

Abstract
Fine-mapping to plausible causal variation may be more effective in multi-ancestry cohorts, particularly in the MHC, which has population-specific structure. To enable such studies, we constructed a large (n = 21,546) HLA reference panel spanning five global populations based on whole-genome sequences. Despite population specific long-range haplotypes, we demonstrated accurate imputation at G-group resolution (94.2%, 93.7%, 97.8% and 93.7% in Admixed African (AA), East Asian (EAS), European (EUR) and Latino (LAT) populations). Applying HLA imputation to genome-wide association study (GWAS) data for HIV-1 viral load in three populations (EUR, AA and LAT), we obviated effects of previously reported associations from population-specific HIV studies and discovered a novel association at position 156 in HLA-B. We pinpointed the MHC association to three amino acid positions (97, 67 and 156) marking three consecutive pockets (C, B and D) within the HLA-B peptide binding groove, explaining 12.9% of trait variance.
The human leukocyte antigen (HLA) genes located within the major histocompatibility complex (MHC) region encode proteins that play essential roles in immune responses, including antigen presentation. They account for more heritability than all other variants together for many diseases1–4. It also has more reported GWAS trait associations than any other locus5. The extended MHC region spans 6 Mb on chromosome 6p21.3 and contains more than 260 genes6. It is striking for its structural diversity and long-range linkage equilibrium (LD). Due to population-specific positive selection, it harbors unusually high sequence variation, longer haplotypes than most of the genome, and haplotypes that are specific to individual ancestral populations7–9. Consequently, the MHC is among the most challenging regions in the genome to analyze. Advances in HLA imputation10–12 have enabled MHC association and fine-mapping studies at single gene and long-range haplotype level2,13–16. But despite large effect sizes, fine-mapping in multiple populations simultaneously is challenging without a single large and high-resolution multi-ancestry reference panel. This has caused confusion in some instances. For example, the Human Immunodeficiency Virus (HIV) resulted in 38.0 million people living with HIV infection in 2019, and led to 770,000 deaths in 2018 alone17. Multiple risk HLA risk alleles have been independently reported in different populations1,14,18, and it remains unclear if they represent truly population-specific signals or are confounded by high LD in the region.
Results
Performance evaluation of inferred classical HLA alleles.
To build a large-scale multi-ancestry HLA imputation reference panel, we used high-coverage whole genome sequencing (WGS) datasets19–23 from the Japan Biological Informatics Consortium22, the BioBank Japan Project20, the Estonian Biobank24, the 1000 Genomes Project (1KG)23 and a subset of studies in the TOPMed program (Supplementary Note and Supplementary Tables 1 and 2). To perform HLA typing using WGS data, we extracted reads mapped to the extended MHC region (chr6:25Mb-35Mb) and unmapped reads from 21,546 individuals. We applied a population reference graph25–27 for the MHC region to infer classical alleles for three HLA class I genes (HLA-A, -B and -C) and five class II genes (HLA-DQA1, -DQB1, -DRB1, -DPA1 and -DPB1) at G-group resolution, which determines the sequences of the exons encoding the peptide binding groove (Extended Data Fig. 1). We required samples to have >20x coverage across all HLA genes (Supplementary Tables 1 and 3). After quality control, our panel included 10,187 EUR, 7,849 AA, 2,069 EAS, 952 LAT and 489 South Asian (SAS) individuals.
To assess the accuracy of the WGS HLA allele calls, we compared the inferred HLA classical alleles to gold standard sequence-based typing (SBT) in 955 1KG individuals and 288 Japanese individuals and quantified concordance. In both cohorts, we observed slightly higher average accuracy for class I genes, obtaining 99.0% (one-field, formally known as two-digit), 99.2% (amino acid) and 96.5% (G-group resolution), than class II genes, obtaining 98.7% (one-field), 99.7% (amino acid) and 96.7% (G-group resolution) (Methods, Supplementary Fig. 1, Supplementary Tables 4 and 5, and Supplementary Data 1).
HLA reference panel construction and evaluation.
Next, we constructed a multi-ancestry HLA imputation reference panel based on classical HLA alleles and 38,398 genomic markers in the extended MHC region using a novel HLA-focused pipeline HLA-TAPAS (HLA-Typing At Protein for Association Studies; see Code availability). Briefly, HLA-TAPAS can handle HLA reference panel construction (MakeReference), HLA imputation (SNP2HLA) and HLA association (HLAassoc) (Fig. 1 and Methods). Compared to a widely used HLA reference panel with European-only individuals (The Type 1 Diabetes Genetics Consortium11, T1DGC), this new reference panel has a six-fold increase in the number of observed HLA alleles and non-HLA genomic markers (Supplementary Table 6). We noted the difference in observed classical HLA alleles is mainly due to the inclusion of diverse populations rather than its size; after downsampling the reference panel to be the same size as T1DGC (n = 5,225), there was still a three-fold increase in observed alleles (Fig. 2a).
Figure 1 |. A schematic showing the overall study design.
We used whole-genome sequences of 21,546 individuals from five global populations to construct an HLA imputation reference panel. We then performed HLA imputation and fine-mapping in HIV-1 viral load jointly in three populations.
Figure 2 |. The multi-ancestry HLA reference panel shows improvement in allele diversity and imputation accuracy.
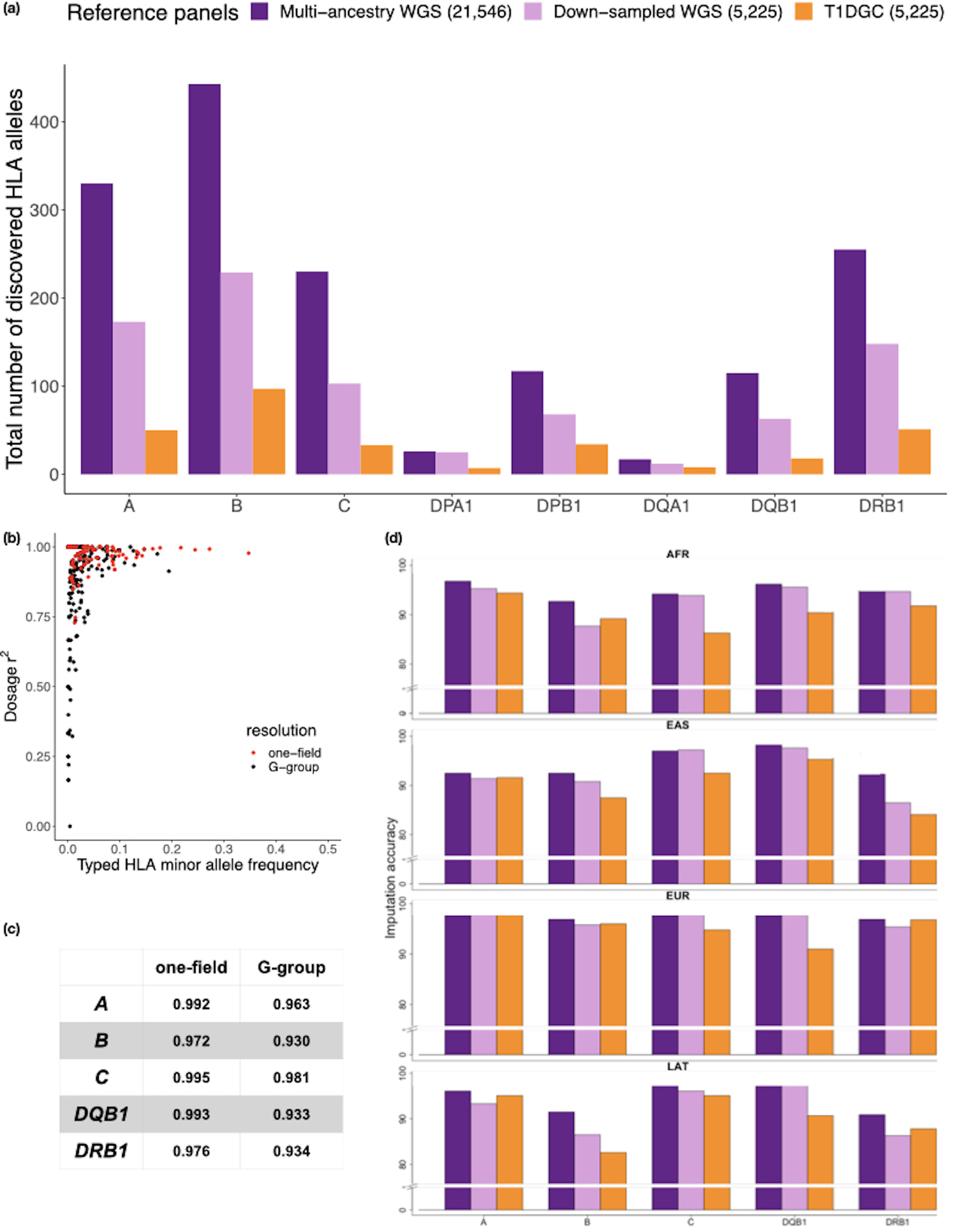
a, The number of HLA alleles at the two-field resolution included in the multi-ancestry HLA reference panel (n = 21,546) compared to the European only Type 1 Diabetes Genetics Consortium (T1DGC) panel (n = 5,225) as well as a subset of the multi-ancestry HLA panel down-sampled to the same size as T1DGC. b, The correlation between imputed and typed dosages of classical HLA alleles using the multi-ancestry HLA reference panel at one-filed (red) and G-group resolution (black) of 955 individuals with SBT HLA typing data from the 1000 Genomes project. c, The imputation accuracy for five classical HLA genes at one-field, two-field and G-group resolution. d, The imputation accuracy at G-group resolution of the 1000 Genomes individuals stratified by four diverse ancestries when using three different imputation reference panels as described in a.
To empirically assess imputation accuracy of our reference panel, we first used the publicly available gold-standard HLA types (HLA-A, -B, -C, -DRB1 and -DQB1) of 1,267 diverse samples from AA, EAS, EUR and LAT included in 1KG. We removed 955 overlapping samples within the reference panel, and to ensure a representative analysis we kept 6,007 markers overlapping with the Global Genotyping Array. Across the five genes, the average G-group resolution accuracies were 94.2%, 93.7%, 97.8% and 93.7% in AA, EAS, EUR and LAT (Figure 2b,c, Supplementary Table 7, Supplementary Data 2, and Methods). Compared to the T1DGC panel, our multi-ancestry reference panel showed the most improvement for individuals of non-European descent; we obtained 4.27%, 2.96%, 2.90% and 1.05% improvement at G-group resolution for AA, EAS, LAT, and EUR individuals, respectively. Increased diversity was responsible for the improvement; downsampling the reference panel be the same size as the T1DGC panel still yielded superior performance (Fig. 2d). To validate our panel further, we imputed HLA alleles into a multi-ancestry cohort of 2,291 individuals from the Genotype and Phenotype (GaP) registry genotyped on the ImmunoChip array. We obtained HLA type information for seven classical class I and class II loci (HLA-A, -B, -C, -DQA1, -DQB1, -DRB1 and -DPB1) in 75 samples with diverse ancestral background (25 EUR, 25 EAS and 25 AA; Supplementary Fig. 2 and Methods). Average accuracies were 99.0%, 95.7% and 97.0% for EUR, EAS and AA, respectively, when comparing SBT HLA alleles at G-group resolution (Methods and Supplementary Data 2). Similar to the 1KG analysis, the multi-ancestry reference panel showed significant improvement for individuals with non-European descent (6.3% and 11.1% improvement for EAS and AA individuals, respectively, at G-group resolution), and a more modest 2% improvement in EUR (Supplementary Fig. 3 and Supplementary Table 8). At the amino acid level, the average accuracies were greater than 99% for all populations, and this accuracy is similar inside and outside the peptide binding groove at six classical HLA genes (Supplementary Table 9).
Fine-mapping causal variants of HIV-1 set point viral load.
Next, we investigated MHC effects within human immunodeficiency virus type 1 (HIV-1) set point viral load. Upon primary infection with HIV-1, the set point viral load is reached after the immune system has developed specific cytotoxic T lymphocytes (CTL) that are able to partially control the virus. It has been well-established that the set point viral load (spVL) varies in the infected population and positively correlates with rate of disease progression28. Previous studies suggested that HIV-1 infection has a strong genetic component, and specific HLA class I alleles explain the majority of genetic risk14,29. The existence of multiple independent, population-specific, risk-associated alleles has been reported in both European1,14 and African American18 populations. However, without a multi-ancestry reference panel, it has not been possible to determine if these signals are consistent across different ancestral groups.
To define the MHC allelic effects shared across multiple populations, we applied our multi-ancestry MHC reference panel to 7,445 EUR, 3,901 AA and 677 LAT HIV-1 infected individuals (Methods and Supplementary Table 10). Imputation resulted in 640 classical HLA alleles, 4,513 amino acids in HLA proteins and 49,321 SNPs in the extended MHC region for association and fine-mapping analysis. We confirmed 94.3% and 99.1% imputation accuracy at G-group and amino acid resolution, respectively, for HLA alleles with a minor allele frequency > 0.5% in this cohort by comparing imputed classical alleles to the SBT alleles in a subset of 1,067 AA individuals18 (Extended Data Fig. 2, Supplementary Table 11, and Supplementary Data 2).
We next tested SNPs, amino acid positions and classical HLA alleles across the MHC for association to spVL. We performed this jointly in EUR, AA and LAT population using a linear regression model with sex, principal components and ancestry as covariates (Methods). In agreement with previous studies, we found the strongest spVL-associated classical HLA allele is B*57 (effect size = −0.84, Pbinary = 8.68 × 10−144). This corresponded to a single residue Val97 in HLA-B that tracks almost perfectly with B*57 (r2 = 0.995) and showed the strongest association of any single residue (effect size = −0.84, Pbinary = 5.99 × 10−145, Extended Data Fig. 3).
To determine which amino acid positions have independent association with spVL, we tested each of the amino acid positions by grouping haplotypes carrying a specific residue at each position in an additive model2,13 (Methods). We found the strongest spVL-associated amino acid variant in HLA-B is as previously reported1,14,18 at position 97 (Fig. 3a,b and Supplementary Table 12), which strikingly explains 9.06% of the phenotypic variance. Position 97 in HLA-B was more significant (Pomnibus = 2.86 × 10−184) than any single SNP or classical HLA allele, including B*57 (Extended Data Fig. 3 and Supplementary Data 3). Of the six allelic variants (Val/Asn/Trp/Thr/Arg/Ser) at this position, the Val residue conferred the strongest protective effect (effect size = −0.88, P = 9.32 × 10−152, Supplementary Fig. 4) relative to the most common residue Arg (frequency = 47.8%). All six amino acid alleles have consistent frequencies and effect sizes across the three population groups (Fig. 4a,b and Extended Data Fig. 4).
Figure 3 |. Stepwise conditional analysis of the allele and amino acid positions of classical HLA genes to HIV-1 viral load.
