

Abstract
Purpose:
Somatic driver mutations in TP53 are associated with triple negative breast cancer (TNBC) and poorer outcomes. Breast cancers in women of African ancestry (AA) are more likely to be TNBC and have somatic TP53 mutations than cancers in non-Hispanic White (NHW) women. Missense driver mutations in TP53 have varied functional impact including loss-of-function (LOF) or gain-of-function (GOF) activity, and dominant negative (DNE) effects. We aimed to determine if there were differences in somatic TP53 mutation types by patient ancestry or TNBC status.
Methods:
We identified breast cancer datasets with somatic TP53 mutation data, ancestry, age, and hormone receptor status. Mutations were classified for functional impact using published data and type of mutation. We assessed differences using Fisher’s exact test.
Results:
From 96 breast cancer studies, we identified 2964 women with somatic TP53 mutations: 715 (24.1%) Asian, 258 (8.7%) AA, 1931 (65.2%) NHW, and 60 (2%) Latina. The distribution of TP53 mutation type was similar by ancestry. However, 35.8% of tumors from NHW individuals had GOF mutations compared to 29% from AA individuals (p=0.04). Mutations with DNE activity were positively associated with TNBC (OR=1.37, p=0.03) and estrogen receptor (ER) negative status (OR=1.38; p=0.005).
Conclusions:
Somatic TP53 mutation types did not differ by ancestry overall, but GOF mutations were more common in NHW women than AA women. ER negative and TNBC tumors are less likely to have DNE+ TP53 mutations which could reflect biological processes. Larger cohorts and functional studies are needed to further elucidate these findings.
INTRODUCTION
Tumor protein 53, encoded by TP53, is a transcription factor with tumor suppressive activity that regulates genes in response to cellular stress. Pathways regulated by TP53 include cell cycle check point, senescence, DNA repair, cell metabolism and apoptosis. Somatic mutations in TP53 are the most common genetic abnormality in multiple cancers. TP53 is mutated in 40–60% of breast cancers [1–3]. Mutated TP53 is a negative prognostic factor and is associated with aggressive triple-negative breast cancers (TNBCs) and basal-like breast cancers [4, 5].
Over eighty percent of TP53 mutations are missense mutations with consequences that differ depending on mutation position and amino acid change [6]. Pathogenic somatic mutations in TP53 often disrupt DNA binding capability, impair transcriptional activity and result in other loss-of-function (LOF) effects. However, a subset of missense somatic variants demonstrate new gain-of-function (GOF) activities. GOF activity is typically mediated by the mutant protein binding to other tumor suppressive or oncogenic proteins or to novel regulatory regions [7]. GOF mutations result in accelerated tumor onset, metastasis, drug resistance and poorer survival outcomes [8, 9]. TP53 missense mutations can also display dominant negative activity (DNE), in which a mutant TP53 protein disrupts the activity of non-mutant protein partners including TP63 and TP73 during tetramerization [10]. DNE is more common of hotspot mutations, sites where approximately 30% of somatic TP53 mutations occur, and may contribute to accelerated loss of heterozygosity and tumor progression [11]. Because the importance of TP53 mutations has been well-established for decades, there are abundant functional studies identifying LOF, GOF, and DNE activity for specific TP53 mutations.
TNBCs, which are negative for estrogen receptor (ER) and progesterone receptor (PR) expression and lack HER2 amplification, have poorer prognoses compared to ER-positive breast cancer subtypes [12]. TNBCs are more likely to have somatic TP53 mutations than other types of breast cancer such as Luminal A and Luminal B subtypes [13]. Interestingly, racial differences are observed between different breast cancer subtypes; TNBCs occur more frequently in women of African Ancestry (AA) (28–30%) or Latina ethnicity (17.5%) compared to non-Hispanic White (NHW) (12–15%) women [14–18]. AA women have a 42% higher breast cancer mortality rate [19–21] and a higher risk of tumor recurrence than NHW women (hazard ratio, 2.22; CI; 1.05–4.67)[22].
As TNBCs are more common in breast tumors from AA women than NHW women, and TP53 mutations are more frequently observed in TNBC than other subtypes, it is not surprising that the proportion of all breast tumors with TP53 mutations is 1.5- to 1.6-fold higher in AA than NHW women [22–24]. While there has been extensive research about overall TP53 somatic mutation frequency by race, there has been little investigation to determine if there are differences by TP53 mutation type. Given that TP53 mutation effects can impact prognosis, mutation type is an important consideration [7, 9, 25]. Because of the differences in clinical outcomes between AA women and NHW women, even after accounting for subtype differences, and the literature supporting different outcomes for GOF versus LOF TP53 mutations, we hypothesized that there would be frequency differences in types of TP53 mutations across racial and ethnic groups. To test this hypothesis, we compared the racial distribution of TP53 mutation type in breast cancer using existing published and unpublished datasets.
METHODS
Summary of data
This study was approved by the Ohio State Cancer Institutional Review Board. Data for this study were ascertained from multiple sources including The Ohio State University Total Cancer Care repository, existing data in publicly accessible databases, existing data in publications, and unpublished data contributed by study authors. A description of all included studies is detailed in Supplementary Table S1.
Study inclusion and exclusion criteria
We included data from women with breast tumors with somatic TP53 mutations and available ancestry information. For inclusion, all studies must have sequenced tumor DNA for TP53 using any method (Sanger sequencing or next-generation targeted, exome, or whole genome sequencing) and at least included exons 5–8 which contain the majority of TP53 mutations [26]. We excluded studies that used immunohistochemistry or other non-DNA-sequencing based methods to infer TP53 mutation genotype. All likely invasive stages, grades, and morphologies of primary breast tumor were considered. Non-invasive ductal carcinoma in-situ tumors were excluded because only ~40% of these lesions progress to invasive cancer which could vary by TP53 mutation status [27]. Data were annotated with race and ethnicity by the original authors or were from homogeneous populations. Studies were excluded if they lacked ancestry data or represented a unique population that was underpowered to detect differences. If available, patient age, tumor grade, stage, receptor status, and morphology data were collected. We considered studies of any design that fit the criteria, including population and clinical-based studies.
