

CONSPECTUS:
This Account summarizes recent findings centered on the role that redox partner binding, allostery, and conformational dynamics plays in cytochrome P450 proton coupled electron transfer. P450s are one of Nature’s largest enzyme families and it is not uncommon to find a P450 wherever substrate oxidation is required in the formation of essential molecules critical to the life of the organism or in xenobiotic detoxification. P450s can operate on a remarkably large range of substrates from the very small to the very large, yet the overall P450 three-dimensional structure is conserved. Given this conservation of structure, it is generally assumed that the basic catalytic mechanism is conserved. In nearly all P450s, the O2 O–O bond must be cleaved heterolytically enabling one oxygen atom, the distal oxygen, to depart as water and leave behind a heme iron-linked O atom as the powerful oxidant that is used to activate the nearby substrate. For this process to proceed efficiently, externally supplied electrons and protons are required. Two protons must be added to the departing O atom while an electron is transferred from a redox partner that typically contains either a Fe2S2 or FMN redox center. The paradigm P450 used to unravel the details of these mechanisms has been the bacterial CYP101A1 or P450cam. P450cam is specific for its own Fe2S2 redox partner, putidaredoxin or Pdx, and it has long been postulated that Pdx plays an effector/allosteric role by possibly switching P450cam to an active conformation. Crystal structures, spectroscopic data, and direct binding experiments of the P450cam–Pdx complex provide some answers. Pdx shifts the conformation of P450cam to a more open state, a transition that is postulated to trigger the proton relay network required for O2 activation. An essential part of this proton relay network is a highly conserved Asp (sometimes Glu) that is known to be critical for activity in a number of P450s. How this Asp and proton delivery networks are connected to redox partner binding is quite simple. In the closed state, this Asp is tied down by salt bridges, but these salt bridges are ruptured when Pdx binds, leaving the Asp free to serve its role in proton transfer. An alternative hypothesis suggests that a specific proton relay network is not really necessary. In this scenario, the Asp plays a structural role in the open/close transition and merely opening the active site access channel is sufficient to enable solvent protons in for O2 protonation. Experiments designed to test these various hypotheses have revealed some surprises in both P450cam and other bacterial P450s. Molecular dynamics and crystallography show that P450cam can undergo rather significant conformational gymnastics that result in a large restructuring of the active site requiring multiple cis/trans proline isomerizations. It also has been found that X-ray driven substrate hydroxylation is a useful tool for better understanding the role that the essential Asp and surrounding residues play in catalysis. Here we summarize these recent results which provide a much more dynamic picture of P450 catalysis.
Graphical Abstract
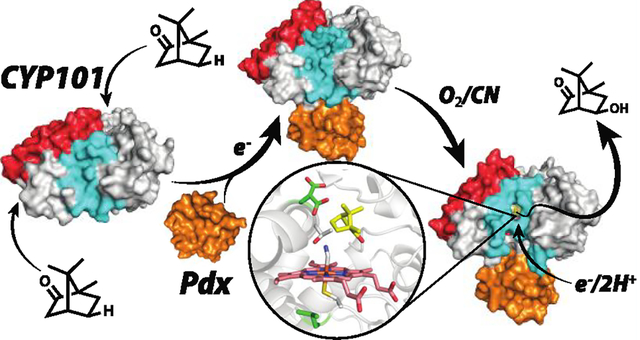
INTRODUCTION
Cytochromes P450 are remarkable catalysts that can selectively oxidize unactivated C–H bonds in a variety of organic compounds. Understanding how P450s do so under physiological conditions with near 100% efficiency in many P450s has been one of the great challenges in modern enzymology. The key to this puzzle is, of course, understanding how the surrounding protein tunes heme reactivity and controls substrate selectivity. Many heme enzymes, with the most well understood being peroxidases and P450s, share similar mechanistic steps utilizing a resting state Fe(III) heme iron and O2 or peroxide to form the reactive ferryl intermediate, Fe(IV)=O (Figure 1). While peroxidases bind H2O2 directly to Fe(III) without the need for an external source of electrons, P450s require two electrons to be externally supplied from redox partners to first give the Fe(II)–oxy complex and then the dihydroperoxy species, Fe(III)–OOH2. The O–O bond of Fe(III)–OOH2 undergoes heterolytic cleavage resulting in the departure of water and generation of the Fe(IV)=O oxidant and porphyrin radical, compound I. Despite the identity of this key mechanistic intermediate being similar between these enzymes, the respective capabilities of their oxidants are quite different. In P450s, but not peroxidases, Fe(IV)=O is able to abstract an H atom from an unactivated C–H bond. The essential difference arises from the nature of heme ligation. Peroxidases use a histidine axial ligand while P450s utilize cysteine. The prevailing view is that the increased electron donating ability of Cys increases the pKa of the ferryl O atom. Since there is a direct correlation between the pKa of the ferryl O atom and the O–H bond strength formed upon H atom abstraction, the higher pKa helps to overcome the thermodynamic barrier of breaking a C–H bond in favor of an O–H bond.5–7 How the enzyme protects itself from such powerful oxidants and how the local structure tunes reactivity are active areas of research and focus primarily on the heme center and its local surroundings.
Figure 1.
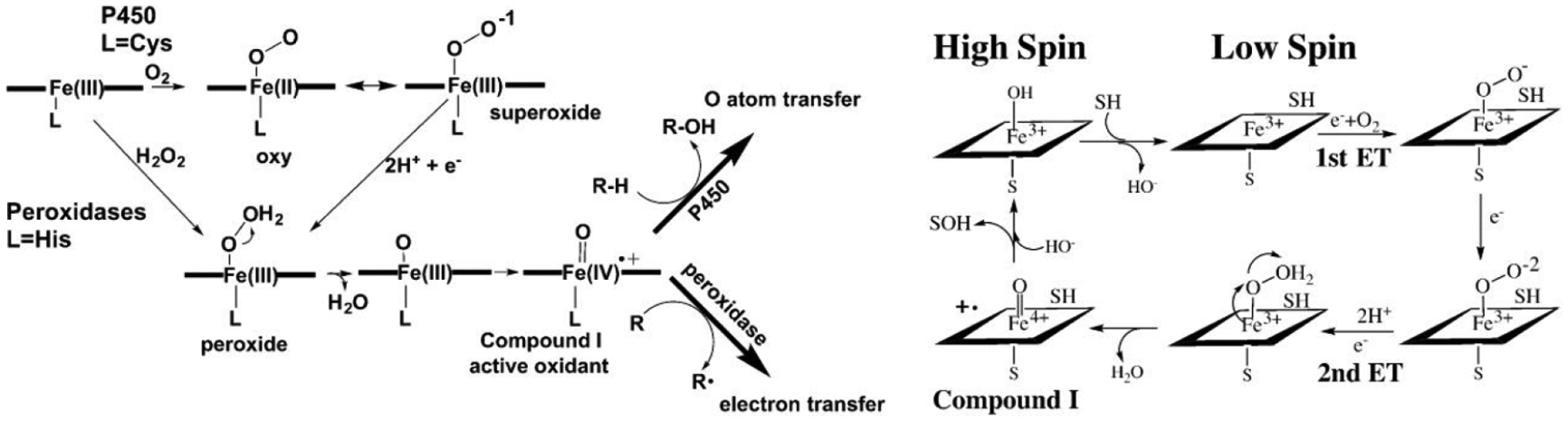
On the left is a comparison of the O2/H2O2 activation by P450s and peroxidases and on the right is a more detailed outline of the P450 mechanism. In P450s, the iron must first be reduced to Fe(II) to enable O2 to bind. The favored resonance form shown for the oxy complex is ferric-superoxide, Fe(III)–OO–. This is followed by a second electron transfer step together with protonation of the iron-linked dioxygen to give the peroxide intermediate. Peroxidases bind H2O2 directly to the Fe(III) iron. The peroxide O–O bond is cleaved heterolytically leaving behind the reactive compound I oxidant.
