

Abstract
Background
Staphylococcus aureus cause diseases both in humans and animals. These diseases range from mild to fatal infections thus necessitating development of a specific molecular method for detection of pathogenic S. aureus.
Objectives
To identify and analyze genetic profile of pathogenic S. aureus using bacteriophage based genetic biomarkers.
Methods
Using culture and biochemical methods, 148 S. aureus (87 %) were isolated from 170 raw milk samples taken from 10 dairy farms in Marsabit and Isiolo counties in Northern Kenya between June 2016 and February 2017. The samples were collected directly from dairy lactating cows previously diagnosed with S. aureus in a follow‐up study. The isolates were analyzed by PCR and sequencing of beta hemolysin (hlb) gene. The genetic relationship between five Kenyan S. aureus isolates and five isolates previously identified was inferred.
Results
From the 96 isolates screened for hlb gene, 75 (78.1%) tested positive. Some of the positive isolates yielded a band size of 975 bp, while others 1100 bp. Through Basic Local Alignment Search Tool (BLAST) search analysis, the two different band sizes (975 bp and 1100 bp) were both confirmed to be hlb gene from S. aureus isolates indicating that the difference in band size may have been due to deletions that were detected in the 975 bp hlb gene. Some S. aureus isolates from Kenya appeared to be closely related to isolates from other parts of the world, while some showed a distant relationship.
Conclusions
Phage‐derived hlb gene is a suitable molecular marker for detection of pathogenic S. aureus.
Keywords: bacteriophages, hlb gene, molecular identification, Staphylococcus aureus
Beta hemolysin gene derived from Staphylococcus Phage 3AJ_2017 genome was used as a molecular marker to detect and characterize pathogenic Staphylococcus aureus.
1. INTRODUCTION
Staphylococcus aureus is one of the major causes of infectious disease both in community and hospitals (Feng et al., 2008; Kimmig et al., 2021). Infections caused by S. aureus ranges from mild superficial skin infection to fatal cases such as toxic shock syndrome (Schmidt et al., 2017). Mortality rate due to S. aureus infection ranges from about 15% to 40% (Schmidt et al., 2017) with an economical loss of about 30%–40% in livestock farming (Sineke et al., 2021). Accurate detection of pathogenic S. aureus is therefore of importance for effective management of infections (Chen et al., 2013; Tadros et al., 2013). Previously bacteriophage (Phage K) has been used to detect S. aureus using liquid chromatography mass‐spectrometry instead of conventional PCR assay (Kurlenda & Grinholc, 2012). Other previous studies have used other genes such as 16S rRNA and nuc for the identification of S. aureus. However, sometimes these genes are not species specific (Ferry et al., 2005; Rees & Barr, 2017); hence, there is a need for a more specific gene. Ability of bacteriophage to aid in horizontal gene transfer between bacteria prompted us to use lysogenic S. aureus 3AJ_2017 bacteriophage in this study. It is known that during infection phages integrate their unique genomic materials into their bacterial host chromosome; thus, they are able to replicate as the bacterial host acquires unique genes. These unique genes can be used in identification of the host bacteria (C. P. Gordon et al., 2013; Hacker & Carniel, 2001; Haveri et al., 2008). Usually, pathogenicity of S. aureus is as a result of virulence factors such as beta hemolysin which aids in biofilm formation for their attachment on the host skin, their invasion and survival in their host by evasion of the host immune defence mechanism (R. J. Gordon & Lowy, 2008). These virulence factors also enable S. aureus to acquire nutrients and to proliferate in mammalian hosts (Ferry et al., 2005; Rees & Barr, 2017).
Apart from beta hemolysin, other virulence factors include coagulase and leukocidine (C. P. Gordon et al., 2013).
