

Abstract
There is of significant interest and importance to develop robust machine learning models to assist organic chemistry synthesis. Typically, task-specific machine learning models for distinct reaction prediction tasks have been developed. In this work, we develop a unified deep learning model T5Chem for a variety of chemical reaction predictions tasks by adapting the ”Text-to-Text Transfer Transformer”(T5) framework in natural language processing (NLP). Based on self-supervised pre-training with PubChem molecules, the T5Chem model can achieve state-of-the-art performances for four distinct types of task-specific reaction prediction tasks using four different open-source datasets, including reaction type classification on USPTO_TPL, forward reaction prediction on USPTO_MIT, single-step retrosynthesis on USPTO_50k and reaction yield prediction on high-throughput C-N coupling reactions. Meanwhile, we introduced a new unified multi-task reaction prediction dataset USPTO_500_MT, which can be used to train and test five different types of reaction tasks, including the above four as well as a new reagent suggestion task. Our results showed that models trained with multiple tasks are more robust and can benefit from mutual learning on related tasks. Furthermore, we demonstrated the use of SHAP (SHapley Additive exPlanations) to explain T5Chem predictions at the functional group level, which provides a way to demystify sequence-based deep learning models in chemistry. T5Chem is accessible through https://yzhang.hpc.nyu.edu/T5Chem
Graphical Abstract
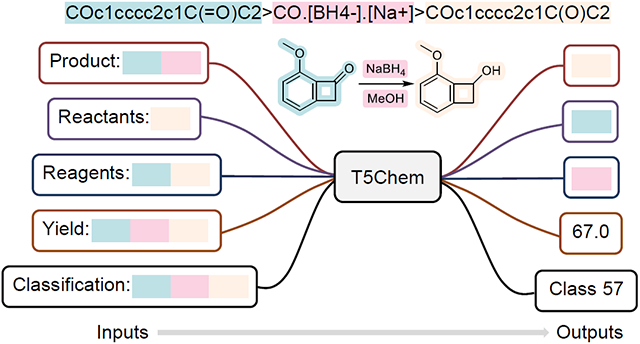
Introduction
Organic synthesis is one of the most fundamental problems in chemistry. With the advances of computing power, data availability and algorithms, there has been of significant interest in developing machine learning (ML) models to assist a variety of organic reaction-related tasks,1-4 including reaction product prediction,5-18 retrosynthesis,9,14,17,19-37 reaction condition optimization,38-41 reaction yield prediction42-54 and reaction type classification. 38,51,55-57 These ML-based data-driven approaches for organic synthesis can be classified into descriptor-based models,5-10,19-26,38-41,51,55,57 graph-based models11-13,27,28,52 and sequence-based models,14-18,29-37,53,54,56 depending on how molecules are represented as input for machine learning. Descriptor-based models use hand-crafted features as molecular representations, and often need feature engineering or template extraction for different reaction prediction tasks, which set limitations to generalizability. 43,58,59 For this reason, people start to turn to end-to-end models. Graph-based models treat molecules as graphs where atoms are viewed as nodes and bonds are viewed as edges. Although molecules can be naturally represented as graphs, stereoisomers, molecular structures with the same graph connectivity but different spatial arrangements, remain underexplored despite few previous works.60,61 Furthermore, most graph-based models suffer from the need of atomic mapping and inability of handling stereoisomers. Currently few attempts have been made to develop graph-based models for reaction yield prediction and reaction type prediction. On the other hand, sequence-based models showcased the feasibility of language modeling strategy on chemical reactions by using text-based representations of molecular graphs. Simplified molecular-input line-entry system (SMILES62) is a commonly used63,64 linear string notation in deep neural networks. Accordingly, reaction prediction can be formulated as machine translation problem, where reactants/reagents SMILES serve as source language and product SMILES serve as target sequences.
The recent key advance that has made for deep learning in language modeling is the transformer,65 which stands out for its outstanding performance and wide applicability. Schwaller et. al.16 adapted the transformer architecture for the forward reaction prediction task, namely molecular transformer, for the first time. After that, many efforts have been made to apply transformer models for the retrosynthesis task.14,17,30-36 Recently, Schwaller et al. developed a new type of molecular fingerprint, rxnfp,56 for reaction type classification. Rxnfp is derived from bidirectional encoder representations from transformers (BERT),66 a transformer-based encoder that converts an input sequence into an internal vector representation. The BERT classifier reached a classification accuracy of 98.2% in an open-sourced dataset. Soon afterwards, they extended the BERT model with a regression layer to predict reaction yield.53
Transformer-based models not only shed light on general-purpose reaction predictions, but also transferable to particular reactions. Pesciullesi et al.18 published a transfer learning approach to predict regio- and stereoselective reactions on carbohydrates with molecular transformer. Moreover, transformers take advantage of pre-training56,67,68 and data augmentation17 with improved performance. In spite of all advances transformers have made, models developed for different reaction prediction tasks are still not interchangeable.
Herein we present a unified deep learning model T5Chem for a variety of chemical reaction predictions tasks by adapting the Text-To-Text Transfer Transformer69 (T5).The T5 model is structurally similar to the original Transformer,65 except that instead of using the Layer Norm bias, it places a layer normalization outside the residual path and uses a different, relative position embedding scheme. The idea of multi-tasking predictions is inspired by natural human learning process that knowledge learned from one task should be helpful to other related tasks. Meanwhile, we introduce a new multi-task reaction prediction dataset USPTO_500_MT, which can be used to train and test five different types of reaction tasks, including a novel reagent suggestion task.
In the following, we show how T5Chem can be tailored to tackle multiple reaction prediction tasks with task-specific prompts, which requires no or minor modifications on output layers. Then we apply this framework to several open-source datasets individually to illustrate its applicability and state-of-the-art performances. After that, we demonstrate this unified T5Chem model can be trained on the new unified multi-task reaction prediction dataset USPTO_500_MT without degrading performance on individual tasks. Finally, we look into specific prediction examples, and shed light on black-box model predictions with SHAP70 (SHapley Additive exPlanations).
Method
Model
T5Chem model is developed based on Text-To-Text Transfer Transformer69 (T5), which is an encoder-decoder model from the transformer family, as illustrated in Figure 1. In comparison with the original transformer model,65 T5 model uses the same self-attention strategy with two modifications: 1. It removes the layer norm bias, and places the normalization outside of the residual path. 2. It uses relative positional embedding scheme instead of sinusoidal position embedding. T5 model is designed to convert all language problems into a text-to-text format so that it is capable to work on a variety of tasks at the same time. To specify which task the model should perform, a task-specific prompt would be prepended to the original input sequence before feeding it to the model.
Figure 1:

Illustration of the general transformer architecture65 used in T569 and T5 Chem
To develop a unified deep learning model T5Chem for a variety of chemical reaction predictions tasks, besides the use of character-level tokenization for the SMILES input and introduction of task-specific prompts for different reaction prediction tasks into the vocabulary as special tokens, we have modified the original T5 output layer (language modeling head) into three different kinds in T5Chem as illustrated in Figure 2: the molecular generation head for all sequence to sequence tasks, i.e., reaction product prediction, single-step retrosynthesis and reagent suggestion; the classification head for reaction type classification; and the regression head for product yield prediction which employs the soft-label strategy.
