

Abstract
Progesterone (P) and prolactin (PRL) fulfill crucial roles during growth and differentiation of the mammary epithelium, and each has been implicated in the pathogenesis of mammary cancer. We previously identified that these hormones synergistically stimulate the proliferation of mouse mammary epithelial cells in vivo, although the mechanism(s) underlying their cooperative effect are unknown. We now report a novel pathway by which P and PRL synergize to activate transcription from the long terminal repeat (LTR) of the mouse mammary tumor virus-LTR (MMTV-LTR) in T47D breast cancer cells. Using serial 5′ and 3′ deletions of the MMTV-LTR, in addition to selective mutations, we identified that a previously uncharacterized inverted palindrome on the distal enhancer (−941/−930), in addition to a signal transducer and activator of transcription 5 site, was essential for the synergistic activation of transcription by P and PRL. Notably, hormone synergy occurred via a mechanism that was independent of the P receptor DNA-binding elements found in the proximal MMTV-LTR hormone-response element. The palindrome specifically recruited a protein complex (herein termed mammary gland-specific complex) that was almost exclusive to normal and cancerous mammary cells. The synergy between P and PRL occurred via a Janus kinase 2 and c-Src/Fyn-dependent signaling cascade downstream of P and PRL receptors. Combined, our data outline a novel pathway in T47D cells that may facilitate the action(s) of P and PRL during mammary development and breast cancer.
Introduction
Numerous hormones and growth factors modulate growth and development of the mammary gland and its cancer (Hovey et al. 2002). Of these, progesterone (P) and prolactin (PRL) are key players. Both hormones are required for normal alveolar development in the mouse mammary gland (Ormandy et al. 1997a, Brisken et al. 1998). PRL is involved in the initiation and/or progression of breast cancer (Muhlbock & Boot 1959, Boot et al. 1962, Oakes et al. 2007), while loss of P receptor (PR) is associated with a protective effect against mammary carcinogenesis (Lydon et al. 1999). In addition to these individual effects, it is well known that normal and neoplastic mammary epithelial cells respond to hormones in combination differently than to each alone, as exemplified by the recognized and pronounced effects of estrogen (E) plus P (Haslam et al. 2002).
Any cooperation between PRL and P during development of the mammary gland (Nagasawa et al. 1985, Imagawa et al. 1990) and its cancer (Leung et al. 1981, Nagasawa et al. 1986) has been subjected to only limited investigation. Our finding that P and PRL synergize to stimulate mammary epithelial proliferation in vivo (Hovey et al. 2001) affirmed the potential for crosstalk between these two hormones, as has been shown for P and epidermal growth factor (Lange 2004). Consistent with such a proposal, PRL receptor (PRLR) gene expression correlates with PR and E receptor gene expression in breast cancer cells, and PRL increases the level of mRNA for PR and its subsequent binding of ligand (Ormandy et al. 1997b). These data also align with our demonstration that PR and PRLR co-localize to a subpopulation of epithelial cells in the normal mouse mammary gland (Hovey et al. 2001). Furthermore, P and PRL synergize to increase receptor activator of nuclear factor (NF)κ-B ligand expression in the mammary epithelium of PRL-deficient mice (Srivastava et al. 2003). Along similar lines, Chilton et al. identified that transcription of the uteroglobin gene in the rabbit uterus is synergistically induced by P+PRL through a novel Janus kinase (Jak)- and RUSH-dependent and signal transducer and activator of transcription 5 (STAT5)-independent mechanism (Daniel et al. 1984, Hewetson et al. 2002, Hewetson & Chilton 2003).
Mouse mammary tumor virus (MMTV) is a milk-borne retrovirus that, upon integration of its long terminal repeat (LTR), promotes the aberrant transcription of adjacent oncogenes (Gunzburg & Salmons 1992, van Leeuwen & Nusse 1995) to stimulate mammary hyperplasia and preneoplasia (Medina 1973). A unique aspect of the MMTV-LTR is its pronounced transcriptional activation in mammary epithelial cells compared with other tissues (Henrard & Ross 1988). The proximal region of the MMTV-LTR includes a well-characterized hormone-response element (HRE) that is able to bind multiple activated steroid hormone receptors (von der Ahe et al. 1985, Ponta et al. 1985, Cato et al. 1987, Ham et al. 1988, Gowland & Buetti 1989). Separately, the 5′ distal enhancer includes two transcriptional moieties, the Ban2 enhancer (−1075/−978) and the mammary-specific enhancer of MMTV (MEM) element (−956/−862) (Grimm & Nordeen 1998), that bind a number of relatively uncharacterized cis-acting transcription factors. Several of these including mammary activating factor (MAF)/F2 (Mink et al. 1992), mp4 (Lefebvre et al. 1991) and STAT5 (Haraguchi et al. 1997) are also PRL inducible. These factors, and possibly others, confer mammary-specific transcription from regions spanning both −1166/−987 (Mok et al. 1992) and −956/−862 (Grimm & Nordeen 1998) of the MMTV-LTR. Given that the distal enhancer of the MMTV-LTR coordinates a synergistic transcriptional response to P+PRL and dexamethasone (DEX) plus PRL in T47D cells (Haraguchi et al. 1992, 1993), it constitutes an excellent model for resolving the mechanism(s) of hormone synergy in the normal mammary gland and breast cancer.
Within this report, we describe a mechanism by which P and PRL synergistically modulate transcription from the MMTV-LTR in T47D breast cancer cells. This effect requires a novel inverted palindrome that is activated via the non-genomic action of PR, a STAT5 consensus site that operates independently of STAT5a and STAT5b, and signaling via Jak2 and c-Src/Fyn. This site also recruits an uncharacterized mammary-specific transcription factor complex, herein termed mammary gland-specific complex (MGSC).
Materials and methods
Cell culture
The T47D human mammary carcinoma cell line was cultured in growth medium (GM) comprising RPMI1640 supplemented with 5% fetal bovine serum (FBS), 100 U penicillin/100 μg streptomycin (P/S) per ml, 1 μg/ml insulin, 2 mg/ml sodium bicarbonate, and 2.42 mg/ml HEPES. Cells for nuclear extracts were grown in standard conditions for each line (Horoszewicz et al. 1980, Westley & Rochefort 1980, Travers et al. 1982, Danielson et al. 1984, Horwitz & Freidenberg 1985, Ball et al. 1988, Soule et al. 1990, Williams et al. 1990, Liao & Jaken 1993, Hua et al. 1995, Dittrich et al. 1996, Trott et al. 2003, Hassan et al. 2008), which included supplementation with FBS (from 2 to 10%) and P/S. The exceptions were nuclear extracts from HeLa, HepG2 and U-937 cells that were purchased from Active Motif (Carlsbad, CA, USA). The origin of each cell line used for nuclear extraction is stated in Table 1.
Table 1.
Origin of cell lines used for screening of mammary gland-specific complex (MGSC) binding to the −941/−930 palindromic site
| Species | |
|---|---|
| Cell line/cell types | |
| Mammary (normal) | |
| HC11 | Mouse |
| MCF10A | Human |
| Comma-1D | Mouse |
| Mammary (neoplastic) | |
| BT-549 | Human |
| MCF-7 | Human |
| SKBR3 | Human |
| T47D | Human |
| T47D-CO | Human |
| Non-mammary (epithelial) | |
| CHO | Chinese Hamster |
| HeLa | Human |
| HepG2 | Human |
| LnCaP | Human |
| SKOV3 | Human |
| THLE3 | Human |
| Non-mammary (nonepithelial) | |
| COS | African green monkey |
| REF52 | Rat |
| U-937 | Human |
Plasmids
All promoter fragments were amplified by PCR from the MMTV-LTR (pMAM-neo; Amersham Bioscience) using Precision Plus polymerase (Stratagene, La Jolla, CA, USA) and were gel purified (Wizard SV Gel Clean-Up System, Promega) before sub-cloning into pCR2.1-TOPO (Invitrogen). Site-directed mutagenesis was performed using QuikChange Site-Directed Mutagenesis (Stratagene). All promoter fragments were sequence verified. Fragments were excised from pCR2.1, gel-purified, and ligated into either the pGL3 Basic or pGL3 Promoter (containing a minimal SV40 promoter) enhancer-less luciferase gene expression vectors (Promega). The plasmid that contained the minimal HRE was generated by excising the MMTV-LTR promoter region upstream of a proximal SstI site at position −108. Dominant-negative plasmids were as follows: the dominant-negative Jak2-829, STAT5a, and STAT5b plasmids were kindly provided by Dr Nelson Horseman, University of Cincinnati, OH; the dominant-negative c-Src and Fyn plasmids were kindly provided by Dr Filippo Giancotti, Memorial Sloan Kettering Cancer Center, NY; and the dominant-negative Map/Erk kinase (MEK)1/2 plasmid was kindly provided by Dr Silvio Gutkind, National Institutes of Health, Bethesda, MD, USA. Plasmids for transfection were purified using the PhenIX midiprep DNA Purification kit (Q-Biogene, Irvine, CA, USA) with two exceptions: the 5′ deletion mutants Fig. 4B and MGSC mutants (Fig. 7C) were prepared using the Qiagen maxiprep kit while the 5′ MMTV-LTR constructs (Fig. 2) were prepared using the Genopure Maxiprep kit (Roche).
Transient transfection and reporter gene assays
T47D cells in GM were plated into 12-well culture plates (Falcon) at 250 000 cells/well. Cells at 70% confluence were transiently transfected with 0·8 μg MMTV-pGL3 plasmid DNA and 0·2 μg of the β-galactosidase reporter plasmid (Invitrogen) using FuGene6 (Roche). For signaling experiments, 0·3 μg dominant-negative plasmid DNA or empty vector DNA was co-transfected with 0·3 μg of −1080/−909_SV40pGL3 plasmid DNA and 0·2 μg of the pEF-6/β-galactosidase reporter plasmid. After 24 h, culture medium was changed to hormone-deficient medium (5% charcoal-stripped serum, 1 μg/ml insulin, P/S, 2 mg/ml sodium bicarbonate, and 2·42 mg/ml HEPES), then 48 h later, the culture medium was changed to hormone-free medium (hormone-deficient medium without insulin). Cells were treated with ethanol vehicle, ovine PRL (500 ng/ml), P (1 μM), DEX (1 μM) or P+PRL. For signaling experiments, cells were also treated with dimethylsulfoxide (DMSO) vehicle, wortmannin (100 nM; Sigma) or LY294 002 (25 μM; Cell Signaling Technology, Beverly, MA, USA). All treatments included an equal concentration of ethanol and DMSO. After 40 h, cells were lysed for the measurement of β-galactosidase and luciferase activity (Bright-Glo Luciferase Assay System, Promega) using a Synergy HT plate reader (BioTek, Winooski, VT, USA). Luciferase values were corrected for transfection efficiency as determined by β-galactosidase activity in a colorimetric assay (Promega). We found that the dominant-negative MEK1/2, c-Src, Fyn, STAT5a, and STAT5b plasmids suppressed β-galactosidase expression; therefore, values for these experiments are reported as uncorrected luciferase activity.