Data from unpublished datasets
Previously unpublished TP53 somatic mutation data and self-reported race and ethnicity were ascertained from The Ohio State University’s Total Cancer Care repository for 143 women (Supplementary Table S2). Other previously or partially unpublished data included 163 individuals with TP53 mutant breast tumors enrolled through The Western New York Exposures and Breast Cancer (WEB) study, 24 through The Sylvester Comprehensive Cancer Center, and 21 through The City of Hope Comprehensive Cancer Center. Patients were enrolled through protocols approved by their respective institutional review boards.
Data from publicly accessible databases
We identified individuals and studies in databases with somatic mutation information that met inclusion criteria. From the International Agency for Research on Cancer (IARC)[28], 1254 individuals from 78 studies met inclusion criteria, as well as 333 from The Cancer Genome Atlas (TCGA), and 637 from the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) [29, 30] (Supplementary Table S1).
Data from literature review
To obtain additional data from previously published work, we conducted a literature search for studies in which individual level TP53 data and race/ethnicity information were available. A PubMed search using “TP53 and race” identified 277 articles. We excluded articles: 1) with samples already captured from database search; 2) of cancers other than breast; 3) on germline variants or polymorphisms in TP53; and 4) where study inclusion were not met. For studies without individual level race and/or ethnicity information, we contacted authors by e-mail to request this information. All studies included are listed in Supplementary Table S1.
Categorizing TP53 missense variants
We used a standardized approach to evaluate findings from IARC and other TP53 literature. All missense variant annotations were based on existing functional studies with cell culture, yeast, or animal experiments; we did not consider in-silico testing alone for inclusion. However, in cases of mutations with uncharacterized function, we utilized PHANTM (Broad Institute) to exclude variants with a predicted function close to wildtype TP53 (maximum PHANTM score <0, ~50 variants)[11]. We excluded well-established germline polymorphisms, such as p.P72R, and mutations with activity comparable to wildtype TP53. TP53 mutations were categorized by function (GOF or LOF) and dominant negative activity (DNE+ or DNE−) as separate criteria.
We described function as GOF or LOF. GOF mutations resulted in significantly different activity from both TP53 null and TP53 WT proteins such as novel transcript activity, TP73 interference, growth advantage, and facilitation of oncogene activity. LOF mutations had evidence of protein truncation, loss of tetramerization, or activity comparable to TP53 null. When we found reports of both LOF and GOF activities, but not direct contradictions for the same TP53 function, we categorized variants as GOF. Variants with limited functional data available and PHANTM prediction scores that differed from wild-type were annotated as unknown. DNE+ variants were those with published evidence that the TP53 mutant protein interfered with TP53 WT function in heterozygous cells. DNE+ mutants formed heterotetramers with TP53 WT units and changed TP53 WT function, causing a dominant GOF or LOF effect. DNE+ mutations may also interfere with TP63 and TP73, and therefore may have unique biological impact beyond GOF [10]. Transcript-truncating mutations, such as nonsense, splicing, frameshifts, and large deletions, were assumed to be LOF without DNE because these mutations will activate nonsense-mediated decay and result in loss of a functional protein. Hotspot codons in the DNA binding domain may have different functional properties than missense mutations elsewhere [26, 31]. These sites were at positions 175, 245, 248, 249, 273 and 282, and CpG hotspot mutations were defined as C to T transitions at those codons plus R158H and P152L as described [7]. CpG hotspot mutations were studied separately because they may be part of a mutational signature [26, 32].
For tumors with multiple TP53 somatic mutations, we considered the sum of multiple predicted effects. If any mutations were DNE+, the tumor was considered DNE+. GOF and LOF mutations were prioritized over unknown or functional mutations. Tumors with both GOF and LOF mutations were called GOF/LOF.
Statistical analyses
A Fisher’s exact test for count data was used for comparisons between mutation categories (GOF/LOF, DNE/not DNE or CpG hotspot/not hotspot) and race, TNBC status, and ER status. For comparisons of mutation categories and age, a Welch Two Sample t-test was used. Analyses were run in R version 3.6.3 (2020-02-29) [33]. A comparison-wise p-value of 0.05 was considered significant.
RESULTS
Characteristics of study population
The study population is summarized in Table 1. We included somatic TP53 mutation data from 2964 breast cancers from 96 studies for analysis (Supplementary Table S1). Patients were categorized into 4 racial/ethnic groups. The study population was 65.2% NHW (n=1931), 24.1% Asian (n=715), and 8.7% AA (n=258). Two percent (n=60) of patients had Hispanic or Latina ethnicity with European or undefined race (n=47 [1.6%] and n=13 [0.4%], respectively). Populations excluded from analysis due to low representation included Pacific Islander, Ashkenazi Jewish, Southwest Asian/North African, Indian Asian, and Latina AA women.
