

Abstract
Precise identification of sites of RNA modification is key to studying the functional role of such modifications in the regulation of gene expression and for elucidating relevance to diverse physiological processes. tRNA reduction and cleavage sequencing (TRAC-Seq) is a chemically based approach for the unbiased global mapping of 7-methylguansine (m7G) modification of tRNAs at single-nucleotide resolution throughout the tRNA transcriptome. m7G TRAC-Seq involves the treatment of size-selected (<200 nt) RNAs with the demethylase AlkB to remove major tRNA modifications, followed by sodium borohydride (NaBH4) reduction of m7G sites and subsequent aniline-mediated cleavage of the RNA chain at the resulting abasic sites. The cleaved sites are subsequently ligated with adaptors for the construction of libraries for high-throughput sequencing. The m7G modification sites are identified using a bioinformatic pipeline that calculates the cleavage scores at individual sites on all tRNAs. Unlike antibody-based methods, such as methylated RNA immunoprecipitation and sequencing (meRIP-Seq) for enrichment of methylated RNA sequences, chemically based approaches, including TRAC-Seq, can provide nucleotide-level resolution of modification sites. Compared to the related method AlkAniline-Seq (alkaline hydrolysis and aniline cleavage sequencing), TRAC-Seq incorporates small RNA selection, AlkB demethylation, and sodium borohydride reduction steps to achieve specific and efficient single-nucleotide resolution profiling of m7G sites in tRNAs. The m7G TRAC-Seq protocol could be adapted to chemical cleavage-mediated detection of other RNA modifications. The protocol can be completed within ~9 d for four biological replicates of input and treated samples.
Introduction
tRNAs are subject to numerous post-transcriptional modifications—including methylation, which can affect tRNA folding, stability, and function in mRNA translation—and have important roles in normal physiology and disease1–4. Although next-generation high-throughput sequencing techniques have been widely used to sequence the mRNA transcriptome and epitranscriptome5–7, there is a lack of available methods for the sequence-specific detection of most tRNA modifications. It remains a challenge to profile tRNA expression by sequencing, because tRNAs are highly structured and typically heavily modified, which can interfere with reverse transcription during cDNA library construction8. New methods that identify the subset of tRNAs that contain a particular modification, as well as identify the exact site of the modification, are therefore required to understand the biological roles of individual tRNA modifications. Mutation of the m7G tRNA methyltransferase complex component WDR4 results in microcephalic primordial dwarfism; however, the molecular mechanism underlying mis-regulated m7G tRNA modifications in disease pathogenesis was unknown9. We recently developed a method, m7G site-specific TRAC-Seq, to globally map the m7G modification of tRNAs at single-nucleotide resolution in mouse embryonic stem cells (mESCs)10. m7G TRAC-Seq allows single-nucleotide-resolution mapping of the tRNA m7G methylome using total RNA samples isolated from various cells and tissues and could be adapted to chemical cleavage-mediated detection of other RNA modifications.
Development of the protocol
To study the role of m7G modification in stem cell self-renewal and differentiation, we developed TRAC-Seq to profile the tRNA m7G methylome in mESCs10. m7G TRAC-Seq incorporates four key stages: (i) the isolated small RNAs are treated with the bacterial demethylase AlkB to remove the majority of tRNA modifications to facilitate efficient reverse transcription during cDNA library construction11,12; (ii) the demethylated small RNAs are treated with sodium borohydride and aniline to induce reduction of m7G-modified sites and cleavage of the RNA backbone13,14, which results in a 3′ fragment with a 5′ phosphate; (iii) a 5′ adaptor is ligated to the 3′ fragment, resulting in an adaptor-RNA junction at the m7G-mediated cleavage sites; and (iv) the cDNA libraries are constructed and sequenced and bioinformatic analysis is performed to identify m7G-modified sites (Fig. 1). Using m7G TRAC-Seq, we identified a subset of 22 m7G-modified tRNAs out of 45 detectable tRNAs in mESCs, suggesting a complex m7G tRNA modification landscape in mammals10.
Fig. 1 |. Partial m7G TRAC-Seq workflow.
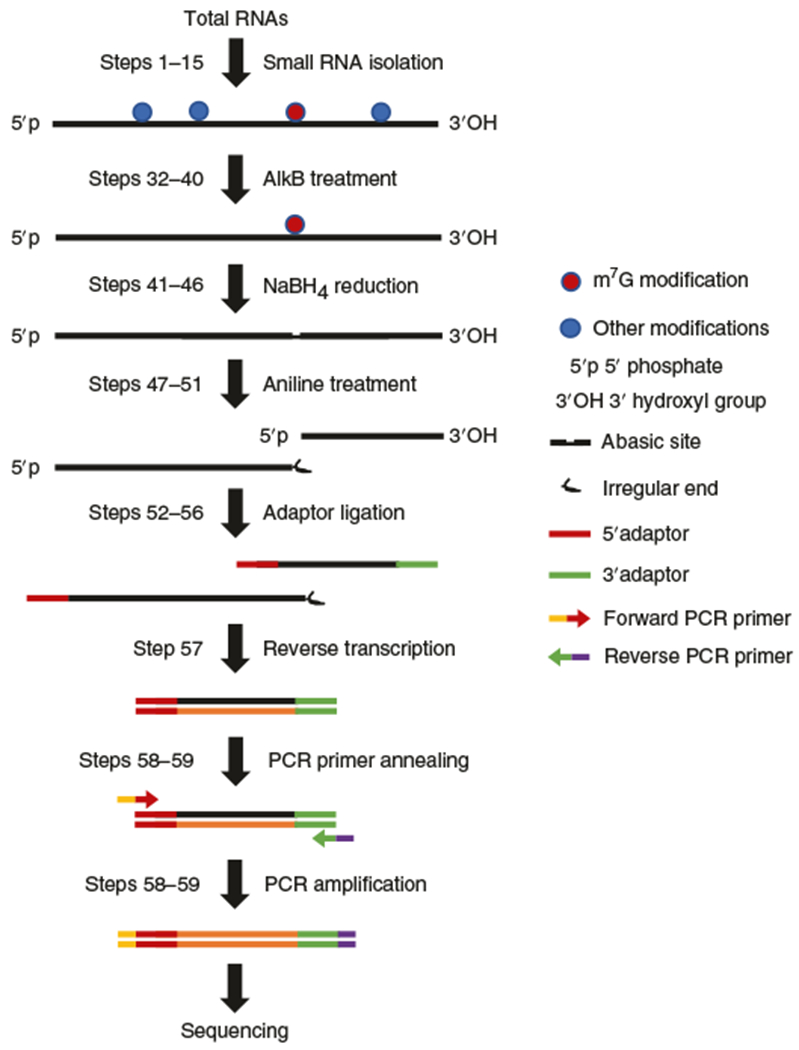
Isolated small RNAs are treated with the bacterial demethylase AlkB to remove the majority of tRNA modifications and then the demethylated small RNAs are treated with NaBH4 and aniline to induce reduction and cleavage of the RNA backbone at m7G-modified sites. Cleavage at the m7G results in two products: a 5′ product with a 3′ irregular end that prevents adaptor ligation and therefore cannot be sequenced, and a 3′ product that has both ends available for adaptor ligation. After that a 5′ adaptor is ligated to the cleaved sites of the 3′ cleavage product for the subsequent cDNA library construction. Libraries are sequenced and bioinformatic analysis is performed to identify m7G-modified sites.
Applications of the method
m7G TRAC-Seq enables the accurate profiling of m7G tRNA methylomes and the quantification of relative tRNA methylation levels in cell lines and is probably also applicable to tissues and organs. tRNA modification regulates tRNA stability and function, and mis-regulation of tRNA modifications often results in neural developmental defects4. Therefore, TRAC-Seq will be useful for the study of the molecular mechanisms underlying tRNA-related diseases. Although m7G TRAC-Seq was initially developed in mouse cells to profile the m7G tRNA landscape at single-nucleotide resolution, this method could also be used to identify m7G modifications in other RNA species and/or organisms. In addition, other RNA modification sites, including dihydrouridine, wybutosine, and 3-methylcytidine (m3C), could be cleaved by chemical treatment and aniline-induced chain scission15. The logic behind TRAC-Seq could be easily extended to other chemical cleavage approaches that generate 5′ phosphate ends for the single-nucleotide resolution profiling of other RNA modifications in different RNA species. These potential developments will have widespread utility in the rapidly developing RNA epitranscriptomics research field and will provide insights into the molecular mechanisms underlying the essential physiological functions of post-transcriptional RNA modifications.
Comparison with other methods
Liquid chromatography with tandem mass spectrometry (LC–MS/MS) is widely used to study the percentage of m7G nucleosides in a tRNA pool or individual tRNAs16. The RNA samples are first digested into mononucleosides and then the m7G level is determined by LC–MS/MS, which provides valuable information on the total m7G modification levels in specific samples16. However, LC–MS/MS could not uncover the m7G-modified sites and modification statuses in individual tRNAs. Because of the extensive modifications and complex secondary structure of tRNAs, there was no efficient high-throughput sequencing method to profile tRNA expression and modifications.
Recently, two independent groups developed efficient tRNA-sequencing methods (DM-tRNA-seq11 and ARM-seq12) using the bacterial AlkB to remove major tRNA modifications, including m1A and m1G but not m7G, therefore allowing efficient reverse transcription during tRNA sequencing library construction. These methods served as a basis for the development of TRAC-Seq, in which we combined the AlkB-mediated tRNA-sequencing approach with sodium borohydride/aniline-induced cleavage at the m7G sites to achieve the single-nucleotide-resolution profiling of m7G modifications. In our recent study, we also developed the anti-m7G antibody–based methylated RNA immuno-precipitation coupled with next-generation sequencing (m7G MeRIP-Seq) technique to profile the tRNA m7G methylome, and we found that both m7G MeRIP-Seq and m7G TRAC-Seq can efficiently profile the tRNA methylome in mESCs. However, m7G MeRIP-Seq could identify only which tRNAs are methylated; it cannot uncover specific m7G modification sites at high resolution, cannot assess the relative stoichiometry of m7G sites, and is highly dependent on antibody quality. Therefore, in contrast to the antibody-based meRIP-Seq, which has potential issues with background nonspecific binding of the antibody and does not provide single-nucleotide-resolution mapping, the TRAC-Seq method instead relies on very specific chemical cleavages at the modified nucleotide and therefore allows precise nucleotide-resolution mapping of the m7G tRNA methylome.
The AlkAniline-Seq method was recently developed to profile m7G and m3C modifications in all types of RNAs in yeast and human cells17. Compared to AlkAniline-Seq, TRAC-Seq incorporates small RNA selection, AlkB demethylation, and sodium borohydride reduction steps to enrich small RNAs and to facilitate the highly efficient reverse transcription of tRNA for cDNA library construction. Both methods identify m7G modifications with nucleotide-level precision. AlkAniline-Seq uses total RNA as starting material; therefore, it can profile m7G and m3C modifications in all types of RNAs. TRAC-Seq starts with small RNAs and incorporates an AlkB demethylation step to facilitate efficient tRNA reverse transcription and sequencing; therefore, TRAC-Seq provides higher specificity and accuracy in tRNA m7G modification profiling. Overall, TRAC-Seq enables specific and efficient single-nucleotide-resolution profiling of m7G sites in tRNAs.
