

ABSTRACT
Carbon ion radiotherapy (CIRT) is an emerging type of radiotherapy for the treatment of solid tumors. In recent years, evidence accumulated that CIRT improves the therapeutic outcome in patients with otherwise poor response to immune checkpoint blockade. Here, we aimed at identifying the underlying mechanisms of CIRT-induced tumor immunogenicity and treatment efficacy. We used human U2OS osteosarcoma cells for the in vitro assessment of immunogenic cell death and established several in vivo models of melanoma in mice. We treated the animals with conventional radiation, CIRT, PD-1-targeting immune checkpoint blockade or a sequential combinations of radiotherapy with checkpoint blockade. We utilized flow cytometry, polyacrylamide gel electrophoresis (PAGE) and immunoblot analysis, immunofluorescence, immunohistochemistry, as well as enzyme-linked immunosorbent assays (ELISA) to assess biomarkers of immunogenic cell death in vitro. Treatment efficacy was studied by tumor growth assessment and the tumor immune infiltrate was analyzed by flow cytometry and immunohistochemistry. Compared with conventional radioimmunotherapy, the combination of CIRT with anti-PD-1 more efficiently triggered traits of immunogenic cell death including the exposure of calreticulin, the release of adenosine triphosphate (ATP), the exodus of high-mobility group box 1 (HMGB1) as well as the induction of type-1 interferon responses. In addition, CIRT plus anti-PD-1 led to an increased infiltration of CD4+, and CD8+ lymphocytes into the tumor bed, significantly decreased tumor growth and prolonged survival of melanoma bearing mice. We herein provide evidence that CIRT-triggered immunogenic cell death, enhanced tumor immunogenicity and improved the efficacy of subsequent anti-PD-1 immunotherapy.
KEYWORDS: Carbon ion radiotherapy, immunogenic cell death, melanoma, anti-PD-1 therapy
Introduction
Both radiotherapy as well as chemotherapy can enhance the immunogenicity of tumor cells through mechanisms such as the induction of immunogenic cell death (ICD).1 ICD is characterized by the emission of a series of damage-associated molecular patterns (DAMPs)2,3 that act on pattern recognition receptors expressed on host immune cells ultimately promoting the immune-mediated elimination of residual tumor cells.3 In the course of ICD the endoplasmic reticulum (ER)-resident chaperon calreticulin (CALR) translocates together with other proteins from the ER lumen to the plasma membrane surface,4 where it serves as an “eat me” signal for dendritic cells (DC).5 The prominent role of CALR was underlined by experiments employing cells silenced for CALR expression showing a lack of immunogenicity that could be restored by absorbing recombinant to their surface.6 Mechanistically membrane exposure of CALR requires cellular cofactors such as PDIA3 (better known as ERp57), and depends on the phosphorylation of eukaryotic translation initiation factor 2 subunit alpha (eIF2α) downstream of eukaryotic translation initiation factor 2 kinase 3 (EIF2AK3, better known as PERK) in the course of ER stress.7 Of note eIF2α is also an apical trigger of macroautophagy (hereafter called autophagy) which in the course of ICD orchestrates the lysosomal secretion of adenosine triphosphate (ATP) by dying tumor cells then serving as “find-me” signal that can be detected by phagocytic cells expressing purinergic receptor P2X 7 (P2RX7).8 It was shown that cells with defective autophagic machinery are unable to trigger full-blown cancer immunity in mice, underlining the importance of this pathway.8,9 Moreover, lack of function mutation of P2RX7 was correlated with poor prognosis in breast cancer patients receiving immunogenic chemotherapy.10 The passive exodus of high mobility group box 1 (HMGB1) in the final stage of ICD leads to the toll-like receptor 4 (TLR4)-dependent maturation of DC which then governs antigen processing and presentation to T cells.
The immune-checkpoint molecule programmed cell death protein 1 (PD-1) is expressed by exhausted T cells that are refractory to stimulation. Targeting this immune checkpoint with therapeutic monoclonal antibodies can (re)activate T cells and boost adaptive immune responses. Although the use of anti-PD-1 antibodies has proven to be a significant success in the clinical treatment of cancer,11 response to checkpoint inhibition with anti-PD-1 therapy as monotherapy is limited to certain patients.12–14 This resistance may be related to a limited abundance of neoantigens from the tumor and/or immuno-exclusion of CD8+ T cells.15,16 Consistently tumors that essentially lack antigen presentation or have fewer T cells that respond to antigens are significantly less likely to respond to PD-1 checkpoint blockade. Therefore, we reason that therapies which increase the immunogenicity of the tumor may augment the effectiveness of anti-PD-1 treatment. Several recent clinical reports now corroborate this hypothesis as the sequential combination of ICD induction by chemo- or radiotherapy with PD-1 checkpoint blockade achieved excellent therapeutic efficacy in several neoplastic indications including breast and small cell lung cancer.17–23
Here we focused on elucidating the mechanisms underlying the antitumor immune responses elicited by ionizing X-ray and carbon ion irradiation. Ionizing radiation induces innate immune response as well as adaptive immune response via the type I interferon pathway. Ionizing radiation can also cause surface exposure or the release of DAMPs from dying cells, both of which aid in the maturation and activation of DCs.24 Recent studies have shown that the combination approach of radiotherapy and immunotherapy could provoke a strong tumor-specific CD8+ T cell response, resulting in systemic tumor regression.25 Higher relative biological effectiveness (RBE) caused by changes in linear energy transfer (LET) along the ion path can be limited to tumor volume, with minimal normal tissue damage along the inlet trajectory, making 12C6+ the best ion for tumor therapy.26 Although the immunomodulatory effects of X-rays in animal models and patients have been extensively studied, preclinical data showing the effects of carbon ions on immune responses are lacking. We demonstrate that carbon ion radiotherapy (CIRT) induced ICD thus exhibiting immunostimulatory effects that in turn led to a remodeling of the tumor immune microenvironment and an enhancement of antitumor efficacy of PD-1 checkpoint blockade. These results provide novel insight into the mechanisms of CIRT-induced immunogenicity and provide a robust theoretical basis for further exploration of therapies combining carbon ion irradiation with immunotherapy.
