

Abstract
Biomaterial-based approaches for a combination of radiotherapy and immunotherapy can improve outcomes in metastatic cancer through local delivery of both therapeutic modalities to the primary tumor to control local tumor growth and distant metastases. This study describes an injectable depot for sustained intratumoral (i.t.) delivery of an iodine-131 (131I) radionuclide and a CpG oligodeoxynucleotide immunostimulant, driven by the thermally sensitive phase transition behavior of elastin-like polypeptides (ELPs). We synthesized and characterized an ELP with an oligolysine tail (ELP-K12) that forms an electrostatic complex with CpG for delivery from an ELP depot and evaluated the ability of the complex to enhance local and systemic tumor control as a monotherapy and in combination with 131I-ELP brachytherapy. I.t delivery of CpG from an ELP-K12 depot dramatically prolongs i.t. retention to more than 21 days as compared to soluble CpG that is only retained within the tumor for <24 h. ELP-K12 also enhances CpG delivery by increasing cellular uptake of CpG to generate greater toll-like receptor 9 (TLR9) activation than CpG alone. I.t. treatment with an ELP-K12/CpG depot slows primary tumor growth and reduces lung metastases in a poorly immunogenic 4T1 syngeneic breast cancer model whereas i.t treatment of CpG alone has no significant effect on primary tumor growth or metastases. Notably, a combination of 131I-ELP brachytherapy and ELP-K12/CpG delivered i.t. inhibited 4T1 tumor growth and strongly decreased the development of lung metastases, leading to a synergistic improvement in mouse survival. These preclinical results demonstrate that injectable ELP depots may provide a useful approach for the delivery of combination radio- and immunotherapy to treat metastatic disease.
Keywords: Elastin-like polypeptide, Cancer immunotherapy, Radiotherapy, Sustained release, Metastatic cancer
Graphical Abstract
Introduction
Metastatic disease persists as a significant barrier to increasing survival in cancer, as it is responsible for the majority of cancer-related deaths1,2. The advent of cancer immunotherapies has led to clear clinical gains in treating metastatic disease3. As a result, much attention has been paid to combinations of immunotherapies and traditional treatments like external beam radiation therapy (EBRT), where early efforts have demonstrated synergy between EBRT and immunotherapies in clinical4–6 and preclinical settings7,8 in improving the response to metastatic cancer. However, a majority of patients still do not respond to combinations currently in clinical trials4,5,9,10, and questions remain as to whether dose-limiting toxicity is exacerbated by the interaction between treatments11,12.
A promising approach to overcome the challenges of combining immunotherapy and radiation therapy utilizes engineered materials to localize the therapies to the primary tumor. Local retention reduces radiation exposure of healthy tissues and mitigates systemic toxicities of potent immunotherapies. Furthermore, a platform that provides spatiotemporal control of radionuclide delivery and immune signaling can deliver a greater radiation dose to the primary tumor and generate a systemic immune response against metastases. Multiple preclinical studies have demonstrated that traditional seed brachytherapy with Iodine-125 has increased antitumor efficacy in combination with either poxvirus vaccination or chimeric-antigen receptor natural killer cells13,14. Yet these strategies rely on invasive implantation of non-degradable titanium seeds and provide no method for sustained release of immunotherapies. A novel biomaterials approach that uses alginate gels for the co-delivery of a radioisotope and an immunostimulant has demonstrated inhibition of distal and metastatic tumors in preclinical models15. However, the poorly defined composition and structure of alginate gels limits control over the retention kinetics of encapsulated therapeutics. The ideal biomaterial platform for the delivery of radiation therapy and immunotherapy would allow for local administration and a mechanism for sustained localization of signals within the tumor with precise control over the kinetics of retention.
Elastin-like polypeptides (ELPs) are a class of biomaterials that are biocompatible with low immunogenicity16,17 and have unique properties for the precise spatiotemporal delivery of biomolecules. ELPs are artificial intrinsically disordered proteins, consisting of repeats of a Val-Pro-Gly-Xaa-Gly motif, where Xaa is any amino acid except Pro, that can be designed with a lower critical solution temperature (LCST) phase transition from a solution to an insoluble coacervate that can be triggered by a small increase in temperature18. The temperature at which coacervation of the ELP into a water-insoluble phase occurs, termed the inverse transition temperature (Tt), can be tuned at the sequence level to occur at any desired temperature between 0–100 °C. ELPs therefore can be designed to be soluble at room temperature and coacervate at body temperature upon intratumoral (i.t.) administration. Because the Tt is inversely proportional to concentration, dilution of the ELP at the margins of the depot reverses its LCST phase transition and leads to a sustained dissolution of the ELP from the depot with zero-order release kinetics19. As ELPs retain their phase behavior when fused to peptides or proteins, and when conjugated to small molecules, ELP depots have been used for sustained release of covalently-attached therapeutics20. Furthermore, ELPs containing positively-charged residues have been demonstrated to electrostatically interact with and deliver nucleic acids21–23. This approach, however, has not been used to deliver electrostatically complexed immunostimulants from an ELP depot, as demonstrated here.
ELPs have also been used to create stable depots that are retained at the site of injection for brachytherapy. In an example of this approach, an Iodine-131 (131I) radionuclide was conjugated to the tyrosine trailer of a micelle-forming ELP24, wherein the ELP micelles were designed to coacervate at a Tt below body temperature so that upon i.t. administration they form a radioactive depot. The depot is then further stabilized by radiation-induced crosslinking of the ELP, leading to prolonged retention of the radioactive ELP within the tumor for over 60 days, with little escape of radioactivity into systemic circulation. ELP brachytherapy has demonstrated curative efficacy in multiple immunocompromised mouse models of cancer25.
The objective of this study was to combine immunotherapy and 131I brachytherapy, each delivered from an ELP depot, to treat metastatic breast cancer (Fig. 1). In combination with ELP-based brachytherapy, an immunostimulant delivered from a depot-forming ELP would provide highly focused immunostimulatory signaling in the tumor, which would enhance the anticancer immune response to radiation-induced tumor neoantigens. Thus, we hypothesized that an ELP-based combination of radiotherapy and immunotherapy would yield greater efficacy than either modality alone in treating metastatic cancer.
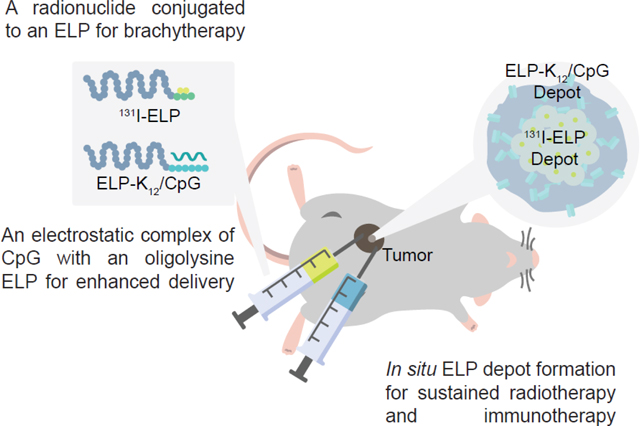
Figure 1: Schematic of combination of 131I-ELP brachytherapy and ELP-K12/CpG immunotherapy.
ELP brachytherapy is formulated as a 131I radionuclide conjugated to Tyr residues at the C-terminus of an ELP (131I-ELP), while CpG is electrostatically complexed to a (Lys)12 peptide at the C-terminus of an ELP (ELP-K12). Each conjugate is injected i.t. and undergoes an LCST phase transition at body temperature to form ELP depots. The 131I is retained within the tumor because of radiation-induced crosslinking of the ELP and irradiates the tumor from the inside-out, while the electrostatically complexed CpG is released from the depot over time to activate immune signaling within the tumor.
To test this hypothesis, we developed a platform for the i.t. delivery of the toll-like receptor 9 (TLR9) agonist CpG oligodeoxynucleotide (CpG). CpG is a potent stimulator of innate immunity that functions to mature and activate dendritic cells, increasing their expression of costimulatory molecules and inflammatory cytokines and leading to enhanced antigen presentation. Through these effects, CpG has been demonstrated to enhance antitumor immunity and lead to modest preclinical and clinical anticancer efficacy as a monotherapy26,27. It has also been shown to improve anticancer efficacy in combination with radiation28,29 and brachytherapy15 by improving the presentation of antigens that are released during radiation-induced apoptosis26. However, CpG is a small molecule with an extremely short half-life that we hypothesize limits its efficacy30. To enhance the activity and retention of CpG within the tumor, we synthesized an ELP with depot-forming phase behavior at body temperature and an oligolysine tail that forms an electrostatic complex with CpG (ELP-K12/CpG). We tested this brachy- and immuno-therapy combination in a syngeneic 4T1 mammary carcinoma mouse model, as it spontaneously metastasizes to the lungs and other organs31 and is difficult to treat with immunotherapy alone7,32. We show that the combination of i.t. delivery of CpG by an ELP depot and 131I-ELP brachytherapy inhibits local and systemic tumor growth and demonstrates synergy in increasing the survival of 4T1-tumor-bearing mice.
Materials and Methods
ELP cloning, production, and purification
The cloning strategy utilized a pet24-a+ vector that was previously modified for seamless cloning of ELPs33. DNA primers (Table S1) were purchased (Integrated DNA Technologies, Coralville, IA) and used to modify the pet24-a+ vector via overhang PCR to insert a DNA sequence encoding for 12 lysines (K12). Using the previously developed seamless cloning strategy33, the vector encoding for K12 was digested with BglI and BseRI, while a vector encoding for an ELP consisting of 60 repeats of the amino acid sequence VPGVG was digested with BglI and AcuI (New England Biolabs, Ipswich, ME). The vector fragments were ligated together to fuse the K12 tag to the C-terminus of the ELP. The vector with the final construct also included a C-terminal tryptophan to facilitate protein quantification via UV-vis spectrophotometry. The DNA sequences for the primers and amino acid sequences are available in the supplemental information (Table S1–S2).