Bacteriophages influence their host pathogenicity either by directly encoding virulence factors or indirectly by enhancing the bacterial fitness during infection (Hacker & Carniel, 2001). Busby et al. (2013) reported that the pathogenic bacteria strains harboured greater portion of phage‐related genes than the non‐pathogenic bacterial strains. Bacteriophages have narrow host range and high host specificity owing to the fact that they identify their host by specific receptor molecules on the host cell (Deghorain & Van Melderen, 2012; Mosier‐Boss et al., 2003; Rastogi et al., 2018). Recently, applications of phages in the areas of biotechnology, treatment of multidrug resistant pathogens, food preservation, aquaculture diseases, pollution remediation and waste water treatment have become an inevitable option of research (Gharieb et al., 2020; Sharma et al., 2017). In this regard, we hypothesize that by using bacteriophage genetic marker shared with S. aureus genome, it is possible to develop a molecular tool for specifically detecting and characterizing the pathogenic bacterium. Therefore, this study has identified hlb gene derived from Staphylococcus Phage 3AJ_2017 genome for specific detection and characterization of pathogenic S. aureus.
2. MATERIALS AND METHODS
2.1. Isolation and identification of S. aureus
S. aureus was isolated from 170 raw milk samples that were collected from domestic farms in Marsabit and Isiolo counties in Northern Kenya. Bacterial cells were revived by mixing 1 ml of sample in 4 ml of sterile buffered peptone water (BPW, Oxoid, England) and incubated at 37°C for 24 h. A loop full of BPW enrichment was streaked on mannitol salt agar (MSA, Oxoid) and incubated for 12–48 h at 37°C. The MSA plates were examined for growth of S. aureus usually seen as yellow colonies. To further confirm S. aureus isolates, the yellow colonies from MSA were inoculated on sheep blood agar (SBA, Oxoid) to confirm haemolysin production.
Suspected S. aureus colonies cultured on SBA were enriched on tryptone soy agar (TSA, HIMEDIA India) (Kateete et al., 2010), and then screened biochemically using catalase and tube coagulase test (Benson et al., 2004; Reiner, 2016).
2.2. DNA extraction from the bacterial cells
Bacterial DNA was extracted using Invitrogen DNA Kits (DNAeasy, USA) according to the manufacturer's protocol. At first, bacterial colonies were harvested from the TSA plates and suspended in a tube containing 180 μl lysozyme digestion buffer (LDB, Sigma‐Aldrich, USA) (lysozyme concentration of 20 mg/ml). The bacteria pellets were resuspended in180 μl of LDB then incubated at 37°C for 30 min for subsequent lysis. To the lysate mixture, 20 μl of proteinase K was added and incubated for 30 min at 55°C. An amount of 200 μl of absolute ethanol was added to the digested mixture, followed by binding of the bacterial DNA to silica gel membrane by a brief centrifuge process. In order to remove the protein impurities, two washing steps were carried out using two wash buffers. The pure bound DNA was then eluted using elution buffer and stored at 4°C for subsequent analysis. Using NanoDrop ND‐1000 full spectrum UV–Vis spectrophotometer (Thermo Fisher, USA), the DNA quantity and purity was assessed at a wavelength ranging from 260 to 280 nm.
2.3. Genetic marker identification and primer design
S. aureus bacteriophage 3AJ_2017 sequences were retrieved from GenBank using a customized R script (Altschul, 1990). The retrieved sequences were queried against the National Centre for Biotechnology Information (NCBI) database using Basic Local Alignment Search Tool (BLAST) algorithm to determine the genes that were shared between the phage and S. aureus. This search was carried out at an E‐value of 1e‐3. The hlb gene was selected from the phage as the suitable genetic marker based on its role in pathogenicity of S. aureus. Primers targeting hlb gene in S. aureus were designed using Primer BLAST tool (Ye et al., 2012). The hlb primer sequences were as follows: forward primer as 5′TGCAGAAGATGGTGGCGTAG3′ and reverse primer as 5′CTGGGGCTATTGGTCTGGTG3′.