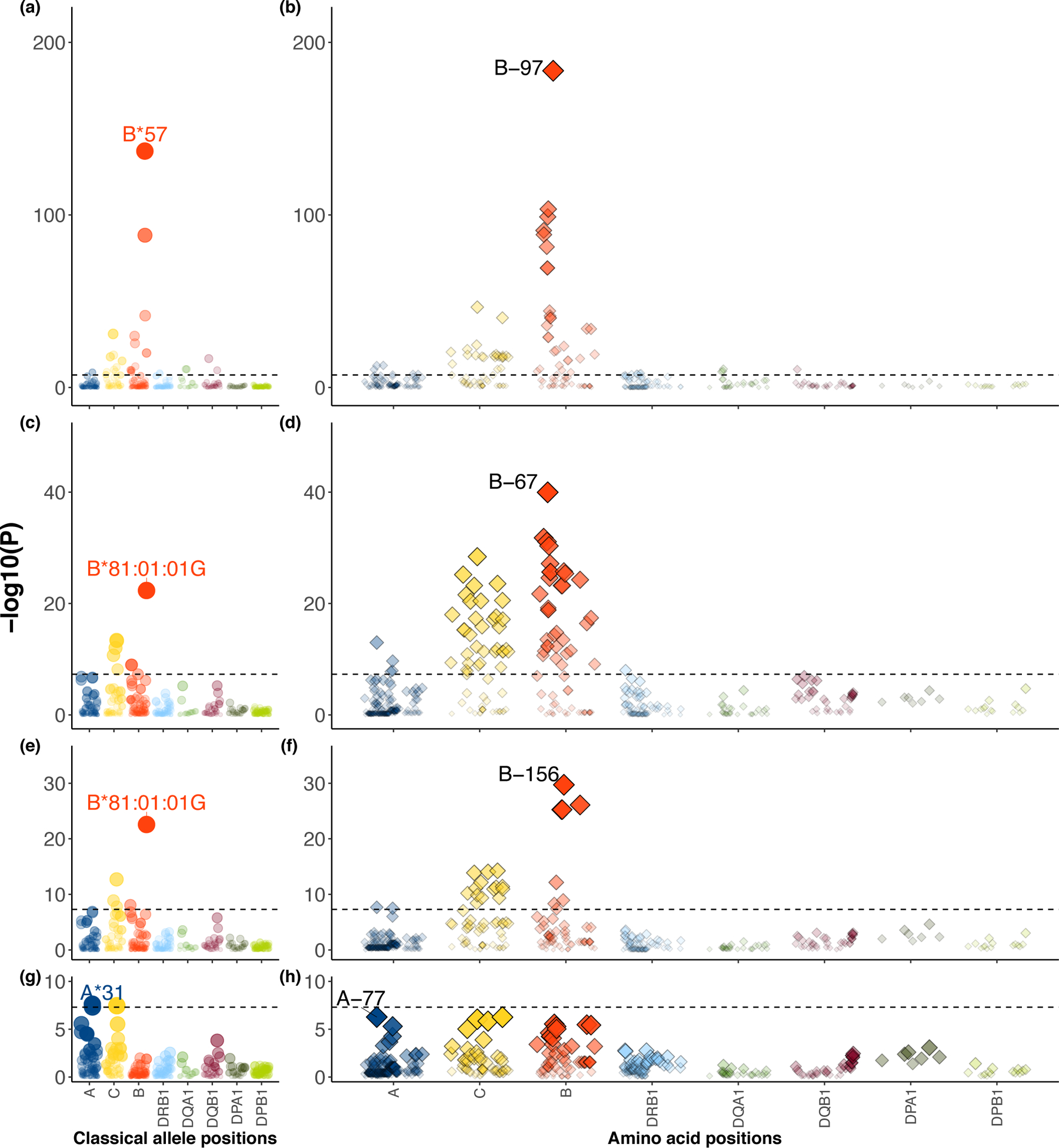
a-h, Each circle point represents the -log10(Pbinary) from two-sided linear regression for all classical HLA alleles. Each diamond point represents -log10(Pomnibus) from one-sided F-test for the tested amino acid positions in HLA (blue, HLA-A; yellow, HLA-C; red, HLA-B; light blue, HLA-DRB1; green, HLA-DQA1; purple, HLA-DQB1, dark green, HLA-DPA1; light green, HLA-DPB1). Association at amino acid positions with more than two alleles was calculated using a multi-degree-of-freedom omnibus test. The dashed black line represents the significance threshold of P = 5 × 10−8 to correct for multiple comparisons (Bonferroni correction). Each panel shows the association plot in the process of stepwise conditional omnibus test. One-field classical allele HLA-B*57 (P = 9.84 × 10−138) (a) and amino acid position 97 in HLA-B (Pomnibus = 1.86 × 10−184) (b) showed the strongest association signal. Results conditioned on position 97 in HLA-B showed a secondary signal at classical allele HLA-B*81:01:01:G (P = 4.53 × 10−23) (c) and position 67 in HLA-B (Pomnibus = 1.08 × 10−40) (d). Results conditioned on position 97 and 67 in HLA-B showed the same classical allele HLA-B*81:01:01G (P = 2.70 × 10−23) (e) and third signal at position 156 in HLA-B (Pomnibus = 1.92 × 10−30) (f). Results conditioned on position 97, 67 and 156 in HLA-B showed a fourth signal at HLA-A*31 (P = 2.45 × 10−8) (g) and position 77 in HLA-A (Pomnibus = 5.35 × 10−7) outside HLA-B (h).
Figure 4 |. Location and effect of three independently associated amino acid positions in HLA-B.
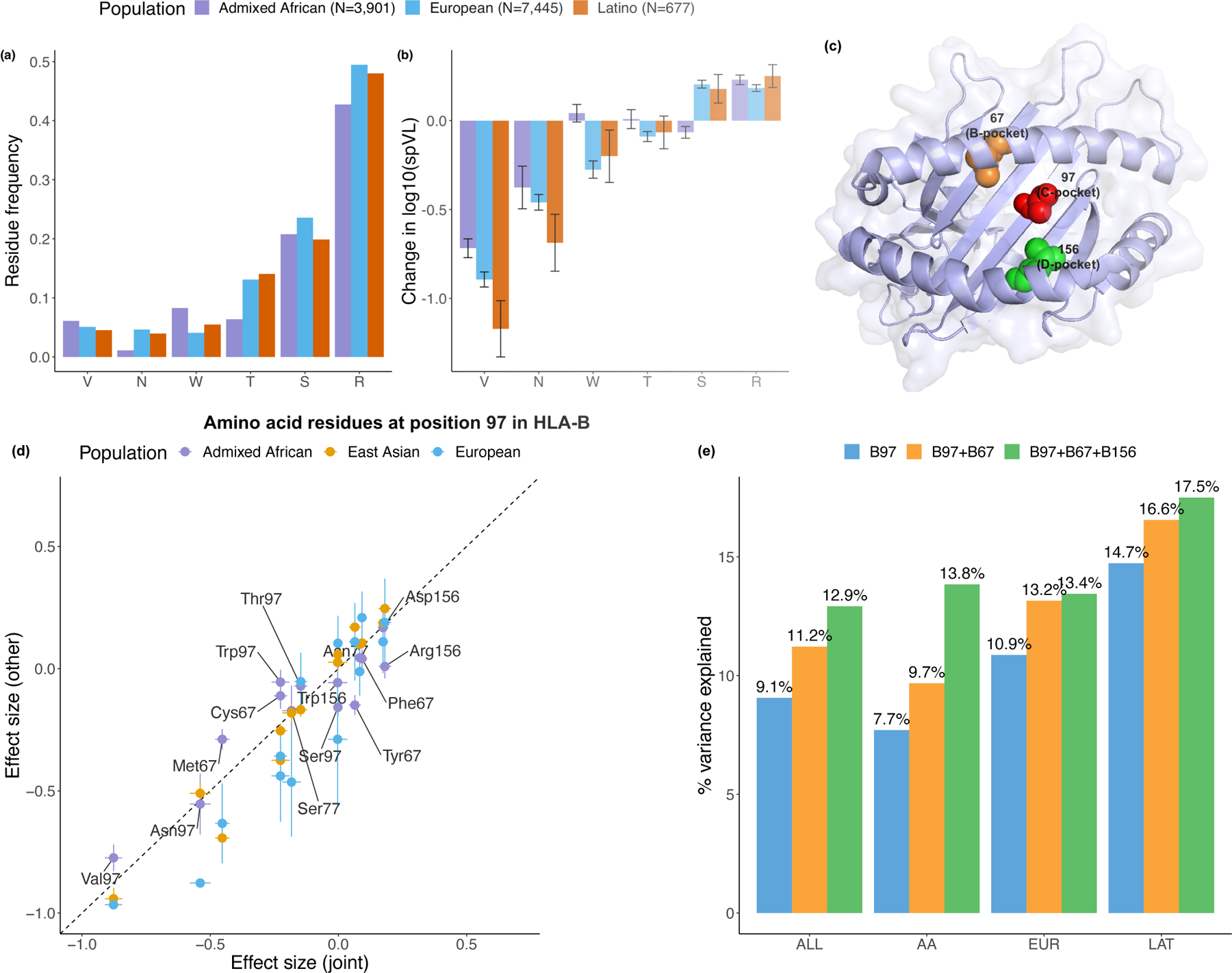
a, Allele frequencies of six residues at position 97 in HLA-B among three populations. b, Effect on set point viral load (spVL) (i.e., change in log10 HIV-1 spVL per allele copy) of individual amino acid residues at position 97 in HLA-B. Results were calculated per allele using linear regression models, including gender and principal components within each ancestry as covariates. There are 3,901 Admixed African (purple), 7,455 European (blue) and 677 Latio (orange) independent samples included in the analysis. Data are presented as mean values (beta) ± standard errors. c, HLA-B (PDB ID code 2bvp) proteins. Omnibus and stepwise conditional analysis identified three independent amino acid positions (positions 97 (red), 67 (orange), and 156 (green) in HLA-B. d, Effect on spVL (i.e., change in log10 HIV-1 spVL per allele copy) of individual amino acid residues at each position reported in this and previous work14,18. Results were calculated per allele using linear regression models. The x-axis shows the effect size and its standard errors in the joint analysis, and the y-axis shows the effect sizes ± standard error in individual populations (purple, Admixed African, n = 3,901; blue, European, n = 7,455; orange, Latino, n = 677). e, Variance of spVL explained by the haplotypes formed by different amino acid positions.
We next wanted to test whether there were other independent effects outside of position 97 in HLA-B. After accounting for the effects of amino acid 97 in HLA-B using a conditional haplotype analysis (Methods), we observed a significant independent association at position 67 in HLA-B (Pomnibus = 2.82 × 10−39, Fig. 3c,d and Supplementary Table 12). Considering this might be an artifact of forward search, we exhaustively tested all possible pairs of polymorphic amino acid positions in HLA-B. Of 7,260 pairs of amino acid positions, none obtained a better goodness-of-fit than the pair of positions 97 and 67, which collectively explained 11.2% variance in spVL (Fig. 4e and Supplementary Table 13). At position 67, Met67 shows the most protective effect (effect size = −0.44, P = 1.19 × 10−59) among the five possible amino acids (Cys/Phe/Met/Ser/Tyr) relative to the most common residue Ser (frequency = 10.0%).
Conditioning on positions 97 and 67 revealed an additional association at position 156 in HLA-B (Pomnibus = 1.92 × 10−30, Fig. 4e,f and Supplementary Table 12). In agreement with the stepwise conditional analysis, when we tested all 287,980 possible combinations of three amino acid positions in HLA-B, the most statistically significant combination of amino acids sites is 67, 97 and 156 (P = 5.68 × 10−244, Supplementary Table 14). These three positions explained 12.9% of the variance (Fig. 4e). At position 156, residue Arg shows the largest risk effect (effect size = 0.180, P = 8.92 × 10−14) among the four possible allelic variants (Leu/Arg/Asp/Trp), relative to the most common residue Leu (frequency = 35.1%).
These amino acid positions mark three consecutive pockets within the HLA-B peptide-binding groove (Fig. 4c). Position 97 is located in the C-pocket and has an important role in determining the specificity of the peptide-binding groove30,31. Position 67 is in the B-pocket, and Met67 side chains occupy the space where larger B-pocket anchors reside in other peptide-MHC structures; its presence limits the size of potential peptide position P2 side chains31. Amino acid position 156 is part of the D-pocket and influences the conformation of the peptide-binding region32. These results are consistent with the observation that in HLA-B*57, the single most protective spVL-associated one-field allele (a single change at position 156 from Leu → Arg or equivalently HLA-B*57:03 → HLA-B*57:02) leads to an increased repertoire of HIV-specific epitope33,34.
Despite differences in the power to detect associations due to differences in allele frequencies (Supplementary Fig. 5), we observed generally consistent effects of individual residues across populations (Fig. 4d and Supplementary Figs. 6 and 7). There are 26 unique haplotypes defined by the amino acids at positions 67, 97 and 156 in HLA-B (Table 1 and Supplementary Table 16). When we tested for effect size heterogeneity by ancestry for each of these haplotypes (Methods), we observed only 2 of 26 haplotypes showed heterogeneity (F-test P < 0.05/26), possibly due to different interplay between genetic and environmental variation at the population level. These results support the concept that these positions mediate HIV-1 viral load in diverse ancestries.
Table 1 |. Effect estimates for the haplotypes defined by the three independent amino acids in HLA-B associated with HIV-1 viral load.
Only haplotypes with >1% frequency in the overall population are listed (Supplementary Table 16). Classical alleles of HLA-B are grouped based on the amino acid residues presented at position 97, 67 and 156 in HLA-B. For each haplotype, the multivariate effect is given as an effect size, taking the most frequent haplotype (97R-67S-156L) as the reference (effect size = 0). Heterogeneity P-value (P(het), two-sided) of each haplotype is calculated using F-statistics with two degrees of freedom (Methods). Effect size and its standard error in each population are listed only for haplotypes that show evidence of heterogeneity (P < 0.05 /26 Bonferroni-corrected for multiple tests, bolded). Unadjusted haplotype frequencies are given in each population.
| HLA-B amino acid at position | Effect size (standard error) | Unadjusted allele frequency | Classical HLA-B allele | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 97 | 67 | 156 | AA | EUR | LAT | Joint | P(het) | AA | EUR | LAT | Joint | |
| V | M | L | −0.921 (0.036) | 0.031 | 0.056 | 0.049 | 0.059 | 0.051 | B*57:01;B*57:03 | |||
| N | C | L | −0.554 (0.041) | 0.257 | 0.012 | 0.046 | 0.037 | 0.035 | B*27:05 | |||
| T | S | L | −0.436 (0.041) | 0.041 | 0.028 | 0.039 | 0.056 | 0.037 | B*13:02;B*52:01 | |||
| W | C | L | −0.397 (0.041) | 0.581 | 0.03 | 0.039 | 0.054 | 0.037 | B*14:01;B*14:02 | |||
| S | S | L | −0.252 (0.066) | 0.013 | 0.002 | 0.014 | 0.07 | 0.013 | B*40:02 | |||
| R | S | W | −0.177 (0.038) | 0.618 | 0.009 | 0.062 | 0.028 | 0.044 | B*15:01;B*15:10;B*15:16 | |||
| T | F | L | −0.125 (0.036) | 0.001 | 0.03 | 0.059 | 0.073 | 0.051 | B*51:01;B*78:01 | |||
| R | M | L | −0.125 (0.045) | 0.375 | 0.061 | 0.014 | 0.028 | 0.029 | B*15:16;B*58:01 | |||
| R | C | L | −0.078 (0.039) | 0.055 | 0.042 | 0.039 | 0.06 | 0.041 | B*15:10;B*15:16;B*39:10 | |||
| R | S | D | 0.165 (0.056) | −0.07 (0.034) | −0.153 (0.173) | −0.019 (0.028) | 0.002 | 0.075 | 0.108 | 0.084 | 0.097 | B*37:01;B*44:02;B*45:01 |
| R | S | L | Reference | 0.536 | 0.191 | 0.176 | 0.197 | 0.18 | B*15:03;B*15:10;B*18:01;B*39:10;B*40:01;B*44:03;B*49:01;B*50:01 | |||
| S | Y | D | 0.015 (0.055) | 0.884 | 0.059 | NA | 0.017 | 0.019 | B*42:01;B*42:02 | |||
| S | Y | R | −0.06 (0.055) | 0.037 (0.033) | −0.002 (0.187) | 0.022 (0.027) | 0.007 | 0.08 | 0.124 | 0.07 | 0.108 | B*07:02;B*07:05 |
| S | F | D | 0.041 (0.031) | 0.218 | 0.034 | 0.095 | 0.042 | 0.074 | B*08:01 | |||
| R | F | L | 0.045 (0.027) | 0.73 | 0.182 | 0.095 | 0.113 | 0.122 | B*35:01;B*53:01 | |||
| W | M | L | 0.098 (0.064) | 0.268 | 0.046 | NA | NA | 0.014 | B*58:02 | |||
| T | Y | L | 0.176 (0.058) | 0.207 | 0.005 | 0.021 | NA | 0.016 | ||||
To assess whether there were other independent MHC associations outside HLA-B, we conditioned on all amino acid positions in HLA-B and observed associations at HLA-A, including at position 77 in HLA-A (Pomnibus = 9.10 × 10−7, Fig. 3g,h and Supplementary Table 12), the classical HLA allele HLA-A*31 (Pbinary = 2.45 × 10−8) and the rs2256919 promoter SNP (Pbinary = 3.10 × 10−16, Extended Data Fig. 3). These associations argue for an effect at HLA-A, but larger studies and functional studies will be necessary to define the driving effects.
We next tested for the presence of non-additive effects with respect to the three observed amino acid associations (Methods). In agreement with the previous study14, we did not observe any single allele amino acid position showing significant departure from additivity after accounting for multiple comparisons (Supplementary Table 17).
HLA diversity.
To quantify MHC diversity, we calculated identity-by-descent (IBD) distances35 between all individuals using 38,398 MHC single nucleotide polymorphisms (SNPs) included in the multi-ancestry HLA reference panel (n = 21,546) and applied principal component analysis (PCA, Methods). PCA distinguished EUR, EAS and AA as well as the admixed LAT and SAS samples (Extended Data Fig. 5 and Supplementary Fig. 8). This reflected widespread HLA allele frequency differences between populations (Extended Data Fig. 6). Of 130 unique common (frequency > 1%) G-group alleles, 129 demonstrated significant differences of frequencies across populations (4 degree-of-freedom Chi-square test, P < 0.05/130, Supplementary Figure 9). The only exception was DQA1*01:01:01G, which was nominally significant (unadjusted P = 0.047). These differences may be related to adaptive selection. For example, the B*53:01:01G allele is enriched in Admixed Africans (11.7% in AA versus 0.3% in others) and it has been previously associated with malaria protection36,37. Consistent with previous reports9,38, we observed that HLA-B had the highest allelic diversity (n = 443) while HLA-DQA1 had the least (n = 17, Extended Data Fig. 7, Supplementary Fig. 10, and Supplementary Table 18). We included the overall and population-specific frequencies of each inferred HLA allele at G-group resolution in Supplementary Data 4. We next tested Hardy-Weinberg equilibrium (HWE) at each of the eight loci using an exact test (Methods). In agreement with previous work39–41, we observed that HLA-DRB1 showed the greatest deviation from HWE, while the least deviation was for HLA-DQA1, which has the least allelic diversity (Supplementary Table 19). Considering five global populations individually, the null hypothesis of HWE is rejected at 3 of 8 loci in AA (HLA-A with P = 0.035; HLA-DRB with P = 1.82 × 10−5; HLA-DPA1 with P = 0.012), 4 of 8 loci in EUR, 6 of 8 loci in EAS, 2 of 8 loci in LAT, and 2 of 8 loci in SAS. This strongly suggests the existence of subpopulation structure within each global population in the HLA region.
