

Abstract
Adaptive designs are gaining popularity in early phase clinical trials because they enable investigators to change the course of a study in response to accumulating data. We propose a novel design to simultaneously monitor several endpoints. These include efficacy, futility, toxicity and other outcomes in early phase, single-arm studies. We construct a recursive relationship to compute the exact probabilities of stopping for any combination of endpoints without the need for simulation, given pre-specified decision rules. The proposed design is flexible in the number and timing of interim analyses. A R Shiny app with user-friendly web interface has been created to facilitate the implementation of the proposed design.
Keywords: Go/no go decisions, multiple endpoints, biomarker endpoints, conditional power, early phase
1. Introduction
Clinical studies to evaluate the toxicity and efficacy of a novel treatment are normally conducted in separate phases. Conventionally, Phase I trials are first-in-human studies aimed at identifying the maximum tolerated dose (MTD) of an experimental agent. In a subsequent Phase II trial, the candidate drug will be evaluated at the MTD to determine whether it has sufficient therapeutic activity to warrant further testing. The majority of Phase II clinical studies are designed as single-arm trials without a control group and are commonly conducted following a two-stage process.1
The sample sizes of most Phase I trials are too small to allow accurate identification of MTD, so patients in Phase II trials might be exposed to sub-therapeutic doses or overly toxic doses resulting in excessive serious adverse events (SAEs). To overcome these issues, Phase I dose-expansion cohorts (DECs) are now frequently used to assess preliminary efficacy and to further characterize toxicity. In the development of immune checkpoint blockade, for example, the use of DECs serves to generate a continuum of the drug development process, blurring the distinctions between Phase I dose finding, Phase II proof of concept and Phase III comparative efficacy trials.2,3 Despite its popularity, the majority of DECs are designed without sample size justification or treated as single arm, Phase II trials without statistically accounting for toxicities.4 Boonstra et al. state: “Regulatory agencies and others have expressed concern about the uncritical use of dose expansion cohorts (DECs) in Phase I oncology trials.”5
Consider a single-arm study to evaluate the efficacy of a novel treatment. We are interested in testing if the response rate to this new treatment is higher than the response rate in historical controls. Hereafter, we refer to this study as our prototype. Based on the commonly used Simon two-stage design, our prototype trial might be conducted in two stages, with the option to terminate accrual after the first stage if the number of responses is below the futility bound. Various extensions to the Simon two-stage design have been developed.6–11 Notably, Mander and Thompson investigated designs optimal under the alternative hypothesis and constructed two-stage designs with the flexibility to stop early for efficacy, which leads to substantial reductions in expected sample size.8 Bryant and Day proposed a two-stage, two-endpoint design by integrating safety considerations into the early stopping rule of Phase II trials.7 Based on curtailed sampling procedures, Chen and Chi introduced two-stage designs with two dependent binary endpoints and demonstrated the effectiveness of their method in reducing the expected sample size when the treatment lacks efficacy or is too toxic.9 Li et al. proposed new two-stage designs including provisions for early termination due to sufficient effectiveness and safety, ineffectiveness and toxicity.10
When the outcome of interest can be quickly observed, continuous monitoring designs are particularly useful. With the use of simulated annealing method, Chen and Lee constructed optimal stopping boundaries for the continuous-monitoring of futility.12 Law et al. proposed curtailed designs allowing a trial to be terminated early for efficacy or futility after evaluating the response of every patient.13 Ivanova et al. constructed a Pocock-type boundary to continuously monitor toxicity in Phase II clinical trials.14 The use of the sequential probability ratio test (SPRT) is not appropriate in this context because the open-ended nature of SPRT.15 The majority of continuous monitoring designs are proposed in a Bayesian framework based on the posterior or the predictive probability of exceeding the historical response rate.16–18 A number of Bayesian designs have been proposed to consider both toxicity and response to treatment in Phase II setting based on the Dirichlet-Multinomial Model.19–22 Sambucini23 developed predictive probability rules for monitoring bivariate binary outcomes; Teramukai et al.24 proposed a Bayesian adaptive design allowing futility stopping with continuous safety monitoring; Zhong et al.25 applied a copula model to describe the joint probability of efficacy and toxicity. These Bayesian designs rely on extensive simulations to search the optimal design parameters and determine trial operating characteristics. Implementing these methods is also challenging because statisticians need to be informed in real time when new data become available. Complicated computations are often required to provide updated estimates on efficacy or toxicity, and user-friendly software for these previous work are often lacking.
Schulz et al. proposed a recursive formula to calculate the exact probability of accepting or rejecting a null hypothesis for multiple stage designs with a binary endpoint26 and similar approaches have been adopted by some of the previously cited continuous monitoring designs.12,13 In this work, we extend the univariate recursive relationship to calculate the exact probabilities of making go/no go decisions in single-arm clinical trials with multiple binary valued endpoints. Based on the extended recursive relationship, we propose the unified exact design, which provides a unified statistical framework for making futility, efficacy and/or toxicity stopping decisions in early phase clinical trials with mutiple-stage or continuous monitoring design. The proposed design provides transparent decision rules and is easy to implement because the cut-off values of stopping for toxicity, efficacy, or futility are spelled out a priori without the need to call upon statistical support mid-trial. Barring any of these endpoints, we continue to enroll patients until a maximum sample size is reached. Unlike the previously cited Bayesian methods, which rely on simulation to determine the operating characteristics of their designs, we can calculate the exact frequentist probability of continuing or stopping a trial for any combination of these causes.
The remainder of this paper is organized as follows. We introduce the recursive relationship in Section 2.1 and construct efficacy and futility stopping bounds in Section 2.2. We extend the recursive relationship to accommodate multiple binary endpoints in Section 2.3. In Section 2.4, we describe the identification of design parameters. We apply the unified exact design to our prototype in Section 3 and discuss the properties of the proposed design in Section 4. In Section 5, we present a Shiny application to facilitate the implementation of the proposed design.
2. Method
2.1. The recursive relationship
We denote by in the number of responses out the first n patients enrolled, where n = 1, 2, …, N and N is the maximum sample size. Conditional on the unknown response rate p, each in has a binomial distribution with parameters n and p in the absence of interim monitoring. We will stop the trial and declare the new treatment worthy of further investigation the first value of n for which in exceeds a pre-specified efficacy cut-off value en. A clinical study can also be terminated for futility. We stop a trial early for futility if the number of responses in observed in the first n patients is less than fn, the pre-specified futility bound. Similar to Bayesian designs where the stopping bounds is often expressed as the lower and upper quantiles of a beta distribution for response rate, here we define the bounds fn and en based on the lower and upper quantiles of the corresponding binomial distribution of in, respectively.