2.4. PCR and sequencing of S. aureus isolates
PCR reaction was carried out as a 25 μl volume reaction in a 0.2 ml PCR tube containing 12.5 μl of X1 Taq Master Mix (Biolabs, England), 1 μl (10 pmol) of reverse primers and forward primers and 9.5 μl of nuclease free water and 1 μl of DNA template. PCR conditions for the cycler Veriti (Applied Biosystems, USA) were as follows: 95°C for 2 min (95°C for 30 s, 56°C for 30 s) × 35 cycles and an extension time of 5 min at 72°C. The PCR products were detected through gel electrophoresis. An agarose gel of 1.5% in concentration was prepared from tris‐acetate‐ethylenediaminetetraacetic acid buffer where ethidium bromide (1 mg/ml) was used as the staining dye. A total of 4 μl of PCR products were loaded into the wells. An amount of 2 μl 100 bp DNA ladder (Biolabs) was used as a molecular size marker. Electrophoresis was finally carried out at 70 V for 45 min. Gel image was viewed under ultra violet illuminator machine (Bio‐Rad, England). ATCC 25923 S. aureus strain was used as the positive control, while for negative control distilled water was added into the reaction tube instead of DNA template. The PCR‐products were purified using QIAquick gel purification kit (QIAGEN GmbH Hilden, Germany). Twelve purified S. aureus DNA were sequenced using the hlb gene primer used in the PCR reaction. The sequenced isolates were confirmed by BLAST analysis accessed through GenBank database of the (NCBI). These sequenced isolates were analyzed for conserved domains and five of them submitted to NCBI through Bankit tool (Pirovano et al., 2017) to obtain the following accession numbers: MH674195, MH674196, MH674197, MH674198 and MH674199.
2.5. Phylogenetic analysis of DNA sequences
The hlb gene sequences from this study were compared with hlb gene of S. aureus from other parts of the world to infer genetic relationship among them. Using a customized R script, hlb gene sequences from other parts of the world were retrieved from NCBI. The sequences were as follows: NC_022604 and NC_016928 from Taiwan, NC_021059 from United Kingdom and NC_017340 from Germany. Seaview tool (Gouy et al., 2010) was used to align the sequences by employing MUSCLE v.3.8.3 Program (Edgar, 2004). Jalview v2.10.46 (Clamp et al., 2004) was then used to edit the aligned sequences. The aligned sequences were further analyzed under maximum likelihood method by PhyML v30 (Guindon et al., 2010). General Time Reversible (Gatto et al., 2007) substitution method was employed, a bootstrap was set at 100 and neighbour‐joining interchange was selected to improve the tree construction.
3. RESULTS
3.1. S. aureus isolated from raw milk
Out of 170 bacterial isolates, 168 (98.8%) were successfully revived in BPW. From the 168 revived isolates, 148 (88.0%) grew on MSA revealing yellow colonies with the media changing colour from pink to yellow. Furthermore, 105 (70.7%) of the isolates that grew on MSA also yielded colonies on SBA revealing translucent zones around them, which was an indication of beta haemolysis. The 148 (100%) isolates were all catalase positive, while 141 (95.2%) isolates were coagulase positive (Table 1). The culture and biochemical findings indicated that the isolates were S. aureus.
TABLE 1.
Proportion of isolates that were revived on culture and positively identified as S. aureus using biochemical test
| Culture and biochemical characterization | Number of positives (%) |
|---|---|
| Growth on MSA | 148 (88.0%) |
| Beta hemolysis | 105 (70.9%) |
| Coagulase | 141 (95.2%) |
| Catalase | 148 (100%) |
Abbreviation: MSA, mannitol salt agar.
3.2. Beta hemolysin gene identified from the phage genome
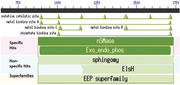
S. aureus 3AJ_2017 bacteriophage (KX232515.1) sequences were retrieved from NCBI database (Mosier‐Boss et al., 2003) using a customized R script. The size of the bacteriophage genome was 43922 bp. BLASTn search analysis of the phage whole genome yielded exclusively S. aureus species as its homologs with an identity of 100%, indicating that this phage specifically infects S. aureus. The hlb gene comprising of 1023 bp was identified in the position 42,899 to 43,922 in the bacteriophage genome and then selected as the ideal genetic marker for the detection of S. aureus. The primers designed from the hlb gene, using primer Blast tool, yielded a predicted product size of 975 bp.
