

ABSTRACT
Autoimmune diseases with hematological manifestations are often characterized by chronicity and relapses despite treatment, and the underlying pathogenetic mechanisms remain unknown. Epigenetic alterations play a vital role in the deregulation of immune tolerance and the development of autoimmune diseases. In recent years, study of epigenetic mechanisms in both adult and childhood autoimmune disorders has been seeking to explain the pathophysiology of these heterogeneous diseases and to elucidate the interaction between genetic and environmental factors. Various mechanisms, including DNA methylation, histone modifications (chromatin remodeling), and noncoding RNAs (ncRNAs), have been studied extensively in the context of autoimmune diseases. This paper summarizes the epigenetic patterns in some of the most common childhood autoimmune disorders with hematological manifestations, based on epigenetic studies in children with primary immune thrombocytopenia (ITP), systemic lupus erythematosus (SLE), and juvenile idiopathic arthritis (JIA). Research findings indicate that methylation changes in genes expressed on T cells, modifications at a variety of histone sites, and alterations in the expression of several ncRNAs are involved in the pathogenesis of these diseases. These mechanisms not only determine the development of these diseases but also affect the severity of the clinical presentation and biochemical markers. Further studies will provide new tools for the prevention and diagnosis of childhood autoimmune disorders, and possible novel treatment options.
Keywords: Autoimmune diseases, Children, DNA methylation, Epigenetic mechanisms, Histone modifications, Noncoding RNAs

INTRODUCTION
The term autoimmune diseases include a wide variety of disorders that are characterized by the lack of immunological tolerance, leading to immune‐mediated damage of healthy cells and tissues. Despite extensive research, much remains to be clarified in the etiology and pathophysiology of these diseases, with recent focus on the study of epigenetics and the effect of environmental factors. 1 One of the main stimuli for this field of research was the incomplete concordance rate of autoimmune diseases in monozygotic twins, which can only be explained by nontraditional genetic mechanisms. 2
Epigenetic mechanisms include alterations in gene expression, related to environmental factors, without changes in the sequence of the bases in the DNA. These potentially heritable and reversible changes in transcription activity and cell functioning have been implicated in the pathophysiology of many childhood diseases, including cancer, allergies, autoimmune diseases, and autoinflammatory conditions. 3 , 4 , 5 , 6
Various epigenetic mechanisms, including DNA methylation, histone modifications (chromatin remodeling), and noncoding RNAs (ncRNAs), appear to play a crucial role in cellular immunity, and in recent years have been under intensive study. The relationship between epigenetic modifications and alterations in the immune system is now being used for the discovery of potential novel therapeutic strategies in autoimmune diseases and other disorders. 7 , 8
In this review, we tried to summarize the recent findings of the main epigenetic mechanisms in the field of some of the most common childhood autoimmune diseases with hematological manifestations, as epigenetic regulators play an important role in hematopoiesis and in the life cycle and survival of peripheral blood cells. 9 We focused on three diseases, namely, primary immune thrombocytopenia (ITP), systemic lupus erythematosus (SLE), and juvenile idiopathic arthritis (JIA). These diseases are three of the most common pediatric autoimmune diseases, and their clinical manifestations include significant hematological manifestations. 10 , 11 , 12 The analysis of epigenetic changes in these diseases can lead to the establishment of new biomarkers for the diagnosis as well as the discovery of new methods of treatment for these conditions that adversely affect the health of children.
EPIGENETIC MECHANISMS AND EPIGENETIC THERAPIES
Epigenetic modifications include three important processes that allow reversible regulation of gene transcription, without altering the DNA sequence. These mechanisms play a key role in cell differentiation and in the development and functioning of the immune system. 13
DNA methylation is probably the most widely studied epigenetic modification in humans. It consists of the addition of a methyl group to the fifth carbon (C5) of cytosine in CpG dinucleotides, and the process is catalyzed by enzymes called DNA methyltransferases (DNMTs). Hypermethylation usually results in gene inactivation, while hypomethylation leads to the activation of gene transcription (Figure 1). 14
FIGURE 1.