Figure 2:

Multi-tasking of T5Chem. Five different tasks are shown above. All models have a general structure of encoder, decoder and output layers. However, they may have different prompts and head types, depending on task types.
Tokenization is the process that breaking a sequence into small chunks. In Natural Language Processing (NLP), tokenization can be performed on either word or sub-word level. Similarly, different tokenization approaches can be applied to molecules. T5Chem employs the character-level tokenization, which simply splits reaction SMILES into single alphabet letter, digit or special symbol. In comparison with atom tokenization which runs a WordPiece tokenization algorithm over SMILES strings using a regular expression,15 and SELFIES which is short for Self-Referencing Embedded Strings,71 one main advantage of the character-level tokenization is its simplification, flexibility and much smaller vocabulary size. In our exploration, neither atom tokenization nor SELFIES is found to outperform character-level tokenization for reaction prediction tasks (Table S1).
In T5Chem, each task-specific prompt is an English word that would not appear in original SMILES, and thus would be added to vocabulary as a special token. Here we use “Product:” for reaction product prediction, “Reactants:” for single-step retrosynthesis, “Reagents:” for reagent suggestion, “Classification:” for reaction type prediction, and “Yield:” for reaction yield prediction. Note that special tokens only work as identifiers for different tasks, and the exact wording of these prompts would not impact our model training.
For chemical reaction predictions, different tasks can have different label formats, and thus the output layer need to be changed accordingly, as illustrated in Figure 2. For sequence-to-sequence tasks like forward reaction prediction, retrosynthesis and reagents prediction, the input and output sequences share the same vocabulary. Correspondingly, the output layer, molecular generation head, shares weights with input embedding layer, and produces a probability distribution among the whole vocabulary space.
For classification tasks such as reaction type classification, a classification head was used as the output layer. This is a new linear layer that outputs a probability distribution among all classification categories. As an example, for a reaction type classification task consisting of 1,000 reaction template classes, the classification head for this task would be a linear layer with an output size to be 1,000.
For regression tasks such as reaction yield prediction, we applied the soft label strategy for min-max normalized training labels: the regression head is a single linear layer with an output dimension to be 2, representing weights for the min and max values. Different from molecular generation and classification heads, the Kullback-Leibler divergence is used as the loss function to minimize the probability distribution between outputs and true labels for regression tasks.
Implementation and Training Details
T5Chem is implemented in Python (version 3.7) by using RDKit (version 2021.03.1)72 for reaction preprocessing and SMILES validation, and employing pytorch (version 1.7.0)73 and huggingface transformers (version 4.10.2)74 for seq2seq modeling. The source code is available online at https://github.com/HelloJocelynLu/t5chem.
Similarly to the original work, a BERT66-like “masked language modeling” objective would be used for model pre-training in a self-supervised manner. 97 million molecules from Pubchem75 were used for pre-training. During the pre-training, tokens of source sequences would be randomly masked, and the goal is to predict correct tokens that have been masked. We applied the same mask rate, 15%, as original T5 model. Masked tokens would be substituted to mask token (<mask>) in 80% of time, or be replaced by another random token in vocabulary for 10% of time, and remain the same for the rest of time. Then the model would be fine-tuned in supervised downstream tasks. During the fine-tuning, various task-specific prompts and output layers would be used for different output styles, as illustrated in Fig. 2.
In T5Chem, the molecular generation head for all sequence to sequence tasks, including reaction product prediction, reactants prediction and reagents prediction, is the same as the output layer of T5ForConditionalGeneration with huggingface transformers74 package. It produces a probability distribution among the same vocabulary space as input sequence. For classification tasks, the classification head is used, which is a linear output layer that generates a probability distribution among all classification categories. Both molecular generation head and classification head use cross entropy loss as the loss function. On the other hand, for regression tasks, the soft label strategy is used for min-max normalized training labels, and the regression head is a single linear layer with an output dimension to be 2, representing the probability distribution between min and max. The Kullback–Leibler divergence is used as the loss function to minimize the probability distribution between outputs and true labels for regression tasks. During the test phase, molecular generation head of T5Chem repeatedly generates molecules token by token, until the ”end of sentence token” is generated or the maximum length of allowed prediction is reached, while both classification and regression heads can be viewed as a one-time generation procedure which has the maximum allowed prediction length to be 1.
For all tasks, T5Chem uses whole encoder and decoder architecture with 4 layers and 8 attention heads. We used 256 as hidden dimension for T5Chem and 2048 for intermediate feed forward layer. The maximum vocabulary size was set to 100, which include 70 tokens from character-level tokenization of SMILES of pubchem and USPTO molecules, 5 special structural tokens (<s>, </s>, <unk>, <pad>, <mask>), 6 prompting tokens and 19 placeholders. The <pad> token is used as the first input token for decoder to initialize decoding process. It is worth mentioning that placeholders can be replaced to other prompting tokens at any time when needed, which makes T5Chem extendable to more tasks without retraining the whole neural network.
Dataset
In this work, we used four open-sourced datasets introduced by previous published work for better comparison. We also introduced one new reaction dataset, USPTO_500_MT for multi-tasking purpose. All chemical reactions were described using SMILES representation as inputs for T5Chem. Table 1 shows an overview of these data sets.
Table 1:
Dataset Splits Used for the Experiments
| Dataset | Train | Valid | Test | Total | Task |
|---|---|---|---|---|---|
| USPTO_TPL56 a | 360,545 | 40,059 | 44,511 | 445,115 | Reaction type classification |
| USPTO_MIT12 | 409,035 | 30,000 | 40,000 | 479,035 | Forward prediction |
| USPTO_50k29 a | 40,029 | 5,004 | 5,004 | 50,037 | Retrosynthesis |
| C-N Coupling44
a, b (Random splits) |
2,767 | – | 1,188 | 3,955 | Reaction yield prediction |
| C-N Coupling44
a, b (Out-of-sample test1) |
3,057 | – | 898 | 3,955 | Reaction yield prediction |
| C-N Coupling44
a, b (Out-of-sample test2, 4 |
3,055 | – | 900 | 3,955 | Reaction yield prediction |
| C-N Coupling44
a, b (Out-of-sample test3) |
3,058 | – | 897 | 3,955 | Reaction yield prediction |
| USPTO_500_MTa | 116,360 | 12,937 | 14,238 | 143,535 | Multi-task prediction |
Contains stereochemical information
With reactants/reagents separation
USTPO_TPL for reaction type classification.
The dataset for reaction type classification task was originally derived from the USPTO database by Lowe76 and introduced by Schwaller et al.56 This strongly imbalanced dataset consists of 445,000 reactions divided into 1,000 classes without reactant-reagent separation. Reaction classes here were defined by SMART reaction templates, and obtained by atom-mapping the USPTO dataset with RXNMapper. Among all USPTO reactions, 1,000 most frequent template hashes were selected as targets. Reactions were randomly split into 90% for training and validation, and 10% for testing.