Nuclear extracts and electrophoretic mobility shift assay (EMSA)
Crude nuclear proteins were extracted on ice from cell lines grown to confluence according to the methods of Jones et al. (2000). Protein concentration was determined using Bradford’s (1976) method. Extracts were flash-frozen in liquid nitrogen and stored at −80 °C.
Complementary oligonucleotides (Table 2) were heated to 95 °C for 5 min and annealed by cooling to room temperature. Oligonucleotides were end-labeled using Moloney’s murine leukemia virus (MMLV) reverse transcriptase to fill in GGG overhangs with (α-32P)dCTP (10 mCi/ml; 6000 Ci/mmol; MP Biomedicals, Irvine, CA, USA) and purified using a DNA G-50 spin column (Roche). Nuclear extracts (10 μg) were incubated with 200 000 c.p.m. of labeled probe in 5× Gel Shift Binding Buffer (Promega) on ice for 30 min. Competition assays included a 100-fold excess of unlabeled competitor DNA that was added 15 min prior to the addition of labeled probe. Protein–DNA interactions were resolved on a 5% polyacrylamide-TBE gel. Gels were dried and exposed to a phosphorimaging screen that was imaged on a Storm Phosphorimager (Molecular Dynamics, Sunnyvale, CA, USA).
Table 2.
Electrophoretic mobility shift assay (EMSA) oligonucleotides
| Direction | 5′ to 3′ | |
|---|---|---|
| Oligo | ||
| −954/−922 | Fwd | GCTCTTCTGTGCAAGATTACAATCTAAACAATT |
| Rev | GGGAATTGTTTAGATTGTAATCTTGCACAGAAGAGC | |
| Palindromea | Fwd | CCACATACTAAGATTACAATCTGTACACGCTTC |
| Rev | GGGGAAGCGTGTACAGATTGTAATCTTAGTATGTGG | |
| −954/−909 | Fwd | GCTCTTCTGTGCAAGATTACAATCTAAACAATTCGGAGAACTCGAC |
| Rev | GGGTCGAGTTCTCCGAATTGTTTAGATTGTAATCTTGCACAGAAGAGC | |
| Palindrome mutb | Fwd | CTCTTCTGTGCAAGCTGACACTCTAAACAATTCGGAGAACTCGAC |
| Rev | GGGGTCGAGTTCTCCGAATTGTTTAGAGTGTCAGCTTGCACAGAAGAG | |
| −997/−909 | Fwd | GCGGTTCGTGCTCGCAGGGCTCTCACCCTTGACTCTTTTAATAGCTCTTCTGTGCAAGATTACAATCTAAACAATTCGGAGAACTCGAC |
| Rev | GGGGTCGAGTTCTCCGAATTGTTTAGATTGTAATCTTGCACAGAAGAGCTATTAAAAGAGTCAAGGGTGAGAGCCCTGCGAGCACGAACCGC | |
| GATA-consensus | Fwd | CACTTGATAACAGAAAGTGATAACTCT |
| Rev | GGGAGAGTTATCACTTTCTGTTATCAAGTG | |
| STAT-consensus | Fwd | GGACTTCTTGGAATTAAGGGAC |
| Rev | GGGGTCCCTTAATTCCAAGAAGTCC | |
Complementary oligonucleotides used in EMSA were annealed and end-labeled using MMLV reverse transcriptase to fill in GGG overhangs with (α-32P) dCTP.
Palindrome sequence is in bold.
Mutant sequence is in bold.
Statistical analysis
Data were analyzed using Statistical Analysis Software (SAS Institute Inc., Cary, NC, USA). The main effects of individual hormones and their interactions were tested for statistical significance using the general linear model procedure. Where indicated, basal transcription levels were compared across different constructs using the ANOVA procedure. Levels of statistical significance are indicated within the results and figure legends.
Results
P and PRL synergistically activate 5′ enhancer elements to initiate transcription from the MMTV-LTR promoter
It is known that PRL+DEX and PRL+P coordinate significant transcriptional synergy from the MMTV-LTR in T47D human breast cancer cells (P<0·01; Fig. 1; consistent with Haraguchi et al. 1992, 1997). Previous work by Haraguchi et al. (1992, 1993, 1997) established that a 5′ distal enhancer region of the MMTV-LTR coordinates the synergistic response to PC+PRL by T47D cells. To resolve the element(s) responsible for this response, we generated 5′ deletions between positions −1080 and −909, and tested the transcriptional response to each hormone alone or in combination (Fig. 2). All constructs demonstrated a significant (P<0·05) transcriptional response to P that is generally ascribed to the recruitment of liganded PR to the proximal (−202/−50) promoter (Ponta et al. 1985, Ham et al. 1988). While PRL alone only had a minor effect on transcription, the combination of P+PRL elicited a positive synergistic response from the three longest promoter fragments (Figs 1 and 2; P<0·1), which was ablated upon a deletion of 25 bp between positions −954 and −929 (Fig. 2; P>0·1). The basal level of transcription (defined in this study as transcription in hormone-free media) also declined significantly upon deletion of the region between −1080 and −1048 bp, and also following deletion of the region between −954 and −929 bp (Fig. 2; P<0·05). These results confirmed previous data indicating that this region regulates basal transcription (Lefebvre et al. 1991). Therefore, elements within a 25 bp region (−954/−929) of the MMTV-LTR distal enhancer are required for the synergistic transcriptional response to P+PRL in T47D breast cancer cells.
Figure 1.
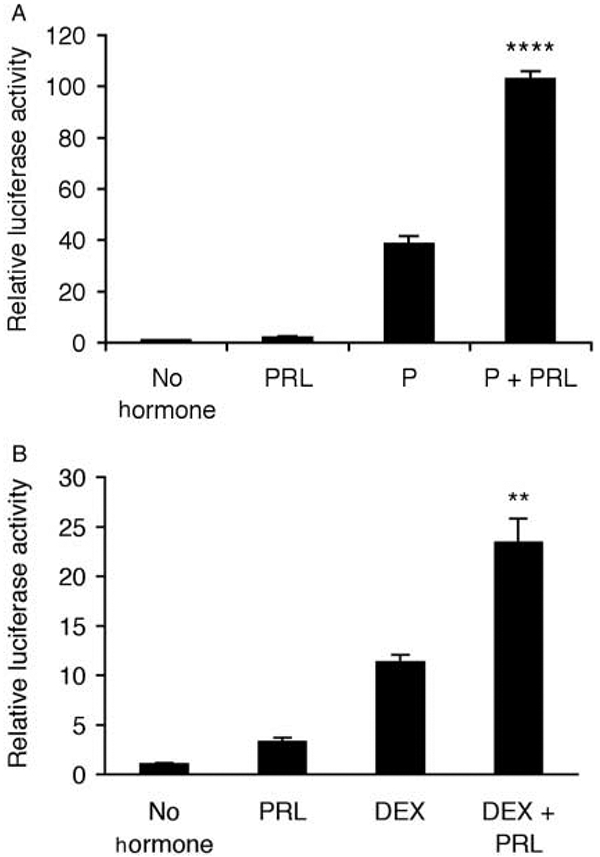
Transcription from the mouse mammary tumor virus-long terminal repeat (MMTV-LTR) is synergistically stimulated by (A) progesterone (P)+prolactin (PRL) and (B) dexamethasone (DEX)+PRL. A full-length (−1227/+177) MMTV-pGL3 construct was transiently transfected into T47D cells prior to treatment with either no hormones, PRL (500 ng/ml), P (1 μM), or DEX (1 μM) alone or in combination for 40 h. Cells were lysed and transcriptional activity was determined using a luciferase assay and normalized for β-galactosidase activity. (A) Data are means±s.e.m. (n=3 independent experiments); (B) data are means±s.e.m. (n=3 replicates) and representative of three independent experiments. A PxPRL-positive interaction is indicated by **P<0·01 or ****P<0·0001.
Figure 2.
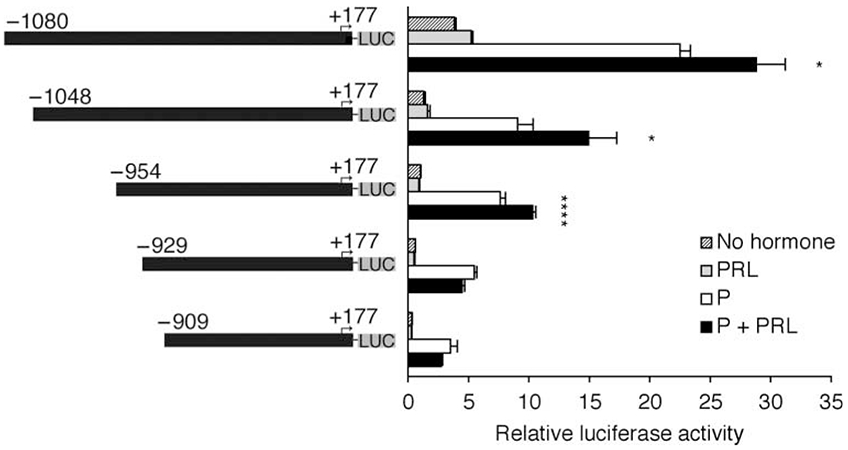
The distal enhancer of the mouse mammary tumor virus-long terminal repeat (MMTV-LTR) facilitates the synergistic transcriptional response to progesterone (P)+prolactin (PRL). Deletions of the (−1227/+177) MMTV-LTR with 5′ termini at positions −1080, −1048, −954, −929, and −909, positioned upstream of a luciferase reporter gene, were transiently transfected into T47D cells prior to their treatment with no hormones, PRL (500 ng/ml), P (1 μM), or P+pRL for 40 h. Cells were lysed and transcriptional activity was determined using a luciferase assay and normalized for β-galactosidase activity. Data are representative of three independent experiments; data are means±s.e.m. (n=4–6 replicates). A PxPRL-positive interaction is indicated by *P<0·1 or ****P<0·0001.
Synergistic activation of MMTV-LTR by P+PRL is independent of the HRE
We further dissected the MMTV-LTR to evaluate the role of the proximal HRE in the synergistic response to P+PRL. Numerous studies have demonstrated that binding of dimerized PR to this region of the MMTV-LTR facilitates P- and DEX-induced transcription (Ponta et al. 1985, Gowland & Buetti 1989). Hypothesizing a role for this HRE in the response to P+PRL, we first sought to examine whether the 5′ distal enhancer (−1080/−840) conferred a synergistic response to P+PRL when it was placed upstream of a minimal MMTV promoter with a truncated HRE (−107/+177 MMTV-LTR). This promoter construct lacked intervening sequence (−839/−108), which includes a transcriptional repressor region (Morley et al. 1987, Hsu et al. 1988, Lefebvre et al. 1991), and also lacked two P-response elements within the HRE in order to reduce the magnitude of the P-induced transcriptional response. As shown in Fig. 3A, deletion of the region −839/−108 did not ablate the synergistic response to P+PRL (P<0·0001), indicating that this region is not required for hormone synergy.
Figure 3.