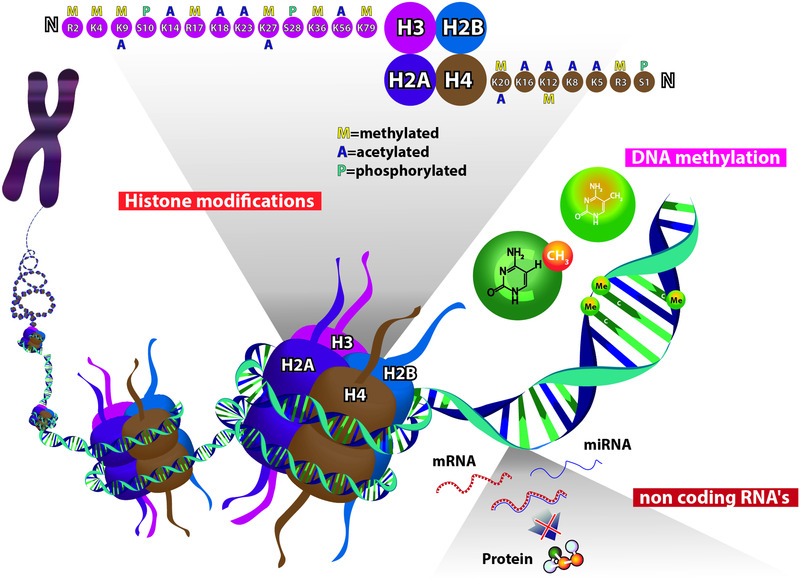
Epigenetic mechanisms. Histone modifications: The histones (H) most widely studied for their modification status are H4 (specific sites: K5, K8, K12, K16, K20, R3, S1), H3 (specific sites: K4, K9, K14, K18, K23, K27, K36, K56, K79, S10, S28, R2, R17), and less conserved sites for H2A and H2B. DNA methylation: Addition of a methyl group (CH3) to the fifth carbon (C5) of cytosine in CpG dinucleotides. The CpG sites are regions of DNA where a cytosine nucleotide is followed by a guanine nucleotide in the linear sequence of bases along its 5'>3' direction. Higher methylation correlates with lower gene expression. Noncoding RNAs: RNA molecules that are not translated into proteins and function as regulators of gene expression. Epigenetic related noncoding RNAs (ncRNAs) include microRNA (miRNA), small interfering RNA (siRNA), piwi‐interacting RNA (piRNA), long intergenic RNA (lincRNA), and others.
Histones, a family of proteins that associate with DNA, compressing it into chromatin, can undergo many post‐translational modifications, including acetylation, methylation and others (e.g., ubiquitination, phosphorylation and SUMOylation). These modifications result in chromatin remodeling, which alters the accessibility of DNA to transcription factors, resulting in either activation or inactivation of genes (Figure 1). 15 , 16
NcRNAs are RNA molecules that are transcribed from DNA but not translated into proteins. They are classified into two groups: long ncRNAs (lncRNAs), consisting of >200 nucleotides, and small ncRNAs, consisting of <200 nucleotides, mainly micro RNAs (miRNAs). In general, ncRNAs function to regulate gene expression at the transcriptional and post‐transcriptional levels (Figure 1). 17 , 18
Recently epigenetic mechanisms have been used for the discovery of novel therapeutic options in autoimmune and other diseases. Such epigenetic therapies can reverse existing changes in DNA methylation and histone acetylation. For instance DNMT's inhibitors like 5‐aza‐20‐deoxycytidine (decitabine) have been studied on systemic sclerosis. Decitabine has been seen to decrease the methylation levels of the promoters of DKK1 and SFRP1 genes and that way it can effectively decrease normal wnt signaling (a conserved pathway that regulates crucial aspects of cell fate decisions and tissue patterning during early embryonic and later development) in systemic sclerosis fibroblasts and effectively ameliorate fibrosis. 19 Additionally histone deacetylase (HDAC) inhibitors like trichostatin A have been studied on rheumatoid arthritis patients and they were able to disrupt inflammatory cytokine production in the synovial cells by reducing the stability of interleukin 6 (IL‐6) mRNA in the fibroblast like synoviocytes and macrophages. 20
EPIGENETIC MODIFICATIONS IN PRIMARY ITP
Primary ITP is an autoimmune hematological disease characterized by low platelet count (<100 × 109/L) with no other causes found, whereas secondary ITP develops in the context of other exogenous diseases and drugs. 21 The etiology of ITP is not completely understood, but it is widely accepted that both genetic and environmental factors play a role in its development. Recent studies indicate that irregularities associated with ITP are related to abnormal T‐cell responses, particularly defective CD4+CD25+ T regulatory cell (Treg) activity. 22 Epigenetic research on ITP is ongoing and includes study of DNA methylation, histone modifications, and ncRNAs (Table 1).
TABLE 1.
