

Abstract
Protein post-translational modifications (PTMs) generate an enormous, but as yet undetermined, expansion of the produced proteoforms. In this Viewpoint, we firstly reviewed the concepts of proteoform and peptidoform. We show that many of the current PTM biological investigation and annotation studies largely follow a PTM site-specific rather than proteoform-specific approach. We further illustrate a potentially useful matching strategy in which a particular “modified peptidoform” is matched to the corresponding “unmodified peptidoform” as a reference for the quantitative analysis between samples and conditions. We suggest this strategy has the potential to provide more directly relevant information to learn the PTM site-specific biological functions. Accordingly, we advocate for the wider use of the nomenclature “peptidoform” in future bottom-up proteomic studies.
Proteins and PTMs.
Currently, it is widely accepted that one gene frequently generates multiple forms of proteins, rather than only one distinctive form. These forms include a variety of e.g., sequence variation, RNA alternative splicing isoforms, and protein post-translational modifications (PTMs). Together, they render the extensive biological variability and phenotypic diversity [1,2]. Protein PTMs, include phosphorylation, glycosylation, acetylation, ubiquitination, and methylation, among a few hundred others [3]. PTMs can alter the protein physicochemical and biological properties – frequently in a modification-site-specific manner. For example, protein phosphorylation can mediate protein-protein interaction [4,5], alter the protein three-dimensional structure, stability, turnover [6,7], and subcellular localization [8].
Top-down and Bottom-up Proteomics: Proteoform and Peptidoform.
There are two general mass spectrometry (MS) based proteomic strategies for analyzing proteins and their PTM isoforms – the bottom-up approach and the top-down approach. In a bottom-up workflow [9], proteins are digested into peptides, which makes resolving of PTM isoforms at the whole-protein level challenging. In contrast, in top-down workflows, intact proteins and protein variants are measured, enabling a comprehensive characterization of the protein level molecular composition. The bottom-up and top-down strategies are not mutually exclusive and assist each other in validating the type and existence of PTMs.
The concept of proteoform is highly relevant for understanding top-down perspectives. The Consortium for Top-down Proteomics has defined the nomenclature “proteoform” to designate all the different molecular forms in which the protein product of a single gene can be found, which include changes due to genetic variations, alternatively spliced RNA transcripts, and posttranslational modifications [10]. In essence, each individual molecular form of expressed proteins is a proteoform [10].
Indeed, the concept of proteoform has greatly sharpened our views on protein diversity. Following the definition of proteoform, protein subpopulations carrying a phosphate moiety at two sites could generate four possible proteoforms – three modified at each or both sites and another unmodified proteoform. Likewise, theoretically, the same or different types of PTMs at different amino acid residues might decorate a given protein in any combinatorial patterns. Therefore, an exceptional increase in the theoretical number of proteoforms might be expected due to PTMs (Figure 1, Upper view). Moreover, certain PTM types such as polyubiquitination and glycosylation can generate additional constitutive variation at the same site. For example, if we consider the variable structural feature of oligosaccharides, the number of proteoforms carrying distinct glycan structure will be fairly large. Very interestingly, despite such a theoretical “proteoform explosion”, the real number of human proteforms seems to be much lower, according to a community-level estimation [3]. A total of ~one million proteoforms were roughly estimated based on e.g., the current practice of histone modification analysis, the technical detection threshold, cell type uniqueness, and the practical cellular constraints in controlling the enzymatic writing and maintenance of PTMs. The authors nevertheless acknowledged that a precise estimate of the number of human proteoforms is difficult to provide [2,3]. Moreover, for a specific protein the pool of all possible proteoforms can be immense [11]. Although top-down studies have proven to be an exceptionally powerful resource for hypothesis-driven research on defined protein targets, there seems to be a quite long way to go for the top-down technique to routinely detect most (e.g., one million) proteoforms in a sample, due to its current limited sensitivity which is still perceived as worse than in the bottom-up approach [12,13].
Figure 1. The top-down and bottom-up views for studying PTM biology.