Materials and methods
Radiotherapy
Carbon ion (12C6+) beam radiation was performed at the treatment terminal of the Heavy Ion Research Facility in Lanzhou (energy, 80 MeV/u; peak LET, 50 KeV/μm, SOBP). X-rays were generated by an X-Rad 225 generator (Precision) (energy, 225 KV/13.3 mA). The RBE value of the carbon ions was 1.7 times higher than for X-rays at an LET of approximately 50 KeV/μm, designating 5 GyE of carbon ions as equivalent to a dose of 2.94 Gy of carbon ions (Fig. S1A,B). The melanoma-bearing mice were placed on the platform after anesthesia (pentobarbital sodium, 50 mg/kg, i.p.), and a tailored lead protection device was used in which only the tumor site was exposed to radiation.
Cell lines
Human osteosarcoma U2OS, murine melanoma B16 cell and S91 cells were purchased from the Chinese Academy of Sciences Cell Bank. U2OS cells were cultured in McCoy’s 5A medium containing 10% fetal bovine serum (FBS), and B16 and S91 cell lines were cultured in Dulbecco’s modified eagle’s medium (DMEM) supplemented with 10% FBS. All cell lines were verified as being free of microbial contamination.
PAGE and immunoblotting
Cells were collected in RIPA buffer (150 mM sodium chloride, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and protease-inhibitor cocktails), ultrasonicated, and mixed with 5x loading buffer. The resulting cell lysate was placed in a metal bath at 98°C and heated for 10 min to allow for the denaturation of proteins. Polyacrylamide gel electrophoresis (PAGE) was employed with a voltage of 80 V for protein concentration and 120 V for separation. The proteins were electrotransfered onto PVDF membranes at 120 V and 0.2 A with specific transfer times depending on the molecular weight of the proteins of interest. The membranes were blocked with 5% BSA at room temperature for 2 h and then incubated with the primary antibody overnight at 4°C. The next day and after washing five times with phosphate-buffered saline (PBST), an HRP-labeled secondary antibody was added, and the membrane was incubated at room temperature for 90 min. Chemiluminescence images were acquired and analyzed with Image J software (National Institutes of Health, Bethesda, MD, USA). Calreticulin (#12238), phospho-eIF2α (#3398) and HMGB1 (#6893) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA); IFN-γ antibody (#Abs119966) was purchased from Absin (Shanghai, China).
Flow cytometric assay
Mice were killed by cervical dislocation and spleen and tumor were collected and erythrocytes were lysed with red blood cell lysis buffer (#07800, Stemcell Technologies, Vancouver, Canada). The tumor tissue was dissociated with Gentle Collagenase/Hyaluronidase (#07919, Stemcell Technologies) and incubated at 37°C in 5% carbon dioxide (CO2) in a constant-temperature incubator with saturated humidity for 30 min. Single-cell suspensions were prepared by gently passing the cells through disposable cell strainer (70 μm) into cold PBS. We then centrifuged the cells at 200 g for 10 min, the supernatant was discarded, and 1 × 105 splenocytes or 1 × 107 tumor tissue cells were resuspended in cluster of differentiation-specific antibody-containing cell-staining buffer (#420201, BioLegend, San Diego, CA, USA) followed by incubation in the dark at 4°C for 30 min. The antibodies FITC-CD11b (#101206), APC-GR1 (#108412), FITC-CD3 (#100204), FITC-CD4 (#100406), and APC-CD8b.2 (#140410) were purchased from BioLegend (San Diego, CA, USA). Flow cytometry was conducted using a Beckman MoFloAstrios EQ flow cytometer (the gating strategies are shown in Fig. S2A,B).
ELISA
The quantification of circulating HMGB1, ATP, and IFN-γ was performed by means of enzyme-linked immunosorbent assay (ELISA), following the manufacturer’s instructions. Absorbance (at 450 nm) was analyzed by means of an i3 Paradigm multimode platereader (Molecular Devices, San Jose, CA, USA). The HMGB1 (#E-EL-M0676c) and IFN-γ (#E-EL-M0048c) ELISA kits were purchased from Elabscience (Wuhan, China); the ENLITEN ATP assay kits (#ff2000) was purchased from Promega (Madison, WI, USA).
Real-time quantitative RT-PCR
Trizol was used to extract RNA, and cDNA was obtained by taking 3 μg of RNA for reverse transcription according to the GeneCopoeia reverse-transcription kit manual. The cDNA was diluted three times, and utilized SYBR Green was used for real-time quantitative detection. The final volume was 20 μL, and a total of 40 cycles were executed. Predenaturation was implemented at 95°C for 10 min, denaturation at 95°C for 10 sec, annealing at 60°C for 20 sec, and primer template extension at 72°C for 15 sec – at a heating rate of 0.5°C/6 sec between 72°C and 95°C. All data collection was completed using the Bio-Rad CFX96 PCR system software (Bio-Rad Laboratories, Hercules, CA, USA). We calibrated the collected Ct values to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as an internal reference, and analyzed target gene expression using the 2-∆∆CT method. Human primer sequences: IFN-α1 (F: CAGAGTCACCCATCTCAGCAAGC R: CACCACCAGGACCATCAGTAAAGC); IFN-α2 (F: CAATCTCTTCAGCACAAAGGAC R: AAGTATTTCCTCACAGCCAG
AA); IFN-β1 (F: TGGCTGGAATGAGACTATTGTT R: GGTAATGCAGAATCCTC
CCATA) Mouse primer sequences: IFN-α1 (F: AGGAGAGGGTGGGAGAAA R:CA
GGCACAAGGGCTGTAT); IFN-α2 (F: TTTCAACCA GTCTAGCAGCA R: CAGA
TCACAGCCCACAGAG); IFN-β1 (F: CCAACAAGTGTCTCCTCCA R: TATTCAA
GCCTCCCATTCA)
Experimental mouse model
Male wild-type C57BL/6 mice at 6–8 weeks of age were obtained from the Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences; and maintained in the animal facility at the Gansu University of Chinese Medicine in specific pathogen-free conditions in a temperature-controlled environment with 12 h light, 12 h dark cycles, and received food and water ad libitum. All animal experiments were approved by the Ethical Committee of the Gansu University of Chinese Medicine and followed the EU Directive 2010/63/EU guidelines. B16 and S91 tumors were established in C57BL/6 hosts by subcutaneously inoculating 5 × 105 cells. When the tumors became palpable (50 mm3), they were locally irradiated with 5 GyE of carbon ion beams or 5 Gy X-rays, and anti-PD-1 i.p. injections were administered on days 2, 4, and 6 after irradiation. Tumor-bearing mice were frequently monitored, and tumor growth was documented regularly. Following the ethical committee’s advice, the mice were killed when the tumor size reached ethical end-points or when we observed obvious signs of discomfort associated with the treatment. Tumor vaccination model: 5 × 105 B16 and S91 cells were irradiated with 5 GyE carbon ion beams or treated with 2 μM MTX for 24 h, respectively, and then inoculated subcutaneously into C57BL/6 mice. Two weeks later, they were rechallenged with 5 × 104 B16 and S91 cells and tumor growth was recorded regularly (Fig. S3A,B).
