

Abstract
Local, sustained drug delivery of potent therapeutics holds promise for the treatment of a myriad of localized diseases while eliminating systemic side effects. However, introduction of drug delivery depots such as viscous hydrogels or polymer-based implants is highly limited in stiff tissues such as desmoplastic tumors. Here, we present a method to create materials-free intratumoral drug depots through Tissue-Reactive Anchoring Pharmaceuticals (TRAPs). TRAPs diffuse into tissue and attach locally for sustained drug release. In TRAPs, potent drugs are modified with ECM-reactive groups and then locally injected to quickly react with accessible amines within the ECM, creating local drug depots. We demonstrate that locally injected TRAPs create dispersed, stable intratumoral depots deep within mouse and human pancreatic tumor tissues. TRAPs depots based on ECM-reactive paclitaxel (TRAP paclitaxel) had better solubility than free paclitaxel and enabled sustained in vitro and in vivo drug release. TRAP paclitaxel induced higher tumoral apoptosis and sustained better antitumor efficacy than the free drug. By providing continuous drug access to tumor cells, this material-free approach to sustained drug delivery of potent therapeutics has the potential in a wide variety of diseases where current injectable depots fall short.
Keywords: sustained release, materials-free delivery, paclitaxel, pancreatic cancer, extracellular matrix, N-hydroxysuccinimide ester
Graphical Abstract
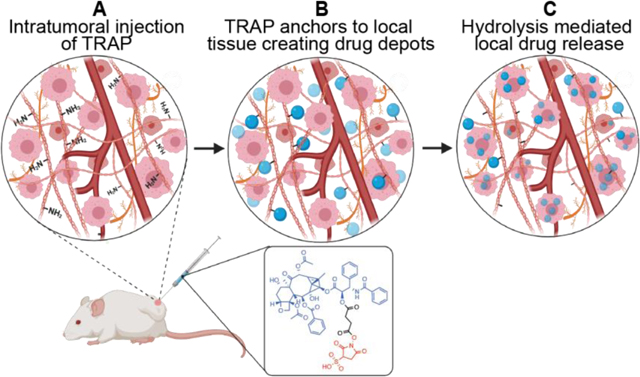
Introduction:
Sustained drug release from local drug depots has the potential to overcome the challenged of systemic toxicity and rapid clearance challenges observed with systemic drug administration [1], including delivery of chemotherapy to locally advanced tumors [2], immunosuppressive agents to transplanted organs [3], antibiotics to localized infections [4], and immune regulators [5] to lymph nodes [6] and arthritic joints [7]. Conventionally, local drug delivery depots strategies rely on implantation or injection of viscous materials such as oils [8], polymers [9] and hydrogels [10] which encapsulate and retain drugs at the administration site. Unfortunately, these materials and viscous solutions often struggle to penetrate stiff or inflamed tissues to deliver drugs deep into the target area [9,11].
In this study, we demonstrate that local depots can be administered without the use of materials. The Tissue Reactive Anchoring Pharmaceuticals (TRAPs) technology creates local drug depots through chemical labelling of tissue extracellular matrix (ECM) and subsequently releases active drug locally (Fig. 1). To create the TRAPs depots, drugs conjugated to ECM-reactive chemical groups are infused into target tissues, where they diffuse throughout the tissue and react with accessible amines in the ECM to anchor drugs through stable amide bonds. For diseases defined by densely packed stroma, TRAPs depots turn the stroma from a hinderance into an asset labeling the abundant primary amines present as lysine chains groups and N-termini protein [12,13]. Moreover, delivery of TRAPs in non-viscous, aqueous vehicles allows radial penetration of drug molecules in the dense stroma, overcoming the primary obstacle for implant-based delivery strategies [14].
Figure 1:

Overview of the Tissue Reactive Anchoring of Pharmaceutics (TRAPs) technology. An injectable prodrug consisting of a drug (red) conjugated to an anchoring motif (blue) through a cleavable linker (green) is injected locally. When injected into tissues, the anchor reacts with tissue extracellular matrix (ECM), anchoring the drug to the tissues. Over time, the linker connecting the drug to the ECM slowly dissolves, releasing drug in the tissue.
The need for improved sustained local drug delivery of potent chemotherapeutics is particularly acute in locally advanced solid tumors [15], including locally advanced pancreatic cancer (LAPC) [16]. LAPC constitutes 30% of diagnosed pancreatic adenocarcinomas and comprises unresectable malignant disease without overt distant metastases [17,18]. LAPC is often treated with systemic administration of Gemcitabine and FOLFIRINOX [17,19] but, effective management of LAPC is hampered by dense desmoplastic stroma surrounding tumor cells [20]. The dense stroma is rich in fibrillar collagen and accounts for almost 70–80% of the total tumor volume. The density, reorganization and alignment of the extracellular matrix contributes to reduced blood perfusion, a hypoxic tumor microenvironment and increased interstitial pressure [21], presenting a barrier for systemic therapeutic agents to reach and accumulate in the tumor tissue at therapeutic concentrations [22]. Due to the inadequacy of systemic therapy for LAPC, attention has focused on local drug delivery solutions for control of unresectable tumors, to debulk the tumor mass, or to provide palliative care [23,24]. These strategies include locally implanted hydrogels, [25–27] polymeric matrices [28,29], patches [30,31] and membranes [32] inserted intratumorally or peritumorally to achieve high local drug concentration for long periods of time. Local drug delivery strategies have shown superior potential in treating locally advanced pancreatic tumors [33], but they rely on injection or implantation of materials into a stiff tissue and struggle to deliver drugs deep into the solid tumors [34]. Therefore, there is an unmet need for effective local drug delivery strategies for management of locally advanced pancreatic cancer that would allow better drug penetration in the dense tumor tissue with minimum patient morbidity.
Paclitaxel (PTX) is a promising candidate against a broad spectrum of solid cancers including in pancreatic cancer, showing promise in improving LAPC resectability [35]. However, paclitaxel’s poor solubility [36] and poor tumor penetrance [37] necessitate improved drug delivery technologies. Previous approaches to paclitaxel delivery, including nanoparticles [38,39], hydrogels [40] and polymeric implants [41], show promise in preclinical pancreatic tumor models. Utilizing the ability of paclitaxel to effectively control pancreatic tumor growth, we sought novel approaches to deliver this potent chemotherapeutic deep within the solid tumor in a sustained manner without a viscous injections or implantation surgeries.
To test the ability of TRAPs depots to distribute widely throughout stiff tumors and sustainably produce anti-tumor therapy, we tested TRAP paclitaxel for the treatment of locally advanced pancreatic cancer (LAPC). In this report, we demonstrate the formation of stable intratumoral paclitaxel depots created by injecting TRAP paclitaxel into the dense pancreatic tumor. We demonstrate that TRAPs can anchor molecules within mouse and human pancreatic tissues. We additionally show that TRAP paclitaxel increases tumor apoptosis and anti-cancer efficacy as compared to free paclitaxel in a mouse model of pancreatic adenocarcinoma. The TRAPs approach to localized drug delivery provides a conceptual framework for local drug release that uses attachment to tissue extracellular matrix, rather than reliance on co-administered excipients (i.e. polymers, hydrogels, oils), for retention at disease sites. Materials-free sustained release of drugs at target tissue sites could be generalized to the clinical treatment of many other diseases benefiting from local drug presentation.