After enrolling and evaluating the first n patients, we can choose to stop for efficacy if the number of responses exceeds en or stop for futility if the number of responses falls below fn. That is, we continue to accrue if fn ≤ in ≤ en. Let θn(in) denote the probability of continuing the study after observing in = fn, …, en responses among the first n patients. We define θn(in) = 0 for other values of in. We define θ0(0) = 1 before any patients are enrolled.
According to Schultz et al.,26 the recursive relationship relating θn to θn+1 is given by
| (1) |
for fn+1 ≤ in+1 ≤ en+1 and zero otherwise. The support for in+1 in equation (1) is different from that of a binomial distribution. To enroll one additional patient after having already observed n patients, the number of responses in these n patients cannot exceed en and cannot fall below fn.
The marginal probability of continuing the trial after observing the n-th patient, θn, is then
2.2. Monitoring efficacy and futility
Consider a study design with pre-specified stopping rules for efficacy (en) and futility (fn). These decision rules can be described as follows:
After enrolling and evaluating n = 1, …, N − 1 patients, and having not stopped the trial earlier,
if in > en, then stop the trial and declare the treatment promising,
if in < fn, then stop the trial for futility,
otherwise, continue to enroll the n + 1-th patient.
After evaluating all the N patients, and having not stopped the trial earlier,
if iN > eN, then reject the null hypothesis,
if iN ≤ eN, then declare the treatment is not worthy of further investigation.
Denote PSEn(p) the conditional probability of stopping for efficacy with the next patient after the n-th patient has been evaluated, given the trial has not been stopped earlier and the true probability of response is p. Then we have
Let PSFn(p) be the conditional probability of stopping for futility with the next patient after the n-th patient has been evaluated, given the trial has not been stopped earlier. Then we have
Figure 1 shows an example of the efficacy and futility stopping bounds. The decision rules sometimes prevent us from stopping at certain points in the trial, regardless of the most recent outcome. Specifically, if en > en−1 and we do not stop after the n − 1-th patient, then it is impossible to stop after the n-th patient regardless of the outcome of that patient. Similarly, it is impossible to stop the trial at the n-th patient for futility if fn = fn−1, regardless of the outcomes of the next patient.
Figure 1.
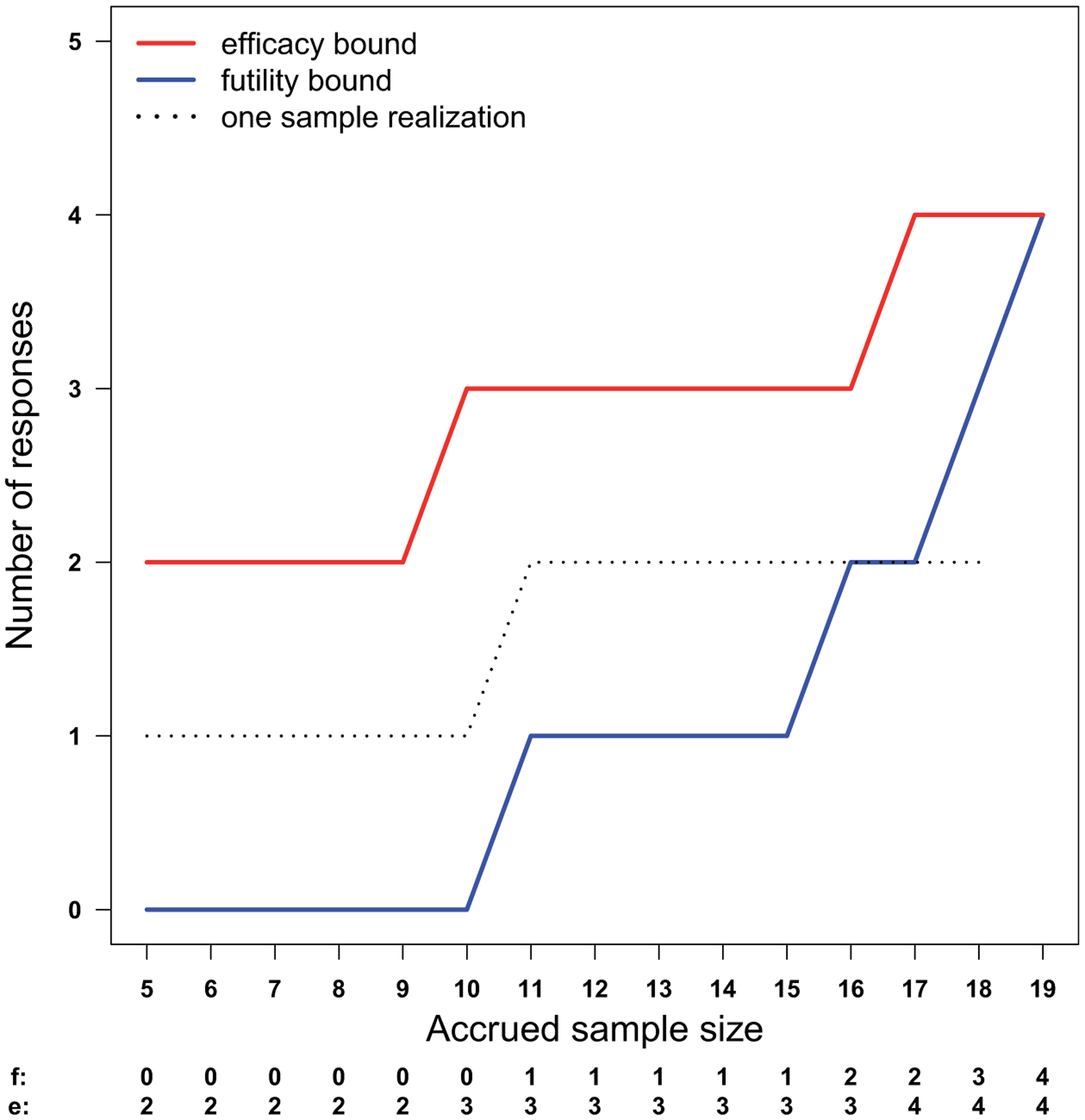
Efficacy and futility stopping bounds for the prototype and a sample path. A study will be stopped for efficacy or futility if the number of responses is above the efficacy bound (e) or below the futility bound (f). The sample path (dotted line) crosses the futility bound when two out of the first 18 patients respond to the treatment, leading to a futility decision. Under the null hypothesis, the response rate is 10%. Under the alternative, the response rate is 35%. This design achieved 90.1% power at a significance level of 0.093.
Denote p0 as the historic, or standard of care response rate. We are interested in testing
with a power of at least 1 − β for all p ≥ p1, where p1 represents a target response rate of interest (p1 > p0). Based on our efficacy and futility stopping rules, the trial-wide statistical significance level is
| (2) |
the trial-wide type II error rate is
| (3) |
and the expected sample size is
| (4) |
where PSn(p) = PSEn(p) + PSFn(p).