For expression, the vector encoding ELP-K12 was transformed into BL21(DE3) E. Coli (New England Biolabs, Ipswich, ME). Bacterial cultures were grown in Terrific Broth (VWR International, Wayne, PA) with kanamycin sulfate (Millipore Sigma, St. Louis, MO) in shake flasks at 37°C and 225 rpm for 6 h before induction of protein expression by the T7 promoter with the addition of 0.5 mM IPTG (Gold Biotechnology, St. Louis, MO) and cultured for an additional 16 h. Cells were pelleted by centrifugation, resuspended in cold PBS, lysed by sonication (Q500 sonicator, QSonica, Newtown, CT), and clarified by centrifugation after the addition of polyethylenimine (VWR International, Wayne, PA) to remove nucleic acids. Inverse transition cycling (ITC) with a modified “bakeout” step was performed as previously described34,35. Briefly, the LCST phase transition was triggered by the addition of 0.2 M ammonium sulfate to clarified cell lysates, and the samples were then centrifuged at 24,000 rcf for 10 min at 35°C. The protein pellets were resuspended in cold PBS and centrifuged at 24,000 rcf for 10 min at 4°C to remove impurities. This was followed by 2–4 “bakeout” rounds in which the sample is heated to 95°C for 10 min, immediately cooled on ice for 10 min, and centrifuged at 24,000 rcf for 10 min at 4°C to remove impurities. The ITC-purified protein was dialyzed to remove residual salts and small molecule contaminants. Finally, ELP-K12 was purified with poly(ε-lysine) endotoxin removal resin (ThermoFisher Scientific, Waltham, MA) and confirmed to have an endotoxin content of <0.05 EU/mg.
An ELP of 60 repeats of VPGVG without a K12 tail and an ELP of 120 repeats of VPGVG with a trailer sequence with 7 tyrosines (Table S2) were expressed and purified using the same protocol. Recombinant ELPs were assessed for molecular weight and purity by SDS-PAGE analysis. To synthesize the 131I-ELP radionuclide conjugate, 131Iodine (International Isotopes, Idaho Falls, ID) was conjugated to the tyrosine tail of the ELP via the iodogen oxidative reaction method, as previously described25,36. Levels of radioactivity of the conjugate were measured with an AtomLab 400 dose calibrator (Biodex, Shirley, NY) and the conjugate was diluted with free ELP to the desired radioactivity dose.
Characterization of CpG binding and LCST phase behavior
The ability of ELP-K12 to electrostatically bind CpG ODN 1826 (AdipoGen Life Sciences, San Diego, CA) was assessed by a gel shift assay. Solutions of 20 μM CpG alone or with ELP-K12 at an N:P ratio of 1:1, 3:1, or 7:1 were prepared and run on a 1% w/v agarose gel. The binding of CpG to ELP-K12 was visualized by the retardation of CpG migration in the gel in the presence of ELP-K12. The LCST phase transition behavior of ELP-K12 was characterized by UV-vis spectrophotometry. A Cary 300 UV-vis spectrophotometer (Agilent Technologies, Santa Clara, CA) was used to measure the optical density at 350 nm (OD350) of solutions of 25 μM, 50 μM, 100 μM, 200 μM, 500 μM, and 1000 μM of ELP-K12 in PBS with or without CpG at an N:P ratio of 7:1. Samples were heated and then cooled at a rate of 0.66°C/min to obtain plots of optical density versus time. The inverse transition temperature (Tt) was defined as the temperature with the maximum rate of increase in OD350.
In vitro assays for activity and cellular uptake
RAW264.7 cells (American Type Culture Collection, Manassas, VA) were cultured in Gibco’s DMEM (ThermoFisher Scientific, Waltham, MA) supplemented with 10% HI-FBS, incubated at 37°C and 5% CO2, and cultured according to the manufacturer’s instructions. To evaluate the effects of ELP-K12 on TLR9 activation by CpG ODN 1826, cells were incubated for 16 h with 5 μg/mL of CpG alone or with ELP-K12 at a 1:1 or 3:1 N:P ratio. After incubation, the nitrite concentrations in the cell supernatants were quantified by a Griess reagent kit (ThermoFisher Scientific, Waltham, MA). Cell uptake was evaluated by incubating RAW264.7 cells for 16 h with 5 μg/μL of a FITC-CpG conjugate (Invivogen, Carlsbad, CA) alone or with ELP-K12 at an N:P ratio of 1:1. Cells were removed by scraping, washed with PBS + 1% w/v BSA, and run on an Accuri C6 flow cytometer (BD Biosciences, Franklin Lakes, NJ). Cell uptake of ELP-K12 was visualized by confocal fluorescence microscopy as follows. ELP-K12 was labeled at its N-terminus with AlexaFluor350 (AF350) NHS-ester (ThermoFisher Scientific, Waltham, MA) and purified by ultrafiltration (Amicon, Millipore Sigma, St. Louis, MO), RAW264.7 cells were then incubated for 16 h with 2.5 μg/μL of FITC-labeled CpG alone or with AF350-labeled ELP-K12 at an N:P ratio of 1:1. After incubation, cells were washed with PBS, fixed with 4% paraformaldehyde, permeabilized with 0.2% Tween20, incubated with an anti-LAMP1 antibody (Millipore Sigma, St. Louis, MO), and were stained with AlexaFluor 594 (AF594) anti-mouse antibody (AbCam, Cambridge, UK). Samples were imaged with an Axios Observer confocal microscope (Zeiss, Oberkochen, Germany) with a 20x objective.
Establishment of the orthotopic mouse model
The 4T1 mammary carcinoma cell line (American Type Culture Collection, Manassas, VA) was cultured in Gibco’s RPMI-1640 (ThermoFisher Scientific, Waltham, MA) supplemented with 10% HI-FBS, incubated at 37°C and 5% CO2, and cultured according to the manufacturer’s instructions. Cells were verified as pathogen-free by IMPACT III murine pathogen analysis performed by IDEXX BioResearch (Columbia, MO). 60% confluent cells were trypsinized, pelleted, and resuspended in RPMI-1640 without FBS at a concentration of 1×106 cells/mL. Female 6–8 week-old BALB/c mice were purchased (Charles River, Wilmington, MA) and housed in the Duke Cancer Center Isolation Facility. Before inoculation, fur was removed from the abdomen of mice, and the mice were then anesthetized with 1.5% vaporized isoflurane, and 1×105 4T1 cells were injected into the 4th mammary fat pad of each mouse.
Intratumoral retention
AlexaFluor 647 (AF647) labeled CpG ODN 1826 was purchased (Integrated DNA Technologies, Coralville, IA) and used to track CpG retention in vivo. CpG was prepared for injections by mixing AF647-labeled CpG and unlabeled CpG at a 1:2400 ratio. BALB/c mice bearing ~100 mm3 orthotopic 4T1 tumors received treatment with either CpG alone, CpG with a control ELP, or CpG with ELP-K12. Each group received a dose of 100 μg of CpG. Control ELP was prepared as 1000 μM of ELP mixed with CpG, while ELP-K12 was prepared a solution of 500 μM ELP-K12, 500 μM of ELP, and CpG to yield a total ELP concentration of 1000 μM and an N:P ratio of 1:1. 50 μL of each CpG solution was injected i.t. through a 27 ½ gauge microsyringe that was kept on ice and actuated with a motorized pump to maintain an injection rate of 120 μL/min. After the injection, mice were imaged on a Fluorescence Molecular Tomography 4000 In Vivo Imaging System (Perkin Elmer, Waltham, MA). Fluorescence at the tumor site was quantified using the TrueQuant software (Perkin Elmer, Waltham, MA). Standards of known concentration were separately imaged, and the fluorescence versus concentration calibration was used to calculate the amount of CpG retained in the tumor from the fluorescence signal of the tumor.
In vivo efficacy
4T1 tumors, orthotopically inoculated in BALB/c mice, were allowed to grow to ~100 mm3, at which time treatment was initiated (day 0). For assessment of 131I-ELP dose, at day 0, mice received 131I-ELP i.t. injections at ⅓ the total tumor volume with 1000 μM of ELP and radioactivity doses of 122.1 kBq/mm3, 244.2 kBq/mm3, and 370 kBq/mm3 of tumor tissue. For the ELP-K12/CpG monotherapy, at day 0, mice received 50 μL i.t. injections of 50 μg or 100 μg of CpG mixed with ELP-K12 at an N:P ratio of 1:1 and a total ELP concentration of 1000 μM. For combination therapy, on day 0, mice received i.t. injections of 100 μg of CpG with or without ELP-K12, and on day 1, mice received i.t. injections of 122.1 kBq/mm3 of 131I-ELP. All treatments were infused into the center of the tumor through a 27 ½ gauge microsyringe that was kept on ice and actuated with a motorized pump at a rate of 120 μL/min. Bodyweight was measured with a scale and total body radioactivity of mice receiving 131I-ELP treatment was measured with an AtomLab 400 dose calibrator. Tumor dimensions were measured over time, and the tumor volume was calculated as length*width2*0.52. Mice were sacrificed when tumors reached a volume of 1650 mm3, when mice sustained a bodyweight loss of >15%, or when mice exhibited moribundity, such as lack of mobility and difficulty breathing.
To quantify lung metastases, 4T1 bearing BALB/c mice were euthanized once 40% of the control mice reached their endpoint, and the abdominal cavity was opened to expose the lungs. 15% India ink was injected into the lungs through the trachea to stain the lungs and provide the necessary contrast to visualize metastatic nodules. Lungs were harvested, washed with PBS, fixed with Fekete’s solution, and imaged with a camera. The total number of tumor nodules in the lungs was counted.
Results
ELP brachytherapy inhibits 4T1 tumor growth but does not extend mouse survival, regardless of dose.