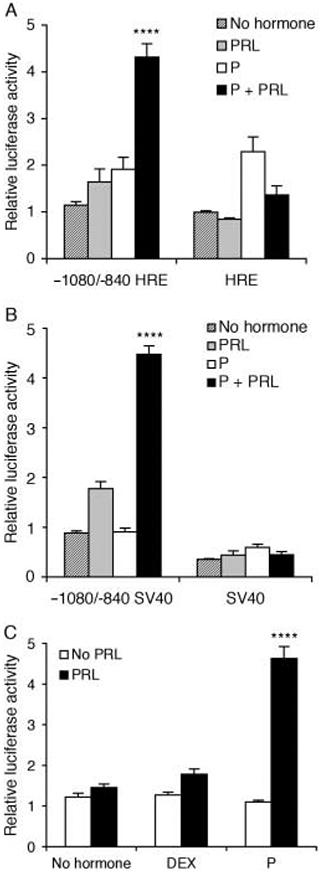
Progesterone (P)+prolactin (PRL) synergistically activates the mouse mammary tumor virus-long terminal repeat (MMTV-LTR) distal enhancer in the absence of proximal progesterone receptor DNA-binding domains. A distal fragment (−1080/−840) of the MMTV-LTR was positioned upstream of either (A) a proximal fragment of the mMtV-LTR (−107/+177) that included a partial steroid hormone-response element (HRE) or (B) a minimal SV40 promoter. Constructs were transiently transfected into T47D cells prior to their treatment with no hormones, PRL (500 ng/ml), P (1 μM), or P+PRL for 40 h. (C) The construct used in panel B was similarly transfected into cells that were then treated with either no hormones, PRL (500 ng/ml), P (1 μM), or DEX (1 μM) alone or in combination. Cells were lysed and transcriptional activity was determined using a luciferase assay and normalized for β-galactosidase activity. Values are expressed as fold induction over no hormone-treated cells. Data are means±s.e.m. (n=3 independent experiments). A PxPRL-positive interaction is indicated by ****P<0·0001.
We next sought to confirm the role of the HRE in the synergistic response to P+PRL by placing the 5′ distal enhancer (−1080/−840) upstream of a minimal SV40 promoter. The 5′ distal enhancer (−1080/−840) still mediated P+PRL synergy (Fig. 3B; P<0·0001), indicating that the effect of P in this response does not involve PR recruitment to the HRE. The 5′ distal enhancer (−1080/−840) clearly acted as an enhancer of basal activity (promoter activity in hormone-free media) when juxtaposed upstream of the SV40 minimal promoter (Fig. 3B; P<0·05), but did not significantly enhance basal activity when upstream of the truncated HRE (Fig. 3A; P>0·05).
Given our previous data (Fig. 1), we next tested the ability of DEX+PRL to synergistically enhance transcription from the −1080/−840 minimal SV40 construct. The fact that this construct did not respond to DEX+PRL in the same way that the full-length MMTV construct did (Fig. 3C; P>0·1) indicates that DEX, but not P, requires sequences between −840 and +1 (which includes the HRE) to coordinate transcriptional synergy with PRL. Combined, these data indicate that the −1080/−840 MMTV-LTR 5′ enhancer is directly activated by a novel progesterone response element (PRE)-independent mechanism that synergizes with the actions of PRL.
Synergy between P and PRL requires elements between −966 and −909 of the distal MMTV-LTR enhancer
The 5′ distal enhancer (−1080/−840) of the MMTV-LTR includes multiple transcription factor-binding domains that have been characterized to varying degrees (Mink et al. 1990, 1992, Lefebvre et al. 1991, Yanagawa et al. 1991, Haraguchi et al. 1993, Mellentin–Michelotti et al. 1994, Kusk et al. 1996, Grimm & Nordeen 1998, 1999). Given the essential requirement for this region in the synergistic response to P+PRL, we sought to more precisely define the contribution of these cis-acting elements. Taking the −1080/−840 MMTV SV40 construct, we first tested 3′ deletions of the enhancer (−1080 to either −874, −909, −928, or −956) that deleted putative sites for NF1 (Kusk et al. 1996), MAF/F2 (Mink et al. 1990), STAT5 (Haraguchi et al. 1997), and an unidentified transcription factor complex (Fp1; Grimm & Nordeen 1999) respectively. As shown in Fig. 4A, deletion of elements downstream of position −909 did not ablate the synergistic response to P+PRL (P<0·001), whereas deletion of sequence containing a consensus STAT5 site (−923/−915) ablated the hormone synergy (P=0·9). Basal transcription also decreased by ~60% upon deletion of the region −909/−928 (data not shown).
Figure 4.
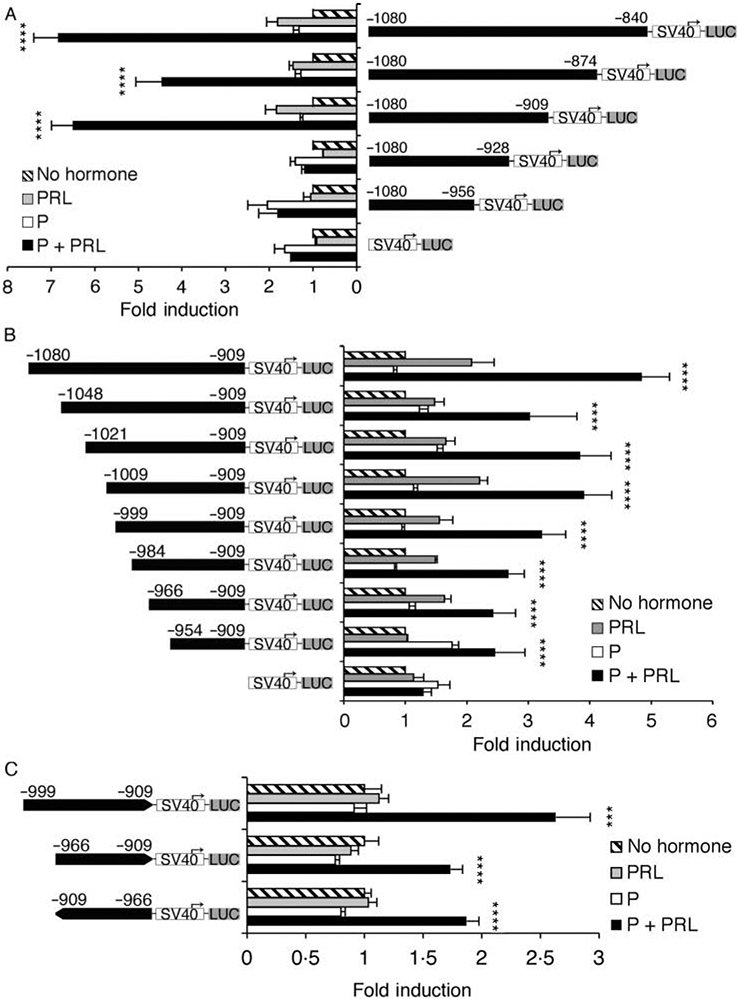
5′ and 3′ deletion analysis of the mouse mammary tumor virus-long terminal repeat (MMTV-LTR) enhancer region that facilitates the synergistic transcriptional response to progesterone (P)+prolactin (PRL). Various (A) 3′ and (B) 5′ deletions of the MMTV-LTR enhancer (−1080/−840) were positioned upstream of a minimal SV40 promoter and a luciferase reporter gene. (C) The minimal enhancer region −966/−909 was positioned in both orientations upstream of a minimal SV40 promoter and luciferase reporter gene. Constructs were transiently transfected into T47D cells prior to their treatment with no hormones, PRL (500 ng/ml), P (1 μM), or P+PRL for 40 h. Cells were lysed and transcriptional activity was determined using a luciferase assay and normalized for β-galactosidase activity. Values are expressed as fold-induction over no hormone-treated cells. (A and B) Data are means±s.e.m. (n=3–6 independent experiments). (C) Data are representative of two independent experiments; means±s.e.m. (n=6 replicates). SV40, minimal SV40 promoter; LUC, luciferase gene. A PxPRL-positive interaction is indicated by ***P<0·001 or ****P<0·0001.
Given these data, we also analyzed 5′ serial deletions of the MMTV-LTR enhancer that were fixed to position −909 at the 3′ end and were positioned upstream of the minimal SV40 promoter. These constructs deleted binding sites for an mp4 factor (−1048) that is PRL inducible (Lefebvre et al. 1991), CCAAT transcription factor (CTF)/NF1 (−1021; Kusk et al. 1996), AP-2/mp5 (−1009; Mellentin-Michelotti et al. 1994), and MAF/F12 (−999; Mink et al. 1990) respectively. Deletion of mp4, CTF/NF1, AP-2/mp5, or MAF/F12 sites did not ablate the synergistic response to P+PRL (P<0·0001; Fig. 4B). Mutation of the CTF/NF1, AP-2/mp5, or MAF/F12 sites in the −1048/+177 MMTV-LTR construct also failed to ablate the P+PRL synergy (data not shown). Further deletions, all of which retained the hypothesized minimal enhancer region −954/−909, also retained the significant interactive response to P and PRL (P<0·0001; Fig. 4B). We did find that the P+PRL synergistic response to P+PRL was more robust when the region −966/−909 was compared with the −954/−909 fragment (data not shown). Taken together, these data suggest that the minimal enhancer region required for the optimal synergistic response to P+PRL is −966/−909. This fragment meets the criteria for classification as an enhancer element given its ability to function in either orientation Fig. 4C.
A palindrome within the distal MMTV-LTR enhancer binds a mammary-specific transcription factor complex
We next used gel shift analysis to evaluate transcription factor recruitment to the MMTV-LTR enhancer region that facilitated the synergistic response to P+PRL. Sequence analysis of the region −954/−929 of the MMTV-LTR distal enhancer identified a previously uncharacterized 12 bp inverted palindrome (−941/−930; AGATTacAATCT) positioned within a transcription factor-binding domain identified by DNase footprinting (Fp1; Grimm & Nordeen 1999). Using probes corresponding to positions −997/−909, −954/−909, and −954/−922, we found that the greatest binding of the largest complex (hereafter referred to as MGSC; see below) was to the longest probe (Fig. 5A). The shorter probes also recruited MGSC at a lower abundance, as well as other smaller complexes. These data indicate that MGSC can bind to the minimal sequence −954/−922 that includes a previously uncharacterized inverted palindrome. Furthermore, the promoter sequence flanking −954/−922 increases MGSC formation, suggesting that a longer probe increases the stability of the complex. To further investigate whether the −941/−930 palindrome bound MGSC, we introduced single base pair substitutions into each position of a minimal palindrome EMSA probe (Table 2). This preliminary substitution analysis suggested the requirement of at least 6 bp (agattacaatct) for MGSC formation (Fig. 5B), thereby further substantiating the recruitment of factors to this site. We also sought to confirm the specific requirement for the palindrome in MGSC binding. As shown in Fig. 5C, deletion of the central 2 bp (−936/−935) within the palindromic site abolished MGSC formation.
Figure 5.
