

Abstract
Objectives:
Obesity and diabetes are associated with an increased incidence of pancreatic cancer. Fatty acid binding protein 4 (FABP4), noted to be higher in patients with severe obesity, is linked to the development and progression of several cancers, and its level in circulation decreases after bariatric surgery. In this paper, we evaluated the role of FABP4 in pancreatic cancer progression.
Methods and results:
When Panc-1 (human) and Pan02 (Mouse) pancreatic cancer cells were treated with FABP4 or R126Q FABP4 (fatty acid binding site mutant), only FABP4 stimulated cellular proliferation. The transcriptional activity of nuclear factor E2-related factor 2 (NRF2) was increased in response to FABP4, but not the R126Q. FABP4 treatment also leaded to downregulation of reactive oxygen species (ROS) activity. Consistent with induced cell propagation by FABP4, the tumor growth of Pan02 tumor was decreased in FABP4 null animals compared to C57BL/6J controls.
Conclusions:
These results suggest that FABP4 increases pancreatic cancer proliferation via activation of Nrf2 and downregulation of ROS activity.
Keywords: Fatty acid binding protein 4, FABP4, pancreatic cancer, Nrf2, cell proliferation
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the third leading cause of cancer related death, with 5-year survival of PDAC estimated at 9% in the United States(1). Most patients with PDAC are diagnosed with metastatic lesions, leaving less than 20% as candidates for surgical resection. Risk factors for PDAC include age, sex, gene mutations, cigarette smoking, obesity, chronic pancreatitis, and diabetes(2).
Recently, obesity has been implicated in the pathogenesis of a variety of cancers. The diagnosis of obesity is associated with not only diagnosis of cancer but also increased hazard of cancer related death, secondary to more advanced stages upon diagnosis(3,4). Fatty infiltration of the pancreas in obesity has been associated with increased inflammation and cancer development(5,6). Further, the incidence of PDAC is associated with obesity and often associated with new onset type 2 diabetes(T2DM), although causality has not been clearly established(7).
Excess triglyceride in obesity leads to adipocyte hypertrophy cell hypoxia and macrophage infiltration into pancreatic tissue(7,8). In this setting, adipokines including leptin, tumor necrosis factor-alpha (TNF-α), interleukins, and monocyte chemoattractant proteins are secreted from both the adipocyte fraction and the stromal-vascular fraction in pancreatic adipose tissue, potentiating inflammation. It is suggested that increased levels of adipokines, altered gut microbiota, changes in bile acid composition, and inflammation are involved in PDAC progression(8,9). The link between obesity and PDAC is not sufficiently elucidated. Studies that clarify the connection of obesity and PDAC might provide important information regarding the pathogenesis of PDAC.
Fatty acid binding proteins are water soluble and small proteins that can affect lipid fluxes, metabolism and signaling within cells. Twelve fatty acid binding proteins family members have been identified, and fatty acid binding protein 4 (FABP4, alternatively called adipocyte protein 2, aP2) has been suggested as a molecule of interest (10). FABP4 is a lipid chaperone that facilitates fatty acid flux as well as an intracellular stabilizer of hydrophobic lipid in hydrophilic cytosol; it is notably the most abundant protein in mature adipocytes(11). Higher levels of circulating FABP4 have been reported in obese subjects, and levels correlates with BMI, metabolic syndrome, and inflammatory markers(12). Despite its first description as a fatty acid chaperone, it has been implicated in a diverse array of cellular processes in addition to simple lipid shuttling function(11). FABP4 has also been linked to the development and progression of a various cancers(10), most recently in breast cancer. Higher intracellular levels of FABP4 in breast cancer specimens is significantly correlated with recurrence, and has demonstrated a negative correlation with disease-free survival(13). Mechanistic studies have shown increased proliferation, invasiveness, and stemness of breast cancer cells in response to exogenous FABP4 treatment(11,14). Additionally, FABP4 is robustly expressed in macrophages, and indirectly promoting cancer progression by altering matrix metalloproteinase (MMP) activity and cytokines from macrophages(15). Tumor progression is significantly decreased in whole body and macrophage specific FABP4 knockout models(10,16).
Bariatric (weight loss) surgery has been associated with reduction in obesity related cancers. and clinical studies suggest that bariatric surgery has also been associated with a protective effect for PDAC development, progression, and mortality (17). We have observed that FABP4 is reduced 12 months after Roux-en-Y gastric bypass surgery but not after intensive lifestyle modification and medical management protocol in patients with T2DM(18). In PDAC patients, higher FABP4 expression in PDAC tumor measured by immunohistochemistry is associated with both disease progression and poor prognosis(19). The transcriptional regulation in the downstream of FABP4 has been linked to cellular redox status, with Nrf2 being an upstream regulator of this balance(20). Alterations in redox signaling are thought to play an important role in the deleterious inflammatory and metabolic effects of FABP4 in both macrophages and adipose tissue(21,22), but a link between FABP4 and PDAC cells has not yet been elucidated. In this study, we investigate the direct effect of FABP4 on PDAC cell proliferation and redox homeostasis, as well as in vivo tumor growth.
Materials and Methods
Cell culture and proliferation analysis
Panc-1 (ATCC CRL-1469, ATCC, Manassas, VA) and Pan02 (DTP, DCTD Tumor Repository, National Cancer Institute, Frederick, MD) cells were maintained in DMEM with 10% FBS. All cells were cultured at 37 ℃ and 5% CO2. Cell suspensions were added to each well (6-well plate, 2×105 cells in 1ml medium/well). Cells were incubated in serum-starved media containing 0.1% FBS and 2uL of insulin/mL of media. in serum-starved media containing 0.1% FBS and 2uL of insulin/mL of media. After 24 hours of culture, the baseline cell counts performed utilizing automated cell counter (Countess, Thermo Fisher). Media was replaced the same composition media containing recombinant FABP4 (20, 40 and100 ng/mL), R126Q (R- single point mutant FABP4, 100ng/mL), or vehicle solvent at the same volume. Cell counts were then performed at time points 24hr and 48hr.
Cell cycle analysis
Panc-1 and Pan02 cells were seeded into 100mm dishes and cultured with phosphate-buffered saline (PBS) as a vehicle control or FABP4 100 ng/ml for 48 h. After incubation, the cells were fixed with 70 % ethanol and stored overnight at −20 °C. The cells were washed and then stained with a solution containing 0.1 % Triton® X-100 (Promega), 0.02 mg/ml propidium iodide (PI; Sigma-Aldrich, St. Louis, MO, USA), and 0.2 mg/ml RNase A (Qiagen, Hilden, Germany) in the dark at 37 °C for 15 min. After staining, the cells were subjected to cellular DNA content examination by a flow cytometer (BD FACS Canto II: BD Biosciences). The data were analyzed by FlowJo software.
Apoptosis analysis by flow cytometry
After harvesting, apoptosis was evaluated with an apoptosis detection kit (Annexin V Apoptosis Detection Kit APC, eBioscience, San Diego, CA, USA) according to the manufacturer’s instructions. After staining, the cells were examined using a flow cytometer (BD FACS Canto II: BD Biosciences). The data were analyzed by FlowJo software.