3.3. S. aureus identified by PCR and sequencing
A pair of oligonucleotide primers targeting hlb gene amplified the fragment yielding a band corresponding to 975 bp. The same primers also yielded amplified products of 1100 bp (Figure 1). The resulting two different band size PCR products suggested that we could have amplified other bacterium apart from S. aureus. Therefore, we sequenced the two PCR products of different sizes and analyzed the corresponding PCR bands by BLASTn. Both PCR products yielded sequences homologous to S. aureus hlb gene revealing a nucleotide identity of 99%. Furthermore, BLASTx analysis revealed that the translated amino acid sequences were homologous to S. aureus hlb protein with amino acid sequence identity of 100%. An alignment of the PCR product sequences corresponding to 975 bp and 1100 bp revealed that they were similar, indicating that the difference in sizes may have been due to deletion of 125 bp in some of the bacterial hlb gene. The results from PCR analyses confirmed that 75 (78%) isolates out of 96 obtained from the milk samples were indeed S. aureus. Five of hlb sequences of the S. aureus isolates obtained from this study have been deposited in GenBank Database under the accession numbers MH674195 and MH674199.
FIGURE 1.
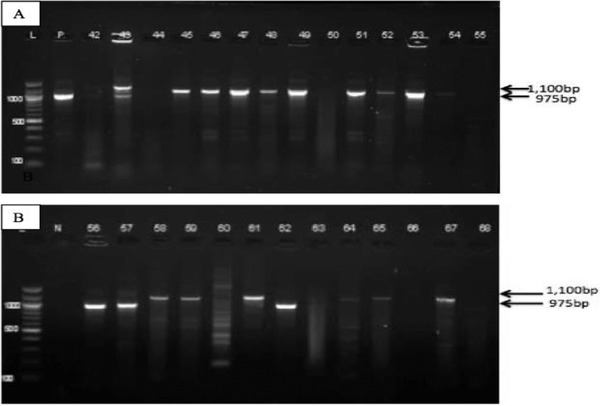
A gel image of PCR‐amplified hlb gene of putative pathogenic S. aureus isolates. The isolates yielded different bands of approximately 975 bp and 1100 bp. Panels a and b represent S. aureus isolates positive for PCR. Lane L is the DNA ladder (100 bp), Lane P is the reference strain for S. aureus ATCC 25923TM and Lane N is the negative control
3.4. Conserved domain contain virulence factor
In order to identify conversed domains and their potential role in virulence, we analyzed amino acid sequences translated from the nucleotide sequences of the 12 S. aureus isolates. The translated amino acid residues contained a conserved domain known as neutral sphingomyelinase (nSMase). This domain consisted of a catalytic site, metal binding site A, B and C and a phosphate binding site (Figure 2). The neutral sphingomyelinase belonged to the superfamily endonuclease exonuclease phosphates, which is known to consist of catalytic enzymes that cleave phosphodiester bonds. Neutral sphingomyelinase is a virulent factor known to be responsible for the pathogenesis of S. aureus. This factor promotes colonization of the host cell by catalyzing the hydrolysis of sphingomyelin on the host cell membrane to ceramide and phosphorylcholine. These findings indicate that the S. aureus isolates could be pathogenic strains.
FIGURE 2.