To understand the haplotype structure of HLA between pairs of HLA genes, we calculated a multiallelic LD linkage disequilibrium (LD) measurement index42–44, ε, which is 0 when there is no LD and 1 when there is perfect LD (Extended Data Fig. 8). We observed higher ε between DQA1, DQB1, and DRB1, between DPA1 and DPB1, and between B and C (Supplementary Fig. 11). The heterogeneity between different populations was underscored by the presence of population-specific common (frequency >1%) high resolution long-range haplotypes (HLA-A~C~B~DRB1~DQA1~DQB1~DPA1~DPB1, Fig. 5, Supplementary Figs. 12–16, Supplementary Data 4, and Methods). The most common within-population haplotype was A24::DP6 (HLA-A*24:02:01G~C*12:02:01G~B*52:01:01G~DRB1*15:02:01G~DQA1*01:03:01G~DQB1*06:01:01G~DPA1*02:01:01G~DPB1*09:01:01G) found at a frequency of 3.61% in EAS (Supplementary Fig. 12). This haplotype is strongly associated with immune-mediated traits such as HIV45 and ulcerative colitis46 in Japanese individuals. The next most common haplotype was the well-described European-specific ancestral haplotype A1::DP1 or 8.147,48 (frequency = 2.76%, HLA-A*01:01:01G~C*07:01:01G~B*08:01:01G~DRB1*03:01:01G~DQA1*05:01:01G~DQB1*02:01:01G~DPA1*02:01:02G~DPB1*01:01:01G, Supplementary Fig. 13). This haplotype is associated with diverse immunopathological phenotypes in the European population, including systemic lupus erythematosus49, myositis50 and several other conditions47. We observed long-range haplotypes in admixed populations including A1::DP4 in SAS (frequency = 1.86%, Supplementary Fig. 14), A30::DP1 in AA (frequency = 1.18%, HLA-A*30:01:01G~C*17:01:01G~B*42:01:01:G~DRB1*03:02:01G~DQA1*04:01:01G~DQB1*04:02:01G~DPA1*02:02:02G~DPB1*01:01:01G, Supplementary Fig. 15), and A29::DP11 in LAT (frequency = 0.74%, HLA-A*29:02:01G~C*16:01:01G~B*44*03:01:G~DRB1*07:01:01G~DQA1*02:01:01G~DQB1*02:01:01G~DPA1*02:01:01G~DPB1*11:01:01G, Supplementary Fig. 16). These haplotypes also have associations with multiple diseases: for example, C*06:02~B*57:01 is associated with psoriasis51 and A*30:01~C*17:01~B*42:01 is associated with HIV33.
Figure 5 |. Pairwise LD and haplotype structure for eight classical HLA genes in five population groups.
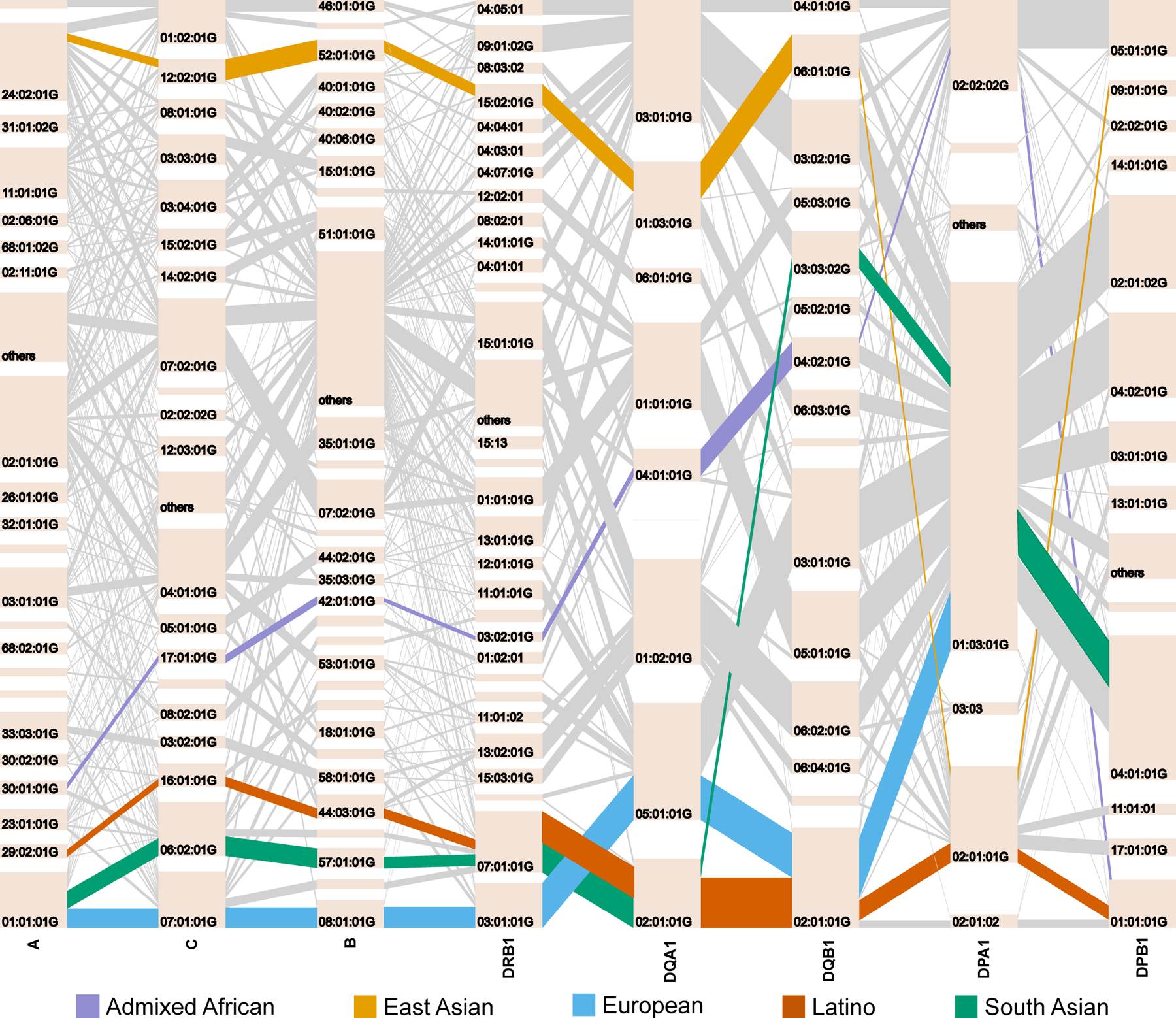
Haplotype structures of the eight classical HLA genes in each population. The tile in a bar represents an HLA allele, and its height corresponds to the frequencies of the HLA allele. The gray lines connecting between two alleles represent HLA haplotypes. The width of these lines corresponds to the frequencies of the haplotypes. The most frequent long-range HLA haplotypes within each population is bolded and highlighted in a color described by the key at the bottom.
HLA selection signature.
Previous studies have suggested that recent natural selection favors African ancestry in the HLA region in admixed populations52–55. To test this hypothesis in our data, we obtained WGS data from a subset of individuals within two admixed populations (1,832 AA and 594 LAT, determined by the first three global principal components, Supplementary Fig. 17 and Supplementary Note). Admixed individuals have genomes that are a mosaic of different ancestries. If genetic variations or haplotypes from an ancestral population are advantageous, then they are under selection and are expected to have higher frequency than by chance. Using ELAI56, we quantified how much the ancestry proportions differed within the MHC from the genome-wide average. In AA, we observed that the average genome-wide proportion of African ancestry was 74.5%, compared to 78.0% in the extended MHC region, corresponding to a 3.42 (95% CI: 3.35–3.49) standard deviation increase. In LAT, we observed 5.76% African ancestry genome-wide versus 16.0% in the extended MHC region, representing an increase of 4.23 (95% CI: 4.14–4.31) standard deviations (Methods). To ensure our results are robust to different local ancestry inference methods, we applied an alternative method called RFMix57 and observed a similarly consistent MHC-specific excess of African ancestry in LAT, and also an excess in AA that was more modest (Extended Data Fig. 9).
Discussion
In our study, we demonstrated accurate imputation with a single large reference panel for HLA imputation. We have shown how this reference panel can be used to impute genetic variation at eight HLA classical genes accurately across a wide range of populations. Accurate imputation in multi-ancestry studies is essential for fine-mapping.
Like previous HLA imputation reference panels11,16,58, our current work has two main limitations. First, our current implementation of the multi-ancestry reference panel is limited to G-group resolution, and amino acids outside the binding groove were taken as its best proximity (Methods). Even though we are currently unable to differentiate alleles belonging to the same G-group, we showed that the imputation accuracy is comparable inside and outside the peptide binding groove (Supplementary Table 9). The third generation sequencers (e.g., Pac-Bio SMRT59) will be able to provide phased, unambiguous and allele-level information at true four-field resolution. We aim to re-evaluate our panel performance, especially for accuracy outside the binding groove, when these data become more available in the future. Second, all imputation accuracy assessment is using a Beagle (v4.1) model integrated in HLA-TAPAS. We observed similar performance between Beagle (v4.1) and Minimac4 (Supplementary Fig. 18), but did not perform extensive comparison among other HLA imputation methods10,12. However, we note that our multi-ancestry reference panel can serve as a useful resource for method evaluation especially in a multi-ancestry setting.
Despite this limitation, we showed the utility of this approach by defining the alleles that best explain HIV-1 viral load in infected individuals. Our work implicates three amino acid positions (97, 67 and 156) in HLA-B in conferring the known protective effect of HLA class I variation on HIV-1 infection. Combining all alleles at these three positions explained 12.9% of the variance in spVL (Fig. 4e). These positions all fall within the peptide-binding groove of the respective MHC protein (Fig. 4c), indicating that variation in the amino acid content of the peptide-binding groove is the major genetic determinant of HIV control. Supported by experimental studies34,60–62, positions highlighted in our work indicated a structural basis for the HLA association with HIV disease progression that is mediated by the conformation of the peptide within the class I binding groove. This result highlights how a study with ancestrally diverse populations can potentially point to causal variation by leveraging linkage disequilibrium differences between genetic ancestry groups.
We note that previous studies have shown that position 97 in HLA-B has the strongest association with HIV-1 spVL or case-control in African American and European populations, but highlighted different additional signals via conditional analysis (position 45, 67 in HLA-B and position 77, 95 in HLA-A in Europeans1,14,18 and position 63, 116 and 245 in HLA-B in African Americans18). These signals do not explain the signals we report here; after conditioning on positions 45, 63, 116, 245 of HLA-B and 95 of HLA-A, the association of the four identified amino acids identified in this study remained significant (P < 5 × 10−8). In contrast, our binding groove alleles explain these other alleles; conditioning on the four amino acid positions identified in this study (positions 67, 97 and 156 in HLA-B), all previously reported positions did not pass the Bonferroni-corrected significance threshold (P > 5 × 10−8, Extended Data Fig. 10).
Furthermore, defining the effect sizes for HLA alleles across different populations is essential for defining risk of a wide-range of diseases in the clinical setting. There is increasing application of genome-wide genotyping by patients both by healthcare providers and direct-to-consumer vendors. The large effects of the MHC region for a wide-range of immune and non-immune traits makes it essential to define HLA allelic effect sizes in multi-ancestry studies in order to build generally applicable clinical polygenic risk scores for many diseases in diverse populations63–66. Resources like the one we present here will be an essential ingredient in such studies.
Methods
Ethics statement.
Study participants included in the reference panel were from the Jackson Heart Study (JHS, n = 3,027), Multi-Ethnic Study of Atherosclerosis (MESA, n = 4,620), Chronic Obstructive Pulmonary Disease Gene (COPDGene) study (n = 10,623), Estonian Biobank (EST, n = 2,244), Japan Biological Informatics Consortium (JPN (JBIC), n = 295), Biobank Japan (JPN (BBJ), n = 1,025) and 1000 Genomes Project (1KG, n = 2,504). Each study was previously approved by respective institutional review boards, including for the generation of WGS data and association with phenotypes. All participants provided written consent. Further details of cohort descriptions and phenotype definitions are described in the Supplementary Note.
All participants included in the HIV host response study were adults, and written informed consent for genetic testing was obtained from all individuals as part of the original study in which they were enrolled (Supplementary Table 10). Ethical approval was obtained from institutional review boards for each of the respective contributing centers.
HLA-TAPAS.
HLA-TAPAS (HLA-Typing At Protein for Association Studies, https://github.com/immunogenomics/HLA-TAPAS) is an HLA-focused pipeline that can handle HLA reference panel construction (MakeReference), HLA imputation (SNP2HLA), and HLA association (HLAassoc). It is an updated version of SNP2HLA11 to build an imputation reference panel and perform HLA classical allele, amino acid and SNP imputation within the extended MHC region. Briefly, major updates include (1) using PLINK1.9 instead of v1.07; (2) using BEAGLE v4.1 instead of v3 for phasing and imputation; and (3) including custom R scripts for performing association and fine-mapping analysis at amino acid level in multiple ancestries. The source code is available for download (Code Availability).
We note that our current implementation of the reference panel is limited to the G-group resolution (DNA sequences that determine the exons 2 and 3 for class I and exon 2 for class II genes, Extended Data Fig. 1), and amino acid positions outside the binding groove were taken as its best approximation (Supplementary Tables 20 and 21). When converting G-group alleles to the two-field resolution, we first approximated G-group alleles to their corresponding allele at the four-field resolution based on the ordered allele list in the distributed IPD-IMGT/HLA database8 (version 3.32.0). We explicitly include exonic information in the HLA-TAPAS output.
Construction of a multi-ancestry HLA reference panel using whole-genome sequences.
To construct a multi-ancestry HLA imputation reference panel, we used 24,338 whole-genome sequences at different depths (Supplementary Table 1). Details of the construction using deep-coverage whole-genome sequencing are described in the Supplementary Note. Briefly, alignment and variant-calling for genomes sequenced by each cohort were performed independently. We performed local realignment and quality recalibration with the Genome Analysis Toolkit67 (GATK; version 3.6) on Chromosome 6:25,000,000–35,000,000. We detected single nucleotide variants (SNV) and indels using GATK with HaplotypeCaller. To eliminate false-positive sites called in the MHC region, we restricted our panel to SNVs reported in 1000 Genomes Project23 only.
We next inferred classical HLA alleles at G-group resolution for eight classical HLA genes (HLA-A, -B, -C, -DQA1, -DQB1, -DRB1, -DPA1 and -DPB1) using a population reference graph26,27. To extend the reference panel versatility, we inferred amino acid variation, one-field and two-field resolution alleles from the inferred G-group alleles. To convert the G-group alleles to two-field and one-field resolution, we first approximated each G-group allele to its corresponding allele at four-field resolution based on the first allele in the ordered allele list in the distributed IPD-IMGT/HLA database8 (version 3.32.0). We then reduced the resolution to one- and two-field based on this approximated four-field allele. After removing samples with low-coverage and failed genome-wide quality control (Supplementary Table 3), we constructed a multi-ancestry HLA imputation reference panel (n = 21,546) using the HLA-TAPAS MakeReference module (Methods).
Sequence-based typing of HLA alleles.
Genomic DNA from the 288 unrelated samples of Japanese ancestry underwent high-resolution allele typing (three-field alleles) of six classical HLA genes (HLA-A, -B and -C for class I; and HLA-DRB1, -DQA1 and -DPB1 for class II)22.
The 1000 Genomes panel consists of 1,267 individuals with information on five HLA genes (HLA-A, -B, -C, -DQB1, and -DRB1) at G-group resolution among four major ancestral groups (AA, EAS, EUR and LAT)7.
Purified DNA from the 75 donors from the GaP registry (at the Feinstein Institute for Medical Research) was sent to NHS Blood and Transplant, UK, where HLA typing was performed. Next-generation sequencing was done for HLA-A, -B, -C, -DQB1, -DPB1 and -DRB1. PCR-sequence-specific oligonucleotide probe sequencing was performed for HLA-DQA1 in all samples. These typing methods yielded classical allele calls for six genes at three-field (HLA-A, -B, -C,-DQB1, -DPB1 and -DRB1) or G-group resolution (HLA-DQA1).