Materials and Methods:
Cell culture
KPC 4662 pancreatic tumor cell vials were received from the laboratory of Dr. Pylayeva-Gupta at UNC Chapel Hill at passage 9. Cells were revived and maintained in 1× high-glucose Dulbecco’s modified Eagle’s culture medium (DMEM) (Hyclone, cat #SH30022.01) supplemented with 10% fetal bovine serum (Gibco, cat# 26140–079) and 100U/mL penicillin/ streptomycin (Fisher, cat#15–140-122) at 37 °C and 5% CO2. To subculture, cells were washed with PBS and detached using 0.05% trypsin-ethylenediaminetetraacetic acid (EDTA) (Fisher, cat#25–300-054).
Formation and retention of intratumoral depot
To study the formation and retention of ECM anchored depots, NHS esters of AlexaFluor647 dye (AF647-NHS esters) (click chemistry tools, cat #1344–1) were injected intratumorally in the ectopic pancreatic tumor bearing mice and imaging was performed at specified time intervals to visualize AF647 signal. Ectopic pancreatic tumor model was developed by subcutaneous inoculation of approximately 1*106 KPC 4662 murine pancreatic tumor cells in 1:1 PBS/matrigel (Corning/ cat# 354234) solution on the dorsal flanks of 8 week old female albino C57BL/6 mice (Charles River). Once the tumors were approximately 100mm3 in volume, animals were randomly divided into two groups to receive intratumoral infusion of activated or hydrolyzed NHS ester. For control experiments using inactivated NHS ester, AF647-NHS was dissolved in sterile saline (pH 7.4) at a concentration of 0.1 mM and incubated at room temperature for 24 hours in the dark to ensure complete hydrolysis. After randomization, each group received intratumoral infusion of 50 μl of 0.1 mM activated AF647-NHS ester (n=3) or hydrolyzed inactivated AF647 (n=3) using 27g winged catheter and a syringe pump (Harvard apparatus, cat #70–4500) at the rate of 10 μL/min for 5 minutes. All animals were imaged at 0, 4, 24, 48 and 72 hours post intratumoral infusion using In Vivo Imaging System (IVIS). ICG BKG / ICG excitation and emission filters were used for all IVIS images presented and no image math in the Living Image software was performed. All animals were euthanized by intra-arterial perfusion of 10 ml of cold PBS under anesthesia followed by 10 ml of 10% neutral buffered formalin (Millipore Sigma, cat # 34172–118) prior to tumor extraction.
To assess the ECM anchored depot formation in the human pancreatic tumors. Excised human pancreatic tumor tissue was received from a Tissue Procurement Facility at UNC Lineberger Comprehensive Cancer Center following pancreatic tumor surgery. The collected human tumor samples were cut into pieces approximately 100 mm3 volume per piece. Each piece was washed with PBS and imaged using IVIS before dye fluorophore-NHS administration. Tumor pieces (n=3) were injected with 50 μL of 0.2 mM AF647 NHS using a 27g winged catheter and a syringe pump at the rate of 5 μL/min over 10 minutes. Control samples were untreated. Each piece was imaged immediately after dye injection and 24 hours post-injection using IVIS. AF 647 signal was further quantified to determine the depot anchoring.
Mouse and human tumors were fixed in 10% neutral buffered formalin and submitted to clearing using iDISCO protocol [42]. Briefly, all samples were dehydrated using increasing concentrations of methanol (20–100%) and kept shaking at room temperature for 1 hour each. Dehydrated samples were shaken three times in dichloromethane (Acros organics, cat# 348465000) for 30min each to wash excess methanol. Finally, tissues were placed in dibenzyl ether (Sigma, cat#33630) for clearing. Clear tumors were imaged using a Lavision Ultramicroscope II and evaluated using IMARIS software version 9.7. Postprocessing using IMARIS included visualizing AF647 signal with respect to tumor autofluorescence signal (AF488) and creating isosurfaces for AF488 and AF647 signals. AF488 isosurface was made transparent to visualize red colored AF647 signal. Quantitative analysis of the created isosurfaces was performed to calculate depot volume.
Synthesis and characterization of TRAP paclitaxel
Highly reactive ECM-anchoring TRAP paclitaxel conjugate was synthesized in a two-step process. Initially, paclitaxel succinic acid was synthesized using a method described in Shan et al. [43] with slight modifications. Briefly, paclitaxel (Medkoo, cat# 100690) (1 equiv.) was dissolved in dichloromethane (DCM) (Acros organics, cat# 348465000) under inert conditions and reacted with succinic anhydride (TCI, cat#TCS0107) (2 equiv.) in the presence of (4-Dimethylamino) pyridine (DMAP) (Aldrich, cat# 107700) (1 equiv.). The reaction was kept stirring overnight at room temperature. The crude mixture was purified using silica gel chromatography with gradient elution of 10%MeOH in DCM. Further, purified paclitaxel succinic acid (1 equiv.) was reacted with 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) (Oakwood chemicals, cat# 024810) (1 equiv.) and 1-Hydroxy-2,5-dioxopyrrolidine-3-sulfonic acid (sNHS) (Combi-Blocks, cat# 82436–78-0) (1 equiv.) dissolved in NN-Dimethylformamide (DMF) (Acros organics, cat# 348435000) under inert conditions. The reaction was kept stirring overnight at room temperature and purified using ether (Fisher, cat#AAL14030AU) precipitation. The final purified paclitaxel-sNHS conjugate was collected and further characterized using LCMS (Fig. S1) and NMR (Fig. S2).
In vitro paclitaxel release
Hydrolytic studies of paclitaxel-succinic acid were carried out at 37°C in PBS containing 0.01% DMSO and 0.1% Tween 80 at pH 7.4 and pH 6.5. 5.33 μM of paclitaxel-succinic acid was dissolved in a 5 ml of medium and kept continuously stirring at 150 rpm. 50 μL of sample was removed at predetermined time intervals of 0, 24, 48, 72 and 96 hours and replaced with 50 μL of fresh media. Removed samples were mixed with 50% of acetonitrile and analyzed using Agilent 1290 HPLC system with Agilent 2.1 X 50 mm C18 column and mobile phase consisting of 60% ammonium formate and 40% acetonitrile. Concentration of paclitaxel succinic acid and free paclitaxel were determined using calibration curves (Fig. S3).
Determination of solubility and stability of TRAP paclitaxel
To assess the solubility of free paclitaxel in saline and injection vehicle consisting of 50:50 NMP: saline, excess paclitaxel was incubated for 30 min in 5 ml of respective solvents at room temperature with constant agitation. At 0.5 hour, samples were centrifuged at 3600 rpm for 20 minutes to separate the supernatant. Collected supernatants were further filtered through a 0.45 μm syringe filter, mixed with an equal volume of acetonitrile and analyzed using the HPLC method described above. Similarly, to determine the solubility of TRAP paclitaxel in saline, excess drug was incubated for 10 min with agitation and centrifuged to separate the supernatant. Filtered supernatant was analyzed using UV-Visible spectroscopy at 230nm. Concentration of dissolved free paclitaxel and TRAP paclitaxel was determined using calibration curves.