As noted, an additional complexity of P450s relative to peroxidases is that the enzyme does not act alone but must receive electrons from a redox partner. It has become increasingly clear that delivery of electrons may not be the only role of the redox partner, but that protein–protein interactions result in structural changes that are required for activity. These changes appear to involve a direct effect on O2 activation by helping to arm the proton relay network required for catalysis (Figure 1).
Much of what we understand about P450 electron transfer derives from the well-studied Pseudomonas putida P450cam and its Fe2S2 redox partner, Pdx, although this effort in our lab is expanding to include other P450s. In this review, we summarize early work attempting to unravel the effector role of Pdx and then discuss more recent contributions to our understanding of the P450cam–Pdx complex as well as a few other select P450s. The picture that emerges from these efforts is that P450s are much more flexible than heretofore thought which raises additional questions on the role such plasticity plays in catalysis.
EFFECT OF REDOX PARTNER BINDING
The first few P450 crystal structures showed that the overall P450 fold is conserved8–12 and that, in the substrate complex, the active site is effectively sealed off from the outside world. In many P450s the F/G helices and the loop connecting these helices undergoes a large open/close motion which provides a path for substrate entry (Figure 2). It is generally thought in all P450s the redox partner binds on the side opposite to the substrate entry channel providing the closest approach to the heme thereby minimizing the electron transfer distance.
Figure 2.
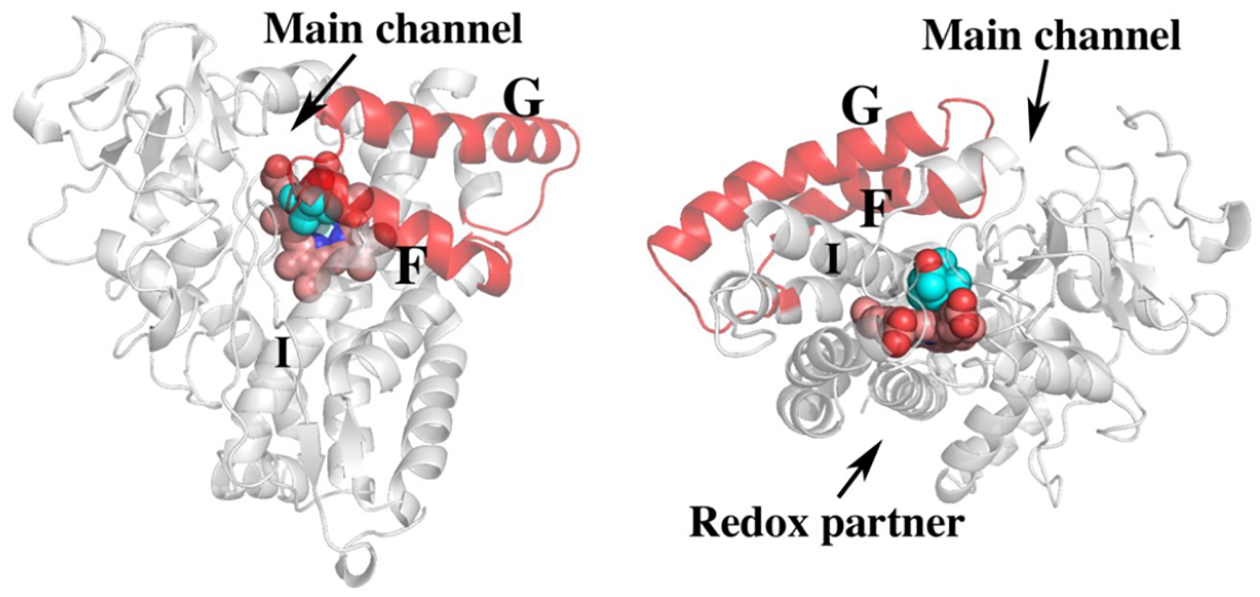
Two views of P450cam highlighting the F and G helices. In many P450s the F/G helices undergo significant motion that opens up the heme pocket providing the primary entry path to the active site in most, but not all, P450s. The I helix runs over the surface of the heme and provides key substrate contact interactions in addition to residues important for O2 activation. The redox partner binds to a concave surface just under the proximal side of the heme. The first indication that Pdx binding results in long-range structural effects is that Pdx binding at the indicated site results in perturbation of NMR resonances on the opposite (F/G helical) side of the protein.13
The mechanism for providing protons for O2 activation involves a conserved Asp/Glu found in many P450s (Figure 3). Asp251 in P450cam (Figure 3) is required for catalysis14 and the corresponding Asp or Glu in three other bacterial P450s have been shown to be critical for activity.15–17 However, Asp251 is tied up in salt bridges with Lys178 and Arg186 (Figure 3) and these ionic interactions must be broken in order for Asp251 to carry out its proposed function in proton transfer. Crystal structures1,18 show that Pdx binding induces a structural change that results in a rupture of these salt bridges which frees Asp251 to serve its function in proton transfer. These Pdx specific structural changes explain why no other redox partner can support P450cam catalysis.19 Prior to determination of the P450cam–Pdx crystal structure, it was well-known that Pdx binding must induce significant structural changes. As noted, it was demonstrated early on that P450cam is selective for Pdx leading to the conclusion that Pdx serves an “effector” role in P450cam catalysis.19 Much later it was found that the binding of oxidized Pdx to the P450cam-substrate complex shifts P450cam from high-spin toward the low-spin state.20 Low-spin P450 is typically associated with water coordinating to the heme iron and when substrate binds, this water is displaced thereby giving a 5-coordinate high-spin state. That Pdx binding shifts the spin state equilibrium back toward low spin implies that water can now gain access to the active site and coordinate the heme iron. When it became possible to apply NMR to a large protein like P450cam, it was determined that Pdx binding perturbs residues in P450cam well removed from the assumed Pdx binding site including the F/G helical regions, which undergo the largest motion in the open/close transition.13 While at the time these observations were not associated with changes resulting in opening of the active site, these earlier results are fully consistent with what we now know about the effect of Pdx binding in shifting P450cam toward a more open state. Another elegant approach that supports the open/close transition in solution are the electron paramagnetic resonance (EPR) spin labeling experiments from the Goodin lab.23,24 Here two spin labels are attached to engineered Cys residues and the distance between the two sites is measured with/without Pdx bound. The increase in distance when Pdx is bound is consistent with opening of the active site. Isothermal titration calorimetry studies found that Pdx binds more tightly to substrate-free than substrate-bound P450cam indicating an increased affinity for the open form.25 Further evidence that Pdx is critical in forming the proton relay network derives from recent molecular dynamics/quantum mechanics studies. Here it was found that the proton relay network depicted in Figure 3 involving both Asp251 and Thr252 forms only when Pdx is bound.21
Figure 3.