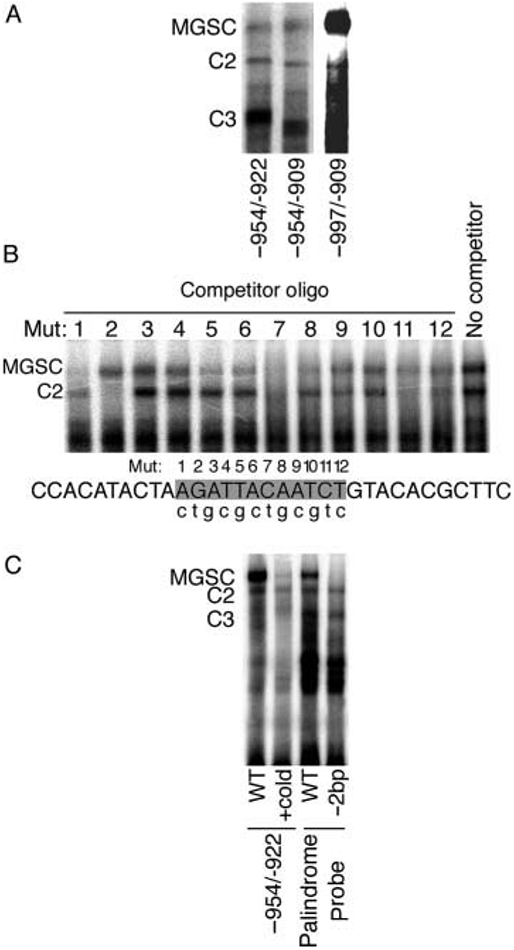
Analysis of protein binding to the distal enhancer of the mouse mammary tumor virus-long terminal repeat (MMTV-LTR) by EMSA. (A) Radiolabeled oligonucleotides corresponding to −997/−909, −954/−909, and −954/−922 (see Table 2 for sequence) were incubated with nuclear proteins from T47D cells prior to electrophoresis. Film exposure time for the −997/−909 probe was reduced due to a greater abundance of MGSC formation compared with levels for the −954/−909 and −954/−922 probes. (B) Transcription factor binding to an inverted palindrome within the MMTV-LTR distal enhancer is modulated by single base pair mutations. EMSA reactions were performed by incubating nuclear extracts from T47D cells with a radiolabeled palindrome probe and competition with a 100-fold excess of unlabeled oligonucleotide bearing a single base pair mutation at the numbered base pair site (lower case letter indicates corresponding base pair substitution). (C) EMSA was performed using T47D nuclear extracts and a radiolabeled oligonucleotide corresponding to −954/−922. Competition was performed using a 100-fold excess of cold −954/−922 probe. T47D nuclear extracts were also incubated with either a radiolabeled palindrome probe that included the entire palindrome (AGATTacAATCT) flanked by non-specific DNA or the palindrome probe lacking the central 2 bp separating the palindrome (−2 bp). C, complex; MGSC, mammary gland-specific complex.
Previously, Grimm & Nordeen (1999) identified binding of a MGSC to −951/−919 of the MMTV-LTR enhancer. We therefore tested whether the −941/−930 palindrome recruited this same complex. First, we extended Grimm & Nordeen’s data by showing that MGSC was present within all cells of mammary (normal and neoplastic) origin (Fig. 6A) while being present at a low level in only two out of nine non-mammary cell lines (Fig. 6A). Given our hypothesis that the −941/−930 palindrome binds MGSC, we used the core 12 bp site flanked by redundant sequence (Table 2) as a probe to further analyze nuclear proteins from the same cell lines. As shown in Fig. 6B, the palindrome probe can also bind the MGSC, albeit more weakly than the −954/−922 probe, as well as several smaller complexes.
Figure 6.
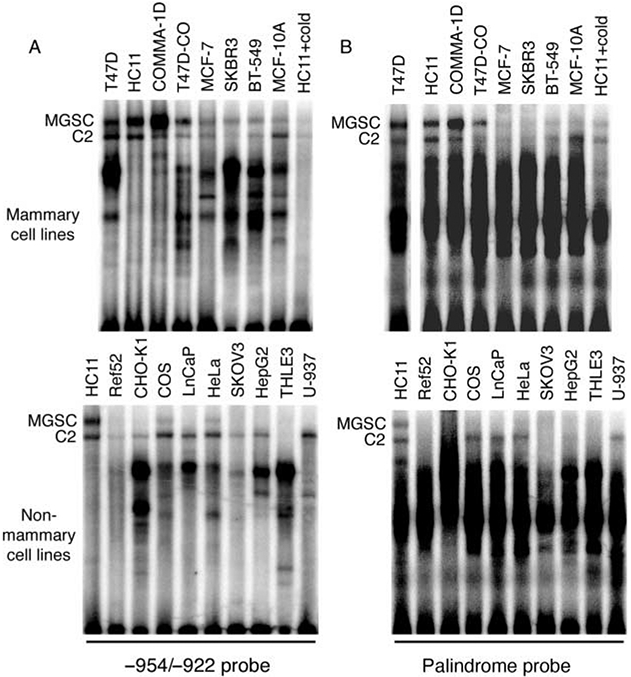
The palindrome site (−941/−930) in the mouse mammary tumor virus-long terminal repeat (MMTV-LTR) binds a mammary-specific transcription factor complex. EMSA experiments were performed using extracts from the indicated mammary and non-mammary cell lines (see Table 1 for cell origins). Nuclear proteins were incubated with either a radiolabeled (A) −954/−922 or (B) palindrome probe (see Table 2 for sequence). C, complex; MGSC, mammary gland-specific complex.
Mutation of either a palindromic DNA-binding domain or the putative STAT5 site on the 5′ MMTV-LTR enhancer abolishes the synergistic effect of P+PRL
Given that deletion of the region −954/−929 in the partial MMTV-LTR (−954/+177) ablated the synergistic effect of P+PRL (Fig. 2), we analyzed the palindrome’s role by introducing successive 4 bp mutations across this site within the −954/+177 MMTV-LTR construct. Notably, mutants 1–4 failed to elicit a synergistic response to P+PRL (P>0·1) while mutant 5 was able to confer the response (P<0·0001). These data indicate that 10 bp of the 12 bp palindromic site, in addition to 6 bp upstream of the palindrome, appear to be required for the synergistic response to P+PRL. We confirmed the role of this palindrome by mutating 3 bp within the palindrome (2 bp corresponding to those within mutant 3 and 1 bp within mutant 4) of the full-length (−1227/+177) MMTV-LTR. As shown in Fig. 7B, mutation of these 3 bp completely abolished the synergistic response to P+PRL (P=0·4), but did not significantly reduce the basal transcriptional activity of the MMTV-LTR (P>0·05).
Figure 7.
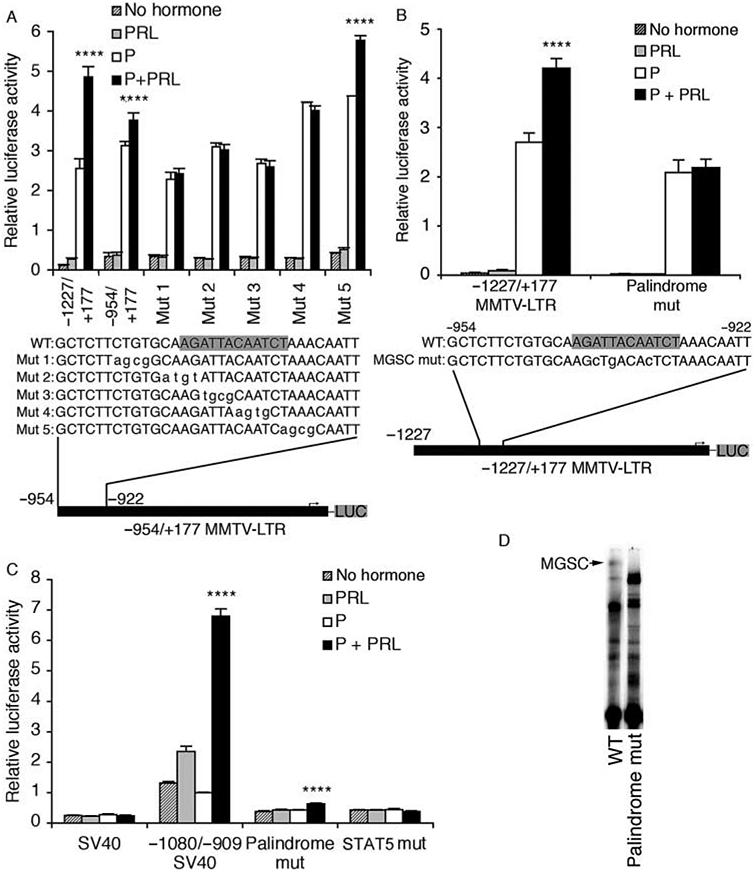
Mutations of either −948/−932 or −923/−915 of the mouse mammary tumor virus-long terminal repeat (MMTV-LTR) abolishes the synergistic transcriptional effect of progesterone (P)+prolactin (PRL). (A) Successive mutations were introduced across the region −948/−927 of the −954/+177 MMTV-LTR construct (see Fig. 2) before positioning them upstream of a luciferase reporter gene in pGL3-Basic. Data are means±s.e.m. (n=2 independent experiments). (B) Site-directed mutagenesis was used to mutate 3 bp of the inverted palindrome within the distal enhancer of the full-length (−1227/+177) MMTV-LTR. Data are means±s.e.m. (n=3 independent experiments). (C) Mutations were introduced into either the inverted palindrome (3 bp, same as panel B) or a putative STAT5 site (TTCGGAGAA changed to cgaGGAtcg) within the minimal distal enhancer (−1080/−909) before positioning them upstream of an SV40 promoter. All constructs were transiently transfected into T47D cells prior to treatment with no hormones, PRL (500 ng/ml), P (1 μM), or P+PRL for an additional 40 h. Cells were lysed and transcriptional activity was determined using a luciferase assay and normalized for β-galactosidase activity. Data are means±s.e.m. (n=3 independent experiments). (D) Complex formation was analyzed by EMSA using either the −954/−909 wild-type probe (WT) or the same probe (see Table 2) bearing a 3 bp mutation that eliminated the synergistic response to P+PRL in panels B and C (palindrome mut). SV40, minimal SV40 promoter; LUC, luciferase gene. The palindrome sequence (−941/−930) is shaded in the WT sequences. A PxPRL-positive interaction is indicated by ****P<0·0001.
Taken together with our earlier deletion analyses (Figs 2 and 4A), we had strong evidence to indicate that both the inverted palindrome (−941/−930) and an adjacent consensus STAT5 site (−923/−915) were essential for the synergistic response to P+PRL. To confirm a role for each of these sites, we performed mutation analysis of the −1080/−909 minimal enhancer positioned upstream of the SV40 minimal promoter. The −1080/− 909_SV40 pGL3 construct conferred a significant synergistic response to P+PRL (P<0·0001) in the absence of a response to P alone (Fig. 7C), consistent with data presented in Fig. 4A and B. Site-directed mutation of the palindrome in the −1080/−909 fragment (same mutations as those illustrated in Fig. 7B) nearly abolished the synergistic effect of P+PRL (reduced from a 418% induction to a 65% induction). In parallel, site-directed mutation of the adjacent downstream STAT5 site completely abolished the positive synergistic response to P+PRL (Fig. 7C). Regarding basal transcriptional activity, the −1080/−909 fragment increased basal transcription by 430% compared with the SV40 promoter alone (P<0·0001) while mutation of either the palindrome or STAT5 sites significantly reduced basal transcription (Fig. 7C; P<0·0001). Combined, these data demonstrate that P and PRL cooperatively stimulate transcription from the MMTV-LTR via a previously uncharacterized inverted palindrome as well as an adjacent STAT5 consensus site contained within the minimal region −966/−909. These data also confirm the importance of this region for enhancing basal transcription from the MMTV-LTR.