We obtained HLA typing of the 1,067 African American individuals included in the HIV-1 viral load study as described previously18,68. Briefly, seven classical HLA genes (HLA-A, -B, -C, -DQA1, -DQB1 -DRB1 and -DPB1) were obtained by sequencing exons 2 and 3 and/or single-stranded conformation polymorphism PCR (i.e., at G-group resolution), and were provided at approximated two-field resolution.
A summary of all SBT of HLA alleles in three different cohorts (1000 Genomes, GaP registry and HIV-1) used for imputation accuracy evaluation are summarized in Supplementary Table 22.
Accuracy measure between inferred and sequence-based typing HLA genotypes.
Allelic variants at HLA genes can be typed at different resolutions: one-field HLA types specify serological activity, two-field HLA types specify the amino acids encoded by the exons of the HLA gene, and three-field types determine the full exonic sequence including synonymous variants. G-group resolution determines the sequences of the exons encoding the peptide binding groove, that is, exons 2 and 3 for class I and exon 2 for class II genes. This means many G-group alleles can map to multiple three-field and two-field HLA alleles.
We calculated the accuracy at each HLA gene by summing across the dosage of each correctly inferred HLA allele or amino acid across all individuals (N), and divided by the total number of observations (2*N). That is,
where Accuracy(g) represents the accuracy at a classical HLA gene (e.g. HLA-B). Di represents the inferred dosage of an allele in individual i, and alleles A1i,g and A2i,g represent the true (SBT) HLA types for an individual i.
To evaluate the accuracy between the inferred and validated HLA types obtained from SBT at G-group resolution, we translated the highest resolution specified by the validation data to its matching G-group resolution based IMGT/HLA database (e.g. HLA-A*01:01 → HLA-A*01:01:01G), and compared it to the primary output from HLA*LA or HLA-TAPAS. We also translated all G-group alleles to their matching amino acid sequences and compared them against the validation alleles; we referred to this as the amino acid level.
To evaluate imputation performance in individual classical HLA alleles and amino acids, we calculated the dosage r2 correlation between imputed and SBT dosage:
where xi and yi represents the inferred and SBT dosage of an allele in individual i. N represents the number of individuals.
Principal component analysis.
We performed a principal component analysis of the MHC region based on the identity-by-descent (IBD) distances between all 21,809 individuals included in the multi-ancestry reference panel. We computed the IBD distance using Beagle (Version 4.1) and averaged over 100 runs with all variants (54,474) included in the HLA reference panel. Due to uneven representation of different genetic ancestry groups (Supplementary Table 2), we applied a weighted PCA approach, where mean and standard deviation of the IBD matrix within an ancestry group are weighted inversely proportional to the sample size.
HLA haplotype frequency estimation and Hardy-Weinberg equilibrium.
We applied an expectation-maximization algorithm approach implemented in Hapl-o-Mat (v1.1)69 to estimate HLA haplotype frequency based on eight classical HLA alleles inferred at G-group resolution. We estimated haplotype counts and frequencies both overall and within five continental populations (Supplementary Data 4).
To assess departure from Hardy-Weinberg Equilibrium (HWE), we used the chi-square test implemented in the BIGDAWG (v.2.3.1)70. We limited typing results to the first field of the nomenclature for this analysis as no ambiguity occurred on this level. We reported P-values obtained from the exact chi-square test on each of the eight HLA loci (HLA-A, -B, -C, -DRB1, -DQA1, -DQB1, -DPA1 and -DPB1) in each of the five populations separately.
Local ancestry inference.
To detect local ancestry in admixed samples, we first applied ELAI56 to chromosome 6 with 1000 Genomes Project23 as the reference panel. We extracted 63,998 common HapMap3 SNPs between the WGS (MESA cohort) and the 1000 Genome reference panel. We used the same set of SNPs for ELAI and RFMix analysis. We applied ELAI56 to 1,832 African Americans and 594 Latinos. For 1,832 African American individuals included in the study, we used genotypes of 99 CEU and 108 YRI in the 1000 Genome Project as reference panel, assuming admixture generation to be seven generations ago. We used two upper-layer clusters and 10 lower-layer clusters in the model. For Latinos, we selected 65 Latinos with Native American (NAT) ancestry > 75% included in the 1000 Genomes Project identified using the ADMIXTURE analysis71 and used these individuals with high NAT, as well as CEU and YRI from 1000 Genomes as reference panels. We assumed that the admixture time was 20 generations ago. For ELAI, we used three upper-layer clusters and 15 lower-layer clusters in the model.
To address the technical concerns that local ancestry methods are biased by the high LD of the MHC region72,73, we performed an alternative method, RFMix57, for local ancestry inference that accounts for high LD and lack of parental reference panels. Similar deviation from genome-wide ancestry was observed using RFMix (Extended Data Fig. 10), indicating that the selection signals we observed here are robust to different inference methods.
HLA imputation in the HIV-1 viral load GWAS data in three population.
We used genome-wide genotyping data from 12,023 HIV-1 infected individuals aggregated across more than 10 different cohorts (Supplementary Table 10). The details of these samples and quality control procedures have been described previously14,74. Using the HIV-1 viral load GWAS data, we extracted the genotypes of SNPs located in the extended MHC region (chr6:28–34Mb, Supplementary Table 10). We conducted genotype imputation of one-field, two-field and G-group classical HLA alleles and amino acid polymorphisms of the eight class I and class II HLA genes using the constructed multi-ancestry HLA imputation reference panel and the HLA-TAPAS pipeline.
After imputation, we obtained the genotypes of 640 classical alleles, 4,513 amino acid positions of the eight classical HLA genes, and 49,321 SNPs located in the extended MHC region. We excluded variants with MAF < 0.5% and imputation r2 < 0.5 for all association studies. In total, we tested 51,358 variants in our association and fine-mapping study.
HLA association analysis.
For the HIV-1 viral loads of EUR, AA and LAT samples, we conducted a joint haplotype-based association analysis using a linear regression model under the assumption of additive effects of the number of HLA haplotypes for each individual. Phased haplotypes at a locus (i.e., HLA amino acid position) were constructed from the phased imputed genotypes of variants in the locus (i.e., amino acid change or SNP) and were converted to a haplotype matrix where each row is observed haplotypes (in the locus), not genotypes.
For each amino acid position, we applied a conditional haplotype analysis. We tested a multiallelic association between the HIV-1 viral load and a haplotype matrix (of the position) with covariates, including sex, study-specific PCs, and a categorical variable indicating a population. That is
where xi is the amino acid haplotype formed by each of the m amino acid residues that occur at that position, and cj are the covariates included in the model.
To get an omnibus P-value for each position, we estimated the effect of each amino acid by assessing the significance of the improvement in fit by calculating the in-model fit, compared to a null model following an F-distribution with m − 1 degrees of freedom. This is implemented using an ANOVA test in R as described previously43,75. The most frequent haplotype was excluded from a haplotype matrix as a reference haplotype for association.
For the conditional analysis, we assumed that the null model consisted of haplotypes as defined by residues at previously defined amino acid positions. The alternative model is in addition of another position with m residues. We tested whether the addition of those amino acid positions, and the creation of k additional haplotypes groups, improved on the previous set. We then assessed the significance of the improvement in the delta deviance (sum of squares) over the previous model using an F-test. We performed stepwise conditional analysis to identify additional independent signals by adjusting for the most significant amino acid position in each step until none met the significance threshold (P = 5 × 10−8). We restricted analysis to haplotypes that have a minimum of 10 occurrences within HLA-B, and removed any individual with rare haplotypes for the conditional analysis.
For the exhaustive search, we tested all possible amino acid pairs and triplets for association. For each set of amino acid positions, we used the groups of residues occurring at these positions to estimate effect size and calculated for each of these models the delta deviance in risk prediction and its P-values compared to the null model.
Analysis of non-additive effects.
To assess non-additive associations of three reported amino acid positions (97, 67 and 156 in HLA-B), we examined disease risk of homozygotes and heterozygotes for each haplotype, using an established linear regression framework14,76:
where β0 is the linear regression intercept, β1i is the additive effect of allele i, xi is the amino acid haplotype formed by each of the amino acid residues that occur that position, di represents the dominance term for each represented haplotype, δxi = 1 if and only if xi = 1 (heterozygous) and 0 otherwise, and cj are the covariates included in the model (sex, study-specific PCs, and a categorical variable indicating the population).
To determine the relative non-additive effect of a specific haplotype with frequency greater than 0.5%, we assessed the change in deviance between the additive model and the non-additive model for each amino acid variant, which follows a chi-square distribution with 1 degrees of freedom. We used a significance threshold of P < 0.05/26 to correct for multiple tests.
Heterogeneity testing of effect sizes.
We used interaction analyses with models that included haplotype-by-ancestry (Haplotype x Ancestry) interaction terms. The fit of nested models was compared to a null model using the F-statistic with two degrees of freedom, for which the association interaction P-value indicated whether the inclusion of the Haplotype x Ancestry interaction terms improved the model fit compared to the null model that did not include the interaction terms. Interaction P-values for all haplotypes formed by positions 97, 67 and 156 in HLA-B are listed in Supplementary Table 16. Haplotypes that had a significant Bonferroni-corrected Haplotype x Ancestry interaction heterogeneity P-value (P < 0.05/26) were considered to show evidence of significant effect size heterogeneity between ancestries.
Code Availability
HLA-TAPAS, https://github.com/immunogenomics/HLA-TAPAS;
GATK version3.6, https://software.broadinstitute.org/gatk/download/archive;
HLA*PRG, https://github.com/AlexanderDilthey/MHC-PRG;
HLA*LA, https://github.com/DiltheyLab/HLA-PRG-LA;
PLINK version1.90, https://www.cog-genomics.org/plink2;
Beagle version4.1, https://faculty.washington.edu/browning/beagle/b4_1.html;
Hapl-o-Mat version1.1, https://github.com/DKMS/Hapl-o-Mat/;
BIGDAWG version2.3.6, https://cran.r-project.org/web/packages/BIGDAWG/index.html
Data Availability
All scripts and data for generating figures presented in the manuscript are available at https://github.com/immunogenomics/HLA-TAPAS. The reference panel can be accessed for imputation at the Michigan Imputation Server, https://imputationserver.sph.umich.edu. IPD-IMGT/HLA database (version 3.32.0), https://www.ebi.ac.uk/ipd/imgt/hla/. 1000 Genomes gold-standard HLA types, http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/HLA_types/.
Extended Data
Extended Data Fig. 1. HLA nomenclature.

Description of a classical HLA allele using current standard nomenclature. The first field corresponds to the serological antigen. The second field distinguishes HLA alleles that differ by one or more missense variants. The third field distinguishes HLA alleles that differ by one or more synonymous variants. The G-group distinguishes HLA alleles that differ by one or more synonymous variants within the exons that encode the peptide binding groove regions (exon 2 and 3 for HLA class I genes and exon 2 for HLA class II genes).
Extended Data Fig. 2. Correlation between imputed and typed dosage (dosage r2) of classical HLA alleles in 1,067 Admixed African HIV-1 samples.
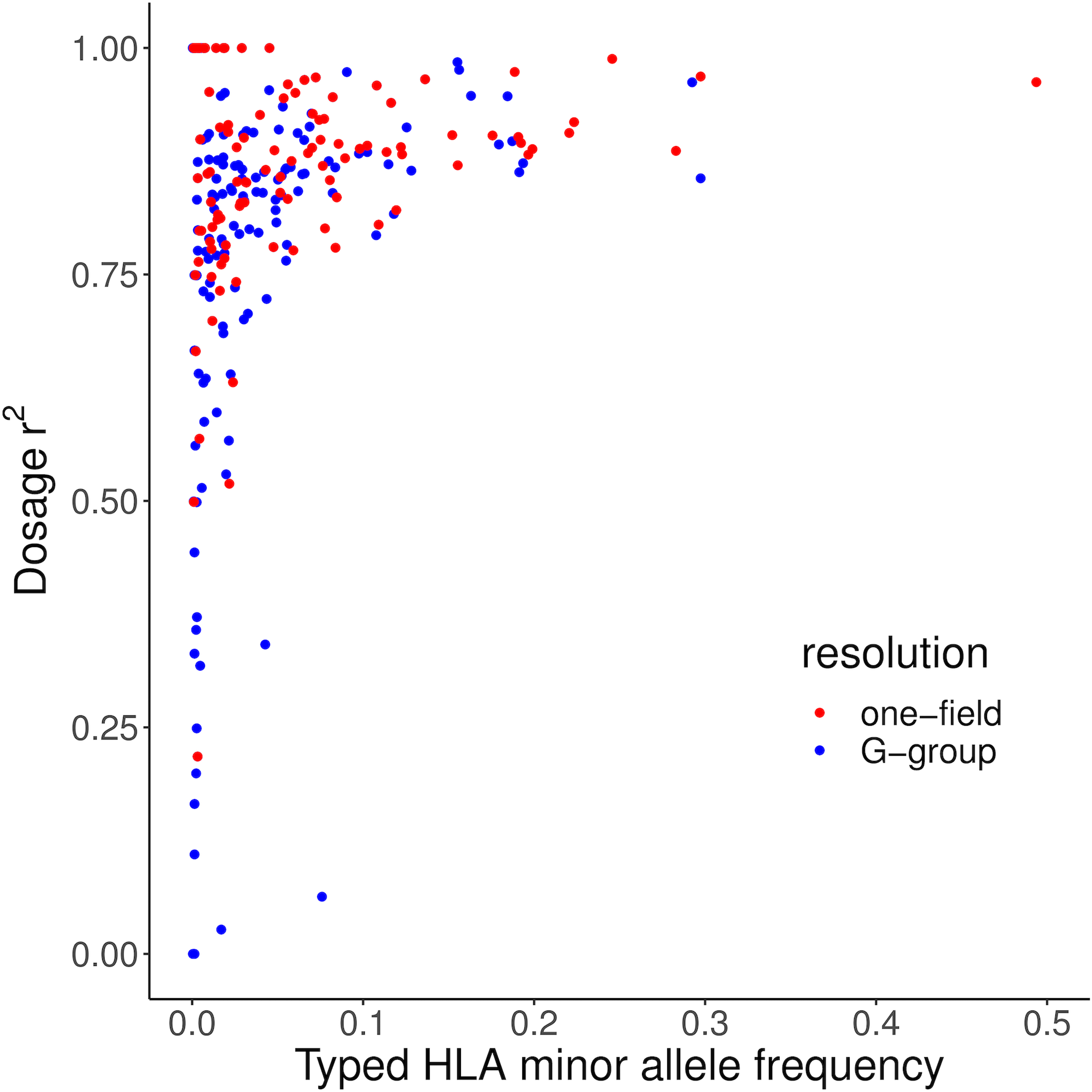
The x-axis shows the minor allele frequency observed in the SBT dataset. Blue points show G-group HLA alleles. Red points show one-field HLA alleles.
Extended Data Fig. 3. Association tests within the MHC to HIV-1 viral load.