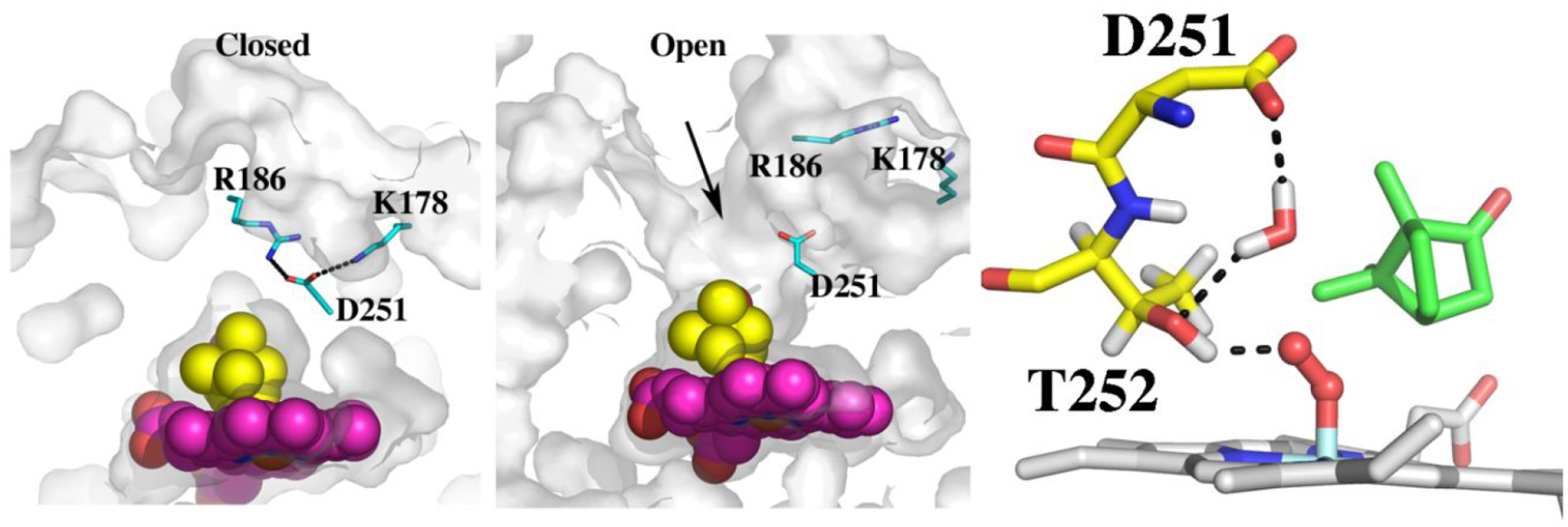
Crystal structures of the closed (2CPP) and open (4JX1) forms of P450cam. In the closed state, Asp251 is tied up with salt bridges to Arg186 and Lys178. When Pdx binds, these salt bridges are broken and the active site opens. The arrow indicates where a second camphor molecule is observed in 3 of the 4 asymmetric units in 4JX1. Also shown is the proposed proton relay network taken after Ugur et al.21 Thr252 is the direct proton donor to the iron-linked dioxygen while a water bridges between Asp251 and Thr252. For Asp251 to shuttle protons into the active site it must be able to adopt different rotamer conformations. The structure of the P450cam–Pdx complex22 in the presence of cyanide which mimics the oxy complex shows that Asp251 can, indeed, adopt the “in” rotamer as shown.
Despite the agreement between crystal structures and solution studies, there has been some controversy on whether or not the open form observed in the crystal structures of the P450cam–Pdx complex occurs in solution based on NMR studies.26 Molecular dynamics (MD) has helped to reconcile some of these discrepancies. Figure 4 shows the results of a principal component analysis of 3 long MD runs: closed, open without Pdx, and open with Pdx. Here, the α carbon backbones from each run were projected on to the average of the open and closed structures and the two principal components represent the two dominant correlated motions of P450cam. As might be expected, snapshots from the closed state MD run cluster around the closed structure while the open structure clusters around the open structure. Note, however, that the snapshots are much more scattered in the open structure run. Most of this scatter is due to flexibility of the F/G helical region. Starting with exactly the same open structure except with Pdx bound, the structure moves toward the more closed state. This is consistent with similar, albeit much longer, MD runs by Ugar et al.21 Despite this movement toward a more closed form, the salt bridges to Asp251 do not re-form2,21 so the proton relay network required for O2 activation remains armed and ready. These MD data also are more consistent with NMR studies.27 The MD simulations help to explain another puzzle on how regio- and stereo-selectivity of camphor hydroxylation is maintained in the more open structure. The MD runs of the open state indicate that camphor is in position for stereoselective hydroxylation at C5 for less that 40% of the time in 5 out of 6 100 ns MD runs and 50% in one run. However, with Pdx bound the camphor remains in position for C5 hydroxylation 60% of the time in 3 of 6 100 ns runs and >90% in 3 other 100 ns runs. Therefore, while an “open” conformation is favored, Pdx binding additionally stabilizes camphor in the proper position for stereo- and regioselective hydroxylation. The conclusion that Pdx binding helps stabilize camphor in the active site was initially proposed using NMR spectroscopy.13 Another factor to consider is that in the P450cam–Pdx crystal structure, a second camphor molecule is bound in the open substrate access channel just above the normal substrate binding site and the electron density maps show that this second camphor is well ordered.1 This second camphor also helps to stabilize the structure and hold the substrate to be oxidized in place which indicates that the fully open structure may well be active if the access channel is occupied with a second camphor molecule.
Figure 4.
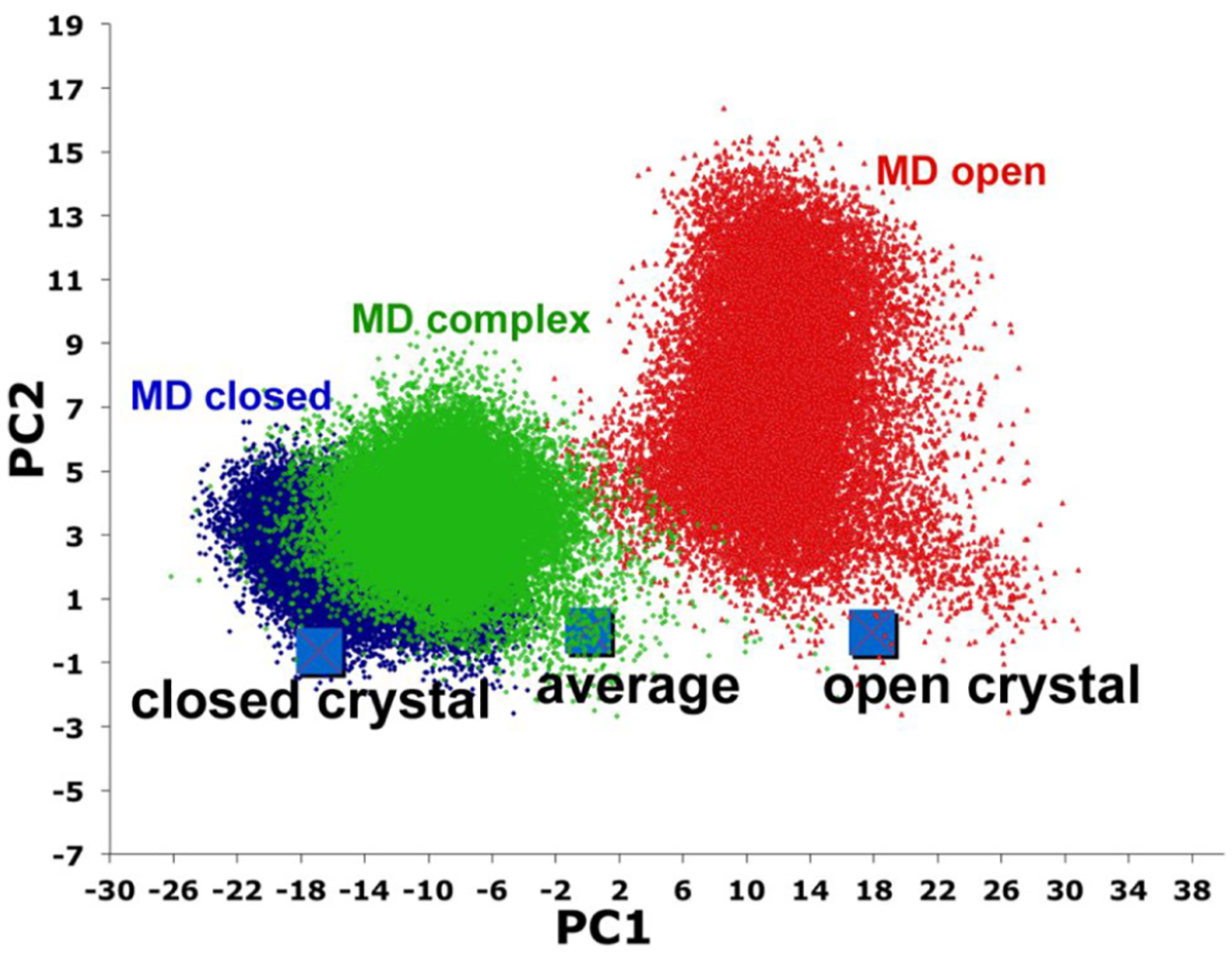
Principal component analysis where each dot is a snapshot taken from ≈500 ns MD runs.2 A plot of the first two primary principal components, PC1 and PC2 (orthogonal to PC1), provides a simple representation on the majority of the correlated motion in going from the average structure to closed and open, where the average structure is midway between the open/close transition. The Cα atoms of each snapshot is a rms fit to the average structure and from the covariance matrix the first two major principal components (PC1 and PC2) that define the dominant correlated motions are computed. Since the average structure is rms fit to itself the average structure sits at PC1 = PC2 = 0 while the magnitude of PC1 and PC2 are identical for the open and closed structures but have opposite signs. The open structure is taken from the P450cam–Pdx complex crystal structure. Without Pdx bound (red) P450cam remains closer to the open state although there is considerable scatter due primarily to large motions in the F/G helical region. The closed MD run (blue) stays close to the closed state. With Pdx bound, the open state moves more toward the closed (green). In the various MD simulations reported both P450cam and Pdx were modeled in the oxidized states. Reproduced with permission from ref 2. Copyright 2017 American Chemical Society.