Table 1:
Study Population Demographics and Tumor Characteristics
| Study Population Demographics | NHW n (%) | AA n (%) | Asian n (%) | Latina n (%) | Total n (%) |
|---|---|---|---|---|---|
| Individuals1 | 1931 (65%) | 258 (9%) | 715 (24%) | 60 (2.0%) | 2964 |
| Breast Cancer dx age available2 | 1461 (76%) | 162 (63%) | 286 (40%) | 60 (100%) | 1969 (66%) |
| Age range (years) | 21– 96 | 24 −84 | 26–90 | 31–72 | 21–90 |
| Median age (years) | 56 | 49 | 47 | 52 | |
| p-value | [Ref] | <0.0001 | <0.0001 | 0.0161 | |
| Tumor Characteristics 2 | |||||
| ER+ | 553 (49%) | 54 (36%) | 60 (42%) | 37 (62%) | 704 (47.5%) |
| ER− | 575 (51%) | 95 (64%) | 84 (58%) | 23 (38%) | 777 (52.5%) |
| No ER data | 803 | 109 | 571 | 0 | 1483 |
| Non-TNBC | 660 (64%) | 47 (51%) | 31 (69%) | 44 (77%) | 782 (64%) |
| TNBC | 366 (36%) | 46 (49%) | 14 (31%) | 13 (23%) | 439 (36%) |
| No TNBC data | 905 | 165 | 670 | 3 | 1743 |
NHW, non-Hispanic White, AA, African ancestry; n, number; % percentage; ref, reference population; dx, diagnosis
Percentages were calculated for participants in the study for each racial/ethnic group category
Percentages for age at diagnosis and tumor characteristics were calculated only including individuals for whom marker information was available within each racial/ethnic group category
Ages at diagnosis were available for 1969 patients. Across the study population, ages ranged from 21 to 96 years, with a median age of 54 years and an average age of 55 years. By racial/ethnic group, median ages were 49 for AAs, 47 for Asians, 56 for NHW, and 52 years for Latina women.
Only a subset of tumors had receptor data available. ER status was available for 1481 tumors, with 47.5% ER+ (n=704) and 52.5% ER− (n=777). A smaller subset had additional tumor information. TNBC status was available for 1221 tumors with 36% classified as TNBC (n=439) and 64% as non-TNBC (n=782). Data were collected for morphology, grade, and stage, but were not used due to low availability across the datasets and the high number of categories.
Characteristics of mutations
Fifty-four tumors had multipleTP53 mutations, for a total of 3024 TP53 mutations across all 2964 patients (Supplementary Tables S3 and S4). A majority of mutations were missense (65%, n=1972). We identified 829 distinct alterations, including 427 missense, 63 nonsense, 209 frameshift, 58 in-frame insertion/deletions, 2 large insertion/deletions, and 56 unique splicing changes (Supplementary Tables S3 and S4). The most frequent missense mutations were p.R175H (n=138, 4.7%), p.R248Q (n=104, 3.4%), p.R273H (n=87, 2.9%), and p.R248W (n=73, 2.5%), all known hotspot mutations (Figure 1). Overall, mutations at reported hotspot sites (R175, G245, R248, R273 and R282) accounted for 20% of mutations (n=616). More specifically, mutations of CpG nucleotides at hotspots accounted for 17.5% of mutations (n=530).
Figure 1: TP53 Gene Structure and Mutation Frequency.
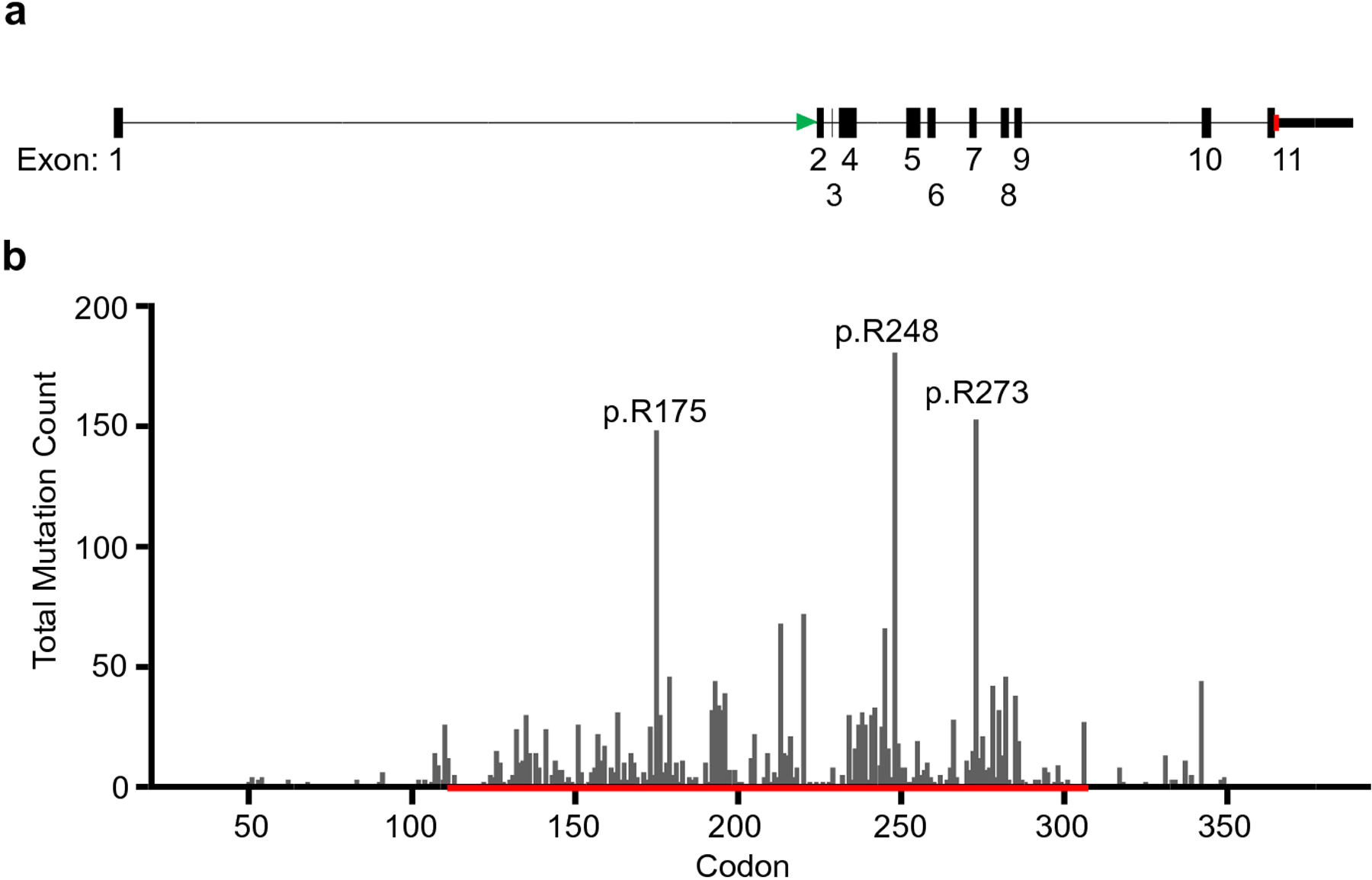
(a). A diagram of the intron/exon structure of TP53 is drawn to scale. A green arrow denotes the exon containing the start codon and a red arrow denotes the exon containing the stop codon.(b). The frequency of somatic TP53 mutations by codon is plotted. Exons 5 through 8 are denoted by a red line. Intronic mutations affecting splicing are not included. The figure was created in GraphPad Prism 8.