Immunoblot analysis
Cells were lysed with radio-immunoprecipitation assay (RIPA) buffer supplemented with protease inhibitors (Calbiochem, San Diego, CA). 30 μg of protein from each sample were separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene fluoride (PVDF) membrane. After blocking with Odyssey blocking buffer (Li-Cor Biosciences, Lincoln, NE), membranes were incubated with primary antibody overnight at 4°C. Membranes were washed and incubated with secondary antibody conjugated to Li-Cor IRDye for 1h and visualized using Odyssey infrared imaging (Li-Cor Biosciences). The primary antibodies used were FABP4 (23) and rabbit monoclonal antibody against beta-actin (#4970; Cell Signaling Technology, Danvers, MA)
Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) Reporter Assay
The antioxidant response element (ARE) luciferase reporter vector and constitutively active Renilla luciferase vector (BPS Bioscience, Madison, WI) were transfected into Panc-1 and Pan02 cells utilizing Lipofectamine (Invitrogen, Waltham, MA). After 24 hours, the media was replaced with media containing 100 ng/mL FABP4. After another 24 hours of incubation, dual luciferase reporter assay (Promega, Madison, WI) was performed utilizing a luminometer per the manufacturer’s instructions, and ARE reporter activity was normalized to Renilla activity.
Reactive Oxygen Species (ROS) Analysis
Hydrogen peroxide level was assayed utilizing an Amplex Red hydrogen peroxide/peroxidase assay kit (Invitrogen, Carlsbad, CA), per manufacturer’s instructions.
Mice
All experimental procedures using animals were approved by the University of Minnesota Institutional Animal Care and Use Committee (IACUC). Whole body FABP4 null (AKO) mice(24), C57BL/6J wild type (WT) and hetero type (Het) mice were bred and maintained in the animal facility of the University of Minnesota with authorized protocol from the IACUC. Mice were fed ad libitum a high-saturated-fat (lard) diet (F3282; BioServe, Flemington, NJ) for 12 weeks after weaning. At week 12–14 of high fat diet, mice were injected with Pan02 cells (5×106) into the flank to assess the effect of FABP4 in the syngeneic tumor model in mice. Tumor volume was measured three times weekly with calipers. Mice were euthanized at endpoints of >2cm3, tumor ulceration, metastases, or the end of study period at 26 days. The mice whose tumor had ulcerations before the endpoint were sacrificed due to IACUC protocol.
Statistical analysis
Values are reported as mean ±standard deviation (SD). Differences were compared using a Student’s t-test or analysis of variance (ANOVA) with Sidak’s post hoc multiple comparisons test. Survival was evaluating utilizing Kaplan-Meier method and log-rank tests. Above statistical analyses were performed using GraphPad Prism version 7.0 (GraphPad Software, La Jolla, California, USA).
Results
FABP4 increased pancreatic cancer cell proliferation.
When we assessed the effect of the exogenous FABP4 on pancreatic cancer cell proliferation, exogenous FABP4 in the culture medium (100ng/mL) resulted in increased Panc-1 and Pan02 cell proliferation while 20 and 40 ng/ml did not increase the proliferation. (Fig. 1A and B). We then utilized an FABP4 mutant at the lipid binding site (R126Q) to evaluate the contribution of FABP4’s lipid binding to the increase in cell proliferation in Panc1 cells. The single residue substitution of arginine for glutamine (R126Q) was reported to alter the hydrophobic cavity of FABP4 in a manner hindering its ability to bind free fatty acids while maintaining the overall protein structure of FABP4(25). Cell proliferation increased only with wild type recombinant FABP4 and was not affected by treatment with mutant R126Q (Fig. 1C). This indicated that extrinsic FABP4 enhanced PDAC cell proliferation, and the free fatty acid binding site was critical for this function.
Figure 1. FABP4 increased the proliferation of pancreatic cancer cells.
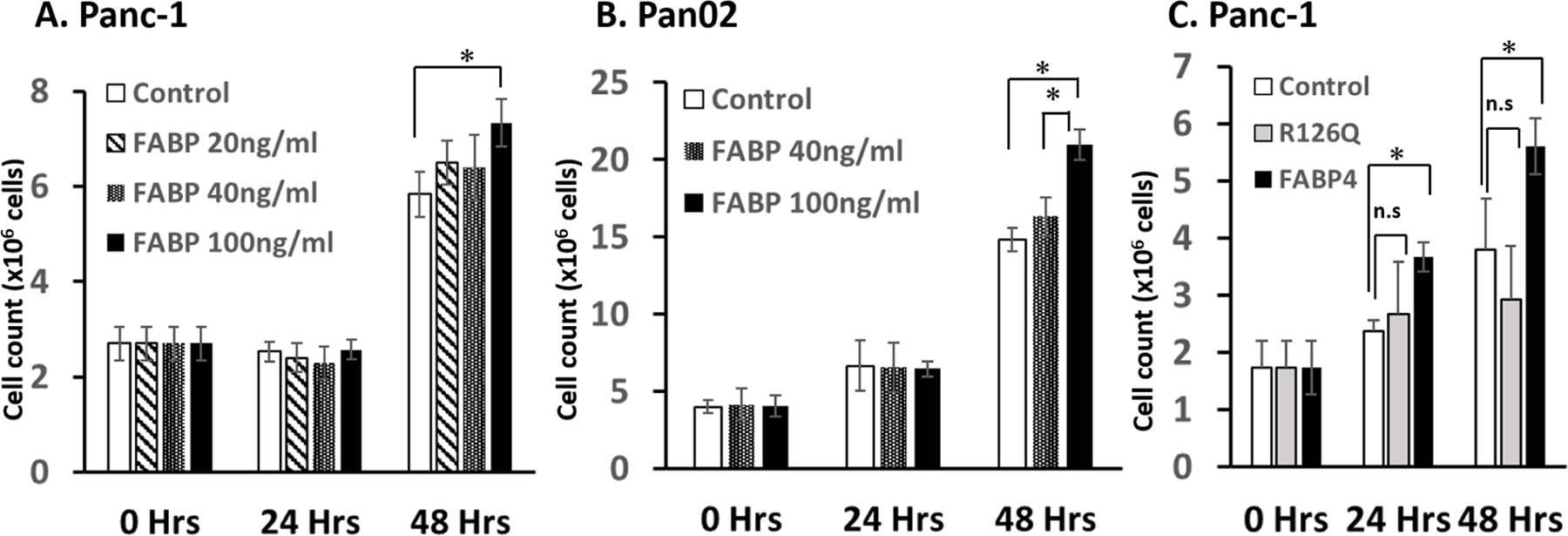
A) Effect of FABP4 on Panc-1 proliferation. Extrinsically added 100 ng/ml FABP4 increased the proliferation of Panc-1 cells while 20 and 40 ng/ml did not increase the proliferation. B) Effect of incubation with FABP4 on Pan02 proliferation. 100 ng/ml FABP4 increased the proliferation of Pan02 cells. C) Effect of FABP4 or R126Q (R- single point mutant FABP4) on Panc-1 proliferation. Extrinsically added R126Q did not increase the proliferation. Results are presented as mean ± SD (n = 3). (n.s.: not significant, *:P < 0.05)
FABP4 facilitated the entry of G1 phase cells into the S and G2 phase.