Main findings of the studies on epigenetic alterations in children with idiopathic thrombocytopenic purpura (ITP), systemic lupus erythematosus (SLE), and juvenile idiopathic arthritis (JIA)
| Disease | DNA methylation | Histone modifications | Noncoding RNAs |
|---|---|---|---|
| ΙΤΡ |
FOXP3 hypermethylation 23 DNMT3B‐rs2424913 SNP increased expression 25 , 26 IL‐1 Ra VNTR polymorphism expression 26 DNMT3B −579 G>T SNP increased expression 28 DNMT3B −149C/T SNP increased expression 29 DNMT3A −448 G/A SNP decreased expression 29 |
Global H3K9 hypomethylation 30 Downregulation of HMTs (SUV39H2 and EZH2) 30 |
Increased MALAT1 and THRIL expression 31 Increased miR‐302c‐3p, miR‐483‐5p, miR‐410, miR‐302a‐3p, miR‐223‐3p, and miR‐597 expression 32 Decreased miR‐544a expression 32 |
| SLE |
FOXP3 hypermethylation 41 Type I INF‐related genes (e.g., MX1, IFI44L, PARP9, and DTX3L) hypomethylation 42 JAK2 hypomethylation 43 SOCS3 hypermethylation 43 HMGB1 hypomethylation 44 |
Global histone H3 and H4 hypoacetylation 45 Global histone H3K9 hypomethylation 45 |
Increased miRNA‐516a‐3p expression 46 Increased miRNA‐629 expression 46 Increased miRNA‐525‐5p expression 46 Increased miRNA‐27a* expression 47 Decreased NKG2D and ULBP2 expression 47 |
| JIA |
MRPL28 hypomethylation 62 IL32 hypomethylation 62 DNMT1 decreased expression 63 DNMT3a decreased expression 63 |
H3K4me3 and H3K27me3 SNPs and indels 64 |
Increased miRNA‐155 expression 66 Increased miRNA‐16 expression 66 Decreased miRNA‐204 expression 66 Increased miRNA‐125a‐5p expression 67 Decreased miR‐19a expression 68 Decreased miR‐21 expression 68 Increased RP11‐340F14.6 expression 69 Increased serum miR‐223 levels 70 |
Abbreviations: FOXP3, forkhead box P3; IL‐1Ra VNTR, the interleukin‐1 receptor antagonist variable‐number tandem repeats; HMTs, histone methyltransferases; DNMTs; DNA methyltransferases; MALAT1, metastasis associated lung adenocarcinoma transcript 1; THRIL, tumor necrosis factor‐α (TNF‐α) and heterogeneous nuclear ribonucleoprotein L (hnRNPL) immune‐regulatory lncRNA; INF, interferon; JAK2, janus kinase 2; SOCS3, suppressor of cytokine signaling 3.
DNA methylation in ITP
Forkhead box P3 (FOXP3), also known as scurfin, is a protein involved in immune system responses that appears to function as a major regulator of the pathway in the development and function of Tregs. 23 Stable Foxp3 expression in Tregs is sustained by selectively activated DNA demethylation at a highly conserved region within the FOXP3 promoter, called TSDR (Treg‐specific demethylated region). 24 The status of DNA methylation in the FOXP3 promoter was investigated in 49 children with newly diagnosed, 18 with persistent, and 15 with chronic ITP. The children with ITP showed a hypermethylation pattern at several CpG sites, indicating an involvement in the pathogenesis of ITP. Higher CpG‐6 (one of the 12 different CpG sites) expression was related to the persistent and chronic types of the disease. 23 Other studies were focused on the DNMTs, the main mediators of DNA methylation. The prevalence of the single‐nucleotide polymorphism (SNP) rs2424913, located in the DNMT3B gene promoter, was investigated in children with chronic ITP. The homozygous pattern of this SNP was higher in patients with ITP than in control subjects, and it was related to chronic ITP through alteration of the methylation phenotype. 25 In a study from Greece, the same DNMT3B SNP, rs2424913, and the interleukin‐1 (IL1) receptor antagonist (IL‐1Ra) variable‐number tandem repeats (VNTR) polymorphism were studied in 30 children with newly diagnosed ITP. No significant differences were found in the genotype distribution of this polymorphism between patients and control subjects, but the frequency of the T‐allele was increased in children with ITP and appeared to be correlated with susceptibility to ITP. In the case of IL‐1Ra polymorphism, children with ITP had a significantly higher frequency of genotype I/II, compared with the control group. 26 IL‐1Ra inhibits the activities of IL1 alpha (IL1A) and IL1 beta (IL1B), and modulates a variety of IL1‐related immune and inflammatory responses. 27 In a prospective study, the DNMT3B −579 G>T SNP promoter polymorphism was shown to be associated with a higher likelihood of ITP in children. 28 Lastly, in a prospective case–control study of patients with ITP, the frequency of the DNMT3A −448 G/A SNP variant A‐allele was significantly lower in the patients with primary ITP than in the control subjects, whereas the DNMT3B −149 C/T SNP variant T‐allele was significantly higher in the patients with almost double the risk of ITP compared with the control. These findings demonstrate that the DNMT3A −448 SNP variant A‐allele might exert a protective effect against ITP, whereas the DNMT3B −149 SNP variant T‐allele could be considered as a molecular risk factor for ITP. 29
From the above studies it appears that hypermethylation of the promoter region of FOXP3 can alter regulatory T‐cell activity, and thus play a crucial role in the development of persistent and chronic ITP in children. Polymorphisms in the promoter of the main regulatory enzymes of DNA methylation, such as DNMT3B rs2424913, DNMT3B −579 G>T, and DNMT3B −149 C/T, can lead to de novo methylation of CpG islands in genes expressed on T cells, thus they appear to constitute a risk factor for ITP in children.
