Stöckler-Ipsiroglu S, Ferreira CR, van Karnebeek CDM. 2021. Treatable inherited metabolic disorders causing intellectual disability: 2021 review and digital app. Orphanet J Rare Dis 16: 170. 10.1186/s13023-021-01727-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp S, Huffnagel IC, Linthorst GE, Wanders RJ, Engelen M. 2016. Adrenoleukodystrophy: neuroendocrine pathogenesis and redefinition of natural history. Nat Rev Endocrinol 12: 606–615. 10.1038/nrendo.2016.90 [DOI] [PubMed] [Google Scholar]
- Kim J, Hu C, Moufawad El Achkar C, Black LE, Douville J, Larson A, Pendergast MK, Goldkind SF, Lee EA, Kuniholm A, et al. 2019. Patient-customized oligonucleotide therapy for a rare genetic disease. N Engl J Med 381: 1644–1652. 10.1056/nejmoa1813279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok G, Tseng L, Schene IF, Dijsselhof ME, Salomons G, Mendes MI, Smith DEC, Wiedemann A, Canton M, Feillet F, et al. 2021. Treatment of ARS deficiencies with specific amino acids. Genet Med 23: 2202–2207. 10.1038/s41436-021-01249-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostanjsek N. 2011. Use of the international classification of functioning, disability and health (ICF) as a conceptual framework and common language for disability statistics and health information systems. BMC Public Health 11: S3. 10.1186/1471-2458-11-S4-S3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotulska K, Kwiatkowski DJ, Curatolo P, Weschke B, Riney K, Jansen F, Feucht M, Krsek P, Nabbout R, Jansen AC, et al. 2021. Prevention of epilepsy in infants with tuberous sclerosis complex in the EPISTOP trial. Ann Neurol 89: 304–314. 10.1002/ana.25956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer LS, Bader DM, Mertes C, Kopajtich R, Pichler G, Iuso A, Haack TB, Graf E, Schwarzmayr T, Terrile C, et al. 2017. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat Commun 8: 15824. 10.1038/ncomms15824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krey I, von Spiczak S, Johannesen KM, Hikel C, Kurlemann G, Muhle H, Beysen D, Dietel T, Møller RS, Lemke JR, et al. 2022. L-serine treatment is associated with improvements in behavior, EEG, and seizure frequency in individuals with GRIN-related disorders due to null variants. Neurotherapeutics 10.1007/s13311-021-01173-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, Wilson KA, Byars A, Sahmoud T, Franz DN. 2010. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med 363: 1801–1811. [DOI] [PubMed] [Google Scholar]
- Lee JJY, Wasserman WW, Hoffmann GF, van Karnebeek CDM, Blau N. 2018. Knowledge base and mini-expert platform for the diagnosis of inborn errors of metabolism. Genet Med 20: 151–158. 10.1038/gim.2017.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills PB, Struys E, Jakobs C, Plecko B, Baxter P, Baumgartner M, Willemsen MAAP, Omran H, Tacke U, Uhlenberg B, et al. 2006. Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat Med 12: 307–309. 10.1038/nm1366 [DOI] [PubMed] [Google Scholar]
- Mueller T, Jerrentrup A, Bauer MJ, Fritsch HW, Schaefer JR. 2016. Characteristics of patients contacting a center for undiagnosed and rare diseases. Orphanet J Rare Dis 11: 81. 10.1186/s13023-016-0467-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller AR, Brands MM, van de Ven PM, Roes KC, Cornel MC, van Karnebeek CD, Wijburg FA, Daams JG, Boot E, van Eeghen AM. 2021. The power of 1: systematic review of N-of-1 studies in rare genetic neurodevelopmental disorders. Neurology 96: 529–540. 10.1212/wnl.0000000000011597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overwater IE, Rietman AB, Mous SE, Bindels-de Heus K, Rizopoulos D, Ten Hoopen LW, van der Vaart T, Jansen FE, Elgersma Y, Moll HA, et al. 2019. A randomized controlled trial with everolimus for IQ and autism in tuberous sclerosis complex. Neurology 93: e200–e209. 10.1212/WNL.0000000000007749 [DOI] [PubMed] [Google Scholar]