Immunofluorescence assay
Paraformaldehyde (4%) was used for approximately 10 min to fix the cells. Then the supernatant was discarded and the cells were incubated with cold methanol for 20 min, and ultimately washed with 75% ice-cold ethanol. After rehydration, 0.5% Triton X-100 was used to permeabilize the cell membranes for approximately 5 min, and the tumors were embedded in paraffin and sectioned at 3 μm. After dewaxing and hydration, the slices were placed in 0.01 M sodium citrate buffer solution (pH 6.0) and heated at 95°C for 10 min. Then 0.5% Triton X-100 was used to permeabilize the cell membranes and 3% H2O2 to remove endogenous peroxides. All samples were placed in goat serum diluted with PBS (1:20) and blocked for 2 h, followed by the addition of primary antibody (dilution ratio, 1:500) and incubation for 2 h. After rinsing five times with PBST, the corresponding fluorescent dye–conjugated secondary antibody (dilution ratio 1:1000) was added, and the samples were incubated in the dark for 1.5 h. We then added 10 μL of DAPI and collected images by means of a ECHO RVL-100-G fluorescence microscopy.
Immunohistochemical assay
For the immunohistochemical staining of CD3 (ab5690, Abcam), CD4 (ab183685, Abcam), CD8 (ab209775, Abcam), granzyme B (ab4059, Abcam), and PDL-1 (ab111101, Abcam), 3 μm sections were sliced from the paraffin blocks and placed on positively charged slides. The primary antibody was used at a dilution of 1:200, and we used the Lab Vision™ UltraVision™ Quanto Detection System (#TL-060-QAL, Thermo Fisher Scientific, Waltham, MA, USA) together with 3,3′-diaminobenzidine tetrahydrochloride (DAB) as the chromogen. Five fields were counted per slide in every sample, and the percentage of phenotypically altered cells was evaluated using ImageJ software (http://imagej.nih.gov/ij/).
Statistical analysis
Unless otherwise specified, we executed our experiments in triplicate and repeated them at least once. The data were analyzed and the histograms generated using GraphPad Prism 7 software. Statistical differences were determined using a two-way analysis of variance (ANOVA) followed by Bonferroni’s test to compare treatments with controls (* p < .05, ** p < .01, and ***p < .001).
Results
Carbon ion radiotherapy–induces immunogenic cell death in vitro
We and others previously showed that radiotherapy induces immunogenic cell death and has immunostimulatory properties that sensitize tumors to the subsequent treatment with immune checkpoint blockade.27,28 Here we used carbon ion radiotherapy (CIRT) to irradiate tumors in vitro and in vivo and analyzed the onset of immunogenic cell death (ICD). The hallmarks of ICD were analyzed in human osteosarcoma U2OS cells and mouse melanoma B16/S91 cells 6 and 24 h after exposure to CIRT (5 GyE) or X-ray (5 Gy) irradiation. The prototype ICD inducer mitoxantrone (MTX) (1 μM) was used as a positive control. Both CIRT and X-ray irradiation induced a time-dependent membrane translocation of calreticulin (CALR) in human osteosarcoma U2OS cells and mouse melanoma cells assessed by flow cytometry and marked by immunostaining, consistent with the results obtained in U2OS and B16/S91 biosensor cells that express CALR coupled with a green fluorescent protein (GFP) assessed by fluorescence microscopy (Figure 1(a,b) S4A-C,E). We further determined CALR protein expression by immunoblotting and showed that both types of irradiation induced CALR expression similar to the prototype ICD inducer mitoxantrone (MTX) (Figure 1(c) S4D,F). Next we checked the upstream signaling and found that exposure of U2OS cells and B16/S91 cells to CIRT and X-ray irradiation increased the phosphorylation of eIF2α using immunofluorescence and immunoblot assays (Figure 1(d,e) S4G-J). When we used quinacrine (a fluorophore that selectively stains ATP-rich cytoplasmic vesicles) to measure ATP release,29 we determined that CIRT as well as X-ray irradiation significantly promoted the release of ATP by U2OS cells and B16/S91 cells into the medium (Figure 1(f,g) S4K-N). Furthermore, we stained irradiated U2OS cells and B16/S91 cells with HMGB1-GFP antibody and found that CIRT and to a lesser extend X-ray induced the release of HMGB1 from the nucleus, which was consistent with immunoblot assay from nuclear extracts that showed that CIRT significantly decreased the abundance of nuclear HMGB1 (Figure 1(h,i) S4O-R). Moreover, we used quantitative real-time polymerase chain reaction (PCR) to analyze the expression of type I interferon at the mRNA level in irradiated U2OS cells as well as B16/S91 cells and showed that CIRT significantly stimulated the expression of IFN-α1, IFN-α2, and IFN-β1 (Figure 1(j) S4S,T). In total we showed that CIRT induced ICD in vitro indicated by CALR exposure, ATP release, the exodus of HMGB1, and the induction of the type-1 interferon response, similar to X-ray irradiation and MTX treatment. Of note CIRT was significantly more potent in inducing HMGB1 release as compared to X-ray irradiation.
Figure 1.