Histone modifications in ITP
In a study on 35 patients with ITP (20 active and 15 in remission), global H3K9 hypomethylation in CD4+ T cells was observed in the patients with active ITP, but no difference was found between patients with ITP in remission and healthy control subjects. The mRNA expression analysis of three histone methyltransferases (HMTs), SUV39H1, SUV39H2, and EZH2, showed significant downregulation of EZH2 and SUV39H2, but no difference in SUV39H1 in patients with active ITP compared with control subjects and patients with ITP in remission. These HMTs are responsible for most of the H3K9 and H3K27 methylation and these findings indicate that their expression is involved in the etiology of ITP. 30
In summary, H3K9 hypomethylation in CD4+ T cells and downregulation of specific HMTs (EZH2 and SUV39H2) may contribute to the dysregulation of immune response by activating or deactivating specific immune‐related genes and are associated with ITP in children.
Noncoding RNAs in ITP
In a study of children with ITP, Ayoub et al. 31 analyzed the expression profiles of two lncRNAs metastasis associated lung adenocarcinoma transcript 1 (MALAT1) and tumor necrosis factor‐α (TNF‐α) and heterogeneous nuclear ribonucleoprotein L (hnRNPL) immune‐regulatory lncRNA (THRIL). The analysis showed that MALAT1 and THRIL were both significantly upregulated in patients with ITP, compared with healthy control subjects. MALAT1 is a highly conserved nuclear‐retained lncRNA that is abundantly expressed in cells and tissues and has been shown to play a role in regulating genes at both the transcriptional and posttranscriptional levels. THRIL lncRNA is involved in the pathogenesis of immune‐related and inflammatory disease through controlling the expression of TNF‐α. 31 In another study, seven miRNAs showed significant differences in patients with ITP, specifically, increased expression of miR‐302c‐3p, miR‐483‐5p, miR‐410, miR‐302a‐3p, miR‐223‐3p, and miR‐597, and decreased expression of miR‐544a. Increased expression of miR‐302c‐3p appeared to increase the risk of the acute form of ITP. These findings indicate that plasma miRNA levels may provide useful information for prediction of the course of disease. 32
As described above, changes in the expression of ncRNAs appear to be associated with the susceptibility to ITP in childhood. In particular, several differentially expressed miRNAs can modulate the functions of T lymphocytes and differentiation of Th1/Th2 cells, and thus increase the risk of ITP appearance in children.
Epigenetic therapies in ITP
Recent research on ITP has led on to the new therapeutic approaches that contribute to reversal of existing epigenetic changes. 5‐Aza‐20‐deoxycytidine (decitabine) is a DNMT inhibitor that has been used for the treatment of myelodysplastic syndrome. 33 In a study of low‐dose decitabine for the treatment of ITP, the therapy has been seen to promote megakaryocyte maturation and platelet release by increasing the expression of tumor necrosis factor‐related apoptosis inducing ligand (TRAIL) via decreasing its promoter methylation. 34 Moreover, Han et al. 35 using low‐dose decitabine for patients with ITP observed a consequent improved function of Treg cells and suppression of Th1 and Th17 cells. Next‐generation RNA sequencing revealed downregulated phosphorylated STAT3 (STAT3 is a member of the STAT protein family that plays a vital role in the maintenance of CD4+ T‐cell homeostasis). The data suggested that in ITP, low‐dose decitabine might restore Treg cells by inhibiting STAT3 activation. 35 Finally, the same researchers reported that decitabine at a low dose can restore the methylation level and expression of the programmed cell death protein 1 (PD‐1) on CD8+ T cells of ITP patients. Reduced expression of PD‐1 was correlated with lower cytotoxic T lymphocytes‐mediated platelet destruction. 36
EPIGENETIC MODIFICATIONS IN SLE
SLE is a prototype of immunological abnormalities, and probably the most widely studied autoimmune disease in relation to epigenetic modifications. It is a rheumatic disease characterized by autoantibodies directed against self‐antigens, immune complex formation, and immune dysregulation, with resultant damage to essentially any organ. Hematological manifestations are common in SLE, both at diagnosis and during the course of the disease, the most common of which are leukopenia, thrombocytopenia, lymphadenopathy, and/or splenomegaly. 11 , 37 Many recent studies have documented the importance of dysregulation of the T‐cell and B‐cell response as a trigger in SLE. 38 Abnormal cell responses in SLE include downregulation of the Th1 and Treg cells, upregulation of Th17 cells, decreased cytotoxicity of CD8+ T cells, and increased B‐cell activation and autoantibody production. 38 , 39 One of the first connections between epigenetic dysregulation and SLE was the lupus‐like disease caused by methylation inhibitor drugs, like procainamide and hydralazine. 40