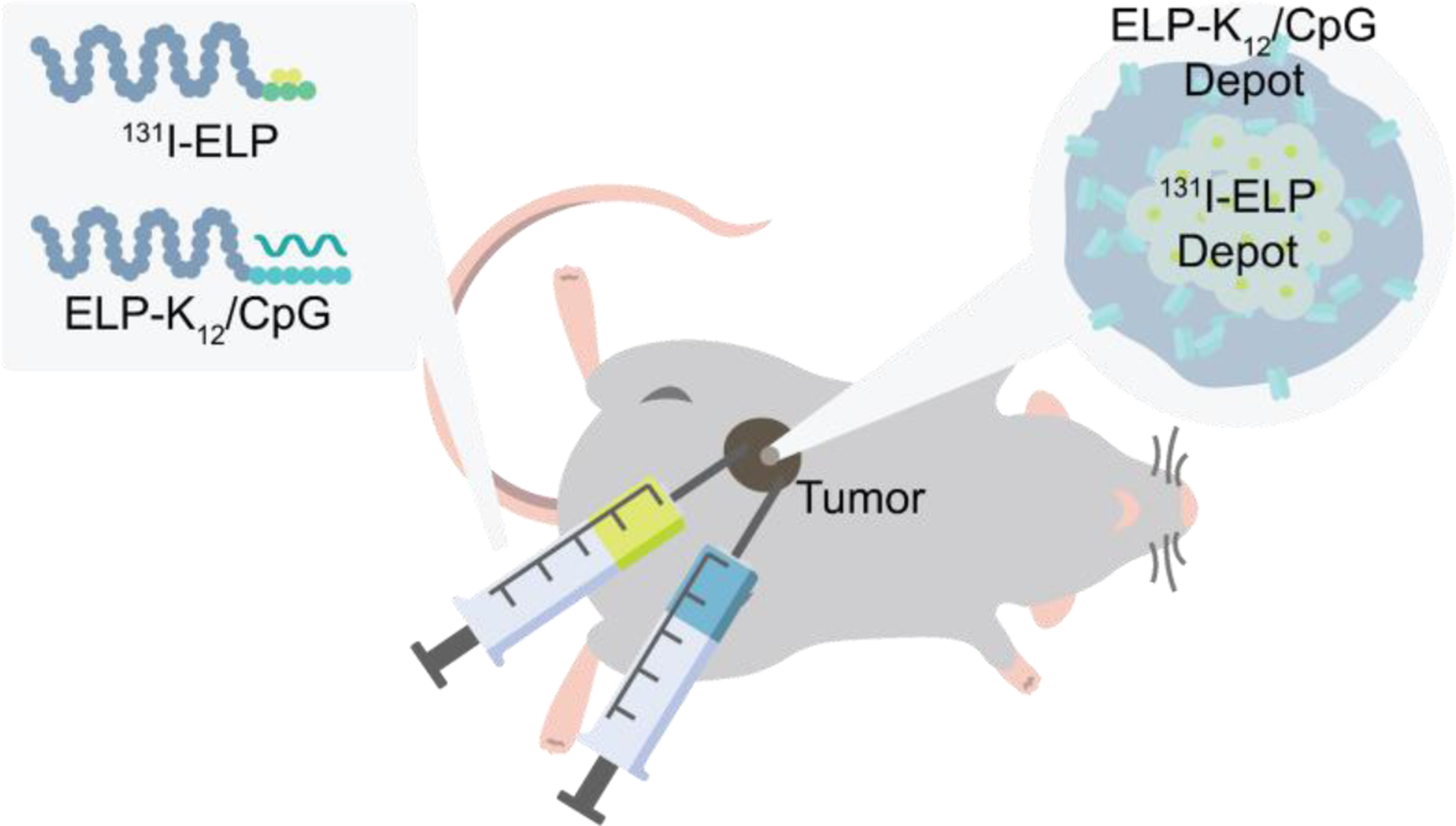
We first tested the antitumor efficacy of brachytherapy as a function of the dose of 131I-ELP in a metastatic 4T1 breast cancer model in immunocompetent mice. We established tumors by orthotopic inoculation of 4T1 cells in 8-week-old female BALB/c mice. Once tumors reached approximately 100 mm3 in size, we injected the depot-forming 131I-ELP conjugate into the core of the tumor in a volume 1/3 of the tumor size and at three radioactivity doses – 122.1 kBq/mm3, 244.2 kBq/mm3, and 370 kBq/mm3 of tumor tissue, which correspondsto 3.3 μCi/mm3, 6.6 μCi/mm3, and 10 μCi/mm3 of tumor tissue. Tumor growth and mouse survival were monitored, and treatment groups were compared to a control group of untreated mice. Consistent with our previous study25, 131I-ELP treatments were well tolerated with only a minimal decrease in body weight, and the radioactivity level in the tumor decayed exponentially over the period of 2–3 weeks (Fig. S1). There was significant inhibition of tumor growth at all three doses of 131I-ELP, as the tumors of treated mice were approximately threefold smaller than untreated mice after 19 days (p<0.05, ANOVA, Tukey). There was no difference in tumor growth inhibition between the three treatment groups (Fig. 2A). Despite significant tumor growth inhibition, there was no improvement in mouse survival, possibly due to metastatic tumor burden as mice had to be sacrificed due to signs of moribundity (Fig. 2B). These findings suggest that while local irradiation with i.t. 131I-ELP was effective at controlling the size of the primary tumor in an immunocompetent breast cancer model, a lack of systemic tumor control limited any survival benefit from 131I-ELP brachytherapy.
Figure 2: Antitumor efficacy of 131I-ELP in orthotopic 4T1 tumors.
(A) Average size of untreated tumors or tumors treated with 122.1 kBq/mm3, 244.2 kBq/mm3, and 370 kBq/mm3 of 131I-ELP through first 19 days post-treatment (n=4). (B) Kaplan-Meier survival curves for treatment groups. (n=4). *p<0.05 (ANOVA, Tukey).
A recombinant ELP fusion with an oligolysine tail binds CpG and exhibits LCST phase behavior.
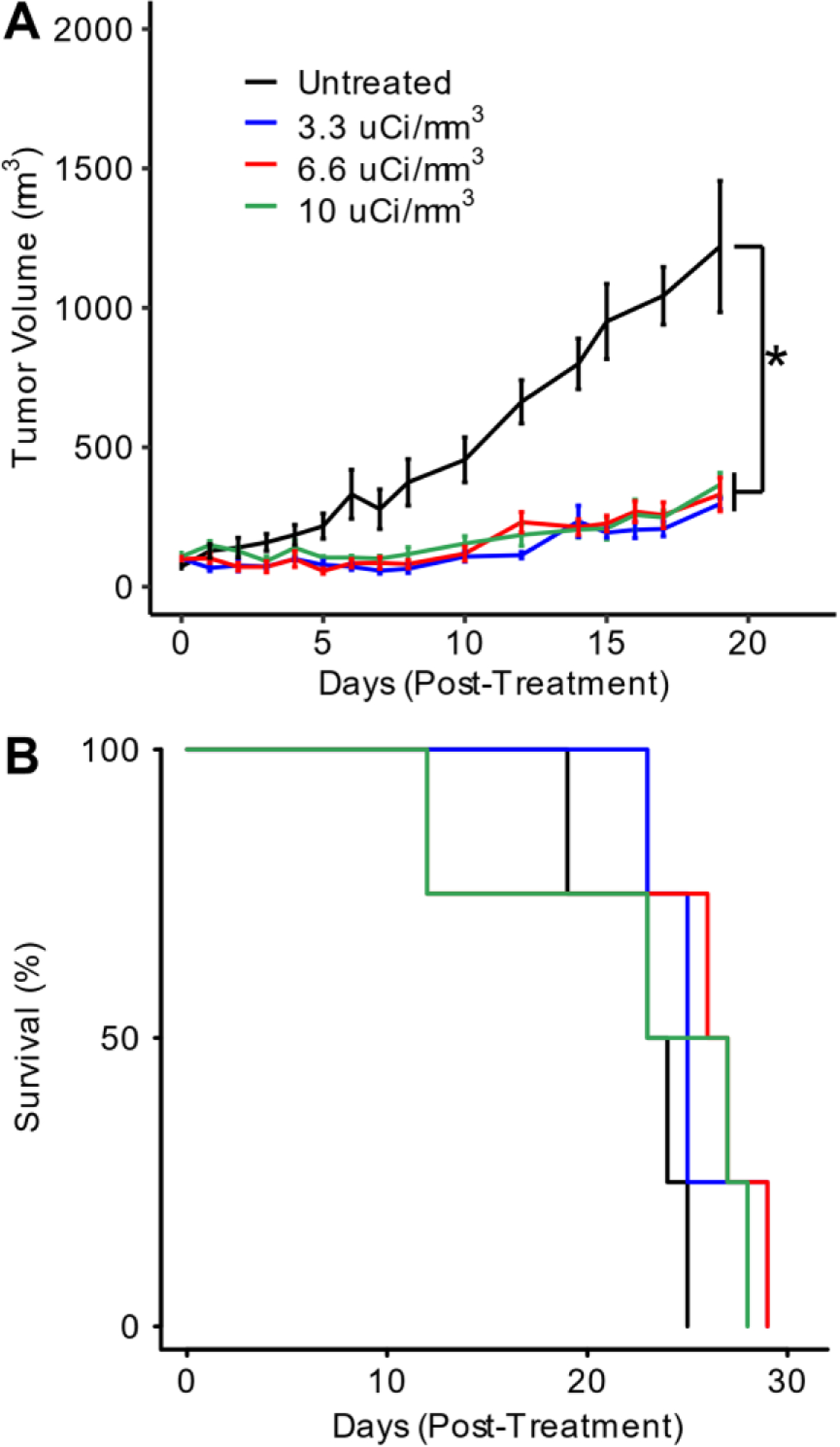
For sustained-release of CpG from an ELP depot, we designed an ELP with a Tt below body temperature for depot formation and a positively charged tail for complexation with CpG. We chose an ELP sequence consisting of 60 repeats of the VPGVG pentamer as it has a Tt of 32°C at 1000 μM, which is the target ELP concentration for i.t. delivery of CpG (Fig. S2). We fused a K12 tail to the C-terminus of the ELP at the gene level to enable electrostatic complexation of the negatively charged CpG to ELP-K12. We chose lysine residues due to the extensive use of oliglysine polymers and lysine-rich peptides to deliver nucleic acids37–39, including CpG in particular40–42. We chose to append 12 contiguous lysine residues to each ELP chain to balance the binding affinity of CpG to the oligolysine tail with the increase in the Tt of the ELP caused by the introduction of ionizable Lys residues. The ELP-K12 fusion was expressed in Escherichia coli from a T7 expression vector and was purified by exploiting the LCST phase behavior of the ELP-K12 fusion using inverse-transition cycling (ITC)34 (Fig. S3). After 2–3 rounds of ITC, ELP-K12 was >95% pure, as verified by SDS-PAGE (Fig. 3A) and had the expected molecular weight, as assessed by mass spectrometry (Fig. S4). A gel shift assay was performed to assess the ability of ELP-K12 to bind CpG through its oligolysine tail. In the presence of ELP-K12, CpG is prevented from migrating in an agarose gel, indicating a strong interaction between the ELP and CpG (Fig. 3B). CpG-binding to ELP-K12 in the gel shift assay was demonstrated at ratios of ELP to CpG that correspond to amine to phosphate (N:P) ratios of 1:1, 3:1, and 7:1 (Fig. S6).