Experimental design
AlkB demethylation
AlkB wild-type (WT) and AlkB D135S proteins are induced and purified from BL21 bacteria. AlkB WT efficiently demethylates m1A and m3C but has little effect on m1G modifications. The AlkB D135S mutant protein has efficient demethylation activity on m1G modification. A combination of the AlkB WT and AlkB D135S proteins achieves efficient demethylation of m1A, m3C, m1G but not m7G modifications, facilitating efficient reverse transcription during the cDNA library construction11.
NaBH4 and aniline treatment
After AlkB demethylation, 60% of the RNA sample is subjected to NaBH4 and aniline treatment to induce reduction and cleavage at m7G-modified sites. This is referred to as the ‘treat’ sample throughout the article, and it stands for AlkB demethylated and NaBH4/aniline-treated. The AlkB-demethylated and NaBH4/aniline-treated RNA samples are purified and subjected to cDNA library construction starting from Step 52. The remaining 40% of the AlkB-demethylated RNA sample, which serves as the non-treated input control, is directly used for cDNA library construction starting from Step 52. This sample is referred to as the ‘non-treat’ or ‘input’ sample throughout the text and stands for AlkB-demethylated only, without NaBH4 and aniline treatment.
Library preparation
The treat and non-treat RNA samples are used for high-throughput sequencing library construction using the NEBNext Multiplex Small RNA Library Prep Set for Illumina. Owing to the secondary structure of tRNA, the reverse transcription temperature is increased to 60 °C to achieve high reverse transcription efficiency.
Bioinformatic analysis
The AlkB treatment facilitates NaBH4- and aniline-mediated cleavage, and adaptor ligation results in adaptor–RNA junctions at m7G sites. After adaptor removal and sequence read mapping, cleavage scores are calculated by comparing the ratio of the number of reads that initiate from a specific site to the number of reads across that site in the treated and control (without chemical treatment) samples to identify the global m7G cleavage sites in tRNAs.
All the bioinformatics analyses use freely available software or in-house-developed programs (https://github.com/rnabioinfor/TRAC-Seq, https://doi.org/10.5281/zenodo.2671795). The chemical treatments in TRAC-Seq generate cleavages at tRNA m7G sites; these are detected by sequencing. Thus, the principle of tRNA m7G site identification is based on the cleavage site (occurring at the + 1 site downstream of m7G; m7G + 1 site) analysis of mature tRNA sequence from TRAC-Seq data. We use the cleavage score ratio between the input (non-treat; AlkB-demethylated) sample and the chemically treated (AlkB-demethylated and NaBH4/aniline-treated) sample to define the cleavage site. The cleavage ratio of site i is defined as the ratio between the number of reads starting at site i and the read depth of site i18. Then the cleavage score of the site is calculated as follows:
where ‘treat’ stands for reads obtained from the AlkB-demethylated and NaBH4/aniline-treated sample and ‘non-treat’ stands for reads obtained from the AlkB-demethylated sample.
The positions with a cleavage score >6 and a cleavage ratio >0.1 are considered to be the candidate m7G sites. The thresholds can be changed in the program by the users to alter the definition of the cleavage site. If users need to obtain higher-confidence candidate modification sites, they should increase these thresholds; if they want to expand the candidate modification list, they should decrease the thresholds. The motif around identified m7G sites is tested by an unbiased motif search using the MEME tool19 to validate the authenticity of the candidate tRNA m7G site (Fig. 2).
Fig. 2 |. Flowchart summarizing the basic bioinformatic analyses.
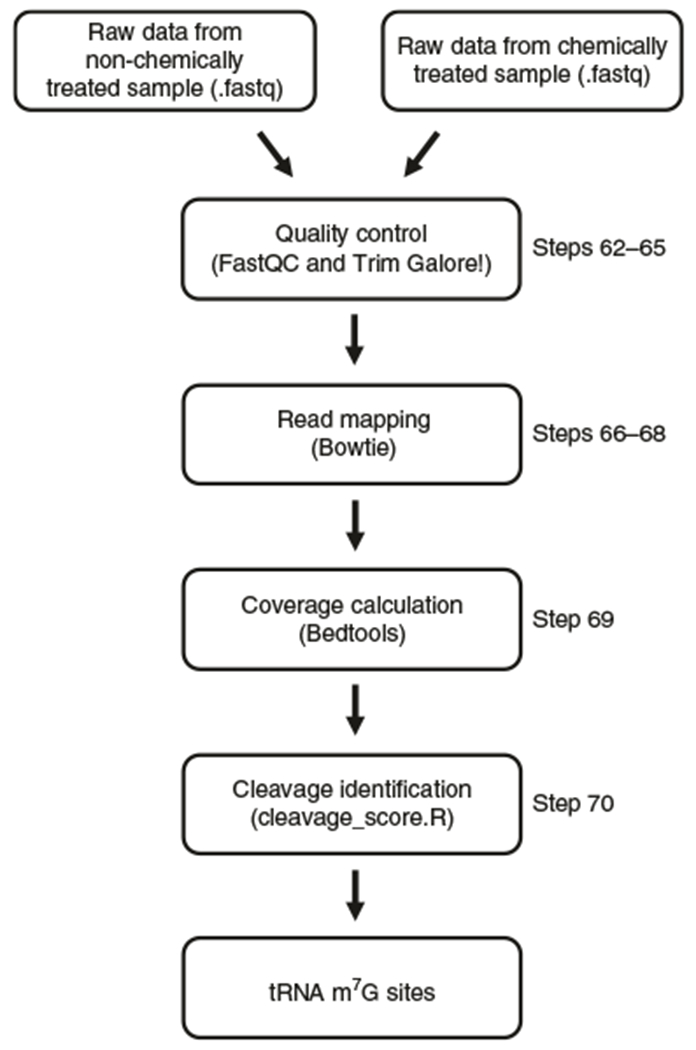
First, a quality check of the .fastq reads is performed with FastQC and then the adaptor and low-quality reads are removed with Trim Galore!. After that, the cleaned reads are aligned to the reference tRNA genome using Bowtie. Read coverage of each site on the tRNAs is calculated with bedtools. Finally, a custom pipeline (cleavage_score.R) is used to calculate the cleavage scores to identify the m7G sites. Non-chemically treated (non-treat): sequencing data from libraries constructed with demethylated RNA sample from Step 40; chemically treated (treat): sequencing data from libraries constructed with demethylated and NaBH4/aniline-treated RNA from Step 51.
Advantages and limitations
We recently developed TRAC-Seq and reported the first global profiling of the tRNA methylome at single-nucleotide resolution10. Although next-generation sequencing has been widely used to profile mRNA expression (RNA-seq) and mRNA modifications (MeRIP-Seq), its application in tRNA biology was largely delayed because the extensive tRNA modifications interfere with reverse transcription during cDNA library construction. On the basis of AlkB-mediated demethylation, which permits the sequencing of tRNAs11,12, we developed a method that combines this ability to sequence tRNAs with the chemically induced cleavage of m7G-modified nucleotides and thereby used high-throughput cDNA-sequencing approaches to map, for the first time (to our knowledge), the m7G tRNA landscape at single-nucleotide resolution in mammalian cells. The primary limitation is that TRAC-Seq is specific to the detection of m7G-modification sites, and other chemical methods would need to be used or developed to adapt this protocol for the detection of other tRNA modifications. Another limitation is that the minimum amount of starting material for TRAC-Seq is 1 μg of small RNAs, and for some special tissues, it might be difficult to obtain enough starting material. In addition, the current protocol focuses on m7G modifications on small RNAs; therefore, it cannot profile the m7G modification globally in all RNA species.
Materials
Biological materials
Cells, tissues, or other biological materials can be used for RNA isolation and the subsequent m7G TRAC-Seq analysis. The biological material used for our TRAC-Seq study was R1/E cells (ATCC SCRC-1036). The R1/E cells were authenticated by short tandem repeat profiling and tested to confirm that there was no mycoplasma infection ! CAUTION The cell lines used should be regularly checked to ensure that they are authentic and are not infected with mycoplasma.
BL21(DE3) competent E. coli (New England Biolabs, cat. no. C2527I)
Reagents
DMEM (Gibco, cat. no. 11965092)
Mouse leukemia inhibitory factor (mLIF; Gemini, cat. no. 400-495)
Stem cell–qualified FBS (Gemini, cat. no. 100-125),
Sodium pyruvate (Gibco, cat. no. 11360070)
Non-essential amino acids (NEAA; Gibco, cat. no. 11140050),
L-Glutamine (Gibco, cat. no. 25030081)
2-Mercaptoethanol (Thermo Fisher Scientific, cat. no. 60-24-2)
Penicillin–streptomycin (Gibco, cat. no. 15140163)
TRIzol reagent (Invitrogen, cat. no. 15-596-018) ! CAUTION TRIzol reagent is a serious health hazard, wear protective clothing, including gloves, goggles and face mask, when handling.
Chloroform (Sigma-Aldrich, cat. no. C2432) ! CAUTION Chloroform is a health hazard. Wear protective clothing, including gloves, goggles and face mask, when handling.
Ethanol (Sigma-Aldrich, cat. no. E7023) ! CAUTION Ethanol is highly flammable; keep flammable liquids away from all sources of ignition.
mirVana miRNA Isolation Kit (Thermo Fisher Scientific, cat. no. AM1561)
tRNA LysCTT probe (5′-AACGTGGGGCTCGAACCCAC-3′; Integrated DNA Technologies (IDT), custom order. Dissolve in H2O to 10 μM and stored at −20 °C for up to 3 years)
pET30a-AlkB and pET30a-AlkB-D135S plasmids (Addgene, cat. nos. 79050 and 79051) ▲ CRITICAL It is crucial that pET30a-AlkB and pET30a-AlkB-D135S be used, because the combination of AlkB and AlkB-D135S results in efficient demethylation of m1A, m3C, and m1G.
IPTG (Sigma-Aldrich, cat. no. I5502)
LB medium (Sigma-Aldrich, cat. no. L3022)
Kanamycin (Sigma-Aldrich, cat. no. K1377)
Phenylmethanesulfonyl fluoride (PMSF; Sigma-Aldrich, cat. no. P7626) ! CAUTION PMSF is toxic to mucous membranes of the lung, eyes, and skin. Wear protective clothing, including gloves, goggles and face mask, when handling.