For functional classification, tumors were analyzed based on the net effect of all TP53 mutations per tumor (n=2964). Overall, we characterized 939 mutations as GOF and 1739 LOF; 286 mutations did not have sufficient information for classification. Evaluating mutations for DNE, there were 1246 DNE+, 1190 DNE−, and 528 uncharacterized mutations.
Association of race, tumor characteristics, and age with mutation type
To determine if there were associations between race and type of mutation, we conducted Fisher’s exact test for racial and ethnic ancestry by mutation categories. No significant associations were identified overall for GOF/LOF status (p = 0.15), DNE (p=0.62), mutation hotspots (p=0.32) or CpG sites (p=0.52) and race (Table 2). However, association of GOF/LOF status and race was significant when comparing GOF versus LOF in NHW and AA patients only, with NHW patients more likely to have GOF mutations (35.8% versus 29.2%, respectively, p=0.04). We additionally tested association between ER or TNBC status and mutation type (Table 3). We identified a significant association between DNE and TNBC (p=0.03) and related ER status (p=0.005). ER− tumors and TNBCs were less likely to have TP53 somatic mutations that were DNE+. We did not identify associations between ER and GOF/LOF (p=0.51), with mutation hotspots (p=0.1514) or with CpG hotspots (p=0.24). Patients with hotspot mutations were slightly younger, with a mean age of 53.6 years versus 55.0 years for patients with non-hotspot mutations, at a level approaching significance (p=0.065) (Figure 2). We did not identify significant associations with age and GOF/LOF, with a mean age of 54.5 years for GOF and 55.0 years for LOF (p=0.52). We also found no significant association between age and DNE; the mean age was 54.5 years for DNE+, and 55.0 years for DNE− (p=0.49).
Table 2:
TP53 Mutation Frequency by Type and Population
| Populations | Total | ||||
|---|---|---|---|---|---|
| Mutation type | NHW (n=1931) | Asian (n=715) | AA (n=258) | Latina (n=60) | n=2964 |
| GOF | 623 (32.2%) | 222 (31%) | 70 (27.1%) | 24 (40%) | 939 (31.7%) |
| LOF | 1116 (57.8%) | 419 (58.6%) | 170 (65.9%) | 34 (56.7%) | 1739 (58.7%) |
| Unknown function | 192 (10%) | 74 (10.3%) | 18 (7%) | 2 (3.3%) | 286 (9.6%) |
| p-value | 0.15 | ||||
| DNE+ | 829 (43%) | 286 (40%) | 105 (40.7%) | 26 (43.3%) | 1246 (42%) |
| DNE− | 767 (39.7%) | 280 (39.2%) | 116 (45%) | 27 (45%) | 1190 (40.1%) |
| Unknown | 335 (17.3%) | 149 (20.2%) | 37 (14.3%) | 7 (11.7%) | 528 (17.8%) |
| p-value | 0.62 | ||||
| Hotspot | 416 (21.5%) | 140 (19.6%) | 44 (17%) | 13 (21.7%) | 613 (20.7%) |
| Non-hotspot | 1515 (78.5%) | 575 (80.4%) | 214 (83%) | 47 (78.3%) | 2351 (79.3%) |
| p-value | 0.32 | ||||
| Hotspot CpG | 353 (18.3%) | 125 (17.5%) | 39 (15.1%) | 13 (21.7%) | 530 (17.9%) |
| Non-hotspot CpG | 1578 (81.7%) | 590 (82.5%) | 219 (84.9%) | 47 (78.3%) | 2434 (82.1%) |
| p-value | 0.52 | ||||
n, number; GOF, gain-of-function mutation; LOF, loss-of-function mutation; DNE+, dominant negative activity present; DNE−, no dominant negative activity; NHW, non-Hispanic White, AA, African Ancestry
Table 3:
TP53 Mutation Frequency by Age and Tumor Characteristics
| Tumor Subtypes | |||||
|---|---|---|---|---|---|
| Mutation type | ER− (n=777) | ER+ (n=704) | TNBC (n=439) | Non-TNBC (n=782) | Mean age |
| GOF | 216 (27.8%) | 216 (30.7%) | 126 (28.7%) | 230 (29.4%) | 54.5 |
| LOF | 514 (66.1%) | 424 (60.2%) | 291 (66.3%) | 485 (62%) | 55.0 |
| Unknown | 47 (6%) | 64 (9.1%) | 22 (5%) | 67 (8.6%) | NA |
| p-value | 0.10 | 0.51 | 0.52 | ||
| DNE+ | 288 (37.1%) | 297 (42.2%) | 162 (36.9%) | 317 (40.5%) | 54.5 |
| DNE− | 384 (49.4%) | 285 (40.5%) | 226 (51.5%) | 331 (42.3%) | 55.0 |
| Unknown | 105 (13.5%) | 122 (17.3%) | 51 (11.6%) | 134 (17.1%) | NA |
| p-value | 0.0045 | 0.029 | 0.49 | ||
| Hotspot | 143 (18.4%) | 151 (21.4%) | 81 (18.5%) | 164 (21%) | 53.6 |
| Non-hotspot | 634 (81.6%) | 553 (78.6%) | 358 (81.5%) | 618 (79%) | 55.0 |
| p-value | 0.15 | 0.20 | 0.065 | ||
| Hotspot CpG | 126 (16.2%) | 131 (18.6%) | 75 (17.1%) | 135 (17.3%) | 54.0 |
| Non-hotspot CpG | 651 (83.8%) | 573 (81.4%) | 364 (82.9%) | 647 (82.7%) | 55.0 |
| p-value | 0.24 | 1.0 | 0.30 | ||
n, number; GOF, gain-of-function mutation; LOF, loss-of-function mutation; DNE+, dominant negative activity present; DNE−, no dominant negative activity; ER, Estrogen receptor; TNBC, Triple negative breast cancer
Figure 2: Association of age of breast cancer diagnosis with TP53 mutation characteristics.