DNA methylation in SLE
Hanaei et al. 41 analyzed the methylation status of FOXP3 in girls with SLE. Promoter methylation of the FOXP3 gene was significantly higher in children with SLE than in healthy control subjects. Also, expression of FOXP3 gene was associated with renal involvement and the levels of anti‐dsDNA in SLE patients. One of the causes of increased immune response in SLE could therefore be the inappropriate activity of Tregs caused by lower expression of FOXP3 gene due to hypermethylation of its promoter. 41 A genome‐wide methylation study of 16 Chinese patients with childhood‐onset SLE and 13 healthy control subjects compared DNA methylation both in whole blood and within each independent cell lineage (CD4+ T cells, CD8+ T cells, B cells, and neutrophils). A consistent pattern of hypomethylation at 21 CpG sites encompassing 15 genes was identified across both myeloid and lymphoid cell lineages. Type I interferon‐related genes (i.e., MX1, IFI44L, PARP9, and DTX3L) were identified as the hypomethylated genes that significantly distinguished between SLE patients and control subjects. This suggests that epigenetic reprogramming of type I interferon‐related genes is a widespread phenomenon in the blood cells of children with SLE. 42 The methylation status of the promoters of Janus kinase 2 (JAK2) and suppressor of cytokine signaling 3 (SOCS3) genes was analyzed in a study of 25 pediatric patients with SLE and 24 healthy control subjects. The results showed that JAK2 promoter was significantly hypomethylated, whereas SOCS3 promoter was significantly hypermethylated in SLE patients compared with the control patients. 43 In an epigenetic study on the T cells of 10 patients with SLE, HMGB1, a gene encoding the homonymous protein with important immunological activity, showed significant hypomethylation. 44
From the above studies, it appears that deregulation of Tregs activity through FOXP3 promoter hypermethylation constitutes a risk factor for SLE in children. Moreover, SOCS3 promoter hypermethylation and JAK2, HMGB1, and type I INF‐related genes hypomethylation, which may alter the activity of T cells and also other cell lineages (B cells and neutrophils), could also increase the likelihood of SLE appearance in children.
Histone modifications in SLE
Hu et al. 45 investigated alterations in CD4+ T‐cell histones in patients with SLE. Global histone H3 and H4 hypoacetylation was found in the patients, and the degree of histone H3 acetylation was negatively correlated with increased disease activity and with laboratory markers. Global histone H3K9 hypomethylation was observed in the CD4+ T cells of the patients with SLE, but not of the control subjects. 45
Therefore, it seems that global histone H3 and H4 hypoacetylation and global histone H3K9 hypomethylation in the T cells can alter cytokine production and so act as potential biomarkers for SLE.
NcRNAs in SLE
In a Chinese study comparing the expression of ncRNAs in children with newly diagnosed active SLE, patients with inactive disease and healthy control subjects, high expression of miRNA‐516a‐3p, miRNA‐629, and miRNA‐525‐5p was recorded in the children with active SLE patients, which was positively correlated with disease activity and the level of C‐reactive protein. 46 The expression of miRNAs that might potentially target an essential activating receptor, NKG2D, and its ligand ULBP2 in peripheral blood mononuclear cells (PBMCs) and natural killer (NK) cells was investigated in 37 patients with childhood‐onset SLE. NKG2D is one of the most significant activating receptors of NK cells. In the PBMCs and NK cells of patients with SLE, miR‐27a* was overexpressed, and was found to have a putative binding site on the NKG2D mRNA. In contrast, NKG2D and ULBP2 mRNA were downregulated in the PBMCs and NK cells of the patients with SLE. 47 Lastly, in a comparison study, lncRNA and mRNA microarrays were performed in children with SLE and paired healthy children. A total of 1042 lncRNAs and 1162 mRNAs were differentially expressed, indicating that ncRNAs are involved in pathways with crucial pathobiological relevance in SLE. 48
From the above studies, it is apparent that ncRNAs are involved in the pathogenesis of childhood‐onset SLE by affecting gene expression on mononuclear and NK cells. Specifically, high expression of miRNA‐516a‐3p, miRNA‐629, miRNA‐525‐5p, and miR‐27a*, and decreased expression of NKG2D and ULBP2 appear to be strongly associated with childhood SLE.