USPTO_MIT for forward reaction prediction.
This benchmark dataset has been prepared by Jin et al.12 based on the USPTO database of Lowe76 and include both separated and mixed versions. The mixed version without reactants/reagents separation is used as a more challenging task which needs to learn to identify “reagents” and “reactants” by itself. In machine-aided reaction prediction, reagents are defined as chemical species that do not appear in (major) products. Therefore, the mixed version reflects a real-world scenario that users do not have prior knowledge about products before predictions were made. This dataset consists of 479 k reactions: 409 k for training, 30 k for validation and 40 k for testing.
USPTO_50K for single-step retrosynthesis.
Retrosynthesis is the process of deconstructing a molecule. More specifically, single-step retrosynthesis prediction task is defined as given a product as input, to find reactants combination that could generate the input compound. The dataset was a filtered version of Lowe’s patent dataset. It contains only 50 k reactions that have been classified into 10 board reaction types.77 In this work, we did not remove stereochemical information and did not provide model with reaction types as we would encounter in real-world situation. We followed the splitting proposed by Liu et al,29 and also has been used by many works.17,19,27,28,30,31 40 k, 5 k and 5 k reactions were used for training, validation and testing, respectively.
C-N coupling dataset for reaction yield prediction.
This is a high-throughput experiment dataset. In 2018, Ahneman et al performed 4,608 Pd-catalyzed Buchwald-Hartwig C-N cross coupling reactions.43 These nanomole-scale experiments were carried out on three 1536-well plates consisting of a full matrix of 15 aryl and heteroaryl halides, 3 bases, 4 ligands and 23 isoxazole additives. 3,955 applicable reactions after removing control groups were used. In their work, a random forest model was used with 120 DFT calculated features of compounds. This strategy gave a promising performance. Recently, Schwaller et al53 proposed a BERT-based reaction encoder enriched with a regression layer (Yield-BERT). This model can directly take reaction SMILES as input and thus is extendable to other reaction types without feature engineering. It outperforms random forest model on many tests except for the most challenging out-of-sample test. To demonstrate the capability of T5Chem on regression task, we followed previous data splitting: ten random splitting (70/30 for training/testing) and four out-of-sample sets. Test samples in out-of-sample sets contain reactions additives which are not included in the training data, which can be used to evaluate the generalizability of machine learning models.
USPTO_500_MT for multi-task reaction prediction.
In order to illustrate the multi-tasking capability of our unified model, we introduced a new dataset that is applicable for multiple reaction prediction tasks, including forward reaction prediction, reactants prediction (single step retrosynthesis), reagents prediction, reaction yield prediction and reaction type classification. The same splitting is expected for all tasks to avoid possible data leakage problem. A more detailed overview for this newly introduced dataset is available in Supporting Information (Figure S1-S2).
USPTO_TPL (in Task 1: Reaction type classification) is a good starting point to construct this multi-tasking dataset, as it contains reactants, reagents, product and reaction type for every reaction instance. In order to recover reaction yield for each reaction, we looked into text-mined records78 by Lowe, and matched reactions based on reactants, reagents and product. In the original record, yields are reported either directly mined from the document or calculated from the isolated product amount or both. However, discrepancies exist between text-mined and calculated values for a same reaction in the same patent,3 and some calculated yields have values > 100%. Thus all yields with values > 100% were removed first. Then for reactions with only one type of yield available, that one is used as the label for reaction yield. When both text-mined and calculated values are available, their average value is used if the difference between two values is less than 10%, otherwise, the text-mining value is used. A successful curation of reaction yield with a complete match is only achieved for about one-third of reactions in the USPTOTPL dataset. We noticed that some reactions classes are very sparse (as many reactions cannot be recovered). Therefore, we kept the top-500 most frequent reaction classes and ended up with 116k reactions for training, 13k and 14k reactions for validation and testing, respectively. We refer this dataset as USPTO_500_MT.
Results and discussion
One of the most intriguing features for transformer models is the effectiveness of its pre-training. T5Chem was pre-trained with masked language modeling (MLM) objective on 97 million PubChem molecules. The pre-trained model shows a steady learning curve and faster convergence (Figure S3), and it has been used to initialize all models used in the following experiments. First, we examined T5Chem for four distinct types of task-specific reaction prediction tasks using four different open-source datasets, and the results indicated that the T5Chem model can achieve state-of-the-art performances. Then we applied to USPTO_500_MT, which demonstrates the multi-tasking capability and robustness of T5Chem. Finally, we used SHAP (SHapley Additive exPlanations) to explain T5Chem predictions at the functional group level, which illustrates how to demystify sequence-based deep learning models in chemistry.
Task 1. Reaction type classification
Here we used the USPTO TPL dataset the same way as the original work, i.e., the BERT classifier,56 in which reactants and reagents are not separated. T5Chem has been trained for 100 epochs, and the test results are summarized in Table 2. Our model outperformed BERT classifier56 and achieved 99.5% accuracy. Looking at the confusion entropy of a confusion matrix (CEN) and the overall Matthews correlation coefficient (MCC) also leads to the same conclusion.
Table 2:
Results for reaction type classification. The lower the confusion entropy of a confusion matrix79 (CEN) and the higher the Matthews correlation coefficient80 (MCC) the better. The bold entries highlight the best-performing approach.
| Model | Accuracy | CEN | MCC |
|---|---|---|---|
| BERT classifier56 | 0.989 | 0.006 | 0.989 |
| T5Chem | 0.995 | 0.003 | 0.995 |
Task 2. Forward reaction prediction
USPTO_MIT has been used for benchmarking forward predictions in many previous works. T5chem was trained on this dataset with a task-specific prompt “Product:” for 30 epochs. The results are summarized in Table 3 with comparison to some previous models. Seq2seq15 is one of the first attention-based sequence-to-sequence model that treats molecules as strings. WLDN513 performed a two-stage model using graph-convolutional neural network. Both models were only evaluated with reactants/reagents separation. We presented results as top-k accuracy. Top-k accuracy takes k model predictions with highest probability. If one of them is a true label, it classifies the prediction as correct. Note that here we just used a single model without any data augmentation. T5Chem shows slight better performance in comparison with molecular transformer,16 and clearly outperforms both Seq2seq and WLDN5.
Table 3:
Results for forward reaction prediction. T5Chem achieved comparable performance with molecular transformer model on this individual task. The bold entries highlight the best-performing approach.
| Model | Mixed | Separated | ||||
|---|---|---|---|---|---|---|
| Top-1 (%) | Top-2 (%) | Top-5 (%) | Top-1 (%) | Top-2 (%) | Top-5 (%) | |
| Seq2seq15 | – | – | – | 80.3 | 84.7 | 87.5 |
| WLDN513 | – | – | – | 80.6 | 90.5 | 93.4 |
| Molecular Transformer16 | 88.6* | 92.4* | 94.2* | 88.8 | 92.6 | 94.4 |
| T5Chem | 88.9 | 92.9 | 95.2 | 90.4 | 94.2 | 96.4 |
It is Unclear whether data augmentation strategy has been used for these results.