To explore how FABP4 upregulate the cell proliferation, Panc-1 and Pan02 were incubated with either the vehicle control (PBS) or 100 ng/ml FABP4 48h, and the cell cycle was examined with flow cytometry using PI staining. The analyzed results are shown in Fig. 2A. The percentage of G1 phase cells are shown in Fig. 2B and 2C. In both cell lines, the percentage of G1 phase cells incubated with 100 ng/ml concentration of FABP4 was decreased. These results demonstrated that 100 ng/ml FABP4 promoted the entry of G1 phase cells into the S and G2 phase.
Figure 2. FABP4 promoted the entry of G1 phase cells into the S and G2 phase.
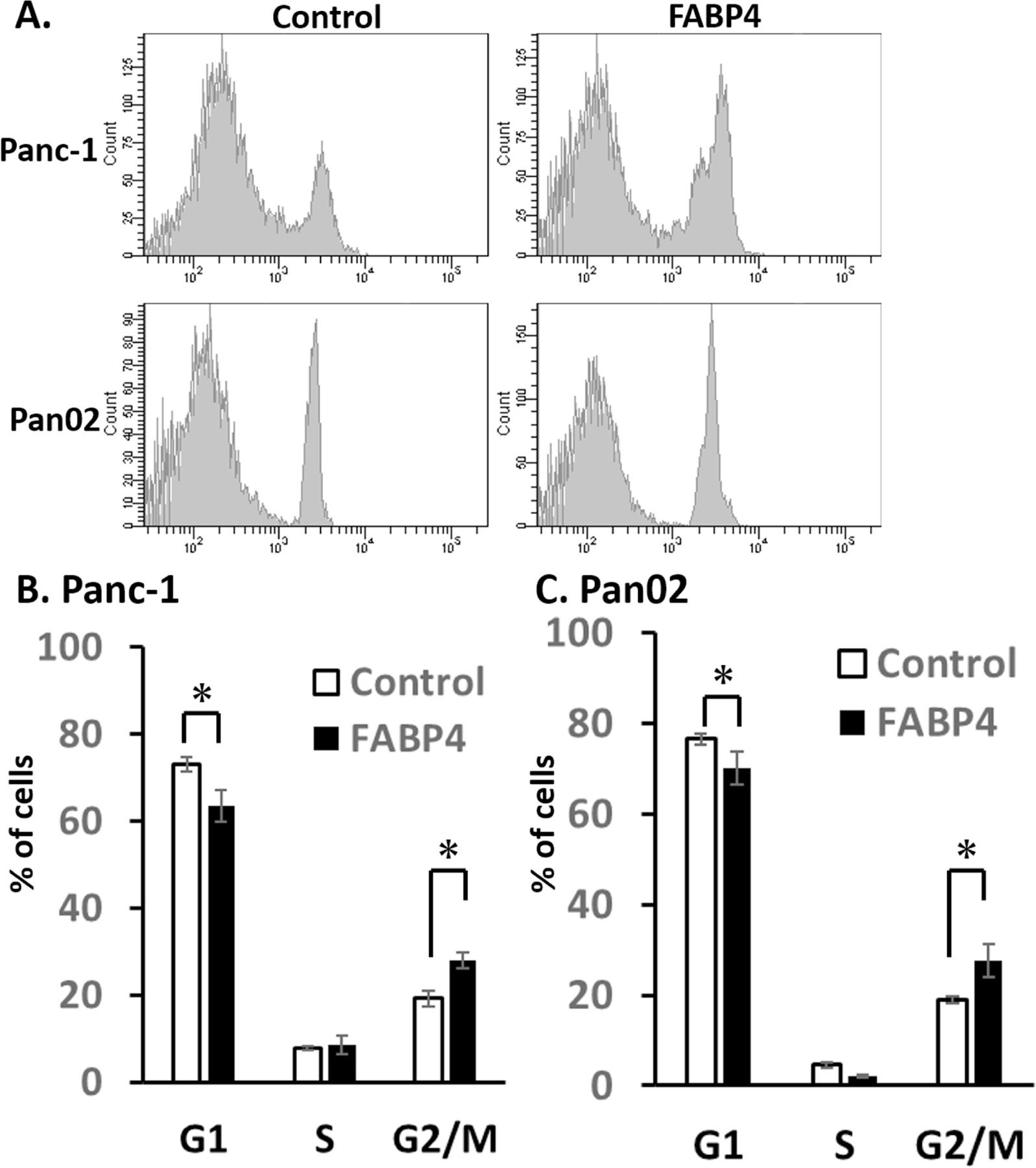
A) Effect of FABP4 on Panc-1 and Pan02 cell cycle. Panc-1 and Pan02 cells were incubated with either the vehicle control (PBS) or 100 ng/ml FABP4 48h, and the cell cycle was examined with flow cytometry using PI staining. B) The percentage of G1 phase Panc-1 cells are shown. In FABP4 group, the percentage of G1 phase cells was decreased. C) The percentage of G1 phase Pan02 cells are shown. Exogenous FABP4 decreased G1 phase Pan02 cells. Results are presented as mean ± SD (n = 3). (*:P < 0.05)
FABP4 did not induce apoptosis in pancreatic cancer cell lines.
To further characterize the mechanisms of FABP4, apoptosis was examined by flow cytometry using PI and Annexin V staining. The results are shown in Fig. 3A. The number of live cells was defined as the number of cells negative for both Annexin V and PI. The number of cells in early apoptosis was defined as cells positive for Annexin V only, whereas late apoptosis was defined as cells positive for both Annexin V and PI. The number of necrotic cells was defined as the cells negative for Annexin V but positive for PI. The percentages of apoptotic cells are shown in Fig. 3B. FABP4 slightly lowered apoptotic cells, however this was not significant. These results demonstrated that reduction of apoptosis was not the main reason of higher proliferation in pancreatic cancer cell line with FABP4.
Figure 3. FABP4 did not cause apoptosis in pancreatic cancer cell lines.