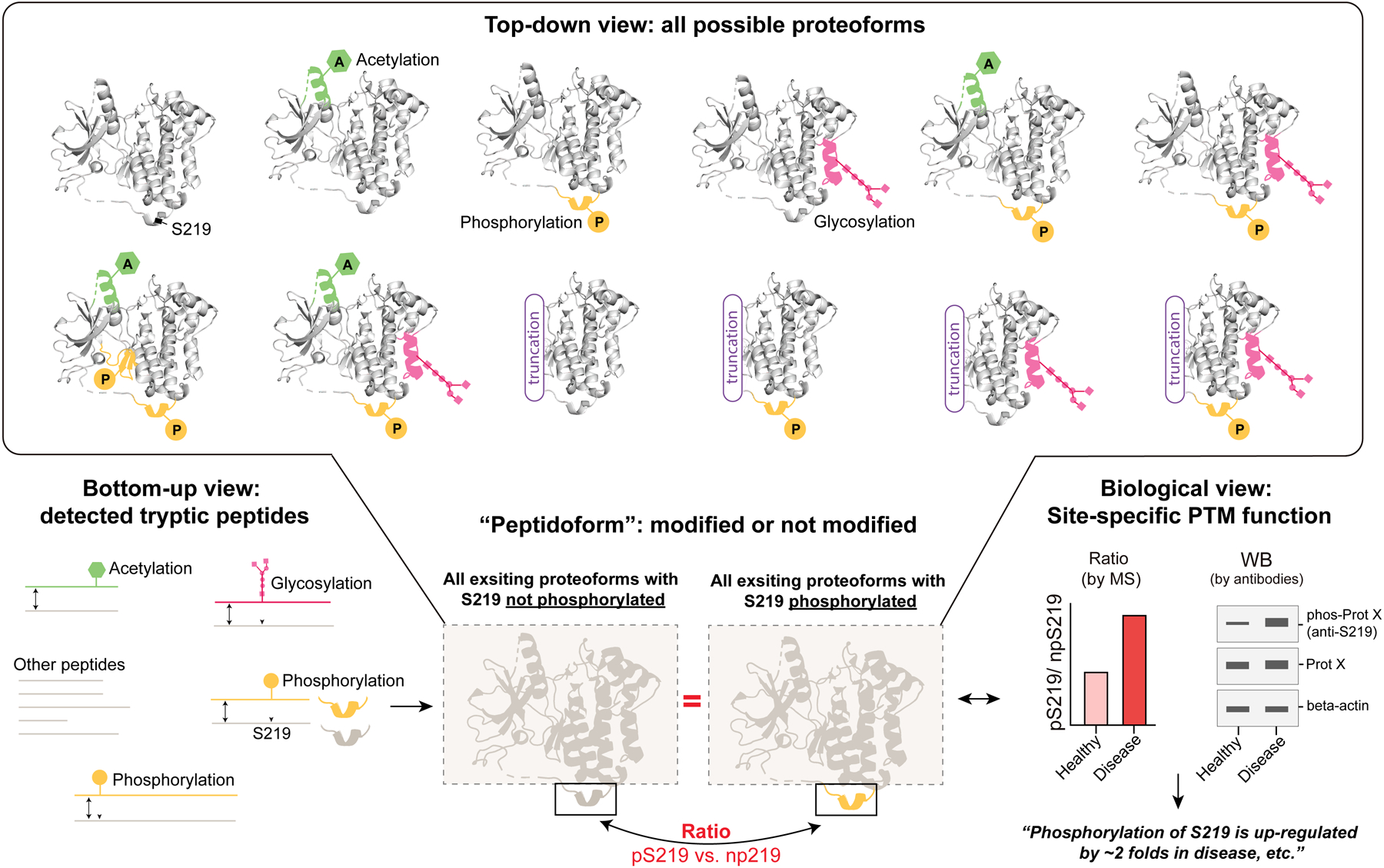
A virtual example of protein X (representing any protein) is shown. This protein X can be modified by different enzymes at different amino acid residues, forming a variety of possible proteoforms carrying acetylated, phosphorylated, or glycosylated sites and even the combinatorial PTMs in the cells. Additionally, the mRNA alternative splicing could create some truncated proteoforms of X. In bottom-up proteomics, the modified peptidoforms can be measured in PTM enriched samples. If we take the non-modified peptidoform counterpart (measured by total proteomics) as a comparative reference, we can extract a pair of PTM/non-PTM peptidoforms, irrespective of the total proteoform pool, for interrogating the impact of a site-specific PTM among samples. Such information can be verified by e.g., western-blot, illustrating site-specific PTM functions. Protein structural regions of different colors denote the location of respective peptidoforms. The phosphorylated S219 represents any PTM site of any type. The red equals sign highlights the proteoform background which is the same in PTM-enriched and non-PTM measurements.