Task 3. Single-step retrosynthesis
In this task, we used the same model architecture as forward reaction prediction. We trained T5Chem on USPTO_50k for 100 epochs without given reaction types, and compared our results with some previous studies with sequence-based models using the same training data. The results for T5Chem without data augmentation were shown as Table 4. Specifically, Seq2seq29 model utilizes LSTM81 architecture. Molecular transformer14 was based on the Transformer architecture65 which was solely based on self-attention mechanisms. SCROP32 is short for self-corrected retrosynthesis predictor, they used a transformer-based retrosynthetic reaction predictor coupling with a neural network-based syntax corrector. We can see that T5Chem is capable to perform well for retrosynthesis as a single task, as it achieves better performance in comparison with other smiles-based sequence-to-sequence models.
Table 4:
Results for single-step retrosynthesis on USPTO_50k with other smiles-based sequence-to-sequence models.
Task 4. Reaction yield prediction
In order to fit our T5Chem to reaction yield prediction, a regression head was added to give number outputs as described in Method section. Task-specific prompt “Yield:” was prepended to input reaction SMILES to specify the task type. To test the proposed approach, the same splits were used as in Sandfort et al,44 and Schwaller et al.53 We trained T5Chem on this cross coupling data for 100 epochs. The results are shown in Table 5.
Table 5:
Results for reaction yield prediction on C-N coupling reactions. The bold entries highlight the best-performing approach.
Since only reaction SMILES were used in our model, this approach can be easily adapted to other reaction types without any modifications or feature engineering. Generally, our model performs best among listed models. For random splitting, our model achieved average R2 of 0.970. For more challenging out-of-sample tests, T5Chem also gets best results except for Test 1. Test 4 is viewed as the most challenging test among all testsets as shown in other models. This might imply that T5Chem would have better generalizability in this task. To further investigate our model predictions on four test sets, we examined the prediction accuracy for each test. In real world problem like singleton batch experiments and THE, there are underlying experimental errors in measurement. To address this issue, we used ±10% as acceptable error range and calculated prediction accuracy within this range as shown in Table 6.
Table 6:
Accuracies for reaction yield prediction on C-N coupling test sets. Predictions within 10% error range are viewed as accurate.
| Test 1 | Test 2 | Test 3 | Test 4 | |
|---|---|---|---|---|
| Accuracy (%) | 75.5 | 83.9 | 64.5 | 59.9 |
Task 5. Multi-task prediction
After showing that T5Chem is able to achieve comparable or better performance with other sequence-based models for individual tasks, we carried out multi-task experiments with the newly prepared USPTO_500_MT dataset to demonstrate multi-tasking capability and transferability of T5Chem. The new dataset USPTO_500_MT contains five objectives: forward reaction prediction, single-step retrosynthesis, reagent prediction, reaction type prediction and reaction yield prediction. Note that the curation of this dataset does rely on atom-mapping for labeling, but T5Chem does not require atom-mapping as inputs. The training/validation/testing sets are well separated to ensure no reaction overlapping across all task types.
We first applied T5Chem in all tasks independently to get baseline results. Though the model has already achieved promising results, we are interested in figuring out whether further performance gains can be obtained by cross-task training. The five tasks are grouped into two subgroups. The first subgroup includes forward reaction prediction, single-step retrosynthesis and reagents prediction, as they are all sequence-to-sequence tasks and can share the same model architecture. The second subgroup consists of reaction classification and reaction yield prediction. Both tasks take the whole reaction sequence as inputs, and we proposed that model can be trained on the two tasks at the same time by combining their loss functions together. A multi-task training using combined loss functions from all five tasks has also been carried out with worse performance. It may due to the fact that the regression and classification tasks are internally less similar than sequence to sequence predictions. When we trained them together, we forced the major part (embedding, encoder and decoder) to share the same weights among all tasks. The molecular generation head also shares weights with embedding layer. The only flexibility components: regression head and classification head are not sufficient to differentiate those tasks. The results can be found in the Supporting Information. (Table S2)
For sequence-to-sequence subgroup, we mixed training sets of three tasks. To distinguish different tasks, task-specific prompts (”Product:”, ”Reactants:”, ”Reagents:”) were used. We used a combined, big training set as shown in Figure 3. In addition, we explored 5-fold data augmentation using non-canonical SMILES, and found that data augmentation did not improve performance on USPTO_500_MT dataset in terms of top-k accuracy (even slightly worse results), but it did improve SMILES validity (Table S3).
Figure 3:

Mix training dataset from different tasks. In multi-task training scheme, we combined forward reaction prediction task, retrosynthesis task and reagents prediction task together. Every input instance starts with a task-specific prompt, then followed by actual input. In forward reaction prediction, the model takes reactants and reagents (without separation) as source sequence. In retrosynthesis, the model takes only product SMILES as source sequence. In reagents prediction, the model takes both reactants and product SMILES as inputs. A reduction reaction is shown above as an example.
In this experiment, we only trained one model with the mixed dataset, and then tested it separately on three test sets from different tasks. We compared this result with individual models that were trained and tested on separate datasets. Two metrics have been reported: The accuracy in top-k predictions and the invalid SMILES rate in top-k predictions. Both metrics are calculated on all predictions. The valid SMILES is defined as SMILES string that can be parsed by RDKit package. Note that one may have a high top-k accuracy and high SMILES invalidity at the same time: For example, if T5Chem gives 5 predictions to a custom input, and the first prediction is correct. Then all top-k accuracy would be 100% because the first prediction is always included. However, if the 2nd-5th predictions are wrong and chemical invalid, we will have top-5 SMILES invalidity as high as 80% (4/5)! The results are summarized as Figure 4.
Figure 4:

Test results on USPTO_500_MT. We evaluated both accuracy and SMILES invalidity for top-k predictions on different tasks. Individual tasks and multi-task training schemes have comparable performance in accuracy for all three tasks. But combined training gives much lower invalid SMILES rate. Note that one may have a high top-k accuracy and high SMILES invalidity at the same time.
T5Chem achieves top-1 accuracy of 97.5%, 72.9% and 24.9% for forward prediction, retrosynthesis and reagents prediction, respectively. It achieves comparable performance as being trained on separate tasks in terms of top-k accuracy. This demonstrates that these three tasks are closely related, and are possible to learn at once. T5Chem also generates much less grammatically invalid molecules when being trained on mixed training data, especially when more molecules are generated (when k > 1 in top-k predictions). It may indicate that leveraging knowledge from different yet related tasks is helpful to build a more robust model.
Upon deeper look into the new task – reagents prediction task, T5Chem actually made some reasonable suggestions for those some seemly ”wrong predictions”. Figure 5 shows one reaction from test set that T5Chem failed to predict. The proposed reagents do not have the exact match and therefore being labelled as ‘incorrect”. But similar transformation (reaction class template 112) under proposed conditions can be found in training dataset.