Based on the recursive relationship, we can calculate the interim conditional power of the study, which is the probability of ever stopping for efficacy after observing x responses out of m patients. We define conditional power as
where θm(x) = 1 if fm ≤ x ≤ em. We define CP = 1 if x > em and CP = 0 if x < fm.
We suggest choosing stopping bounds e = (e1, …, eN)T and f = (f1, …, fN)T based on an upper or lower quantile of the binomial distribution. The selection of suitable stopping bounds can be accomplished with the use of a grid search to explore the possible combinations of these quantiles. Among all the pairs of vectors e and f defined by quantiles on the grid, we select the set (e, f) minimizing μ(p0) as the efficacy and futility stopping boundaries for our design, subject to the pre-specified constraints on α(p0), β(p1) and N. We consider N fixed, which is determined by accrual rate, budget and other logistic factors. In the case when N is too small, we will need to gradually increase N until a solution is found.
Let us give an example of setting efficacy and futility monitoring bounds based on our prototype. Consider p0 = 0.10 and p1 = 0.35. Figure 1 shows the stopping bounds (e and f) and one realized sample path. The efficacy cutoff values en are the 93 percentiles of the binomial (n, p0) distribution. We stop for futility if the number of responses in out of n patients is less than the 1.1 percentile of the corresponding binomial (n, p1) distribution, or if it is impossible to reach the efficacy bound, i.e. in < n + en − N. The futility bound fn is the maximum of these two values.
The cumulative probabilities of stopping for efficacy and futility using these bounds are shown in Figure 2. The trial-wide probability of ever crossing the efficacy bound is 0.0932 under p0. This is the statistical significance of the design. With a maximum sample size of 19, this design yields 90.12% power if the true response rate is 35%. Continuous monitoring is conducted after five patients have been accrued and evaluated. The futility cutoff values fn of this design is 0 for the first 10 patients, that is, futility stopping is only possible when the outcome for the 11th patient becomes available. Under the null hypothesis, the probability of stopping for futility is 0.3138 when the interim sample size is 11 and 0.5403, when the interim sample size is 16. On the other hand, the proposed design allows earlier decisions on efficacy. If the true response rate is 35%, then the probability of stopping the treatment for efficacy after enrolling nine patients is 0.6627.
Figure 2.
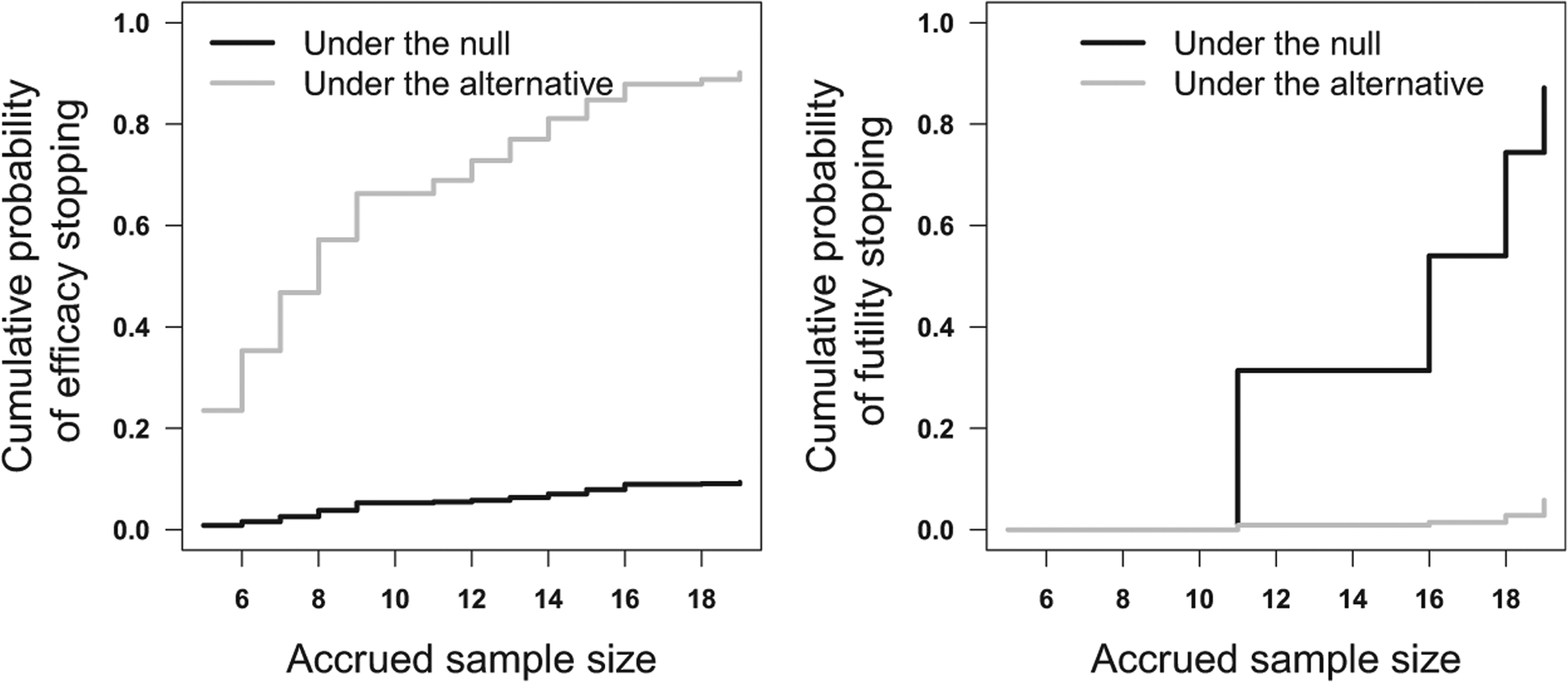
The cumulative probabilities of stopping for efficacy (left panel) and futility (right panel). Under the null hypothesis, the response rate is 10%. Under the alternative, the response rate is 35%. This design achieved 90.1% power at a significance level of 0.093.
Early stopping for efficacy is not always preferred in early phase clinical trials, so the proposed design provides the option to ignore early efficacy stopping by specifying en = n for n = 1, …, N − 1.
2.3. The joint monitoring of efficacy, futility and toxicity
In addition to the binary indicator for response, let us assume each patient has a binary valued indication of a SAE. Let π = (π00, π01, π10, π11) denote the probability of a patient in the following categories: no response and no SAE, no response but have SAE, response without SAE, response with SAE. The probability of each category is shown in Table 1. Let y = (y00, y01, y10, y11) be the multinomial distributed random variable with index 1 and probability vector π representing the outcomes for the next patient.
Table 1.