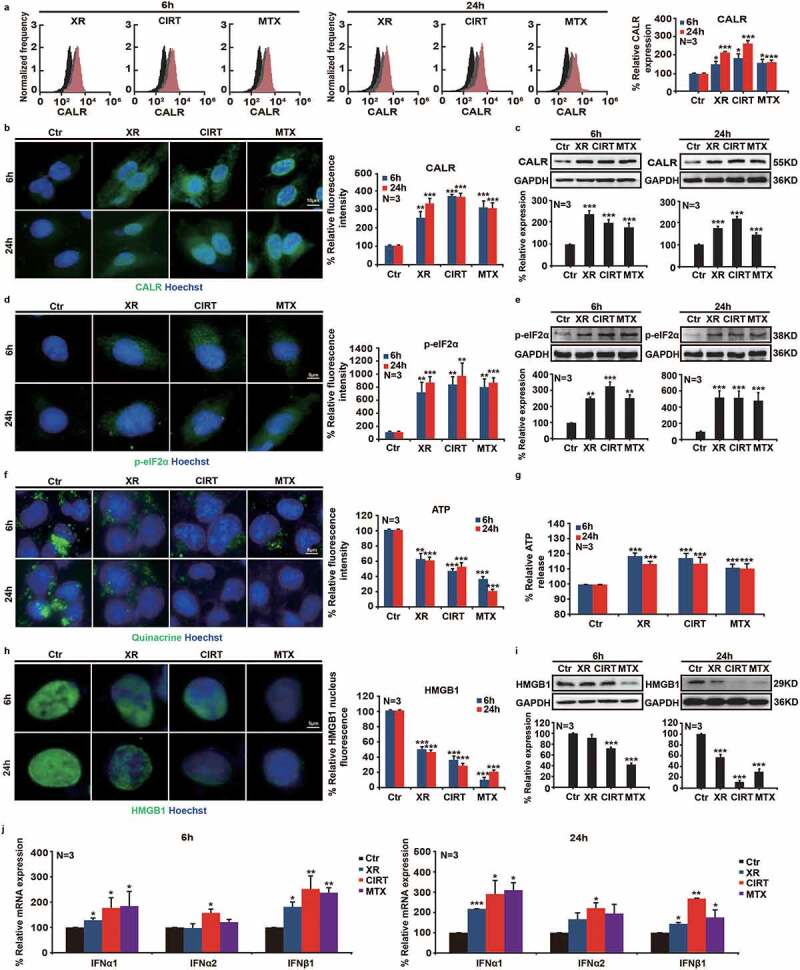
CIRT-induced immunogenic cell death in vitro. Human osteosarcoma U2OS cells were irradiated by X-ray and carbon ion radiotherapy (CIRT), and biomarkers of immunogenic cell death were evaluated at 6 h and 24 h after irradiation. Mitoxantrone (MTX, 1 µM) was used as a prototype immunogenic cell death inducer Flow cytometry, immunofluorescence, and immunoblot assays showed that CIRT induced CALR exposure and elevated the phosphorylation level of eIF2α, enhanced ATP release by quinacrine staining and ELISA, augmented HMGB1 exodus from the nucleus, and significantly up-regulated the expression of type I interferon at the mRNA level. Representative images and quantification are shown (mean ± SD of triplicate assessments, Student’s t test, **p < .01, ***p < .001).
CIRT-induced immunogenic cell death in vivo
We next explored whether CIRT exerts immunogenic effects in vivo. For this purpose, we established the tumor vaccination model by using CIRT (5 GyE) and MTX (2 μM) treated B16/S91 cells2 (Fig. S3A,B). And the results showed that vaccination protected mice against a subsequent challenge with live tumor cells of the same type (Figure 2(a-f)). When we analyzed the serum collected from mice using enzyme-linked immunosorbent assay (ELISA), we observed a significant elevation in the serum levels of HMGB1 and IFN-γ in vaccinated mice (Figure 2(g,h)). In addition, we used B16 and S91 melanoma-bearing mice and irradiated tumors with either CIRT (5 GyE) or X-ray (5 Gy). Eight days later the tumors were harvested and paraffin embedded to prepare sections. Using immunofluorescence staining, we found that CIRT, similar to X-ray irradiation and MTX treatment induced a significant de novo expression of CALR in tumors consistent with the increasing phosphorylation of eIF2α (Figure 2(i-n)). We also noted that HMGB1, stained by immunofluorescence, was markedly released from the nucleus after CIRT and to a lesser extend in response to X-ray irradiation (Figure 2(o-q)). Consistently, we analyzed the serum collected from treated mice using ELISA and observed a significant elevation of HMGB1 and ATP (Figure 2(r,s)) strongly suggesting that CIRT triggers immunogenic cell death in vivo.
Figure 2.