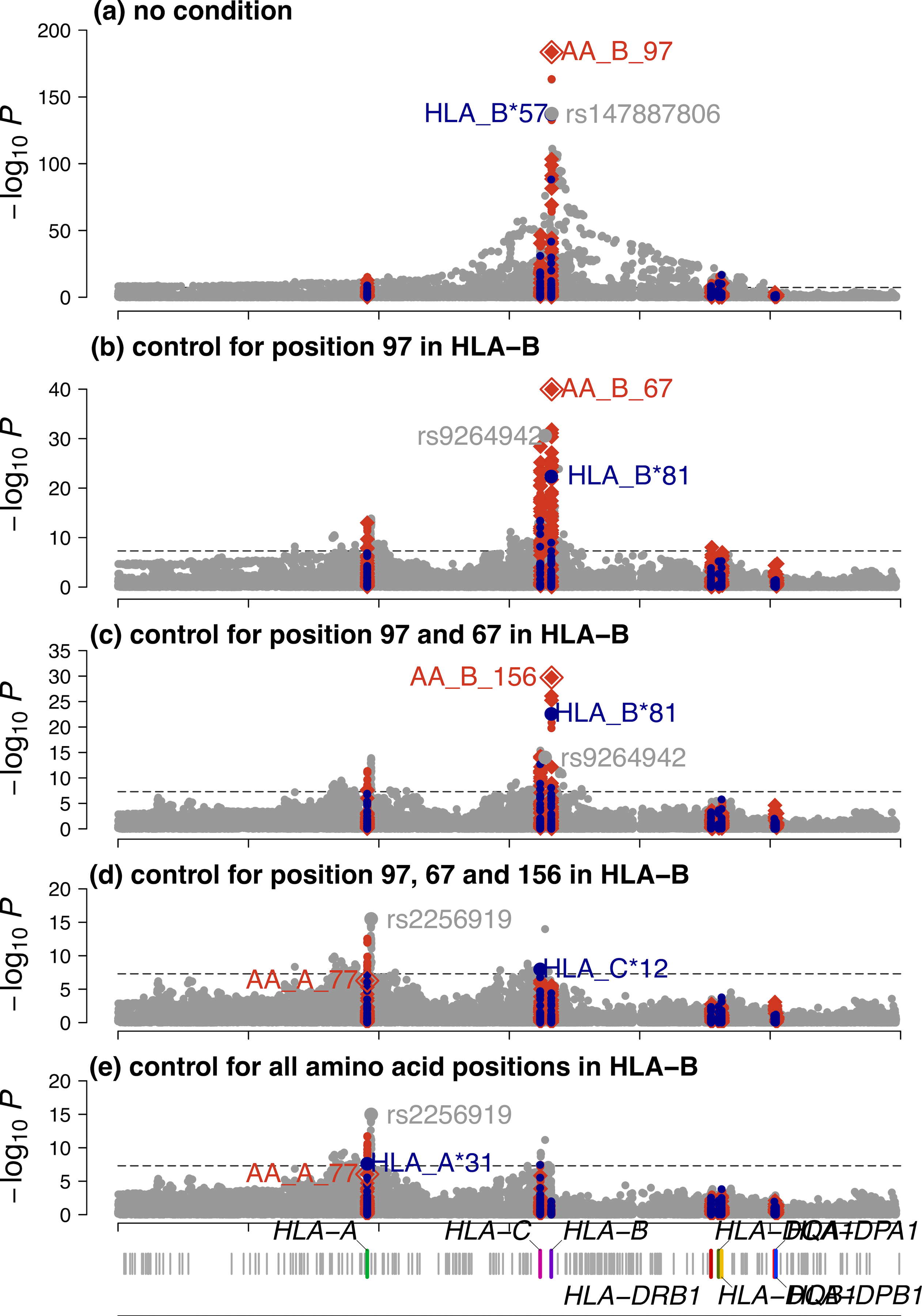
The x-axis shows the genomic positions of chromosome 6 (build 37), and the y-axis is the -log10 (P-value) obtained from two-sided regression analyses for SNPs (gray), classical HLA alleles (blue) and amino acids (red). The dashed black line indicates the genome-wide significance threshold (P = 5 × 10−8). For biallelic markers, results were calculated by a linear regression model including sex, cohort-specific principal components and ancestry indicator as covariates (circle). Association at amino acid positions with more than two residues was calculated using a multi-degree-of-freedom omnibus test (one-sided F-test) including the same covariates (diamond). The top associated amino acid, classical HLA allele and SNPs are annotated in the figure. a, Of all variants tested, the top hit maps to amino acid position 97 in HLA-B. b, Subsequent conditional analysis controlling for all residues at position 97 in HLA-B revealed an independent association at position 67 in HLA-B. c, Results conditioned on position 97 and 67 in HLA-B showed a third signal at position 156 in HLA-B. d, Results conditioned on position 97, 67 and 156 in HLA-B showed position 77 in HLA-A has the strongest association signal outside HLA-B among all amino acid positions. e, Results conditioned on all amino acid positions in HLA-B. Notably, amino acid positions were more significant than any single SNP or classical HLA allele in each conditional analysis for the three amino acid positions in HLA-B.
Extended Data Fig. 4. Effect on set point viral load of individual residues at position 97 in HLA-B.
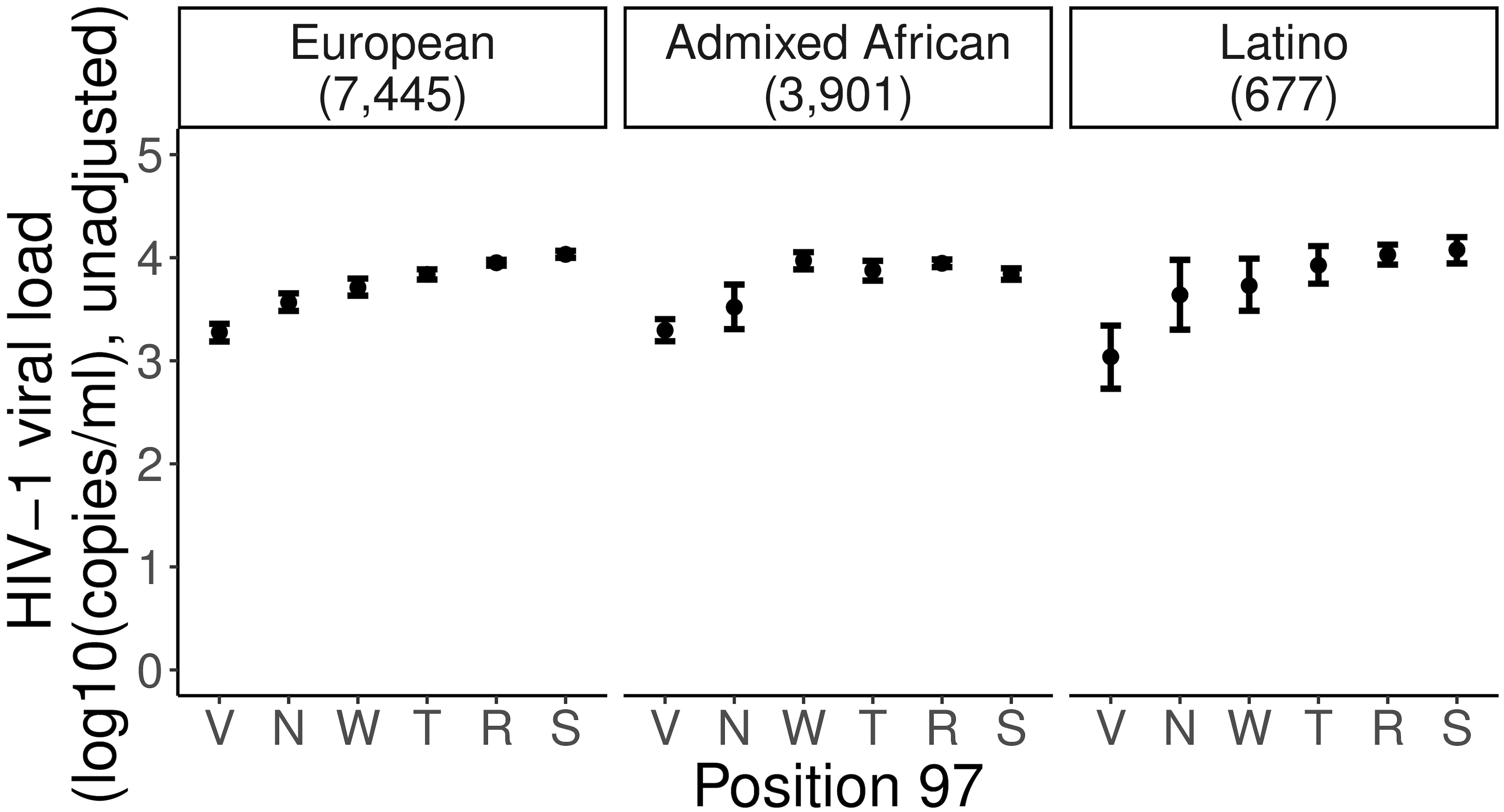
Mean set point viral load (spVL, RNA copies per milliliter) and its standard error of all six residues at position 97 in HLA-B in three populations independently. Data are presented as mean values ± standard errors. Residues are ranked from the most protective to the riskiest in the overall population. There are 3,901 Admixed African, 7,455 European, and 677 Latino independent samples included in the analysis.
Extended Data Fig. 5. Global diversity of the MHC region.
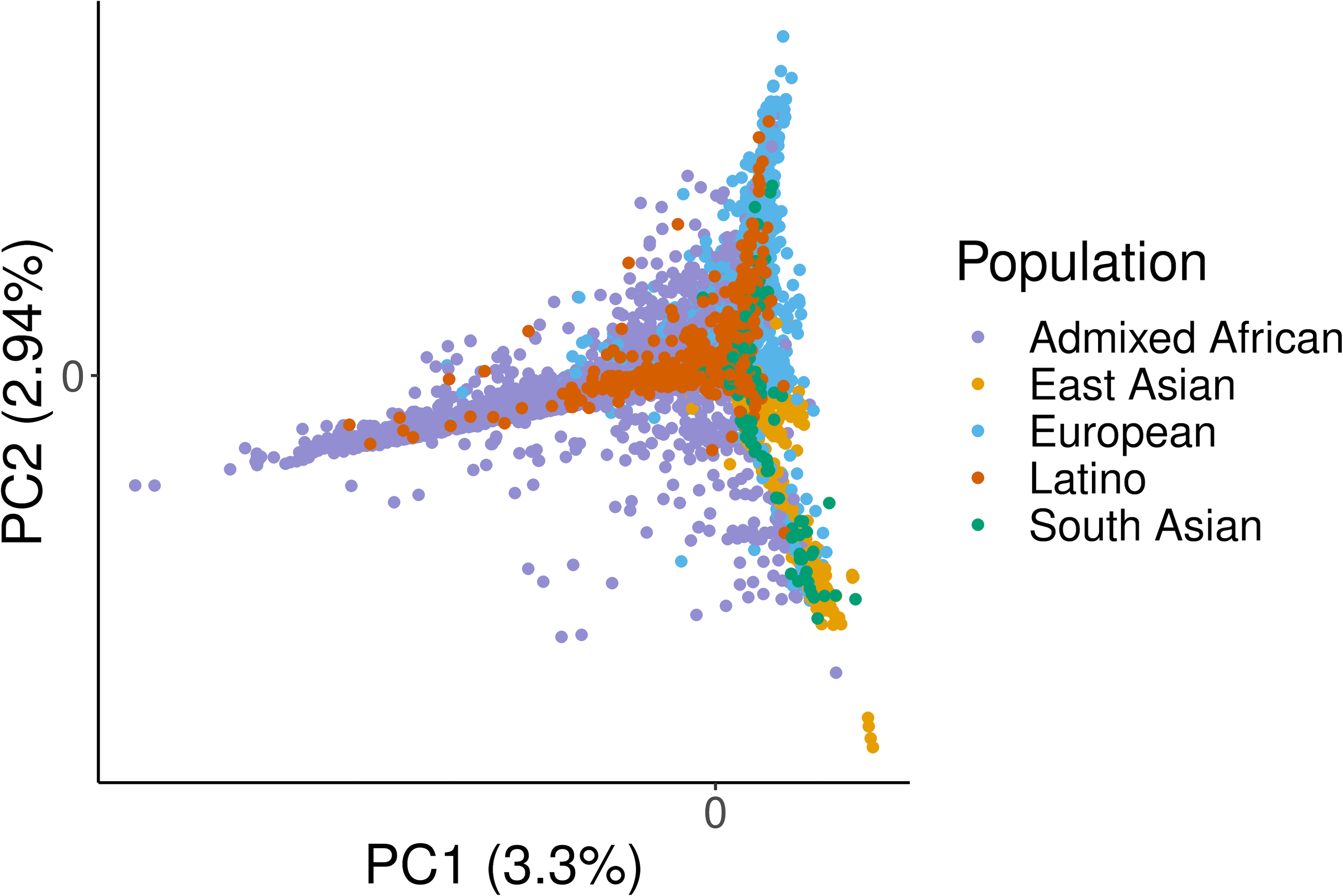
Principal component analysis of the pairwise IBD distance between 21,546 samples using MHC region markers. The first two principal components show separation of continental groups.
Extended Data Fig. 6. Diversity of eight classical HLA genes in the constructed multi-ancestry MHC reference panel.
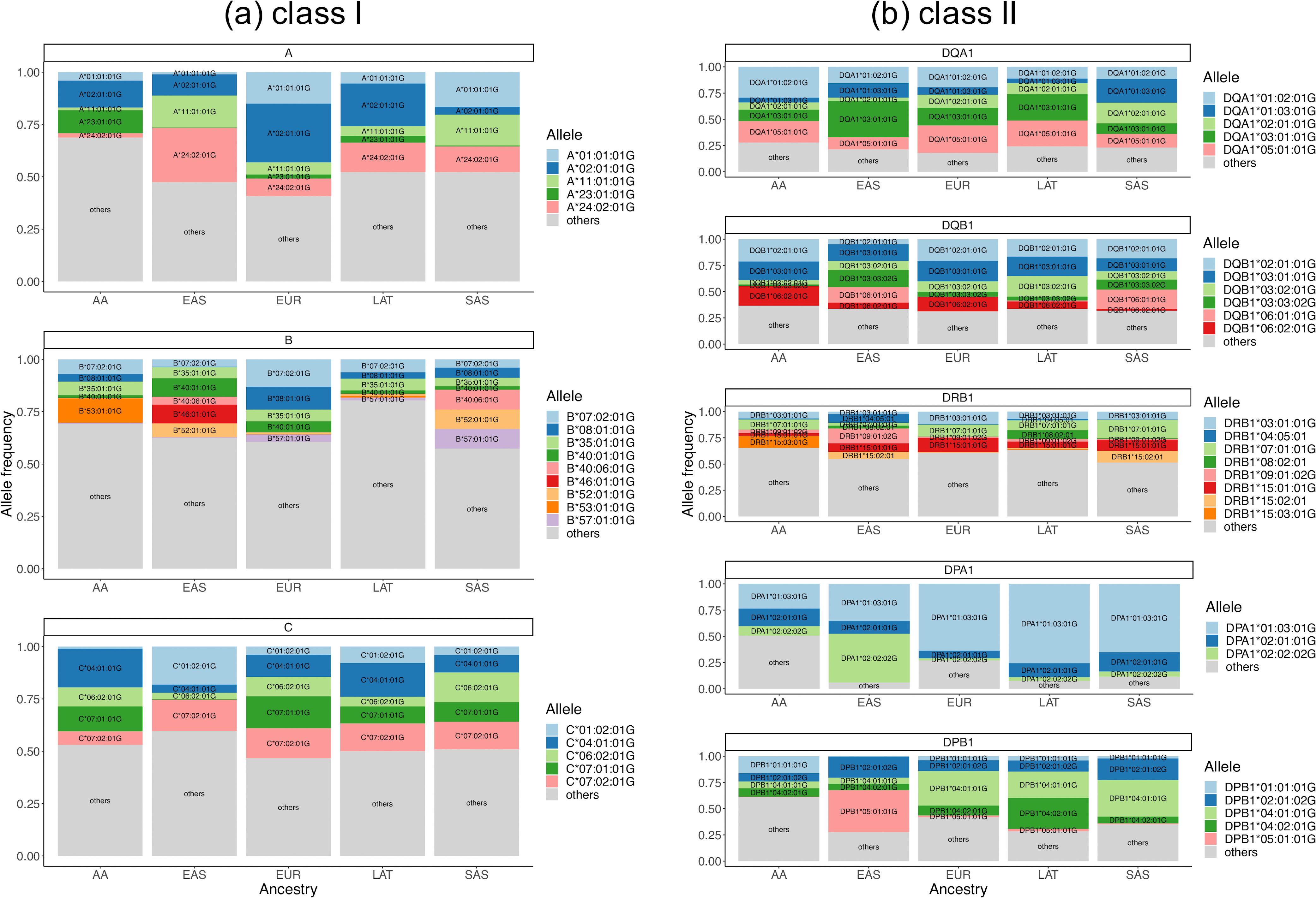
Each gene is stratified by six populations (AA, Admixed African; EAS, East Asian; EUR, European; LAT, Latino; SAS, South Asian). The top two most common alleles within each classical gene of each population are plotted across all panels. Alleles that have frequencies greater than 1% are also labelled in the bar plots. a, Class I genes. b, Class II genes.
Extended Data Fig. 7. Allele diversity of eight classical HLA genes in global populations.