Imidazole (Sigma-Aldrich, cat. no. I5513)
PBS (Thermo Fisher Scientific, cat. no. 10010023)
Trizma base (Sigma-Aldrich, cat. no. T6791)
NaCl (Sigma-Aldrich, cat. no. T6791)
Glycerol (Sigma-Aldrich, cat. no. G5516)
KCl (Sigma-Aldrich, cat. no. P9333)
Ni-NTA agarose (Qiagen, cat. no. 30210)
Agarose (Denville Scientific, cat. no. CA3510-8)
Colloidal Blue Staining Kit (Invitrogen, cat. no. LC6025)
MgCl2 (Sigma-Aldrich, cat. no. M8266)
Ammonium iron(II) sulfate hexahydrate [(NH4)2Fe(SO4)2·6H2O] (Sigma-Aldrich, cat. no. 215406)
2-Ketoglutarate (2-KG; Sigma-Aldrich, cat. no. 75890)
L-Ascorbic acid (Sigma-Aldrich, cat. no. A0278)
BSA (Sigma-Aldrich, cat. no. 05470)
NuPAGE 4–12% Bis–Tris protein gels (Invitrogen, cat. no. NP0322BOX)
2-(N-Morpholino)ethanesulfonic acid (MES; Sigma-Aldrich, cat. no. M3671)
NaOH (Sigma-Aldrich, cat. no. S8045)
RNasin ribonuclease inhibitors (Promega, cat. no. N2111)
EDTA (0.5 M, pH 8.0; Thermo Fisher Scientific, cat. no. 15575020)
Acid phenol/chloroform/isoamyl alcohol (Ambion, cat. no. AM9720) ! CAUTION Phenol is corrosive and will severely burn the skin. Wear protective clothing, including gloves, goggles and face mask, when handling.
Sodium acetate (3 M; pH 5.2; Sigma-Aldrich, cat. no. S7899)
7-Methylguanosine 5′-triphosphate sodium salt (m7GTP; Sigma-Aldrich, cat. no. M6133)
Sodium borohydride (NaBH4; Sigma-Aldrich, cat. no. 452882)
Aniline (Sigma-Aldrich, cat. no. 242284) ! CAUTION Aniline is toxic if swallowed, upon contact with skin or if inhaled. Wear protective clothing, including gloves, goggles and face mask, when handling.
NEBNext Multiplex Small RNA Library Prep Set for Illumina (Set 1; New England Biolabs, cat. no. E7300S)
QIAquick PCR Purification Kit (Qiagen, cat. no. 28104)
Glacial acetic acid (Thermo Fisher Scientific, cat. no. A38-212)
TAE buffer (50×, Thermo Fisher Scientific, cat. no. B49)
RNaseZap RNase decontamination solution (Thermo Fisher Scientific, cat. no. AM9780)
Tris/Glycine/SDS Electrophoresis Buffer (10×, Bio-Rad, cat. no. 1610772EDU)
Equipment
Microcentrifuge tubes (1.7 ml; Axygen, cat. no. MCT-175-C)
CO2 incubators (Thermo Fisher Scientific)
Microbiological incubator (Thermo Fisher Scientific)
MaxQ 4000 benchtop orbital shaker (Thermo Fisher Scientific)
Econo-Pac chromatography columns (Bio-Rad, cat. no. 7321010)
Slide-A-Lyzer (10k MWCO, MINI dialysis devices (Thermo Fisher Scientific, cat. no. PI88401)
Microcentrifuge (Eppendorf, model no. 5424)
Refrigerated microcentrifuge (Eppendorf, model no. 5424R)
Ultracentrifuge (Beckman Coulter)
Spectrophotometer (NanoDrop Technologies, model no. ND-1000)
Agilent 2100 Bioanalyzer or High Sensitivity D1000 ScreenTape (Agilent Technologies)
Spectrophotometer (Bio-Rad, model no. SmartSpec 3000)
Sonicator (Misonix, model no. 3000)
Centrifuge (Beckman Coulter)
Sequencing system (Illumina, model no. NextSeq 500)
Software and data
Test data set: TRAC-Seq of mESCs (GEO accession no. GSE112670)
Mouse mature tRNA sequence and tRNA genomic loci annotation (mm10), downloaded from the outputs of tRNAscan-SE software in the GtRNAdb database20 (http://gtrnadb.ucsc.edu/genomes/eukaryota/Mmusc10)
Mouse reference genome sequence (mm10), downloaded from UCSC database (http://hgdownload.soe.ucsc.edu/goldenPath/mm10/bigZips)
SRA toolkit (NCBI, v.2.3.5) for .fastq extraction from SRA files (https://www.ncbi.nlm.nih.gov/sra/docs/toolkitsoft)
FastQC (Babraham Bioinformatics, v.0.11.8) for visually checking the sequencing quality of TRAC-Seq data (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/)
Bowtie (v.1.2.2) for mapping short reads to tRNA sequences to call modification sites21 (http://bowtie-bio.sourceforge.net/index.shtml)
Bowtie2 (v2.3.4) for mapping short reads in tRNA expression analysis22 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml)
Samtools (v.0.1.19) for converting, sorting and indexing the SAM/BAM mapping result files23 (https://www.htslib.org/)
bedtools (v.2.27.1) for read depth calculation24 (https://bedtools.readthedocs.io/en/latest/)
MEME25 (University of Nevada, Reno, v.4.6.1) for m7G site motif searching19 (http://meme-suite.org/tools/meme)
ARM-Seq pipeline (v.1.0) for tRNA expression analysis12 (https://github.com/UCSC-LoweLab/tRAX)
R package data.table (v.1.12.2) (https://cran.r-project.org/web/packages/data.table/index.html)
R package cowplot (v.0.9.4) (https://cran.r-project.org/web/packages/cowplot/index.html)
R package gridExtra (v.2.3) (https://cran.r-project.org/web/packages/gridExtra/index.html)
R package seqinr (v.3.4-5) (https://cran.r-project.org/web/packages/seqinr/index.html)
Bioconductor package GenomicAlignments (v.1.20.0) (https://bioconductor.org/packages/release/bioc/html/GenomicAlignments.html)
Bioconductor package GenomicFeatures (v.1.36.0) (https://bioconductor.org/packages/release/bioc/ html/GenomicFeatures.html)
Bioconductor package DESeq2 (v.1.24.0) for tRNA differential expression analysis (https://bioconductor.org/packages/release/bioc/html/DESeq2.html)
R program cleavage_score.R (v.1.0.0) for cleavage site calling (https://github.com/rnabioinfor/TRAC-Seq, https://doi.org/10.5281/zenodo.2671795)
Reagent setup
100 mM PMSF
Dissolve 174 mg of PMSF in 10 ml of ethanol. The prepared solution can be stored at −20 °C for up to 3 months.
Bacteria lysis buffer
This solution is 20 mM imidazole, 100 μM PMSF in 1× PBS. Add 10 ml of 1 M imidazole and 500 μl of 100 mM PMSF to 500 ml of PBS; mix and keep on ice. The solution should be made up fresh each time.
1 M imidazole
Dissolve 68.08 g of imidazole in 1 liter of H2O. The prepared solution can be stored at room temperature (20–25 °C) for up to 6 months when protected from direct light.
1 M Tris, pH 7.8
Dissolve 121.14 g of Tris in 800 ml of H2O. Adjust pH to 7.8 with the appropriate volume of concentrated HCl. Bring final volume to 1 liter with H2O. The prepared solution can be stored at room temperature for up to a year when protected from direct light.
1 M Tris, pH 8.2
Dissolve 121.14 g of Tris in 800 ml of H2O. Adjust pH to 8.2 with the appropriate volume of concentrated HCl. Bring final volume to 1 liter with H2O. The prepared solution can be stored at room temperature for up to a year when protected from direct light.
5 M NaCl
Dissolve 146.1 g of NaCl in 500 ml of H2O. The prepared solution can be stored at room temperature for up to a year when protected from direct light.
Low-imidazole buffer
This solution is 10 mM Tris, 50 mM imidazole, 500 mM NaCl, and 100 μM PMSF. Mix 2 ml of 1 M Tris, pH 7.8, 10 ml of 1 M imidazole, 20 ml of 5 M NaCl, 200 μl of 100 mM PMSF, and 167.8 ml of H2O. Mix and keep on ice. The solution should be made up fresh each time.
Elution buffer
This solution is 10 mM Tris, 500 mM imidazole, 500 mM NaCl, and 100 μM PMSF. Mix 0.1 ml of 1 M Tris, pH 7.8, 5 ml of 1 M imidazole, 1 ml of 5 M NaCl, 10 μl of 100 mM PMSF, and 3.89 ml of H2O. Mix and keep on ice. The solution should be made up fresh each time.
BC100 low-salt buffer
This solution is 20 mM Tris, 10% (vol/vol) glycerol, and 100 mM KCl. Mix 20 ml of 1M Tris, pH 7.8, 100 ml of glycerol, 100 ml of 1 M KCl, and 780 ml of H2O. Precool in cold room for at least 2 h before using for dialysis. The solution should be made up fresh each time.
1 M KCl
Dissolve 37.3 g of KCl in 500 ml of H2O. The prepared solution can be stored at room temperature for up to a year when protected from direct light.
1 M MgCl2
Dissolve 4.76 g of MgCl2 in 50 ml of H2O ! CAUTION Dissolving MgCl2 in H2O generates heat; add to water a little at a time to avoid splashing. The prepared solution can be stored at room temperature for up to a year when protected from direct light.
100 mM (NH4)2Fe(SO4)2·6H2O
Dissolve 1.96 g of (NH4)2Fe(SO4)2·6H2O in 50 ml H2O. The solution should be made up fresh each time.
300 mM 2-KG
Dissolve 2.19 g of 2-KG in 50 ml of H2O. The prepared solution can be stored at 4 °C for a month.
0 2 M L-ascorbic acid
Dissolve 1.76 g of l-ascorbic acid in 50 ml of H2O. The prepared solution can be stored at 4 °C for up to a month.
50 mg/ml BSA
Dissolve 2.5 g of BSA in 50 ml of H2O. Prepare aliquots and store at −20 °C for up to 1 month. Do not reuse thawed aliquots.
0.5 M MES, pH 5.0
Dissolve 4.88 g of MES in 40 ml of H2O; add 40% (wt/vol) NaOH to adjust pH to 5.0 and add H2O to a final volume of 50 ml. ! CAUTION 40% (wt/vol) NaOH is extremely hazardous; use safety glasses, gloves, and coat. The prepared solution can be stored at 4 °C for up to a month.
2× Demethylation buffer
This solution is 600 mM KCl, 4 mM MgCl2, 0.1 mM (NH4)2Fe(SO4)2·6H2O, 0.6 mM 2-KG, 4 mM L-ascorbic acid, 0.1 mg/ml BSA, and 0.1 M MES (pH 5.0). To prepare 5 ml, mix 3 ml of 1 M KCl, 20 μl of 1 M MgCl2, 5 μl of 100 mM (NH4)2Fe(SO4)2·6H2O, 10 μl of 300 mM 2-KG, 100 μl of 0.2 M L-ascorbic acid, 10 μl of 50 mg/ml BSA, 1 ml of 0.5 M MES (pH 5.0), and 855 μl of H2O to a final volume of 5 ml (final concentrations for 2× buffer). The solution should be made up fresh each time.