Figure 3: Expression and characterization of ELP-K12 fusion.
(A) SDS-PAGE gel of purified ELP-K12 fusion. (B) Gel-shift assay for CpG with and without ELP-K12. (C) Optical turbidity at 350 nm as a function of temperature of ELP-K12 and ELP-K12:CpG complex at an ELP concentration of 200μM.
We next characterized the LCST phase behavior of the ELP-K12 fusion by temperature-programmed turbidimetry. ELP-K12 exhibits a small gradual change in turbidity around body temperature (37°C) that is characteristic of micelle formation (Fig. 3C). Dynamic light scattering (DLS) and cryo-transmission electron microscopy (Cryo-TEM) confirmed that ELP-K12 forms spherical micelles with a hydrodynamic radius (Rh) of 22 nm at a temperature greater than 37°C (Fig. S7 & S8). When mixed with CpG, electrostatic complexation of the negatively charged CpG with the positively charged K12 tail of ELP-K12 abolishes micelle formation, and the complex displays the classic soluble to coacervate phase behavior, as seen by the sharp and large increase in optical turbidity above its Tt of 27 °C (Fig. 3C) These data also show that the turbidity of the complex is thermally reversible, which is indicative of thermodynamic phase behavior. We also quantified the Tt as a function of ELP-K12 concentration and determined that at all concentrations between 25 μM and 1000 μM the Tt is below body temperature. At the proposed injection concentration of 1000 μM, ELP-K12/CpG has a Tt of ~22 °C – a suitable temperature for depot formation upon administration in vivo. (Fig. S5).
ELP-K12 complexation enhances TLR9 activation by increasing cellular uptake of CpG.
Activation of the innate immune system by CpG was measured by a Griess reagent assay that quantified nitric oxide (NO) production from RAW264.7 macrophage cells incubated for 24 h with CpG. The endocytosis of CpG ODN 1826 by RAW264.7 murine macrophages results in TLR9 activation and endosomal maturation, leading to the MyD88-dependent activation of NF-κB expression and the production of NO by inducible NO synthase43. The ELP-K12/CpG complex increased innate immune activation over CpG alone, leading to a two-fold increase in NO production (p<0.05, ANOVA, Tukey) (Fig. 4A). This enhancement of activity occurs at an N:P ratio as low as 1:1, which we selected as the N:P ratio for i.t. administration of ELP-K12/CpG. We hypothesized that the enhanced activity of ELP-K12/CpG in vitro is due to an increase in cellular uptake of the charge-neutralized, ELP-K12/CpG complex, leading to greater activation of the intracellular TLR9 receptor compared to CpG alone. To measure cellular uptake of the CpG, RAW264.7 macrophage cells were cultured overnight with FITC-labeled CpG with and without ELP-K12. Flow cytometry analysis confirmed that ELP-K12 led to a significant increase in cellular uptake of CpG (p<0.05, Student t-test) (Fig. 4B). To visualize intracellular localization of the CpG, FITC-labeled CpG was added to RAW264.7 cells with or without AlexaFluor350 (AF350)-labeled ELP-K12, and the cells were cultured overnight, fixed, and permeabilized. Cells were stained for LAMP1 with AlexaFluor594 (AF594) to determine CpG localization within the early endosome, where TLR9 is expressed. Confocal fluorescence microscopy was used to visualize and compare the difference in uptake and intracellular localization of CpG between groups (Fig. 4C–D). For each group, FITC fluorescence within the cells was quantified and the colocalization of FITC and AF594 in cells was calculated as the Pearson’s correlation coefficient for the two signals. This analysis revealed an increase in the intracellular CpG and the colocalization of CpG with LAMP1 in cells incubated with ELP-K12/CpG compared to cells incubated with CpG alone (p<0.05, student t-test) (Fig. S9). These results indicate that the complexation of CpG with ELP-K12 increases the innate immune stimulation through improved cellular uptake and trafficking of CpG to the endosome where it can activate TLR9.
Figure 4: CpG complexed with ELP-K12 shows greater cellular uptake and immune stimulation than CpG alone.
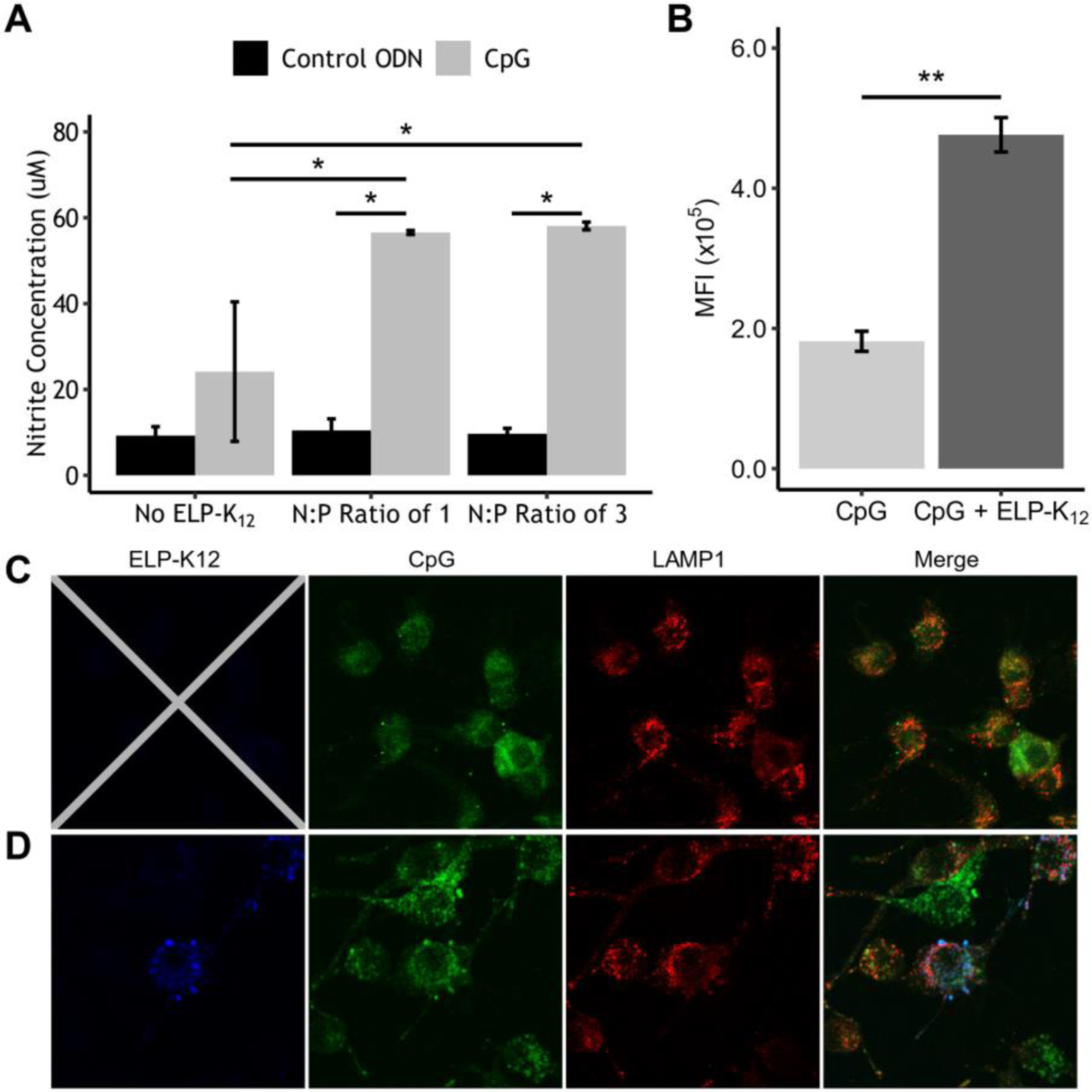
(A) Production of NO by Raw264.7 cells induced by ELP-K12/CpG-ODN complex at multiple N:P ratios as measured by a colorimetric Griess assay. (B) Flow cytometry analysis of cellular uptake of fluorescently labeled CpG by RAW264.7 cells treated with ELP-K12/CpG or CpG alone. Confocal fluorescence microscopy of cellular uptake of FITC-labeled CpG (green) alone (C) and with AlexaFluor350-labeled ELP-K12 (blue) (D). Secondary labeling with AlexaFluor594 marks LAMP1, designating the early endosome (red). *p<0.05 (ANOVA, Tukey), **p<0.05 (student t-test)
Intratumoral delivery by ELP-K12 dramatically prolongs the retention of CpG.
We next investigated the effect of an i.t. ELP-K12 depot on the tumor localization and release kinetics of CpG from the depot. AlexaFluor647 (AF647)-labeled CpG was diluted 1:2400 with unlabeled CpG. 100 μg of the labeled CpG mixture was formulated alone, with 1000 μM of ELP, or with 500 μM of ELP and 500 μM of ELP-K12 to yield an N:P ratio of 1:1. The CpG formulations were injected i.t. into orthotopic 4T1 tumors in BALB/c mice, which were subsequently imaged at multiple time points to quantify the fluorescence intensity within the tumor. Images of fluorescence standards and the ratio of AF350-labeled CpG to total CpG were used to calibrate the fluorescence intensity to CpG dose within the tumor at each time point (Fig. S10). When administered alone or in combination with a control ELP depot lacking a K12 tail, CpG rapidly diffused out of the tumor within the first 24 h (Fig 5A–B). In stark contrast, CpG delivered from an ELP-K12 depot resulted in sustained retention of CpG within the tumor with 25% (25 μg) of CpG remaining within the tumor 21 days after injection (Fig. 5A–B). A comparison of the calculated area under the curve (AUC) between the different groups demonstrates a significant increase in the CpG exposure of the tumor for the ELP-K12/CpG complex relative to the other groups (p<0.05, ANOVA, Tukey) (Fig. 5C).
Figure 5: Intratumoral retention of CpG delivered by an ELP depot.