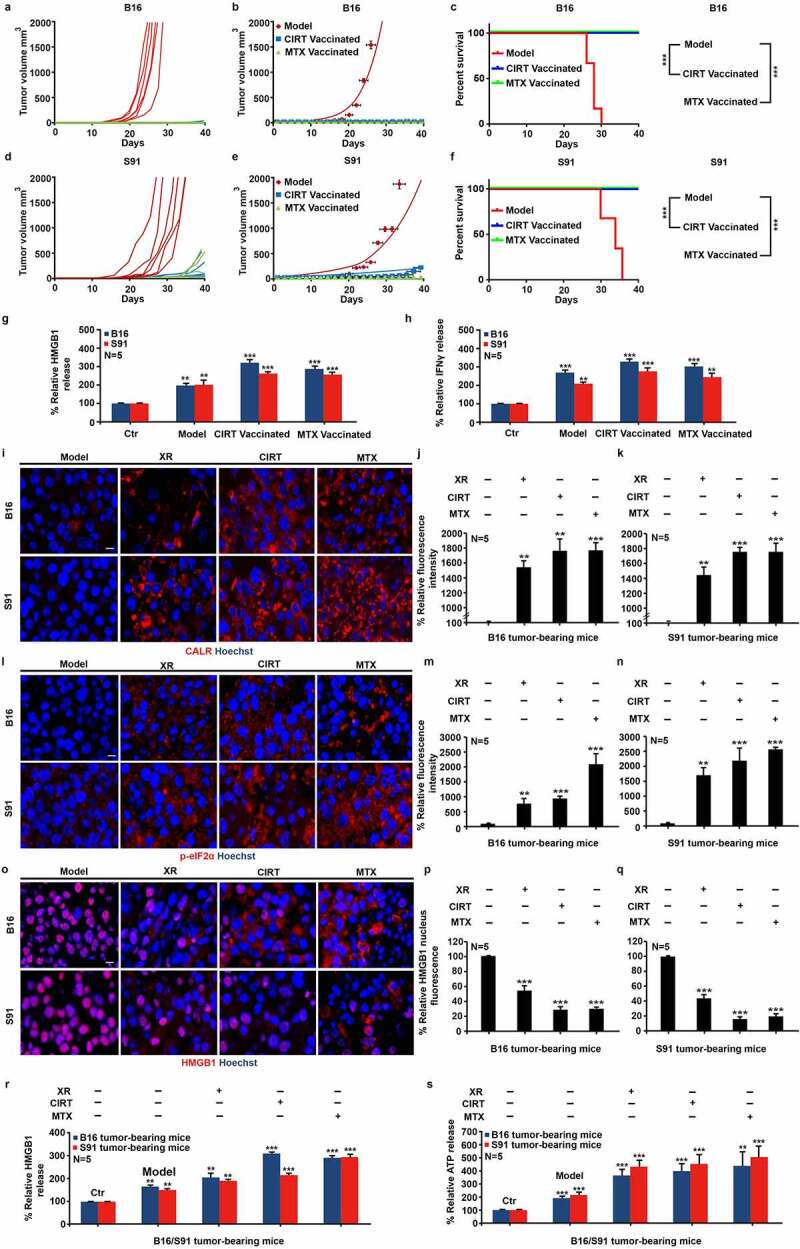
CIRT-induced immunogenic cell death in vivo. 5 × 105 B16 and S91 cells were irradiated with 5 GyE of carbon ion beams or treated with 2 μM MTX for 24 h respectively, then subcutaneously inoculated in C57BL/6 mice. 2 weeks after, rechallenged with 5 × 104 B16 and S91 cells and the tumor growth was documented regularly. C57BL/6 mice bearing subcutaneous B16 or S91 melanoma were locally irradiated with X-rays (XR, 5 Gy) or carbon ions (CIRT, 5 GyE) at the tumor site. Mitoxantrone (MTX, 2.0 mg/Kg)-treated animals were used as positive control. Eight days after irradiation, CALR exposure, eIF2α phosphorylation, and HMGB1 exodus were examined by immunofluorescence. The secretion of HMGB1, ATP, and IFN-γ in the serum was determined by ELISA; and infiltration of IFN-γ expressing cell in the tumor bed was assessed by immunohistocheistry. Representative images and quantification are shown (mean ± SD of triplicate assessments, Student’s t test, ***p < .001).
CIRT induces TME remodeling
We further observed a significant increase in the serum levels of IFN-γ (Figure 3(a)). Intrigued by this observation we analyzed the tumor microenvironment (TME) by immunohistochemistry, and showed that CIRT significantly induced the infiltration of IFN-γ-expressing cells into the tumor of B16 and S91 melanoma-bearing mice (Figure 3(b-d)). Based on this we reasoned that CIRT can enhance the immunogenicity of tumors and therefore explored the effects of CIRT combined with anti-PD-1 immune-checkpoint blockade. Following irradiation, anti-PD-1 antibodies (10 mg/kg) were injected every other day for a total of three times and 8 days after irradiation, the samples were harvested. Consistent with previous findings CIRT induced a rise in circulating IFN-γ levels and this was further increased by combination with immune checkpoint blockade (Figure 3(e)). In similar ways checkpoint blockade enhanced the infiltration of the TME by IFN-γ-expressing cells (Figure 3(f-h)). We thus concluded that CIRT significantly induced TME infiltration by immune cells which was further enhanced by combination with anti-PD-1.
Figure 3.
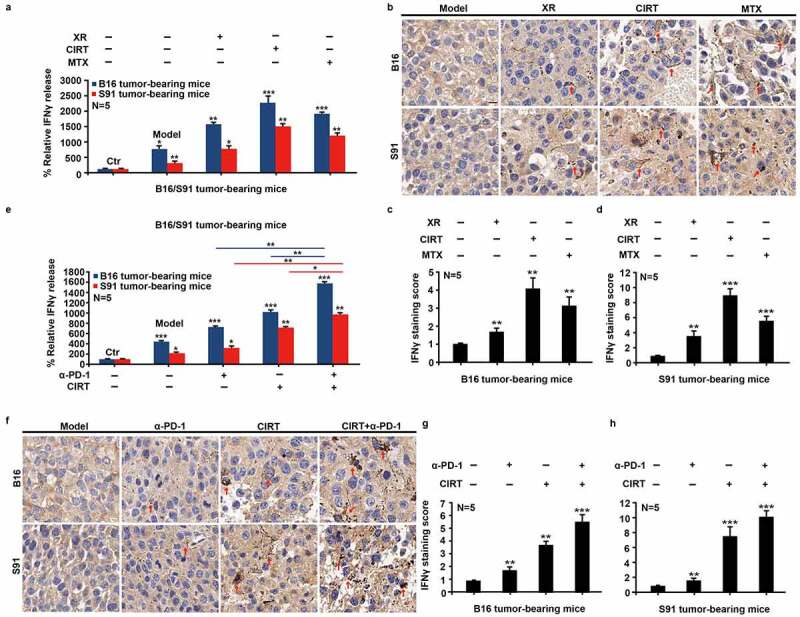
Combination therapy with CIRT and anti-PD-1 stimulates immunogenic cell death. C57BL/6 mice bearing subcutaneous B16 and S91 melanoma (Model) were injected intraperitoneally with anti-PD-1 (α-PD-1) or locally irradiated with 5 GyE of carbon ions (CIRT) at tumor sites, or received treatment with 5 Gy CIRT plus anti-PD-1 (CIRT+α-PD-1). Eight days after irradiation, we examined CALR exposure, eIF2α phosphorylation, and HMGB1 exodus by immunofluorescence; secretion of IFN-γ in serum by ELISA; and IFN-γ infiltration in the tumor by immunohistochemical assay. Representative images and quantification are shown (mean ± SD of triplicate assessments, Student’s t test, ***p < .001).