The probability π of a patient with and without response and/or SAE.
| Without SAEs | With SAEs | Marginal | |
|---|---|---|---|
| No Response | π 00 | π 01 | 1 − p |
| Response | π 10 | π 11 | p |
| Marginal | 1 − q | q | 1 |
Toxicity and efficacy are not generally independent.27 In Immuno-oncology trials, for example, we can expect to see the patient developing a rash as a sign the treatment is taking effect. Define the efficacy-toxicity odds ratio as
Define q as the marginal probability of a patient developing a SAE. The joint probability π can be calculated given the marginal probabilities p = π10 + π11 and q = π01 + π11 and a pre-specified λ based on the global cross-ratio model.28
Let jn be the number of patients with SAEs after n patients have been accrued and evaluated. Let bn denote the toxicity bound, which is the maximum number of SAE’s we are willing to tolerate among the first n patients and continue to accrue to the trial. Let θn(in, jn) denote the probability of continuing to accrue after observing in responses and jn SAEs following the outcomes of the first n patients, for in = fn, …, en responses and jn = 0, …, bn SAEs. For other values of in and jn, we define θn(in, jn) = 0. Then we can write
| (5) |
where Multi is the multinomial probability density function, in+1 − in = y10 + y11 and jn+1 − jn = y01 + y11.
Intuitively the sequences en, fn, bn are non-decreasing.
After observing the outcomes of each patient, we can take one of four actions: stop for toxicity, stop for efficacy, stop for futility, or continue to accrue based on observed data up to the maximum sample size N. Specifically, a study proceeds as follows according to our design:
After n = 1, …, N − 1 patients have been enrolled and evaluated, and having not stopped the trial earlier,
if jn > bn, then terminate accrual for excessive SAEs,
if jn ≤ bn and in < fn, then stop the trial for futility,
if jn ≤ bn and in > en, then stop the trial for efficacy, otherwise, continue to the next stage.
After all the N patients have been evaluated, and having not stopped the trial earlier,
if jN > bN, then conclude the treatment is too toxic,
if jN ≤ bN and iN ≤ eN, then conclude the treatment is not promising, but not toxic,
if jN ≤ bN and iN > eN, then conclude the treatment is worthy of further investigation.
Given the pre-specified decision rules, the conditional probability of stopping the trial for toxicity after n patients have been enrolled and evaluated, given the trial has not been stopped earlier, is
for jn−1 = 0, …, bn−1, in−1 = fn−1, …, en−1.
Likewise, the conditional probability of stopping for efficacy after n patients have been enrolled and evaluated, given the trial has not been stopped earlier, is
and the conditional probability of stopping the trial for futility after n patients have been enrolled and evaluated, given the trial has not been stopped earlier, is
where in − in−1 = y10 + y11 and jn − jn−1 = y01 + y11.
2.4. Selection of design parameters
Bryant and Day considered different approaches to determine the design parameters for the two-stage two-end-point design and recommended the approach assuming independence (λ = 1) between response and safety. This approach is preferred for practical use as it maintains desirable operating characteristics and is easy to convey to investigators.7 Chen and Chi also demonstrated that different levels of response-safety correlation have little effect on the selection of design parameters.9 Based on these work, we assume λ = 1 and find suitable stopping boundaries for monitoring response and safety separately using the marginal probability of response and safety.
Let q0 denote the maximum toxicity rate we are willing to tolerate in early phase trials. The value of q0 can be set based on the maximum-tolerated toxicity rate in Phase I trials or chosen to satisfy specific regulatory requirement. We define the safety stopping bound bn based on the upper quantiles of the binomial (n, q0) distribution. Denote γ(q0) as the probability of falsely stopping for safety when q = q0, which can be calculated based on the recursive formula given by equation (1). Unlike previous work,7,9,11 which aimed to establish safety by formally testing an acceptable versus an unacceptable toxicity rate, our objective is to safeguard against excessively toxic agents by controlling the false alarm rate γ(q0) without overly inflating the maximum sample size (N) of a trial. Similar to Ivanova et al.,14 we accomplish this objective by choosing values of bn such that γ(q0) is as close to the pre-specified value (e.g. 0.1) as possible, but not exceeding it. Figure 3 shows an example of the safety stopping boundary and the cumulative probabilities of safety stopping associated with this boundary, assuming q0 = 0.10 and γ(q0) = 0.10.
Figure 3.
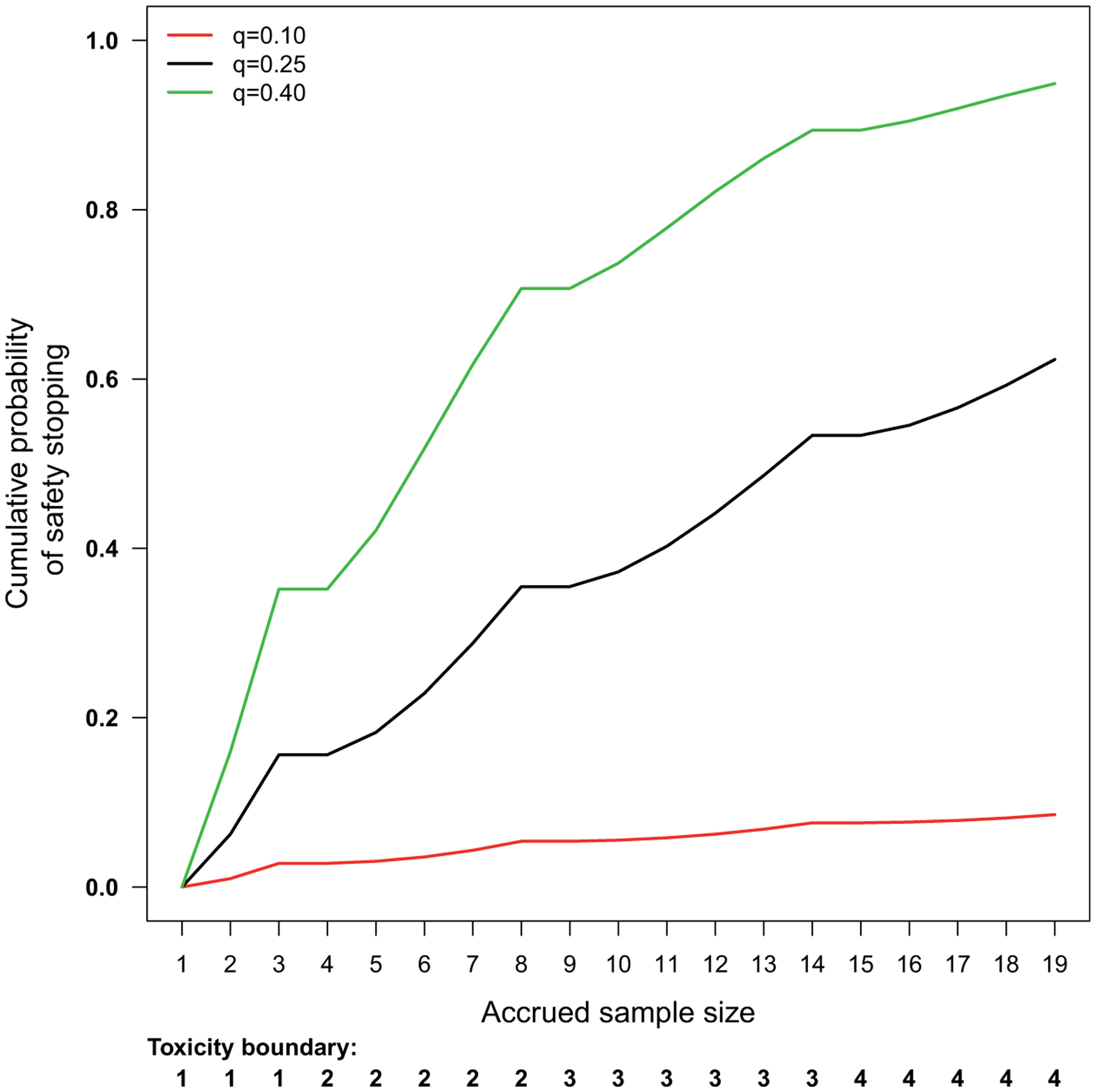
The toxicity bound and the cumulative probability of safety stopping for the prototype. The trial-wide probability of safety stopping is 0.0855, 0.6231 and 0.9489, assuming the true probability of a patient developing SAEs is 0.10, 0.25, and 0.40, respectively. We stop for safety if the number of patients with SAEs exceeds the toxicity bound. The highest SAE rate considered safe is 0.10 and the false alarm rate we are willing to tolerate is 0.10.