On the other hand, the concept of peptidoform has been used in several studies [14–25], but not widely. Peptidoform stands for specifically modified or mutated peptides with the same backbone amino acid sequence [20]. In the early days, peptidoform was mentioned always together with proteoform to deliver the corresponding isoform complexity at the peptide level. More recently, peptidoform was independently used in bottom-up studies, such as in a few data-independent acquisition (DIA) MS-based [26,27] quantitative PTM investigations [14,16,20]. For example, we and colleagues have developed Inference of Peptidoforms (IPF), a computational algorithm for confident, systematic identification and quantification of peptidoforms in DIA-MS datasets [20] and applied it in analyzing plasma peptidoforms of human twin individuals [20,28]. We will further discuss the concept of peptidoform below.
Studying the PTM function: a Proteoform-specific approach or PTM Site-specific approach?
With the concepts of proteoform and peptidoform reviewed above, it becomes inspiring to revisit how we study PTM biology currently. To learn the function of a protein PTM, the researchers normally perform experiments to measure the abundance and other properties of it (e.g., localization, stability, etc.) in a biological process such as drug-induced perturbation or disease development. The researchers would then refer to published literature or PTM databases and sometimes perform new validation experiments to fully establish a functional link between the PTM site and the biological question. Although bottom-up and top-down approaches both support well the broad, relative proteomic quantification between samples and conditions, they measure the de facto individual peptides (digested) and proteoforms, respectively. In an ideal scenario, if distinctive proteoforms are precisely identified and quantified between biological/clinical samples (see a nice example in Ref. [29]), and with such knowledge being accrued over time, the top-down measurement will pinpoint the different functions of every detectable proteoform. This type of proteoform-specific knowledge would also nicely fit the structural view of protein complexity, because actions taken by the each intact proteoform species in the cellular processes should be anyway depending on their unique molecular structure.
However, it is crucial to stress that, different proteoforms of the same gene can share the same site-specific PTM, whereas lots of PTMs are currently studied and annotated (in various databases) with a site-specific, rather than a proteoform-specific, manner. For a virtual example, please see Figure 1. Herein, the S219 of protein X (can be any protein) is able to be phosphorylated by a particular kinase Y, resulting in phosphorylated S219 (i.e., pS219, highlighted in orange color). All the proteoform species carrying pS219 (n=7 in this case) might co-exist in the cell after the enzymatic kinase-substrate reaction. Currently, most functional studies are performed for pS219 itself, but not for one of the seven proteoforms carrying pS219 (which would otherwise need the protein-level separation or purification [30]). Also, the resultant knowledge and annotations are built on pS219 (e.g., it is a substrate site of kinase Y) [31], but not on each of the seven proteoforms.
Furthermore, it is intriguing to ask what is really being measured in PTM analyses performed using classic non-MS methods, such as western blotting (WB), immunohistochemistry (IHC), and enzyme-linked immunosorbent assay (ELISA). For these assays, antibodies have to be developed targeting proteins carrying particular PTMs. During antibody production, the immunogen—the part of the protein that the antibody recognizes (e.g., a continuous stretch of amino acids) or the full-length protein, is the key. In the first production of phosphorylation-dependent antibodies twenty years ago, benzyl phosphonate was injected into rabbits to generate antibodies detecting phosphotyrosine-containing proteins [32][WEB1]. Nowadays, phosphosite-specific antibodies are typically generated by the immunization using synthetic phosphopeptide surrounding the phosphosite of interest. A further selection is normally performed to remove antibodies detecting the non-phosphorylated version. Therefore, whether WB, IHC, or ELISA measure a specific proteoform or multiple proteoforms carrying the same PTM site largely depends on the particular antibody and the sample. Although in WB the molecular weight (MW) of the protein target is obtained by referring to MW markers, the MW information can be lacking in IHC and ELISA assays. Even in WB, due to the limited MW resolution of electrophoresis, a detected protein band might still represent multiple proteoforms with varied molecular compositions (but sharing a PTM site) that are just too close in MW. In this regard, using full-length proteoforms as antigens for production of high-quality affinity reagents using methods like phage display might be helpful to increase the proteoform-level specificity in WB, IHC, and ELISA analyses [3].