EXPERIMENTAL EVIDENCE THAT LOOSENING OF THE STRUCTURE IS REQUIRED FOR ACTIVITY: X-RAY ENZYMOLOGY
Reducing equivalents generated during X-ray exposure have the potential for driving P450 reactions in crystallo. In P450cam there is no indication that product is formed in crystallo while in the P450cam–Pdx complex product clearly is formed.1 This implies that X-rays can drive the P450cam reaction in a more open state but not closed. Whether or not in crystallo substrate hydroxylation uses the same mechanism as in solution remains an open question. During data collection crystals are maintained in a liquid N2 stream although it is very doubtful that O2 is totally excluded, in which case the normal O2 activation process can proceed. Alternatively, potent X-ray generated reducing power could conceivably initiate redox chemistry resulting in compound I, Fe(IV)=O, where the O atom derives from water. In either case, a more open conformation enabling solvent entry is required. Further evidence that a “looser” structure is required for activity derives from mutagenesis studies where one of the salt bridges to Asp251 (Figure 3) was removed by replacing Arg186 with Ala. The mutant retains substantial activity and is highly coupled, but product is formed in the X-ray beam.2 CYP101D1, a bacterial P450 that carries out exactly the same reaction as P450cam,28 also forms product in the X-ray beam.15 CYP101D1 has the same essential Asp as P450cam (Asp259 in CYP101D1) but is missing one of the salt bridges since the homologue to Lys178 is Gly. These various studies indicate that the critical Asp salt bridges must be loosened in order to allow proton transfer required for O2 activation. While CYP101D1 is very similar to P450cam, the binding of its Fe2S2 redox partner does not shift CYP101D1 more toward low spin and CYP101D1 is not specific for its own redox partner.29 It appears that because the critical Asp is not tightly locked down by two salt bridges, redox assisted conformational changes are not required for activity. CYP101D1 is armed and ready while P450cam needs help from its redox partner.
The Asp251Glu mutant in P450cam and the corresponding Asp259Glu mutant in CYP101D1 were both expected to have modest effects. However, this mutant eliminated all activity in P450cam while with CYP101D1 about 28% wild-type activity is retained and is about 83% coupled.4 Single turnover experiments showed that in CYP101D1 the second electron transfer step (Figure 1), reduction of the oxy complex, is slowed and has an increased kinetic solvent isotope effect indicating that in the Asp259Glu mutant proton transfer required for O2 activation is slowed and, possibly, rate limiting. Why P450cam is inactive remained a puzzle although a close examination around the site of mutation revealed a subtle but important difference between P450cam and CYP101D1. In P450cam, the Glu251 mutant side chain is within H-bonding distance to Asp182 (Figure 5). One does not normally consider two carboxyl groups H-bonding with one another yet when the H-bond donor/acceptor pair have a matched or similar pKa, the H-bond is especially strong. Molecular dynamics simulations indicate that this H-bond together with the Glu251-Lys178 salt bridge is significantly more stable in the D251E mutant than in wild-type P450cam. This raised the possibility that the D251E is “locked” down and cannot readily adopt the more open active conformation. Indeed, it was found that Pdx is unable to shift the D251E mutant toward low spin as in the wild-type enzyme. The double mutant, D251E/K178G, recovers about 18% wild-type activity presumably because the double mutant is no longer locked in a closed conformation. It thus appears that the lack of activity in the D251E mutant is due to the inability of Pdx binding to break the stronger H-bonding interactions involving the mutant Glue251 side chain. None of this is an issue with CYP101D1 since the residue corresponding to Asp182 is an Asn.
Figure 5.
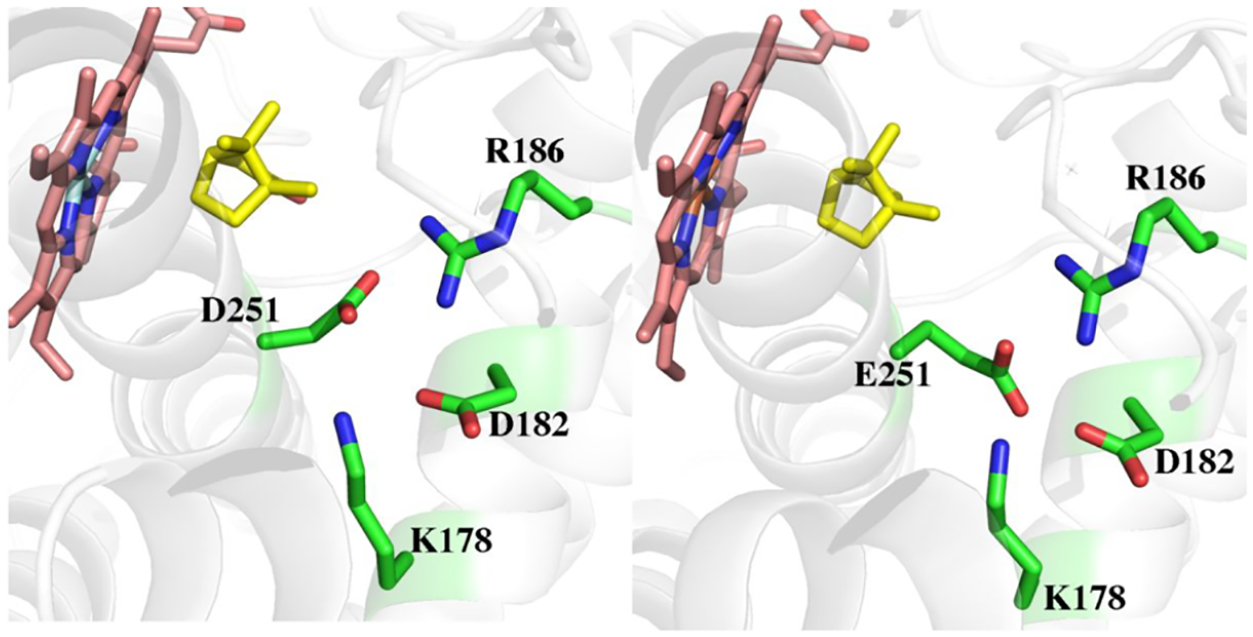
Structure of wild-type (2CPP) and the D251E P450cam mutant (6WE6). The mutant side chain is close enough to Asp182 for H-bonding interactions. The corresponding residue in CYP101D1 is Asn.