10 mM m7GTP
Dissolve 5 mg of m7GTP in 930.7 μl of H20. Prepare aliquots and store at −80 °C for up to a year.
0.2 M NaBH4
Dissolve 0.0757 g of NaBH4 in cold 0.5 M Tris (pH 8.2) buffer. The solution should be made up fresh each time.
Cleavage buffer
This solution is 7:3:1 H2O/glacial acetic acid/aniline. Mix 700 μl of H2O, 300 μl of glacial acetic acid, and 100 μl of aniline in a 1.5-ml tube. The solution should be made up fresh each time.
R1/E mESC culture medium
This medium is DMEM supplemented with 1,000 U/ml mLIF, 15% (vol/vol) stem cell-qualified FBS, 1× sodium pyruvate, 1× NEAA, 1× L-glutamine, 50 mM 2-mercaptoethanol, and 1% (vol/vol) penicillin-streptomycin
Equipment setup
▲ CRITICAL All the analysis commands in the protocol can be executed on a UNIX/Linux operating system. A ‘$’ character indicates commands executed from the shell prompt. All the software required for the analysis needs to be defined in a PATH system environment variable if you do not want to input the system path of the software. The in-house-developed program for candidate modification site calling (cleavage_score.R) was written in R (https://www.r-project.org), which needs to be installed in your UNIX/Linux system.
R installation
Download the most recent source code R-x.y.z.tar.gz (x.y.z represents the version of R; v.3.5.0 was used in this protocol) from https://CRAN.R-project.org/mirrors.html
Unpack it with the following command:
$tar -xzvf R-x.y.z.tar.gz
3 Install R with the following commands:
$cd R-x.y.z
$ ./configure & make & make install
Installation of R dependencies
For the R packages cowplot, gridExtra, and seqinr, use the following R command (‘>‘ character indicates the R shell prompt):
>install.packages(“the pack name”)
2 For the Bioconductor packages GenomicAlignments, GenomicFeatures, and DESeq2, use one of the following commands:
>source(“https://bioconductor.org/biocLite.R“)
>biocLite(“the Bioconductor package name”)
or:
>if (!requireNamespace(“BiocManager”, quietly = TRUE)) install. packages(“BiocManager”)
>BiocManager::install(“the Bioconductor package name”)
Procedure
Isolation of small RNAs ● Timing 4 h
-
Culture R1/E mESCs with mESC culture medium to ~50% confluency (~1 × 107 cells) at 37 °C in cell culture incubators supplied with 5% CO2. Isolate total RNA samples with TRIzol reagent (1 ml of TRIzol for 107 cells), according to the manufacturer’s instructions. Resuspend the RNA pellet with 100 μl of H2O. The amount of isolated total RNA should be ~100–200 μg.
▲ CRITICAL STEP Use TRIzol reagent to isolate RNAs of all sizes. If using an RNA purification column for RNA isolation, make sure that the RNA purification column method can isolate small RNAs.
Measure total RNA concentration with a spectrophotometer; the expected concentration is ~1–2 μg/μl. Then isolate small RNAs (<200 nt) by using the mirVana miRNA Isolation Kit and following the manufacturer’s ‘Isolation of Small RNAs from Total RNA Samples’ procedures as described in the Steps 3–14.
Mix 50–100 μg of total RNA (in 40 μl of H2O) with 5 volumes (200 μl) of lysis/binding buffer.
Add 24 μl of miRNA homogenate additive (from the kit), mix well, and incubate 10 min on ice.
Add 88 μl of 100% (vol/vol) ethanol to the 264 μl of RNA mixture from the previous step. Mix thoroughly by vortexing or inverting the tube several times.
Pipette the lysate–ethanol mixture from the previous step onto the filter cartridge provided with the kit. Centrifuge at 5,000g for 1 min at 25 °C. Collect the filtrate.
Add 235 μl of 100% (vol/vol) ethanol to the filtrate (i.e., flow-through). Mix thoroughly.
Pipette the lysate–ethanol mixture from the previous step onto a new filter cartridge. Centrifuge at 5,000g for 1 min at 25 °C. Discard the flow-through.
Apply 700 μl of miRNA wash solution 1 (working solution mixed with ethanol, from the kit) to the filter cartridge and centrifuge for 1 min at 5,000g at 25 °C. Discard the flow-through from the collection tube and replace the filter cartridge into the same collection tube.
Apply 500 μl of wash solution 2/3 (working solution mixed with ethanol, from the kit) and draw it through the filter cartridge as in the previous step.
Repeat Step 10 with a second 500-μl aliquot of wash solution 2/3.
After discarding the flow-through from the last wash, place the filter cartridge back into the same collection tube and spin the assembly for 1 min at 10,000g at 25 °C to remove residual fluid from the filter.
-
Transfer the filter cartridge to a fresh collection tube. Apply 30 μl of 95 °C elution solution and close the cap.
▲ CRITICAL STEP Be sure to preheat the elution solution provided with the kit to 95 °C before eluting the small RNA from the filter.
Incubate at room temperature for 2 min. Spin at 10,000g for 1 min at 25 °C and collect the flow-through that contains the small RNA.
-
Measure the small RNA concentration with a spectrophotometer; the expected yield is 10% of the total input RNA. Validate RNA size by 1% agarose gel electrophoresis with TAE buffer (Fig. 3) or by Agilent 2100 Bioanalyzer.
? TROUBLESHOOTING
■ PAUSE POINT Isolated RNA can be stored at −80 °C for up to 1 year.
Fig. 3 |. Isolation of small RNAs and total RNA from R1/E mESCs.
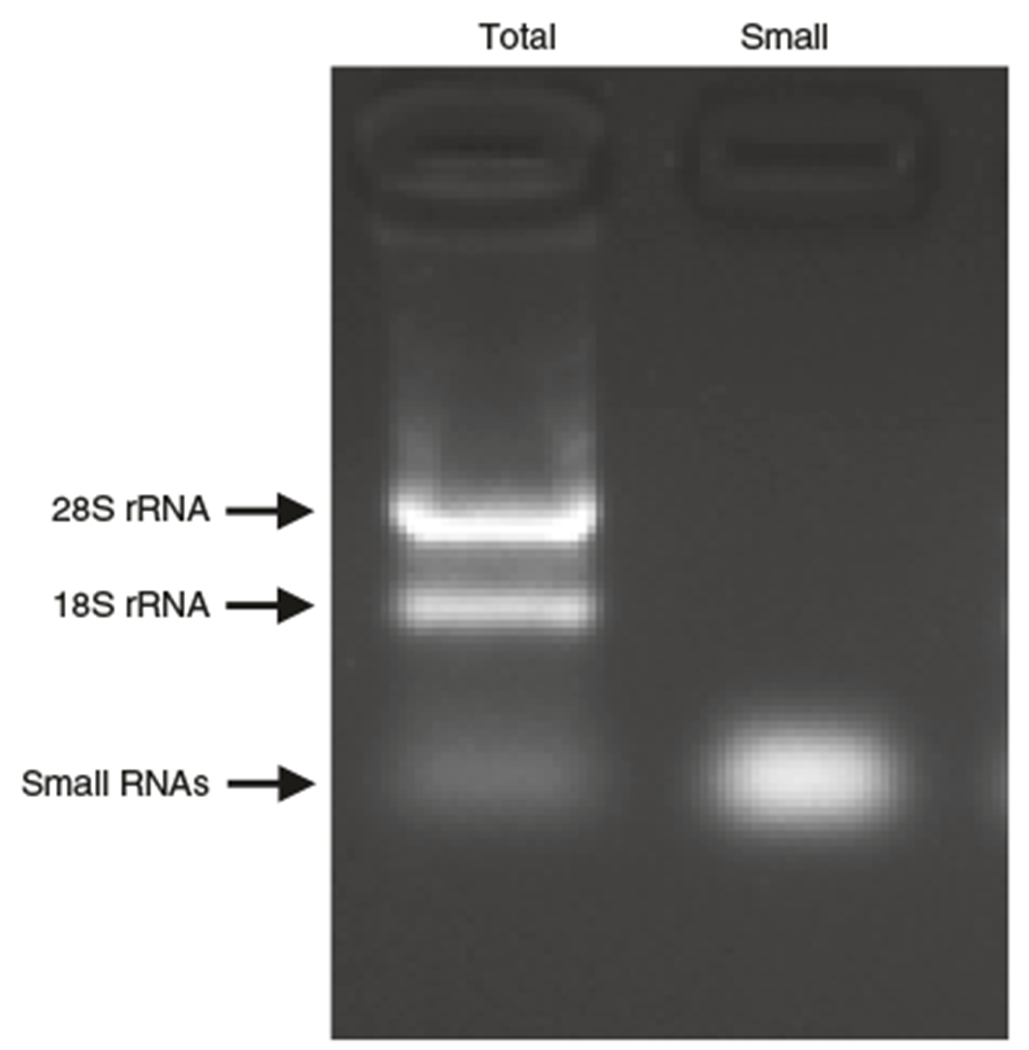
A representative image of the total RNA (Total) and the isolated small RNAs (Small). The RNA samples were analyzed by 1.5% agarose gel electrophoresis using 120 V for 15 min.
Purification of recombinant AlkB and AlkB-D135S proteins ● Timing 5 d
▲ CRITICAL The combination of the AlkB WT and AlkB D135S proteins in the demethylation step achieves efficient demethylation of m1A, m3C, and m1G while preserving the m7G modification11, which facilitates efficient reverse transcription during the cDNA library construction. The following steps describe the parallel purification of recombinant AlkB WT and AlkB D135S proteins expressed in BL21 (DE3) competent cells.
16 Transform 50 ng each of pET30a-AlkB and pET30a-AlkB-D135S plasmids into two separate tubes of BL21(DE3) competent cells. Plate the transformed competent cells onto 10-cm LB-coated plates supplemented with 20 μg/ml kanamycin and incubate overnight in a 37 °C incubator.
17 Inoculate a single BL21 clone from each transformation plate into 2 ml of LB medium with 20 μg/mL kanamycin starter culture each. Grow overnight at 37 °C.
18 Inoculate 500 ml of LB medium with 20 μg/ml kanamycin with the 2 ml of starter culture. Grow at 37 °C in shaker at 250 r.p.m. until OD600 value is 0.4–0.6.
19 Add IPTG to final concentration of 200 μM (1:5,000 of 1 M stock) and grow overnight in a shaker at 250 r.p.m. at 20°C.
20 Spin down bacteria at 5,000g for 15 min at room temperature and resuspend each pellet in 15 ml of cold bacteria lysis buffer.
-
21 Sonicate five times at level 4.0 for 30 s with 1-min break after each sonication (we use a Misonix Sonicator 3000 with the standard-size probe).
▲ CRITICAL STEP Keep the sample on ice during the sonication and the 1-min breaks to prevent heating-induced protein degradation.