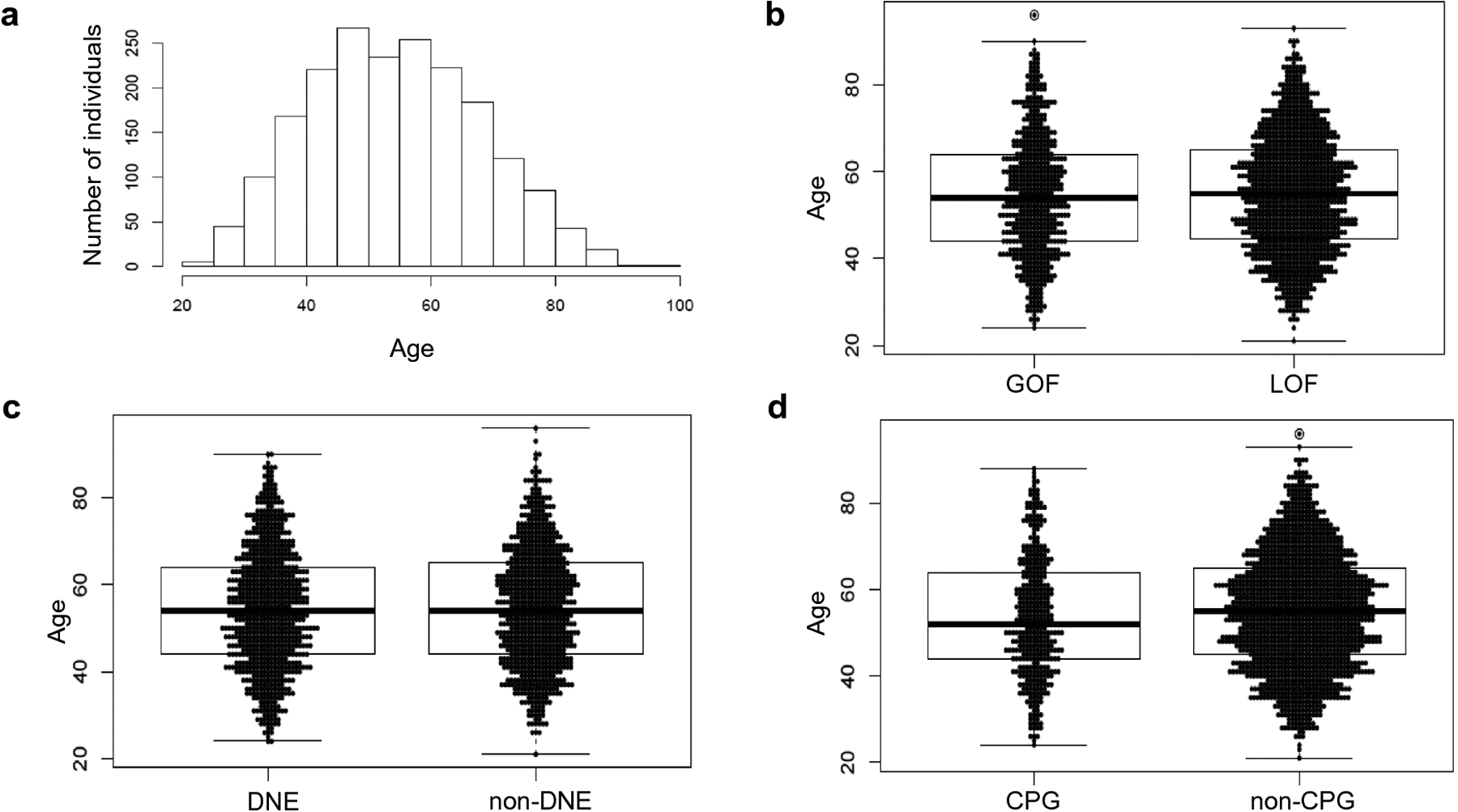
The frequency of age of breast cancer diagnosis of all individuals included in the study was plotted by histogram (a). Age of diagnosis was not significantly associated with TP53 GOF versus LOF, p-value =0.5 (b) or dominant negative effect, p-value 0.49 (c). Individuals with a TP53 mutation at a CpG hotspot were slightly, but not significantly younger at age of diagnosis (53.6 years versus 55.0 years), p-value 0.065 (d). P-values were calculated using a Welch two sample T-test. P-values < 0.05 were considered significant. The figure was created in R.
DISCUSSION
The goal of our study was to determine if the type of TP53 somatic mutation (GOF or LOF, DNE− or DNE+, hotspot status, and CpG nucleotide position) varied in frequency between patients of different ancestry. Considering that the overall rate of somatic TP53 mutations in breast cancer differs by race, this is an important concern for study of TP53-mutant breast tumors and differences in outcomes and treatment response by race [14–18]. We identified a modest difference between AA and NHW individuals, with NHWs slightly more likely to have GOF mutations.