Although many current research tools and annotating frameworks largely follow the PTM site-centric assumption, we want to point out that there is no doubt that the link between proteoform species and their functional significance would be a major advance in the future. This will catalyze the fundamental knowledge drift from PTM site-centric to proteoform-centric investigation, because eventually, proteoforms are the real biological molecules in the cell carrying respective functions. Furthermore, the crosstalk between PTM sites of the same protein (i.e., the coordinated function between PTM sites) will be more straightforwardly measured by the proteoform-level analysis. functions. Ultimately, the complete primary structures of proteins on a proteome scale will be useful. [13,33] Before that, however, corresponding experimental and informatic paradigms have to be established and widely applied. Recently, interesting workflows have been applied to infer proteoform-dependent functions from e.g., peptide co-varying analysis across multiple samples and comparisons [34–36]. Emerging bioinformatic annotation tools, such as PTMsigDB, just started to drift towards PTM site-specific annotation following PTM proteomic profiling [37,38] (which already presented a major conceptual advance compared to the conventional, widely used, gene-specific annotating frameworks [39,40]). Yet, the proteoform-specific annotation databases have not been configured proteome-wide, mainly due to the lack of data. In this regard, the recent initiative of The Human Proteoform Project is very timely and extremely important for assembling an atlas with more detailed knowledge by creating a comprehensive proteoform index [33].
Peptidoform: the concept revisited.
Due to the above challenges, we reason that the concept of peptidoform may facilitate studying PTMs using bottom-up proteomics (Figure 1, and below). Compared to proteoform analysis, peptidoform profiling in fact provides a closer analysis to which is being measured in most antibody-based assays. In particular, herein, we propose that, the usage of peptidoform should clearly embrace both unmodified and differentially modified peptides that share the same backbone amino acid sequence. The peptidoforms can be generated by trypsin digestion or by other proteases. We further propose that the previously used terms such as phosphomodiform [6] can be unified under the nomenclature “peptidoform”, because phosphomodiform essentially means phosphorylated peptidoform. In essence, although a peptidoform is generated by protease digestion and obviously not really existing in a cell, it contains qualitative and quantitative information of a collection of proteoforms sharing a particular modified or unmodified amino acid sequence.
A modified peptide can emerge from all isoforms of one gene but can also be derived from multiple isoforms on multiple genes, due to the well-known protein inference problem in bottom-up proteomics [41]. As a common strategy widely used in shotgun proteomics, we propose the term proteotypic peptidoforms can be used to present those distinct representative peptides using direct mapping or after the proper consideration of the protein inference problem [42]. Most importantly, compared to using the terms such as “modified peptides”, the broader usage of peptidoform would emphasize the site-specific PTM biology in bottom-up proteomics, considering the enormous number of proteoforms in cells. In this regard, taking phosphorylation as an example, the phosphosite abundance profiling experiments in most previous phosphoproteomic studies, the phosphomodiform thermal stability analysis in Huang et al. and others [6,43,44], and our previous phosphomodiform lifetime study [7] all belong to the peptidoform profiling.
A peptidoform matching strategy for relative quantification between samples.
In the present Viewpoint, we would like to suggest that a peptidoform matching strategy could be a powerful approach to study PTM site-specific functions. To elaborate, in our recent study [7], by using the pulse experiment of stable isotope-labeled amino acids in cells (pSILAC) [45], we performed a pilot phosphoproteome turnover analysis. Particularly, we adopted a peptidoform matching strategy that directly interrogates the impact of individual phosphosites on protein turnover. This particular method is referred to as DeltaSILAC (delta determination of turnover rate for modified proteins by SILAC) [7]. In DeltaSILAC, for each site-specific phosphorylation, we determined the lifetime difference between the phosphorylated peptidoform (measured by pSILAC in enriched phosphoproteomes) and the non-phosphorylated peptidoform counterpart (measured by pSILAC in the same peptide samples without phospho-enrichment). This strategy successfully revealed that phosphorylation of the majority of sites increased protein stability in growing HeLa cells, which was not apparent without the matching strategy [7].