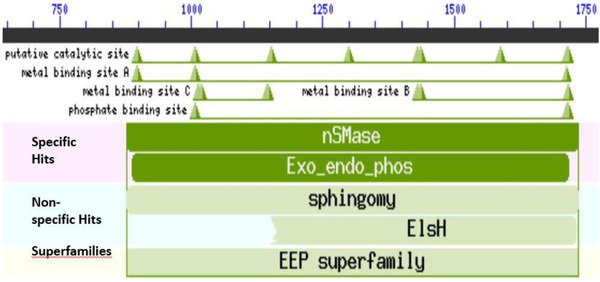
Bioinformatics analysis of translated hlb polypeptide of putative pathogenic S. aureus isolates. The conserved domain contains neutral sphingomyelinase (nMSase) with a catalytic site, phosphate binding site and metal binding site A, B and C
3.5. Phylogenetic analysis of S. aureus isolates
The next task was to determine whether the Kenyan S. aureus were genetically distinct. Subsequently, we compared the hlb sequences of the Kenyan isolates with the hlb sequences of other isolates previously identified in the different parts of the world. We found that the isolates from Kenya clustered on the same clade and hence appeared to share a common ancestry. The sequences from other parts of the world namely Denmark, Taiwan, UK and Germany also shared the same ancestry. However, one isolate from Taiwan (NC_016928.2) was an outlier as it did not share ancestry with any other isolates (Figure 3).
FIGURE 3.
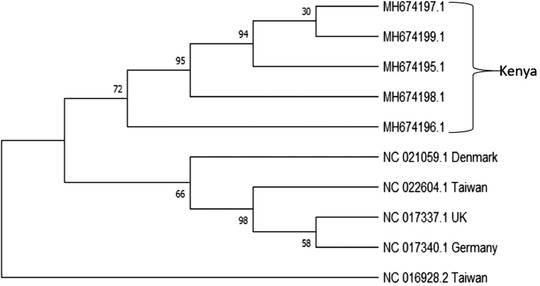
Phylogenetic analysis based on the hlb gene fragments of S. aureus isolates from Kenya and other regions of the world
4. DISCUSSION
S. aureus exist both in humans and animals either as a commensal or pathogenic bacteria causing diseases (Sineke et al., 2021) ranging from mild superficial skin infection to fatal cases such as toxic shock syndrome (Schmidt et al., 2017). Therefore, there is a need for the development of a specific and sensitive molecular method for detection and characterization of pathogenic S. aureus. This study used a bacteriophage genetic marker common to S. aureus to develop a molecular tool for specific detection and characterization of pathogenic S. aureus. The temperate phages are known to integrate their genetic materials into their host chromosome resulting in exchange of unique genes between the phages and their host. We have confirmed that phages are vectors for horizontal gene transfer in the bacterium as many unique genes were found to be shared between 3AJ_2917 bacteriophage and S. aureus. Among these unique genes were the hlb gene, which was chosen as the ideal marker for S. aureus identification as it codes for beta hemolysin protein, which plays a key role in S. aureus pathogenicity (Srinivasan et al., 2015). The betahemolysin proteins aids in the colonization of the host skin by S. aureus during infection (Katayama et al., 2013). In addition, beta hemolysin has sphingomyelinase (SMase) activities and therefore, during infection, S. aureus is able to break the sphingomyelin on the cell membrane to ceramide and phosphocholine resulting in cell destruction (Ma et al., 2017). Apart from erythrocytes, beta hemolysin affects other cells such as leukocytes and keratinocytes. The beta hemolysin is responsible for recurrence of some infection such as furunculosis and respiratory infection. Although the primer targeting hlb gene yielded two bands of different sizes, the products were confirmed to be S. aureus. The difference in amplicons band size may have been due to deletion in the hlb gene as was seen in the alignment that showed the two sequences to be identical except one was 125 bp shorter than the other. The findings of this study indicated that hlb gene is a specific genetic marker for detection of pathogenic S. aureus. This is the first study where phage genetic marker has been used to identify and confirm S. aureus by PCR assay. In another study where phage was used to detect S. aureus, liquid chromatography was used instead of molecular genetic based tools (Rees & Barr, 2017). Neutral sphingomyelinase (nSMase) domain was found to be present across all the S. aureus isolated in this study. This enzyme is a virulence factor that hydrolyses sphingomyelin on the host cell membrane, thus promoting host colonization by S. aureus. The sphingomyelin is broken down into ceramide, which is responsible for adhesion, differentiation, proliferation and cell death, indicating that these isolates were pathogenic S. aureus. Neutral sphingomyelinase also acts as a hemolysin, which is responsible for host red blood cell lysis. Indeed, the S. aureus isolated from this study hemolyzed sheep blood agar, indicating that these sequences encode a functional domain. This study also revealed that Kenyan S. aureus isolates shared the same ancestry even though they did not show any phylogenetic relationship with S. aureus from other parts of the world. The phylogenetic analysis, therefore, gave a satisfactory insight into the evolutionary relationship among these isolates as shown form the bootstraps, which have significant values ranging from 30% to 95%. In summary, this study gave a solid platform and relevant information on molecular detection of pathogenic S. aureus based on the Staphylococcus Phage 3AJ_2017 genome. This information is further critical in promoting future phage‐based research for the development of specific diagnostic methods for specific detection of bacterial infections. The results of this study have demonstrated that Staphylococcus Phage 3AJ_2017 genome can be used in developing PCR kit for molecular identification and characterization of pathogenic S. aureus.