The efficacy and futility stopping bounds (en, fn) are determined as described in Section 2.2 by minimizing the expected sample size under H0 : p ≤ p0 subject the pre-specified constraints on the marginal error rates α(p0) and β(p1).
Given a specific response-toxicity odds ratio λ, the trial-wide type I error rate and false alarm rate considering efficacy, futility and toxicity stopping are defined as
| (6) |
and
| (7) |
The statistical power of a study is given by
| (8) |
Note the false alarm rate γ(p, q0, λ) is less than the marginal false alarm rate γ(q0) in those trials which would be stopped for efficacy or futility, for p > 0. Likewise, there is
for q > 0. The statistical power, that is, α(p1, q, λ) of a study will also be reduced due to the competition of safety stopping and setting γ(q0) to a small value (0.05 or 0.10) helps to maintain power at an acceptable level.
Yin et al. developed two-stage and multiple-stage designs by identifying the parameter values at which the maximum type I error rate and the minimum power are achieved when safety and response rate are jointly tested.11 In practice, implementation of this approach is difficult due to the intensive computations required to find the stopping boundaries. As detailed in this section, the approach proposed here sacrifices statistical sophistication for practical advantage such that the joint monitoring of response and safety can be implemented in studies with limited sample sizes while reducing the computational complexity in boundary identification.
2.5. Generalize to multiple endpoints
The joint monitoring of multiple endpoints is of special interest in certain disease areas. Monitoring multiple endpoints allows clinical trialists to comprehensively capture complex efficacy or toxicity profiles when a single binary endpoint is not adequate to account for the complexities of outcomes. Consider a clinical trial of prostate cancer with multiple binary endpoints including objective response per RECIST (OR), SAE, and reduction in Prostate-specific antigen (PSA). PSA is a widely used biomarker of tumor burden for patients with prostate cancer. PSA response (PR) is defined as at least 50% reduction in the level of PSA and is known to be a good predictor of overall survival.29 We will use this hypothetical prostate cancer trial to illustrate how to jointly monitor multiple endpoints.
Denote y = (y000, y010, …, y111) as the random variable with eight different categories, representing all the possible combinations of OR (yes/no), SAE(yes/no) and PR(yes/no). For example, y000, y010, y100 and y110 represent the following categories: no response and no SAE, no response but have SAE, response without SAE, response with SAE, all in the absence of PSA response. We assume y follows a multinomial distribution
where π = (π000, π010, …, π111) denotes the probability of each combinational category.
Denote θn the probability of continuing the study after observing in ORs, jn SAEs and kn PRs after n patients have been accrued and evaluated, for in = fn, …, en, jn = 0, …, bn and kn = ln, …, un, with ln and un denoting the lower and upper bound of PSA responses. We define θn = 0 for other values of in, jn and kn. Then the recursive relationship for these endpoints is given by
| (9) |
where , , and are the marginal sums of y.
The conditional probability of stopping for safety after n patients have been enrolled and evaluated, given the trial has not been stopped earlier, is given by
The conditional probability of stopping for efficacy or futility after n patients have been enrolled and evaluated, given the trial has not been stopped earlier, is given by
and
if jn ≤ bn and kn = ln, …, un
Similarly, we can calculate the conditional probability of stopping for PSA responses when its upper (un) or lower bound (ln) is crossed, given the trial has not been stopped earlier. Specifically, we can write
and
if jn ≤ bn and in = fn, …, en.
The expansion from (1) to (9) demonstrates how we can accommodate an arbitrary number of endpoints and their combinations for early stopping. The recursive relationship can be further generalized to allow interim analyses at pre-specified intervals. Let ng denote the number of patients enrolled and evaluated up to stage g. The recursive formula relating the g + 1-th stage of the trial to the g-th stage is
| (10) |
where y~Multi(ng+1 − ng, π), with the constrains , , and .
We would like to test the following hypothesis on PR
with at least 1 − β(pPR,1) power for pPR ≥ pPR,1, where pPR,1 is a promising PSA response rate worthy of further investigation (pPR,1 > pPR,0).
The stopping boundaries for PR are determined in a similar manner as the stopping boundaries for response rate. Specifically, we find the suitable upper (un) and lower (ln) bound for PR stopping subject to the constraints on the marginal error rates α(pPR,0) and β(pPR,1), while minimizing the expected sample size under the null HPR,0. Due to the inclusion of PR stopping, the amount of significance level allocated to objective response per RECIST and PSA response rate need to be carefully adjusted based on the clinical context to maintain the trial-wide significance level.
3. Application
In this section, we retrospectively apply the unified exact design to a Phase II study of Olaparib proposed at our institution. This study aims at evaluating the response rate to Olaparib in patients with acute myeloid leukemia carrying isocitrate dehydrogenase mutations. The MTD has been determined in a Phase I trial. Previous experience suggests the response rate for patients with these tumors is approximately 10%. We are interested in testing
with at least 90% power for all p ≥ 0.35. This trial was originally designed using an optimal Simon two-stage design. Initially, 11 patients will be enrolled. If no more than one response is observed, the expansion cohort will be terminated early. If two or more patients respond, an additional eight patients will be enrolled for a total of 19 patients. The null hypothesis will be rejected if four or more responses are observed in these 19 patients. This design yields 90.86% power at an α level of 0.0988, with 0.6974 probability of early negative stopping and an expected sample size of 13.4 under the null. This design can be expressed using our notation with e11 = 11, f11 = 2 for the first stage and e19 = 3, f19 = 3 for the second stage.