The underlying, critical assumption of this peptidoform matching strategy lies in the total “proteoform pool” that must remain always the same in both PTM-enriched and non-PTM measurements. Taking the example of Protein X again (Figure 1, bottom panel), no matter what and how many proteoform species of X exist in the cells, these proteoforms will generate a common peptide mixture after protease digestion. The bottom-up measurements on the peptidoforms of pS219 and npS2I9 (non-phosphorylated S219) will only extract two peptidoforms, compare their difference between samples/conditions, and directly infer the impact of S219 phosphorylation. Similarly, this comparison can be applied to all the other PTM sites and types (such as acetylation and glycosylation, see green and pink sites in Figure 1) or even to a simultaneous analysis of multiple PTMs. In the case of PTMs on Lysine and Arginine residues, alternative proteases other than trypsin might be used to generate the PTM/non-PTM pair of peptidoforms.
Of note, the relative quantitative comparison between samples is crucial because the pS219 and npS219 peptidoforms may have different physicochemical properties, ending up with different flyability and responsiveness in the mass spectrometer. The relative fold-change of the ratio of pS219/npS219 between samples, rather than the pS219/npS219 ratio itself, can provide valuable and relevant information to the “PTM site-specific” biological functions. In this regard, the recent consistent proteomic methods such as the MS2-based quantification afforded by DIA-MS will facilitate the confident and reproducible detection of unique modified Peptidoforms by using unique ion signatures [20], allowing for the high-resolute PTM site-specific quantification between multiple samples.
Conceivably, the extensive detection and quantification of non-PTM peptidoforms for each PTM site seem to be crucial for this conceptualized matching strategy, which can be difficult for PTM sites of high stoichiometry. Indeed, in the DeltaSILAC study, despite the fact that we estimated lifetimes for ~13,000 phosphorylated peptidoforms, we only measured lifetimes for 2,100 phos-/non-phos- peptidorm pairs [7]. Fortunately, the technical barrier in identifying non-PTM peptides currently is lower than the PTM analysis. Normally, the amounts of the non-PTM samples are less limited in proteomics experiments, allowing for e.g., peptide-level fractionation to increase the coverage of unmodified peptidoform. As the final relevant note, our studies have suggested that, if the non-PTM protein-level reference is used to match the quantitative results of the PTM peptidoform, ~three times more PTM sites could be matched. This solution is of course less ideal, which impaired the turnover measurement [7] but seemed to be acceptable for the abundance profiling of the PTM sites among steady-state cells [46].
The blind man feels an elephant – to take a part for the whole.
Previously, a variety of experimental and bioinformatic strategies have been developed with the purpose of properly normalizing PTM proteomic data using the protein-level results (or analyzing the two jointly) for which we regret that we do not have space to cite and discuss here. The Viewpoint summarizes some of our understanding on the important concepts about peptide, protein, and PTM analyses by MS, rather than a comprehensive review on the relevant topics.
To conclude, it seems that, even with the high resolving power provided by modern mass spectrometers, the complete, high-throughput quantification of all or most proteoforms might be still formidable in the near future. While the field is waiting for improvements in the technology and informatic paradigms for studying proteoforms, a good stand-in is the peptidoform. We deduce that the peptidoform measurement and a peptidoform matching strategy currently can provide direct and relevant information to study the “PTM site-specific” biological functions, even if one is somehow (unfortunately) “blind” to the “proteoform universe” in the sample. This “taking part for the whole” strategy will likely work well for quantitative PTM analysis for a fairly long time. We advocate the bottom-up community should consider using the nomenclature “peptidoform” more often.
In essence, we argue that “bottom-up” strategy pioneered by Eng and Yates [47] about 25 years ago does not just measure the peptide surrogates, but the peptidoforms collapsed from the huge pool of proteoforms in a given sample. In fact, the relative quantification of a “peptidoform” between different conditions and samples provides quantitative information for understanding the PTM site-specific function, which many biologists care about and study for many years.
Acknowledgement.
Y.L. thanks Dr. Barbora Salovska, Dr. George Rosenberger, and Dr. Ben C. Collins for their careful proof reading and critical comments on the manuscript. Y.L. thanks the support from the National Institute of General Medical Sciences (NIGMS), National Institutes of Health (NIH) through Grant R01GM137031 to Y.L, as well as a Pilot Grant from Yale Cancer Center and a Career Enhancement Program Grant from the Yale SPORE in Lung Cancer (1P50CA196530).