SUBSTRATE ALLOSTERY
Related to the open/close transitions modulated by redox partner binding is substrate allostery and associated structural changes. Simple spectral titration studies showed that at high camphor concentrations, P450cam shifts back toward low spin and it was concluded that there may be a second camphor binding site.30,31 Based on what we now know, binding to this presumed second site should open up the active site similar to the effects of Pdx. The first hint as to the location of this site derived from NMR studies where it was found that P450cam has a second much weaker binding site about 15 Å from the iron and that this site is very near a Thr residue32 although which of the many Thr residues in P450cam remained unknown. Another NMR study33 also showed that at high camphor concentrations, Leu166 is perturbed either due to direct interaction with camphor or via longer range structural changes. The location of an allosteric site near Leu166 is consistent with both NMR studies. This site is ≈15 Å from the iron and has a nearby Thr residue, Thr217, although there was no direct structural data until recent MD simulations indicated that a second camphor can bind near Leu166.3 In these simulations three camphor molecules were randomly placed in a solvent box surrounding substrate-free P450cam. The goal was to see if camphor can find its way into the active site, but quite surprisingly several microsecond simulations showed that the site near Leu166 is highly populated with camphor (Figure 6). A reexamination of the crystal structure of the open P450cam crystal structure showed that crystals of substrate free P450cam soaked in camphor34 had a large lobe of unaccounted for electron density at this site. Thus, the computationally identified second camphor site is consistent with the NMR and crystallographic data. The MD simulations also showed that a new active site access channel opens when camphor binds to the “allosteric” site (Figure 6). Well before these more recent MD studies, steered MD simulations of P450cam indicated possible alternate channels.35 The opening of this second channel additionally offers an explanation of early spectroscopic titrations and why binding to the second camphor site is associated with a switch to low spin. In this new “open” conformation, solvent can readily access the active site enabling water to coordinate the heme iron. This study was followed by crystallographic confirmation of this new access channel. Crystals of the P450cam–Pdx complex were soaked in cyanide since CN– is an excellent mimic of the oxy complex.36,37 Quite surprisingly something as seemingly simple as CN– coordination to the heme iron results in a very large structural change that opens up the same new active site channel identified in the MD simulations.22 This change requires a major restructuring of the active site and B/C loop region including the cis to trans isomerization of two proline residues, Pro89 and Pro105. How such large rearrangements occur within the confines of the crystal lattice suggests that the energetic barriers to such conformational gymnastics are fairly low. As noted, what is especially remarkable is the cis/trans isomerization of peptide bonds, which were suggested to occur upon Pdx binding by NMR studies38 but had never been crystallographically observed. An additional surprising result was that owing to rearrangement of the B/C loop induced by CN–, hydrogen bonds to the 7-propionate carboxylate break and allow for the reorientation of the 7-propionate group. While there are more than 100 crystallographic structures of P450cam and its associated mutants and complexes deposited in the Protein Data Bank, none display both heme propionate groups in the same dihedral orientation. This pseudo-cis orientation of the propionate groups is common in many other P450s but had not been observed in P450cam. This result supports work where P450cam was reconstituted with a “one-legged” heme that suggested the interacting residues, Asp297, Arg299, and Gln322 serve to prevent solvent access to the active site.39 This reorientation of the heme was also noted in the MD studies that postulated the opening of channel 2.22
Figure 6.
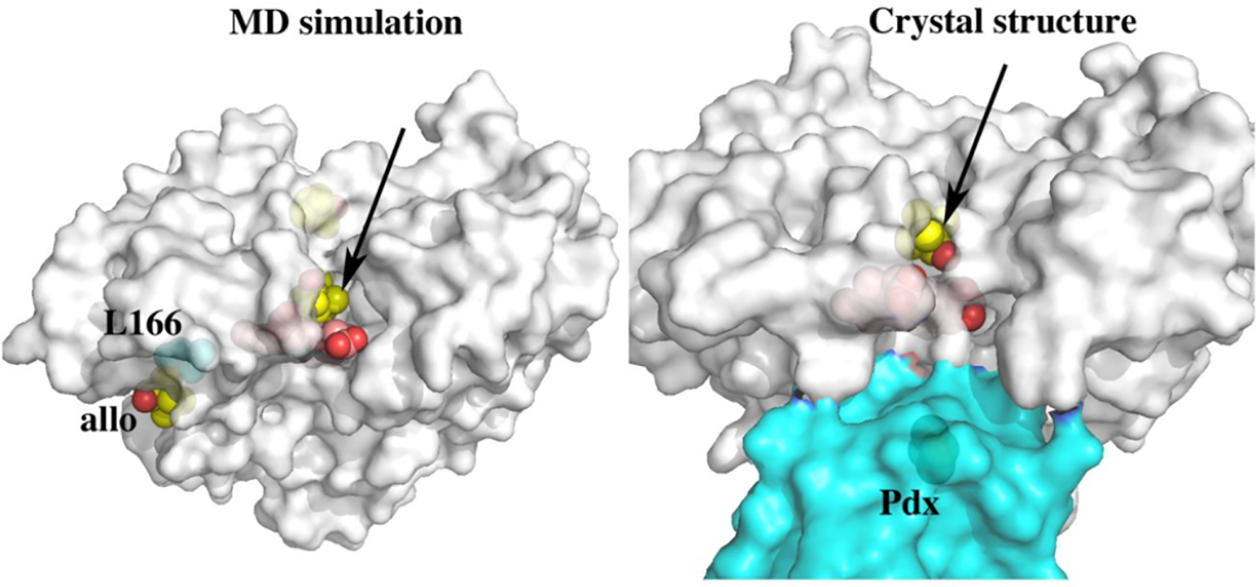
On the left and indicated by the arrow is a new active site access channel that opens up when substrate binds in the allosteric (allo) site. The camphor molecules are indicated as yellow spheres. The “allo” site is where Leu166 is located. The dynamic picture that emerges from these simulations is that camphor binding to the allosteric site is favored when the new channel is open but, once camphor binds in the active site, the structure closes down and the allosteric camphor dissociates. On the right is the crystal structure of the P450cam–Pdx complex soaked in CN– (6NBL). The same new access channel identified in the MD simulations opens up when CN– binds. When CN– binds, the I helix that is close to both the CN– and camphor must move. This motion is communicated to the surface resulting in major restructuring of the active site, cis/trans isomerization of two proline residues, and opening of the new channel.
Further evidence that CN– causes major changes derives from recent DEER (double electron–electron resonance) measurements coupled with molecular dynamics studies. Cyanide binding to the P450cam–Pdx complex results in P450cam adopting an intermediate state between open and closed and that the I helix undergoes a large conformational rearrangement as well.40 These changes result in water being positioned into the active similar to that shown in Figure 3. The agreement between the solution experiments,40 molecular dynamics,3,21,40 crystallography,22 and NMR13,27 is exceptional and strengthens the argument that Pdx binding is essential in forming the proton relay network required for O2 activation.
These results offer a dynamic picture of P450cam catalysis. The identification of channel 2 and the large restructuring of the active site suggests the possibility that substrate enters via channel 1 while product egress occurs via channel 2. While in the P450cam–Pdx structure the substrate access channel is in the open conformation and contains 2 molecules of camphor, the CN–P450cam–Pdx has channel 2 open, while channel 1 is closed. The cis–trans isomerization of the proline residues on the B/C loop allows for the B′-helix to unfurl and remain in contact with the F/G loop. This allows the F/G helices to move into an open conformation, Asp251 salt bridges to break, and for the Asp to adopt a conformation that assists with proton shuttling, all without opening the original substrate access channel. In this way, camphor migration occurs in a unidirectional fashion ensuring that substrate entry and product exit do not limit catalysis.