- Petrikin JE, Willig LK, Smith LD, Kingsmore SF. 2015. Rapid whole genome sequencing and precision neonatology. Semin Perinatol 39: 623–631. 10.1053/j.semperi.2015.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, Coban Akdemir ZH, Walkiewicz M, Bi W, Xiao R, Ding Y, et al. 2017. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med 376: 21–31. 10.1056/nejmoa1516767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaio CRDAC, Moreira CM, Novo-Filho GM, Sacramento-Bobotis PR, Groenner Penna M, Perazzio SF, Dutra AP, da Silva RA, Santos MNP, de Arruda VYN, et al. 2020. Diagnostic power and clinical impact of exome sequencing in a cohort of 500 patients with rare diseases. Am J Med Genet C Semin Med Genet 184: 955–964. 10.1002/ajmg.c.31860 [DOI] [PubMed] [Google Scholar]
- Richmond PA, van der Kloet F, Vaz FM, Lin D, Uzozie A, Graham E, Kobor M, Mostafavi S, Moerland PD, Lange PF, et al. 2020. Multi-omic approach to identify phenotypic modifiers underlying cerebral demyelination in X-linked adrenoleukodystrophy. Front Cell Dev Biol 8: 520. 10.3389/fcell.2020.00520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rode J. 2005. Rare diseases: understanding this public health priority. EURORDIS, Paris. [Google Scholar]
- Rosso M, Fremion E, Santoro SL, Santoro SL, Oreskovic NM, Chitnis T, Skotko BG, Skotko BG, Santoro JD, Santoro JD. 2020. Down syndrome disintegrative disorder: a clinical regression syndrome of increasing importance. Pediatrics 145: e20192939. 10.1542/peds.2019-2939 [DOI] [PubMed] [Google Scholar]
- Sadikovic B, Levy MA, Kerkhof J, Aref-Eshghi E, Schenkel L, Stuart A, McConkey H, Henneman P, Venema A, Schwartz CE, et al. 2021. Clinical epigenomics: genome-wide DNA methylation analysis for the diagnosis of Mendelian disorders. Genet Med 23: 1065–1074. 10.1038/s41436-020-01096-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin M, Sur M. 2015. Genes, circuits, and precision therapies for autism and related neurodevelopmental disorders. Science 350: 10.1126/science.aab3897.aab3897. 10.1126/science.aab3897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, Peng M, Collins R, Grove J, Klei L, et al. 2020. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180: 568–584.e23. 10.1016/j.cell.2019.12.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkel LC, Kernohan KD, McBride A, Reina D, Hodge A, Ainsworth PJ, Rodenhiser DI, Pare G, Bérubé NG, Skinner C, et al. 2017. Identification of epigenetic signature associated with alpha thalassemia/mental retardation X-linked syndrome. Epigenetics Chromatin 10: 10. 10.1186/s13072-017-0118-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senn S. 2016. Mastering variation: variance components and personalised medicine. Stat Med 35: 966–977. 10.1002/sim.6739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senn S. 2019. Sample size considerations for n-of-1 trials. Stat Methods Med Res 28: 372–383. 10.1177/0962280217726801 [DOI] [PubMed] [Google Scholar]
- Slade A, Isa F, Kyte D, Pankhurst T, Kerecuk L, Ferguson J, Lipkin G, Calvert M. 2018. Patient reported outcome measures in rare diseases: a narrative review. Orphanet J Rare Dis 13: 61. 10.1186/s13023-018-0810-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snoeijen-Schouwenaars FM, van Ool JS, Verhoeven JS, van Mierlo P, Braakman HMH, Smeets EE, Nicolai J, Schoots J, Teunissen MWA, Rouhl RPW, et al. 2019. Diagnostic exome sequencing in 100 consecutive patients with both epilepsy and intellectual disability. Epilepsia 60: 155–164. 10.1111/epi.14618 [DOI] [PubMed] [Google Scholar]
- Soden SE, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, LePichon JB, Miller NA, Thiffault I, Dinwiddie DL, et al. 2014. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med 6: 265ra168. 10.1126/scitranslmed.3010076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepien KM, Kieć-Wilk B, Lampe C, Tangeraas T, Cefalo G, Belmatoug N, Francisco R, del Toro M, Wagner L, Lauridsen AG, et al. 2021. Challenges in transition from childhood to adulthood care in rare metabolic diseases: results from the first multi-center European survey. Front Med 8: 652358. 10.3389/fmed.2021.652358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker S, Potter BK, Yuskiv N, Tingley K, Patterson M, van Karnebeek C. 2020. Developments in evidence creation for treatments of inborn errors of metabolism. J Inherit Metabol Dis 44: 88–98. 