Our finding that TP53 mutations without DNE activity were associated with TNBC (p=0.03) and ER− status (p=0.005) is novel. This association could be due to complex interactions and shared regulation of apoptotic genes between TP53 and ER. In ER+ tumors, estrogen receptor-beta (ESR2) activity has a pro-proliferative effect on TP53-wildtype tumors, but an anti-proliferative effect on TP53-mutant tumors [34]. DNE+ TP53 mutations may have unique interactions with ESR2 in ER+ tumors that drives higher DNE+ frequency. In this study, that only includes TP53-mutant tumors, we observed a higher proportion of ER− and TNBC tumors overall compared to unselected populations. This is consistent with previous studies that identified TP53 somatic mutations in ~85% of TNBC versus 40–60% of unselected breast tumors [1–3, 35]. There has been some debate about the significance of mutant TP53 DNE versus GOF activity, as many common somatic mutations, including hotspot mutations, are both DNE+ and GOF[36]. It is thus of great interest that the association with receptor status was only significant for DNE, not GOF/LOF, though functional studies are needed to better understand this phenomenon.
Our cohort included somatic TP53 mutation data from TCGA, METABRIC, and IARC data-bases, studies identified for inclusion from literature, and 351 previously unpublished cases (Supplementary Table S1). The frequency of hotspot mutations observed in our study (20%) was slightly lower than previous studies finding that 28% of TP53 mutations occurred at mutation hotspots [26]. We observed that 36% of tumors from NHW individuals had GOF mutations compared to 29% in AA individuals (p=0.04). This is opposite of what we expected to find as GOF variants have been associated with poorer prognosis or worse outcomes and breast cancers in AA women have worse outcomes [7]. We considered that this effect may be an artifact of more NHW patients sequenced with earlier technology, such as Sanger, which could bias the TP53 mutation detected to the exons more likely to have GOF mutations. However, there was no difference in use of Sanger vs NGS between these population groups, with 43.6% of NHW patients sequenced with Sanger, compared to 43.8% of AA patients. There also was no difference in the number of exons sequenced; 67.3% of NHW patients had at least exons 2–11 sequenced, compared to 68.6% of AA patients. Additionally, there was no difference in the percentage of unclassified variants between groups (7% in AA versus 9.9% in NHW for GOF/LOF, 14.3% in AA versus 17.3% in NHW for DNE). Thus, this difference is not likely due to mutation detection or classification. This paradox could be due to factors other than the TP53 mutations or those that influence aggressiveness in addition to the TP53 mutations that vary between ancestral groups such as differences in somatic mutation of other key driver genes or methylation pattern differences [23, 37]. Further studies of larger numbers of AA and NHW women are warranted to confirm this finding.
Participants with hotspot mutations were younger than those with non-hotspot mutations, with a mean age of 53.61 in hotspots versus 55.04 in non-hotspots, but this was not statistically significant (p=0.065). Age did not correlate with DNE or GOF/LOF. This finding is somewhat unexpected. Susceptibility to hotspot mutations is likely due to properties of the genetic sequence being vulnerable to mutation, rather than purely selective growth advantage of tumor cells [26]. A high proportion of hotspot mutations are CpG sites, a feature of mutation signature 1, which correlates with age, so it would seem more likely for somatic hotspot mutations at CpG sites to be associated with later age at diagnosis [32, 38]. However, a correlation for breast cancer has not yet been reported in the literature of which we are aware. Studies of TP53 hotspot mutations, mutational signatures and breast cancer age of onset may reveal additional insight.
Strengths of this study include the large number of women included from multiple sources, including previously unpublished data. Previous studies characterizing TP53 mutation types have not focused on race or ancestry. We limited the dataset to only include tumors with TP53 somatic mutations and only included participants with race or ancestry data. There are a number of limitations to this study. Many of the studies used self-reported race and ethnicity information, which may not reflect genetic ancestry, and may have been categorized differently by study, such as distinguishing NHW and Ashkenazi Jewish ethnicity. There may be differences in TP53 mutation types between ethnic groups within a racial group, such as between NHW individuals from Greece versus Finland. For studies in countries that are predominantly one racial group and for which detailed racial information was not available, we assumed that the individuals were of that racial group (e.g. Norway and European ancestry; China and Asian ancestry). Few studies only performed analyses of exons 4 through 8 which could miss more LOF variants that occur in other exons compared to GOF or DNE-associated missense variants that predominantly map to these exons. Because of the mixed data sources, this study did not include large TP53 copy number changes or loss of heterozygosity data. There may be undetected effects by gene copy number or loss of heterozygosity, either acting alone or modifying point mutation effects which could impact our findings. Finally, classifications of variants as GOF/ LOF and DNE were made based on literature. For some variants there was discordant information; we used the classifications from studies that tested more variants or included a larger number of assays. It is possible that some of the rarer missense variants were misclassified or have different effects in humans than in the system tested (e.g. yeast).
Summary
In this study, we found that somatic TP53 mutation types did not differ by race overall, but GOF mutations were more common in NHW women when compared to AA women. We uncovered a modest association between DNE− and tumor receptor status. Functional studies are needed to understand this phenomenon. Additional tumor sequencing data from more racially diverse cohorts are needed to follow-up on these findings.
Supplementary Material
Supplementary File 5: Supplementary References. A list of references for supplemental information.
Supplementary Table S1: Studies included. A table of studies for which data are included
Supplementary Table S2: OSU Total Cancer Care Participants. A table of demographic and tumor characteristics for the OSU Total Cancer Care study participants.
Supplementary Table S3: Master list of TP53 coding mutations in study and classifications. A table of all coding mutations that were included in this study.