-
22 Transfer lysates to round-bottom plastic bottles with push-on tops. Centrifuge at 25,000g for 1 h at 4 °C.
▲ CRITICAL STEP Perform Steps 23–31 in a cold room.
23 During the spin, vortex the Ni-NTA agarose slurry bottle to resuspend the 50% (vol/vol) Ni-NTA agarose and pipette 1 ml of 50% (vol/vol) Ni-NTA agarose slurry into two 20-ml Bio-Rad Econo-Pac chromatography columns. Let drain.
24 Wash the Ni-NTA agarose with 20 ml of cold bacteria lysis buffer. Let the column empty. Cap the bottom of the column.
25 When the spin in Step 22 is finished, add all the lysates (~14 ml of supernatant) to the equilibrated, capped column. Incubate while rocking or rotating at 4 °C for 3 h.
26 Uncap the bottom of the tube and let the supernatant flow through.
27 Add 20 ml of cold low-imidazole buffer to the uncapped column and let the cold low-imidazole buffer flow through to wash out the nonspecific binding proteins. Repeat the wash two times for a total of three washes.
28 Elution. For elution 1, add 500 μl of cold elution buffer and allow the column to empty into a collection tube. For elution 2, cap the column and add 500 μl of cold elution buffer. Incubate for 30 min in a cold room. Uncap the column and collect the eluted fraction into a new collection tube. Perform another elution the same way as elution 2 for a total of three elutions. Do not pool the fractions.
29 Because the fractions are eluted in a high-salt buffer, dialyze the eluted proteins with 1 liter of cold BC100 low-salt buffer overnight. Prepare 100-μl aliquots of the dialyzed proteins and store at −80 °C.
30 To analyze the purified recombinant AlkB WT and AlkB D135S proteins by SDS-PAGE with a 4–20% polyacrylamide gel, load 5 μl of each elution together with different amounts (0.1, 0.2, 0.5, 0.8, and 1 μg) of BSA as standard. Perform electrophoresis in 1× Tris/Glycine/SDS electrophoresis buffer.
-
31 Stain the SDS–PAGE gel with the Colloidal Blue Staining Kit to check the purity and calculate protein concentration (expected concentration, 0.2–0.5 μg/μl), using BSA as standard. The molecular weights of the recombinant AlkB WT and AlkB D135S are ~25 kDa (Fig. 4). Use the elution containing the purest recombinant protein for the subsequent demethylation assay.
? TROUBLESHOOTING
■ PAUSE POINT Purified recombinant proteins can be stored at −80 °C for up to 1 year.
Fig. 4 |. SDS-PAGE analysis of purified recombinant AlkB WT and AlkB D135S.
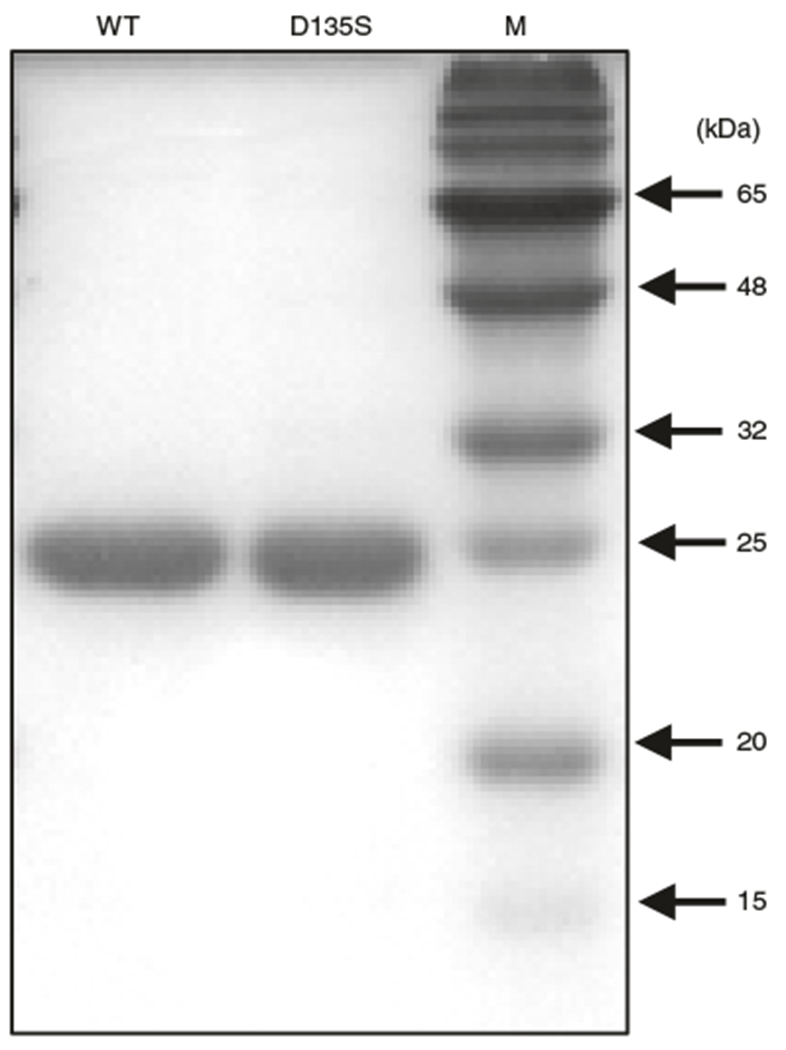
Purified recombinant proteins were separated on a 15% polyacrylamide gel and stained with the Colloidal Blue Staining kit. Their molecular weights are ~25 kDa. M, protein marker.
Demethylation of small RNAs using recombinant AlkB and AlkB-D135S ● Timing 4 h
- 32 Setup the demethylation assay as outlined in the table below:
Component Amount Final concentration 2× Demethylation buffer 50 μl 1× AlkB WT 2 μg 0.02 μg/μl AlkB D135S 4 μg 0.04 μg/μl RNasin (40 U/μl) 5 μl 2 U/μl Small RNAs (Step 15) 2.5 μg 0.025 μg/μl H2O To a final volume of 100 μl Total 100 μl 33 Incubate at room temperature for 2 h.
34 Quench the reaction with 1 μl of 0.5 M EDTA to a final concentration of 5 mM EDTA.
35 Add 300 μl of H2O to the reaction to make a final volume of 400 μl. Add 400 μl of acid phenol/chloroform/isoamyl alcohol to the tube. Vortex for 1 min and centrifuge for 5 min at 16,000g at room temperature.
36 Transfer the upper, aqueous phase to a fresh tube.
37 Add 0.1 volume of 3 M sodium acetate (pH 5.2) and 2.5 volumes of 100% (vol/vol) ethanol. Mix and keep at −20 °C for at least 1 h.
38 Spin at 16,000g in a microcentrifuge at 4 °C for 15 min.
39 Carefully remove and discard the supernatant; then add 1 ml of 75% (vol/vol) ethanol to the tube, vortex the tube briefly, spin at 16,000g in a microcentrifuge at room temperature for 5 min, and discard the 75% (vol/vol) ethanol.
-
40 Dry the pellet at room temperature for 5 min and resuspend the RNA sample in 16 μl of H2O.
■ PAUSE POINT Isolated RNA can be stored at −80 °C for up to 1 year. ~40% of this sample will be used as a non-chemically treated control (non-treat sample) in Step 52.
NaBH4/aniline-mediated reduction and cleavage of m7G sites ● Timing 4 h
- 41 In a 1.7-ml Eppendorf tube, mix demethylated small RNAs, m7GTP (as methylation carrier), and Tris buffer as outlined in the table below, then put the tube on ice.
Component Amount (μl) Final concentration Demethylated small RNAs (from Step 40) 9 10 mM m7GTP 10 2 mM 1 M Tris, pH 8.2 25 0.5 M H2O 6 Total 50 -
42 Add 50 μl of freshly made 0.2 M NaBH4 in 0.5 M Tris, pH 8.2, to the mixture from Step 41, mix, and incubate the reaction on ice in the dark for 30 min.
▲ CRITICAL STEP Dissolving NaBH4 in cold 0.5 M Tris (pH 8.2) releases a lot of hydrogen gas and produces vigorous bubbles; use the freshly made NaBH4 buffer immediately.
43 Add 10 μl of 3 M sodium acetate (pH 5.2) and 400 μl of 100% (vol/vol) ethanol, mix, and keep at −20 °C for at least 1 h.
44 Centrifuge at 16,000g for 15 min at 4 °C.
45 Remove and discard the supernatant; wash the pellet with 75% (vol/vol) ethanol. Spin at 12,000g for 5 min at room temperature.
46 Remove and discard the 75% (vol/vol) ethanol; air dry for 5 min.
-
47 Resuspend the pellet with 100 μl of cleavage buffer (7:3:1 H2O/glacial acetic acid/aniline); store at room temperature in the dark for 2 h.
? TROUBLESHOOTING
48 Add 10 μl of 3 M sodium acetate (pH 5.2) and 400 μl of 100% ethanol, mix, and keep at −20 °C for at least 1 h. Centrifuge at 16,000g for 15 min at 4 °C.
49 Remove and discard the supernatant; wash the pellet with 75% (vol/vol) ethanol. Spin at 12,000g for 5 min at room temperature.
50 Remove and discard the 75% (vol/vol) ethanol; air dry for 5 min.
-
51 Resuspend the RNA pellet in 6 μl of H2O.
▲ CRITICAL STEP It is highly recommended to first optimize the NaBH4/aniline-mediated reduction and cleavage procedure (Steps 41–51) before treating the demethylated small RNAs. For optimization, treat 2.5 μg of total RNAs from Step 1 as described in Steps 41–51 and then analyze the treated and untreated samples on a northern blot using a probe against tRNA LysCTT10. The NaBH4/aniline treatment results in specific cleavage of m7G sites in tRNAs (Fig. 5). A successful cleavage should result in two bands corresponding to the full-length and the cleaved 3′ fragment of tRNA LysCTT in the northern blot analysis.
? TROUBLESHOOTING
■ PAUSE POINT Isolated RNA can be stored at −80 °C for up to 1 year.
Fig. 5 |. Cleavage of an m7G site in tRNA.