OTHER P450S
While our understanding on the structural underpinnings of how redox partner binding relates to conformational changes required for O2 activation has advanced considerably with P450cam, a major unanswered question is whether or not P450cam is unique or do other P450s display such redox partner dependent complexities. It is well-known that many, if not most, P450s can be supported by non-natural redox partners. There is, however, one feature that a small handful of P450s do share in common. The binding of oxidized Pdx to the oxy-P450cam complex dramatically destabilizes the oxy complex (Table 1).41
Table 1.
Half-Life of Oxy Complexes with and without Redox Partner
Since electron transfer is not involved, then binding alone somehow destabilizes the oxy complex. We have found that the same destabilization of the oxy complex occurs in two other P450s, P450cin43 and CYP101D129 (Table 1), both of which can be supported by non-natural redox partners. Since we know that the CYP101D1 redox partner does not shift CYP101D1 more toward low spin, it is unlikely that the cause of oxy-complex destabilization is the result of a switch to a more open conformation. There also is an interesting mutant of P450cam, L358P, that exhibits properties similar to the P450cam–Pdx complex.42,44 As shown in Figure 7, L358 is just under the heme and, when Pdx binds, Leu358 adopts a rotamer that pushes up against the heme. Pro358 in the mutant also packs tighter against the proximal surface of the heme and it was concluded that this “push” on the proximal surface of the heme mimics the effects of Pdx binding. Most important for the present discussion, the L358P oxy complex exhibits decreased stability (Table 1).42 If redox partners all bind to the same region as observed in the three known crystal structures of P450 redox complexes,1,18,45,46 then destabilization of the oxy complex very likely has to do with this redox partner “push” on the proximal side of the heme. Resonance Raman studies show that there is a small increase in the S–Fe stretch frequency when Pdx binds, indicating a slightly stronger (shorter) S–Fe bond. This could then increase electron density distribution to the iron-linked dioxygen which might be the basis for a decrease in stabilization and could potentially be important for O2 activation. Molecular dynamics simulations also noted the importance of this rotameric conformation and were utilized to reconcile pseudocontact shift (PCS) NMR data that were unable to back predict the location of several residues.27 The NMR data were much better fit to these new MD simulations and suggested that L358 oscillates between two conformations: gauche and trans that represent the normal and “packed” confirmations, respectively.27
Figure 7.
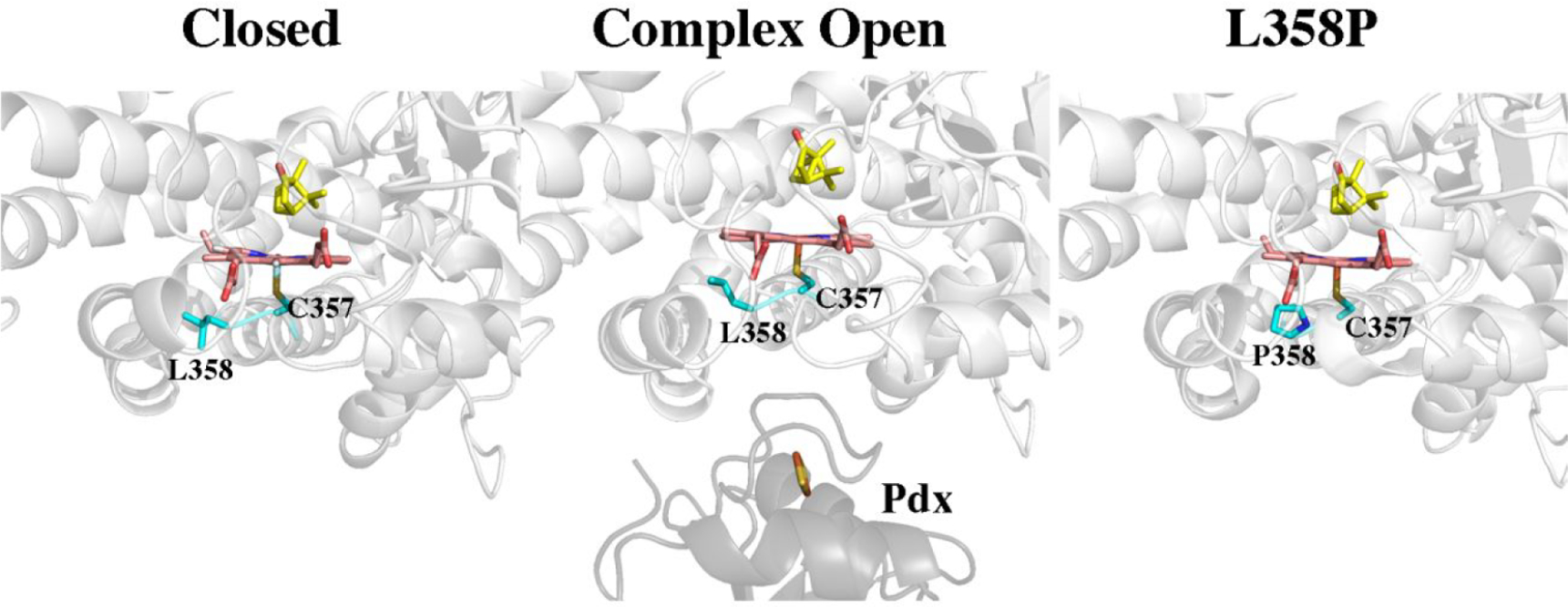
Structures of the closed (2CPP), P450cam–Pdx (open, 4JX1), and L358P mutant (1T86). When Pdx binds, Leu358 adopts a rotamer conformation which is closer to the heme thereby “pushing” on the proximal face of the heme. Replacement of Leu258 with Pro provides a similar push effect. As a result, the L358P mutant exhibits properties similar to P450cam when complexed with Pdx.42
CONCLUSIONS
The recent work on P450cam summarized in this review shows that P450cam can adopt a number of conformational states some of which result in a dramatic restructuring of the active site and the opening and closing of multiple active site access routes. Such flexibility could enable substrate binding via one route and substrate egress via a second which would promote high substrate turnover. The effector role of Pdx is to promote structural changes which enable the proton relay network required for O2 activation to function. One common feature shared by a few other P450s is that redox partner binding destabilizes the P450–oxy complex. This could be due to structural changes that “push” on the proximal face of the heme that potentially increase the electron donating ability of the Cys ligand thereby destabilizing the oxy complex. Thus, redox partner binding may play a more direct role in O2 activation. A major unresolved question, however, is why P450cam is so tightly regulated while other well studied P450s are not, although it is most unlikely that P450cam is alone in exhibiting redox partner selectivity and conformational dynamics. To understand the potential reason underlying P450cam’s complexity requires appreciation of the biological advantage of these allosteric effects. P450cam and the two additional proteins required for camphor oxidation, Pdx and the flavoprotein, PdR, are controlled by a camphor-dependent repressor on the cam operon in Pseudomonas putida.47 The cam operon is leaky48 so even at camphor concentrations too low to support life, P450cam is present. It would be an idle waste of reducing equivalents if P450cam were active at such low levels of camphor and if other potential redox partners were able to support P450cam catalysis. Therefore, Nature selected a more complex level of control to ensure that P450cam is active only when camphor concentrations are at a level compatible with life. While speculative, this explanation provides a point of departure in looking for other P450s that exhibit similar biological requirements.
KEY REFERENCES.
Tripathi, S.; Li, H.; Poulos, T. L. Structural basis for effector control and redox partner recognition in cytochrome P450. Science 2013, 340, 1227–1230.1 The crystal structure of the P450cam–Pdx complex shows that Pdx promotes a swtich from the closed to open form of P450cam. This frees Asp251 from ionic interactions so it can serve its role in proton coupled electron transfer.