Figure 5:

A alcohol protection reaction with an acetate group. The ground true answer is colored in red. T5Chem failed to predict the exact match, but the proposed predictions are also reasonable despite they are identified as ”wrong predictions”. Similar transformation (reaction class template 112) under the top-2 proposed conditions can be found in training dataset.
Previously, people needed to train models separately for different tasks. By using mixed dataset, we can obtain one common model that is able to do three tasks at the same time without further fine-tuning.
The other two tasks, which include reaction type classification and reaction yield prediction, take whole reaction sequence as inputs, we hypothesized that the model may benefit from transfer learning. We first trained a reaction type classification model as an individual task, and got an accuracy of 99.6% in reaction type classification with USPTO_500_MT. This result is close to that for USPTO_TPL as shown in Table 2. The classification head of T5Chem was then replaced by a regression head for reaction yield prediction. This transfer learning model achieves better performance on yield prediction than directly training in terms of both R2(0.22 v.s. 0.20) and MAE(17.5 vs. 17.8) in percentage yield. Furthermore, we build a combined model to train both tasks together. During training, we calculated summation of losses from individual tasks and selected the best checkpoint based on validation loss. The combined model got an accuracy of 99.4% in reaction type classification, R2 of 0.22 and MAE of 17.8 in reaction yield prediction. The results are comparable to its counterpart that be trained individually. The prediction accuracy within ±10(%) absolute error range is 39.7% for this combined model.
Table 7 summarized the results of T5Chem on USPTO_500_MT. We observed promising results on this multi-task dataset. Note that reaction yield prediction in USPTO has always been a challenging task as there are many noises in this dataset. The best model till now applies yield prediction at different mass scales, and only get R2 of 0.117 (gram) and 0.195 (sub-gram).53
Table 7:
Multi-task testing results with T5Chem on USPTO_500_MT. The five tasks are grouped into two subgroups. The first subgroup includes forward reaction prediction, single-step retrosynthesis and reagents prediction, as they are all sequence-to-sequence tasks and can share the same model architecture. The second subgroup consists of reaction classification and reaction yield prediction. Both tasks take the whole reaction sequence as inputs, and can be trained at the same time by combining their loss functions together. Only one model was trained per group with the mixed dataset.
| Task Type | Forward | Retrosynthesis | Reagents | Classification | Yield |
|---|---|---|---|---|---|
| Metrics | Top-1 Accuracy | Top-1 Accuracy | Top-1 Accuracy | Accuracy | R |
| Results | 97.5% | 72.9% | 24.9% | 99.4% | 0.46 |
Model interpretation with SHAP
In order to explain predictions given by our T5Chem, we applied SHAP70 (https://github.com/slundberg/sl to explain T5Chem predictions at the functional group level. SHAP is short for SHapley Additive exPlanations, which has been one of the most popular tools to decipher machine learning models via game theoretic approach. It measures the contributions to the final outcome (i.e., prediction) from each player (i.e., feature) separately among the coalition. For a particular prediction, every input token was assigned a SHAP value, which can be viewed as its contribution to that prediction. For a better visualization to reveal chemical insights, tokens are grouped as functional groups.
Figure 6 shows one reaction example from USPTO_500_MT testset. It is a Pt catalyzed reduction. Input compounds for each task are colored based on their SHAP values using bwr colormap from matplotlib.82 Generally, SHAP values are calculated for each output token with regard to every input token. Here we map tokens back into atoms and clustered them into functional groups with EFGs package (https://github.com/HelloJocelynLu/EFGs).83 In forward prediction and retrosynthesis, we summed the SHAP values over those changed atoms, as we would like to reveal contributions from inputs that lead to the key transformation. In reagents prediction, we examined on all output atoms since we want to have an overview for whole reaction environment.
Figure 6:

Visualize SHAP values in multi-task predictions. The intensity of color stands for the magnitude of SHAP values. Positive contributions are drew in red and negative contributions are drew in blue.
As expected, the nitro group, as well as its reduced species – amine group, have strong positive contributions to this transformation in all three tasks. Interestingly, the aromatic ring also shows strong positive contributions in all tasks even though it is not directly involved in this reaction. Recall that nitro groups in alkyl and aryl nitro compounds are in different chemical environments, and behave differently, it may imply that our model learned to pay attention to key substructures even though they are not directly involved in a transformation. In forward prediction, the catalyst Pt also have positive contribution to product as expected. In retroaynthesis, we noticed that chloride and another side chains also showed some SHAP intensities. It may due to the fact that this compound is also potentially synthesizable by chlorination or ether synthesis, but with much lower probability, as indicated by more predictions if we increased the beam size.
Figure 7 A) shows most influential reaction components in C-N coupling reactions.43 The SHAP values indicate that the selections of aryl halides, additives and catalysts contribute most to all yield predictions. Among which 4-chloromethoxybenzene plays the most significant role in reaction yield prediction. We noticed that the benzene ring and chloride substitution have strong negative contributions. This observation can be demonstrated by the fact that the average reaction yield with 4-chloromethoxybenzene as one reactant is 30% lower than total average.
Figure 7:

Visualize SHAP values in reaction yield predictions. A) SHAP values show that the selections of aryl halides, additives and catalysts contribute most to yield predictions. Among all compounds, 4-chloromethoxybenzene is the most influential compound with negative contributions. B) Two reactions that only differ in reactant A have distinguished reaction yields. The benzene and chloride in 4-chloromethoxybenzene have negative contributions in low yield reaction prediction while the iodide and pyridine ring in 2-iodopyridine show positive contributions in high yield reaction prediction.
To show how SHAP value explanation can help us optimize reactions, we select two reactions with distinguished reaction yields as illustrated in Figure 7 B). These two reactions are only different on reactant A, aryl halides, and have the same reaction conditions. T5Chem predicts both reaction yields successfully. We noticed that the benzene and chloride in 4-chloromethoxybenzene have negative contributions to T5Chem predictions while the iodide and pyridine ring in 2-iodopyridine show positive contributions. This example implies that one may modify negative contributed substructures in reaction components to get a higher predicted yield.
Conclusion
In this work, we presented an explainable and unified transformer model T5Chem for multiple machine learning tasks related to organic chemistry synthesis. Our T5Chem shows state-of-the-art performances for four distinct types of reaction prediction tasks using four different open-source datasets. Furthermore, we introduced a new dataset USPTO_500_MT for multi-task machine learning of chemical reactions, including forward reaction prediction, retrosynthesis, reagents prediction, reaction type classification (500 classes) and reaction yield prediction. Our results showed that T5Chem models trained with multiple tasks are more robust and can benefit from mutual learning on related tasks. Finally, we demonstrated the applicability of SHAP to explain T5Chem predictions at the functional group level, which provides a way to demystify sequence-based deep learning models in chemistry. This sets the stage to develop T5Chem into a widely applicable and versatile machine learning framework for a variety of prediction tasks in molecular science.
Supplementary Material
Figure S1-S2. Data distribution on USPTO_500_MT.
Figure S3. Learning curve for pre-trained model vs. model trained from scratch.