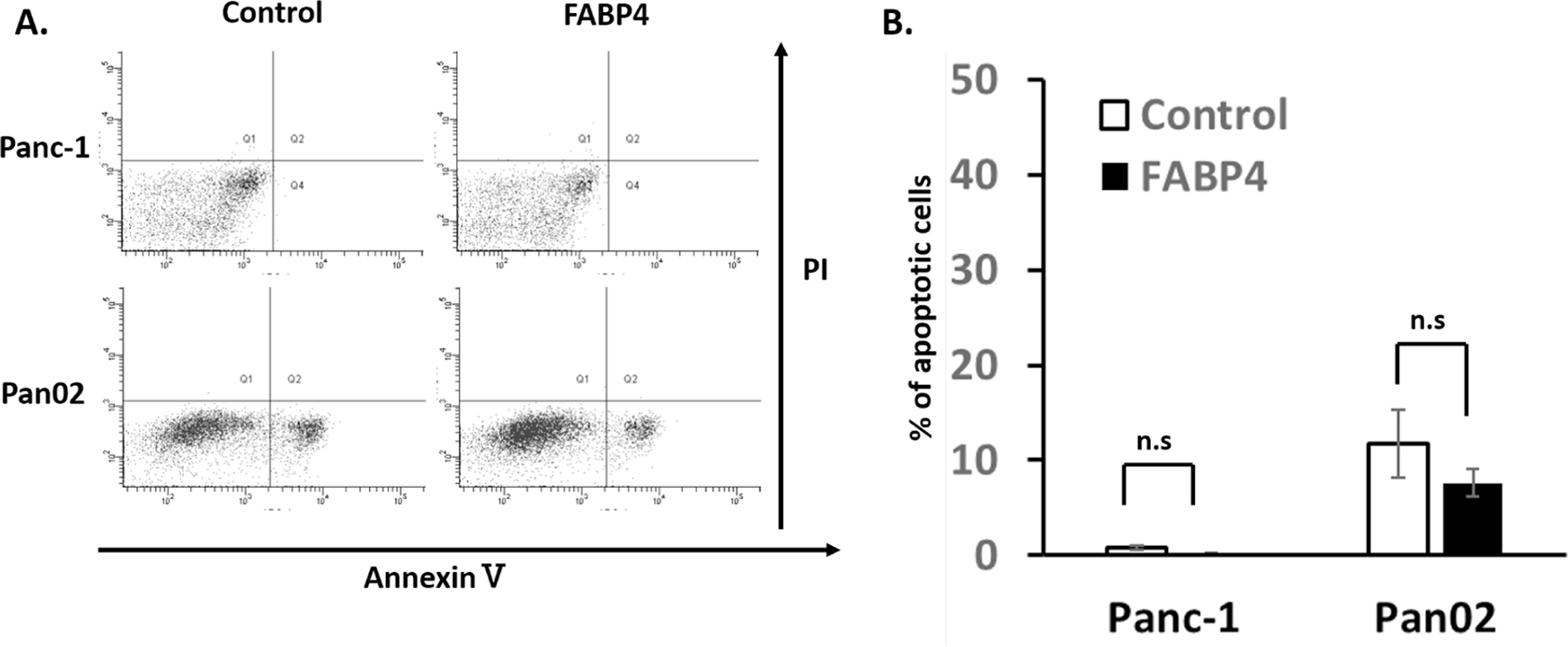
A) Effect of FABP4 on Panc-1 and Pan02 cell apoptosis. Cells were incubated with the vehicle control (PBS) or FABP4 for 48 h. Cells were stained with Annexin V/PI and examined by flow cytometry. B) It was not statistically significant, but FABP4 demonstrated lower population of apoptotic cells. The percentages of live and apoptotic cells are presented as mean ± SD (n = 3). (n.s.: not significant)
Exogenous FABP4 increased endogenous FABP4 protein expression.
Endogenous (intracellular) FABP4 expression was assessed after addition of FABP4 extracellularly since the different role of overexpressed has been suggested(15). Endogenous FABP4 protein levels in Panc-1 (Fig. 4A, B and Supplemental Fig. 1) and Pan02 cells (Fig. 4A, C and Supplemental Fig. 2) were assessed by western blot. In both cell lines, the endogenous FABP4 protein expression levels were significantly upregulated in the cells treated with exogenous FABP4.
Figure 4. Exogenous FABP4 increased endogenous FABP4 protein expression.
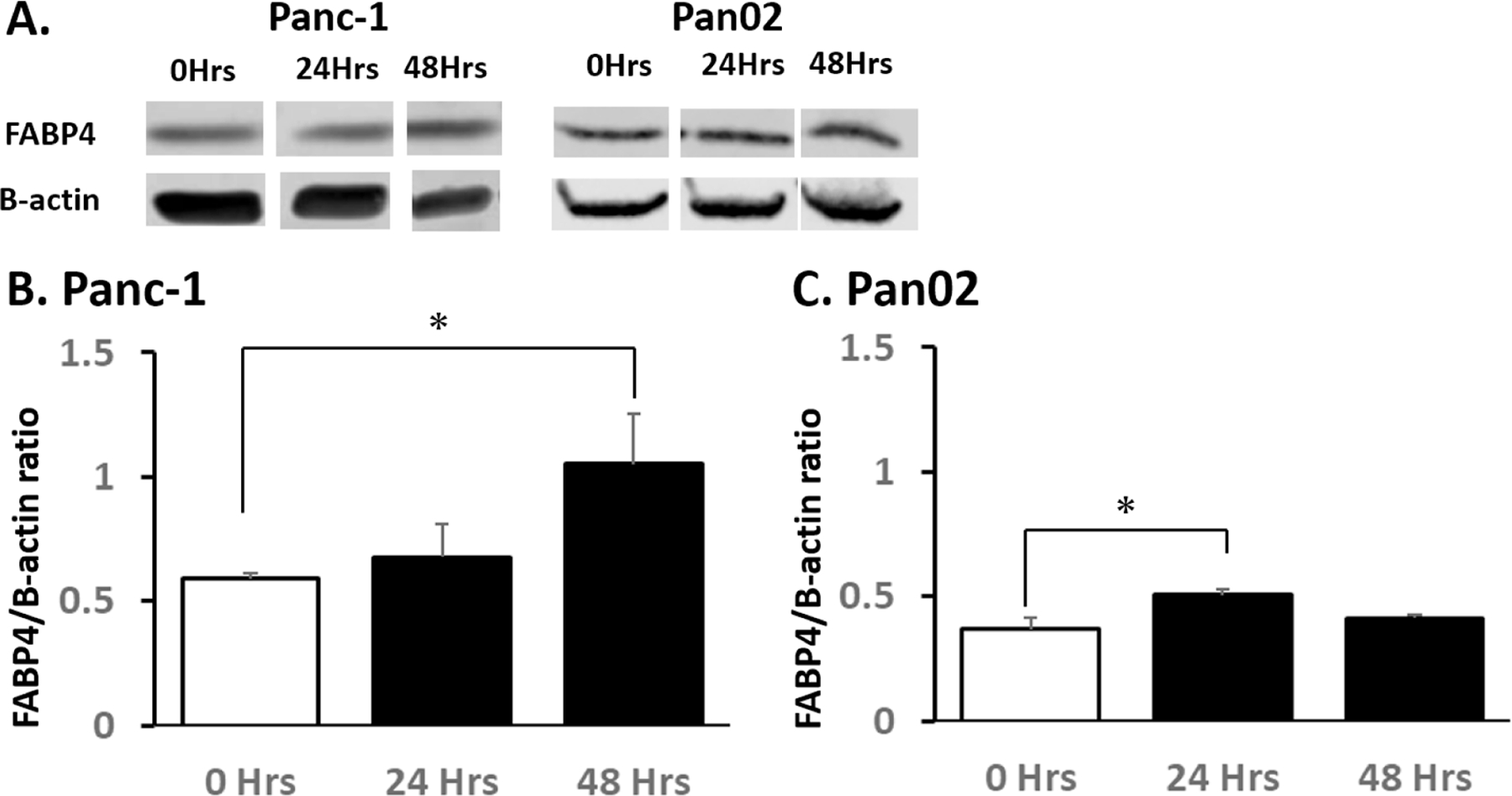
A) Expression FABP4 and β-actin in Panc-1 and Pan02 cells following addition of 100 ng/ml FABP4 for 24 hours and 48 hours. B) The quantitation of protein band intensity shown as the FABP4/B-actin ratio in Panc-1 cells following addition of 100 ng/ml FABP4. Endogenous FABP4 expression levels of Panc-1 were upregulated in the cells 48 hours after FABP4 treatment. C) The average protein band intensity of the FABP4/B-actin in Pan02 cells following addition of 100 ng/ml FABP4. Endogenous FABP4 expression levels of Pan02 were upregulated in the cells 24 hours after FABP4 addition. Results are presented as mean ± SD (n = 3). (*: P < 0.05)
FABP4 treatment increased Nrf2 Activity, downregulated reactive oxygen species.