The proposed design has the flexibility to include efficacy stopping. Consider a design with interim analysis planed after the first 5, 10, 15, and 20 patients have been accrued and evaluated. The corresponding efficacy bound is e = (2, 2, 3, 4) and the corresponding futility bound is f = (0, 1, 3, 4). We provide the cumulative probabilities of stopping for efficacy and futility assuming different response rates in Table 2. With a maximum sample size of 20, the prototype trial has 90.11% power at a one-sided significance level of 0.0968. Assuming a true response rate of 35%, the probability of stopping the treatment for efficacy after enrolling 10 patients is 0.7384.
Table 2.
The cumulative probabilities of efficacy (CPE) and futility (CPF) stopping for our prototype using the unified exact design.
| p = 0.10 | p = 0.20 | p = 0.30 | p = 0.35 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | f n | e n | CPF | CPE | CPF | CPE | CPF | CPE | CPF | CPE |
| 5 | 0 | 2 | 0.0000 | 0.0086 | 0.0000 | 0.0579 | 0.0000 | 0.1631 | 0.0000 | 0.2352 |
| 10 | 1 | 2 | 0.3487 | 0.0702 | 0.1074 | 0.3222 | 0.0282 | 0.6172 | 0.0135 | 0.7384 |
| 15 | 3 | 3 | 0.8189 | 0.0893 | 0.4042 | 0.4171 | 0.1314 | 0.7471 | 0.0649 | 0.8558 |
| 20 | 4 | 4 | 0.8731 | 0.0968 | 0.4628 | 0.4640 | 0.1518 | 0.8044 | 0.0741 | 0.9011 |
Note: In the prototype, we aim to test if the response rate to a novel agent is greater than 10%, assuming a target response rate of 35%. Interim analyses are conducted after 5, 10, 15, and 20 patients have been accrued and evaluated. For each interim sample size n, en is the maximum number of treatment successes without stopping for efficacy; and fn is the minimum number of treatment successes without stopping for futility. The true probability of response rate is assumed to be p = 0.10, p = 0.20, p = 0.30 and p = 0.35.
Consider the continuous monitoring of efficacy and futility after enrolling five patients, and the continuous monitoring of toxicity outcomes for all the patients, with a maximum sample size of 19. To implement a unified exact design for this trial, we specify
The efficacy and futility cutoff values for this design are labeled in Figure 1. To include safety monitoring, we consider q0 = 0.10. That is, the treatment is considered overly toxic if the SAE rate is above 10% and the false alarm rate of stopping for safety when the treatment is safe (q ≤ q0) is maintained at 0.10. In this example, we assume an efficacy–toxicity odds ratio of 1.5, corresponding to a weak positive correlation coefficient of approximately 0.10. A weak correlation of this magnitude has been suggested by studies of targeted agents30 and adopted by other studies.31 The safety cutoff values for this study are labeled in Figure 3. The unified exact design allows users to specify other levels of correlations as well as independence.
This trial can be stopped as early as after two patients are accrued, if the first two patients both develop SAEs. We can also choose to stop this trial if more than two of the first five patients respond to the treatment. We continue to accrue if the number of SAEs is below the toxicity bound, and the number of responses is between the efficacy and the futility bound. If the trial reaches its planned maximum sample size of 19 patients, we conclude the treatment is promising if at least five patients have responded and at most four patients have SAEs.
The probabilities of early stopping using continuous monitoring under different scenarios are shown in Figure 4. Scenarios A and C represent cases when the treatment is safe, whereas scenarios B and D correspond to situations when the treatment is too toxic. The trial-wide probability of safety stopping is less than 0.10 in both scenario A (q = 0.10, p = 0.10) and C (q = 0.10, p = 0.35). When the treatment if safe but not efficacious (Figure 4(a)), the trial-wide probability of stopping for futility and efficacy is 0.8091 and 0.0868, respectively. The trial-wide probability of efficacy stopping is 0.8577 if the treatment is safe and efficacious (Figure 4(c)). When the treatment is overly toxic, the trial-wide probabilities of stopping for toxicity are 0.8608 and 0.6781 in B (q = 0.40, p = 0.10) and D (q = 0.40, p = 0.35), respectively. The trial-wide probabilities of stopping for efficacy are 0.0210 and 0.3126 under scenario B and D, respectively. Table 3 shows the operating characteristics of this design, when interim analysis is planed after 5, 10, 15, and 20 patients have been accrued and evaluated.
Figure 4.