Epigenetic therapies in SLE
Nowadays, new epigenetic‐based drugs are candidates for the treatment of SLE. Such drugs seem to be able to reverse hypoacetylation of specific histone sites and alter expression of dysregulated ncRNAs. 49 Trichostatin A (TSA) is an HDAC inhibitor that selectively inhibits the class I and II histone deacetylase families of enzymes. 50 TSA was found to be able to inhibit interferon‐α production by dendritic cells that were activated in vitro in the presence of serum obtained from SLE patients. 51 Moreover, treatment with TSA has been observed to increase Foxp3 gene expression, as well as the production and suppressive function of Tregs. 52 Vorinostat, also known as suberanilohydroxamic acid (SAHA), is another HDAC inhibitor that has been used for the treatment of cutaneous manifestations in patients with cutaneous T‐cell lymphoma. 53 SAHA has been observed to inhibit TNF‐α, IL‐6, NO, and inducible NO synthase expression and to reverse proteinuria and pathologic renal disease in MRL/lpr mice (mice that develop an autoimmune disease resembling SLE). 54 Administration of anti‐miR‐155 in MRL/lpr mice led to subsequent decrease of serum IgG anti‐dsDNA autoantibodies that resulted in the reduction of kidney inflammation. 55
EPIGENETIC MODIFICATIONS IN JIA
JIA is the most common chronic rheumatic disease in children. Children with JIA may exhibit a variety of hematological abnormalities associated with active disease, including anemia of chronic disease, thrombocytosis, sometimes a mild leukocytosis, and less often, eosinophilia, leukopenia/neutropenia, thrombocytopenia, or pancytopenia. A rare inflammatory hematological complication is the macrophage‐activation syndrome, which occurs most commonly in systemic arthritis subtype. 12 , 56 As the word “idiopathic” suggests, the cause of JIA is unclear, although the current understanding is that it arises from a combination of genetic and environmental factors. 57 Predominance of CD4+ T cells in the synovial infiltrates of JIA patients indicates the importance of T cells involvement in the disease. 58 Studies on patients with JIA have shown that children with active disease have elevated levels of Th17 and Treg cells, and moreover, the higher Th17 levels predicted a longer period to reach disease inactive stage. 59 Several studies have already linked epigenetic research with the treatment of JIA. It has been shown that the treatment of rheumatoid arthritis patients with methotrexate acts through restoring defective Tregs function by demethylation of the FOXP3 locus, leading to a subsequent increase in FOXP3 expression. 60 Epigenetic research on JIA is ongoing and the findings are promising.
DNA methylation in JIA
In a case–control study of CD4+ T cells from 56 pediatric patients with oligoarticular JIA, no evidence of substantial common alterations to the DNA methylome was found in children with oligoarticular JIA, in contrast to the findings for adult‐onset rheumatoid arthritis. 61 In another genome‐scale analysis of CD4+ T cells from 14 children with oligoarticular and polyarticular JIA, a total of 91 genes were found to be more highly methylated in the children with JIA, although the data suggested a possible impact of methotrexate treatment on the methylome. Methylation at MRPL28 and IL32 appeared reduced in the T cells of children with JIA, and these findings were robust to methotrexate exposure. MRPL28 encodes mitochondrial ribosomal protein L28, and therefore, reduced MRPL28 methylation could be associated with mitochondrial dysfunction. 62 Ghavidel et al. 63 found expression of DNMT1 and DNMT3a was significantly lower in the patients with JIA than in the healthy control subjects.
From the above, it appears that MRPL28 and IL32 hypomethylation can alter T‐cell activity and cause mitochondrial dysfunction and increase the risk of development of both oligoarticular and polyarticular JIA. Underexpression of DNMT1 and DNMT3a in PBMCs causes reduction in DNA methylation and appears to be associated with an increased risk of JIA.
Histone modifications in JIA
In a deep whole‐genome sequencing study of 48 children with the polyarticular form of JIA, the researchers searched for novel JIA variants in histone modification peaks. The results revealed significant enrichment of JIA novel SNPs within H3K4me3 and H3K27me3 marks in CD20+ B cells and novel JIA indels were enriched in H3K4me3 peaks of CD4+ T cells. 64 Therefore, it seems that specific novel SNPs in H3K4me3 and H3K27me3 marks of CD20+ B cells and several indels in H3K4me3 peaks of CD4+ T cells may serve as potential biomarkers for JIA appearance.
Noncoding RNAs in JIA
Hu et al. 65 studied 35 children with newly diagnosed, untreated polyarticular, rheumatoid factor (RF)‐negative JIA. Among the dysregulated miRNAs with the largest effects were miR‐15/16, miR‐384, and miR‐223, which are positively correlated with innate immunity, whereas miR‐320 and miR‐185 are negatively correlated with innate immunity. 65 In another study, the plasma levels of four candidate miRNAs (miRNA‐16, miRNA‐155, miRNA‐204, and miRNA‐451), which are known to be associated with autoimmunity, were compared. Plasma miRNA‐155 levels were higher in the patients with JIA than in the healthy control subjects. Raised miRNA‐16 levels and reduced miRNA‐204 levels were recorded in patients with oligoarticular or polyarticular JIA, and the miRNA‐16 levels were correlated with disease activity. In a comparison of the JIA subtypes, the miRNA‐204 levels were higher in polyarticular JIA, whereas the miRNA‐451 levels were higher in enthesitis‐related arthritis. 66 Schulert et al. 67 found 110 differentially expressed microRNAs in PBMCs from patients with active systemic JIA. MiRNA‐125a‐5p was highly upregulated from children with active systemic JIA, compared to children with clinically inactive JIA and those with active polyarticular JIA, and its expression was correlated with systemic features of the disease. In another study, the expression levels of miR‐19a and miR‐21 were significantly lower in the systemic JIA group compared with the control group. 68 In another study, lncRNA RP11‐340F14.6 was upregulated in patients with JIA and also negatively correlated with Foxp3 expression. These findings suggest that this lncRNA plays a pivotal role in stimulating Th17 differentiation and simultaneously suppressing Treg distribution. 69 Kamiya et al. 70 evaluated serum levels of seven miRNAs (miR‐16, miR‐132, miR‐146a, miR‐155, and miR‐223) in 24 JIA patients (eight systemic onset, 16 polyarthritis) and eight healthy controls. MiR‐223 levels were significantly higher in patients in the active phase of systemic‐onset JIA than in controls. Moreover, levels of miR‐223 and miR‐16 correlated with erythrocyte sedimentation rate and matrix metalloproteinase‐3 in both systemic‐onset JIA and polyarthritis patients. 70
In summary, it appears that alterations in miRNA's expression of the PBMCs can alter cytokine production and increase the likelihood of development of JIA. Specifically, increased miRNA‐16, miRNA‐125a‐5p, miRNA‐155, and RP11‐340F14.6 expression and decreased miR‐19a, miR‐21, and miRNA‐204 expression constitute a risk factor for JIA. Also, increased serum miR‐223 levels seem to also increase the risk for JIA appearance.