5. CONCLUSION
We conclude that the S. aureus bacteriophage 3AJ_2017 has unique genetic markers that can be exploited to identify and characterize pathogenic S. aureus infecting animals and possibly humans. We further conclude that the phage‐based PCR technique developed in this study could provide a platform for specific molecular detection of pathogenic S. aureus. Nevertheless, large‐scale analysis of S. aureus isolates will still be required to validate the PCR analysis.
CONFLICT OF INTEREST
The authors declare no conflict of interests.
AUTHOR CONTRIBUTIONS
Gabriel O. Aboge, Rosaline W. Macharia and George O. Obiero conceived and designed the study, and Silviane A. Miruka performed the experiments. Isaac M. Omwenga acquired the study samples. Silviane A. Miruka, Rosaline W. Macharia and Gabriel O. Aboge analyzed data and wrote the manuscript. All the authors read and approved the final version of manuscript.
EITHICS STATEMENT
No ethical approval was required as the study did not involve any animal experiment.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/vms3.676
ACKNOWLEDGEMENTS
We would like to thank the director for CEBIB of the University of Nairobi for the great support. The authors would like to thank Kevin Ochwedo and Kelvin Muteru for assistance with Bioinformatics analysis. We would also like to extend our gratitude to Ann Owiti, Tiberias Aboge, Catherin Ngwaya of CEBIB and members of molecular laboratory, Department of Public Health Pharmacology and Toxicology, University of Nairobi for assistance with molecular work.
Miruka, S. A. , Aboge, G. O. , Macharia, R. W. , Obiero, G. O. , & Omwenga, I. M. (2022). Beta hemolysin gene of Staphylococcus phage 3AJ_2017 genome is a suitable molecular marker for identification and characterization of pathogenic Staphylococcus aureus . Veterinary Medicine and Science, 8, 845–851. 10.1002/vms3.676
DATA AVAILABILITY STATEMENT
The data used to support the findings of this study are included in the manuscript after the references.