One of the major downstream changes of FABP4 is the cellular redox status(20), with the NRF2/ARE axis as the major regulatory pathway controlling ROS(26). Nrf2 activity measured by ARE reporter assay was significantly increased with exogenous FABP4 treatment, and was not affected by treatment with mutant R126Q (Fig. 5A and B). This data suggested that FABP4 treatment regulated NRF2 activity. This increase in Nrf2 is concurrently associated with a significant downregulation of ROS activity, as measured by Amplex Red hydrogen peroxide/peroxidase assay (Fig. 5C and D). This experiment indicated that Nrf2 increased only with wild type recombinant FABP4, not with mutant R126Q and that activation of Nrf2 activation significantly decreased the level of ROS.
Figure 5. FABP4 treatment increased Nrf2 Activity and downregulated reactive oxygen species.
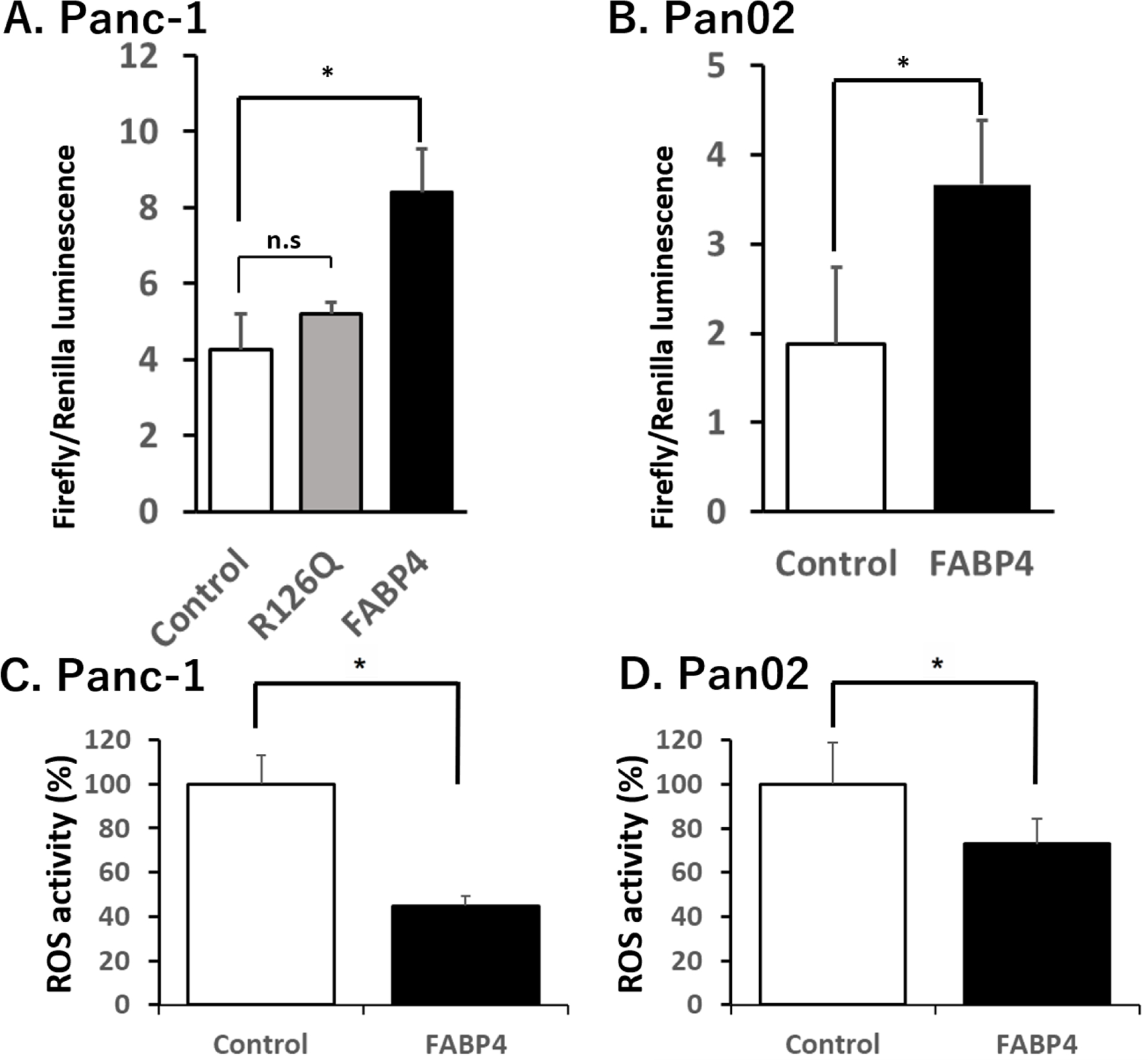
A) Extrinsically added FABP4 increased Nrf2 activity of Panc-1 cells while R126Q did not affect the Nrf2 activity. Panc-1 cells were treated with 100 ng/ml FABP4 or 100 ng/ml R126Q for 24 hours and the Nrf2 activity evaluated using the antioxidant response element (ARE) luciferase reporter gene system. B) Exogenous FABP4 increased Nrf2 activity of Pan02 cells. Pan02 cells were treated with 100 ng/ml FABP4 for 24 hours and the Nrf2 activity evaluated using the ARE luciferase reporter gene system. C) Exogenous FABP4 decreased ROS activity of Panc-1 cells. The Panc-1 cells treated with 100 ng/ml FABP4 showed suppression of ROS level after 48 hours. D) Exogenous FABP4 decreased ROS activity of Pan02 cells. Pan02 cells treated with 100 ng/ml FABP4 for 48 hours also showed reduction of ROS production. Results are presented as mean ± SD (n = 3). (n.s.: not significant, *: P < 0.05)
Host FABP4 knockout decreased in vivo tumor growth
To assess the in vivo effect of FABP4 in the syngeneic tumor model in mice, Pan02 cells were injected subcutaneously into the backs of the AKO, WT and Het mice. The average tumor volumes of the AKO mice and WT/Het mice were 35.2 ± 38.5 and 94.2 ± 94.78 mm3, respectively at Day 26. In a C57BL/6J high fat diet mouse model, growth of the murine pancreatic cancer cell line Pan02 was significantly decreased in the AKO mice compared to growth in WT/Het mice (Fig. 6A). The body weight of AKO mice has no significant difference with the body weight of WT/Het mice (Fig. 6B). These results suggested that exogenous FABP4 was critical for the tumor growth in the syngeneic mouse PDAC model.