To assess the stability of TRAP paclitaxel in the injection vehicle, paclitaxel-sNHS was dissolved in 50:50 NMP:saline at the concentration of 500 μM (pH 6) (n=3), aliquoted and incubated at room temperature for several hours. After predetermined time points, aliquots were removed and analyzed using UV-Visible spectroscopy at 260 nm to assess the presence of free sNHS. To further analyze the reactivity of TRAP paclitaxel in the injection vehicle, intentional hydrolysis was performed by adding 2 μL of 1M NaOH and changes in sNHS absorption were measured at 260 nm.
In vivo assessment of TRAP paclitaxel induced apoptosis
To assess the ability of intratumoral depots to release free paclitaxel and induce apoptotic tumor cell death, ectopic pancreatic tumor model was developed by subcutaneous inoculation of approximately 1*106 KPC 4662 murine pancreatic tumor cells in 1:1 PBS/matrigel (Corning/ cat# 354234) solution on the dorsal flanks of 8 week old female albino C57BL/6 mice (Charles River). Once the tumors were approximately 100mm3 in volume, animals were randomly divided into four groups to receive either no treatment or single intratumoral injection of vehicle (50% NMP: alfa aesar, #AA44063-K2, 50% Saline: cat#470302–026, VWR), free paclitaxel (Medkoo, cat# 100690, 20 mg/kg) and TRAP paclitaxel (n=3/group, 20 mg/kg). All animals were euthanized to collect tumors 72 hours post injection. Tumors were fixed in 10%NBF and sectioned to stain for cleaved caspase 3 and hematoxylin. Stained tumor slides were scanned, and analysis of apoptosis was performed using Image J (version 2.1.0/1.53c). For quantification of apoptosis, CC-3-stained vs hematoxylin-stained areas were measured with respect to total tumor area [44,45].
Evaluation of antitumor efficacy of TRAP paclitaxel in ectopic model
The antitumor effect of TRAP paclitaxel was studied in a syngeneic subcutaneous KPC 4662 tumor model. Briefly, 5*105 KPC 4662 murine pancreatic tumor cells in 1:1 PBS/matrigel (Corning/ 354234) suspension were injected in the dorsal flank of female C57BL/6J (Charles River) mice. Tumor growth was monitored using vernier calipers every alternate day and tumor volume was calculated using a formula (length*width*width)/2. When tumors were approximately 100mm3 in volume, all animals were randomly divided into four groups to receive single intratumoral administration of either free paclitaxel (20 mg/kg), TRAP paclitaxel-low dose (20 mg/kg), TRAP paclitaxel- high dose (50 mg/kg) or vehicle control. Each mouse was administered 50 μL of intratumoral injection of respective treatment at the rate of 5 μL/min over 10 min using 27g winged catheter and syringe pump. Tumor growth was monitored every 2 days until one of the animals reached the experimental end point. On Day-30, all animals were euthanized, and tumors and organs (heart, liver, kidneys, lungs, and spleen) were collected to evaluate local and systemic immune response. Collected tissues were immediately fixed in 10% neutral buffered formalin and submitted for further histological processing and H&E staining (Histology core at NC State College of Veterinary Medicine). Stained sections were imaged and were sent to a blinded, certified pathologist for evaluation.
Statistical analysis
All data was presented and analyzed for statistical significance using Prism software (version 9.0.1). To evaluate the statistical significance, student t test was used with Holm-Sidak correction for multiple comparison.
Results and discussion:
TRAP depots distribute throughout and are retained within mouse and human pancreatic tumors.
Effective clinical responses for local depots require widely distributed drugs that are “anchored” to their location, preventing fast drug clearance from the tissues. We hypothesized that the activated N-hydroxysuccinimide (NHS) esters of carboxylic acids, amine-reactive chemical groups commonly used to label proteins [12], could be used to anchor molecules to tissue extracellular matrix. To study depot distribution after intratumoral introduction, we chose the KPC 4662 adenocarcinoma pancreatic tumors. Tumor created by KPC 4662 demonstrates very high stiffness and fibrosis [21], making intratumoral injections of viscous hydrogels nearly impossible. In contrast, the collagen rich nature of these tumors provides numerous accessible amine sites for TRAPs reactivity. To study intratumoral depot formation, distribution and retention, we infused the NHS esters of AlexaFluor647 dye (AF647-NHS) into subcutaneous KPC 4662 pancreatic tumors (Fig. 2A). Inactivated AF647-NHS ester was used as a negative control, created through NHS ester hydrolysis in saline. Animals were imaged immediately after intratumoral injection and then daily for 72 hours using in vivo fluorescence imaging (IVIS) to visualize AF647 retention (Fig. 2B). 24 hours after intratumoral injection, the majority of the initial fluorescence signal remained in the tumors and this fraction was robustly maintained over the next 72 hours, with no further decrease in signal. In sharp contrast to the results seen with intratumoral injection of intact AF647-NHS, inactivated AF647-NHS rapidly cleared from the tumor, with only 3% of the signal detectable at 24 hours, consistent with tumor clearance in the absence of ECM anchoring (Fig. 2C).
Figure 2: In vivo anchoring and distribution of TRAPS within ectopic pancreatic tumors.

A) Schematics showing experiment methodology to assess formation and retention of intratumoral AF647 depots. B) Representative IVIS images and C) Quantitation of tumor fluorescence after intratumoral injection of intact AF647-NHS or AF647-NHS inactivated by hydrolysis. D) Isosurface visualization of AF647 signal in extracted cleared tumors 72 hours post intratumoral injection of intact or inactivated AF647-NHS. E) Volumetric quantification of reacted AF647 visualized within the mouse tumors. F) Schematics showing experimental approach to testing TRAPs anchoring in excised human pancreatic tumor. G) Visualization of AF647 signal in human pancreatic tumors injected with AF647-NHS. Scale bar: 200μm, *p < 0.05, by Student’s t-test.
Drug anchoring inside the tumor is dependent on the infusion rate as well as post-infusion diffusion. A computational model presented in our previous publication [14] predicted that anchoring directly labels tissues within a 3–4mm radius of the infusion needle. The model further predicted that the concentration of anchored depots decreases with increased distance from the infusion needle. To analyze the distribution of anchored AF647-NHS, we submitted tumors to tissue clearing and analyzed fluorescence spread by confocal imaging. 72 hours after injection, tumors were excised, cleared using the iDISCO protocol and imaged by light sheet microscopy. Confocal images were processed with IMARIS to visualize the volumetric representation of the AF647-NHS. No distinguishable fluorescence was observed in tumors infused with inactivated AF647-NHS. In sharp contrast, all tumors receiving intact AF647-NHS demonstrated distinct volumes of fluorescence signal (Fig. 2D). Some variability was noted, with some tumors demonstrating homogeneous signal throughout the entirety of the tumors, while in one tumor, a distinct localization at the tumor margins was noted. Volumetric analysis of the images showed an approximate spread volume of 22 μL, which is in good agreement with the in vivo imaging data (Fig. 2E). The decrease in observed spread volume (22 μL) vs infused volume (50 μL) may be explained by reaction and retention of NHS esters at the infusion front. Alternatively, it is possible that a fraction of injected material escapes the tumor.