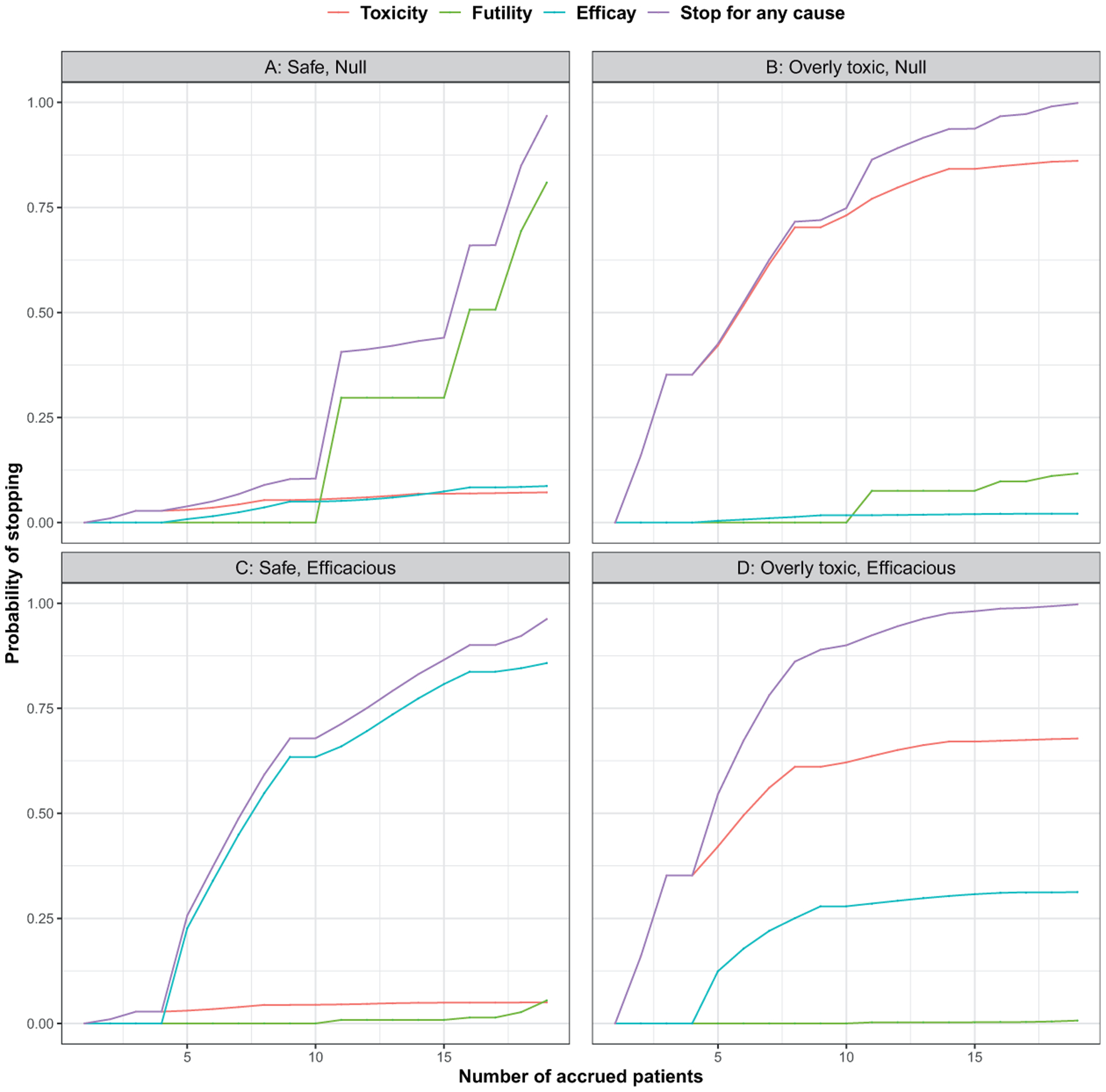
The cumulative probabilities of stopping the prototype for efficacy, futility, toxicity, or any cause in different scenarios. (a) and (c) represent treatment with acceptable toxicity profiles, assuming the probability of SAE is 0.10, whereas (b) and (d) represent treatment with unacceptable toxicity levels, assuming the probability of SAE is 0.40. The probability of response is 0.10 and 0.35 under the null and efficacious scenarios, respectively.
Table 3.
The cumulative probabilities of efficacy (CPE), futility (CPF), and toxicity (CPT) stopping for our prototype using the unified exact design.
| A : q = 0.10, p = 0.10 | B : q = 0.40, p = 0.10 | C : q = 0.10, p = 0.35 | D : q = 0.40, p = 0.35 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | f n | e n | b n | CPF | CPE | CPT | CPF | CPE | CPT | CPF | CPE | CPT | CPF | CPE | CPT |
| 5 | 0 | 2 | 2 | 0.0000 | 0.0084 | 0.0086 | 0.0000 | 0.0051 | 0.3174 | 0.0000 | 0.2325 | 0.0086 | 0.0000 | 0.1484 | 0.3174 |
| 10 | 1 | 2 | 2 | 0.3266 | 0.0648 | 0.0696 | 0.0641 | 0.0133 | 0.8287 | 0.0128 | 0.6996 | 0.0540 | 0.0031 | 0.2299 | 0.7182 |
| 15 | 3 | 3 | 3 | 0.7543 | 0.0819 | 0.0809 | 0.0984 | 0.0144 | 0.8814 | 0.0606 | 0.8076 | 0.0585 | 0.0082 | 0.2394 | 0.7452 |
| 20 | 4 | 4 | 4 | 0.8027 | 0.0886 | 0.0819 | 0.1001 | 0.0146 | 0.8844 | 0.0690 | 0.8490 | 0.0593 | 0.0087 | 0.2413 | 0.7489 |
Note: The prototype tests if the response rate to a novel agent is greater than 10%, assuming a target response rate of 35%. Any SAE rate greater than 10% is considered overly toxic. For each interim sample size n, en is the maximum number of treatment successes without stopping for efficacy; and fn is the minimum number of treatment successes without stopping for futility; bn is the maximum number of SAEs without stopping for toxicity. The CPF, CPE and CPT after n patients have been evaluated are calculated in different scenarios, where p and q denote the marginal probability of response and SAE, respectively.
4. Discussion
In this paper, we proposed the unified exact design to simultaneously monitor the efficacy, futility and toxicity outcomes of a single-arm clinical trial. We developed a recursive relationship to calculate the exact probabilities of stopping for any combinations of these outcomes. Compared to the Simon two-stage design, which only allows one futility check and has no provisions for safety monitoring, the unified exact design provides the flexibility to stop the trial early for all the necessary causes. Although we choose stopping bounds based on the tail areas of a binomial distribution in this paper, Bayesian methods can also be used to set the stopping bounds. It is beyond the scope of this paper to discuss other types of stopping bounds; however, it should be noted that the proposed recursive method is valid as long as the stopping bounds is a non-decreasing function of sample size. Our future work will focus on computationally efficient methods to optimize the stopping bounds.
When results from previous studies are available, we can specify the joint probability of efficacy, futility and toxicity based on historical data. Given a correlation structure, it is possible to find stopping bounds producing smaller expected sample sizes compared to the stopping bounds found assuming the independence between response and toxicity. However, these data are not always available and the performance of the unified exact design can be negatively affected if the toxicity-efficacy relationship is mis-specified. To deal with this problem, an adaptive two-stage design similar to Zang and Lee31 can be used to learn the efficacy–toxicity relationship in the first stage, assuming independence of these outcomes, and jointly model efficacy toxicity outcomes in the next stage.
The unified exact design allows the stopping boundaries to be completely specified prior to the start of a trial, saving the need for complex computations in the midst of a trial. All possible decision rules can be tabulated, which helps to convey the statistical design to trial practitioners. Continuous monitoring provides clinical trialists the advantage of altering the course of a trial in response to real time data; however, continuous monitoring is difficult to implement if the outcome of interest is not quickly available. The proposed method is flexible in the number and timing of interim analyses, with the flexibility to allow for multiple stage design as well as continuous monitoring design.