Supplementary Table S4: Master list of TP53 confirmed and suspected splicing mutations in study and classifications
Acknowledgements:
We thank Lara Sucheston-Campbell, Philip Tsichlis, and Michael Freitas for the initial ideas leading to this study. The Ohio State University Comprehensive Cancer Center (OSU CCC) Total Cancer Care from the Biospecimen Services Shared Resource provided mutational data on breast cancer cases. The results published here are in whole or part based upon data generated by The Cancer Genome Atlas (TCGA) Research Network: https://www.cancer.gov/tcga.
Funding
The OSU CCC TCC is supported in part by National Cancer Institute grant P30 CA016058. This work was supported by R01 CA215151. JR was supported by a Pelotonia Fellowship. KCD was supported by The UNC Breast Cancer SPORE Grant CA58223 from the National Cancer Institute. OIO and DH were supported by grants from National Institutes of Health (P20 CA233307, R01 MD013452) and Breast Cancer Research Foundation. For the City of Hope Hispanic breast cancer cases: sample collection and data were collected under support from NIH R01 CA184585 (SLN and EZ); tumor sequencing was supported by the National Institute on Minority Health and Health Disparities; and tumor blocks were pulled and slides cut in the Pathology Lab Core supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviation Term
- AA
African ancestry
- DNE
Dominant negative
- ER
Estrogen receptor
- ESR2
Estrogen receptor-beta
- GOF
Gain of function
- LOF
Loss of function
- NHW
Non-Hispanic White
- OR
Odds ratio
- PR
Progesterone receptor
- TCC
Total Cancer Care
- TNBC
Triple-negative breast cancer
- TP53
Tumor protein 53
Footnotes
Publisher's Disclaimer: This AM is a PDF file of the manuscript accepted for publication after peer review, when applicable, but does not reflect post-acceptance improvements, or any corrections. Use of this AM is subject to the publisher’s embargo period and AM terms of use. Under no circumstances may this AM be shared or distributed under a Creative Commons or other form of open access license, nor may it be reformatted or enhanced, whether by the Author or third parties. See here for Springer Nature’s terms of use for AM versions of subscription articles: https://www.springernature.com/gp/open-research/policies/accepted-manuscript-terms
Conflicts of Interest
HH is on the scientific advisory board for Invitae Genetics, Promega, and Genome Medical and has stock/stock options in Genome Medical and GI OnDemand. None of these are direct conflicts with this study of somatic TP53 mutations in breast cancer.
Ethics Approval
This study was approved by the OSU Cancer Institutional Review Board. The City of Hope Institutional Review Board and the University of Chicago IRB approved study for participants enrolled at their respective sites.
Consent to Participate
Participants with previously unpublished data provided informed consent for this study. The remainder of data was de-identified and from publicly available databases or publications.
Availability of data
All data generated or analyzed during this study are included in this published article (Supplementary Tables S1–S4) or in the original publications (Supplementary File S5).
References
- 1.Olivier M, Langerød A, Carrieri P, et al. (2006) The clinical value of somatic TP53 gene mutations in 1,794 patients with breast cancer. Clin Cancer Res 12:. 10.1158/1078-0432.CCR-05-1029 [DOI] [PubMed] [Google Scholar]
- 2.Pitt JJ, Riester M, Zheng Y, et al. (2018) Characterization of Nigerian breast cancer reveals prevalent homologous recombination deficiency and aggressive molecular features. Nat Commun. 10.1038/s41467-018-06616-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang G, Wang Y, Chen B, et al. (2019) Characterization of frequently mutated cancer genes in Chinese breast tumors: a comparison of Chinese and TCGA cohorts. Ann Transl Med. 10.21037/atm.2019.04.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller LD, Smeds J, George J, et al. (2005) An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci U S A. 10.1073/pnas.0506230102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prat A, Adamo B, Cheang MCU, et al. (2013) Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist 18:123–133. 10.1634/theoncologist.2012-0397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leroy B, Fournier JL, Ishioka C, et al. (2013) The TP53 website: an integrative resource centre for the TP53 mutation database and TP53 mutant analysis. Nucleic Acids Res 41:D962–9. 10.1093/nar/gks1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamamoto S, Iwakuma T (2018) Regulators of Oncogenic Mutant TP53 Gain of Function. Cancers (Basel) 11:. 10.3390/cancers11010004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alexandrova EM, Mirza SA, Xu S, et al. (2017) p53 loss-of-heterozygosity is a necessary prerequisite for mutant p53 stabilization and gain-of-function in vivo. Cell Death Dis 8:e2661. 10.1038/cddis.2017.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou X, Hao Q, Lu H (2019) Mutant p53 in cancer therapy-the barrier or the path. J Mol Cell Biol 11:293–305. 10.1093/jmcb/mjy072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Billant O, Léon A, Le Guellec S, et al. (2016) The dominant-negative interplay between p53, p63 and p73: A family affair. Oncotarget 7:69549–69564. 10.18632/oncotarget.11774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giacomelli AO, Yang X, Lintner RE, et al. (2018) Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet 50:1381–1387. 10.1038/s41588-018-0204-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott LC, Mobley LR, Kuo T-M, Il’yasova D (2019) Update on triple-negative breast cancer disparities for the United States: A population-based study from the United States Cancer Statistics database, 2010 through 2014. Cancer 125:3412–3417. 10.1002/cncr.32207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dumay A, Feugeas JP, Wittmer E, et al. (2013) Distinct tumor protein p53 mutants in breast cancer subgroups. Int J Cancer. 10.1002/ijc.27767 [DOI] [PubMed] [Google Scholar]