Having demonstrated that TRAP AF647 efficiently labels a stiff, highly desmoplastic mouse pancreatic tumor, we tested the TRAPs anchoring paradigm in human tumors. Surgically resected primary human pancreatic tumors were cut into ~100 mm3 pieces and each piece was infused with AF647-NHS and imaged immediately after and 24 and 96 hours after infusion in the IVIS to quantify retention of fluorescent signal (Fig. 2F). Non-injected 100 mm3 pieces were used as controls. Approximately 80% of the signal was retained after 24 hours and no significant change was seen in the signal intensity up to 96 hours. The efficient retention of fluorescence in the human tumors suggests efficient anchoring and intratumoral AF647 depot formation in these tumor tissues. To evaluate the depot distribution throughout the human pancreatic tumor tissue, all pieces were submitted to iDISCO tissue clearing followed by confocal light sheet imaging. Evaluation of the AF647 signal suggests diffuse distribution of AF647 depots throughout two of the tumors and a more centralized localization of the signal for one tumor (Fig. 2G).
Taken together, our data demonstrates that TRAPs depot can be introduced into stiff pancreatic tumors of both humans and mice with wide distribution and good anchoring. The majority of injected materials is stably anchored and distributed throughout the tumor. A fraction of the fluorescence was lost within 24 hours, likely due to a combination of NHS ester hydrolysis and reaction with soluble factors that clear from the site.
Although AF647-NHS provides a good demonstration for the feasibility of intratumoral injection and depot formation in vivo, drug distribution throughout the tumor will depend on multiple factors including the solubility, hydrodynamic radius and tissue interactions of the TRAP drugs after injections. It is possible that the fluorescent drug surrogate used in these studies (AF647) may not adequately recapitulate the behavior of hydrophobic drugs such as paclitaxel. To further expand the understanding and application of intratumoral drug depots with a wide range of drugs, future studies will focus on validation of drug anchoring to tumor through mass spectroscopy-based imaging [46,47] or fluorescent drug surrogates validated to have similar PK/PD and diffusion profiles as parent therapeutics [48].
Synthesis and in vitro characterization of TRAP paclitaxel
In order to function within the TRAPs system, drugs must be conjugated to reactive NHS esters through cleavable linkers. In the case of paclitaxel, the ester modification to the 2’ hydroxyl has previously been described to be hydrolyzed with sustained kinetics [49]. Thus, TRAP paclitaxel was synthesized from paclitaxel over two steps with 61% overall yield (Fig. 3A). First, paclitaxel was reacted with succinic anhydride in the presence of DMAP to give the 2’-O-succinyl paclitaxel 2 in 80% yield. Then, 2 was coupled to sNHS in the presence of EDC to yield paclitaxel-sNHS esters in 77% yield. Purified paclitaxel succinic acid and paclitaxel sNHS esters were evaluated using LC-MS and NMR (Fig. S1, S2). The purity of synthesized compounds was approximately 92% for paclitaxel succinate and 87% for paclitaxel sNHS.
Figure 3: Synthesis and in vitro hydrolysis of TRAP paclitaxel.

A) Paclitaxel (1 eq.), succinic anhydride (2 eq.) and DMAP (1 eq.) reacted in anhydrous DCM to get paclitaxel succinic acid. Purified paclitaxel succinic acid (1 eq.), EDC (1 eq.) and sNHS (1 eq.) reacted in anhydrous DCM to get paclitaxel sNHS (TRAP paclitaxel). B) Kinetics of paclitaxel succinate hydrolysis at pH 6.5 and 7.4 (solid lines) and release of free paclitaxel (dashed lines) over time.
Effective delivery and distribution of paclitaxel to solid tumors is significantly limited by its solubility in aqueous solvents [36],[37]. Due to the charged sulfo group, we suspected that TRAP paclitaxel would have better aqueous solubility than free paclitaxel. Solubility testing demonstrated that the solubility of TRAP paclitaxel was roughly 9-fold higher than free paclitaxel (6.48μM vs 0.48 μM) in pure saline (Table S1).
We next assessed the stability and reactivity of TRAP paclitaxel conjugates in an aqueous/organic vehicle. NHS esters are known to hydrolyze in water, but are significantly more stable in organic solvents as well as in acidic pH [50]. We hypothesized that an injection vehicle consisting of 50:50 NMP saline with pH=6 would reduce hydrolysis prior to administration. To test this hypothesis, we evaluated the hydrolysis of TRAP paclitaxel in an injection vehicle at room temperature. Free sNHS absorbs strongly at 260 nm (Fig. S4A,B) [51], so hydrolysis was quantified by UV-VIS spectroscopy at the 260nm absorbance wavelength. Our results show no significant hydrolysis of the sNHS over 6 hours (Fig. S4C), suggesting stability of the TRAP paclitaxel in the injection vehicle.
The hydroxyl group at the C-2’ position is necessary for paclitaxel’s anticancer activity. Therefore, the succinate ester must hydrolyze to release paclitaxel and achieve desired antitumor responses [49]. After intratumoral injection, the reaction between TRAP paclitaxel and amine groups present in the tumor ECM yields a stable amide bond, creating anchored drug depots and releasing sNHS (Fig. S5). Upon hydrolysis of the ester bond, active paclitaxel is released in the tumor microenvironment. To assess the release of paclitaxel, we analyzed the hydrolysis kinetics of paclitaxel-succinate in PBS at 37° in both neutral pH and the slightly acidic conditions (pH ~6.5) commonly observed in the tumor microenvironments. Disappearance of the paclitaxel succinate peak and appearance of paclitaxel peak was quantified using HPLC. paclitaxel-succinate demonstrated a half-life of ~37 hours at neutral pH and ~56 hours at pH 6.5 (Fig. 3B). After 24 hours, nearly 82% of paclitaxel-succinate remained intact in acidic conditions, while 54% remained in the neutral condition. Interestingly, formation of free paclitaxel over time did not perfectly mirror disappearance of paclitaxel-succinate. At neutral pH, paclitaxel concentrations reached a maximum of 32% release at 96 hours, but did not increase past this point, despite continued decrease in the concentration of paclitaxel succinate. In contrast at the more acidic pH, paclitaxel continued its accumulation in the release media, with 60% of initial dose observed at 96 hours. The observed loss of some paclitaxel-succinate without a concomitant increase in paclitaxel suggests that at neutral pH, other side reactions were consuming paclitaxel, likely through hydrolysis of the acetyl groups on C4 or C10. The lower presence of paclitaxel in neutral conditions further suggests that accidental introduction of TRAP paclitaxel into healthy surrounding tissues would lead to less paclitaxel release and, thus, lower unintended toxicity.
Taken together, our data demonstrate that TRAP paclitaxel has improved aqueous solubility and stability in the injection vehicle. Moreover, TRAP paclitaxel slowly releases the parent drug at lowered pH, suggesting that TRAPs is a strong candidate for intratumoral delivery. This hydrolysis-mediated release of active therapeutics from anchored depots provides a proof of concept for the potential of the TRAP system in sustained drug release. More work will be needed to increase TRAP drug solubility, which limits delivery dosage for intratumoral injection of TRAP drugs. Improved solubility could be achieved with charged linkers or by starting with more soluble parent drugs. Future work could focus on tuning the drug release by incorporation of other linkers, including hydrazones [52–54], and aryl sulfonyls [55,56].