Epigenetic therapies in JIA
Recent articles investigated new treatment options that interfere with existing epigenetic changes in patients with JIA. Givinostat (originally ITF2357) is a class I and II HDAC inhibitor with potential anti‐inflammatory, antiangiogenic, and antineoplastic activities. 71 It can reverse histone hypoacetylation and reduce production and release of several proinflammatory cytokines like TNF‐α, IL‐1β, IL‐6, and IL‐12. 72 In a study of orally given Givinostat on JIA patients, the treatment exhibited significant therapeutic benefit in patients with systemic‐onset JIA, particularly with regard to the arthritic component. 73
CONCLUSION
Childhood autoimmune diseases with hematological manifestations account for a large part of pediatric morbidity with profound effects on the health of children, both physical and mental. The link between epigenetics and autoimmunity is now widely accepted in the scientific community. This article summarized some of the most important epigenetic mechanisms involved in childhood autoimmune diseases with hematological manifestations. Normal hematopoiesis and the survival of blood cells are under the tight epigenetic regulation. 74
Primary ITP appears to be associated with methylation alterations in specific genes of the CD4+ T cells and SNPs in DNMTs. Deregulation of Tregs through FOXP3 promoter hypermethylation seems to be of particular importance. Hypomethylation in particular histone sites and changes in the regulation of HMTs and specific miRNAs have also been implicated in the pathogenesis of the disease. Regarding SLE, methylation changes in genes expressed in CD4+ T cells (especially FOXP3 promoter hypermethylation leading to inappropriate activity of Tregs), hypoacetylation/methylation at specific histone sites, and changes in expression of some miRNAs have been associated with the onset of the disease in children. Studies on children with JIA have indicated the importance of hypomethylation of genes expressed in T cells, decreased expression of some DNMTs, and changes in expression of several miRNAs as factors associated with the appearance of the disease.
Epigenetic study in these diseases not only has great potential for both the diagnosis and the prevention of these heterogeneous diseases, but also for the discovery of novel therapeutic strategies. It is of note that epigenetic events are potentially reversible, and they may disappear after a variable number of generations. 75 There is a need for better understanding of these autoimmune epigenetic changes; we are just beginning to understand the impact of epigenetic mechanisms on autoimmunity and therefore further research is required in this area.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
Gkoutsias A, Makis A. The role of epigenetics in childhood autoimmune diseases with hematological manifestations. Pediatr Investig. 2022;6:36–46. 10.1002/ped4.12309
REFERENCES
- 1. Ohkura N, Sakaguchi S. Transcriptional and epigenetic basis of Treg cell development and function: its genetic anomalies or variations in autoimmune diseases. Cell Res. 2020;30:465‐474. DOI: 10.1038/s41422-020-0324-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Deapen D, Escalante A, Weinrib L, Horwitz D, Bachman B, Roy‐Burman P, et al. A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis Rheum. 1992;35:311‐318. DOI: 10.1002/art.1780350310 [DOI] [PubMed] [Google Scholar]
- 3. Ota R, Sawada T, Tsuyama S, Sasaki Y, Suzuki H, Kaizaki Y, et al. Integrated genetic and epigenetic analysis of cancer‐related genes in non‐ampullary duodenal adenomas and intramucosal adenocarcinomas. J Pathol. 2020;252:330‐342. DOI: 10.1002/path.5529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Acevedo N, Benfeitas R, Katayama S, Bruhn S, Andersson A, Wikberg G, et al. Epigenetic alterations in skin homing CD4+CLA+ T cells of atopic dermatitis patients. Sci Rep. 2020;10:18020. DOI: 10.1038/s41598-020-74798-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dai R, Wang Z, Ahmed SA. Epigenetic contribution and genomic imprinting Dlk1‐Dio3 miRNAs in systemic lupus erythematosus. Genes (Basel). 