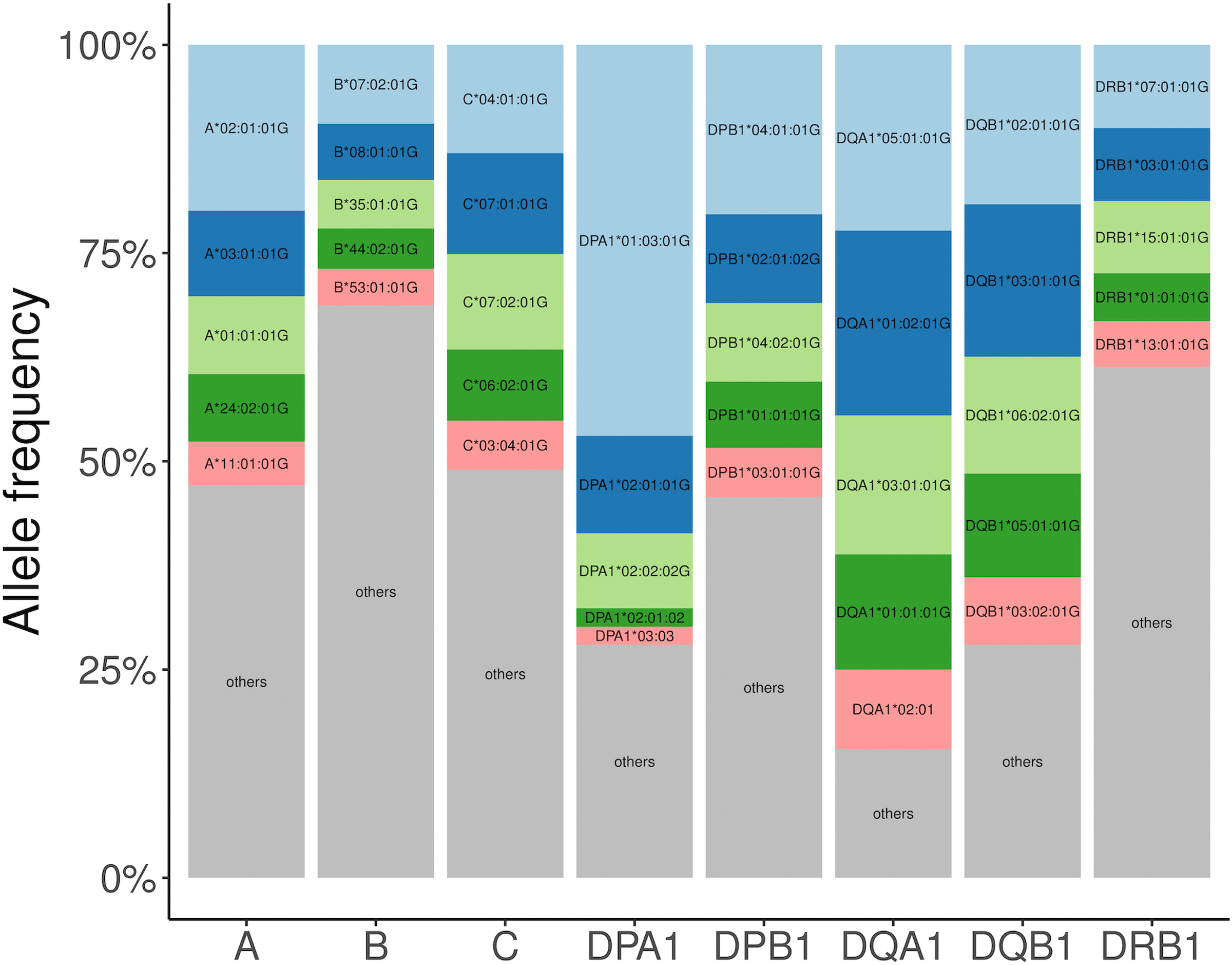
For each gene, the top five most frequent alleles across all populations are shown (light blue, most frequent; dark blue, second frequent; light green, third frequent; dark green, fourth frequent; red, fifth frequent; gray, all other alleles).
Extended Data Fig. 8. Pairwise normalized entropy (ε) among all population groups.
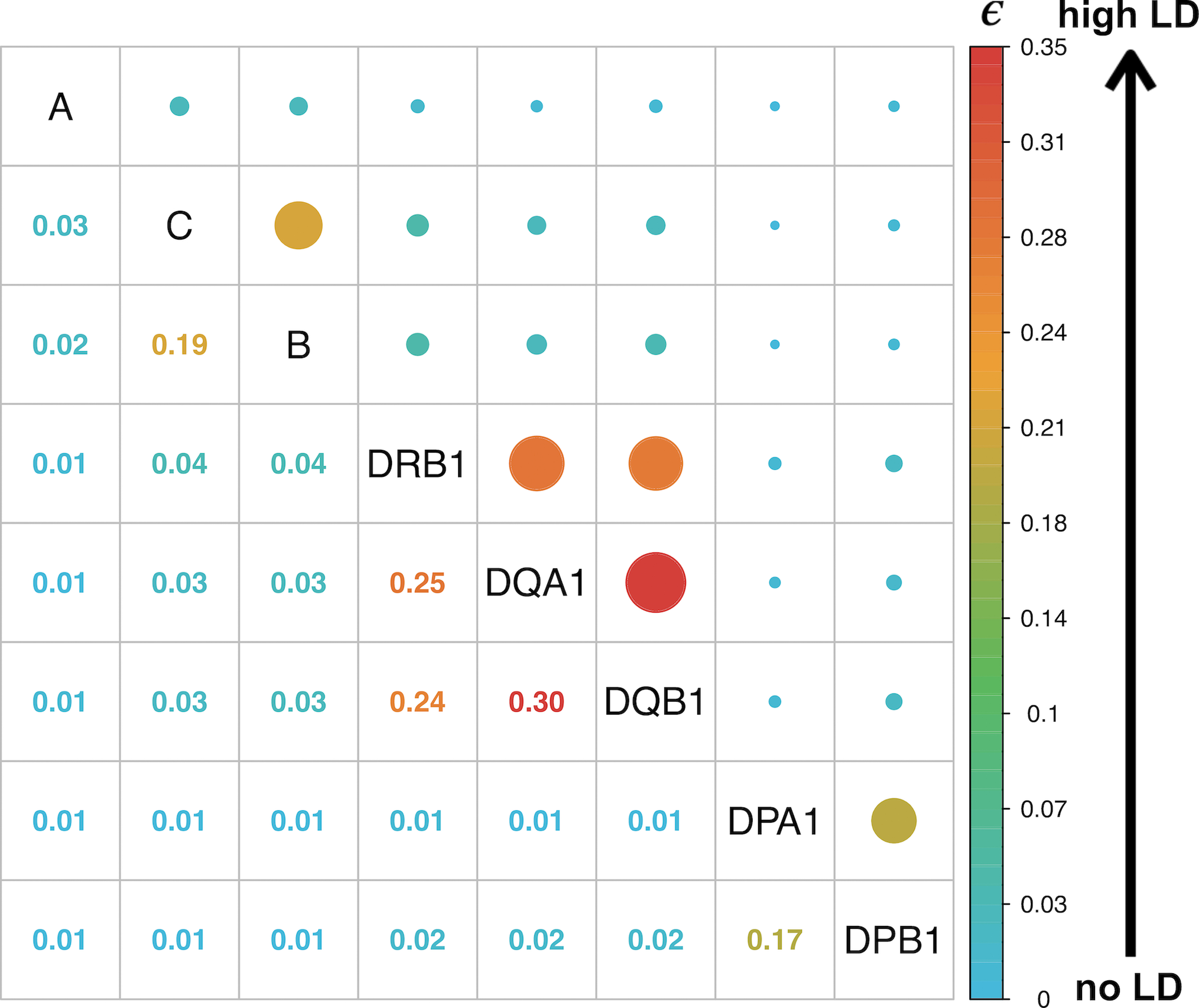
The normalized entropy (ε) measures the difference of the haplotype frequency distribution for linkage disequilibrium and linkage equilibrium, and takes values between 0 (no LD) to 1 (perfect LD).
Extended Data Fig. 9. Deviation from average genome-wide ancestry in Admixed African and Latino populations.
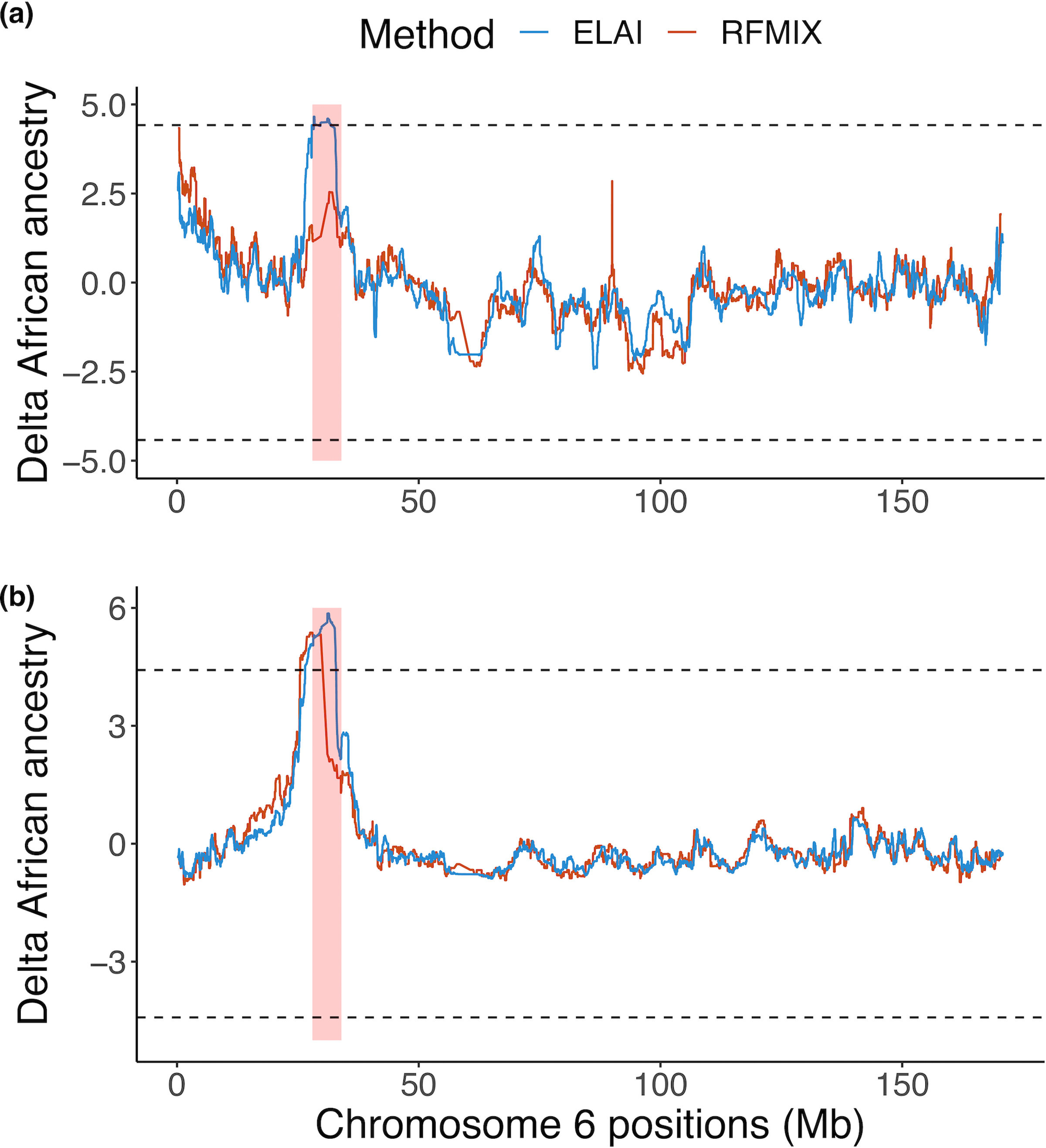
a,b, The x-axis is the genomic position of chromosome 6. The y-axis shows the local African ancestry deviation measure inferred at a given position for Admixed Africans (a) and Latinos (b). The MHC region (chr6:28Mb-34Mb) is highlighted in red shading. Local ancestries were estimated using RFMix (red) and ELAI (blue). The ancestry deviation measure is the difference between African ancestry at a given genomic position with respect to the genome-wide average estimated by ADMIXTURE with K = 3, normalized by the standard deviation of the ancestry estimate. The dashed line indicates the genome-wide significance threshold at ±4.42 standard deviation of the ancestry estimate deviated from the genome-wide average.
Extended Data Fig. 10. Conditional analysis of other previously reported independently associated amino acid positions.
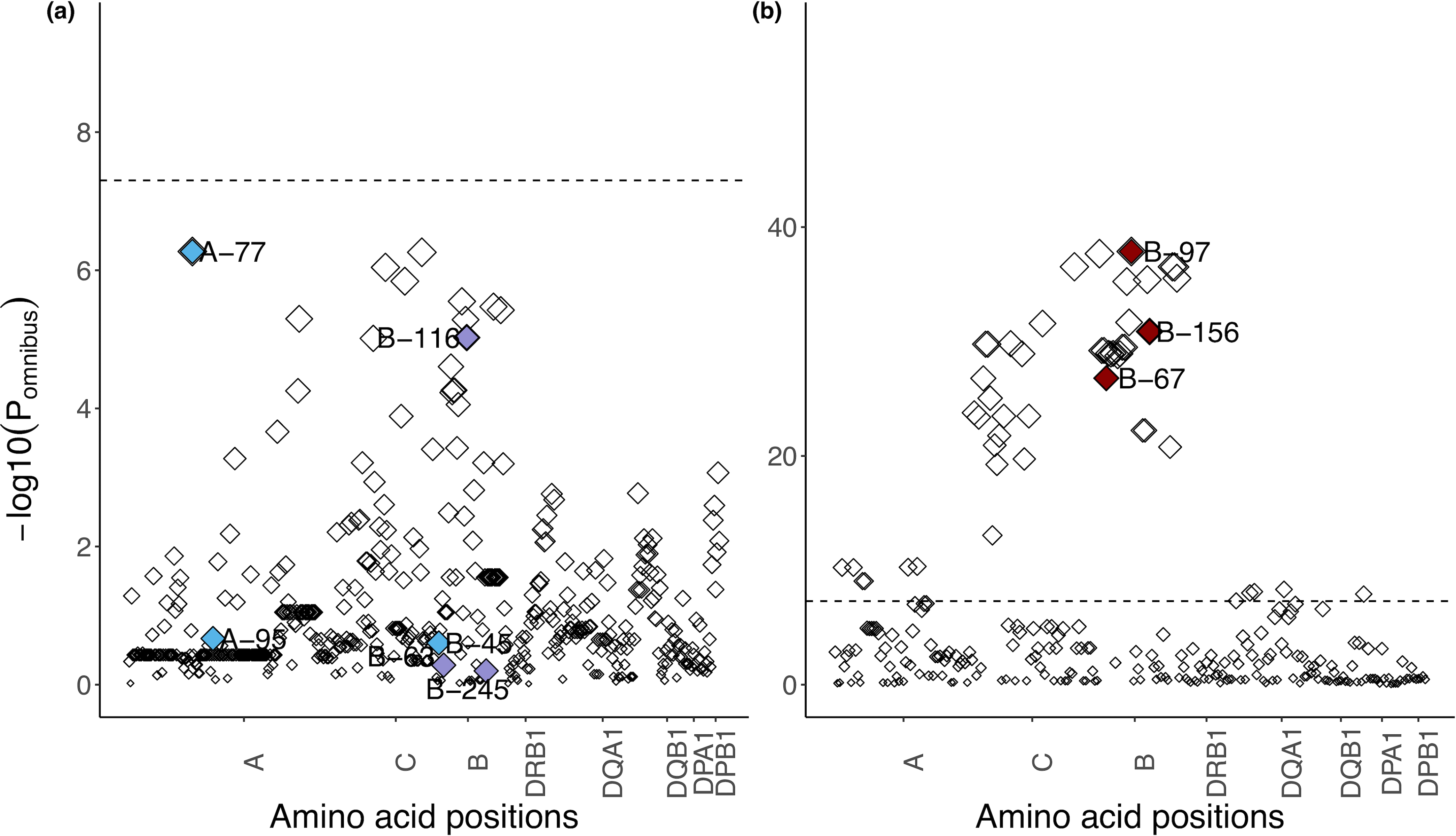
a,b, Manhattan plots of amino acid positions in the six classical HLA genes. Each point shows a single amino acid position and its omnibus P-value after controlling for independent positions that are associated with spVL in this study (position 97, 67 and 156 in HLA-B) (a) and independent positions that are only reported in previous studies14,18 and not in the presented work (position 45, 63 and 116 in HLA-B and position 77, 95 in HLA-A) (b). Independently associated amino acid positions that are only reported in the European population14 are shown in blue. Independently associated amino acid positions that are only reported in the African American population18 are shown in purple. Independently associated amino acid positions identified in this study are shown in red.
Supplementary Material
Summary of inferred HLA alleles at G-group resolution. For each allele observed in the reference panel, we listed its overall frequency (Freq); P-value (Pval) for difference in frequencies across populations (a two-sided chi-square test with 4 degree-offreedom); frequency within each continental population (European, EUR; AA, Admixed African; LAT, Latino; SAS, South Asian; EAS, East Asian) and its accuracy in two validation cohorts (JPN (n = 288 independent samples) and 1000 Genomes Project (n = 955 independent samples)).
Imputation dosage r2 of each allele. Each row shows each imputed classical HLA allele and its imputation accuracy (measure in dosage r2) in three cohorts with validation data (1000 Genomes Project (G1K, n = 955), GaP registry (GAP, n = 75) and HIV (n = 1,067)). We also report the allelic frequency using the gold standard and the inferred alleles.
Association study within the MHC to HIV-1 viral load. For each row, we list the results of association testing for each of the binary markers that we imputed across the extended MHC region. For each marker, its unique identifier (ID), genome basepair position in Grch37 (BP), reference allele (REF), alternative allele (ALT), minor allele frequency (MAF), effect size (BETA) and standard error (SE) in each conditional analysis are listed.
HLA haplotype frequency based on eight classical HLA alleles inferred at G-group resolution. All haplotype with count > 1 in each and overall population are listed. AA represents individuals of Admixed African ancestry; EAS represents individuals of East Asian ancestry; EUR represents individuals of European ancestry; LAT represents individuals of Native American ancestry; SAS represents individuals of South Asian ancestry.