TRAP paclitaxel induces strong apoptosis in vivo
To investigate whether intratumoral injection of TRAP paclitaxel has therapeutic efficacy in vivo, we studied paclitaxel-induced apoptosis in the fibrous, syngeneic KPC 4662 pancreatic adenocarcinoma mouse model. Once tumors reached approximately 100mm3, animals were injected intratumorally with free paclitaxel, TRAP paclitaxel or vehicle control. Although TRAP paclitaxel showed significantly better aqueous solubility as compared to paclitaxel, a vehicle consisting of 50% NMP [57,58] was used to allow direct comparison to the poorly soluble paclitaxel. Vehicle-treated and untreated tumors were used as negative controls. 72 hours after a single intratumoral injection, tumors were collected, fixed, sectioned, and stained for cleaved caspase 3 (CC-3), a marker for apoptosis that plays a central role in early events of cell death [59]. For quantification of apoptosis, whole tissue sections were scanned to detect CC-3 positive areas and quantified to assess the percent of whole tumor area undergoing apoptosis. Tumor sections from treated groups showed large areas of diffuse CC-3 stain mixed with secondary necrosis. Sections from untreated controls showed central CC-3 positive region and minimal necrosis. Quantification of apoptosis was performed by computationally separating CC-3 stained-from hematoxylin-stained areas [44]. Roughly 10% of total area from untreated tumors showed apoptotic signals. While both vehicle and free paclitaxel treatment increased the apoptotic area (24% and 27%, respectively) neither reached statistical significance compared to untreated group. In contrast, tumors treated with TRAP paclitaxel demonstrated a significant increase in apoptotic staining (38% of total tumor) as compared to both the untreated control and vehicle control groups (Fig. S6). To further assess the paclitaxel induced apoptosis and to separate apoptosis from vehicle-induced necrosis, all sections were quantified for viable areas showing uniform cell staining and architecture, apoptotic areas showing CC-3 positive staining, and necrotic areas showing presence of cellular debris and loss of tumor architecture (Fig. 4, S7) by measuring the areas based on color saturations [45]. In this analysis, over 90% of total tumor area in the untreated controls was viable, with less than 5% of apoptotic and necrotic, each. Treatment with either vehicle, free paclitaxel, or TRAP paclitaxel, induced roughly similar levels of necrosis (~30% of tumor area), demonstrating the contribution of vehicle-ablation to induce local necrotic cell death. However, intratumoral administration of TRAP paclitaxel increased the percent apoptotic area by 3–5 fold over administration of either vehicle or free paclitaxel.
Figure 4: TRAP paclitaxel provides long-term apoptosis induction in ectopic pancreatic tumors.

A) Schematics showing experiment methodology. B) Quantification of cleaved caspase 3 stained tumor sections to determine extent of apoptosis. C) Representative images of scanned slides showing the whole tissue section and 10X magnification. Scale bar: 200μm. *p < 0.05, **p < 0.01, ***p < 0.005 by Student’s t-test with Holm Šidák correction for multiple comparisons.
Taken together, significantly higher apoptosis and significantly lower viability within the tumors treated with TRAP paclitaxel demonstrate the contribution of TRAP-mediated controlled paclitaxel release to maintain higher paclitaxel concentrations in the tumor ECM for sustained therapeutic benefits.
TRAP paclitaxel promotes anti-cancer efficacy
Since TRAP paclitaxel induced significantly more apoptotic tumor cell death than free paclitaxel control, we tested its antitumor efficacy in comparison to injection of free paclitaxel and vehicle in a syngeneic pancreatic tumor model. Animals bearing KPC 4662 murine pancreatic tumors (~100mm3 in volume) were randomly divided between four groups to receive intratumoral treatment of vehicle, free paclitaxel, or TRAP paclitaxel. Free paclitaxel was administered at 20mg/kg (50 μL, 8 mg/mL), just above its solubility limit of 7.46 mg/mL. Since TRAP paclitaxel is more soluble than free paclitaxel, it could be given at two doses, a lower dose of 20mg/kg (50 μL, 8 mg/mL) that serves as a direct comparison to free paclitaxel and at a higher dose of 50 mg/kg (50 μL, 20 mg/mL) to demonstrate the potential of TRAP paclitaxel without reference to the solubility limit of the parent drug. The 8mg/mL paclitaxel concentration used here is slightly above those achieved in clinical formulations of Taxol™ (6mg/mL) [60] and Abraxane™ (5mg/mL) [61]. Tumor volume and animal weight were monitored over 30 days (Fig. 5A). Animals receiving either dose of TRAP paclitaxel showed significant tumor growth suppression as compared to vehicle and free paclitaxel control groups. In animals receiving the “low” TRAP paclitaxel dose significance lasted through day 15, whereas the higher dosage of TRAP paclitaxel showed continuous tumor inhibition compared to both negative control and free paclitaxel groups (Fig. 5B), highlighting the advantage of TRAP paclitaxel to provide sustained intratumoral paclitaxel at therapeutic concentrations to reduce tumor burden. Animal weight monitoring showed no apparent body weight reduction (Fig. 5C), suggesting all animals were able to tolerate intratumoral injections and treatments well. On day 30, all animals were euthanized to collect tumors and main organs. Mice treated with the “high” dose of TRAP paclitaxel carried lower tumor weight as compared to other groups (Fig. 5D). A blinded histological analysis by a veterinary histopathologist was performed to evaluate presence of systemic toxicity and suggested no apparent differences in the organs (lungs, hearts, spleens, livers, kidneys and skin) among different treated groups (Fig. 5E, Fig. S8). Since vehicle was observed to induce local necrosis, the anti-tumor impact of vehicle was compared directly to saline controls. A small, but not statistically significant difference in tumor growth was observed in animals receiving intratumoral vehicle versus saline, suggesting little contribution of NMP in the vehicle to tumor growth inhibition (Fig. S9). In addition, intratumoral NMP was tested for systemic toxicity by comparing mice receiving intratumoral vehicle injections to untreated control mice. Histological analysis by a blinded veterinary histopathologist found no differences between the vehicle-injected and untreated groups. (Fig. S10).
Figure 5: Therapeutic efficacy of TRAP paclitaxel following an intratumoral anti-cancer therapy.

A) Schematics showing timeline of an experiment B) Excellent antitumor efficacy of TRAP paclitaxel following single intratumoral administration in an ectopic pancreatic tumor bearing mice. Inset graph showing therapeutic efficacy of two different concentrations of TRAP paclitaxel and free paclitaxel. C) Animal weight monitoring throughout the treatment duration D) Tumor weights of extracted tumors E) Histological sections of organs and tumors. Scale bar: 200μm F) Representative images of excised tumors. *p < 0.05, ****p < 0.0001 by Student’s t-test.
Taken together, continuous paclitaxel presentation at the tumor made possible by intratumorally anchored paclitaxel depots provided superior antitumor efficacy without off-target toxicity. Further improvements to the TRAPs technology, including optimization of intratumoral infusion parameters, identification of alternative TRAPs chemistries, and use of additional intratumoral infusion sites could provide even better anti-tumor efficacy and animal survival.
Conclusion
The Tissue Reactive Anchoring Pharmaceuticals (TRAPs) technology provides sustained local delivery of potent drugs to local tissues, including in stiff and fibrous tumors such as those seen in LAPC. These TRAPs can be readily introduced into stiff tissues like pancreatic tumors of both humans and mice with wide distribution and stable anchoring. Further, we developed a potent, clinically relevant TRAP paclitaxel which demonstrates improved solubility and sustained drug presentation throughout the tumor, improving anti-tumor efficacy as compared to unanchored drugs in ectopic pancreatic tumor models. The TRAPs technology could be used to create local drug depots for small molecule drugs and biologics as a general approach for diseases where localized drug presentation could benefit clinical therapy.