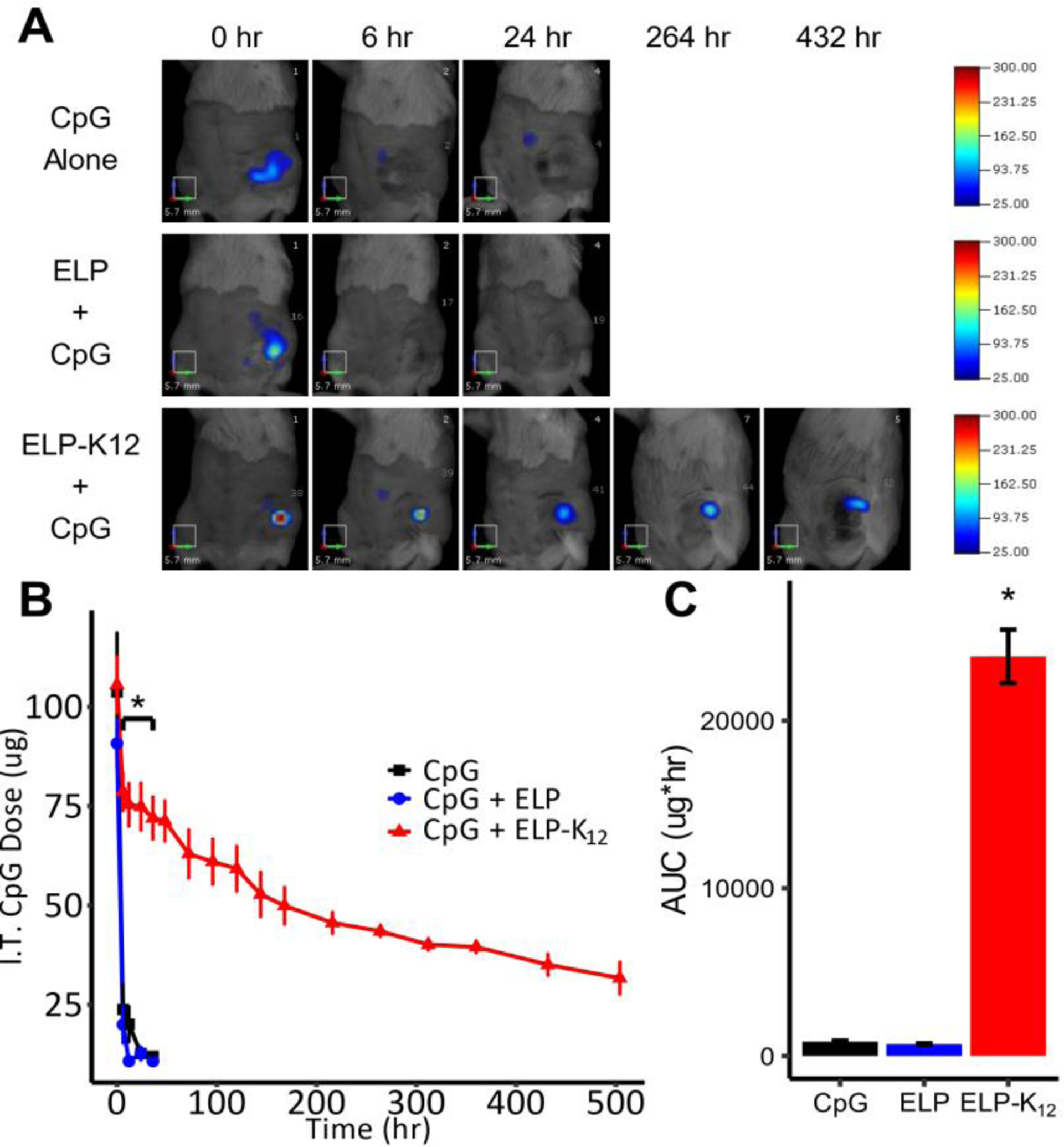
(A) Representative fluorescence molecular tomography (FMT) images of mice bearing orthotopic 4T1 tumors injected with fluorescently labeled CpG alone (n=3), an ELP depot (n=3), or an ELP-K12 depot (n=6), or (B) I.t. retention of CpG delivered with or without an ELP depot over time. (C) Area under the curve (AUC) for the i.t. retention of CpG from the three different treatments. *p < 0.05 (ANOVA, Tukey).
ELP-K12/CpG immunotherapy demonstrates both local and systemic tumor control in a 4T1 breast cancer model.
To determine the efficacy of CpG delivered from a depot of ELP-K12 in generating local and systemic antitumor immunity, we first tested it as a monotherapy in the 4T1 tumor model. Female BALB/c mice bearing 100 mm3 4T1 tumors were injected i.t. with either: (1) 50 μg or (2) 100 μg of CpG complexed with ELP-K12 at an N:P ratio of 1:1. Untreated mice comprised the control group. Primary tumor growth was monitored for 13 days post-treatment, at which point mice were sacrificed to remove the lungs and assess metastatic tumor burden. Both doses of CpG were well tolerated as mice in both treatment groups experienced only transient weight loss of less than 10% (Fig. S11). Treatment with 50 μg of CpG with ELP-K12 led to a reduction in tumor growth that was not statistically different from untreated mice (ANOVA, Tukey). Treatment with 100 μg of CpG complexed with ELP-K12 led to a greater reduction in tumor growth and significantly smaller primary tumors than in the control group at day 13 post-treatment (p<0.05, ANOVA, Tukey) (Fig. 6A). Furthermore, treatment with ELP-K12 at both doses of CpG resulted in a significant reduction of lung metastases (p<0.05, ANOVA, Tukey) (Fig. 6B). These results demonstrate that at a 100 μg dose, immunotherapy with ELP-K12/CpG leads to both local and systemic control in a poorly immunogenic tumor model.
Figure 6: Antitumor efficacy of intratumoral ELP-K12/CpG.
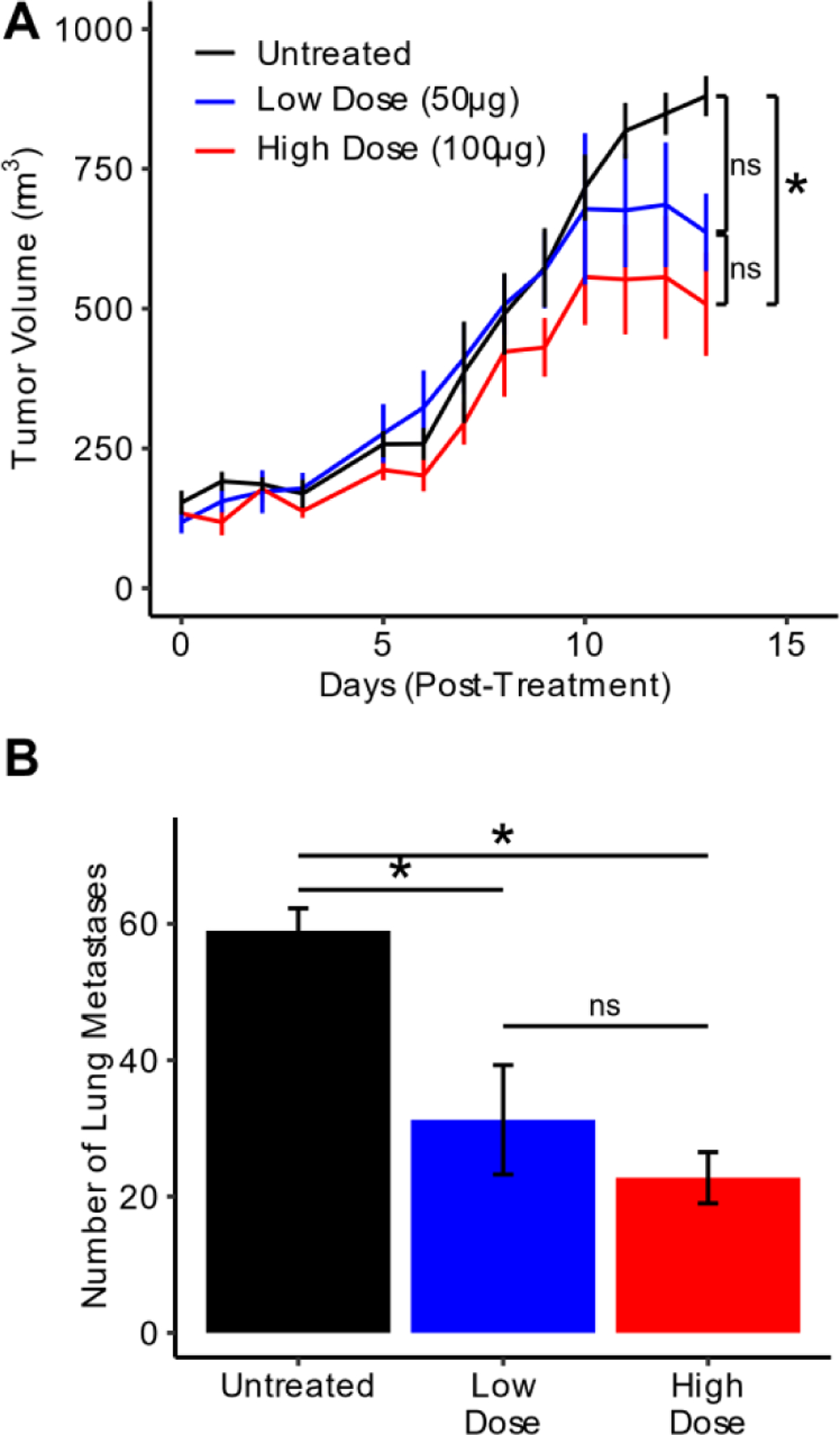
(A) Average size of 4T1 untreated tumors or tumors treated i.t. with 50 μg or 100 μg CpG complexed with ELP-K12 through the first 13 days post-treatment (n=4). (B) Number of metastases in dissected lungs from mice for each treatment group at day 13 post-treatment (n=4). *p<0.05 (ANOVA, Tukey)
A combination of ELP brachytherapy and CpG immunotherapy from an ELP depot significantly improves local tumor control and enhances survival of 4T1 tumor-bearing mice.
Having established the efficacy of 131I-ELP and ELP-K12/CpG monotherapies, we assessed the ability of the combination of ELP radio- and immuno-therapy to act in tandem for the treatment of 4T1 mammary carcinoma. We set out to evaluate the efficacy against 4T1 tumors with treatment groups including the combination therapy, 131I-ELP and ELP-K12/CpG monotherapies, free CpG, and untreated controls. When tumors reached 100 mm3, mice receiving CpG, ELP-K12/CpG, or the combination therapy were injected i.t. with 100 μg of CpG with or without ELP-K12. 24 h later, mice receiving 131I-ELP or the combination therapy were injected i.t. with 122.1 kBq/mm3 of 131I-ELP.