Figure 6. Suppression of mouse syngeneic subcutaneous tumor volume in FABP4 knockout (AKO) mice.
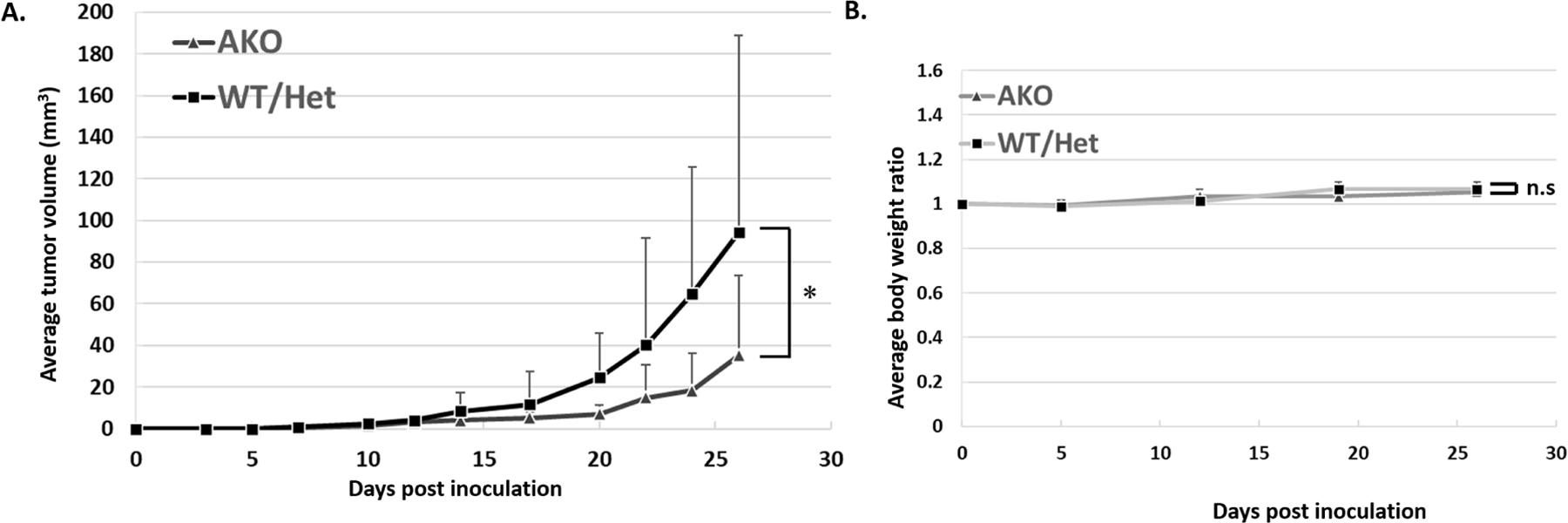
To assess the effect of FABP4 in syngeneic immunocompetent model in mice, Pan02 cells were injected subcutaneously into the backs of the AKO mice and C57BL/6J wild type (WT) and heterozygous (Het) mice which were maintained with high fat diet. The average tumor volumes of the AKO mice and WT/Het mice were 35.2 ± 38.5 and 94.2 ± 94.78 mm3, respectively at Day26. AKO mice significantly showed a smaller tumor growth. The cases with early stage ulceration due to the inoculation failure were excluded. B) The body weight of AKO mice has no significant difference with the body weight of WT/Het mice. Data are presented as mean ± SD. (AKO: N=10, WT: N=18, *:P<0.05)
Discussion
Obesity and diabetes have been reported as the factors corelating with cancer incidence and decreased overall survival(7). Potential mechanisms linking these factors have mainly evaluated alternative metabolic pathways and reprogramming, such as alterations of glucose and glutamine metabolism, i.e. the Warburg effect(25). Of interest here, high fat diet induced obesity in mouse models has been correlated with an increase of peri-pancreatic fat mass, and increased inflammation in the KRASG12D murine model of PDAC(27). This, along with data outlined earlier, highlights the importance of further delineating the role of fatty acid metabolism in cancer pathogenesis(28).
Dysregulation of FABP4 is associated with several disease states in human including obesity, diabetes and insulin resistance. FABP4 released from adipose tissue is also reported to act on adjacent or distant organs to alter cell functions and metabolic status as an adipokine-like paracrine and endocrine signaling molecule(29). Clinical evidence has also demonstrated the potential role of FABP4 in progression of diseases in some cancers. For example, ovarian cancer patients with high FABP4 expression shows significantly shortened overall and progression-free survival(30). While the correlation between FABP4 and cancer progression may be casual as both of them can be induced by obesity, mounting evidence of direct involvement of FABP4 to cancer progression has been reported recently(31). FABP4 may be involved in transporting FAs to cancer cells to facilitate tumor growth(32). FABP4 can influence both tumor cell proliferation and in vivo tumor progression(16,33). It should be noted, however, this data has only showed an indirect effect via macrophages, and direct effect of FABP4 on cancer cell is not clear. Further, the role of overexpressed endogenous (intracellular) FABP4 is also unclear.
In this paper, we provided novel evidence that FABP4 can influence tumor cell proliferation directly. We also indicated that exogenous FABP4 significantly upregulated the endogenous FABP4 protein expression levels in PDAC cells. This data is consistent with that from hepatocellular carcinoma in vitro models, demonstrating increased proliferation when challenged with exogenously administered FABP4(34). The endogenous FABP4 protein expression levels were upregulated by adding the exogenous FABP4 in our study. However, 100 ng/ml FABP4 is higher than the physiological range of serum FABP4 level, and the difference between control group and FABP4 treated group is not so big, especially in Pan02 cells. This indicated that there might be other factors, and further research is needed.
Clinically, we have already reported that FABP4 is reduced after Roux-en-Y gastric bypass surgery in patients with type 2 diabetes mellitus(19). Taking these results into consideration, bariatric surgery that can efficiently lower the circulating FABP4 level could be a promising approach to fight against the cancer progression, particularly in the patients with high FAPB4 in circulation.