Supplementary Material
Highlights.
Tissue-Reacting Anchoring Pharmaceutical (TRAP) enables sustained drug release
TRAP drugs attach to extracellular matrix (ECM) and don’t need a material carrier
Intratumoral TRAP infusion creates dispersed, stable drug depots in stiff tumors
TRAP paclitaxel promotes local apoptosis and improves paclitaxel antitumor efficacy
TRAP technology could be of use in many clinical application for sustained release
Acknowledgement
This work was supported National Cancer Institute through grant R21-CA246414 and R37-CA260223, by the National Center for Advancing Translational Sciences (NCATS) through Grant Award Number UL1TR002489, by the UNC Lineberger Comprehensive Cancer Center’s University Cancer Research Fund, by a Faculty Research and Professional Development Grant from North Carolina State University and by start-up funds from the University of North Carolina and North Carolina State University. Authors thank North Carolina State University College of Veterinary Medicine staff for proper care of animals used in experiments and valuable resources on training. We also thank the Microscopy Services Laboratory, Department of Pathology and Laboratory Medicine, supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center and by the North Carolina Biotech Center Institutional Support Grant 2016-IDG-1016. Mass spectrometry data and NMR data were obtained at the NC State Molecular, Education, Technology and Research Innovation Center (METRIC). For the work in this research, we thank Dr. Pablo Ariel for his help with light sheet imaging and IMARIS training. In addition, we thank CVM histology lab and Pathology Services Core at the University of North Carolina-Chapel Hill, which is supported in part by an NCI Center Core Support Grant (5P30CA016080–42). Schematic images were created using BioRender.com.
Footnotes
The authors declare no competing financial interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Wolinsky JB, Colson YL, Grinstaff MW, Local drug delivery strategies for cancer treatment: gels, nanoparticles, polymeric films, rods, and wafers, J. Control. Release. 159 (2012) 14–26. 10.1016/j.jconrel.2011.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wadee A, Pillay V, Choonara YE, du Toit LC, Penny C, Ndesendo VMK, Kumar P, Murphy CS, Recent advances in the design of drug-loaded polymeric implants for the treatment of solid tumors, Expert Opin. Drug Deliv. 8 (2011) 1323–1340. 10.1517/17425247.2011.602671. [DOI] [PubMed] [Google Scholar]
- [3].Kim H-S, Yang J, Kim K, Shin US, Biodegradable and injectable hydrogels as an immunosuppressive drug delivery system, Mater. Sci. Eng. C Mater. Biol. Appl. 98 (2019) 472–481. 10.1016/j.msec.2018.11.051. [DOI] [PubMed] [Google Scholar]
- [4].Szulc M, Zakrzewska A, Zborowski J, Local drug delivery in periodontitis treatment: A review of contemporary literature, Dent Med Probl. 55 (2018) 333–342. 10.17219/dmp/94890. [DOI] [PubMed] [Google Scholar]
- [5].Stewart JM, Keselowsky BG, Combinatorial drug delivery approaches for immunomodulation, Adv. Drug Deliv. Rev. 114 (2017) 161–174. 10.1016/j.addr.2017.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Azzi J, Yin Q, Uehara M, Ohori S, Tang L, Cai K, Ichimura T, McGrath M, Maarouf O, Kefaloyianni E, Loughhead S, Petr J, Sun Q, Kwon M, Tullius S, von Andrian UH, Cheng J, Abdi R, Targeted Delivery of Immunomodulators to Lymph Nodes, Cell Rep. 15 (2016) 1202–1213. 10.1016/j.celrep.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pandey S, Rai N, Rawat P, Ahmad FJ, Talegaonkar S, Nanofacilitated synergistic treatment for rheumatoid arthritis: A ‘three-pronged’ approach, Medical Hypotheses. 92 (2016) 44–47. 10.1016/j.mehy.2016.04.026. [DOI] [PubMed] [Google Scholar]
- [8].Rahnfeld L, Luciani P, Injectable Lipid-Based Depot Formulations: Where Do We Stand?, Pharmaceutics. 12 (2020). 10.3390/pharmaceutics12060567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].De Souza R, Zahedi P, Allen CJ, Piquette-Miller M, Polymeric drug delivery systems for localized cancer chemotherapy, Drug Deliv. 17 (2010) 365–375. 10.3109/10717541003762854. [DOI] [PubMed] [Google Scholar]
- [10].Sun Z, Song C, Wang C, Hu Y, Wu J, Hydrogel-Based Controlled Drug Delivery for Cancer Treatment: A Review, Mol. Pharm. 17 (2020) 373–391. 10.1021/acs.molpharmaceut.9b01020. [DOI] [PubMed] [Google Scholar]
- [11].Mohtashami Z, Esmaili Z, Vakilinezhad MA, Seyedjafari E, Akbari Javar H, Pharmaceutical implants: classification, limitations and therapeutic applications, Pharm. Dev. Technol. 25 (2020) 116–132. 10.1080/10837450.2019.1682607. [DOI] [PubMed] [Google Scholar]
- [12].Lim CY, Owens NA, Wampler RD, Ying Y, Granger JH, Porter MD, Takahashi M, Shimazu K, Succinimidyl ester surface chemistry: implications of the competition between aminolysis and hydrolysis on covalent protein immobilization, Langmuir. 30 (2014) 12868–12878. 10.1021/la503439g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kalkhof S, Sinz A, Chances and pitfalls of chemical cross-linking with amine-reactive N- hydroxysuccinimide esters, Anal. Bioanal. Chem. 392 (2008) 305–312. 10.1007/s00216-008-2231-5. [DOI] [PubMed] [Google Scholar]
- [14].Adams MR, Moody CT, Sollinger JL, Brudno Y, Extracellular-matrix-anchored click motifs for specific tissue targeting, Mol. Pharm. 17 (2020) 392–403. 10.1021/acs.molpharmaceut.9b00589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Durymanov MO, Rosenkranz AA, Sobolev AS, Current Approaches for Improving Intratumoral Accumulation and Distribution of Nanomedicines, Theranostics. 5 (2015) 1007–1020. 10.7150/thno.11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Oberstein PE, Olive KP, Pancreatic cancer: why is it so hard to treat?, Therap. Adv. Gastroenterol. 6 (2013) 321–337. 10.1177/1756283X13478680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].van Veldhuisen E, van den Oord C, Brada LJ, Walma MS, Vogel JA, Wilmink JW, Del Chiaro M, van Lienden KP, Meijerink MR, van Tienhoven G, Hackert T, Wolfgang CL, van Santvoort H, Groot Koerkamp B, Busch OR, Molenaar IQ, van Eijck CH, Besselink MG, Dutch Pancreatic Cancer Group and International Collaborative Group on Locally Advanced Pancreatic Cancer, Locally Advanced Pancreatic Cancer: Work-Up, Staging, and Local Intervention Strategies, Cancers. 11 (2019). 10.3390/cancers11070976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hammel P, Huguet F, van Laethem J-L, Goldstein D, Glimelius B, Artru P, Borbath I,Bouché O, Shannon J, André T, Mineur L, Chibaudel B, Bonnetain F, Louvet C, Effect of chemoradiotherapy vs chemotherapy on survival in patients with locally advanced pancreatic cancer controlled after 4 months of gemcitabine with or without erlotinib, JAMA. 315 (2016) 1844. 10.1001/jama.2016.4324. [DOI] [PubMed] [Google Scholar]