Footnotes
The author has no conflict of interest to declare.
References
- [1].Pruitt KD, Tatusova T, Brown GR, & Maglott DR (2012). NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res, 40(Database issue), D130–135. doi: 10.1093/nar/gkr1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Smith LM, & Kelleher NL (2018). Proteoforms as the next proteomics currency. Science, 359(6380), 1106–1107. doi: 10.1126/science.aat1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Aebersold R, Agar JN, Amster IJ, Baker MS, Bertozzi CR, Boja ES, … Zhang B (2018). How many human proteoforms are there? Nat Chem Biol, 14(3), 206–214. doi: 10.1038/nchembio.2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Betts MJ, Wichmann O, Utz M, Andre T, Petsalaki E, Minguez P, … Russell RB (2017). Systematic identification of phosphorylation-mediated protein interaction switches. PLoS Comput Biol, 13(3), e1005462. doi: 10.1371/journal.pcbi.1005462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Floyd BM, Drew K, & Marcotte EM (2021). Systematic Identification of Protein Phosphorylation-Mediated Interactions. J Proteome Res, 20(2), 1359–1370. doi: 10.1021/acs.jproteome.0c00750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Huang JX, Lee G, Cavanaugh KE, Chang JW, Gardel ML, & Moellering RE (2019). High throughput discovery of functional protein modifications by Hotspot Thermal Profiling. Nat Methods, 16(9), 894–901. doi: 10.1038/s41592-019-0499-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wu C, Ba Q, Lu D, Li W, Salovska B, Hou P, … Liu Y (2021). Global and Site-Specific Effect of Phosphorylation on Protein Turnover. Dev Cell, 56(1), 111–124 e116. doi: 10.1016/j.devcel.2020.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Krahmer N, Najafi B, Schueder F, Quagliarini F, Steger M, Seitz S, … Mann M (2018). Organellar Proteomics and Phospho-Proteomics Reveal Subcellular Reorganization in Diet-Induced Hepatic Steatosis. Dev Cell, 47(2), 205–221 e207. doi: 10.1016/j.devcel.2018.09.017 [DOI] [PubMed] [Google Scholar]
- [9].Mann M, & Jensen ON (2003). Proteomic analysis of post-translational modifications. Nat Biotechnol, 21(3), 255–261. doi: 10.1038/nbt0303-255 [DOI] [PubMed] [Google Scholar]
- [10].Smith LM, Kelleher NL, & Consortium for Top Down, P. (2013). Proteoform: a single term describing protein complexity. Nat Methods, 10(3), 186–187. doi: 10.1038/nmeth.2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Murray-Zmijewski F, Slee EA, & Lu X (2008). A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev Mol Cell Biol, 9(9), 702–712. doi: 10.1038/nrm2451 [DOI] [PubMed] [Google Scholar]
- [12].Aebersold R, & Mann M (2016). Mass-spectrometric exploration of proteome structure and function. Nature, 537(7620), 347–355. doi: 10.1038/nature19949 [DOI] [PubMed] [Google Scholar]
- [13].Chait BT (2006). Chemistry. Mass spectrometry: bottom-up or top-down? Science, 314(5796), 65–66. doi: 10.1126/science.1133987 [DOI] [PubMed] [Google Scholar]
- [14].Goetze S, Frey K, Rohrer L, Radosavljevic S, Krutzfeldt J, Landmesser U, … Wollscheid B (2021). Reproducible Determination of High-Density Lipoprotein Proteotypes. J Proteome Res. doi: 10.1021/acs.jproteome.1c00429 [DOI] [PubMed] [Google Scholar]
- [15].Dingess KA, Gazi I, van den Toorn HWP, Mank M, Stahl B, Reiding KR, & Heck AJR (2021). Monitoring Human Milk beta-Casein Phosphorylation and O-Glycosylation Over Lactation Reveals Distinct Differences between the Proteome and Endogenous Peptidome. Int J Mol Sci, 22(15). doi: 10.3390/ijms22158140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Robinson AE, Binek A, Venkatraman V, Searle BC, Holewinski RJ, Rosenberger G, … Van Eyk JE (2020). Lysine and Arginine Protein Post-translational Modifications by Enhanced DIA Libraries: Quantification in Murine Liver Disease. J Proteome Res, 19(10), 4163–4178. doi: 10.1021/acs.jproteome.0c00685 [DOI] [PubMed] [Google Scholar]