Acknowledgements
The study was supported by the National Institutes of Health (NIH) TB Research Unit Network, Grant U19 AI111224-01.
We thank Cristen Willer, Brett Vanderwerff and Bethany Klunder from the University of Michigan for help facilitating getting the constructed reference panel on the Michigan Imputation Server.
The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the U.S. Department of Health and Human Services.
The Genotype and Phenotype (GaP) Registry at The Feinstein Institute for Medical Research provided fresh, de-identified human plasma; blood was collected from control subjects under an IRB-approved protocol (IRB# 09-081) and processed to isolate plasma. The GaP is a sub-protocol of the Tissue Donation Program (TDP) at Northwell Health and a national resource for genotype-phenotype studies. https://www.feinsteininstitute.org/robert-s-boas-center-for-genomics-and-human-genetics/gap-registry/
For some HIV cohort participants, DNA and data collection was supported by NIH/NIAID AIDS Clinical Trial Group (ACTG) grants UM1 AI068634, UM1 AI068636 and UM1 AI106701, and ACTG clinical research site grants A1069412, A1069423, A1069424, A1069503, AI025859, AI025868, AI027658, AI027661, AI027666, AI027675, AI032782, AI034853, AI038858, AI045008, AI046370, AI046376, AI050409, AI050410, AI050410, AI058740, AI060354, AI068636, AI069412, AI069415, AI069418, AI069419, AI069423, AI069424, AI069428, AI069432, AI069432, AI069434, AI069439, AI069447, AI069450, AI069452, AI069465, AI069467, AI069470, AI069471, AI069472, AI069474, AI069477, AI069481, AI069484, AI069494, AI069495, AI069496, AI069501, AI069501, AI069502, AI069503, AI069511, AI069513, AI069532, AI069534, AI069556, AI072626, AI073961, RR000046, RR000425, RR023561, RR024156, RR024160, RR024996, RR025008, RR025747, RR025777, RR025780, TR000004, TR000058, TR000124, TR000170, TR000439, TR000445, TR000457, TR001079, TR001082, TR001111, and TR024160.
Molecular data for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the National Heart, Lung and Blood Institute (NHLBI). See the TOPMed Omics Support Table (Supplementary Table 23) for study specific omics support information. Core support including centralized genomic read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Core support including phenotype harmonization, data management, sample-identity QC, and general program coordination were provided by the TOPMed Data Coordinating Center (R01HL-120393; U01HL-120393; contract HHSN268201800001I). We gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed.
The COPDGene project was supported by Award Number U01 HL089897 and Award Number U01 HL089856 from the National Heart, Lung, and Blood Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health. The COPDGene project is also supported by the COPD Foundation through contributions made to an Industry Advisory Board comprised of AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Novartis, Pfizer, Siemens and Sunovion. A full listing of COPDGene investigators can be found at http://www.copdgene.org/directory.
The Jackson Heart Study (JHS) is supported and conducted in collaboration with Jackson State University (HHSN268201800013I), Tougaloo College (HHSN268201800014I), the Mississippi State Department of Health (HHSN268201800015I) and the University of Mississippi Medical Center (HHSN268201800010I, HHSN268201800011I and HHSN268201800012I) contracts from the National Heart, Lung, and Blood Institute (NHLBI) and the National Institute on Minority Health and Health Disparities (NIMHD). The authors also wish to thank the staffs and participants of the JHS.
MESA and the MESA SHARe project are conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with MESA investigators. Support for MESA is provided by contracts 75N92020D00001, HHSN268201500003I, N01-HC-95159, 75N92020D00005, N01-HC-95160, 75N92020D00002, N01-HC-95161, 75N92020D00003, N01-HC-95162, 75N92020D00006, N01-HC-95163, 75N92020D00004, N01-HC-95164, 75N92020D00007, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-000040, UL1-TR-001079, UL1-TR-001420. MESA Family is conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with MESA investigators. Support is provided by grants and contracts R01HL071051, R01HL071205, R01HL071250, R01HL071251, R01HL071258, R01HL071259, by the National Center for Research Resources, Grant UL1RR033176. The provision of genotyping data was supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR001881, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center. This project has been funded in whole or in part with federal funds from the Frederick National Laboratory for Cancer Research, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This Research was supported in part by the Intramural Research Program of the NIH, Frederick National Lab, Center for Cancer Research.
D.H.S. was supported by R01 HL92301, R01 HL67348, R01 NS058700, R01 AR48797, R01 DK071891, R01 AG058921, the General Clinical Research Center of the Wake Forest University School of Medicine (M01 RR07122, F32 HL085989), the American Diabetes Association, and a pilot grant from the Claude Pepper Older Americans Independence Center of Wake Forest University Health Sciences (P60 AG10484). A.M. is supported by Gentransmed grant 2014-2020.4.01.15-0012. D.W.H. is supported by NIH grants AI110527, AI077505, TR000445, AI069439, and AI110527. J.T.E. and P.E.S. were supported by NIH/NIAMS R01 AR042742, R01 AR050511, and R01 AR063611. Y.O. was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (19H01021, 20K21834), and AMED (JP20km0405211, JP20ek0109413, JP20ek0410075, JP20gm4010006, and JP20km0405217), Takeda Science Foundation, and Bioinformatics Initiative of Osaka University Graduate School of Medicine, Osaka University.
Competing Interests
M.H.C. has received consulting or speaking fees from Illumina and AstraZeneca, and grant support from GSK and Bayer. The remaining authors declare no competing interests.
Footnotes
NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium
Namiko Abe40, Gonçalo Abecasis41, Francois Aguet42, Christine Albert43, Laura Almasy44, Alvaro Alonso45, Seth Ament46, Peter Anderson47, Pramod Anugu48, Deborah Applebaum-Bowden49, Kristin Ardlie42, Dan Arking50, Donna K. Arnett51, Allison Ashley-Koch52, Stella Aslibekyan53, Tim Assimes54, Paul Auer55, Dimitrios Avramopoulos50, Najib Ayas56, Adithya Balasubramanian57, John Barnard58, Kathleen Barnes59, R. Graham Barr60, Emily Barron-Casella50, Lucas Barwick61, Terri Beaty50, Gerald Beck58, Diane Becker50, Lewis Becker50, Rebecca Beer62, Amber Beitelshees46, Emelia Benjamin63, Takis Benos64, Marcos Bezerra65, Larry Bielak41, Joshua Bis47, Thomas Blackwell41, John Blangero66, Eric Boerwinkle67, Donald W. Bowden68, Russell Bowler69, Jennifer Brody47, Ulrich Broeckel70, Jai Broome47, Deborah Brown67, Karen Bunting40, Esteban Burchard71, Carlos Bustamante54, Erin Buth47, Brian Cade72, Jonathan Cardwell73, Vincent Carey72, Julie Carrier74, Cara Carty75, Richard Casaburi76, Juan P. Casas Romero72, James Casella50, Peter Castaldi72, Mark Chaffin42, Christy Chang46, Yi-Cheng Chang77, Daniel Chasman72, Sameer Chavan73, Bo-Juen Chen40, Wei-Min Chen78, Yii-Der Ida Chen26, Michael H. Cho33, Seung Hoan Choi42, Lee-Ming Chuang77, Mina Chung58, Ren-Hua Chung79, Clary Clish42, Suzy Comhair58, Matthew Conomos47, Elaine Cornell80, Adolfo Correa29, Carolyn Crandall76, James Crapo69, L. Adrienne Cupples81, Joanne Curran66, Jeffrey Curtis41, Brian Custer82, Coleen Damcott46, Dawood Darbar83, Sean David84, Colleen Davis47, Michelle Daya73, Mariza de Andrade85, Lisa de las Fuentes86, Paul de Vries67, Michael DeBaun87, Ranjan Deka88, Dawn DeMeo72, Scott Devine46, Huyen Dinh57, Harsha Doddapaneni57, Qing Duan89, Shannon Dugan-Perez57, Ravi Duggirala66, Jon Peter Durda80, Susan K. Dutcher86, Charles Eaton90, Lynette Ekunwe48, Adel El Boueiz91, Patrick Ellinor92, Leslie Emery47, Serpil Erzurum58, Charles Farber78, Jesse Farek57, Tasha Fingerlin69, Matthew Flickinger41, Myriam Fornage67, Nora Franceschini89, Chris Frazar47, Mao Fu46, Stephanie M. Fullerton47, Lucinda Fulton86, Stacey Gabriel42, Weiniu Gan62, Shanshan Gao73, Yan Gao48, Margery Gass93, Heather Geiger40, Bruce Gelb94, Mark Geraci64, Soren Germer40, Robert Gerszten95, Auyon Ghosh72, Richard Gibbs57, Chris Gignoux54, Mark Gladwin64, David Glahn96, Stephanie Gogarten47, Da-Wei Gong46, Harald Goring66, Sharon Graw59, Kathryn J. Gray97, Daniel Grine73, Colin Gross41, C. Charles Gu86, Yue Guan46, Xiuqing Guo26, Namrata Gupta42, David M. Haas98, Jeff Haessler93, Michael Hall48, Yi Han57, Patrick Hanly99, Daniel Harris46, Nicola L. Hawley100, Jiang He101, Ben Heavner47, Susan Heckbert47, Ryan Hernandez71, David Herrington68, Craig Hersh72, Bertha Hidalgo53, James Hixson67, Brian Hobbs72, John Hokanson73, Elliott Hong46, Karin Hoth102, Chao (Agnes) Hsiung79, Jianhong Hu57, Yi-Jen Hung103, Haley Huston104, Chii Min Hwu105, Marguerite Ryan Irvin53, Rebecca Jackson106, Deepti Jain47, Cashell Jaquish62, Jill Johnsen104, Andrew Johnson62, Craig Johnson47, Rich Johnston45, Kimberly Jones50, Hyun Min Kang41, Robert Kaplan107, Sharon Kardia41, Shannon Kelly71, Eimear Kenny94, Michael Kessler46, Alyna Khan47, Ziad Khan57, Wonji Kim108, John Kimoff109, Greg Kinney73, Barbara Konkle104, Charles Kooperberg93, Holly Kramer110, Christoph Lange111, Ethan Lange73, Leslie Lange73, Cathy Laurie47, Cecelia Laurie47, Meryl LeBoff72, Jiwon Lee72, Sandra Lee57, Wen-Jane Lee105, Jonathon LeFaive41, David Levine47, Dan Levy62, Joshua Lewis46, Xiaohui Li112, Yun Li89, Henry Lin112, Honghuang Lin81, Xihong Lin111, Simin Liu90, Yongmei Liu52, Yu Liu54, Ruth J. F. Loos94, Steven Lubitz92, Kathryn Lunetta81, James Luo62, Ulysses Magalang113, Michael Mahaney66, Barry Make50, Ani Manichaikul78, Alisa Manning114, JoAnn Manson72, Lisa Martin115, Melissa Marton40, Susan Mathai73, Rasika Mathias50, Susanne May47, Patrick McArdle46, Merry-Lynn McDonald53, Sean McFarland108, Stephen McGarvey90, Daniel McGoldrick47, Caitlin McHugh47, Becky McNeil116, Hao Mei48, James Meigs92, Vipin Menon57, Luisa Mestroni59, Ginger Metcalf57, Deborah A. Meyers117, Emmanuel Mignot54, Julie Mikulla62, Nancy Min48, Mollie Minear118, Ryan L. Minster64, Braxton D. Mitchell46, Matt Moll72, Zeineen Momin57, May E. Montasser46, Courtney Montgomery119, Donna Muzny57, Josyf C. Mychaleckyj78, Girish Nadkarni94, Rakhi Naik50, Take Naseri120, Pradeep Natarajan42, Sergei Nekhai121, Sarah C. Nelson47, Bonnie Neltner73, Caitlin Nessner57, Deborah Nickerson47, Osuji Nkechinyere57, Kari North89, Jeff O’Connell46, Tim O’Connor46, Heather Ochs-Balcom122, Geoffrey Okwuonu57, Allan Pack123, David T. Paik54, Nicholette D. Palmer27, James Pankow124, George Papanicolaou62, Cora Parker116, Gina Peloso81, Juan Manuel Peralta66, Marco Perez54, James Perry46, Ulrike Peters93, Patricia Peyser41, Lawrence S. Phillips45, Jacob Pleiness41, Toni Pollin46, Wendy Post50, Julia Powers Becker73, Meher Preethi Boorgula73, Michael Preuss94, Bruce Psaty47, Pankaj Qasba62, Dandi Qiao72, Zhaohui Qin45, Nicholas Rafaels73, Laura Raffield89, Mahitha Rajendran57, Vasan S. Ramachandran81, D. C. Rao86, Laura Rasmussen-Torvik125, Aakrosh Ratan78, Susan Redline72, Robert Reed46, Catherine Reeves40, Elizabeth Regan69, Alex Reiner126, Muagututi’a Sefuiva Reupena127, Ken Rice47, Stephen S. Rich28, Rebecca Robillard128, Nicolas Robine40, Dan Roden87, Carolina Roselli42, Jerome I. Rotter26, Ingo Ruczinski50, Alexi Runnels40, Pamela Russell73, Sarah Ruuska104, Kathleen Ryan46, Ester Cerdeira Sabino129, Danish Saleheen60, Shabnam Salimi46, Sejal Salvi57, Steven Salzberg50, Kevin Sandow112, Vijay G. Sankaran130, Jireh Santibanez57, Karen Schwander86, David Schwartz73, Frank Sciurba64, Christine Seidman131, Jonathan Seidman131, Frédéric Sériès132, Vivien Sheehan45, Stephanie L. Sherman45, Amol Shetty46, Aniket Shetty73, Wayne Hui-Heng Sheu105, M. Benjamin Shoemaker87, Brian Silver133, Edwin Silverman72, Robert Skomro134, Albert V. Smith17,18, Jennifer Smith41, Josh Smith47, Nicholas Smith47, Tanja Smith40, Sylvia Smoller107, Beverly Snively68, Michael Snyder54, Tamar Sofer72, Nona Sotoodehnia47, Adrienne M. Stilp47, Garrett Storm73, Elizabeth Streeten46, Jessica Lasky Su72, Yun Ju Sung86, Jody Sylvia72, Adam Szpiro47, Daniel Taliun41, Hua Tang54, Margaret Taub50, Kent D. Taylor26, Matthew Taylor59, Simeon Taylor46, Marilyn Telen52, Timothy A. Thornton47, Machiko Threlkeld47, Lesley Tinker93, David Tirschwell47, Sarah Tishkoff123, Hemant Tiwari53, Catherine Tong47, Russell Tracy80, Michael Tsai124, Dhananjay Vaidya50, David Van Den Berg135, Peter VandeHaar41, Scott Vrieze124, Tarik Walker73, Robert Wallace102, Avram Walts73, Fei Fei Wang47, Heming Wang136, Jiongming Wang41, Karol Watson76, Jennifer Watt57, Daniel E. Weeks64, Joshua Weinstock41, Bruce Weir47, Scott T. Weiss72, Lu-Chen Weng92, Jennifer Wessel98, Cristen Willer41, Kayleen Williams47, L. Keoki Williams137, Carla Wilson72, James Wilson95, Lara Winterkorn40, Quenna Wong47, Joseph Wu54, Huichun Xu46, Lisa Yanek50, Ivana Yang73, Ketian Yu41, Seyedeh Maryam Zekavat42, Yingze Zhang64, Snow Xueyan Zhao69, Wei Zhao41, Xiaofeng Zhu138, Michael Zody40, and Sebastian Zoellner41 40New York Genome Center, New York, NY, USA. 41University of Michigan, Ann Arbor, MI, USA. 42Broad Institute, Cambridge, MA, USA. 43Brigham and Women’s Hospital, Cedars Sinai Boston, MA, USA. 44Children’s Hospital of Philadelphia, University of Pennsylvania, Philadelphia, PA, USA. 45Emory University, Atlanta, GA, USA. 46University of Maryland, Baltimore, MD, USA. 47University of Washington, Seattle, WA, USA. 48University of Mississippi, Jackson, MS, USA. 49National Institutes of Health, Bethesda, MD, USA. 50Johns Hopkins University, Baltimore, MD, USA. 51University of Kentucky, Lexington, KY, USA. 52Duke University, Durham, NC, USA. 53University of Alabama, Birmingham, AL, USA. 54Stanford University, Stanford, CA, USA. 55University of Wisconsin Milwaukee, Milwaukee, WI, USA. 56Providence Health Care Research Institute, Vancouver, BC, Canada. 57Baylor College of Medicine Human Genome Sequencing Center, Houston, TX, USA. 58Cleveland Clinic, Cleveland, OH, USA. 59University of Colorado Anschutz Medical Campus, Aurora, CO, USA. 60Columbia University, New York, NY, USA. 61The Emmes Corporation, Rockville, MD, USA. 62National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD, USA. 63Boston University, Massachusetts General Hospital, Boston, MA, USA. 64University of Pittsburgh, Pittsburgh, PA, USA. 65Fundação de Hematologia e Hemoterapia de Pernambuco – Hemope, Recife, Brazil. 66University of Texas Rio Grande Valley School of Medicine, Brownsville, TX, USA. 67University of Texas Health at Houston, Houston, TX, USA. 68Wake Forest Baptist Health, Winston-Salem, NC, USA. 69National Jewish Health, Denver, CO, USA. 70Medical College of Wisconsin, Milwaukee, WI, USA. 71University of California, San Francisco, San Francisco, CA, USA. 72Brigham & Women’s Hospital, Boston, MA, USA. 73University of Colorado at Denver, Denver, CO, USA. 74University of Montreal, Montreal, QC, Canada. 75Washington State University, Seattle, WA, USA. 76University of California, Los Angeles, Los Angeles, CA, USA. 77National Taiwan University, Taipei, Taiwan. 78University of Virginia, Charlottesville, VA, USA. 79National Health Research Institutes, Zhunan, Taiwan. 80University of Vermont, Burlington, VT, USA. 81Boston University, Boston, MA, USA. 82Vitalant Research Institute, San Francisco, CA, USA. 83University of Illinois at Chicago, Chicago, IL, USA. 84University of Chicago, Chicago, IL, USA. 85Mayo Clinic, Rochester, MN, USA. 86Washington University in St Louis, St Louis, MO, USA. 87Vanderbilt University, Nashville, TN, USA. 88University of Cincinnati, Cincinnati, OH, USA. 89University of North Carolina, Chapel Hill, NC, USA. 90Brown University, Providence, RI, USA. 91Channing Division of Network Medicine, Harvard University, Boston, MA, USA. 92Massachusetts General Hospital, Boston, MA. 93Fred Hutchinson Cancer Research Center, Seattle, WA, USA. 94Icahn School of Medicine at Mount Sinai, New York, NY, USA. 95Beth Israel Deaconess Medical Center, Boston, MA, USA. 96Boston Children’s Hospital, Harvard Medical School, Boston, MA, USA. 97Mass General Brigham, Boston, MA, USA. 98Indiana University, Indianapolis, IN, USA. 99University of Calgary, Calgary, AB, Canada. 100Yale University, New Haven, CT, USA. 101Tulane University, New Orleans, LA, USA. 102University of Iowa, Iowa City, IA, USA. 103Tri-Service General Hospital National Defense Medical Center, Taipei, Taiwan. 104Bloodworks Northwest, Seattle, WA, USA. 105Taichung Veterans General Hospital Taiwan, Taichung, Taiwan. 106Oklahoma State University Medical Center, Tulsa, OK, USA. 107Albert Einstein College of Medicine, Bronx, NY, USA. 108Harvard University, Cambridge, MA, USA. 109McGill University, Montreal, QC, Canada. 110Loyola University, Chicago, IL, USA. 111Harvard School of Public Health, Boston, MA, USA. 112Lundquist Institute, Torrence, CA, USA. 113Ohio State University, Columbus, OH, USA. 114Broad Institute, Harvard University, Massachusetts General Hospital, Cambridge, MA, USA. 115George Washington University, Washington, D.C., USA. 116RTI International, Durham, NC, USA. 117University of Arizona, Tucson, AZ, USA. 118National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD, USA. 119Oklahoma Medical Research Foundation, Oklahoma City, OK, USA. 120Ministry of Health, Government of Samoa, Apia, Samoa. 121Howard University, Washington, D.C., USA. 122University at Buffalo, Buffalo, NY, USA. 123University of Pennsylvania, Philadelphia, PA, USA. 124University of Minnesota, Minneapolis, MN, USA. 125Northwestern University, Chicago, IL, USA. 126Fred Hutchinson Cancer Research Center, University of Washington, Seattle, WA, USA. 127Lutia I Puava Ae Mapu I Fagalele, Apia, Samoa. 128University of Ottawa, Ottawa, ON, Canada. 129Universidade de Sao Paulo, Sao Paulo, Brazil. 130Division of Hematology/Oncology, Broad Institute, Harvard University, Boston, MA, USA. 131Harvard Medical School, Boston, MA, USA. 132Université Laval, Quebec City, QC, Canada. 133University of Massachusetts Memorial Medical Center, Worcester, MA, USA. 134University of Saskatchewan, Saskatoon, SK, Canada. 135University of Southern California, Los Angeles, CA, USA. 136Brigham and Women’s Hospital, Partners, Boston, MA, USA. 137Henry Ford Health System, Detroit, MI, USA. 138Case Western Reserve University, Cleveland, OH, USA.