To determine whether recruitment of MGSC to the MMTV-LTR occurs concurrently with the synergistic response to P+PRL, we analyzed transcription factor binding to a −954/− 909 probe bearing the 3 bp mutation that abolished the synergistic response in our site-directed full-length MMTV-LTR (Fig. 7B). As anticipated, the wild-type probe (−954/−909) recruited the same profile of complexes as the palindrome probe, whereas the 3 bp mutation specifically prevented MGSC formation and altered abundance and migration of complexes C2 and C3 (Fig. 7D). These results, combined with our reporter assay data, indicate that MGSC binds to one of the same sites that are required for synergistic activation of the MMTV-LTR by P+PRL.
Jak2 and Src family kinases mediate the synergistic effect of P+PRL on MMTV-LTR transcription
We used dominant-negative plasmids and pharmacologic inhibitors to evaluate the role of PRL-activated signaling molecules in the synergistic activation of the distal MMTV-LTR by P+PRL. Co-expression of dominant-negative Jak2, c-Src, and Fyn kinases prevented any synergistic effect of P+PRL on the −1080/−909 SV40 pGL3 construct (P>0·1; Fig. 8) while co-expression of dominant-negative MEK1/2, STAT5a, or STAT5b did not prevent P+PRL synergy (P<0·01; data not shown). Separately, the P+PRL synergy was still observed (P<0·01) in the presence of LY 294 002 and wortmannin, both chemical inhibitors of phosphatidylinositol-3 (PI3) kinase (data not shown). We also recorded the same results when using these dominant-negative plasmids and pharmacologic inhibitors against the full-length MMTV-LTR pGL3 construct (data not shown). Therefore, transduction of the synergistic effect of P+PRL to the distal MMTV-LTR in T47D cells is facilitated via a Jak2-, c-Src-, and Fyn-dependent signaling pathway.
Figure 8.
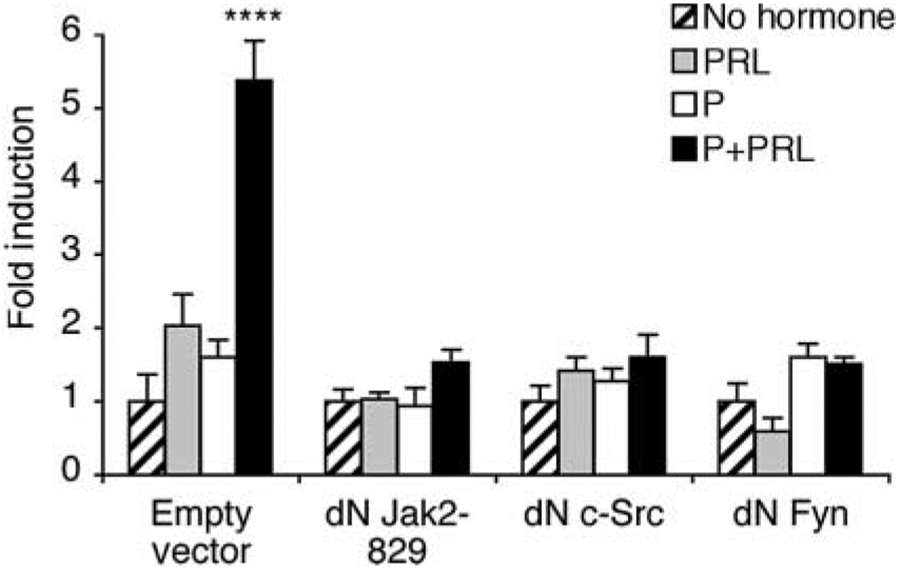
Synergistic activation of transcription from the MMTV-LTR −1080/−909 enhancer in response to P+PRL is mediated by Jak2, Fyn, and c-Src. T47D cells transfected with the −1080/−909 SV40 pGL3 reporter plasmid were co-transfected with dominant-negative (dN) Jak2-829, c-Src, or Fyn. Cells were then treated with either no hormones, PRL (500 ng/ml), P (1 μM), or P+PRL for 40 h. Cells were lysed and transcriptional activity was determined using a luciferase assay and normalized for β-galactosidase activity. Data are representative of at least three independent experiments; data are means±s.e.m. (n=3–4 replicates). A PxPRL-positive interaction is indicated by ****P<0·0001.
The MGSC transcription factor complex involves GATA-like factors
Grimm & Nordeen (1999) proposed that MGSC comprised a transcription factor(s) belonging to the CCAAT/enhancer binding protein (C/EBP) transcription factor family, based on the sequence tgcAgatta-CAATCTaaAca between positions −945 and −925. However, we were unable to validate this proposal using C/EBP-β competitor oligonucleotides in gel shift assays (data not shown). We identified that the adjacent palindromic MGSC-binding site includes a putative consensus sequence for members of the GATA transcription factor family (AGATtA), prompting us to test for the presence of these in MGSC using a consensus GATA oligonucleotide competitor (Table 2). Specific competition for MGSC, but not C2 or C3, was realized using this unlabeled probe (Fig. 9A).
Figure 9.
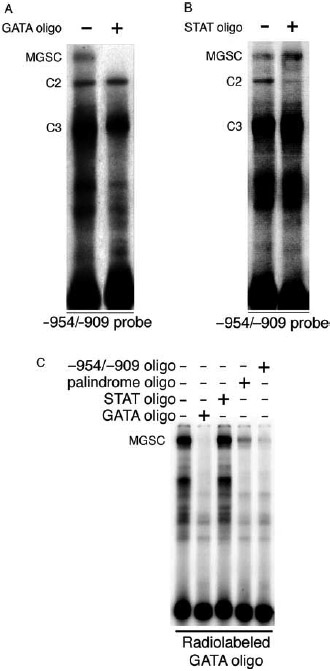
Transcription factor complexes recruited to the −954/−909 mouse mammary tumor virus-long terminal repeat (MMTV-LTR) enhancer region are putative GATA-like proteins. (A and B) eMsA analyses were performed by incubating a radiolabeled −954/−909 probe with T47D nuclear protein extracts (10 μg). Complex binding was competed by a 100-fold excess of either unlabeled (A) GATA or (B) STAT5 consensus-binding sequence (see Table 2). (C) EMSA experiments were performed by incubating a radiolabeled consensus GATA oligonucleotide with nuclear protein extracts from T47D cells in the presence or absence of a 100-fold excess of unlabeled GATA, StAt5, or MMTV-LTR (−954/−909 and palindrome) oligonucleotides. C, complex; MGSC, mammary gland-specific complex.
The MMTV-LTR minimal enhancer region (−1080/−909) includes a consensus STAT5 site (−923/−915) that is essential for P+PRL synergy (Fig. 7C) and binds a PRL-inducible factor (Haraguchi et al. 1997, Biola et al. 2001). We therefore tested the ability of an unlabeled consensus STAT5 oligonucleotide to compete for transcription factor complex binding to the −954/−909 probe. An excess of unlabeled STAT5 oligonucleotide specifically competed for C2 but not for MGSC binding (Fig. 9B). To investigate the presence of GATA-like transcription factors in these complexes, we used a radiolabeled consensus GATA oligonucleotide in EMSA and competed this with MMTV-LTR enhancer sequences. The GATA probe bound a complex that corresponded to the size of MGSC (Fig. 9C). The palindrome probe and −954/−909 enhancer fragments both competed for all complexes recruited to the consensus GATA probe (Fig. 9C). The −954/−909 oligonucleotide competed more effectively than did the palindrome oligonucleotide, consistent with our earlier finding that MGSC binds more effectively to a longer sequence (Fig. 5A). These results support data from Fig. 9A suggesting that GATA-like proteins are recruited to the region −954/−909. We were unable to show competition of unlabelled STAT5 for the complexes recruited to the consensus GATA probe, despite the ability for STAT5-like proteins to bind GATA target sequences or interact with DNA-bound GATA proteins (Wang et al. 2005). Taken together, these data strongly suggest that GATA-like transcription factors are recruited to the −941/−930 palindrome of the MMTV-LTR enhancer.
Discussion
Individually, both P and PRL are essential for normal mammary development and function (Lydon et al. 1995, Ormandy et al. 1997a), while several lines of evidence suggest that they may also impart some of their effects cooperatively. For example, P+PRL synergistically induces mammary epithelial proliferation (Hovey et al. 2001) in ovariectomized mice as well as activation of IRS-1 and IRS-2 (Hovey et al. 2003). Conversely, P antagonizes the lactogenic effect of PRL, either by suppressing glucocorticoid receptor transactivation (Buser et al. 2007) or by downregulating PRLR (Djiane & Durand 1977). Given our interest in determining how these two hormones interactively regulate mammary gland function, we have extended the findings of Haraguchi et al. (1993) who showed that P+PRL synergistically activated the MMTV-LTR (Haraguchi et al. 1992) via its distal enhancer. Using this model, we have identified that at least two adjacent sites, −941/−930 and −923/−915, within the MMTV-LTR distal enhancer are required for this response in T47D cells via a PRE-independent pathway that utilizes Jak2, c-Src, and Fyn.
In these studies, we found that the palindrome (−941/−930) and STAT5 (−923/−915) sites not only facilitated a synergistic response to P+PRL but also enhanced basal transcription. This region of the MMTV-LTR was previously shown to confer tissue-specific basal expression in both promyelocytic progenitor (a known site of MMTV-LTR activity; Reuss & Coffin 2000) and mammary cells (Grimm & Nordeen 1998), resulting in its classification as the MEM region (Grimm & Nordeen 1998). However, the cis-acting site(s) responsible for tissue-specific expression from this region have remained undefined. Our site-directed mutagenesis and single base pair mutation EMSA firmly establish that the inverted palindrome AGATTacAATCT recruits the MGSC in addition to enhancing basal transcription. Although Grimm & Nordeen (1999) previously suggested that an overlapping site bound a C/EBP-like factor (tgcaAgatta-CAATCTaaAca, putative C/EBP site in caps), we were unable to confirm these data. Interestingly, Gross & Garrard (1988) also found that the MEM region contained a DNase hypersensitive site in the vicinity of the palindrome that would agree with it being able to assume an altered conformation. Such a proposal is consistent with the slight retardation of C2 and C3 migration that we observed in EMSA when the mutated −954/−909 and wild-type probes were compared.