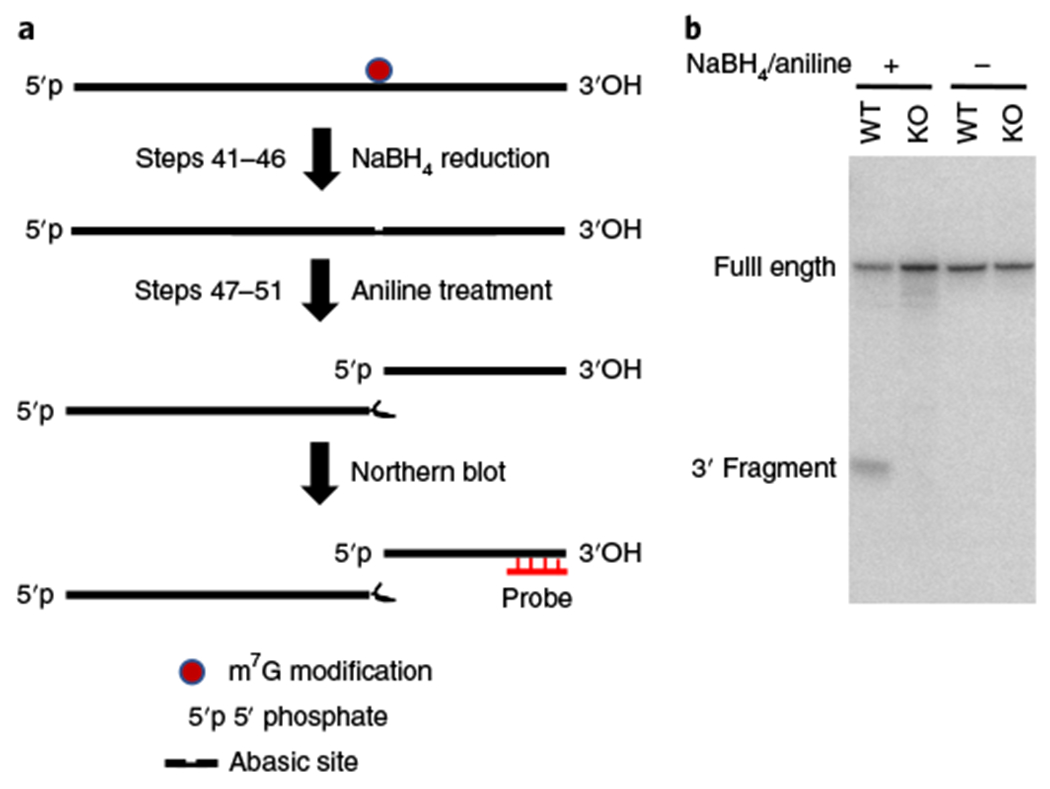
a, Schematic diagram of tRNA reduction and cleavage at m7G sites. RNA samples isolated from R1/E mESCs are treated with NaBH4 and aniline to induce the site-specific cleavage of RNA at m7G-modified guanosines. b, Northern blot of the chemically treated RNA samples with tRNA LysCTT probe. probe, northern blot probe against tRNA.
Small RNA sequencing library construction and high-throughput sequencing ● Timing ~1 d
▲ CRITICAL Perform cDNA library construction with the NEBNext Small RNA Library Prep Set. For the non-chemically treated control (non-treat), use 6 μl of demethylated RNA sample from Step 40; for the chemically treated sample (treat), use 6 μl of demethylated and NaBH4/aniline-treated RNA from Step 51. Proceed to library construction with Steps 52–61, generating the control and treated libraries in parallel.
▲ CRITICAL A demethylated and aniline-treated RNA sample (without NaBH4 treatment) could be used for library construction as an additional control. Without NaBH4 treatment, it is expected that no specific cleavage will be detected.
- 52 Dilute the 3′ SR Adaptor for Illumina (provided in the NEBNext Small RNA Library Prep Set kit) 1:2 with H2O. Mix the following components (in two separate tubes, one for the non-treat control and one for the chemically treated sample) and incubate in a preheated thermal cycler for 2 min at 70 °C. Transfer the tube to ice.
Component Amount (μl) RNA sample (from Step 40 or Step 51) 6 1:2 diluted 3′ SR Adaptor for Illumina 1 Total 7 - 53 Mix the 3′ ligation reaction buffer (2×, provided in the NEBNext Small RNA Library Prep Set kit) and 3′ ligation enzyme mix (provided in the NEBNext Small RNA Library Prep Set kit) as outlined in the table below and incubate for 1 h at 25 °C in a thermal cycler.
Component Amount (μl) Sample from Step 52 7 3′ Ligation reaction buffer (2×) 10 3′ Ligation enzyme mix 3 Total 20 - 54 Add H2O and 1:2 diluted SR RT Primer for Illumina (provided in the NEBNext Small RNA Library Prep Set kit) as outlined below, mix and place in a thermocycler with a heated lid set to >85 °C, and run the following program: 5 min at 75 °C; 15 min at 37 °C; 15 min at 25 °C; hold at 4 °C
Component Amount (μl) Sample from Step 53 20 1:2 Diluted SR RT Primer for Illumina 1 H2O 4.5 Total 25.5 55 With 5 min remaining, resuspend the 5′ SR Adaptor (provided in the NEBNext Small RNA Library Prep Set kit) in 240 μl of H2O. Add a sufficient amount of 5′ SR Adaptor (for the number of samples in the experiment plus an excess of 10%) to a separate, nuclease-free 200-μl PCR tube. Incubate the adaptor in the thermal cycler at 70 °C for 2 min and then immediately place the tube on ice. Keep the tube on ice and use the denatured adaptor within 30 min of denaturation. Aliquot and store the remaining 5′ SR Adaptor at −80 °C.
- 56 Add denatured 5′ SR Adaptor for Illumina, 5′ ligation reaction buffer (10×, provided in the NEBNext Small RNA Library Prep Set kit), and 5′ ligation enzyme mix (provided in the NEBNext Small RNA Library Prep Set kit) as outlined below, mix, and incubate for 1 h at 25 °C in a thermal cycler.
Component Amount (μl) Sample from Step 54 25.5 Denatured 5′ SR Adaptor for Illumina from Step 55 1 5′ Ligation reaction buffer (10×) 1 5′ Ligation enzyme mix 2.5 Total 30 -
57 Mix the following components (provided in the NEBNext Small RNA Library Prep Set kit) as outlined below. Perform reverse transcription at 60 °C for 1 h.
Component Amount (μl) Sample from Step 56 30 First strand synthesis reaction buffer 8 Murine RNase inhibitor 1 ProtoScript II reverse transcriptase 1 Total 40 ▲ CRITICAL STEP Owing to the secondary structure of tRNA, incubation at 60 °C can increase the reverse transcription efficiency.
- 58 Add and mix the following components (provided in the NEBNext Small RNA Library Prep Set kit) to the RT reaction mix from Step 57 as outlined below. The NEBNext Multiplex Small RNA Library Prep Set for Illumina allows the pooling of different libraries by using different index primers. For each reaction, only one index primer is used during the PCR step.
Component Amount (μl) Sample from Step 57 40 LongAmp Taq 2× Master Mix 50 SR primer for Illumina 2.5 Index (×) primer 2.5 H2O 5 Total 100 - 59 Perform PCR amplification using the program below:
Cycle step Temperature (°C) Time Cycles Initial denaturation 94 30 s 1 Denaturation 94 15 s Annealing 62 30 s 15 Extension 70 15 s Final extension 70 5 min 1 Hold 4 ∞ 60 Purify the PCR production with a QIAquick PCR Purification Kit according to the manufacturer’s instructions and elute with 30 μl of H2O. Measure the concentration of the libraries using a spectrophotometer (expected concentration should be >10 ng/μl).
-
61 Perform quality control of the libraries using an Agilent 2100 Bioanalyzer or High Sensitivity D1000 ScreenTape to check the size of the libraries (Fig. 6). Sequence the libraries using single-end 75 bp with Illumina NextSeq 500.
▲ CRITICAL STEP Here, because of the short length of the library, sequencing of the libraries with single-end 75 bp (SE75) is sufficient for the calculation of tRNA expression and tRNA cleavage score.
Fig. 6 |. Size distribution of the TRAC-Seq library.
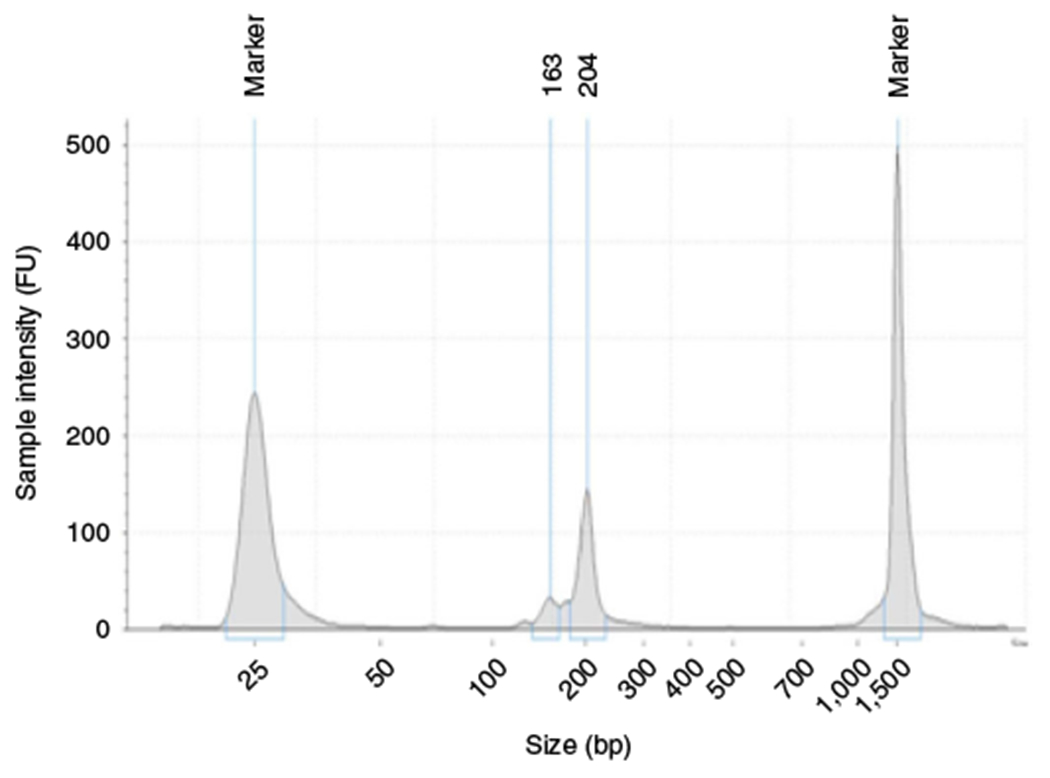
The size of the TRAC-Seq library was determined by High Sensitivity D1000 ScreenTape. The size of the small RNA library is ~140-210 bp.