Table S1. Results for different tokenizations on USPTO_500_MT.
Table S2. Results for multi-task training using combined loss functions.
Table S3. Results for data augmentation for sequence-to-sequence tasks.
Acknowledgement
The authors thank the support by NIH (R35- GM127040) and computing resources provided by NYU-ITS.
Footnotes
Data and Software Availability
T5Chem source codes and datasets are accessible through: https://yzhang.hpc.nyu.edu/T5Chem.
References
- (1).Schwaller P; Laino T Machine Learning in Chemistry: Data-Driven Algorithms, Learning Systems, and Predictions; Chapter 4, pp 61–79. [Google Scholar]
- (2).Coley CW; Green WH; Jensen KF Machine Learning in Computer-Aided Synthesis Planning. Acc Chem Res 2018, 51, 1281–1289. [DOI] [PubMed] [Google Scholar]
- (3).de Almeida AF; Moreira R; Rodrigues T Synthetic Organic Chemistry Driven by Artificial Intelligence. Nat Rev Chem 2019, 3, 589–604. [Google Scholar]
- (4).Struble TJ; Alvarez JC; Brown SP; Chytil M; Cisar J; DesJarlais RL; Engkvist O; Frank SA; Greve DR; Griffin DJ; Hou X; Johannes JW; Kreatsoulas C; Lahue B; Mathea M; Mogk G; Nicolaou CA; Palmer AD; Price DJ; Robinson RI; Salentin S; Xing L; Jaakkola T; Green WH; Barzilay R; Coley CW; Jensen KF Current and Future Roles of Artificial Intelligence in Medicinal Chemistry Synthesis. J Med Chem 2020, 63, 8667–8682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kayala MA; Azencott C-A; Chen JH; Baldi P Learning to Predict Chemical Reactions. J Chem Inf Model 2011, 51, 2209–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kayala MA; Baldi P Reactionpredictor: Prediction of Complex Chemical Reactions at the Mechanistic Level Using Machine Learning. J Chem Inf Model 2012, 52, 2526–2540. [DOI] [PubMed] [Google Scholar]
- (7).Fooshee D; Mood A; Gutman E; Tavakoli M; Urban G; Liu F; Huynh N; Van Vranken D; Baldi P Deep Learning for Chemical Reaction Prediction. Mol Syst Des Eng 2018, 3, 442–452. [Google Scholar]
- (8).Wei JN; Duvenaud D; Aspuru-Guzik A Neural Networks for the Prediction of Organic Chemistry Reactions. ACS Cent Sci 2016, 2, 725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Segler MH; Waller MP Neural-Symbolic Machine Learning for Retrosynthesis and Reaction Prediction. Chem Eur J 2017, 23, 5966–5971. [DOI] [PubMed] [Google Scholar]
- (10).Coley CW; Barzilay R; Jaakkola TS; Green WH; Jensen KF Prediction of Organic Reaction Outcomes Using Machine Learning. ACS Cent Sci 2017, 3, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bradshaw J; Kusner MJ; Paige B; Segler MHS; Hernández-Lobato JM A Generative Model for Electron Paths. arXiv preprint arXiv:1805.10970 2018, [Google Scholar]
- (12).Jin W; Coley CW; Barzilay R; Jaakkola T Predicting Organic Reaction Outcomes With Weisfeiler-Lehman Network. arXiv preprint arXiv:1709.04555 2017, [Google Scholar]
- (13).Coley CW; Jin W; Rogers L; Jamison TF; Jaakkola TS; Green WH; Barzilay R; Jensen KF A Graph-Convolutional Neural Network Model for the Prediction of Chemical Reactivity. Chem Sci 2019, 10, 370–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lee AA; Yang Q; Sresht V; Bolgar P; Hou X; Klug-McLeod JL; Butler CR Molecular Transformer Unifies Reaction Prediction and Retrosynthesis Across Pharma Chemical Space. Chem Commun (Camb) 2019, 55, 12152–12155. [DOI] [PubMed] [Google Scholar]
- (15).Schwaller P; Gaudin T; Lanyi D; Bekas C; Laino T “Found in Translation”: Predicting Outcomes of Complex Organic Chemistry Reactions Using Neural Sequence-to-Sequence Model. Chem Sci 2018, 9, 6091–6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Schwaller P; Laino T; Gaudin T; Bolgar P; Hunter CA; Bekas C; Lee AA Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction. ACS Cent Sci 2019, 5, 1572–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tetko IV; Karpov P; Van Deursen R; Godin G State-of-the-Art Augmented NLP Transformer Models for Direct and Single-Step Retrosynthesis. Nat Commun 2020, 11, 5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Pesciullesi G; Schwaller P; Laino T; Reymond JL Transfer Learning Enables the Molecular Transformer to Predict Regio- and Stereoselective Reactions on Carbohydrates. Nat Commun 2020, 11, 4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Coley CW; Rogers L; Green WH; Jensen KF Computer-Assisted Retrosynthesis Based on Molecular Similarity. ACS Cent Sci 2017, 3, 1237–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hasic H; Ishida T Single-Step Retrosynthesis Prediction Based on the Identification of Potential Disconnection Sites Using Molecular Substructure Fingerprints. J Chem Inf Model 2021, 61, 641–652. [DOI] [PubMed] [Google Scholar]
- (21).Badowski T; Gajewska EP; Molga K; Grzybowski BA Synergy Between Expert and Machine-Learning Approaches Allows for Improved Retrosynthetic Planning. Angew Chem Int Ed Engl 2020, 59, 725–730. [DOI] [PubMed] [Google Scholar]
- (22).Molga K; Dittwald P; Grzybowski BA Navigating around Patented Routes by Preserving Specific Motifs along Computer-Planned Retrosynthetic Pathways. Chem 2019, 5, 460–473. [Google Scholar]
- (23).Baylon JL; Cilfone NA; Gulcher JR; Chittenden TW Enhancing Retrosynthetic Reaction Prediction with Deep Learning Using Multiscale Reaction Classification. J Chem Inf Model 2019, 59, 673–688. [DOI] [PubMed] [Google Scholar]
- (24).Thakkar A; Kogej T; Reymond JL; Engkvist O; Bjerrum EJ Datasets and Their Influence on the Development of Computer Assisted Synthesis Planning Tools in the Pharmaceutical Domain. Chem Sci 2020, 11, 154–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Schreck JS; Coley CW; Bishop KJM Learning Retrosynthetic Planning through Simulated Experience. ACS Cent Sci 2019, 5, 970–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Segler MH; Preuss M; Waller MP Planning Chemical Syntheses With Deep Neural Networks and Symbolic AI. Nature 2018, 555, 604–610. [DOI] [PubMed] [Google Scholar]