Batabyal, D.; Richards, L. S.; Poulos, T. L. Effect of Redox Partner Binding on Cytochrome P450 Conformational Dynamics. J. Am. Chem. Soc. 2017, 139, 13193–13199.2 It is possible that X-ray generated reducing equivalents can drive P450 chemistry in crystallo. This is not observed in wild-type P450cam, but in mutants that free Asp251 from salt brdiges, X-ray driven product formation is found in crystallo.
Follmer, A. H.; Mahomed, M.; Goodin, D. B.; Poulos, T. L. Substrate-Dependent Allosteric Regulation in Cytochrome P450cam (CYP101A1). J. Am. Chem. Soc. 2018, 140, 16222–16228.3 Multiple microsecond molecular dynamics simulations predict an allosteric binding site for camphor that is consistent with previous NMR and crystallographic studies. Binding to the allosteric site is coupled with the opening of a new access channel to the active site
Amaya, J. A.; Batabyal, D.; Poulos, T. L. Proton Relay Network in the Bacterial P450s: CYP101A1 and CYP101D1. Biochemistry 2020, 59, 2896–2902.4 The Asp251Glu mutation results in a totally inactive P450cam. Molecular dynamics and spectral studies indicate that the mutant Glu251 is tied up in an especially strong set of H-bonds that prevents the Pdx induced structural change to the more open and active conformation.
ACKNOWLEDGMENTS
We thank former lab colleagues Drs. Dipanwita Batabyal, Jose Amaya, Sarvind Tripathi, Yarrow Madronna, and Shingo Nagano and current lab colleagues Jessica Gable, Vidhi Murarka, and Drs. Huiying Li and Irina Sevrioukova for their contributions to P450 research at UCI. This work was supported by NIH grant R35-GM131920.
Biographies
Thomas L. Poulos earned a B.A. degree in Zoology at the University of California, Santa Barbara, followed by a Ph.D. in Biology at the University of California at San Diego (UCSD). He then moved to the Chemistry Department at UCSD for postdoctoral work in the protein crystallography lab of Joe Kraut. While at UCSD he solved the first heme enzyme crystal structure, cytochrome c peroxidase, and initiated work on P450s. In 1983 he was recruited to Genex Corp. in Gaithersburg, MD, where he held the position of Principal Research Scientist and then Director of Protein Engineering. It was during this time that he solved the first cytochrome P450 structure. In 1987 he moved to the University of Maryland where he was a Professor of Chemistry and Director of the Center for Advanced Research in Biotechnology. In 1992 he moved to the Department of Molecular Biology and Biochemistry at UCI where he now holds the title of Distinguished Professor and joint appointments in the Departments of Chemistry and Pharmaceutical Sciences.
Alec H. Follmer obtained his B.Sc in Biochemistry from University of the Pacific in 2014 where he began his research career in biophysical chemistry. Alec then earned a Ph.D. in Chemistry from University of California, Irvine, in 2019 under the supervision of Prof. Thomas Poulos. His doctoral work utilized molecular dynamics and X-ray crystallography to propose and characterize novel allosterically induced conformational dynamics in cytochrome P450cam. As a postdoctoral scholar at Caltech, his research focuses on the application of ultrafast time-resolved spectroscopies to P450’s and transition metal complexes.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.accounts.1c00632
The authors declare no competing financial interest.
Contributor Information
Thomas L. Poulos, Departments of Molecular Biology & Biochemistry, Pharmaceutical Sciences, and Chemistry, University of California, Irvine, Irvine, California 92697-3900, United States.
Alec H. Follmer, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, United States
REFERENCES
- (1).Tripathi S; Li H; Poulos TL Structural basis for effector control and redox partner recognition in cytochrome P450. Science 2013, 340, 1227–1230. [DOI] [PubMed] [Google Scholar]
- (2).Batabyal D; Richards LS; Poulos TL Effect of Redox Partner Binding on Cytochrome P450 Conformational Dynamics. J. Am. Chem. Soc 2017, 139, 13193–13199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Follmer AH; Mahomed M; Goodin DB; Poulos TL Substrate-Dependent Allosteric Regulation in Cytochrome P450cam (CYP101A1). J. Am. Chem. Soc 2018, 140, 16222–16228. [DOI] [PubMed] [Google Scholar]
- (4).Amaya JA; Batabyal D; Poulos TL Proton Relay Network in the Bacterial P450s: CYP101A1 and CYP101D1. Biochemistry 2020, 59, 2896–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Behan RK; Hoffart LM; Stone KL; Krebs C; Green MT Evidence for basic ferryls in cytochromes P450. J. Am. Chem. Soc 2006, 128, 11471–11474. [DOI] [PubMed] [Google Scholar]
- (6).Yosca TH; Ledray AP; Ngo J; Green MT A new look at the role of thiolate ligation in cytochrome P450. JBIC, J. Biol. Inorg. Chem 2017, 22, 209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Yosca TH; Rittle J; Krest CM; Onderko EL; Silakov A; Calixto JC; Behan RK; Green MT Iron(IV)hydroxide pK(a) and the role of thiolate ligation in C-H bond activation by cytochrome P450. Science 2013, 342, 825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Cupp-Vickery JR; Poulos TL Structure of cytochrome P450eryF involved in erythromycin biosynthesis. Nat. Struct. Mol. Biol 1995, 2, 144–153. [DOI] [PubMed] [Google Scholar]
- (9).Hasemann CA; Ravichandran KG; Peterson JA; Deisenhofer J Crystal structure and refinement of cytochrome P450terp at 2.3 A resolution. J. Mol. Biol 1994, 236, 1169–1185. [DOI] [PubMed] [Google Scholar]
- (10).Li H; Poulos TL Modeling protein-substrate interactions in the heme domain of cytochrome P450(BM-3). Acta Crystallogr., Sect. D: Biol. Crystallogr 1995, 51, 21–32. [DOI] [PubMed] [Google Scholar]
- (11).Poulos TL; Finzel BC; Howard AJ High-resolution crystal structure of cytochrome P450cam. J. Mol. Biol 1987, 195, 687–700. [DOI] [PubMed] [Google Scholar]
- (12).Ravichandran KG; Boddupalli SS; Hasermann CA; Peterson JA; Deisenhofer J Crystal structure of hemoprotein domain of P450BM-3, a prototype for microsomal P450’s. Science 1993, 261, 731–736. [DOI] [PubMed] [Google Scholar]
- (13).Pochapsky SS; Pochapsky TC; Wei JW A model for effector activity in a highly specific biological electron transfer complex: the cytochrome P450(cam)-putidaredoxin couple. Biochemistry 2003, 42, 5649–5656. [DOI] [PubMed] [Google Scholar]
- (14).Gerber NC; Sligar SG A role for Asp-251 in cytochrome P450cam oxygen activation. J. Biol. Chem 1994, 269, 4260–4266. [PubMed] [Google Scholar]
- (15).Batabyal D; Poulos TL Crystal structures and functional characterization of wild-type CYP101D1 and its active site mutants. Biochemistry 2013, 52, 8898–8906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Stok JE; Yamada S; Farlow AJ; Slessor KE; De Voss JJ Cytochrome P450(cin) (CYP176A1) D241N: investigating the role of the conserved acid in the active site of cytochrome P450s. Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834, 688–696. [DOI] [PubMed] [Google Scholar]
- (17).Yeom H; Sligar SG Oxygen activation by cytochrome P450BM-3: effects of mutating an active site acidic residue. Arch. Biochem. Biophys 1997, 337, 209–216. [DOI] [PubMed] [Google Scholar]
- (18).Hiruma Y; Hass MA; Kikui Y; Liu WM; Olmez B; Skinner SP; Blok A; Kloosterman A; Koteishi H; Lohr F; Schwalbe H; Nojiri M; Ubbink M The structure of the cytochrome p450cam-putidaredoxin complex determined by paramagnetic NMR spectroscopy and crystallography. J. Mol. Biol 2013, 425, 4353–4365. [DOI] [PubMed] [Google Scholar]