Combination therapy with CIRT and anti-PD-1 enhances anticancer immune responses
Next, we characterized the nature of the immune response by analyzing the proportions of CD3+CD4+ and CD3+CD8+ lymphocytes in the spleen (Figure 4(a-f)) and the tumor (Figure 4(g-t)) upon mono- and combination treatments of B16 and S91 melanoma-bearing mice by flow cytometry and immunohistochemistry. We found that all treatments alone and in combination with anti-PD-1 (10 mg/Kg)) increased the abundance of CD4+ and CD8+ lymphocytes in both spleen and tumor (Figure 4(a-r)) and that treated tumors exhibited increased levels of PD-L1 (Fig S5A-C). Of note tumors treated with a combination of CIRT plus anti-PD-1 depicted an infiltration with CD8+ lymphocytes significantly higher than any other tested mono- or combination treatment (Figure 4(g-r)) which was consistent with an increase in granzyme B+ cells (Figure 4(s,t)). Furthermore, the infiltration of intratumoral myeloid-derived suppressor cells (MDSCs) in the CIRT plus anti-PD-1 combinations was significantly decreased relative to other groups (Fig. S6A-D).
Figure 4.

Combination therapy with CIRT and anti-PD-1 enhances immune response. C57BL/6 mice bearing subcutaneous B16 and S91 melanom were injected intraperitoneally with anti-PD-1 (α-PD-1) or locally irradiated with 5 Gy of X-rays (XR) or 5 GyE of carbon ions (CIRT) at tumor sites or received combined treatment with 5 Gy of X-rays and anti-PD-1 (XR+α-PD-1) or 5 GyE of carbon ions and anti-PD-1 (CIRT+α-PD-1). Eight days after irradiation, we analyzed the proportions of CD3+CD4+ and CD3+CD8+ T lymphocytes in the spleen and tumor, and infiltrating MDSCs in the tumor by flow cytometric analysis. CD3+, CD4+, CD8+, and granzymeB+ infiltration in tumors were ascertained by immunohistochemistry. Representative images and quantification are shown (mean ± SD of triplicate assessments, Student’s t test, ***p < .001).
Combination therapy with CIRT and anti-PD-1 increases overall survival of melanoma-bearing mice
As the combination of CIRT plus anti-PD-1 immune checkpoint blockade effectively activated antitumor immune responses in tumor-bearing mice, we next assessed the therapeutic efficacy of this treatment. To this aim B16 and S91 melanoma-bearing mice were treated with CIRT or X-ray irradiation, alone or in combination with anti-PD-1 as follows. Mice were intraperitoneally injected with anti-PD-1 on days 2, 4, and 6 after irradiation, and the tumor was removed eight days after irradiation which resulted in all treatments slowing tumor growth (Fig. S7A,B). Continuous monitoring until endpoint showed that the combination of CIRT plus anti-PD-1 therapy dramatically slowed the growth of melanoma and prolonged mouse survival (Figure 5(a-f)).
Figure 5.
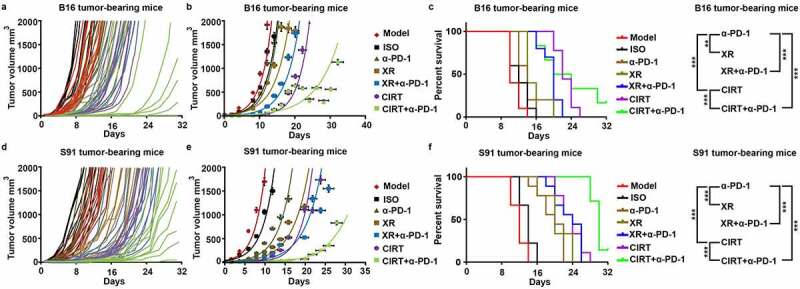
Combination therapy with CIRT and anti-PD-1 increases overall survival of melanoma-bearing mice. C57BL/6 mice bearing subcutaneous B16 and S91 melanoma (Model) were injected intraperitoneally with anti-PD-1 (α-PD-1) or locally irradiated with 5 Gy of X-rays (XR) or 5 GyE of carbon ions (CIRT) at tumor sites or underwent a combined treatment with 5 Gy of X-rays and anti-PD-1 (XR+α-PD-1) or 5 GyE of carbon ions and anti-PD-1 (CIRT+α-PD-1). Anti-PD-1 was injected intraperitoneally on days 2, 4, and 6 after irradiation. All treatments slowed the tumor growth rate. Tumor volume and survival ratio of the mice were measured every second day from the day of irradiation until the tumor volume reached the endpoint (2000 mm3). Representative images and quantification are shown (mean ± SD of triplicate assessments, Student’s t test, ***p < .001).
Discussion and conclusions
PD-1 checkpoint blockade can reactivate immunosurveillance by inhibiting the PD-1/PD-L1 axis thus enabling T cell-mediated to killing. Nevertheless, monoimmunotherapy with anti-PD-1 antibodies is only effective in a subset of patients and does not achieve therapeutic effects in others which might be related to a low degree of tumor immune infiltration or active immunosuppression.30 ICD effectively attracts immune cells into the tumor bed by the release of DAMPs thus enhancing the activation of T cells and the transformation of tumors from “cold” to “hot”, altogether setting the stage for immune-checkpoint inhibitors.31 Consistently the combination of ICD induction by plus immune-checkpoint inhibition has been proven superior over monotherapy in multiple preclinical model and as well as in clinical settings. Radiotherapy can induce ICD and has been widely used for treating various malignancies, either as monotherapy or as part of a multimodal approach, and a large body of preclinical data suggests that radiotherapy is enhancing the activity of immunotherapy.32,33 A growing number of studies suggest that radiation triggers “abscopal effects” nevertheless the induced immune response is often insufficient to fully eradicate primary tumors together with metastases.34,35 Therefore, radiotherapy is often combined with systemic immunotherapy to enhance treatment effects, and to simultaneously eradicated primary and metastatic lesions.36 As a pitfall conventional radiotherapy usually exploits a method of low-dose irradiation in multiple fractions, although repeated irradiation may eliminate the infiltrated lymphocytes in the tumor, thus negating the immunogenic benefits of the treatment. Consistently high-dose irradiation has been shown to efficiently enhance the effect of immunotherapy such as a single dose of 12 Gy of X-ray irradiation for breast or colorectal cancer that led to a reduction in myeloid-derived suppressor cells in tumors, which was further enhanced by the combination with anti-PD-1 therapy.37 In the present study, we found that a biologically effective dose of CIRT similar to the one of X-ray irradiation induced a more robust immune response, which might be related to the focused Bragg peak of CIRT, and the optimal delivery of ions to the tumor foci with minimal damage to normal tissues. This allowed us to use large doses of CIRT to irradiate the tumor and induce an optimal immune response with only a single fraction. In our study, we demonstrated that a dose of 5 GyE of carbon ion irradiation induced ICD characterized by the exposure of calreticulin on the plasma membrane surface, the phosphorylation of eIF2a, the release of ATP into the extracellular space, the exodus of HMGB1 from the nucleus, and the induction of the type-1 interferon response in vitro and in vivo. We established that CIRT precipitated the hallmarks of ICD ultimately leading to IFN-γ secretion. It is important to note that despite the lower energy level 5 GyE of CIRT induced tumor immunogenicity more effectively than 5 Gy of X-rays. The combination of irradiation plus anti-PD-1 therapy further improved the effect of CIRT and enhanced markers of immune response in blood, spleen, and tumors in vivo; increased the expression of PD-L1; and significantly prolonged the survival of treated animals. Altogether it appears that CIRT-induced remodeling of the TME together with PD-1 immune checkpoint blockade provides a favorable option for the treatment of malignant melanoma.