The combination therapy was well tolerated, leading to a transient weight loss in mice of less than 10%, and radioactivity in the tumor exponentially decayed over 2 weeks (Fig. S12). When mice were monitored for tumor growth and survival (Fig. 7A–B), we found that both ELP-K12/CpG and the combination of 131I-ELP and ELP-K12/CpG, but not CpG alone, led to significant tumor growth inhibition over untreated mice by day 19. Thus, i.t. delivery of CpG via ELP depot provides improved anticancer efficacy. The combination of 131I-ELP and ELP-K12/CpG resulted in further tumor growth inhibition. Furthermore, the combination therapy greatly enhanced survival, leading to an increase in median survival by 11 days over untreated mice and significantly outperforming all other treatment groups. This extended survival time in the metastatic model suggests that the 131I-ELP and ELP-K12/CpG therapies improve systemic tumor control. A synergy test was performed by comparing the observed survival to a prediction of survival calculated from the survival curves of 131I-ELP and ELP-K12/CpG treatment groups, assuming a Bliss definition of independence44. The observed survival for the combination therapy was significantly greater than the predicted survival, (Fig. S13), indicating that the combination of 131I-ELP and ELP-K12/CpG act synergistically to generate an antitumor response for improved local and systemic control and prolonged survival in a metastatic tumor model.
Figure 7: Combination of 131I-ELP and ELP-K12/CpG significantly inhibits tumor growth and extends mouse survival.
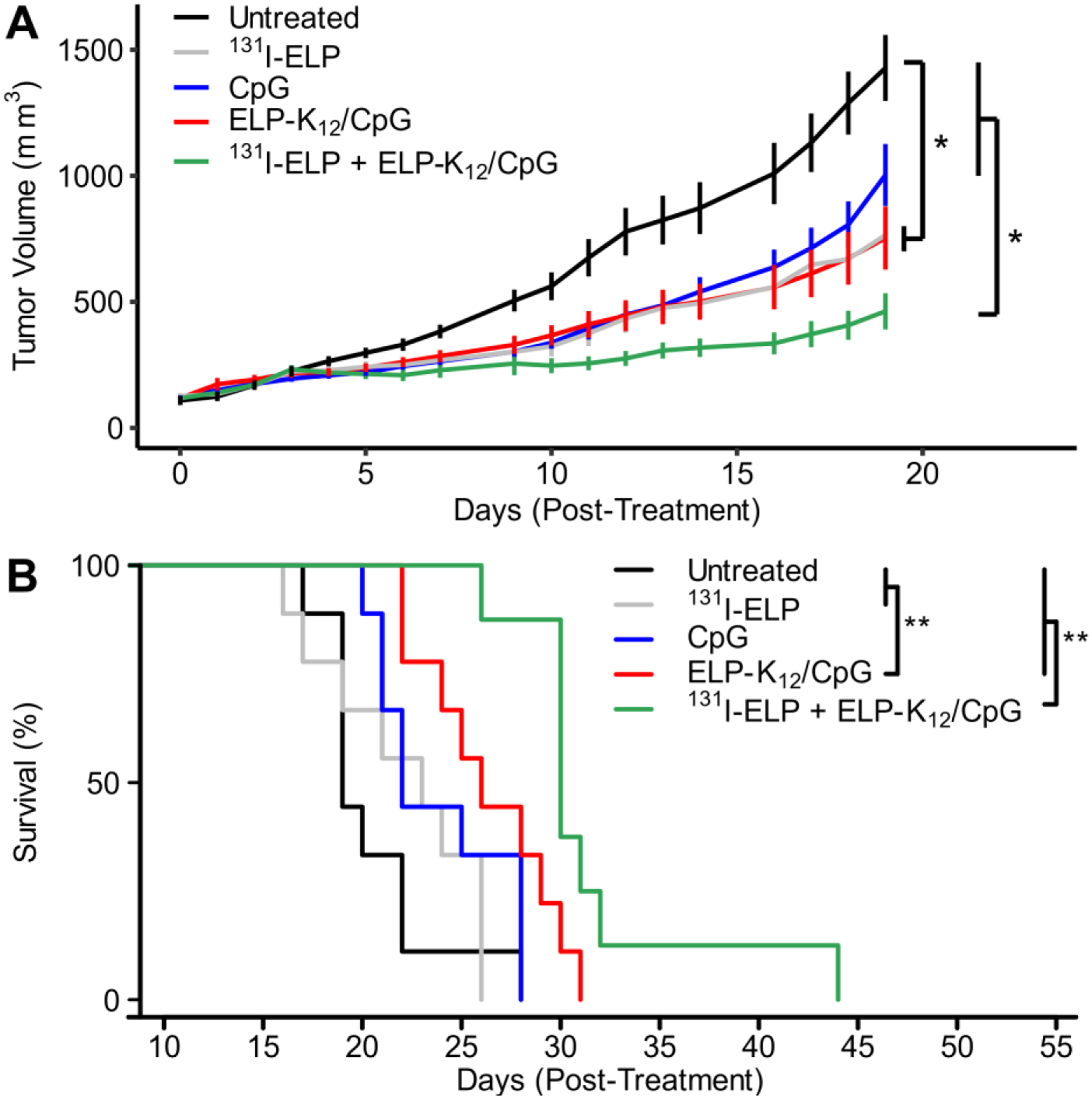
(A) Average size of 4T1 tumors untreated or treated with 131I-ELP monotherapy, CpG monotherapy, ELP-K12/CpG monotherapy, or a combination of 131I-ELP and ELP-K12/CpG. CpG alone or in complex with ELP-K12 was administered i.t. at a dose of 100 μg; for combination therapy 131I-ELP was administered intratumorally one day later at a dose of 122.1 kBq/mm 3. The same radioactivity dose was used for 131I-ELP monotherapy. *p<0.05 (ANOVA, Tukey). (B) Kaplan-Meier survival curves for treatment groups (n=8–9). **p<0.05 (Mantel-Cox test)
Treatment with ELP brachytherapy and ELP-bound CpG leads to significant systemic tumor control.
To investigate whether improved control of distant metastases may have influenced the increase in survival seen for the combination of 131I-ELP brachytherapy and CpG delivery from an i.t. ELP depot, we quantified the lung metastases for the different treatments. 4T1-bearing mice were treated with a combination of 131I-ELP and ELP-K12/CpG, monotherapy controls, or an ELP-only control treatment. Once 40% of ELP-control mice reached their endpoint, mice were sacrificed and lungs were harvested, fixed, and stained. Images of lungs and quantification of lung metastases (Fig. 8A–B) revealed that ELP-K12/CpG as a monotherapy was able to significantly decrease the number of lung metastases in comparison to the control group, showing that ELP-delivery of CpG leads to a systemic antitumor effect (Fig. 8A). Furthermore, the combination of ELP-K12/CpG and 131I-ELP significantly decreased the number of metastases in comparison to 131I-ELP monotherapy and the ELP-only control (Fig. 8A).
Figure 8: Combination of 131I-ELP and ELP-K12/CpG reduces lung metastases.
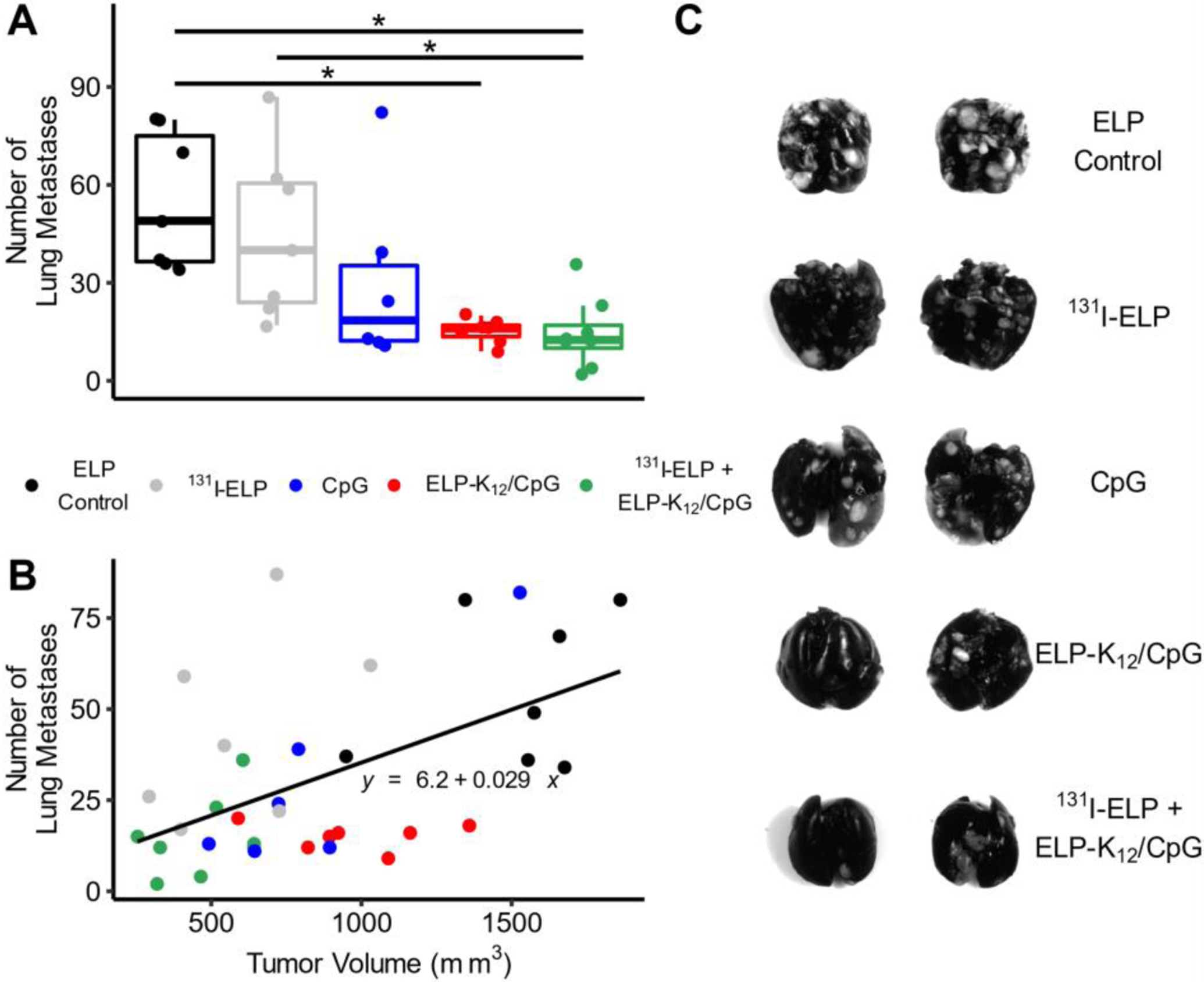
(A) Representative images of lungs resected from mice bearing 4T1 tumors treated with ELP control, CpG monotherapy, ELP-K12/CpG monotherapy, 131I-ELP monotherapy, or a combination of ELP-K12/CpG and 131I-ELP. Treatments of CpG alone or in complex with ELP-K12 were administered intratumorally at a dose of 100μg and one day later treatments of 131I-ELP were administered intratumorally at a dose of 122.1 kBq/mm3 (n=7). Once 40% of the control group reached their endpoint, mice were sacrificed, and lungs were resected and stained to quantify lung metastases. (B) Quantification of metastases. (C) Plot of the number of lung metastases versus tumor volume at day of sacrifice (n=6–7). *p<0.05 (ANOVA, Tukey), R2= 0.2856
These results demonstrate the improved systemic tumor control by CpG immunotherapy from an ELP depot and support the hypothesis that the enhanced survival benefit of the combination therapy is due to systemic control of metastases. As metastatic burden can correlate with primary tumor volume, we compared lung metastases and tumor volume for each mouse to determine whether the treatment controlled metastases indirectly by reducing the size of the primary tumor or directly through inhibition of the formation or dissemination of metastases (Fig. 8C). A linear regression of the data revealed a weak (R2=0.2856), but significant (p<0.05) positive correlation between the number of lung metastases and tumor volume. While 131I-ELP treatment led to smaller primary tumors, the number of lung metastases was greater than predicted by the correlation between metastases and tumor size, as the average of the residuals for 131I-ELP-treated mice was significantly greater than zero (p=0.05, student T-test). In contrast, the number of lung metastases in ELP-K12 treated mice was significantly lower than predicted by the correlation with tumor size (p<0.05, student T-test). These results suggest that ELP-K12/CpG directly reduces metastatic tumor burden and that the combination therapy, which resulted in small tumor sizes and low numbers of metastases, benefitted from the additive effects of both therapies.