- (27).Dai H; Li C; Coley CW; Dai B; Song L Retrosynthesis Prediction With Conditional Graph Logic Network. arXiv preprint arXiv:2001.01408 2020, [Google Scholar]
- (28).Shi C; Xu M; Guo H; Zhang M; Tang J A Graph to Graphs Framework for Retrosynthesis Prediction. International Conference on Machine Learning. pp 8818–8827. [Google Scholar]
- (29).Liu B; Ramsundar B; Kawthekar P; Shi J; Gomes J; Luu Nguyen Q; Ho S; Sloane J; Wender P; Pande V Retrosynthetic Reaction Prediction Using Neural Sequence-to-Sequence Models. ACS Cent Sci 2017, 3, 1103–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Karpov P; Godin G; Tetko IV A Transformer Model for Retrosynthesis. International Conference on Artificial Neural Networks. 2019; pp 817–830. [Google Scholar]
- (31).Chen B; Shen T; Jaakkola TS; Barzilay R Learning to Make Generalizable and Diverse Predictions for Retrosynthesis. arXiv preprint arXiv:1910.09688 2019, [Google Scholar]
- (32).Zheng S; Rao J; Zhang Z; Xu J; Yang Y Predicting Retrosynthetic Reactions Using Self-corrected Transformer Neural Networks. J Chem Inf Model 2019, 60, 47–55. [DOI] [PubMed] [Google Scholar]
- (33).Schwaller P; Petraglia R; Zullo V; Nair VH; Haeuselmann RA; Pisoni R; Bekas C; Iuliano A; Laino T Predicting Retrosynthetic Pathways Using Transformer-Based Models and a Hyper-Graph Exploration Strategy. Chem Sci 2020, 11, 3316–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lin K; Xu Y; Pei J; Lai L Automatic Retrosynthetic Route Planning Using Template-Free Models. Chem Sci 2020, 11, 3355–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wang XR; Li YQ; Qiu JZ; Chen GY; Liu HX; Liao BB; Hsieh CY; Yao XJ RetroPrime: A Diverse, Plausible and Transformer-Based Method for Single-Step Retrosynthesis Predictions. Chem Eng J 2021, 420, 129845. [Google Scholar]
- (36).Mao K; Xiao X; Xu T; Rong Y; Huang J; Zhao P Molecular Graph Enhanced Transformer for Retrosynthesis Prediction. Neurocomputing 2021, 457, 193–202. [Google Scholar]
- (37).Nam J; Kim J Linking the Neural Machine Translation and the Prediction of Organic Chemistry Reactions. arXiv preprint arXiv:1612.09529 2016, [Google Scholar]
- (38).Marcou G; Aires de Sousa J; Latino DA; de Luca A; Horvath D; Rietsch V; Varnek A Expert System for Predicting Reaction Conditions: The Michael Reaction Case. J Chem Inf Model 2015, 55, 239–250. [DOI] [PubMed] [Google Scholar]
- (39).Maser MR; Cui AY; Ryou S; DeLano TJ; Yue Y; Reisman SE Multilabel Classification Models for the Prediction of Cross-Coupling Reaction Conditions. J Chem Inf Model 2021, 61, 156–166. [DOI] [PubMed] [Google Scholar]
- (40).Segler M; Waller M Modelling Chemical Reasoning to Predict and Invent Reactions. Chem Eur J 2016, 23, 6118–28. [DOI] [PubMed] [Google Scholar]
- (41).Gao H; Struble TJ; Coley CW; Wang Y; Green WH; Jensen KF Using Machine Learning to Predict Suitable Conditions for Organic Reactions. ACS Cent Sci 2018, 4, 1465–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Kite S; Hattori T; Murakami Y Estimation of Catalytic Performance by Neural Network—Product Distribution in Oxidative Dehydrogenation of Ethylbenzene. Appl Catal A: Gen 1994, 114, L173–L178. [Google Scholar]
- (43).Ahneman DT; Estrada JG; Lin S; Dreher SD; Doyle AG Predicting Reaction Performance in C–N Cross-Coupling Using Machine Learning. Science 2018, 360, 186–190. [DOI] [PubMed] [Google Scholar]
- (44).Sandfort F; Strieth-Kalthoff F; Kuhnemund M; Beecks C; Glorius F A Structure-Based Platform for Predicting Chemical Reactivity. Chem 2020, 6, 1379–1390. [Google Scholar]
- (45).Granda JM; Donina L; Dragone V; Long D-L; Cronin L Controlling an Organic Synthesis Robot With Machine Learning to Search for New Reactivity. Nature 2018, 559, 377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Fu Z; Li X; Wang Z; Li Z; Liu X; Wu X; Zhao J; Ding X; Wan X; Zhong F; Wang D; Luo X; Chen K; Liu H; Wang J; Jiang H; Zheng M Optimizing Chemical Reaction Conditions Using Deep Learning: A Case Study for the Suzuki–Miyaura Cross-Coupling Reaction. Org Chem Front 2020, 7, 2269–2277. [Google Scholar]
- (47).Eyke NS; Green WH; Jensen KF Iterative Experimental Design Based on Active Machine Learning Reduces the Experimental Burden Associated With Reaction Screening. React Chem Eng 2020, 5, 1963–1972. [Google Scholar]
- (48).Nielsen MK; Ahneman DT; Riera O; Doyle AG Deoxyfluorination with Sulfonyl Fluorides: Navigating Reaction Space with Machine Learning. J Am Chem Soc 2018, 140, 5004–5008. [DOI] [PubMed] [Google Scholar]
- (49).Sato A; Miyao T; Funatsu K Prediction of Reaction Yield for Buchwald-Hartwig Cross-coupling Reactions Using Deep Learning. Mol Inform 2021, e2100156. [DOI] [PubMed] [Google Scholar]
- (50).Zhu X-Y; Ran C-K; Wen M; Guo G-L; Liu Y; Liao L-L; Li Y-Z; Li ML; Yu D-G Prediction of Multicomponent Reaction Yields Using Machine Learning. Chin J Chem 2021, [Google Scholar]
- (51).Probst D; Schwaller P; Reymond J-L Reaction Classification and Yield Prediction using the Differential Reaction Fingerprint DRFP. ChemRxiv 2021, [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Saebi M; Nan B; Herr J; Wahlers J; Guo Z; Zurański A; Kogej T; Norrby P-O; Doyle A; Wiest O; Chawla N On the Use of Real-World Datasets for Reaction Yield Prediction. ChemRxiv 2021, [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Schwaller P; Vaucher AC; Laino T; Reymond J-L Prediction of Chemical Reaction Yields Using Deep Learning. Mach Learn: Sci Technol 2021, 2, 015016. [Google Scholar]
- (54).Jiang S; Zhang Z; Zhao H; Li J; Yang Y; Lu B-L; Xia N When SMILES smiles, Practicality Judgment and Yield Prediction of Chemical Reaction via Deep Chemical Language Processing. IEEE Access 2021, [Google Scholar]
- (55).Raccuglia P; Elbert KC; Adler PD; Falk C; Wenny MB; Mollo A; Zeller M; Friedler SA; Schrier J; Norquist AJ Machine-Learning-Assisted Materials Discovery Using Failed Experiments. Nature 2016, 533, 73–76. [DOI] [PubMed] [Google Scholar]