10.1002/jimd.12315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockler-Ipsiroglu S, Potter BK, Yuskiv N, Tingley K, Patterson M, van Karnebeek C. 2021. Developments in evidence creation for treatments of inborn errors of metabolism. J Inherit Metab Dis 44: 88–98. 10.1002/jimd.12315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Zheng W, Simeonov A. 2017. Drug discovery and development for rare genetic disorders. Am J Med Genet A 173: 2307–2322. 10.1002/ajmg.a.38326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarailo-Graovac M, Shyr C, Ross CJ, Horvath GA, Salvarinova R, Ye XC, Zhang L-H, Bhavsar AP, Lee JJY, Drögemöller BI, et al. 2016. Exome sequencing and the management of neurometabolic disorders. N Engl J Med 374: 2246–2255. 10.1056/nejmoa1515792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate RL, Perdices M. 2019. Single-case experimental designs for clinical research and neurorehabilitation settings. Taylor and Francis, London. [Google Scholar]
- van Karnebeek CDM. 2014. Diagnosis and discovery of treatable inborn errors of metabolism causing intellectual disability. Mol Genet Metab 111. [DOI] [PubMed] [Google Scholar]
- van Karnebeek CDM, Jaggumantri S. 2015. Current treatment and management of pyridoxine-dependent epilepsy. Curr Treat Options Neurol 17: 335. 10.1007/s11940-014-0335-0 [DOI] [PubMed] [Google Scholar]
- van Karnebeek CDM, Bonafé L, Wen XY, Tarailo-Graovac M, Balzano S, Royer-Bertrand B, Ashikov A, Garavelli L, Mammi I, Turolla L, et al. 2016. NANS-mediated synthesis of sialic acid is required for brain and skeletal development. Nat Genet 48: 777–784. 10.1038/ng.3578 [DOI] [PubMed] [Google Scholar]
- van Karnebeek CDM, Wortmann SB, Tarailo-Graovac M, Langeveld M, Ferreira CR, van de Kamp JM, Hollak CE, Wasserman WW, Waterham HR, Wevers RA, et al. 2018. The role of the clinician in the multi-omics era: are you ready? J Inherit Metab Dis 41: 571–582. 10.1007/s10545-017-0128-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kuilenburg ABP, Tarailo-Graovac M, Richmond PA, Drögemöller BI, Pouladi MA, Leen R, Brand-Arzamendi K, Dobritzsch D, Dolzhenko E, Eberle MA, et al. 2019. Glutaminase deficiency caused by short tandem repeat expansion in GLS. N Engl J Med 380: 1433–1441. 10.1056/nejmoa1806627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Os J, Verhagen S, Marsman A, Peeters F, Bak M, Marcelis M, Drukker M, Reininghaus U, Jacobs N, Lataster T, et al. 2017. The experience sampling method as an mHealth tool to support self-monitoring, self-insight, and personalized health care in clinical practice. Depress Anxiety 34: 481–493. 10.1002/da.22647 [DOI] [PubMed] [Google Scholar]
- Vaz FM, McDermott JH, Alders M, Wortmann SB, Kölker S, Pras-Raves ML, Vervaart MAT, van Lenthe H, Luyf ACM, Elfrink HL, et al. 2019. Mutations in PCYT2 disrupt etherlipid biosynthesis and cause a complex hereditary spastic paraplegia. Brain 142: 3382–3397. 10.1093/brain/awz291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers LELM, Gilissen C, Veltman JA. 2016. Genetic studies in intellectual disability and related disorders. Nat Rev Genet 17: 9–18. 10.1038/nrg3999 [DOI] [PubMed] [Google Scholar]
- Wagstaff PE, ten Asbroek ALMA, ten Brink JB, Jansonius NM, Bergen AAB. 2021. An alternative approach to produce versatile retinal organoids with accelerated ganglion cell development. Sci Rep 11: 1101. 10.1038/s41598-020-79651-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanders RJA, Vaz FM, Ferdinandusse S, van Kuilenburg ABP, Kemp S, van Karnebeek CD, Waterham HR, Houtkooper RH. 2019. Translational metabolism: a multidisciplinary approach towards precision diagnosis of inborn errors of metabolism in the omics era. J Inherit Metab Dis 42: 197–208. 10.1002/jimd.12008 [DOI] [PubMed] [Google Scholar]
- Warmerdam HAG, Termeulen-Ferreira EA, Tseng LA, Lee JY, van Eeghen AM, Ferreira CR, van Karnebeek CDM. 2020. A scoping review of inborn errors of metabolism causing progressive intellectual and neurologic deterioration (PIND). Front Neurol 10: 1369. 10.3389/fneur.2019.01369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapata-Pérez R, Wanders RJA, van Karnebeek CDM, Houtkooper RH. 2021. NAD+ homeostasis in human health and disease. EMBO Mol Med 13: e13943. 10.15252/emmm.202113943 [DOI] [PMC free article] [PubMed] [Google Scholar]