- [19].Conroy T, Bachet J-B, Ayav A, Huguet F, Lambert A, Caramella C, Maréchal R, Van Laethem J-L, Ducreux M, Current standards and new innovative approaches for treatment of pancreatic cancer, Eur. J. Cancer. 57 (2016) 10–22. 10.1016/j.ejca.2015.12.026. [DOI] [PubMed] [Google Scholar]
- [20].Miao L, Lin CM, Huang L, Stromal barriers and strategies for the delivery of nanomedicine to desmoplastic tumors, J. Control. Release. 219 (2015) 192–204. 10.1016/j.jconrel.2015.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Drifka CR, Loeffler AG, Mathewson K, Keikhosravi A, Eickhoff JC, Liu Y, Weber SM, Kao WJ, Eliceiri KW, Highly aligned stromal collagen is a negative prognostic factor following pancreatic ductal adenocarcinoma resection, Oncotarget. 7 (2016) 76197–76213. 10.18632/oncotarget.12772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Adiseshaiah PP, Crist RM, Hook SS, McNeil SE, Nanomedicine strategies to overcome the pathophysiological barriers of pancreatic cancer, Nat. Rev. Clin. Oncol. 13 (2016) 750–765. 10.1038/nrclinonc.2016.119. [DOI] [PubMed] [Google Scholar]
- [23].Brunner M, Wu Z, Krautz C, Pilarsky C, Grützmann R, Weber GF, Current Clinical Strategies of Pancreatic Cancer Treatment and Open Molecular Questions, Int. J. Mol. Sci. 20 (2019). 10.3390/ijms20184543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shah R, Ostapoff KT, Kuvshinoff B, Hochwald SN, Ablative Therapies for Locally Advanced Pancreatic Cancer, Pancreas. 47 (2018) 6–11. 10.1097/MPA.0000000000000948. [DOI] [PubMed] [Google Scholar]
- [25].Shi K, Xue B, Jia Y, Yuan L, Han R, Yang F, Peng J, Qian Z, Sustained co-delivery of gemcitabine and cis-platinum via biodegradable thermo-sensitive hydrogel for synergistic combination therapy of pancreatic cancer, Nano Res. 12 (2019) 1389–1399. 10.1007/s12274-019-2342-7. [DOI] [Google Scholar]
- [26].Okino H, Maeyama R, Manabe T, Matsuda T, Tanaka M, Trans-tissue, sustained release of gemcitabine from photocured gelatin gel inhibits the growth of heterotopic human pancreatic tumor in nude mice, Clin. Cancer Res. 9 (2003) 5786–5793. https://www.ncbi.nlm.nih.gov/pubmed/14654564. [PubMed] [Google Scholar]
- [27].Smith JP, Stock E, Orenberg EK, Yu NY, Kanekal S, Brown DM, Intratumoral chemotherapy with a sustained-release drug delivery system inhibits growth of human pancreatic cancer xenografts, Anticancer Drugs. 6 (1995) 717–726. 10.1097/00001813-199512000-00002. [DOI] [PubMed] [Google Scholar]
- [28].Ramot Y, Rotkopf S, Gabai RM, Zorde Khvalevsky E, Muravnik S, Marzoli GA, Domb AJ, Shemi A, Nyska A, Preclinical Safety Evaluation in Rats of a Polymeric Matrix Containing an siRNA Drug Used as a Local and Prolonged Delivery System for Pancreatic Cancer Therapy, Toxicol. Pathol. 44 (2016) 856–865. 10.1177/0192623316645860. [DOI] [PubMed] [Google Scholar]
- [29].Indolfi L, Ligorio M, Ting DT, Xega K, Tzafriri AR, Bersani F, Aceto N, Thapar V, Fuchs BC, Deshpande V, Baker AB, Ferrone CR, Haber DA, Langer R, Clark JW, Edelman ER, A tunable delivery platform to provide local chemotherapy for pancreatic ductal adenocarcinoma, Biomaterials. 93 (2016) 71–82. 10.1016/j.biomaterials.2016.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yi H-G, Choi Y-J, Kang KS, Hong JM, Pati RG, Park MN, Shim IK, Lee CM, Kim SC, Cho D-W, A 3D-printed local drug delivery patch for pancreatic cancer growth suppression, J. Control. Release. 238 (2016) 231–241. 10.1016/j.jconrel.2016.06.015. [DOI] [PubMed] [Google Scholar]
- [31].Jun E, Kim SC, Lee CM, Oh J, Lee S, Shim IK, Synergistic effect of a drug loaded electrospun patch and systemic chemotherapy in pancreatic cancer xenograft, Sci. Rep. 7 (2017) 12381. 10.1038/s41598-017-12670-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bang S, Jang SI, Lee SY, Baek Y-Y, Yun J, Oh SJ, Lee CW, Jo EA, Na K, Yang S, Lee DH, Lee DK, Molecular mechanism of local drug delivery with Paclitaxel-eluting membranes in biliary and pancreatic cancer: new application for an old drug, Gastroenterol. Res. Pract. 2015 (2015) 568981. 10.1155/2015/568981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Smith JP, Kanekal S, Patawaran MB, Chen JY, Jones RE, Orenberg EK, Yu NY, Drug retention and distribution after intratumoral chemotherapy with fluorouracil/epinephrine injectable gel in human pancreatic cancer xenografts, Cancer Chemother. Pharmacol. 44 (1999) 267–274. 10.1007/s002800050977. [DOI] [PubMed] [Google Scholar]
- [34].Exner AA, Saidel GM, Drug-eluting polymer implants in cancer therapy, Expert Opin. Drug Deliv. 5 (2008) 775–788. 10.1517/17425247.5.7.775. [DOI] [PubMed] [Google Scholar]
- [35].Miyasaka Y, Ohtsuka T, Kimura R, Matsuda R, Mori Y, Nakata K, Kakihara D, Fujimori N, Ohno T, Oda Y, Nakamura M, Neoadjuvant chemotherapy with gemcitabine plus nab-paclitaxel for borderline resectable pancreatic cancer potentially improves survival and facilitates surgery, Ann. Surg. Oncol. 26 (2019) 1528–1534. 10.1245/s10434-019-07309-8. [DOI] [PubMed] [Google Scholar]
- [36].Panchagnula R, Pharmaceutical aspects of paclitaxel, Int. J. Pharm. 172 (1998) 1–15. 10.1016/S0378-5173(98)00188-4. [DOI] [Google Scholar]
- [37].Kyle AH, Huxham LA, Yeoman DM, Minchinton AI, Limited tissue penetration of taxanes: a mechanism for resistance in solid tumors, Clin. Cancer Res. 13 (2007) 2804–2810. 10.1158/1078-0432.CCR-06-1941. [DOI] [PubMed] [Google Scholar]