- 14.Newman LA, Stark A, Chitale D, et al. (2017) Association Between Benign Breast Disease in African American and White American Women and Subsequent Triple-Negative Breast Cancer. JAMA Oncol 3:1102–1106. 10.1001/jamaoncol.2016.5598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sineshaw HM, Gaudet M, Ward EM, et al. (2014) Association of race/ethnicity, socioeconomic status, and breast cancer subtypes in the National Cancer Data Base (2010–2011). Breast Cancer Res Treat 145:753–763. 10.1007/s10549-014-2976-9 [DOI] [PubMed] [Google Scholar]
- 16.Martínez ME, Gomez SL, Tao L, et al. (2017) Contribution of clinical and socioeconomic factors to differences in breast cancer subtype and mortality between Hispanic and non-Hispanic white women. Breast Cancer Res Treat 166:185–193. 10.1007/s10549-017-4389-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sturtz LA, Melley J, Mamula K, et al. (2014) Outcome disparities in African American women with triple negative breast cancer: a comparison of epidemiological and molecular factors between African American and Caucasian women with triple negative breast cancer. BMC Cancer 14:62. 10.1186/1471-2407-14-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hines LM, Risendal B, Byers T, et al. (2011) Ethnic disparities in breast tumor phenotypic subtypes in Hispanic and non-Hispanic white women. J Womens Health (Larchmt) 20:1543–1550. 10.1089/jwh.2010.2558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doepker MP, Holt SD, Durkin MW, et al. (2018) Triple-Negative Breast Cancer: A Comparison of Race and Survival. Am Surg 84:881–888 [PubMed] [Google Scholar]
- 20.Troester MA, Sun X, Allott EH, et al. (2018) Racial Differences in PAM50 Subtypes in the Carolina Breast Cancer Study. J Natl Cancer Inst 110:176–182. 10.1093/jnci/djx135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeSantis CE, Fedewa SA, Goding Sauer A, et al. (2016) Breast cancer statistics, 2015: Convergence of incidence rates between black and white women. CA Cancer J Clin 66:31–42. 10.3322/caac.21320 [DOI] [PubMed] [Google Scholar]
- 22.Keenan T, Moy B, Mroz EA, et al. (2015) Comparison of the genomic landscape between primary breast cancer in African American versus white women and the association of racial differences with tumor recurrence. J Clin Oncol. 10.1200/JCO.2015.62.2126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huo D, Hu H, Rhie SK, et al. (2017) Comparison of Breast Cancer Molecular Features and Survival by African and European Ancestry in The Cancer Genome Atlas. JAMA Oncol. 10.1001/jamaoncol.2017.0595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carrot-Zhang J, Chambwe N, Damrauer JS, et al. (2020) Comprehensive Analysis of Genetic Ancestry and Its Molecular Correlates in Cancer. Cancer Cell 37:639–654.e6. 10.1016/j.ccell.2020.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang C, Liu J, Xu D, et al. (2020) Gain-of-function mutant p53 in cancer progression and therapy. J Mol Cell Biol 12:674–687. 10.1093/jmcb/mjaa040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baugh EH, Ke H, Levine AJ, et al. (2018) Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cowell CF, Weigelt B, Sakr RA, et al. (2013) Progression from ductal carcinoma in situ to invasive breast cancer: revisited. Mol Oncol 7:859–869. 10.1016/j.molonc.2013.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bouaoun L, Sonkin D, Ardin M, et al. (2016) TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum Mutat. 10.1002/humu.23035 [DOI] [PubMed] [Google Scholar]
- 29.Pereira B, Chin S-F, Rueda OM, et al. (2016) The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun 7:11479. 10.1038/ncomms11479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rueda OM, Sammut S-J, Seoane JA, et al. (2019) Dynamics of breast-cancer relapse reveal late-recurring ER-positive genomic subgroups. Nature 567:399–404. 10.1038/s41586-019-1007-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kato S, Han S-Y, Liu W, et al. (2003) Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci U S A 100:8424–8429. 10.1073/pnas.1431692100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alexandrov LB, Jones PH, Wedge DC, et al. (2015) Clock-like mutational processes in human somatic cells. Nat Genet 47:1402–1407. 10.1038/ng.3441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.R Core Team (2020) (2020) R: A language and environment for statistical computing. R A Lang. Environ. Stat. Comput R Found. Stat. Comput. Vienna, Austria [Google Scholar]
- 34.Mukhopadhyay UK, Oturkar CC, Adams C, et al. (2019) TP53 Status as a Determinant of Pro- vs Anti-Tumorigenic Effects of Estrogen Receptor-Beta in Breast Cancer. J Natl Cancer Inst 111:1202–1215. 10.1093/jnci/djz051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prat A, Adamo B, Cheang MCU, et al. (2013) Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist 18:123–133. 10.1634/theoncologist.2012-0397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stein Y, Aloni-Grinstein R, Rotter V (2020) Mutant p53 oncogenicity – dominant-negative or gain-of-function? Carcinogenesis. 10.1093/carcin/bgaa117 [DOI] [PubMed] [Google Scholar]
- 37.Mehrotra J, Ganpat MM, Kanaan Y, et al. (2004) Estrogen receptor/progesterone receptor-negative breast cancers of young African-American women have a higher frequency of methylation of multiple genes than those of Caucasian women. Clin cancer Res an Off J Am Assoc Cancer Res 10:2052–2057. 10.1158/1078-0432.ccr-03-0514 [DOI] [PubMed] [Google Scholar]
- 38.Tomkova M, McClellan M, Kriaucionis S, Schuster-Boeckler B (2016) 5-hydroxymethylcytosine marks regions with reduced mutation frequency in human DNA. Elife. 10.7554/eLife.17082 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary File 5: Supplementary References. A list of references for supplemental information.
Supplementary Table S1: Studies included. A table of studies for which data are included
Supplementary Table S2: OSU Total Cancer Care Participants. A table of demographic and tumor characteristics for the OSU Total Cancer Care study participants.
Supplementary Table S3: Master list of TP53 coding mutations in study and classifications. A table of all coding mutations that were included in this study.
Supplementary Table S4: Master list of TP53 confirmed and suspected splicing mutations in study and classifications
Data Availability Statement
All data generated or analyzed during this study are included in this published article (Supplementary Tables S1–S4) or in the original publications (Supplementary File S5).