Quality control for raw sequence data ● Timing ~30 min
62 The TRAC-seq sequencing data used as an example to demonstrate the analysis steps have been uploaded into the GEO (accession no. GSE112670)10 of NCBI. Download the TRAC-Seq raw sequencing reads from the NCBI FTP site using the wget command in a terminal window:
$ wget ftp://ftp-trace.ncbi.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR693/SRR6936910/SRR6936910.sra
$ wget ftp://ftp-trace.ncbi.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR693/SRR6936911/SRR6936911.sra
$ wget ftp://ftp-trace.ncbi.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR693/SRR6936912/SRR6936912.sra
$ wget ftp://ftp-trace.ncbi.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR693/SRR6936913/SRR6936913.sra
Here, the TRAC-Seq data set (GEO accession no. GSE112670) was used as test data to illustrate the stepwise analysis for TRAC-Seq data bioinformatic analysis. The features of the four samples that were sequenced as part of the dataset are summarized in the table below. The downloaded .sra files will be in the current working directory. Users can use the pwd command to show the path of current working directory. Users can also use prefetch program in SRA Toolkit to download the data.
| GSE ID | SRA ID | Cell type | Sample type | Chemical treatment | Sample information |
|---|---|---|---|---|---|
| GSM3076142 | SRR6936910 | R1/E mESCs | Wild type | Non-treat | WT_Alkb_Treated_Input_for_TRAC-Seq (Step 40) |
| GSM3076143 | SRR6936911 | R1/E mESCs | Mettl1 knockout | Non-treat | KO_Alkb_Treated_Input_ for_TRAC-Seq (Step 40) |
| GSM3076144 | SRR6936912 | R1/E mESCs | Wild type | Treat | WT_Alkb_and_NaBH4_Treated_TRAC-Seq (Step 51) |
| GSM3076145 | SRR6936913 | R1/E mESCs | Mettl1 knockout | Treat | KO_Alkb_and_NaBH4_Treated_TRAC-Seq (Step 51) |
63 Extract .fastq-format files from SRA files using fastq–dump in the SRA Toolkit (https://www.ncbi.nlm.nih.gov/sra/docs/toolkitsoft) by entering the following commands:
$ fastq–dump SRR6936910.sra
$ fastq–dump SRR6936911.sra
$ fastq–dump SRR6936912.sra
$ fastq–dump SRR6936913.sra
The .fastq files will be extracted into current working directory.
64 Visually check the sequencing quality, such as quality distribution and adaptor contamination, using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc).
$fastqc SRR6936910.fastq
$fastqc SRR6936911.fastq
$fastqc SRR6936912.fastq
$fastqc SRR6936913.fastq
The quality reports (such as SRR6936910_fastqc.html), which are in .html format, will be generated in the current working directory.
65 Perform adaptor cleaning and low-quality read filtering using Trim Galore! (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore) if the raw sequencing data contain adaptors and/or low-quality sequences.
$mkdir clean_outdir
$trim_galore SRR6936910.fastq --phred33 -a AGATCGGAAGAGCACA -length 25 -q 20 --stringency 1 -o clean_outdir
$trim_galore SRR6936911.fastq --phred33 -a AGATCGGAAGAGCACA -length 25 -q 20 --stringency 1 -o clean_outdir
$trim_galore SRR6936912.fastq --phred33 -a AGATCGGAAGAGCACA -length 25 -q 20 --stringency 1 -o clean_outdir
$trim_galore SRR6936913.fastq --phred33 -a AGATCGGAAGAGCACA -length 25 -q 20 --stringency 1 -o clean_outdir
▲ CRITICAL STEP Here, after adaptor cleaning and low-quality filtering, the clean sequences (such as SRR6936910_trimmed.fq), which have >25-bp length and >Q20 quality, can be found in the clean_outdir directory created using the mkdir command. --phred33 indicates the encode type of sequence quality (the probability of sequencing error) (https://en.wikipedia.org/wiki/Phred_quality_score), which can be identified in the FastQC report generated in Step 64.
Short-read mapping ● Timing ~6 h
66 Map the cleaned sequence generated in Step 65 to the reference tRNA using a short-read aligner tool such as Bowtie25, BWA26, Tophat27, or STAR28, which can generate mapping results in SAM/ BAM format. Here, use Bowtie (v.1.2.2) to align the cleaned sequences to the mm10 tRNA sequence index built by bowtie-build.
$bowtie -a -m 50 -v 3 --best --strata -S bowtie_index/mm10_tRNA clean_outdir/SRR693_6910_trimmed.fq SRR693_6910_trimmed.sam
$bowtie -a -m 50 -v 3 --best --strata -S bowtie_index/mm10_tRNA clean_outdir/SRR693_6911_trimmed.fq SRR693_6911_trimmed.sam
$bowtie -a -m 50 -v 3 --best --strata -S bowtie_index/mm10_tRNA clean_outdir/SRR693_6912_trimmed.fq SRR693_6912_trimmed.sam
$bowtie -a -m 50 -v 3 --best --strata -S bowtie_index/mm10_tRNA clean_outdir/SRR693_6913_trimmed.fq SRR693_6913_trimmed.sam
▲ CRITICAL STEP Here a ‘CCA’ sequence is added to the 3′ end of all tRNAs downloaded from GtRNAdb. Then the small RNA sequences are mapped to all mature mouse tRNA sequences.
The tRNA sequences and Bowtie indexes of human and mouse are also available at https://github.com/rnabioinfor/TRAC-Seq/tree/master/bowtie_index. During the mapping process, a maximum of three mismatches (-v 3) is allowed, and the best alignment of reads with no more than 50 hits (-a -m 50 --best --strata) are reported. mm10_tRNA is the name of the tRNA sequence index.
67 (Optional) If the output of the aligner is in SAM format, such as the SAM outputs from Bowtie25, BWA26, and HISAT29, convert SAM to BAM using Samtools (v.0.1.19).
$samtools view -bS SRR6936910_trimmed.sam > SRR6936910_trimmed.bam
$samtools view -bS SRR6936911_trimmed.sam > SRR6936911_trimmed.bam
$samtools view -bS SRR6936912_trimmed.sam > SRR6936912_trimmed.bam
$samtools view -bS SRR6936913_trimmed.sam > SRR6936913_trimmed.bam
68 Sort and index the BAM files using Samtools (v.0.1.19).
$samtools sort SRR6936910_trimmed.bam SRR6936910_trimmed.sort
$samtools index SRR6936910_trimmed.sort.bam
$samtools sort SRR6936911_trimmed.bam SRR6936911_trimmed.sort
$samtools index SRR6936911_trimmed.sort.bam
$samtools sort SRR6936912_trimmed.bam SRR6936912_trimmed.sort
$samtools index SRR6936912_trimmed.sort.bam
$samtools sort SRR6936913_trimmed.bam SRR6936913_trimmed.sort
$samtools index SRR6936913_trimmed.sort.bam
▲ CRITICAL STEP Here, the covered BAM files are further sorted and indexed to convert them to the input format required by the program cleavage_score.R in Step 70. The sorted and indexed files can also be used to visually check the cleavage peaks in the Integrative Genomics Viewer (IGV)30.
69 Process the alignments to record the read depth of each site on tRNAs using bedtools (v.2.27.1;ref. 31).
$bedtools genomecov -ibam SRR6936910_trimmed.sort.bam -bg > SRR6936910_trimmed.sort.bam.bg
$bedtools genomecov -ibam SRR6936911_trimmed.sort.bam -bg > SRR6936911_trimmed.sort.bam.bg
$bedtools genomecov -ibam SRR6936912_trimmed.sort.bam -bg > SRR6936912_trimmed.sort.bam.bg
$bedtools genomecov -ibam SRR6936913_trimmed.sort.bam -bg > SRR6936913_trimmed.sort.bam.bg
▲ CRITICAL STEP Make sure that the ‘.bg’ suffix is appended to the end of each BAM file name and put the output file in the same directory as the BAM files.
m7G site identification ● Timing ~30 min
70 Calculate the TRAC-seq cleavage score and cleavage score ratio between input and chemically treated sample using the program ‘cleavage_score.R’, which is available at https://github.com/rnabioinfor/TRAC-Seq (https://doi.org/10.5281/zenodo.2671795).
$Rscript cleavage_score.R mm10_tRNA.fa mm10_tRNA_structure.gff SRR6936910_trimmed.sort.bam SRR6936912_trimmed.sort.bam
$Rscript cleavage_score.R mm10_tRNA.fa mm10_tRNA_structure.gff SRR693 6911_trimmed.sort.bam SRR6936913_trimmed.sort.bam
▲ CRITICAL STEP The program depends on several R (v3.5.0 in this protocol) and Bioconductor packages, including data.table (v.1.20.0), cowplot (v.0.9.4), gridExtra (v.2.3), seqinr (v.3.4-5), GenomicAlignments (v.1.12.2), and GenomicFeatures (v.1.36.0). In addition, the program automatically recognizes the accompanying .bg coverage file for each input BAM file by searching files with a .bg extension in the same directory. The tRNA sequences and structure files for human and mouse (such as mm10_tRNA.fa and mm10_tRNA_structure.gff here) are available at https://github.com/rnabioinfor/TRAC-Seq/tree/master/bowtie_index. The outputs of this program include a tab-delimited text file (trimvalue.fc.lt6.list.txt in this protocol) containing the cleavage location on tRNA sequences, cleavage scores for both treat and non-treat samples, cleavage ratio between treat and non-treat samples and 7 nt (3 bp both upstream and downstream) around the cleavage sites. Meanwhile, the program also generates PDF files, which visually show the cleavage score distributions for each type of tRNA. Furthermore, a group of 21-nt sequences around the cleavage sites (10 bp both upstream and downstream) are generated, which can be used to perform motif analysis in Step 71. Here, the positions with a cleavage ratio >6 and cleavage score >0.1 in both non-treat (input) and chemically treated samples were considered to be the candidate m7G sites.
Motif search ● Timing ~30 min
71 Use the MEME tool32 to identify m7G site motifs based on the 21-bp sequences around m7G sites (10 bp both upstream and downstream).
$meme for_motif.fasta -mod zoops -dna -nmotifs 3 -maxw 7 -revcomp -o motif_out
for_motif.fasta is a .fasta format file for 21-bp sequences around m7G sites, which are generated in Step 70. The top three enriched motifs (sorted by the motif enrichment (E) values) with a maximum 7-bp width are reported in a HTML-format file in the output directory motif_out by MEME.
tRNA expression analysis ● Timing ~8 h
72 Use the programs based on the ARM-Seq data analysis pipeline12 to conduct tRNA expression analysis for input (non-treat) samples. The key files and parameters of the analysis are summarized in the table below.
$sh ARM-Seq/maketrnadb.bash mm10 mm10-tRNAs.out mm10.fa
$sh ARM-Seq/mapreads.bash output_prefix mm10 samplefile.txt 6
| Parameter or file | Function |
|---|---|
| mm10 | Name of constructed database |
| mm10-tRNAs.out | The output of the trnascan-SE software20 run on the genome, which is available in gtrnadb database for many species |
| mm10.fa | The reference genome obtained from the UCSC database |
| output_prefix | The name prefix of tRNA expression output |
| samplefile.txt | A sample information file, the example for which can be found at https://github.com/rnabioinfor/TRAC-Seq |
| 6 | The number of CPU cores used in the analysis |
By changing the parameters according to the above table, the script maketrnadb.bash is used to construct a sequence database, whereas the modified mapreads.bash script, which contains only mapping and counting steps (https://github.com/rnabioinfor/TRAC-Seq/tree/master/ARM-Seq), is used to perform expression analysis. Bowtie2 (ref. 22) is used as the mapping software in this step. The output file of tRNA read counts ‘output_prefix-counts.txt’ will be generated in current working directory.