The palindromic MGSC-binding site shares homology with a consensus GATA-binding site and recruits various protein complexes, including MGSC, that were competed by a GATA consensus sequence. These data lead us to propose that a mammary-specific GATA-like factor or GATA-associated complex is a component of MGSC. The GATA family of transcription factors has already been implicated in the regulation of tissue-specific function in a variety of species (Fluck & Miller 2004, Dusing & Wiginton 2005, Pauli et al. 2006). The finding that GATA3 is overexpressed in ER-positive breast cancers (Hoch et al. 1999) while GATA2 expression in the uterus is regulated by P (Lydon et al. 1996) supports a potential role for a GATA-like factor during the hormonal regulation of mammary function. Perhaps, most significant is the recent report by two groups (Kouros-Mehr et al. 2006, Asselin-Labat et al. 2007) that GATA-3 fulfils a critical role during the growth and differentiation of the mammary epithelium, where its deletion prevents cellular progression to a differentiated alveolar phenotype. At present, the identity of any GATA factors in MGSC and their role in hormone synergy remains to be established given that we have not been able to supershift this complex with antibodies against GATA1-6 (data not shown).
An adjacent consensus STAT5 site at position −923/−915 is also essential for the transcriptional synergistic response to P+PRL by T47D cells. Although the function of this site has received limited investigation, it is required for both basal and interleukin-2-induced MMTV transcription in cytotoxic T lymphocyte line (CTLL)-2 T lymphocytes (Biola et al. 2001) and by itself can bind an interleukin-2 (Biola et al. 2001) and PRL-inducible (Haraguchi et al. 1997) factor that can be super-shifted with anti-STAT5 antiserum (Biola et al. 2001). However, this site does not participate in the STAT5-mediated suppression of glucocorticoid receptor activity at the proximal HRE (Stoecklin et al. 1999, Biola et al. 2001), consistent with our finding that the distal enhancer functions independently of the proximal HRE during the synergistic effect of P+PRL. Interestingly, while a STAT5 consensus oligonucleotide specifically competed for the C2 complex that was bound by the palindrome, the larger MGSC apparently does not comprise STAT5, and extension of the palindromic probe to include the adjacent STAT5 site did not alter DNA-protein complex formation. Furthermore, our dominant-negative experiments suggest that neither STAT5a nor STAT5b participate in facilitating the synergistic response to P+PRL. Our further attempts to demonstrate a direct interaction between STAT5-like factors and proteins binding to the MGSC site through a GATA-STAT interaction yielded only limited support for such a relationship despite the fact that cooperative interactions between GATA- and STAT-like factors are known to regulate tissue-specific gene expression. For example, synergistic activation of the atrial natriuretic factor promoter by cytokines and growth factors in cardiomyocytes is directed by an interaction between GATA-4 and STAT1 (Wang et al. 2005). Separately, interferon-1α induction of the HLA-E gene requires an interaction between STAT1α and GATA-1 that converge via p300/CBP. Clearly, the role of STAT proteins in the activity of the MMTV-LTR is complex given that others have implicated this proximal STAT5 site in directing lymphoid and lactation-specific expression (Mok et al. 1992) despite the fact that it is activated in the absence of STAT5a in vivo (Stegalkina et al. 1999).
Generally, it is assumed that P- and DEX-induced transcription from the MMTV-LTR in various cell types, including breast cancer cells, is conferred by the ability of their respective receptors to activate four canonical steroid hormone receptor-binding sites within the HRE (Ham et al. 1988). Our present findings indicate that P, but not DEX, can also interact with PRL to induce transcription from a distal enhancer in the absence of any consensus PRE. Along these lines, it is noteworthy that while the transcription of int genes in mammary tumors is directed by tissue-specific elements in the distal enhancer (Grimm & Nordeen 1998), their expression is not induced by glucocorticoids (Nusse 1988). Interestingly, Grimm & Nordeen concluded that the MEM element within the MMTV-LTR did not mediate hormone action. However, their conclusion was based solely on the lack of any effect of DEX, thereby ratifying our finding that this region specifically mediates the action of P in concert with PRL; whether it also mediates the cooperative action of other hormones remains to be tested.
Our findings are consistent with data showing that the extra-nuclear effects of PR in various breast cancer cells often involve c-Src (Edwards et al. 2002, Migliaccio et al. 2002). For example, activated PR-B interacts with ligand-free ER to induce an ER/c-Src interaction that stimulates proliferation via the c-Src/p21ras/Erk signaling pathway (Migliaccio et al. 1998). Separately, several groups have demonstrated that P induces the expression and phosphorylation of STAT3 and STAT5 (Richer et al. 1998, Proietti et al. 2005), where P-induced activation of Jak1 and Jak2 and phosphorylation of STAT1 and STAT3 is Src dependent (Zhang et al. 2000, Proietti et al. 2005). More recently, c-Src was shown to phosphorylate P-induced caveolin-1 that mediates a proliferative effect of P on mammary tumor cells (Salatino et al. 2006). Although several groups have implicated the MAP and PI3 kinases as downstream effectors of crosstalk between PR and c-Src (Lange 2004), our present results indicate that these pathways do not participate in the synergistic effect of P+PRL on the MMTV-LTR. It is also well established that Jak2 (Campbell et al. 1994, Rui et al. 1994) and Src family kinases (Clevenger & Medaglia 1994, Al-Sakkaf et al. 1997, Fresno Vara et al. 2000, 2001) can converge during PRL signaling, where the Src family kinase p59 (Fyn) can preform with all PRLR isoforms (Clevenger & Medaglia 1994). While a role for STAT1 or STAT3 in the synergistic response by the MMTV-LTR to P+PRL cannot be ruled out, our data do indicate that STAT5a and STAT5b are not required. Similar to our findings, Chilton et al. found that the synergistic effect of P+PRL on the uteroglobin promoter occurred downstream of Jak2, but did not require STAT5, prior to signaling via RUSH1α (Hewetson et al. 2002). These findings contrast to the role of STAT5 in the positive effect of P on subsequent PRL-induced transcription from the β-casein promoter (Richer et al. 1998). Interestingly, STAT5 undergoes differential translocation and phosphorylation following its activation by c-Src compared with the typical post-PRLR pathway, which may explain how P and PRL differentially regulate target gene transcription.
Our finding that a novel region of the MMTV-LTR mediates mammary-specific synergy between P and PRL may be significant in understanding the basis of mammary cancer. MMTV specifically induces alveolar hyperplasia within the mammary gland, and both P and PRL promote alveologenesis (Lydon et al. 1995, Ormandy et al. 1997a), suggesting that these hormones may cooperatively activate oncogene expression in a subpopulation of alveolar progenitor cells. Consistent with this proposal, we previously showed that PR and PRLR co-localize to a subpopulation of epithelial cells in the mouse mammary gland, where we (Hovey & Vonderhaar 2000) and others (Wagner et al. 2001) have also shown that MMTV or its LTR are often only expressed in an epithelial subpopulation. Moreover, recent reports of MMTV sequences in human breast tumors (Wang et al. 1995, 2004, Acha-Orbea et al. 1999, Etkind et al. 2000, Liu et al. 2001, Ford et al. 2003, Indik et al. 2005) have invigorated the debate surrounding a possible viral basis for the disease. Among these sequences is the palindrome within the enhancer that binds MGSC (Wang et al. 2004). The finding that PR expression correlates with the presence of MMTV sequences in breast tumors (Faedo et al. 2004) raises the interesting question of whether the MGSC-binding site is a potential enhancer of P+PRL action in human breast cancer in addition to its role in T47D cells as shown herein.
Acknowledgements
We declare that there is no conflict of interest that would prejudice the impartiality of the research presented in this manuscript. This work was supported in part by The University of Vermont Agricultural Experiment Station, the USDA Cooperative State Research, Education and Extension Service (Grant no. 2004-35206-14140) and a core grant to the Vermont Cancer Center (P30 CA22435). We wish to thank Dr Barbara Vonderhaar and Nicholas Heintz for helpful discussion.
References
- Acha-Orbea H, Finke D, Attinger A, Schmid S, Wehrli N, Vacheron S, Xenarios I, Scarpellino L, Toellner KM, MacLennan IC et al. 1999. Interplays between mouse mammary tumor virus and the cellular and humoral immune response. Immunological Reviews 168 287–303. [DOI] [PubMed] [Google Scholar]
- von der Ahe D, Janich S, Scheidereit C, Renkawitz R, Schutz G & Beato M 1985. Glucocorticoid and progesterone receptors bind to the same sites in two hormonally regulated promoters. Nature 313 706–709. [DOI] [PubMed] [Google Scholar]
- Al-Sakkaf KA, Dobson PR & Brown BL 1997. Prolactin induced tyrosine phosphorylation of p59fyn may mediate phosphatidylinositol 3-kinase activation in Nb2 cells. Journal of Molecular Endocrinology 19 347–350. [DOI] [PubMed] [Google Scholar]
- Asselin-Labat ML, Sutherland KD, Barker H, Thomas R, Shackleton M, Forrest NC, Hartley L, Robb L, Grosveld FG, van der Wees J et al. 2007. Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nature Cell Biology 9 201–209. [DOI] [PubMed] [Google Scholar]
- Ball RK, Friis RR, Schoenenberger CA, Doppler W & Groner B 1988. Prolactin regulation of beta-casein gene expression and of a cytosolic 120-kd protein in a cloned mouse mammary epithelial cell line. EMBO Journal 7 2089–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biola A, Lefebvre P, Perrin-Wolff M, Sturm M, Bertoglio J & Pallardy M 2001. Interleukin-2 inhibits glucocorticoid receptor transcriptional activity through a mechanism involving STAT5 (signal transducer and activator of transcription 5) but not AP-1. Molecular Endocrinology 15 1062–1076. [DOI] [PubMed] [Google Scholar]
- Boot LM, Muhlbock O & Ropcke G 1962. Prolactin and the induction of mammary tumors in mice. General and Comparative Endocrinology 2 601–602. [Google Scholar]
- Bradford MM 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry 72 248–254. [DOI] [PubMed] [Google Scholar]