Discussion
Persistent challenges in treating metastatic cancer have driven the search for new combination therapies to control systemic disease. Significant effort has been devoted to the combination of radiotherapy and immunotherapy because of the ability of these therapies to work in tandem to incite immunogenic cell death and generate cancer-specific T cells to mediate an abscopal response9. For example, Chao et al. demonstrated that 131I brachytherapy could achieve remarkable therapeutic effects in combination with local CpG immunotherapy15. However, their choice of material for a brachytherapy depot – alginate – is a structurally and compositionally variable biomaterial45, which can suffer from burst release kinetics and restricted control of delivery. These adverse characteristics are highlighted by the limited retention of CpG for less than 72 h15. Further advances in the therapeutic efficacy of combinations of radio- and immuno-therapy require a novel platform with precise control of spatial distribution and kinetics for both radiotherapy and immunotherapy. Motivated by these results, we sought to improve upon the combination of radiotherapy with immunotherapy, using an ELP platform.
ELPs have several important properties that are ideal for local radiotherapy and immunotherapy: (1) their depot-forming phase behavior allows tunable local retention of therapeutic agents, (2) the genetically encoded fusion of leader and trailer peptide sequences to an ELP enables the delivery of a diverse range of biologics, and (3) the ability to tune the Tt of the ELP enables precise control of release kinetics19,46,47. We have previously performed extensive optimization of the radiolabeled 131I-ELP to provide superior retention for a potent cancer radiotherapy. The coacervation of an ELP with a Tt below body temperature allows the ELP to form a depot upon injection, while the addition of a tyrosine trailer sequence provides a reactive site for conjugation of 131I and promotes micelle formation to stabilize the depot24. Optimization of the Tt through sequence modification and lengthening of the tyrosine tail led to a 131I-ELP that rapidly forms a depot upon injection in vivo and is retained in the tumor for more than 60 days. These optimized properties enable sustained tumor irradiation with low radiation exposure to healthy tissues and lead to effective tumor control in multiple human xenograft tumor models25. Investigation in an immunocompetent 4T1 mouse model demonstrated that 131I-ELP at doses as low as 122.1 kBq/mm3 leads to significant inhibition of primary tumor growth, but brachytherapy alone is – as expected – insufficient to control systemic metastases and prolong mouse survival.
To improve ELP brachytherapy through the generation of anticancer immunity, we engineered an ELP that has: (1) Tt of 22 °C so that it forms a depot in vivo, and (2) an oligolysine trailer to electrostatically complex CpG to the ELP. Compared to CpG alone, the ELP-K12/CpG complex has two-fold greater cellular uptake. A similar increase in cellular uptake has been observed with other carriers for DNA in general and CpG DNA, in particular, has been attributed to the ability of positively charged amino acids to facilitate endocytosis48–50. Further investigation, however, is needed to determine the exact mechanism of cellular uptake of ELP-K12/CpG. Enhanced cellular uptake corresponded to a two-fold increase in innate immune activation through intracellular TLR9 signaling, highlighting the potential for ELP-K12 delivery to improve the potency of CpG immunotherapy.
Upon i.t. injection in vivo, free CpG is cleared from the tumor within 24 h, whereas ELP-K12/CpG forms a depot within the tumor that releases CpG for over 3 weeks. This increase in the intratumoral retention of CpG represents a significant improvement in CpG delivery over other biomaterials like alginate and DNA hydrogels that only retain CpG for a few days15,51,52. Increased CpG retention should reduce systemic exposure of CpG, hence minimizing any systemic, off-target toxicity, such as lymphoid follicle degradation and immunosuppression that is seen with repeated CpG administration53. The ELP platform allows for the precise control of the Tt19and thereby the release kinetics of the ELP/CpG complex. Our previous work has demonstrated how sequence modifications can be used to tune the ELP Tt and propensities for micelle formation and achieve local retention of i.t. and subcutaneous depots19,24. Furthermore, tuning the binding capacity of ELP-K12 by adjusting the N:P ratio would provide an additional level of control over CpG retention time. Further exploration of ELP sequence modifications to tune and optimize the duration of CpG retention has the potential to provide new insights into the interactions of local radiotherapy and immunotherapy and lead to a further improved combination therapy.
In vivo assessment of ELP-K12/CpG immunotherapy revealed that i.t. injection of 100 μg of CpG in complex with ELP-K12 led to inhibition of local tumor growth and a 2.5-fold reduction in lung metastases. One plausible explanation for this efficacy is the generation of systemic anticancer immunity. Furthermore, the addition of ELP-delivered CpG to i.t. 131I-ELP brachytherapy further inhibited tumor growth and successfully extended median survival by 11 days over untreated mice. In the poorly immunogenic31,54 4T1 mouse model, for which radiation alone does not improve survival7, the significant increase in survival reveals the synergistic effects of the combination of radiotherapy and immunotherapy delivered from an i.t. ELP depot. Analysis of lung metastases showed that the combination treatment resulted in a four-fold reduction of metastases over the ELP control treatment and a three-fold reduction over brachytherapy alone. These findings strengthen the case for the augmented anticancer immunity by CpG delivered from an ELP depot as well as its synergistic effects in combination with 131I-ELP brachytherapy. ELP-K12/CpG controls systemic disease, while 131I-ELP controls local disease, leading to a significant improvement in mouse survival for the combination therapy relative to each monotherapy. Overall, our findings demonstrate a preclinical proof-of-concept of the potential utility of an ELP depot for the sustained, local delivery of brachytherapy and immunotherapy for effective cancer treatment.
Conclusions
With surging interest in novel combinations of immunotherapy and radiotherapy, biomaterials for localized, sustained delivery can improve both local and systemic control of cancer. Here we demonstrate the development of a depot forming ELP fusion to enhance the activity and prolong the retention of CpG immunotherapy within the primary tumor. We show that CpG immunotherapy delivered from an i.t. ELP depot works in tandem with ELP brachytherapy to significantly improve the treatment of poorly immunogenic, metastatic cancer by controlling local tumor growth and reducing metastatic tumor burden. This work establishes a new platform for tunable delivery of immunotherapies and highlights the advantages of sustained, local delivery for a combination of radiotherapy and immunotherapy.
Supplementary Material
Highlights.
Complexation of CpG with an oligolysine functionalized biopolymer enhances immune stimulation
Phase transition of biopolymer into depot upon intratumoral injection retains CpG within tumor for more than 3 weeks
Brachytherapy and immunotherapy synergistically improve survival of mice with 4T1 tumors
Intratumoral CpG immunotherapy reduces metastatic burden in the lungs
Acknowledgements
Mass spectrometry was performed on shared equipment in the Duke Mass Spectrometry facility. Confocal microscopy was performed on shared equipment in the Duke Light Microscopy Core Facility. FMT imaging was performed with shared equipment managed by the Optical Molecular Imaging and Analysis core facility under the Duke Cancer Institute. Flow cytometry was performed on equipment supported by the NSF Research Triangle Materials Research Science and Engineering Center [grant number DMR-1121107]. Transmission electron microscopy was performed at the Duke University Shared Materials Instrumentation Facility (SMIF), a member of the North Carolina Research Triangle Nanotechnology Network (RTNN), which is supported by the National Science Foundation [grant number ECCS-2025064] as part of the National Nanotechnology Coordinated Infrastructure (NNCI). This work was supported by the Duke University Pratt School of Engineering and Duke School of Medicine through a MedX grant. G.K. was funded through the NIH University Training Program in Biomolecular and Tissue Engineering [grant number T32GM008555]. The funding sources had no involvement in study design or preparation of the article.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
A.C. is the founder of and member of the scientific advisory board for PhaseBio Pharmaceuticals Inc., which has licensed the ELP drug delivery technology from Duke University.