- [17].Mallikarjun V, Richardson SM, & Swift J (2020). BayesENproteomics: Bayesian Elastic Nets for Quantification of Peptidoforms in Complex Samples. J Proteome Res, 19(6), 2167–2184. doi: 10.1021/acs.jproteome.9b00468 [DOI] [PubMed] [Google Scholar]
- [18].Zhang S, Raedschelders K, Santos M, & Van Eyk JE (2017). Profiling B-Type Natriuretic Peptide Cleavage Peptidoforms in Human Plasma by Capillary Electrophoresis with Electrospray Ionization Mass Spectrometry. J Proteome Res, 16(12), 4515–4522. doi: 10.1021/acs.jproteome.7b00482 [DOI] [PubMed] [Google Scholar]
- [19].Moyer TB, Heil LR, Kirkpatrick CL, Goldfarb D, Lefever WA, Parsley NC, … Hicks LM (2019). PepSAVI-MS Reveals a Proline-rich Antimicrobial Peptide in Amaranthus tricolor. J Nat Prod, 82(10), 2744–2753. doi: 10.1021/acs.jnatprod.9b00352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rosenberger G, Liu Y, Rost HL, Ludwig C, Buil A, Bensimon A, … Aebersold R (2017). Inference and quantification of peptidoforms in large sample cohorts by SWATH-MS. Nat Biotechnol, 35(8), 781–788. doi: 10.1038/nbt.3908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Labas V, Spina L, Belleannee C, Teixeira-Gomes AP, Gargaros A, Dacheux F, & Dacheux JL (2015). Analysis of epididymal sperm maturation by MALDI profiling and top-down mass spectrometry. J Proteomics, 113, 226–243. doi: 10.1016/j.jprot.2014.09.031 [DOI] [PubMed] [Google Scholar]
- [22].Labas V, Spina L, Belleannee C, Teixeira-Gomes AP, Gargaros A, Dacheux F, & Dacheux JL (2014). Data in support of peptidomic analysis of spermatozoa during epididymal maturation. Data Brief, 1, 79–84. doi: 10.1016/j.dib.2014.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bailleul G, Kravtzoff A, Joulin-Giet A, Lecaille F, Labas V, Meudal H, … Lalmanach AC (2016). The Unusual Resistance of Avian Defensin AvBD7 to Proteolytic Enzymes Preserves Its Antibacterial Activity. PLoS One, 11(8), e0161573. doi: 10.1371/journal.pone.0161573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Schulze S, Igiraneza AB, Kosters M, Leufken J, Leidel SA, Garcia BA, … Pohlschroder M (2021). Enhancing Open Modification Searches via a Combined Approach Facilitated by Ursgal. J Proteome Res, 20(4), 1986–1996. doi: 10.1021/acs.jproteome.0c00799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Rolfs Z, Frey BL, Shi X, Kawai Y, Smith LM, & Welham NV (2021). An atlas of protein turnover rates in mouse tissues. Nat Commun, 12(1), 6778. doi: 10.1038/s41467-021-26842-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Venable JD, Dong MQ, Wohlschlegel J, Dillin A, & Yates JR (2004). Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat Methods, 1(1), 39–45. doi: 10.1038/nmeth705 [DOI] [PubMed] [Google Scholar]
- [27].Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, … Aebersold R (2012). Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics, 11(6), O111 016717. doi: 10.1074/mcp.O111.016717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Liu Y, Buil A, Collins BC, Gillet LC, Blum LC, Cheng LY, … Aebersold R (2015). Quantitative variability of 342 plasma proteins in a human twin population. Mol Syst Biol, 11, 786. doi: 10.15252/msb.20145728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tucholski T, Cai W, Gregorich ZR, Bayne EF, Mitchell SD, McIlwain SJ, … Ge Y (2020). Distinct hypertrophic cardiomyopathy genotypes result in convergent sarcomeric proteoform profiles revealed by top-down proteomics. Proc Natl Acad Sci U S A, 117(40), 24691–24700. doi: 10.1073/pnas.2006764117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Regnier FE, & Kim J (2018). Proteins and Proteoforms: New Separation Challenges. Anal Chem, 90(1), 361–373. doi: 10.1021/acs.analchem.7b05007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, & Skrzypek E (2015). PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res, 43(Database issue), D512–520. doi: 10.1093/nar/gku1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ross AH, Baltimore D, & Eisen HN (1981). Phosphotyrosine-containing proteins isolated by affinity chromatography with antibodies to a synthetic hapten. Nature, 294(5842), 654–656. doi: 10.1038/294654a0 [DOI] [PubMed] [Google Scholar]