The transcriptional regulation in the downstream of FABP4 is known to be linked to cellular redox status, with Nrf2 being an upstream regulator of this balance(20). Nrf2, one of the most critical defense mechanisms against oxidative stress, is known to promote the cell proliferation. A major mechanism by which Nrf2 controls cell proliferation and differentiation is the regulation of ROS (35). To understand the mechanism of increased proliferation in response to FABP4, the redox status of cancer cells in an FABP4 rich environment has been evaluated. Previous data in macrophages demonstrated that deletion of FABP results in lower ROS production, an increase in antioxidant protein expression, and decreased inflammatory cytokine production(36). Data from 3TL-31 adipocytes and tumor associated endothelial cells found contrasting results, an increase in ROS levels after treatment with small interfering RNA against FABP4 or FABP4 knockdown(37,38). In this study, exogenous FABP4 treatment leads to significantly increased levels of intracellular Nrf2 activity and decreased levels of intracellular ROS in PDAC cells. Previously, we found that the level of fatty acids stearic acid and palmitic acid were selectively increased in FABP4 treated breast cancer cells, and the change in cellular lipid saturation was correlated with an increase in endoplasmic reticular (ER) stress (Data not shown). Consistent with increased ER stress, the transcriptional activity of Nrf2 was increased in response to FABP4, but not R126Q.
To use the FABP4 as a target in cancer, testing the FABP4 inhibitors may be the logical next step. BMS309403 and HTS01037, both are small molecule FABP4 inhibitors, are commonly used on animal models of obesity, diabetes mellitus and cardiovascular studies(39,40). Some groups reported that BMS309403 significantly reduced tumor burden in a syngeneic orthotopic high-grade serous ovarian cancer mouse model(41). Investigations to further reveal the clinical benefits of FABP4 are awaited.
In this study, there is some limitation. FABP4 knockout mice shows lower levels of circulating insulin and reduced glucose levels(24). We consider that glucose metabolism also plays an important role when FABP4 indirectly effect to the tumor growth. However, there is limitation to distinguish the glucose metabolism from fat metabolism in vivo study. What we tried to demonstrate in this experiment is that exogenous FABP4 was critical for the tumor growth in the syngeneic mouse PDAC model. How glucose metabolism related to the tumor growth suppression in vivo experiment is not well understanded yet in this paper, and we considered it as a next challenge.
Conclusion
In conclusion, exogenous FABP4 increased pancreatic cancer cell proliferation. FABP4 treatment leaded to activation of Nrf2 and downregulation of ROS activity. In vivo experimentation demonstrated a significant decrease in pancreatic cancer cell growth in FABP4 knockout mice, compared to growth in wild type and Het type mice. FABP4 may contribute to understand the linkage of obesity and pancreatic cancer progression, and further analyses are planned. FABP4-directed therapies may have potential in managing pancreatic cancer progression.
Supplementary Material
Supplemental Figure 1. Single image of Figure.4A-Panc-1.
This is original immunoblot photo of Figure.4A-Panc-1. Expression FABP4 and β-actin in Panc-1 cells following addition of 100 ng/ml FABP4 for 24 hours and 48 hours are showed in this figure. The recombinant FABP4 (rFABP4) contains His-tag and that is why media and rFABP4 group showed heavier molecular weight.
Supplemental Figure 2. Single image of Figure.4A-Pan02.
This is original immunoblot photo of Figure.4A-Pan02. Expression FABP4 and β-actin in Pan02 cell following addition of 40 and 100 ng/ml FABP4 for 24 hours and 48 hours are showed in this figure.
FABP4 stimulated cellular proliferation.
Nuclear factor E2-related factor 2 (NRF2) was increased in response to FABP4.
The tumor growth was decreased in FABP4 null animals compared to controls.
FABP4 increases pancreatic cancer proliferation via activation of Nrf2.
Acknowledgments
This project was partly funded by NIH T32DK108733 (KW, MY), DK053189 (DB), R21DK122832 (SI) and R01DK128325 (SI).
Funding: This project was partly funded by NIH T32DK108733 and NIH R01DK053189, R21DK122832.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest: The authors declare that they have no conflicts of interest.
References
- [1].Siegel Rebecca L, Miller Kimberly D, Ahmedin Jemal. Cancer statistics, 2019. CA Cancer J Clin 2019; 69 (1): 7–34. [DOI] [PubMed] [Google Scholar]
- [2].Hidalgo M Pancreatic cancer. N Engl J Med 2010; 362 (17): 1605–17. [DOI] [PubMed] [Google Scholar]
- [3].Neuhouser ML, Aragaki AK, Prentice RL, et al. Overweight, Obesity, and Postmenopausal Invasive Breast Cancer Risk. JAMA Oncol 2015; 1 (5): 611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Calle Eugenia E., Rodriguez Carmen, Kimberly Walker-Thurmond, B.A., Michael J. Thun. Overweight, Obesity, and Mortality from Cancer in a Prospectively Studied Cohort of U.S. Adults. N. Engl. J. Med 2003; 348: 1625–638. [DOI] [PubMed] [Google Scholar]
- [5].Smits MM, van Geenen EJ. The clinical significance of pancreatic steatosis. Nat Rev Gastroenterol Hepatol 2011; 8 (3): 169–77. [DOI] [PubMed] [Google Scholar]
- [6].Lesmana Cosmas R A, Gani Rino A, Lesmana Laurentius A. Non-alcoholic fatty pancreas disease as a risk factor for pancreatic cancer based on endoscopic ultrasound examination among pancreatic cancer patients: A single-center experience. JGH Open 2017; 2 (1): 4–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pothuraju R, Rachagani S, Junker WM, et al. Pancreatic cancer associated with obesity and diabetes: An alternative approach for its targeting. J Exp Clin Cancer Res 2018; 37 (1): 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Berger NA. Obesity and cancer pathogenesis. Ann N Y Acad Sci 2014; 1311: 57–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Khandekar MJ, Cohen P, Spiegelman BM. Molecular mechanisms of cancer development in obesity. Nat Rev Cancer 2011; 11 (12): 886–95. [DOI] [PubMed] [Google Scholar]
- [10].Amiri M, Yousefnia S, Seyed Forootan F, Peymani M, Ghaedi K, Nasr Esfahani MH. Diverse roles of fatty acid binding proteins (FABPs) in development and pathogenesis of cancers. Gene 2018; 676: 171–183. [DOI] [PubMed] [Google Scholar]
- [11].Hotamisligil GS, Bernlohr DA. Metabolic functions of FABPs--mechanisms and therapeutic implications. Nat Rev Endocrinol 2015; 11 (10): 592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cabré A, Lázaro I, Girona J, et al. Fatty acid binding protein 4 is increased in metabolic syndrome and with thiazolidinedione treatment in diabetic patients. Atherosclerosis 2007; 195 (1): e150–8. [DOI] [PubMed] [Google Scholar]