- (19).Lipscomb JD; Sligar SG; Namtvedt MJ; Gunsalus IC Autooxidation and hydroxylation reactions of oxygenated cytochrome P-450cam. J. Biol. Chem 1976, 251, 1116–1124. [PubMed] [Google Scholar]
- (20).Unno M; Christian JF; Benson DE; Gerber NC; Sligar SG; Champion PM Resonance Raman Investigations of Cytochrome P450cam Complexed with Putidaredoxin. J. Am. Chem. Soc 1997, 119, 6614–6620. [Google Scholar]
- (21).Ugur I; Chandrasekhar P Proton relay network in P450cam formed upon docking of putidaredoxin. Proteins: Struct., Funct., Genet 2020, 88, 558–572. [DOI] [PubMed] [Google Scholar]
- (22).Follmer AH; Tripathi S; Poulos TL Ligand and Redox Partner Binding Generates a New Conformational State in Cytochrome P450cam (CYP101A1). J. Am. Chem. Soc 2019, 141, 2678–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Liou SH; Myers WK; Oswald JD; Britt RD; Goodin DB Putidaredoxin Binds to the Same Site on Cytochrome P450cam in the Open and Closed Conformation. Biochemistry 2017, 56, 4371–4378. [DOI] [PubMed] [Google Scholar]
- (24).Myers WK; Lee YT; Britt RD; Goodin DB The conformation of P450cam in complex with putidaredoxin is dependent on oxidation state. J. Am. Chem. Soc 2013, 135, 11732–11735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Hollingsworth SA; Batabyal D; Nguyen BD; Poulos TL Conformational selectivity in cytochrome P450 redox partner interactions. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 8723–8728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Skinner SP; Liu WM; Hiruma Y; Timmer M; Blok A; Hass MA; Ubbink M Delicate conformational balance of the redox enzyme cytochrome P450cam. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 9022–9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Skinner SP; Follmer AH; Ubbink M; Poulos TL; Houwing-Duistermaat JJ; Paci E Partial Opening of Cytochrome P450cam (CYP101A1) Is Driven by Allostery and Putidaredoxin Binding. Biochemistry 2021, 60, 2932–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yang W; Bell SG; Wang H; Zhou W; Hoskins N; Dale A; Bartlam M; Wong LL; Rao Z Molecular characterization of a class I P450 electron transfer system from Novosphingobium aromaticivorans DSM12444. J. Biol. Chem 2010, 285, 27372–27384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Batabyal D; Lewis-Ballester A; Yeh SR; Poulos TL A Comparative Analysis of the Effector Role of Redox Partner Binding in Bacterial P450s. Biochemistry 2016, 55, 6517–6523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Lange R; Bonfils C; Debey P The low-spin/high-spin transition equilibrium of camphor-bound cytochrome P-450. Effects of medium and temperature on equilibrium data. Eur. J. Biochem 1977, 79, 623–628. [DOI] [PubMed] [Google Scholar]
- (31).Marden MC; Hoa GH P-450 binding to substrates camphor and linalool versus pressure. Arch. Biochem. Biophys 1987, 253, 100–107. [DOI] [PubMed] [Google Scholar]
- (32).Yao H; McCullough CR; Costache AD; Pullela PK; Sem DS Structural evidence for a functionally relevant second camphor binding site in P450cam: model for substrate entry into a P450 active site. Proteins: Struct., Funct., Genet 2007, 69, 125–138. [DOI] [PubMed] [Google Scholar]
- (33).Colthart AM; Tietz DR; Ni Y; Friedman JL; Dang M; Pochapsky TC Detection of substrate-dependent conformational changes in the P450 fold by nuclear magnetic resonance. Sci. Rep 2016, 6, 22035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lee Y-T; Wilson RF; Rupniewski I; Goodin DB P450cam visits an open conformation in the absence of substrate. Biochemistry 2010, 49, 3412–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Lüdemann SK; Lounnas V; Wade RC How do substrates enter and products exit the buried active site of cytochrome P450cam? 2. Steered molecular dynamics and adiabatic mapping of substrate pathways. J. Mol. Biol 2000, 303, 813–830. [DOI] [PubMed] [Google Scholar]
- (36).Fedorov R; Ghosh DK; Schlichting I Crystal structures of cyanide complexes of P450cam and the oxygenase domain of inducible nitric oxide synthase-structural models of the short-lived oxygen complexes. Arch. Biochem. Biophys 2003, 409, 25–31. [DOI] [PubMed] [Google Scholar]
- (37).Nagano S; Poulos TL Crystallographic study on the dioxygen complex of wild-type and mutant cytochrome P450cam. Implications for the dioxygen activation mechanism. J. Biol. Chem 2005, 280, 31659–31663. [DOI] [PubMed] [Google Scholar]
- (38).OuYang B; Pochapsky SS; Dang M; Pochapsky TC A functional proline switch in cytochrome P450cam. Structure 2008, 16, 916–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Hayashi T; Harada K; Sakurai K; Shimada H; Hirota S A role of the heme-7-propionate side chain in cytochrome P450cam as a gate for regulating the access of water molecules to the substrate-binding site. J. Am. Chem. Soc 2009, 131, 1398–1400. [DOI] [PubMed] [Google Scholar]
- (40).Chuo SW; Wang LP; Britt RD; Goodin DB An Intermediate Conformational State of Cytochrome P450cam-CN in Complex with Putidaredoxin. Biochemistry 2019, 58, 2353–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Glascock MC; Ballou DP; Dawson JH Direct observation of a novel perturbed oxyferrous catalytic intermediate during reduced putidaredoxin-initiated turnover of cytochrome P450-CAM: probing the effector role of putidaredoxin in catalysis. J. Biol. Chem 2005, 280, 42134–42141. [DOI] [PubMed] [Google Scholar]
- (42).Tosha T; Yoshioka S; Ishimori K; Morishima I L358P mutation on cytochrome P450cam simulates structural changes upon putidaredoxin binding: the structural changes trigger electron transfer to oxy-P450cam from electron donors. J. Biol. Chem 2004, 279, 42836–42843. [DOI] [PubMed] [Google Scholar]
- (43).Madrona Y; Hollingsworth SA; Tripathi S; Fields JB; Rwigema JC; Tobias DJ; Poulos TL Crystal structure of cindoxin, the P450cin redox partner. Biochemistry 2014, 53, 1435–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Nagano S; Tosha T; Ishimori K; Morishima I; Poulos TL Crystal structure of the cytochrome p450cam mutant that exhibits the same spectral perturbations induced by putidaredoxin binding. J. Biol. Chem 2004, 279, 42844–42849. [DOI] [PubMed] [Google Scholar]
- (45).Sevrioukova IF; Li H; Zhang H; Peterson JA; Poulos TL Structure of a cytochrome P450-redox partner electron-transfer complex. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 1863–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Strushkevich N; MacKenzie F; Cherkesova T; Grabovec I; Usanov S; Park HW Structural basis for pregnenolone biosynthesis by the mitochondrial monooxygenase system. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 10139–10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Fujita M; Aramaki H; Horiuchi T; Amemura A Transcription of the cam operon and camR genes in Pseudomonas putida PpG1. J. Bacteriol 1993, 175, 6953–6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Koga H; Yamaguchi E; Matsunaga K; Aramaki H; Horiuchi T Cloning and nucleotide sequences of NADH-putidaredoxin reductase gene (camA) and putidaredoxin gene (camB) involved in cytochrome P-450cam hydroxylase of Pseudomonas putida. J. Biochem 1989, 106, 831–836. [DOI] [PubMed] [Google Scholar]