References
- 1.Dillekås H, Rogers MS & Straume O Are 90% of deaths from cancer caused by metastases? Cancer Med 8, 5574–5576 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaffer CL & Weinberg RA A Perspective on Cancer Cell Metastasis. Science 331, 1559–1564 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD & Jemal A Cancer statistics, 2020. CA. Cancer J. Clin 70, 7–30 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Theelen WSME et al. Effect of Pembrolizumab After Stereotactic Body Radiotherapy vs Pembrolizumab Alone on Tumor Response in Patients With Advanced Non–Small Cell Lung Cancer: Results of the PEMBRO-RT Phase 2 Randomized Clinical Trial. JAMA Oncol 5, 1276–1282 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ho AY et al. A phase 2 clinical trial assessing the efficacy and safety of pembrolizumab and radiotherapy in patients with metastatic triple-negative breast cancer. Cancer 126, 850–860 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Twyman-Saint Victor C et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520, 373–377 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Demaria S et al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 11, 728–734 (2005). [PubMed] [Google Scholar]
- 8.Pilones KA et al. Radiotherapy Cooperates with IL15 to Induce Antitumor Immune Responses. Cancer Immunol. Res 8, 1054–1063 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mondini M, Levy A, Meziani L, Milliat F & Deutsch E Radiotherapy–immunotherapy combinations – perspectives and challenges. Mol. Oncol 14, 1529–1537 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwon ED et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol 15, 700–712 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delaunay M et al. Immune-checkpoint inhibitors associated with interstitial lung disease in cancer patients. Eur. Respir. J 50, (2017). [DOI] [PubMed] [Google Scholar]
- 12.Louvel G et al. Immunotherapy and pulmonary toxicities: can concomitant immune-checkpoint inhibitors with radiotherapy increase the risk of radiation pneumonitis? Eur. Respir. J 51, (2018). [DOI] [PubMed] [Google Scholar]
- 13.Xia N et al. Robo1-specific CAR-NK Immunotherapy Enhances Efficacy of 125I Seed Brachytherapy in an Orthotopic Mouse Model of Human Pancreatic Carcinoma. Anticancer Res 39, 5919–5925 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Hodge JW, Sharp HJ & Gameiro SR Abscopal Regression of Antigen Disparate Tumors by Antigen Cascade After Systemic Tumor Vaccination in Combination with Local Tumor Radiation. Cancer Biother. Radiopharm 27, 12–22 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chao Y et al. Combined local immunostimulatory radioisotope therapy and systemic immune checkpoint blockade imparts potent antitumour responses. Nat. Biomed. Eng 2, 611–621 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Urry DW et al. Elastic protein-based polymers in soft tissue augmentation and generation. J. Biomater. Sci. Polym. Ed 9, 1015–1048 (1998). [DOI] [PubMed] [Google Scholar]
- 17.Urry DW, Parker TM, Reid MC & Gowda DC Biocompatibility of the Bioelastic Materials, Poly(GVGVP) and Its γ-Irradiation Cross-Linked Matrix: Summary of Generic Biological Test Results. J. Bioact. Compat. Polym 6, 263–282 (1991). [Google Scholar]
- 18.MacEwan SR & Chilkoti A Applications of elastin-like polypeptides in drug delivery. J. Controlled Release 190, 314–330 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luginbuhl KM et al. One-week glucose control via zero-order release kinetics from an injectable depot of glucagon-like peptide-1 fused to a thermosensitive biopolymer. Nat. Biomed. Eng 1, 0078 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jenkins IC, Milligan JJ & Chilkoti A Genetically Encoded Elastin‐Like Polypeptides for Drug Delivery. Adv. Healthc. Mater 10, 2100209 (2021). [DOI] [PubMed] [Google Scholar]
- 21.Yi A, Sim D, Lee Y-J, Sarangthem V & Park R-W Development of elastin-like polypeptide for targeted specific gene delivery in vivo. J. Nanobiotechnology 18, 15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piña MJ et al. Elastin-like recombinamers with acquired functionalities for gene-delivery applications. J. Biomed. Mater. Res. A 103, 3166–3178 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Chen T-HH, Bae Y & Furgeson DY Intelligent Biosynthetic Nanobiomaterials (IBNs) for Hyperthermic Gene Delivery. Pharm. Res 25, 683–691 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Liu W et al. Brachytherapy using injectable seeds that are self-assembled from genetically encoded polypeptides in situ. Cancer Res 72, 5956–5965 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schaal JL et al. Injectable polypeptide micelles that form radiation crosslinked hydrogels in situ for intratumoral radiotherapy. J. Control. Release Off. J. Control. Release Soc 228, 58–66 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jahrsdörfer B & Weiner GJ CpG oligodeoxynucleotides as immunotherapy in cancer. Update Cancer Ther 3, 27–32 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bode C, Zhao G, Steinhagen F, Kinjo T & Klinman DM CpG DNA as a vaccine adjuvant. Expert Rev. Vaccines 10, 499–511 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Milas L et al. CpG Oligodeoxynucleotide Enhances Tumor Response to Radiation. Cancer Res 64, 5074–5077 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Mason KA & Hunter NR CpG plus radiotherapy: a review of preclinical works leading to clinical trial. Front. Oncol 2, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mutwiri GK, Nichani AK, Babiuk S & Babiuk LA Strategies for enhancing the immunostimulatory effects of CpG oligodeoxynucleotides. J. Controlled Release 97, 1–17 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Pulaski BA & Ostrand-Rosenberg S Mouse 4T1 Breast Tumor Model. in Current Protocols in Immunology (eds. Coligan JE, Bierer BE, Margulies DH, Shevach EM & Strober W) (John Wiley & Sons, Inc., 2001). doi: 10.1002/0471142735.im2002s39. [DOI] [PubMed] [Google Scholar]
- 32.Chen L et al. Tumor-Targeted Drug and CpG Delivery System for Phototherapy and Docetaxel-Enhanced Immunotherapy with Polarization toward M1-Type Macrophages on Triple Negative Breast Cancers. Adv. Mater 31, 1904997 (2019). [DOI] [PubMed] [Google Scholar]
- 33.McDaniel JR, MacKay JA, Quiroz FG & Chilkoti A Recursive Directional Ligation by Plasmid Reconstruction Allows Rapid and Seamless Cloning of Oligomeric Genes. Biomacromolecules 11, 944–952 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hassouneh W, Christensen T & Chilkoti A Elastin-Like Polypeptides as a Purification Tag for Recombinant Proteins. in Current Protocols in Protein Science (eds. Coligan JE, Dunn BM, Speicher DW & Wingfield PT) 6.11.1–6.11.16 (John Wiley & Sons, Inc., 2010). doi: 10.1002/0471140864.ps0611s61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yousefpour P et al. Conjugate of Doxorubicin to Albumin-Binding Peptide Outperforms Aldoxorubicin. Small 15, 1804452 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wood WG, Wachter C & Scriba PC Experiences Using Chloramine-T and 1, 3, 4, 6-Tetrachloro-3α, 6α-diphenylglycoluril (lodogen®) for Radioiodination of Materials for Radioimmunoassay. Clin. Chem. Lab. Med 19, (1981). [PubMed] [Google Scholar]
- 37.Mislick KA, Baldeschwieler JD, Kayyem JF & Meade TJ Transfection of Folate-Polylysine DNA Complexes: Evidence for Lysosomal Delivery. Bioconjug. Chem 6, 512–515 (1995). [DOI] [PubMed] [Google Scholar]
- 38.Mandal H et al. ε-Poly-l-Lysine/plasmid DNA nanoplexes for efficient gene delivery in vivo. Int. J. Pharm 542, 142–152 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Nishikawa M et al. Pharmacokinetics and In Vivo Gene Transfer of Plasmid DNA Complexed with Mannosylated Poly(L-Lysine) in Mice. J. Drug Target 8, 29–38 (2000). [DOI] [PubMed] [Google Scholar]
- 40.Kozlowska AK et al. Functionalized bioengineered spider silk spheres improve nuclease resistance and activity of oligonucleotide therapeutics providing a strategy for cancer treatment. Acta Biomater 59, 221–233 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun J et al. Uniform Small Graphene Oxide as an Efficient Cellular Nanocarrier for Immunostimulatory CpG Oligonucleotides. ACS Appl. Mater. Interfaces 6, 7926–7932 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Pressnall MM, Huayamares SG & Berkland CJ Immunostimulant Complexed With Polylysine Limits Transport and Maintains Immune Cell Activation. J. Pharm. Sci 109, 2836–2846 (2020). [DOI] [PubMed] [Google Scholar]
- 43.UTAISINCHAROEN P, ANUNTAGOOL N, CHAISURIYA P, PICHYANGKUL S & SIRISINHA S CpG ODN activates NO and iNOS production in mouse macrophage cell line (RAW 264·7). Clin. Exp. Immunol 128, 467–473 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Demidenko E & Miller TW Statistical determination of synergy based on Bliss definition of drugs independence. PLOS ONE 14, e0224137 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reig-Vano B, Tylkowski B, Montané X & Giamberini M Alginate-based hydrogels for cancer therapy and research. Int. J. Biol. Macromol 170, 424–436 (2021). [DOI] [PubMed] [Google Scholar]
- 46.Amiram M, Luginbuhl KM, Li X, Feinglos MN & Chilkoti A A depot-forming glucagon-like peptide-1 fusion protein reduces blood glucose for five days with a single injection. J. Controlled Release 172, 144–151 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gilroy CA, Roberts S & Chilkoti A Fusion of fibroblast growth factor 21 to a thermally responsive biopolymer forms an injectable depot with sustained anti-diabetic action. J. Controlled Release 277, 154–164 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iyer A et al. Stapling monomeric GCN4 peptides allows for DNA binding and enhanced cellular uptake. Org. Biomol. Chem 13, 3856–3862 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Nishihara S & Kawasaki K Enhanced cellular uptake of CpG DNA by α-helical antimicrobial peptide Kn2-7: Effects on macrophage responsiveness to CpG DNA. Biochem. Biophys. Res. Commun 530, 100–106 (2020). [DOI] [PubMed] [Google Scholar]
- 50.Yang S et al. Cellular uptake of self-assembled cationic peptide–DNA complexes: Multifunctional role of the enhancer chloroquine. J. Controlled Release 135, 159–165 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Nishikawa M et al. Injectable, self-gelling, biodegradable, and immunomodulatory DNA hydrogel for antigen delivery. J. Controlled Release 180, 25–32 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Hori Y, Winans AM & Irvine DJ Modular injectable matrices based on alginate solution/microsphere mixtures that gel in situ and co-deliver immunomodulatory factors. Acta Biomater 5, 969–982 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heikenwalder M et al. Lymphoid follicle destruction and immunosuppression after repeated CpG oligodeoxynucleotide administration. Nat. Med 10, 187–192 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Ravindranathan S et al. Tumor-derived granulocyte colony-stimulating factor diminishes efficacy of breast tumor cell vaccines. Breast Cancer Res 20, 126 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.