- Brisken C, Park S, Vass T, Lydon JP, O’Malley BW & Weinberg RA 1998. A paracrine role for the epithelial progesterone receptor in mammary gland development. PNAS 95 5076–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buser AC, Gass-Handel EK, Wyszomierski SL, Doppler W, Leonhardt SA, Schaack J, Rosen JM, Watkin H, Anderson SM & Edwards DP 2007. Progesterone receptor repression of prolactin/signal transducer and activator of transcription 5-mediated transcription of the beta-casein gene in mammary epithelial cells. Molecular Endocrinology, 21 106–125. [DOI] [PubMed] [Google Scholar]
- Campbell GS, Argetsinger LS, Ihle JN, Kelly PA, Rillema JA & Carter-Su C 1994. Activation of JAK (JAK2) tyrosine kinase by prolactin receptors in NB (NB2) cells and mouse mammary gland explants. PNAS 91 5232–5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cato AC, Henderson D & Ponta H 1987. The hormone response element of the mouse mammary tumour virus DNA mediates the progestin and androgen induction of transcription in the proviral long terminal repeat region. EMBO Journal 6 363–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevenger CV & Medaglia MV 1994. The protein tyrosine kinase p59fyn is associated with prolactin (PRL) receptor and is activated by PRL stimulation of T-lymphocytes. Molecular Endocrinology 8 674–681. [DOI] [PubMed] [Google Scholar]
- Daniel JC Jr, Jetton AE & Chilton BS 1984. Prolactin as a factor in the uterine response to progesterone in rabbits. Journal of Reproduction and Fertility 72 443–452. [DOI] [PubMed] [Google Scholar]
- Danielson KG, Oborn CJ, Durban EM, Butel JS & Medina D 1984. Epithelial mouse mammary cell line exhibiting normal morphogenesis in vivo and functional differentiation in vitro. PNAS 81 3756–3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittrich E, Haft CR, Muys L, Heinrich PC & Graeve L 1996. A di-leucine motif and an upstream serine in the interleukin-6 (IL-6) signal transducer gp130 mediate ligand-induced endocytosis and downregulation of the IL-6 receptor. Journal of Biological Chemistry 271 5487–5494. [DOI] [PubMed] [Google Scholar]
- Djiane J & Durand P 1977. Prolactin-progesterone antagonism in self regulation of prolactin receptors in mammary gland. Nature 266 641–643. [DOI] [PubMed] [Google Scholar]
- Dusing MR & Wiginton DA 2005. Epithelial lineages of the small intestine have unique patterns of GATA expression. Journal of Molecular Histology 36 15–24. [DOI] [PubMed] [Google Scholar]
- Edwards DP, Wardell SE & Boonyaratanakornkit V 2002. Progesterone receptor interacting coregulatory proteins and cross talk with cell signaling pathways. Journal of Steroid Biochemistry and Molecular Biology 83 173–186. [DOI] [PubMed] [Google Scholar]
- Etkind P, Du J, Khan A, Pillitteri J & Wiernik PH 2000. Mouse mammary tumor virus-like ENV gene sequences in human breast tumors and in a lymphoma of a breast cancer patient. Clinical Cancer Research 6 1273–1278. [PubMed] [Google Scholar]
- Faedo M, Ford CE, Mehta R, Blazek K & Rawlinson WD 2004. Mouse mammary tumor-like virus is associated with p53 nuclear accumulation and progesterone receptor positivity but not estrogen positivity in human female breast cancer. Clinical Cancer Research 10 4417–4419. [DOI] [PubMed] [Google Scholar]
- Fluck CE & Miller WL 2004. GATA-4 and GATA-6 modulate tissue-specific transcription of the human gene for P450c17 by direct interaction with Sp1. Molecular Endocrinology 18 1144–1157. [DOI] [PubMed] [Google Scholar]
- Ford CE, Tran D, Deng Y, Ta VT, Rawlinson WD & Lawson JS 2003. Mouse mammary tumor virus-like gene sequences in breast tumors of Australian and Vietnamese women. Clinical Cancer Research 9 1118–1120. [PubMed] [Google Scholar]
- Fresno Vara JA, Carretero MV, Geronimo H, Ballmer-Hofer K & Martin-Perez J 2000. Stimulation of c-Src by prolactin is independent of Jak2. Biochemical Journal 345 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fresno Vara JA, Caceres MA, Silva A & Martin-Perez J 2001. Src family kinases are required for prolactin induction of cell proliferation. Molecular Biology of the Cell 12 2171–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowland PL & Buetti E 1989. Mutations in the hormone regulatory element of mouse mammary tumor virus differentially affect the response to progestins, androgens, and glucocorticoids. Molecular and Cellular Biology 9 3999–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm SL & Nordeen SK 1998. Mouse mammary tumor virus sequences responsible for activating cellular oncogenes. Journal of Virology 72 9428–9435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm SL & Nordeen SK 1999. A composite enhancer element directing tissue-specific expression of mouse mammary tumor virus requires both ubiquitous and tissue-restricted factors. Journal of Biological Chemistry 274 12790–12796. [DOI] [PubMed] [Google Scholar]
- Gross DS & Garrard WT 1988. Nuclease hypersensitive sites in chromatin. Annual Review of Biochemistry 57 159–197. [DOI] [PubMed] [Google Scholar]
- Gunzburg WH & Salmons B 1992. Factors controlling the expression of mouse mammary tumour virus. Biochemical Journal, 283 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ham J, Thomson A, Needham M, Webb P & Parker M 1988. Characterization of response elements for androgens, glucocorticoids and progestins in mouse mammary tumour virus. Nucleic Acids Research 16 5263–5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraguchi S, Good RA, Engelman RW & Day NK 1992. Human prolactin regulates transfected MMTV LTR-directed gene expression in a human breast-carcinoma cell line through synergistic interaction with steroid hormones. International Journal of Cancer 52 928–933. [DOI] [PubMed] [Google Scholar]
- Haraguchi S, Good RA & Day NK 1993. Prolactin acts on the extreme 5′ portion of MMTV LTR involving a mammary cell-specific enhancer. Molecular and Cellular Endocrinology 96 R1–R6. [DOI] [PubMed] [Google Scholar]
- Haraguchi S, Good RA, Engelman RW, Greene S & Day NK 1997. Prolactin, epidermal growth factor or transforming growth factor-α activate a mammary cell-specific enhancer in mouse mammary tumor virus-long terminal repeat. Molecular and Cellular Endocrinology 129 145–155. [DOI] [PubMed] [Google Scholar]
- Haslam SZ, Osuch JR, Raafat AM & Hofseth LJ 2002. Postmenopausal hormone replacement therapy: effects on normal mammary gland in humans and in a mouse postmenopausal model. Journal of Mammary Gland Biology and Neoplasia 7 93–105. [DOI] [PubMed] [Google Scholar]
- Hassan MH, Khatoon N, Curiel DT, Hamada FM, Arafa HM & Al-Hendy A 2008. Toward gene therapy of uterine fibroids: targeting modified adenovirus to human leiomyoma cells. Human Reproduction 23 514–524. [DOI] [PubMed] [Google Scholar]
- Henrard D & Ross SR 1988. Endogenous mouse mammary tumor virus is expressed in several organs in addition to the lactating mammary gland. Journal of Virology 62 3046–3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewetson A & Chilton BS 2003. An Sp1-NF-Y/progesterone receptor DNA binding-dependent mechanism regulates progesterone-induced transcriptional activation of the rabbit RUSH/SMARCA3 gene. Journal of Biological Chemistry 278 40177–40185. [DOI] [PubMed] [Google Scholar]
- Hewetson A, Hendrix EC, Mansharamani M, Lee VH & Chilton BS 2002. Identification of the RUSH consensus-binding site by cyclic amplification and selection of targets: demonstration that RUSH mediates the ability of prolactin to augment progesterone-dependent gene expression. Molecular Endocrinology 16 2101–2112. [DOI] [PubMed] [Google Scholar]
- Hoch RV, Thompson DA, Baker RJ & Weigel RJ 1999. GATA-3 is expressed in association with estrogen receptor in breast cancer. International Journal of Cancer 84 122–128. [DOI] [PubMed] [Google Scholar]
- Horoszewicz JS, Leong SS, Chu TM, Wajsman ZL, Friedman M, Papsidero L, Kim U, Chai LS, Kakati S, Arya SK et al. 1980. The LNCaP cell line – a new model for studies on human prostatic carcinoma. Progress in Clinical and Biological Research 37 115–132. [PubMed] [Google Scholar]
- Horwitz KB & Freidenberg GR 1985. Growth inhibition and increase of insulin receptors in antiestrogen-resistant T47DCO human breast cancer cells by progestins: implications for endocrine therapies. Cancer Research 45 167–173. [PubMed] [Google Scholar]
- Hovey RC & Vonderhaar BK 2000. Application of in situ PCR to studies of the mammary gland. In Methods in Mammary Gland Biology and Breast Cancer Research, pp 223–232. Eds Ip MM & Asch BB. New York: Kluwer Academic. [Google Scholar]
- Hovey RC, Trott JF, Ginsburg E, Goldhar A, Sasaki MM, Fountain SJ, Sundararajan K & Vonderhaar BK 2001. Transcriptional and spatiotemporal regulation of prolactin receptor mRNA and cooperativity with progesterone receptor function during ductal branch growth in the mammary gland. Developmental Dynamics 222 192–205. [DOI] [PubMed] [Google Scholar]
- Hovey RC, Trott JF & Vonderhaar BK 2002. Establishing a framework for the functional mammary gland: from endocrinology to morphology. Journal of Mammary Gland Biology and Neoplasia 7 17–38. [DOI] [PubMed] [Google Scholar]
- Hovey RC, Harris J, Hadsell DL, Lee AV, Ormandy CJ & Vonderhaar BK 2003. Local insulin-like growth factor-II mediates prolactin-induced mammary gland development. Molecular Endocrinology 17 460–471. [DOI] [PubMed] [Google Scholar]
- Hsu CL, Fabritius C & Dudley J 1988. Mouse mammary tumor virus proviruses in T-cell lymphomas lack a negative regulatory element in the long terminal repeat. Journal of Virology 62 4644–4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua W, Christianson T, Rougeot C, Rochefort H & Clinton GM 1995. SKOV3 ovarian carcinoma cells have functional estrogen receptor but are growth-resistant to estrogen and antiestrogens. Journal of Steroid Biochemistry and Molecular Biology 55 279–289. [DOI] [PubMed] [Google Scholar]
- Imagawa W, Bandyopadhyay GK & Nandi S 1990. Regulation of mammary epithelial cell growth in mice and rats. Endocrine Reviews 11 494–523. [DOI] [PubMed] [Google Scholar]
- Indik S, Gunzburg WH, Salmons B & Rouault F 2005. Mouse mammary tumor virus infects human cells. Cancer Research 65 6651–6659. [DOI] [PubMed] [Google Scholar]
- Jones EA, Conover J & Symes AJ 2000. Identification of a novel gp130-responsive site in the vasoactive intestinal peptide cytokine response element. Journal of Biological Chemistry 275 36013–36020. [DOI] [PubMed] [Google Scholar]
- Kouros-Mehr H, Slorach EM, Sternlicht MD & Werb Z 2006. GATA-3 maintains the differentiation of the luminal cell fate in the mammary gland. Cell 127 1041–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusk P, John S, Fragoso G, Michelotti J & Hager GL 1996. Characterization of an NF-1/CTF family member as a functional activator of the mouse mammary tumor virus long terminal repeat 5′ enhancer. Journal of Biological Chemistry 271 31269–31276. [DOI] [PubMed] [Google Scholar]
- Lange CA 2004. Making sense of cross-talk between steroid hormone receptors and intracellular signaling pathways: who will have the last word? Molecular Endocrinology 18 269–278. [DOI] [PubMed] [Google Scholar]