Supplementary Material
Acknowledgments
We would like to thank Qingfeng Wu and Dan Xu from the Institute of Modern Physics, CAS, for their assistance with flow cytometry and radiotherapy. This work was supported by the Hundred-Talent Program of the Chinese Academy of Sciences (NO. 29Y763050, Heng Zhou), the National Nature Science Foundation of China (NO. 11905264, Heng Zhou and NO. 82003356, Chen Tu and NO. 82004094, Liying Zhang), the Science and Technology Research Project of Gansu Province (NO. 17JR5RA307 and NO. 145RTSA012, Jufang Wang).
Funding Statement
This work was supported by the the Hundred-Talent Program of the Chinese Academy of Sciences [29Y763050]; the National Nature Science Foundation of China [11905264]; the Science and Technology Research Project of Gansu Province [145RTSA012]; the National Nature Science Foundation of China [82004094]; the Science and Technology Research Project of Gansu Province [17JR5RA307]; the National Nature Science Foundation of China [82003356].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All data generated and analyzed during this study are included in this article. Each experiment was performed at least three times independently.
Ethics statement
All the animal experiments were approved by the Ethical Committee of the Gansu University of Chinese Medicine and followed EU Directive 2010/63/EU guidelines.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Garg AD, Dudek-Peric AM, Romano E, Agostinis P.. Immunogenic cell death. Int J Dev Biol. 2015;59(1–2–3):131–11. doi: 10.1387/ijdb.150061pa. [DOI] [PubMed] [Google Scholar]
- 2.Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31(1):51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- 3.Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and damps in cancer therapy. Nat Rev Cancer. 2012;12(12):860–875. doi: 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- 4.Liu P, Zhao L, Kepp O, Kroemer G. Quantitation of calreticulin exposure associated with immunogenic cell death. Methods Enzymol. 2020;632:1–13. [DOI] [PubMed] [Google Scholar]
- 5.Feng M, Chen JY, Weissman-Tsukamoto R, Volkmer JP, Ho PY, McKenna KM, Cheshier S, Zhang M, Guo N, Gip P, et al. Macrophages eat cancer cells using their own calreticulin as a guide: roles of tlr and btk. Proc Natl Acad Sci U S A. 2015;112(7):2145–2150. doi: 10.1073/pnas.1424907112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu P, Zhao L, Loos F, Marty C, Xie W, Martins I, Lachkar S, Qu B, Waeckel-Énée E, Plo I, et al. Immunosuppression by mutated calreticulin released from malignant cells. Mol Cell. 2020;77(4):748–760 e9. doi: 10.1016/j.molcel.2019.11.004. [DOI] [PubMed] [Google Scholar]
- 7.Bezu L, Sauvat A, Humeau J, Leduc M, Kepp O, Kroemer G. Eif2alpha phosphorylation: a hallmark of immunogenic cell death. Oncoimmunology. 2018;7(6):e1431089. doi: 10.1080/2162402X.2018.1431089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garg AD, Krysko DV, Vandenabeele P, Agostinis P. Extracellular atp and p(2)x(7) receptor exert context-specific immunogenic effects after immunogenic cancer cell death. Cell Death Dis. 2016;7(2):e2097. doi: 10.1038/cddis.2015.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Draganov D, Gopalakrishna-Pillai S, Chen YR, Zuckerman N, Moeller S, Wang C, Ann D, Lee PP. Modulation of p2x4/p2x7/pannexin-1 sensitivity to extracellular atp via ivermectin induces a non-apoptotic and inflammatory form of cancer cell death. Sci Rep. 2015;5(1):16222. doi: 10.1038/srep16222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15(10):1170–1178. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 11.Vaddepally RK, Kharel P, Pandey R, Garje R, Chandra AB. Review of indications of fda-approved immune checkpoint inhibitors per nccn guidelines with the level of evidence. Cancers (Basel). 2000;12(3): 738. doi: 10.3390/cancers12030738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sui H, Ma N, Wang Y, Li H, Liu X, Su Y, Yang J. Anti-pd-1/pd-l1 therapy for non-small-cell lung cancer: toward personalized medicine and combination strategies. J Immunol Res. 2018;2018:6984948. doi: 10.1155/2018/6984948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng M, Xiong G, Cao Z, Yang G, Zheng S, Song X, You L, Zheng L, Zhang T, Zhao Y, et al. Pd-1/pd-l1 and immunotherapy for pancreatic cancer. Cancer Lett. 2017;407:57–65. doi: 10.1016/j.canlet.2017.08.006. [DOI] [PubMed] [Google Scholar]
- 14.Mooradian MJ, Sullivan RJ. What to do when anti-pd-1 therapy fails in patients with melanoma. Oncology (Williston Park). 2019;33:141–148. [PubMed] [Google Scholar]
- 15.Verma V, Shrimali RK, Ahmad S, Dai W, Wang H, Lu S, Nandre R, Gaur P, Lopez J, Sade-Feldman M, et al. Pd-1 blockade in subprimed cd8 cells induces dysfunctional pd-1(+)cd38(hi) cells and anti-pd-1 resistance. Nat Immunol. 2019;20(9):1231–1243. doi: 10.1038/s41590-019-0441-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Yang X, Zhang C, Wang Y, Cheng T, Duan L, Tong Z, Tan S, Zhang H, Saw PE, et al. Tumor cell-intrinsic pd-1 receptor is a tumor suppressor and mediates resistance to pd-1 blockade therapy. Proc Natl Acad Sci U S A. 2020;117(12):6640–6650. doi: 10.1073/pnas.1921445117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Zhang H, Li Q, Zou P, Huang X, Wu C, Tan L. Cdk12/13 inhibition induces immunogenic cell death and enhances anti-pd-1 anticancer activity in breast cancer. Cancer Lett. 2020;495:12–21. doi: 10.1016/j.canlet.2020.09.011. [DOI] [PubMed] [Google Scholar]