- (56).Schwaller P; Probst D; Vaucher AC; Nair VH; Kreutter D; Laino T; Reymond JL Mapping the Space of Chemical Reactions Using Attention-Based Neural Networks. Nat Mach Intell 2021, 3, 144–152. [Google Scholar]
- (57).Schneider N; Lowe DM; Sayle RA; Landrum GA Development of a Novel Fingerprint for Chemical Reactions and Its Application to Large-Scale Reaction Classification and Similarity. J Chem Inf Model 2015, 55, 39–53. [DOI] [PubMed] [Google Scholar]
- (58).Chuang KV; Keiser MJ Comment on “Predicting Reaction Performance in C–N Cross-Coupling Using Machine Learning”. Science 2018, 362. [DOI] [PubMed] [Google Scholar]
- (59).Estrada JG; Ahneman DT; Sheridan RP; Dreher SD; Doyle AG Response to Comment on “Predicting Reaction Performance in C–N Cross-Coupling Using Machine Learning”. Science 2018, 362. [DOI] [PubMed] [Google Scholar]
- (60).Liu S; Wang H; Liu W; Lasenby J; Guo H; Tang J Pre-training Molecular Graph Representation with 3D Geometry. arXiv preprint arXiv:2110.07728 2021, [Google Scholar]
- (61).Pattanaik L; Ganea O-E; Coley I; Jensen KF; Green WH; Coley CW Message Passing Networks for Molecules with Tetrahedral Chirality. arXiv preprint arXiv:2012.00094 2020, [Google Scholar]
- (62).Weininger D SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules. J Chem Inform Comput Sci 1988, 28, 31–36. [Google Scholar]
- (63).Jastrzebski S; Lesniak D; Czarnecki WM Learning to Smile(s). arXiv preprint arXiv:1602.06289 2016, [Google Scholar]
- (64).Segler MH; Kogej T; Tyrchan C; Waller MP Generating Focused Molecule Libraries for Drug Discovery With Recurrent Neural Networks. ACS Cent Sci 2018, 4, 120–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Vaswani A; Shazeer N; Parmar N; Uszkoreit J; Jones L; Gomez AN; Kaiser L; Polosukhin I Attention Is All You Need. Advances in neural information processing systems. pp 5998–6008. [Google Scholar]
- (66).Devlin J; Chang M-W; Lee K; Toutanova K Bert: Pre-training of Deep Bidirectional Transformers for Language Understanding. arXiv preprint arXiv:1810.04805 2018, [Google Scholar]
- (67).Fabian B; Edlich T; Gaspar H; Segler M; Meyers J; Fiscato M; Ahmed M Molecular Representation Learning With Language Models and Domain-Relevant Auxiliary Tasks. arXiv preprint arXiv:2011.13230 2020, [Google Scholar]
- (68).Irwin R; Dimitriadis S; He J; Bjerrum EJ Chemformer: A Pre-Trained Transformer for Computational Chemistry. Mach Learn: Sci Technol 2021, [Google Scholar]
- (69).Raffel C; Shazeer N; Roberts A; Lee K; Narang S; Matena M; Zhou Y; Li W; Liu PJ Exploring the Limits of Transfer Learning With a Unified Text-to-Text Transformer. arXiv preprint arXiv:1910.10683 2019, [Google Scholar]
- (70).Lundberg SM; Lee S-I In Advances in Neural Information Processing Systems 30; Guyon I, Luxburg UV, Bengio S, Wallach H, Fergus R, Vishwanathan S, Garnett R, Eds.; Curran Associates, Inc., 2017; pp 4765–4774. [Google Scholar]
- (71).Krenn M; Häse F; Nigam A; Friederich P; Aspuru-Guzik A Self-referencing Embedded Strings (SELFIES): A 100% Robust Molecular String Representation. Mach Learn: Sci Technol 2020, 1, 045024. [Google Scholar]
- (72).Landrum G RDKit: Open-Source Cheminformatics. 2006, [Google Scholar]
- (73).Paszke A; Gross S; Massa F; Lerer A; Bradbury J; Chanan G; Killeen T; Lin Z; Gimelshein N; Antiga L; Desmaison A; Kopf A; Yang E; DeVito Z; Raison M; Tejani A; Chilamkurthy S; Steiner B; Fang L; Bai J; Chintala S In Advances in Neural Information Processing Systems 32; Wallach H, Larochelle H, Beygelzimer A, d'Alché-Buc F, Fox E, Garnett R, Eds.; Curran Associates, Inc., 2019; pp 8024–8035. [Google Scholar]
- (74).Wolf T; Debut L; Sanh V; Chaumond J; Delangue C; Moi A; Cistac P; Rault T; Louf R; Funtowicz M; Davison J; Shleifer S; von Platen P; Ma C; Jernite Y; Plu J; Xu C; Le Scao T; Gugger S; Drame M; Lhoest Q; Rush A Transformers: State-of-the-Art Natural Language Processing. Proceedings of the 2020 Conference on Empirical Methods in Natural Language Processing: System Demonstrations. Online, 2020; pp 38–45. [Google Scholar]
- (75).Kim S; Chen J; Cheng T; Gindulyte A; He J; He S; Li Q; Shoemaker BA; Thiessen PA; Yu B; Zaslavsky L; Zhang J; Bolton EE PubChem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res 2021, 49, D1388–D1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Lowe DM Extraction of Chemical Structures and Reactions From the Literature. PhD Thesis, 2012. [Google Scholar]
- (77).Schneider N; Stiefl N; Landrum GA What’s What: The (Nearly) Definitive Guide to Reaction Role Assignment. J Chem Inf Model 2016, 56, 2336–2346. [DOI] [PubMed] [Google Scholar]
- (78).Lowe D Chemical reactions from US patents (1976-Sep2016). 2017; https://figshare.com/articles/dataset/Chemical_reactions_from_US_patents_1976-Sep2016_/5104873/1.
- (79).Wei JM; Yuan XJ; Hu QH; Wang SQ A Novel Measure for Evaluating Classifiers. Expert Syst Appl 2010, 37, 3799–3809. [Google Scholar]
- (80).Gorodkin J Comparing Two K-Category Assignments by a K-Category Correlation Coefficient. Comput Biol Chem 2004, 28, 367–74. [DOI] [PubMed] [Google Scholar]
- (81).Hochreiter S; Schmidhuber J Long Short-term Memory. Neural Comput 1997, 9, 1735–80. [DOI] [PubMed] [Google Scholar]
- (82).Hunter JD Matplotlib: A 2D Graphics Environment. Comput Sci Eng 2007, 9, 90–95. [Google Scholar]
- (83).Lu J; Xia S; Lu J; Zhang Y Dataset Construction to Explore Chemical Space with 3D Geometry and Deep Learning. J Chem Inf Model 2021, 61, 1095–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1-S2. Data distribution on USPTO_500_MT.
Figure S3. Learning curve for pre-trained model vs. model trained from scratch.
Table S1. Results for different tokenizations on USPTO_500_MT.
Table S2. Results for multi-task training using combined loss functions.
Table S3. Results for data augmentation for sequence-to-sequence tasks.