- van Leeuwen F & Nusse R 1995. Oncogene activation and oncogene cooperation in MMTV-induced mouse mammary cancer. Seminars in Cancer Biology 6 127–133. [DOI] [PubMed] [Google Scholar]
- Lefebvre P, Berard DS, Cordingley MG & Hager GL 1991. Two regions of the mouse mammary tumor virus long terminal repeat regulate the activity of its promoter in mammary cell lines. Molecular and Cellular Biology 11 2529–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung BS, Potter AH & Qureshi S 1981. Interaction of prolactin, estrogen and progesterone in a human mammary carcinoma cell line, CAMA-1-I cell growth and thymidine uptake. Journal of Steroid Biochemistry 15 421–427. [DOI] [PubMed] [Google Scholar]
- Liao L & Jaken S 1993. Effect of alpha-protein kinase C neutralizing antibodies and the pseudosubstrate peptide on phosphorylation, migration, and growth of REF52 cells. Cell Growth and Differentiation 4 309–316. [PubMed] [Google Scholar]
- Liu B, Wang Y, Melana SM, Pelisson I, Najfeld V, Holland JF & Pogo BG 2001. Identification of a proviral structure in human breast cancer. Cancer Research 61 1754–1759. [PubMed] [Google Scholar]
- Lydon JP, DeMayo FJ, Funk CR, Mani SK, Hughes AR, Montgomery CA Jr, Shyamala G, Conneely OM & O’Malley BW 1995. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes and Development 9 2266–2278. [DOI] [PubMed] [Google Scholar]
- Lydon JP, DeMayo FJ, Conneely OM & O’Malley BW 1996. Reproductive phenotypes of the progesterone receptor null mutant mouse. Journal of Steroid Biochemistry and Molecular Biology 56 67–77. [DOI] [PubMed] [Google Scholar]
- Lydon JP, Ge G, Kittrell FS, Medina O & O’Malley BW 1999. Murine mammary gland carcinogenesis is critically dependent on progesterone receptor function. Cancer Research 59 4276–4284. [PubMed] [Google Scholar]
- Medina D 1973. Preneoplastic lesions in mouse mammary tumorigenesis. In Methods in Cancer Research, pp 3–53. Ed Busch H. New York: Academic Press. [Google Scholar]
- Mellentin-Michelotti J, John S, Pennie WD, Williams T & Hager GL 1994. The 5′ enhancer of the mouse mammary tumor virus long terminal repeat contains a functional AP-2 element. Journal of Biological Chemistry 269 31983–31990. [PubMed] [Google Scholar]
- Migliaccio A, Piccolo D, Castoria G, Di Domenico M, Bilancio A, Lombardi M, Gong W, Beato M & Auricchio F 1998. Activation of the Src/p21ras/Erk pathway by progesterone receptor via cross-talk with estrogen receptor. EMBO Journal 17 2008–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio A, Castoria G, Di Domenico M, de Falco A, Bilancio A, Lombardi M, Bottero D, Varricchio L, Nanayakkara M, Rotondi A et al. 2002. Sex steroid hormones act as growth factors. Journal of Steroid Biochemistry and Molecular Biology 83 31–35. [DOI] [PubMed] [Google Scholar]
- Mink S, Ponta H & Cato AC 1990. The long terminal repeat region of the mouse mammary tumour virus contains multiple regulatory elements. Nucleic Acids Research 18 2017–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mink S, Hartig E, Jennewein P, Doppler W & Cato AC 1992. A mammary cell-specific enhancer in mouse mammary tumor virus DNA is composed of multiple regulatory elements including binding sites for CTF/NFI and a novel transcription factor, mammary cell-activating factor. Molecular and Cellular Biology 12 4906–4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok E, Golovkina TV & Ross SR 1992. A mouse mammary tumor virus mammary gland enhancer confers tissue-specific but not lactation-dependent expression in transgenic mice. Journal of Virology 66 7529–7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley KL, Toohey MG & Peterson DO 1987. Transcriptional repression of a hormone-responsive promoter. Nucleic Acids Research 15 6973–6989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlbock O & Boot LM 1959. Induction of mammary cancer in mice without the mammary tumor agent by isografts of hypophyses. Cancer Research 19 402–412. [PubMed] [Google Scholar]
- Nagasawa H, Miur K, Niki K & Namiki H 1985. Interrelationship between prolactin and progesterone in normal mammary gland growth in SHN virgin mice. Experimental and Clinical Endocrinology 86 357–360. [DOI] [PubMed] [Google Scholar]
- Nagasawa H, Ohta K, Nakajima K, Noguchi Y, Miura K, Niki K & Namiki H 1986. Interrelationship between pituitary and ovarian hormones in normal and neoplastic growth of mammary glands of mice. Annals of the New York Academy of Sciences 464 301–315. [DOI] [PubMed] [Google Scholar]
- Nusse R 1988. The int genes in mammary tumorigenesis and in normal development. Trends in Genetics 4 291–295. [DOI] [PubMed] [Google Scholar]
- Oakes SR, Robertson FG, Kench JG, Gardiner-Garden M, Wand MP, Green JE & Ormandy CJ 2007. Loss of mammary epithelial prolactin receptor delays tumor formation by reducing cell proliferation in low-grade preinvasive lesions. Oncogene, 26 543–553. [DOI] [PubMed] [Google Scholar]
- Ormandy CJ, Binart N & Kelly PA 1997a. Mammary gland development in prolactin receptor knockout mice. Journal of Mammary Gland Biology and Neoplasia 2 355–364. [DOI] [PubMed] [Google Scholar]
- Ormandy CJ, Hall RE, Manning DL, Robertson JFR, Blamey RW, Kelly PA, Nicholson RI & Sutherland RL 1997b. Coexpression and cross-regulation of the prolactin receptor and sex steroid hormone receptors in breast cancer. Journal of Clinical Endocrinology and Metabolism 82 3692–3699. [DOI] [PubMed] [Google Scholar]
- Pauli F, Liu Y, Kim YA, Chen PJ & Kim SK 2006. Chromosomal clustering and GATA transcriptional regulation of intestine-expressed genes in C. elegans. Development 133 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponta H, Kennedy N, Skroch P, Hynes NE & Groner B 1985. Hormonal response region in the mouse mammary tumor virus long terminal repeat can be dissociated from the proviral promoter and has enhancer properties. PNAS 82 1020–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proietti C, Salatino M, Rosemblit C, Carnevale R, Pecci A, Kornblihtt AR, Molinolo AA, Frahm I, Charreau EH, Schillaci R et al. 2005. Progestins induce transcriptional activation of signal transducer and activator of transcription 3 (Stat3) via a Jak- and Src-dependent mechanism in breast cancer cells. Molecular and Cellular Biology 25 4826–4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuss FU & Coffin JM 2000. The mouse mammary tumor virus transcription enhancers for hematopoietic progenitor and mammary gland cells share functional elements. Journal of Virology 74 8183–8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richer JK, Lange CA, Manning NG, Owen G, Powell R & Horwitz KB 1998. Convergence of progesterone with growth factor and cytokine signaling in breast cancer. Progesterone receptors regulate signal transducers and activators of transcription expression and activity. Journal of Biological Chemistry 273 31317–31326. [DOI] [PubMed] [Google Scholar]
- Rui H, Kirken RA & Farrar WL 1994. Activation of receptor-associated tyrosine kinase JAK2 by prolactin. Journal of Biological Chemistry 269 5364–5368. [PubMed] [Google Scholar]
- Salatino M, Beguelin W, Peters MG, Carnevale R, Proietti CJ, Galigniana MD, Vedoy CG, Schillaci R, Charreau EH, Sogayar MC et al. 2006. Progestin-induced caveolin-1 expression mediates breast cancer cell proliferation. Oncogene 25 7723–7739. [DOI] [PubMed] [Google Scholar]
- Soule HD, Maloney TM, Wolman SR, Peterson WD Jr, Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF & Brooks SC 1990. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Research 50 6075–6086. [PubMed] [Google Scholar]
- Srivastava S, Matsuda M, Hou Z, Bailey JP, Kitazawa R, Herbst MP & Horseman ND 2003. Receptor activator of NF-kappaB ligand induction via Jak2 and Stat5a in mammary epithelial cells. Journal of Biological Chemistry 278 46171–46178. [DOI] [PubMed] [Google Scholar]
- Stegalkina SS, Guerrero A, Walton KD, Liu X, Robinson GW & Hennighausen L 1999. Transcription originating in the long terminal repeats of the endogenous mouse mammary tumor virus MTV-3 is activated in Stat5a-null mice and picks up hitchhiking exons. Journal of Virology 73 8669–8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoecklin E, Wissler M, Schaetzle D, Pfitzner E & Groner B 1999. Interactions in the transcriptional regulation exerted by Stat5 and by members of the steroid hormone receptor family. Journal of Steroid Biochemistry and Molecular Biology 69 195–204. [DOI] [PubMed] [Google Scholar]
- Travers PJ, Arklie JL, Trowsdale J, Patillo RA & Bodmer WF 1982. Lack of expression of HLA-ABC antigens in choriocarcinoma and other human tumor cell lines. National Cancer Institute Monographs 60 175–180. [PubMed] [Google Scholar]
- Trott JF, Hovey RC, Koduri S & Vonderhaar BK 2003. Alternative splicing to exon 11 of human prolactin receptor gene results in multiple isoforms including a secreted prolactin binding protein. Journal of Molecular Endocrinology 30 31–47. [DOI] [PubMed] [Google Scholar]
- Wagner KU, McAllister K, Ward T, Davis B, Wiseman R & Hennighausen L 2001. Spatial and temporal expression of the Cre gene under the control of the MMTV-LTR in different lines of transgenic mice. Transgenic Research 10 545–553. [DOI] [PubMed] [Google Scholar]
- Wang Y, Holland JF, Bleiweiss IJ, Melana S, Liu X, Pelisson I, Cantarella A, Stellrecht K, Mani S & Pogo BG 1995. Detection of mammary tumor virus env gene-like sequences in human breast cancer. Cancer Research 55 5173–5179. [PubMed] [Google Scholar]
- Wang Y, Jiang JD, Xu D, Li Y, Qu C, Holland JF & Pogo BG 2004. A mouse mammary tumor virus-like long terminal repeat superantigen in human breast cancer. Cancer Research, 64 4105–4111. [DOI] [PubMed] [Google Scholar]
- Wang J, Paradis P, Aries A, Komati H, Lefebvre C, Wang H & Nemer M 2005. Convergence of protein kinase C and JAK-STAT signaling on transcription factor GATA-4. Molecular and Cellular Biology 25 9829–9844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westley B & Rochefort H 1980. A secreted glycoprotein induced by estrogen in human breast cancer cell lines. Cell 20 353–362. [DOI] [PubMed] [Google Scholar]
- Williams CJ, Major PP & Dion AS 1990. Enhanced expression and secretion of an epithelial membrane antigen (MA5) in a human mucinous breast tumor line (BT549). Tumour Biology 11 145–157. [DOI] [PubMed] [Google Scholar]
- Yanagawa S, Tanaka H & Ishimoto A 1991. Identification of a novel mammary cell line-specific enhancer element in the long terminal repeat of mouse mammary tumor virus, which interacts with its hormone-responsive element. Journal of Virology 65 526–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Turkson J, Carter-Su C, Smithgall T, Levitzki A, Kraker A, Krolewski JJ, Medveczky P & Jove R 2000. Activation of Stat3 in v-Src-transformed fibroblasts requires cooperation of Jak1 kinase activity. Journal of Biological Chemistry 275 24935–24944. [DOI] [PubMed] [Google Scholar]