- 18.Voloshin T, Kaynan N, Davidi S, Porat Y, Shteingauz A, Schneiderman RS, Zeevi E, Munster M, Blat R, Tempel Brami C, et al. Tumor-treating fields (ttfields) induce immunogenic cell death resulting in enhanced antitumor efficacy when combined with anti-pd-1 therapy. Cancer Immunol Immunother. 2020;69(7):1191–1204. doi: 10.1007/s00262-020-02534-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hossain DMS, Javaid S, Cai M, Zhang C, Sawant A, Hinton M, Sathe M, Grein J, Blumenschein W, Pinheiro EM, et al. Dinaciclib induces immunogenic cell death and enhances anti-pd1-mediated tumor suppression. J Clin Invest. 2018;128(2):644–654. doi: 10.1172/JCI94586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kepp O, Zitvogel L, Kroemer G. Clinical evidence that immunogenic cell death sensitizes to pd-1/pd-l1 blockade. Oncoimmunology. 2019;8(10):e1637188. doi: 10.1080/2162402X.2019.1637188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rompre-Brodeur A, Shinde-Jadhav S, Ayoub M, Piccirillo CA, Seuntjens J, Brimo F, Mansure JJ, Kassouf W. PD-1/PD-L1 immune checkpoint inhibition with radiation in bladder cancer: in situ and abscopal effects. Mol Cancer Ther. 2020;19(1):211–220. doi: 10.1158/1535-7163.MCT-18-0986. [DOI] [PubMed] [Google Scholar]
- 22.Hong S, Bi M, Yu H, Yan Z, Wang H. Radiation therapy enhanced therapeutic efficacy of anti-pd1 against gastric cancer. J Radiat Res. 2020;61(6):851–859. doi: 10.1093/jrr/rraa077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sato H, Okonogi N, Nakano T. Rationale of combination of anti-pd-1/pd-l1 antibody therapy and radiotherapy for cancer treatment. Int J Clin Oncol. 2020;25(5):801–809. doi: 10.1007/s10147-020-01666-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, et al. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy[J]. Nat Med. 2007;13(9):1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 25.Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C, Vanpouille-Box C, Friedman K, Ferrari de Andrade L, Wucherpfennig KW, et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat Med. 2018;24(12):1845–1851. doi: 10.1038/s41591-018-0232-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumari S, Mukherjee S, Sinha D, Abdisalaam S, Krishnan S, Asaithamby A. Immunomodulatory effects of radiotherapy. Int J Mol Sci. 2020;21(21):8151. doi: 10.3390/ijms21218151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vacchelli E, Vitale I, Tartour E, Eggermont A, Sautès-Fridman C, Galon J, Zitvogel L, Kroemer G, Galluzzi L. Trial Watch: anticancer radioimmunotherapy. Oncoimmunology. 2013;2(9):e25595. doi: 10.4161/onci.25595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma Y, Conforti R, Aymeric L, Locher C, Kepp O, Kroemer G, Zitvogel L. How to improve the immunogenicity of chemotherapy and radiotherapy. Cancer Metastasis Rev. 2011;30(1):71–82. doi: 10.1007/s10555-011-9283-2. [DOI] [PubMed] [Google Scholar]
- 29.Forveille S, Humeau J, Sauvat A, Bezu L, Kroemer G, Kepp O. Quinacrine-mediated detection of intracellular ATP. Methods Enzymol. 2019;629:103–113. [DOI] [PubMed] [Google Scholar]
- 30.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17(2):97–111. doi: 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- 32.Uribe-Herranz M, Rafail S, Beghi S, Gil-de-gómez L, Verginadis I, Bittinger K, Pustylnikov S, Pierini S, Perales-Linares R, Blair IA, et al. Gut microbiota modulate dendritic cell antigen presentation and radiotherapy-induced antitumor immune response. J Clin Invest. 2020;130(1):466–479. doi: 10.1172/JCI124332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karam SD, Raben D. Radioimmunotherapy for the treatment of head and neck cancer. Lancet Oncol. 2019;20(8):e404–e416. doi: 10.1016/S1470-2045(19)30306-7. [DOI] [PubMed] [Google Scholar]
- 34.Rodriguez-Ruiz ME, Vanpouille-Box C, Melero I, Formenti SC, Demaria S. Immunological mechanisms responsible for radiation-induced abscopal effect. Trends Immunol. 2018;39(8):644–655. doi: 10.1016/j.it.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yilmaz MT, Elmali A, Yazici G. Abscopal effect, from myth to reality: from radiation oncologists’ perspective. Cureus. 2019;11(1):e3860. doi: 10.7759/cureus.3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buchwald ZS, Wynne J, Nasti TH, Zhu S, Mourad WF, Yan W, Gupta S, Khleif SN, Khan MK. Radiation, immune checkpoint blockade and the abscopal effect: a critical review on timing, dose and fractionation. Front Oncol. 2018;8:612. doi: 10.3389/fonc.2018.00612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, Fu YX. Irradiation and anti-pd-l1 treatment synergistically promote antitumor immunity in mice. J Clin Invest. 2014;124(2):687–695. doi: 10.1172/JCI67313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated and analyzed during this study are included in this article. Each experiment was performed at least three times independently.