REFERENCES
- Benson, D. A. , Karsch‐Mizrachi, I. , Lipman, D. J. , Ostell, J. , & Wheeler, D. L. (2004). GenBank: Update. Nucleic Acids Research, 32, D23–D26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busby, B. , Kristensen, D. M. , & Koonin, E. V. (2013). Contribution of phage‐derived genomic islands to the virulence of facultative bacterial pathogens. Environmental Microbiology, 15(2), 307–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, P.‐W. , Jheng, T. T. , Shyu, C.‐L. , & Mao, F. C. (2013). Synergistic antibacterial efficacies of the combination of bovine lactoferrin or its hydrolysate with probiotic secretion in curbing the growth of meticillin‐resistant Staphylococcus aureus . Journal of Medical Microbiology, 62(Pt 12), 1845–1851. [DOI] [PubMed] [Google Scholar]
- Clamp, M. , Cuff, J. , Searle, S. M. , & Barton, G. J. (2004). The Jalview Java alignment editor. Bioinformatics, 20(3), 426–427 [DOI] [PubMed] [Google Scholar]
- Deghorain, M. , & Van Melderen, L. (2012). The staphylococci phages family: An overview. Viruses, 4(12), 3316–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R. C. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32(5), 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Y. , Chen, C.‐J. , Su, L.‐H. , Hu, S. , Yu, J. , & Chiu, C.‐H. (2008). Evolution and pathogenesis of Staphylococcus aureus: Lessons learned from genotyping and comparative genomics. FEMS Microbiology Reviews, 32(1), 23–37. [DOI] [PubMed] [Google Scholar]
- Ferry, T. , Perpoint, T. , Vandenesch, F. , & Etienne, J. (2005). Virulence determinants in Staphylococcus aureus and their involvement in clinical syndromes. Current Infectious Disease Reports, 7(6), 420–428 [DOI] [PubMed] [Google Scholar]
- Gatto, L. , Catanzaro, D. , & Milinkovitch, M.C. (2007). Assessing the applicability of the GTR nucleotide substitution model through simulations. Evolutionary Bioinformatics Online, 2, 145–55. [PMC free article] [PubMed] [Google Scholar]
- Gharieb, R. M. A. , Saad, M. F. , Mohamed, A. S. , & Tartor, Y. H. (2020). Characterization of two novel lytic bacteriophages for reducing biofilms of zoonotic multidrug‐resistant Staphylococcus aureus and controlling their growth in milk. LWT, 124, 109145. [Google Scholar]
- Gordon, R. J. , & Lowy, F. D. (2008). Pathogenesis of methicillin‐resistant Staphylococcus aureus infection. Clinical Infectious Diseases, 46(Suppl_5), S350–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, C. P. , Williams, P. , & Chan, W. C. (2013). Attenuating Staphylococcus aureus virulence gene regulation: A medicinal chemistry perspective. Journal of Medicinal Chemistry, 56(4), 1389–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouy, M. , Guindon, S. , & Gascuel, O. (2010). SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Molecular Biology and Evolution, 27, 221–224. [DOI] [PubMed] [Google Scholar]
- Guindon, S. , Dufayard, J. F. , Lefort, V. , Anisimova, M. , Hordijk, W. , & Gascuel, O. (2010). New algorithms and methods to estimate maximum‐likelihood phylogenies: Assessing the performance of PhyML 3.0. Systematic Biology, 59, 307–321. [DOI] [PubMed] [Google Scholar]
- Hacker, J. , & Carniel, E. (2001). Ecological fitness, genomic islands and bacterial pathogenicity. A Darwinian view of the evolution of microbes. EMBO Reports, 2(5), 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haveri, M. , Hovinen, M. , Roslof, A. , & Pyorala, S. (2008). Molecular types and genetic profiles of Staphylococcus aureus strains isolated from bovine intramammary infections and extramammary sites. Journal of Clinical Microbiology, 46(11), 3728–3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kateete, D. P. , Kimani C. N., Katabazi F. A., Okeng, A. , Okee, M. S. , Nanteza, A. , Joloba, M. L. , & Najjuka, F. C. (2010). Identification of Staphylococcus aureus: DNase and Mannitol salt agar improve the efficiency of the tube coagulase test. Annals of Clinical Microbiology and Antimicrobials, 9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama, Y. , Baba, T. , Sekine, M. , Fukuda, M. , & Hiramatsu, K. (2013). Beta‐hemolysin promotes skin colonization by Staphylococcus aureus . Journal of Bacteriology, 195(6), 1194–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmig, A. , Hagel, S. , Weis, S. , Bahrs, C. , Löffler, B. , & Pletz, M. W. (2021). Management of Staphylococcus aureus bloodstream infections. Frontiers in Medicine, 7, 1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurlenda, J. , & Grinholc, M. (2012). Alternative therapies in Staphylococcus aureus diseases. Acta Biochimica Polonica, 59(2), 171–184. [PubMed] [Google Scholar]