- [33].Smith LM, Agar JN, Chamot-Rooke J, Danis PO, Ge Y, Loo JA, … Consortium for Top-Down, P. (2021). The Human Proteoform Project: Defining the human proteome. Sci Adv, 7(46), eabk0734. doi: 10.1126/sciadv.abk0734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bludau I, Frank M, Dorig C, Cai Y, Heusel M, Rosenberger G, … Aebersold R (2021). Systematic detection of functional proteoform groups from bottom-up proteomic datasets. Nat Commun, 12(1), 3810. doi: 10.1038/s41467-021-24030-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Rosenberger G, Heusel M, Bludau I, Collins BC, Martelli C, Williams EG, … Califano A (2020). SECAT: Quantifying Protein Complex Dynamics across Cell States by Network-Centric Analysis of SEC-SWATH-MS Profiles. Cell Syst, 11(6), 589–607 e588. doi: 10.1016/j.cels.2020.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dermit M, Peters-Clarke TM, Shishkova E, & Meyer JG (2021). Peptide Correlation Analysis (PeCorA) Reveals Differential Proteoform Regulation. J Proteome Res, 20(4), 1972–1980. doi: 10.1021/acs.jproteome.0c00602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Krug K, Mertins P, Zhang B, Hornbeck P, Raju R, Ahmad R, … Mani DR (2019). A Curated Resource for Phosphosite-specific Signature Analysis. Mol Cell Proteomics, 18(3), 576–593. doi: 10.1074/mcp.TIR118.000943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Seymour RW, van der Post S, Mooradian AD, & Held JM (2021). ProteoSushi: A Software Tool to Biologically Annotate and Quantify Modification-Specific, Peptide-Centric Proteomics Data Sets. J Proteome Res, 20(7), 3621–3628. doi: 10.1021/acs.jproteome.1c00203 [DOI] [PubMed] [Google Scholar]
- [39].Huang da W, Sherman BT, & Lempicki RA (2009). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res, 37(1), 1–13. doi: 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, … Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun, 10(1), 1523. doi: 10.1038/s41467-019-09234-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Nesvizhskii AI, & Aebersold R (2005). Interpretation of shotgun proteomic data: the protein inference problem. Mol Cell Proteomics, 4(10), 1419–1440. doi: 10.1074/mcp.R500012-MCP200 [DOI] [PubMed] [Google Scholar]
- [42].Li YF, Arnold RJ, Li Y, Radivojac P, Sheng Q, & Tang H (2009). A bayesian approach to protein inference problem in shotgun proteomics. J Comput Biol, 16(8), 1183–1193. doi: 10.1089/cmb.2009.0018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Potel CM, Kurzawa N, Becher I, Typas A, Mateus A, & Savitski MM (2021). Impact of phosphorylation on thermal stability of proteins. Nat Methods, 18(7), 757–759. doi: 10.1038/s41592-021-01177-5 [DOI] [PubMed] [Google Scholar]
- [44].Smith IR, Hess KN, Bakhtina AA, Valente AS, Rodriguez-Mias RA, & Villen J (2021). Identification of phosphosites that alter protein thermal stability. Nat Methods, 18(7), 760–762. doi: 10.1038/s41592-021-01178-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, … Selbach M (2011). Global quantification of mammalian gene expression control. Nature, 473(7347), 337–342. doi: 10.1038/nature10098 [DOI] [PubMed] [Google Scholar]
- [46].Gao E, Li W, Wu C, Shao W, Di Y, & Liu Y (2021). Data-independent acquisition-based proteome and phosphoproteome profiling across six melanoma cell lines reveals determinants of proteotypes. Mol Omics, 17(3), 413–425. doi: 10.1039/d0mo00188k [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Eng JK, McCormack AL, & Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom, 5(11), 976–989. doi: 10.1016/1044-0305(94)80016-2 [DOI] [PubMed] [Google Scholar]