- [38].Massey AE, Sikander M, Chauhan N, Kumari S, Setua S, Shetty AB, Mandil H, Kashyap VK, Khan S, Jaggi M, Yallapu MM, Hafeez BB, Chauhan SC, Next-generation paclitaxel-nanoparticle formulation for pancreatic cancer treatment, Nanomedicine. 20 (2019) 102027. 10.1016/j.nano.2019.102027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Min SY, Byeon HJ, Lee C, Seo J, Lee ES, Shin BS, Choi H-G, Lee KC, Youn YS, Facile one-pot formulation of TRAIL-embedded paclitaxel-bound albumin nanoparticles for the treatment of pancreatic cancer, Int. J. Pharm. 494 (2015) 506–515. 10.1016/j.ijpharm.2015.08.055. [DOI] [PubMed] [Google Scholar]
- [40].Mao Y, Li X, Chen G, Wang S, Thermosensitive hydrogel system with paclitaxel liposomes used in localized drug delivery system for in situ treatment of tumor: Better antitumor efficacy and lower toxicity, J. Pharm. Sci. 105 (2016) 194–204. 10.1002/jps.24693. [DOI] [PubMed] [Google Scholar]
- [41].Matthes K, Mino-Kenudson M, Sahani DV, Holalkere N, Fowers KD, Rathi R, Brugge WR, EUS-guided injection of paclitaxel (OncoGel) provides therapeutic drug concentrations in the porcine pancreas (with video), Gastrointest. Endosc. 65 (2007) 448–453. 10.1016/j.gie.2006.06.030. [DOI] [PubMed] [Google Scholar]
- [42].Renier N, Wu Z, Simon DJ, Yang J, Ariel P, Tessier-Lavigne M, iDISCO: a simple, rapid method to immunolabel large tissue samples for volume imaging, Cell. 159 (2014) 896–910. 10.1016/j.cell.2014.10.010. [DOI] [PubMed] [Google Scholar]
- [43].Shan L, Cui S, Du C, Wan S, Qian Z, Achilefu S, Gu Y, A paclitaxel-conjugated adenovirus vector for targeted drug delivery for tumor therapy, Biomaterials. 33 (2012) 146–162. 10.1016/j.biomaterials.2011.09.025. [DOI] [PubMed] [Google Scholar]
- [44].Janke LJ, Ward JM, Vogel P, Classification, Scoring, and Quantification of Cell Death in Tissue Sections, Vet. Pathol. 56 (2019) 33–38. 10.1177/0300985818800026. [DOI] [PubMed] [Google Scholar]
- [45].Krajewska M, Smith LH, Rong J, Huang X, Hyer ML, Zeps N, Iacopetta B, Linke SP, Olson AH, Reed JC, Krajewski S, Image Analysis Algorithms for Immunohistochemical Assessment of Cell Death Events and Fibrosis in Tissue Sections, Journal of Histochemistry & Cytochemistry. 57 (2009) 649–663. 10.1369/jhc.2009.952812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Aichler M, Walch A, MALDI Imaging mass spectrometry: current frontiers and perspectives in pathology research and practice, Lab. Invest. 95 (2015) 422–431. 10.1038/labinvest.2014.156. [DOI] [PubMed] [Google Scholar]
- [47].Morosi L, Spinelli P, Zucchetti M, Pretto F, Carrà A, D’Incalci M, Giavazzi R, Davoli E, Determination of paclitaxel distribution in solid tumors by nano-particle assisted laser desorption ionization mass spectrometry imaging, PLoS One. 8 (2013) e72532. 10.1371/journal.pone.0072532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lee MM, Gao Z, Peterson BR, Synthesis of a Fluorescent Analogue of Paclitaxel That Selectively Binds Microtubules and Sensitively Detects Efflux by P-Glycoprotein, Angew. Chem. Int. Ed Engl. 56 (2017) 6927–6931. 10.1002/anie.201703298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Fu Q, Wang Y, Ma Y, Zhang D, Fallon JK, Yang X, Liu D, He Z, Liu F, Programmed Hydrolysis in Designing Paclitaxel Prodrug for Nanocarrier Assembly, Sci. Rep. 5 (2015) 12023. 10.1038/srep12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Mattson G, Conklin E, Desai S, Nielander G, Savage MD, Morgensen S, A practical approach to crosslinking, Mol. Biol. Rep. 17 (1993) 167–183. 10.1007/BF00986726. [DOI] [PubMed] [Google Scholar]
- [51].Miron T, Wilchek M, A spectrophotometric assay for soluble and immobilized N-hydroxysuccinimide esters, Anal. Biochem. 126 (1982) 433–435. 10.1016/0003-2697(82)90540-1. [DOI] [PubMed] [Google Scholar]
- [52].Zhang X, Huang Y, Ghazwani M, Zhang P, Li J, Thorne SH, Li S, Tunable pH-Responsive Polymeric Micelle for Cancer Treatment, ACS Macro Lett. 4 (2015) 620–623. 10.1021/acsmacrolett.5b00165. [DOI] [PubMed] [Google Scholar]
- [53].Sanchez E, Li M, Wang C, de Castro Abeger A, Li Z-W, Chen H, Berenson JR, Anti- myeloma and anti-angiogenic effects of the novel anthracycline derivative INNO-206, Blood. 116 (2010) 4065–4065. 10.1182/blood.v116.21.4065.4065. [DOI] [Google Scholar]
- [54].Mane S, Advances of hydrazone linker in polymeric drug delivery, J. Crit. Rev. (2019) 1–4. 10.22159/jcr.2019v6i2.31833. [DOI] [Google Scholar]
- [55].Ashley GW, Henise J, Reid R, Santi DV, Hydrogel drug delivery system with predictable and tunable drug release and degradation rates, Proc. Natl. Acad. Sci. U. S. A. 110 (2013) 2318–2323. 10.1073/pnas.1215498110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Santi DV, Schneider EL, Reid R, Robinson L, Ashley GW, Predictable and tunable half-life extension of therapeutic agents by controlled chemical release from macromolecular conjugates, Proc. Natl. Acad. Sci. U. S. A. 109 (2012) 6211–6216. 10.1073/pnas.1117147109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sanghvi R, Narazaki R, Machatha SG, Yalkowsky SH, Solubility improvement of drugs using N-methyl pyrrolidone, AAPS PharmSciTech. 9 (2008) 366–376. 10.1208/s12249-008-9050-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Jouyban A, Fakhree MAA, Shayanfar A, Review of pharmaceutical applications of N-methyl-2-pyrrolidone, J. Pharm. Pharm. Sci. 13 (2010) 524–535. 10.18433/j3p306. [DOI] [PubMed] [Google Scholar]
- [59].Liu P-F, Hu Y-C, Kang B-H, Tseng Y-K, Wu P-C, Liang C-C, Hou Y-Y, Fu T-Y, Liou H-H, Hsieh I-C, Ger L-P, Shu C-W, Expression levels of cleaved caspase-3 and caspase-3 in tumorigenesis and prognosis of oral tongue squamous cell carcinoma, PLoS One. 12 (2017) e0180620. 10.1371/journal.pone.0180620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].HQ Specialty Pharma. Taxol [package insert]. U.S. Food and Drug Administration; website, (n.d.). https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=020262 (accessed November 29, 2021). [Google Scholar]
- [61].Abraxis Bioscience. Abraxane [package insert]. U.S. Food and Drug Administration, (n.d.). https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=021660 (accessed November 29, 2021). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.