- Ma, J. , Gulbins, E. , Edwards M. J., Caldwell, C. C. , Fraunholz, M. , & Becker, K. A. (2017). Staphylococcus aureus α‐toxin induces inflammatory cytokines via lysosomal acid sphingomyelinase and ceramides. Cellular Physiology and Biochemistry, 43(6), 2170–2184. [DOI] [PubMed] [Google Scholar]
- Mosier‐Boss, P. A. , Lieberman, S. H. , Andrews, J. M. , Rohwer, F. L. , Wegley, L. E. , & Breitbart, M. (2003). Use of Fluorescently Labeled Phage in the Detection and identification of bacterial species. Applied Spectroscopy, 57, 1138–1344. [DOI] [PubMed] [Google Scholar]
- National center for biotechnology information , Altschul (1990). http://www.ncbi.nlm.nih.gov
- Pirovano, W. , Boetzer, M. , Derks, M. F. L. , & Smit, S. (2017). NCBI‐compliant genome submissions: Tips and tricks to save time and money. Briefings in Bioinformatics, 18(2), 179–182. [DOI] [PubMed] [Google Scholar]
- Rastogi, V. , Pragya, Verma, N. , Mishra, A. K. , Nath, G. , Gaur, P. K. , & Verma, A. (2018). An overview on bacteriophages: A natural nanostructured antibacterial agent. Current Drug Delivery, 15(1), 3–20. [DOI] [PubMed] [Google Scholar]
- Rees, J. C. , & Barr, J. R. (2017). Detection of methicillin‐resistant Staphylococcus aureus using phage amplification combine with matrix‐assisted laser desorption/ionization mass spectrometry. Analytical and Bioanalytical Chemistry, 409, 1379–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner, K. (2010). Catalase test protocol. American Society for Microbiology, 1–6. [Google Scholar]
- Schmidt, T. , Kock, M. M. , & Ehlers, M. M. (2017). Molecular characterization of Staphylococcus aureus isolated from bovine mastitis and close human contacts in South African dairy herds: Genetic diversity and inter‐species host transmission. Frontiers in Microbiology, 8, 511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, S. , Chatterjee, S. , Datta, S. , Prasad, R. , Dubey, D. , Prasad, R. K. , & Vairale, M. G. (2017). Bacteriophages and its applications: An overview. Folia Microbiologica, 62(1), 17–55. [DOI] [PubMed] [Google Scholar]
- Sineke, N. , Asante, J. , Amoako, D. G. , Abia, A. L. K. , Perrett, K. , Bester, L. A. , & Essack, S. Y. (2021). Staphylococcus aureus in intensive pig production in South Africa: Antibiotic resistance, virulence determinants and clonality. Pathogens, 10(3), 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan, R. , Karaoz, U. , Volegova, M. , MacKichan, J. , Kato‐ Maeda, M. , Miller, S. , Nadarajan, R. , Brodie, E. L. , & Lynch, S. V. (2015). Use of 16S rRNA gene for identification of a broad range of clinically relevant bacterial pathogens. PloS One, 10(2), e0117617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadros, M. , Williams, V. , Coleman, B. L. , McGeer, A. J. , Haider, S. , Lee, C. , Iacovides, H. , Rubinstein, E. , John, M. , Johnston, L. , McNeil, S. , Katz, K. , Laffin, N. , Suh, K. N. , Powis, J. , Smith, S. , Taylor, G. , Watt, C. , & Simor, A. E. (2013). Epidemiology and outcome of pneumonia caused by methicillin‐resistant Staphylococcus aureus (MRSA) in Canadian hospitals. PloS One, 8(9), e75171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, J. , Coulouris, G. , Zaretskaya, I. , Cutcutache, I. , Rozen, S. , & Madden, T. L. (2012). Primer‐BLAST: A tool to design target specific primers for polymerase chain reaction. BMC Bioinformatics, 13, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are included in the manuscript after the references.