73 To study the effects of tRNA modifications on tRNA stability, differential expression analysis can be then performed based on the output count file in Step 72 by using DESeq2 (https://bioconductor.org/packages/release/bioc/html/DESeq2.html).
Troubleshooting
Troubleshooting advice can be found in Table 1.
Table 1 |.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 15 | RNA concentration is low (<1% of input RNA) | RNA was degraded | Repeat RNA purification. Before starting, clean your work area, pipettes, and gloves with RNaseZap RNase decontamination solution |
| RNA was not fully eluted from the column | Repeat elution, making sure the elution solution is preheated to 95 °C | ||
| RNA size is not correct | Ethanol ratio was not correct | Add correct volume of ethanol to ensure that small RNAs are precipitated | |
| 31 | Multiple protein bands in the SDS-PAGE gel | Recombinant proteins were degraded | Induce recombinant protein expression at a lower temperature |
| Nonspecific protein binding to beads | Increase washing duration and perform more washes | ||
| 47 | Few (<1%) reads map to tRNA | Demethylation was not successful | Repeat recombinant protein purification |
| Add more recombinant protein in the demethylation reaction | |||
| 51 | No specific cleavage detected | Reduction efficiency was low | Use freshly made NaBH4 buffer |
| Cleavage efficiency was low | Increase aniline treatment time |
Timing
Steps 1–15 (day 1), isolation of small RNAs: 4 h
Step 16 (day 1), plasmid transformation: 1 h
Steps 17–31 (days 2–6), purification of recombinant protein expression and dialysis: 4 d
Steps 32–51 (day 6), RNA demethylation, reduction and cleavage: 8 h
Steps 52–61 (day 7), small RNA sequencing library construction: ~1 d
Steps 62–73 (day 8), data analysis: ~2 d
Anticipated results
Small RNA isolation
The mirVana miRNA Isolation Kit enables the efficient isolation of small RNAs from total RNA samples (Steps 1–15, Fig. 3). The yield of small RNAs is ~10% of total RNA.
AlkB and AlkB-D135S purification
SDS-PAGE analysis of the recombinant proteins purified from Steps 17 to 31 revealed the strong induction and high purity of the recombinant AlkB and AlkB-D135S proteins (Fig. 4).
NaBH4/aniline-mediated reduction and cleavage of m7G sites
The NaBH4/aniline treatment (Steps 32–51) results in specific cleavage of the m7G site in tRNA. Northern blotting analysis using the tRNA LysCTT probe reveals a cleaved product of smaller size (Fig. 5).
Small RNA sequencing library construction
The size of the small RNA library is ~140–210 bp (Fig. 6). If the library contains adaptor dimers (127-bp peak) or excess primers (70–80 bp), we recommend using 6% polyacrylamide gel to perform size selection by following the instructions from the NEBNext Multiplex Small RNA Library Prep Set.
Quality control
The sequence reads should have an average quality above Q20 and there should be no adaptor sequences detected. FastQC reports for the cleaned test data in this protocol are available at https://github.com/rnabioinfor/TRAC-Seq/tree/master/quality_control_trac_seq.
Read mapping and cleavage score calculation
The mapping results are in BAM format, sorted and indexed by Samtools (v.0.1.19), which can be used to calculate the read depth for each location on the tRNA. The read depth files (.bg files) generated in Step 69 should be put in the same directory as the other BAM files. The BAM files sorted and indexed by Samtools can be used to visually show the cleavage site peaks on tRNAs in the IGV software30. NaBH4/aniline treatment results in specific cleavage of RNA at m7G sites; mapping and visualization of the reads reveals a specific enrichment of reads initiating from the m7G + 1 site (Fig. 7a) and a sharp increase of cleavage score (Fig. 7b) in the WT R1/E cells. Knockout (KO) of the tRNA m7G methyltransferase Mettl1 results in depletion of the m7G modification; therefore, after NaBH4/aniline treatment, there is no m7G-dependent cleavage in the KO cells.
Fig. 7 |. Anticipated result of TRAC-Seq.
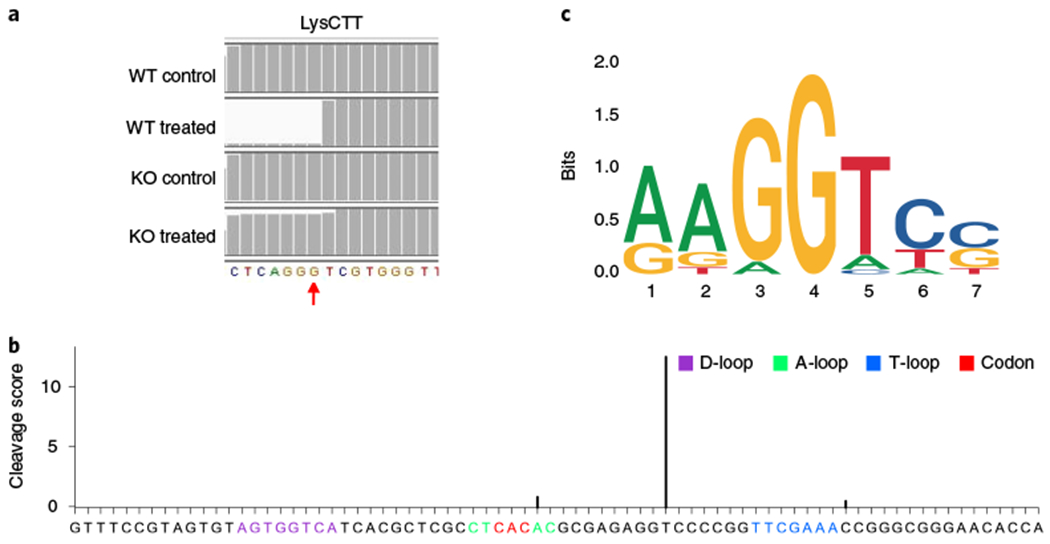
a, A representative image of read alignment to LysCTT tRNA in the IGV using m7G TRAC-Seq data from R1/E mESCs. Red arrow: m7G site. b, Representative cleavage scores calculated by program ‘cleavage_score.R’ at individual sites on ValTAC tRNA. c, Sequence motif identified surrounding m7G sites using MEME. Control, AlkB-demethylated samples; treated, AlkB-demethylated and chemically treated samples.
m7G sites and motif identification
We previously revealed that a subset of 22 tRNAs with a ‘RAGGU’ motif within the variable loop are modified by m7G in mESCs10 (Fig. 7c). MEME should always report RAGGU-like motifs around the m7G sites identified by the users. There should be no cleavage peaks in the tRNAs isolated from the Mettl1 KO mESCs.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The TRAC-Seq data were deposited into the Gene Expression Omnibus database (GEO accession no. GSE112670).
Code availability
The TRAC-seq data analysis source code is available via GitHub (https://github.com/rnabioinfor/ TRAC-Seq, https://doi.org/10.5281/zenodo.2671795) and is for research purposes only.
Supplementary Material
Acknowledgements
S.L. was supported by grants from the National Natural Science Foundation of China (81772999), the Guangzhou People’s Livelihood Science and Technology Project (201903010006), and a Young Investigator grant from the Alex’s Lemonade Stand Foundation (GR-000000296). R.I.G. was supported by grants from the US National Institute of General Medical Sciences (R01GM086386) and the National Institute of Mental Health (R21MH118594).
Footnotes
Supplementary information is available for this paper at https://doi.org/10.1038/s41596-019-0226-7.
Competing interests
The authors declare no competing interests.
References
- 1.Lyons SM, Fay MM & Ivanov P, The role of RNA modifications in the regulation of tRNA cleavage. FEBS Lett. 592, 2828–2844 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sokolowski M, Klassen R, Bruch A, Schaffrath R & Glatt S, Cooperativity between different tRNA modifications and their modification pathways. Biochim Biophys. Acta Gene Regul. Mech 1861, 409–418 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Kawarada L et al. ALKBH1 is an RNA dioxygenase responsible for cytoplasmic and mitochondrial tRNA modifications. Nucleic Acids Res. 45, 7401–7415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Torres AG, Batlle E & Ribas de Pouplana L Role of tRNA modifications in human diseases. Trends Mol. Med 20, 306–314 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Dominissini D, Moshitch-Moshkovitz S, Salmon-Divon M, Amariglio N & Rechavi G Transcriptome-wide mapping of N(6)-methyladenosine by m(6)A-seq based on immunocapturing and massively parallel sequencing. Nat. Protoc 8, 176–189 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Lin S, Choe J, Du P, Triboulet R & Gregory RI The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell 62, 335–345 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choe J et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature 561, 556–560 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boccaletto P et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 46, D303–D307 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shaheen R et al. Mutation in WDR4 impairs tRNA m(7)G46 methylation and causes a distinct form of microcephalic primordial dwarfism. Genome Biol. 16, 210 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin S et al. Mettl1/Wdr4-mediated m(7)G tRNA methylome is required for normal mRNA translation and embryonic stem cell self-renewal and differentiation. Mol. Cell 71, 244–255.e245 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng G et al. Efficient and quantitative high-throughput tRNA sequencing. Nat. Methods 12, 835–837 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cozen AE et al. ARM-seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat. Methods 12, 879–884 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zueva VS, Mankin AS, Bogdanov AA & Baratova LA Specific fragmentation of tRNA and rRNA at a 7-methylguanine residue in the presence of methylated carrier RNA. Eur. J. Biochem 146, 679–687 (1985). [DOI] [PubMed] [Google Scholar]
- 14.Wintermeyer W & Zachau HG Tertiary structure interactions of 7-methylguanosine in yeast tRNA Phe as studied by borohydride reduction. FEBS Lett. 58, 306–309 (1975). [DOI] [PubMed] [Google Scholar]
- 15.Kellner S, Burhenne J & Helm M Detection of RNA modifications. RNA Biol. 7, 237–247 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Helm M & Motorin Y Detecting RNA modifications in the epitranscriptome: predict and validate. Nat. Rev. Genet 18, 275–291 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Marchand V et al. AlkAniline-Seq: profiling of m(7) G and m(3) C RNA modifications at single nucleotide resolution. Angew. Chem 57, 16785–16790 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Schwartz S et al. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 159, 148–162 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bailey TL & Elkan C Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol 2, 28–36 (1994). [PubMed] [Google Scholar]
- 20.Lowe TM & Eddy SR tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langmead B Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinforma 32, 11.7.1–14 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langmead B, Trapnell C, Pop M & Salzberg SL Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H & Durbin R Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trapnell C, Pachter L & Salzberg SL TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim D, Langmead B & Salzberg SL HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robinson JT et al. Integrative genomics viewer. Nat. Biotechnol 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quinlan AR BEDTools: the Swiss-army tool for genome feature analysis. Curr. Protoc. Bioinforma. 47, 11.12.1–34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bailey TL et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The TRAC-Seq data were deposited into the Gene Expression Omnibus database (GEO accession no. GSE112670).