5. Software implementation
To provide a more accessible user-interface, we created a web application using Shiny, which can be accessed at https://weiwei-study-design.shinyapps.io/unified_exact_design/. The web interface of our design gives users the option to choose the types of early stopping (efficacy, futility, toxicity, or their combinations), as well as the timing and number of interim looks expressed in terms of interim sample sizes. To monitor response to treatment, users need to specify α, β, p0 and p1. To include safety monitoring, users need to provide the values for the efficacy toxicity odds ratio (λ), the highest toxicity level consider safe (q0) and the chance of falsely stopping for safety (γ) when treatment is safe. After users input their design parameters, the Shiny app generates the stopping bounds and the operating characteristics of the design. A recommended write up summarizing the statistical design will also be generated for use in a protocol.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health, CTSA Grant Number UL1TR001863; National Institutes of Health Grant Number P30-CA16359, P50-CA196530,P50-CA121974,R01-CA177719,R01-ES005775, R01-CA223481, R41-A120546, U48-DP005023, U01-CA235747, R35CA197574, R01-CA168733, UM1 CA186689, P50 DE030707-01, P50 CA121974, and a Pilot Grant from Yale Cancer Center.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Rubinstein L Phase II design: history and evolution. Chin Clin Oncol 2014; 3: 48. [DOI] [PubMed] [Google Scholar]
- 2.Iasonos A and O’Quigley J. Design considerations for dose-expansion cohorts in phase I trials. J Clin Oncol 2013; 31: 4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Theoret MR, Pai-Scherf LH, Chuk MK, et al. Expansion cohorts in first-in-human solid tumor oncology trials. Clin Cancer Res 2015; 21: 4545–4551. [DOI] [PubMed] [Google Scholar]
- 4.Manji A, Brana I, Amir E, et al. Evolution of clinical trial design in early drug development: systematic review of expansion cohort use in single-agent phase I cancer trials. J Clin Oncol 2013; 31: 4260–4267. [DOI] [PubMed] [Google Scholar]
- 5.Boonstra PS, Shen J, Taylor JMG, et al. A statistical evaluation of dose expansion cohorts in phase I clinical trials. J Natl Cancer Inst 2015; 107: dju429. DOI: 10.1093/jnci/dju429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jung SH, Lee T, Kim K, et al. Admissible two-stage designs for phase II cancer clinical trials. Stat Med 2004; 23: 561–569. [DOI] [PubMed] [Google Scholar]
- 7.Bryant J and Day R. Incorporating toxicity considerations into the design of two-stage phase II clinical trials. Biometrics 1995; 52: 1372–1383. [PubMed] [Google Scholar]
- 8.Mander A and Thompson S. Two-stage designs optimal under the alternative hypothesis for phase ii cancer clinical trials. Contemporary Clin Trials 2010; 31: 572–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen CM and Chi Y. Curtailed two-stage designs with two dependent binary endpoints. Pharmaceut Stat 2012; 11: 57–62. [DOI] [PubMed] [Google Scholar]
- 10.Li S, Jin H and Chen W. Two-stage phase ii trials with early stopping for effectiveness and safety as well as ineffectiveness or harm. Commun Stat-Theory Meth 2018; 47: 5626–5638. [Google Scholar]
- 11.Yin H, Wang W and Zhang Z. On construction of single-arm two-stage designs with consideration of both response and toxicity. Biometr J 2019; 61: 1462–1476. [DOI] [PubMed] [Google Scholar]
- 12.Chen N and Lee JJ. Optimal continuous-monitoring design of single-arm phase ii trial based on the simulated annealing method. Contemporary Clin Trials 2013; 35: 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Law M, Grayling MJ and Mander AP. Optimal curtailed designs for single arm phase II clinical trials. arXiv preprint. 2019; arXiv:1909.03017. https://arxiv.org/abs/1909.03017. [Google Scholar]
- 14.Ivanova A, Qaqish BF and Schell MJ. Continuous toxicity monitoring in phase II trials in oncology. Biometrics 2005; 61: 540–545. [DOI] [PubMed] [Google Scholar]
- 15.Wald A Sequential tests of statistical hypotheses. Ann Math Stat 1945; 16: 117–186. [Google Scholar]
- 16.Thall PF and Simon R. Practical Bayesian guidelines for phase IIb clinical trials. Biometrics 1994; 50: 337–349. [PubMed] [Google Scholar]
- 17.Lee JJ and Liu DD. A predictive probability design for phase II cancer clinical trials. Clin Trial 2008; 5: 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yin G, Chen N and Jack Lee J. Phase II trial design with bayesian adaptive randomization and predictive probability. J Royal Stat Soc: Ser C (Appl Stat) 2012; 61: 219–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thall PF, Simon RM and Estey EH. Bayesian sequential monitoring designs for single-arm clinical trials with multiple outcomes. Stat Med 1995; 14: 357–379. [DOI] [PubMed] [Google Scholar]
- 20.Thall PF and Sung HG. Some extensions and applications of a bayesian strategy for monitoring multiple outcomes in clinical trials. Stat Med 1998; 17: 1563–1580. [DOI] [PubMed] [Google Scholar]
- 21.Zhou H, Lee JJ and Yuan Y. Bop2: Bayesian optimal design for phase II clinical trials with simple and complex endpoints. Stat Med 2017; 36: 3302–3314. [DOI] [PubMed] [Google Scholar]
- 22.Lin R, Coleman RL and Yuan Y. Top: time-to-event bayesian optimal phase II trial design for cancer immunotherapy. JNCI: J Natl Cancer Inst 2020; 112: 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sambucini V. Bayesian predictive monitoring with bivariate binary outcomes in phase II clinical trials. Computat Stat Data Analys 2019; 132: 18–30. [Google Scholar]
- 24.Teramukai S, Daimon T and Zohar S. An extension of bayesian predictive sample size selection designs for monitoring efficacy and safety. Stat Med 2015; 34: 3029–3039. [DOI] [PubMed] [Google Scholar]
- 25.Zhong W, Koopmeiners JS and Carlin BP. A trivariate continual reassessment method for phase I/II trials of toxicity, efficacy, and surrogate efficacy. Stat Med 2012; 31: 3885–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schultz J, Nichol F, Elfring G, et al. Multiple-stage procedures for drug screening. Biometrics 1973; 29: 293–300. [PubMed] [Google Scholar]
- 27.Brahmer JR, Lacchetti C, Schneider BJ, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American society of clinical oncology clinical practice guideline. J Clin Oncol 2018; 36: 1714. [DOI] [PubMed] [Google Scholar]
- 28.Dale JR. Global cross-ratio models for bivariate, discrete, ordered responses. Biometrics 1986; 42: 909–917. [PubMed] [Google Scholar]
- 29.Halabi S, Armstrong AJ, Sartor O, et al. Prostate-specific antigen changes as surrogate for overall survival in men with metastatic castration-resistant prostate cancer treated with second-line chemotherapy. J Clin Oncol 2013; 31: 3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dienstmann R, Braña I, Rodon J, et al. Toxicity as a biomarker of efficacy of molecular targeted therapies: focus on EGFR and VEGF inhibiting anticancer drugs. Oncologist 2011; 16: 1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zang Y and Lee JJ. A robust two-stage design identifying the optimal biological dose for phase I/II clinical trials. Stat Med 2017; 36: 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]