- [13].Cui Y Prognostic significance of fatty acid binding protein-4 in the invasive ductal carcinoma of the breast. Pathol Int 2019; 69 (2): 68–75. [DOI] [PubMed] [Google Scholar]
- [14].Guaita-Esteruelas S, Bosquet A, Saavedra P, et al. Exogenous FABP4 increases breast cancer cell proliferation and activates the expression of fatty acid transport proteins. Mol Carcinog 2017; 56 (1): 208–217. [DOI] [PubMed] [Google Scholar]
- [15].Iain H, et al. Role of fatty acid binding proteins (FABPs) in cancer development and progression. Cell Signal 2019; 62: 109336. [DOI] [PubMed] [Google Scholar]
- [16].Hao J, Yan F, Zhang Y, et al. Expression of Adipocyte/Macrophage Fatty Acid-Binding Protein in Tumor-Associated Macrophages Promotes Breast Cancer Progression. Cancer Res 2018; 78 (9): 2343–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Schauer Daniel P, et al. Bariatric Surgery and the Risk of Cancer in a Large Multisite Cohort. Ann Surg 2019; 269 (1): 95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jahansouz Cyrus, et al. Serum FABP4 concentrations decrease after Rouxen-Y gastric bypass but not after intensive medical management. Surgery 2019; 165: 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Luo Y, Yang Z, Li D, et al. LDHB and FABP4 are Associated With Progression and Poor Prognosis of Pancreatic Ductal Adenocarcinomas. Appl Immunohistochem Mol Morphol 2017; 25 (5): 351–357. [DOI] [PubMed] [Google Scholar]
- [20].Hotamisligil Gökhan S. and Bernlohr David A.. Metabolic functions of FABPs- mechanisms and therapeutic implications. Nat Rev Endocrinol 2015; 11 (10): 592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Xu H, Hertzel AV., Steen KA, Bernlohr DA. Loss of Fatty Acid Binding Protein 4/aP2 Reduces Macrophage Inflammation Through Activation of SIRT3. Mol Endocrinol 2016; 30 (3): 325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Xu H, Hertzel A V., Steen KA, Wang Q, Suttles J, Bernlohr DA. Uncoupling Lipid Metabolism from Inflammation through Fatty Acid Binding Protein-Dependent Expression of UCP2. Mol Cell Biol 2015; 35 (6): 1055–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hertzel AV, Bennaars-Eiden A, Bernlohr DA. Increased lipolysis in transgenic animals overexpressing the epithelial fatty acid binding protein in adipose cells. J Lipid Res 2002; 43: 2105–2111. [DOI] [PubMed] [Google Scholar]
- [24].Hertzel AV, Smith LA, Berg AH, et al. Lipid metabolism and adipokine levels in fatty acid-binding protein null and transgenic mice. Am J Physiol Endocrinol Metab 2006; 290: E814–E823. [DOI] [PubMed] [Google Scholar]
- [25].Hertzel AV, Hellberg K, Reynolds JM, et al. Identification and characterization of a small molecule inhibitor of Fatty Acid binding proteins. J Med Chem 2009. Oct 8; 52 (19): 6024–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jaramillo Melba C 1, Zhang Donna D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev 2013; 27(20): 2179–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hertzer KM, Xu M, Moro A, et al. Robust Early Inflammation of the Peripancreatic Visceral Adipose Tissue During Diet-Induced Obesity in the KrasG12D Model of Pancreatic Cancer. Pancreas 2016; 45 (3): 458–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Röhrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer 2016; 16 (11): 732–749. [DOI] [PubMed] [Google Scholar]
- [29].Cao H, Xu M, Dong W, Deng B, Wang S, Zhang Y, et al. Secondary bile acid induced dysbiosis promotes intestinal carcinogenesis, Int J Cancer 2017. Jun 1; 140 (11): 2545–2556. [DOI] [PubMed] [Google Scholar]
- [30].Gharpure KM, Pradeep S, Sans M, Rupaimoole R, Ivan C, Wu SY, et al. FABP4 as a key determinant of metastatic potential of ovarian cancer. Nat Commun 2018. Jul 26; 9 (1): 2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cheng QZ, Xiu PZ, Ning M, et al. FABP4 suppresses proliferation and invasion of hepatocellular carcinoma cells and predicts a poor prognosis for hepatocellular carcinoma. Cancer Med 2018; 7(6): 2629–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth, Nat Med 2011. Oct 30; 17 (11): 1498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hao J, Zhang Y, Yan X, et al. Circulating Adipose Fatty Acid Binding Protein Is a New Link Underlying Obesity-Associated Breast/Mammary Tumor Development. Cell Metab 2018; 28 (5): 689–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Thompson KJ, Austin RG, Nazari SS, et al. Altered fatty acid-binding protein 4 (FABP4) expression and function in human and animal models of hepatocellular carcinoma. Liver Int 2018; 38: 1074–1083. [DOI] [PubMed] [Google Scholar]
- [35].Murakami Shohei, Motohashi Hozumi. Roles of Nrf2 in cell proliferation and differentiation. Free Radic Biol Med 2015; 88: 168–178. [DOI] [PubMed] [Google Scholar]
- [36].Steen KA, Xu H, Bernlohr DA. FABP4/aP2 Regulates Macrophage Redox Signaling and Inflammasome Activation via Control of UCP2. Mol Cell Biol 2017; 37 (2): 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kajimoto K, Minami Y, Harashima H. Cytoprotective role of the fatty acid binding protein 4 against oxidative and endoplasmic reticulum stress in 3T3-L1 adipocytes. FEBS Open Bio 2014; 4: 602–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Harjes U, Bridges E, Gharpure KM, et al. Antiangiogenic and tumour inhibitory effects of downregulating tumour endothelial FABP4. Oncogene 2017; 36 (7): 912–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Xu Hongliang, Hertzel Ann V., et al. Uncoupling lipid metabolism from inflammation through fatty acid binding protein-dependent expression of UCP2. Mol Cell Biol 2015. Mar;35(6):1055–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Masato F, Gürol T, Cem Z. et al. Treatment of diabetes and atherosclerosis by inhibiting fatty-acid-binding protein aP2. Nature 2007; 447(7147): 959–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Abir M, Chun-Yi C, Helen AD, et al. Adipocyte-Induced FABP4 Expression in Ovarian Cancer Cells Promotes Metastasis and Mediates Carboplatin Resistance. Cancer Res 2020; 80(8): 1748–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Single image of Figure.4A-Panc-1.
This is original immunoblot photo of Figure.4A-Panc-1. Expression FABP4 and β-actin in Panc-1 cells following addition of 100 ng/ml FABP4 for 24 hours and 48 hours are showed in this figure. The recombinant FABP4 (rFABP4) contains His-tag and that is why media and rFABP4 group showed heavier molecular weight.
Supplemental Figure 2. Single image of Figure.4A-Pan02.
This is original immunoblot photo of Figure.4A-Pan02. Expression FABP4 and β-actin in Pan02 cell following addition of 40 and 100 ng/ml FABP4 for 24 hours and 48 hours are showed in this figure.