References
- 1.International HIV Controllers Study et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 330, 1551–1557 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raychaudhuri S et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet. 44, 291–296 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Evans DM et al. Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat. Genet. 43, 761–767 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Snyder A et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 371, 2189–2199 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buniello A et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horton R et al. Gene map of the extended human MHC. Nat. Rev. Genet. 5, 889–899 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Gourraud P-A et al. HLA diversity in the 1000 genomes dataset. PLoS One 9, e97282 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robinson J et al. IPD-IMGT/HLA Database. Nucleic Acids Res. 48, D948–D955 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonzalez-Galarza FF et al. Allele frequency net database (AFND) 2020 update: gold-standard data classification, open access genotype data and new query tools. Nucleic Acids Res. 48, D783–D788 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dilthey AT, Moutsianas L, Leslie S & McVean G HLA*IMP—an integrated framework for imputing classical HLA alleles from SNP genotypes. Bioinformatics 27, 968–972 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jia X et al. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS One 8, e64683 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng X et al. HIBAG—HLA genotype imputation with attribute bagging. Pharmacogenomics J. 14, 192–200 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu X et al. Additive and interaction effects at three amino acid positions in HLA-DQ and HLA-DR molecules drive type 1 diabetes risk. Nat. Genet. 47, 898–905 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McLaren PJ et al. Polymorphisms of large effect explain the majority of the host genetic contribution to variation of HIV-1 virus load. Proc. Natl. Acad. Sci. U. S. A. 112, 14658–14663 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tian C et al. Genome-wide association and HLA region fine-mapping studies identify susceptibility loci for multiple common infections. Nat. Commun. 8, 599 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Onengut-Gumuscu S et al. Type 1 diabetes risk in African-ancestry participants and utility of an ancestry-specific genetic risk score. Diabetes Care 42, 406–415 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.HIV/AIDS. https://www.who.int/news-room/fact-sheets/detail/hiv-aids.
- 18.McLaren PJ et al. Fine-mapping classical HLA variation associated with durable host control of HIV-1 infection in African Americans. Hum. Mol. Genet. 21, 4334–4347 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taliun D et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okada Y et al. Deep whole-genome sequencing reveals recent selection signatures linked to evolution and disease risk of Japanese. Nat. Commun. 9, 1631 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitt M et al. Improved imputation accuracy of rare and low-frequency variants using population-specific high-coverage WGS-based imputation reference panel. Eur. J. Hum. Genet. 25, 869–876 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirata J et al. Genetic and phenotypic landscape of the major histocompatibilty complex region in the Japanese population. Nat. Genet. 51, 470–480 (2019). [DOI] [PubMed] [Google Scholar]
- 23.1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nelis M et al. Genetic structure of Europeans: a view from the north-east. PLoS One 4, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dilthey A, Cox C, Iqbal Z, Nelson MR & McVean G Improved genome inference in the MHC using a population reference graph. Nat. Genet. 47, 682–688 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dilthey AT et al. High-accuracy HLA type inference from whole-genome sequencing data using population reference graphs. PLoS Comput. Biol. 12, e1005151 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dilthey AT et al. HLA*LA-HLA typing from linearly projected graph alignments. Bioinformatics 35, 4394–4396 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mellors JW et al. Quantitation of HIV-1 RNA in plasma predicts outcome after seroconversion. Ann. Intern. Med. 122, 573–579 (1995). [DOI] [PubMed] [Google Scholar]
- 29.Bartha I et al. Estimating the respective contributions of human and viral genetic variation to HIV control. PLoS Comput. Biol. 13, e1005339 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blanco-Gelaz MA et al. The amino acid at position 97 is involved in folding and surface expression of HLA-B27. Int. Immunol. 18, 211–220 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Stewart-Jones GBE et al. Structures of three HIV-1 HLA-B*5703-peptide complexes and identification of related HLAs potentially associated with long-term nonprogression. J. Immunol. 175, 2459–2468 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Archbold JK et al. Natural micropolymorphism in human leukocyte antigens provides a basis for genetic control of antigen recognition. J. Exp. Med. 206, 209–219 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kløverpris HN et al. HIV control through a single nucleotide on the HLA-B locus. J. Virol. 86, 11493–11500 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaiha GD et al. Structural topology defines protective CD8+ T cell epitopes in the HIV proteome. Science 364, 480–484 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Browning BL & Browning SR A fast, powerful method for detecting identity by descent. Am. J. Hum. Genet. 88, 173–182 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hill AV et al. Common west African HLA antigens are associated with protection from severe malaria. Nature 352, 595–600 (1991). [DOI] [PubMed] [Google Scholar]
- 37.Sanchez-Mazas A et al. The HLA-B landscape of Africa: Signatures of pathogen-driven selection and molecular identification of candidate alleles to malaria protection. Mol. Ecol. 26, 6238–6252 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Maiers M, Gragert L & Klitz W High-resolution HLA alleles and haplotypes in the United States population. Hum. Immunol. 68, 779–788 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Chen JJ et al. Hardy-Weinberg testing for HLA class II (DRB1, DQA1, DQB1, AND DPB1) loci in 26 human ethnic groups. Tissue Antigens 54, 533–542 (1999). [DOI] [PubMed] [Google Scholar]
- 40.Tshabalala M et al. Human Leukocyte Antigen-A, B, C, DRB1, and DQB1 allele and haplotype frequencies in a subset of 237 donors in the South African Bone Marrow Registry. J. Immunol. Res. 2018, 2031571 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hagenlocher Y et al. 6-Locus HLA allele and haplotype frequencies in a population of 1075 Russians from Karelia. Hum. Immunol. 80, 95–96 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Nothnagel M, Fürst R & Rohde K Entropy as a measure for linkage disequilibrium over multilocus haplotype blocks. Hum. Hered. 54, 186–198 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Okada Y et al. Construction of a population-specific HLA imputation reference panel and its application to Graves’ disease risk in Japanese. Nat. Genet. 47, 798–802 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Okada Y eLD: entropy-based linkage disequilibrium index between multiallelic sites. Hum. Genome Var. 5, 29 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chikata T et al. Host-specific adaptation of HIV-1 subtype B in the Japanese population. J. Virol. 88, 4764–4775 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nomura E et al. Mapping of a disease susceptibility locus in chromosome 6p in Japanese patients with ulcerative colitis. Genes Immun. 5, 477–483 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Price P et al. The genetic basis for the association of the 8.1 ancestral haplotype (A1, B8, DR3) with multiple immunopathological diseases. Immunol. Rev. 167, 257–274 (1999). [DOI] [PubMed] [Google Scholar]
- 48.Horton R et al. Variation analysis and gene annotation of eight MHC haplotypes: the MHC Haplotype Project. Immunogenetics 60, 1–18 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Graham RR et al. Visualizing human leukocyte antigen class II risk haplotypes in human systemic lupus erythematosus. Am. J. Hum. Genet. 71, 543–553 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller FW et al. Genome-wide association study identifies HLA 8.1 ancestral haplotype alleles as major genetic risk factors for myositis phenotypes. Genes Immun. 16, 470–480 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haapasalo K et al. The psoriasis risk allele HLA-C*06:02 shows evidence of association with chronic or recurrent Streptococcal tonsillitis. Infect. Immun. 86, e00304–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salter-Townshend M & Myers S Fine-scale inference of ancestry segments without prior knowledge of admixing groups. Genetics 212, 869–889 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou Q, Zhao L & Guan Y Strong Selection at MHC in Mexicans since Admixture. PLoS Genet. 12, e1005847 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meyer D, C Aguiar VR, Bitarello BD, C Brandt DY & Nunes K A genomic perspective on HLA evolution. Immunogenetics 70, 5–27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Norris ET et al. Admixture-enabled selection for rapid adaptive evolution in the Americas. Genome Biol. 21, 29 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guan Y Detecting structure of haplotypes and local ancestry. Genetics 196, 625–642 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maples BK, Gravel S, Kenny EE & Bustamante CD RFMix: a discriminative modeling approach for rapid and robust local-ancestry inference. Am. J. Hum. Genet. 93, 278–288 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Degenhardt F et al. Construction and benchmarking of a multi-ethnic reference panel for the imputation of HLA class I and II alleles. Hum. Mol. Genet. 28, 2078–2092 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ambardar S & Gowda M High-resolution full-length HLA typing method using third generation (Pac-Bio SMRT) sequencing technology. Methods Mol. Biol. 1802, 135–153 (2018). [DOI] [PubMed] [Google Scholar]
- 60.Macdonald WA et al. A naturally selected dimorphism within the HLA-B44 supertype alters class I structure, peptide repertoire, and T cell recognition. J. Exp. Med. 198, 679–691 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kloverpris HN et al. HLA-B*57 micropolymorphism shapes HLA allele-specific epitope immunogenicity, selection pressure, and HIV immune control. J. Virol. 86, 919–929 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Carrington M & Walker BD Immunogenetics of spontaneous control of HIV. Annu. Rev. Med. 63, 131–145 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Khera AV et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Khera AV et al. Polygenic prediction of weight and obesity trajectories from birth to adulthood. Cell 177, 587–596.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Torkamani A & Topol E Polygenic risk scores expand to obesity. Cell 177, 518–520 (2019). [DOI] [PubMed] [Google Scholar]
- 66.Martin AR et al. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 51, 584–591 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Van der Auwera GA et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10.1–33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Julg B et al. Possession of HLA class II DRB1*1303 associates with reduced viral loads in chronic HIV-1 clade C and B infection. J. Infect. Dis. 203, 803–809 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schäfer C, Schmidt AH & Sauter J Hapl-o-Mat: open-source software for HLA haplotype frequency estimation from ambiguous and heterogeneous data. BMC Bioinformatics 18, 284 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pappas DJ, Marin W, Hollenbach JA & Mack SJ Bridging ImmunoGenomic Data Analysis Workflow Gaps (BIGDAWG): An integrated case-control analysis pipeline. Hum. Immunol. 77, 283–287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Alexander DH, Novembre J & Lange K Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Price AL et al. Long-range LD can confound genome scans in admixed populations. Am. J. Hum. Genet. 83, 132–135 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pasaniuc B et al. Analysis of Latino populations from GALA and MEC studies reveals genomic loci with biased local ancestry estimation. Bioinformatics 29, 1407–1415 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McLaren PJ et al. Association study of common genetic variants and HIV-1 acquisition in 6,300 infected cases and 7,200 controls. PLoS Pathog. 9, e1003515 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Okada Y et al. Contribution of a non-classical HLA gene, HLA-DOA, to the risk of rheumatoid arthritis. Am. J. Hum. Genet. 99, 366–374 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lenz TL et al. Widespread non-additive and interaction effects within HLA loci modulate the risk of autoimmune diseases. Nat. Genet. 47, 1085–1090 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of inferred HLA alleles at G-group resolution. For each allele observed in the reference panel, we listed its overall frequency (Freq); P-value (Pval) for difference in frequencies across populations (a two-sided chi-square test with 4 degree-offreedom); frequency within each continental population (European, EUR; AA, Admixed African; LAT, Latino; SAS, South Asian; EAS, East Asian) and its accuracy in two validation cohorts (JPN (n = 288 independent samples) and 1000 Genomes Project (n = 955 independent samples)).
Imputation dosage r2 of each allele. Each row shows each imputed classical HLA allele and its imputation accuracy (measure in dosage r2) in three cohorts with validation data (1000 Genomes Project (G1K, n = 955), GaP registry (GAP, n = 75) and HIV (n = 1,067)). We also report the allelic frequency using the gold standard and the inferred alleles.
Association study within the MHC to HIV-1 viral load. For each row, we list the results of association testing for each of the binary markers that we imputed across the extended MHC region. For each marker, its unique identifier (ID), genome basepair position in Grch37 (BP), reference allele (REF), alternative allele (ALT), minor allele frequency (MAF), effect size (BETA) and standard error (SE) in each conditional analysis are listed.
HLA haplotype frequency based on eight classical HLA alleles inferred at G-group resolution. All haplotype with count > 1 in each and overall population are listed. AA represents individuals of Admixed African ancestry; EAS represents individuals of East Asian ancestry; EUR represents individuals of European ancestry; LAT represents individuals of Native American ancestry; SAS represents individuals of South Asian ancestry.
Data Availability Statement
All scripts and data for generating figures presented in the manuscript are available at https://github.com/immunogenomics/HLA-TAPAS. The reference panel can be accessed for imputation at the Michigan Imputation Server, https://imputationserver.sph.umich.edu. IPD-IMGT/HLA database (version 3.32.0), https://www.ebi.ac.uk/ipd/imgt/hla/. 1000 Genomes gold-standard HLA types, http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/HLA_types/.