2021;12:680. DOI: 10.3390/genes12050680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Akkaya‐Ulum YZ, Akbaba TH, Tavukcuoglu Z, Chae JJ, Yilmaz E, Ozen S, et al. Familial Mediterranean fever‐related miR‐197‐3p targets IL1R1 gene and modulates inflammation in monocytes and synovial fibroblasts. Sci Rep. 2021;11:685. DOI: 10.1038/s41598-020-80097-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ren J, Catalina MD, Eden K, Liao X, Read KA, Luo X, et al. Selective histone deacetylase 6 inhibition normalizes B cell activation and germinal center formation in a model of systemic lupus erythematosus. Front Immunol. 2019;10:2512. DOI: 10.3389/fimmu.2019.02512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Romanelli A, Stazi G, Fioravanti R, Zwergel C, Di Bello E, Pomella S, et al. Design of first‐in‐class dual EZH2/HDAC inhibitor: biochemical activity and biological evaluation in cancer cells. ACS Med Chem Lett. 2020;11:977‐983. DOI: 10.1021/acsmedchemlett.0c00014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kunimoto H, Nakajima H. TET2: a cornerstone in normal and malignant hematopoiesis. Cancer Sci. 2021;112:31‐40. DOI: 10.1111/cas.14688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hefney H, Bardis D, Aboalwafa H, Ahmed H. New concepts in pathogenesis of primary immune thrombocytopenia. Sohag Med J. 2021;25:106‐111. DOI: 10.21608/smj.2021.78054.1255 [DOI] [Google Scholar]
- 11. Rekvig OP. Autoimmunity and SLE: factual and semantic evidence‐based critical analyses of definitions, etiology, and pathogenesis. Front Immunol. 2020;11:569234. DOI: 10.3389/fimmu.2020.569234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zaripova LN, Midgley A, Christmas SE, Beresford MW, Baildam EM, Oldershaw RA. Juvenile idiopathic arthritis: from aetiopathogenesis to therapeutic approaches. Pediatr Rheumatol Online J. 2021;19:135. DOI: 10.1186/s12969-021-00629-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu H, Sun YE. Epigenetic regulation of stem cell differentiation. Pediatr Res. 2006;59:21R‐25R. DOI: 10.1203/01.pdr.0000203565.76028.2a [DOI] [PubMed] [Google Scholar]
- 14. Greenberg MVC, Bourc'his D. The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol. 2019;20:590‐607. DOI: 10.1038/s41580-019-0159-6 [DOI] [PubMed] [Google Scholar]
- 15. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381‐395. DOI: 10.1038/cr.2011.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang L, Gao Y, Zheng X, Liu C, Dong S, Li R, et al. Histone modifications regulate chromatin compartmentalization by contributing to a phase separation mechanism. Mol Cell. 2019;76:646‐659. DOI: 10.1016/j.molcel.2019.08.019 [DOI] [PubMed] [Google Scholar]
- 17. Hombach S, Kretz M. Non‐coding RNAs: classification, biology and functioning. Adv Exp Med Biol. 2016;937:3‐17. DOI: 10.1007/978-3-319-42059-2_1 [DOI] [PubMed] [Google Scholar]
- 18. Dahariya S, Paddibhatla I, Kumar S, Raghuwanshi S, Pallepati A, Gutti RK. Long non‐coding RNA: classification, biogenesis and functions in blood cells. Mol Immunol. 2019;112:82‐92. DOI: 10.1016/j.molimm.2019.04.011 [DOI] [PubMed] [Google Scholar]
- 19. Dees C, Schlottmann I, Funke R, Distler A, Palumbo‐Zerr K, Zerr P, et al. The Wnt antagonists DKK1 and SFRP1 are downregulated by promoter hypermethylation in systemic sclerosis. Ann Rheum Dis. 2014;73:1232‐1239. DOI: 10.1136/annrheumdis-2012-203194 [DOI] [PubMed] [Google Scholar]
- 20. Grabiec AM, Korchynskyi O, Tak PP, Reedquist KA. Histone deacetylase inhibitors suppress rheumatoid arthritis fibroblast‐like synoviocyte and macrophage IL‐6 production by accelerating mRNA decay. Ann Rheum Dis. 2012;71:424‐431. DOI: 10.1136/ard.2011.154211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cines DB, Liebman H, Stasi R. Pathobiology of secondary immune thrombocytopenia. Semin Hematol. 2009;46:S2‐S14. DOI: 10.1053/j.seminhematol.2008.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Semple JW, Rebetz J, Maouia A, Kapur R. An update on the pathophysiology of immune thrombocytopenia. Curr Opin Hematol. 2020;27:423‐429. DOI: 10.1097/MOH.0000000000000612 [DOI] [PubMed] [Google Scholar]
- 23. Chen Z, Guo Z, Ma J, Ma J, Liu F, Wu R. Foxp3 methylation status in children with primary immune thrombocytopenia. Hum Immunol. 2014;75:1115‐1119. DOI: 10.1016/j.humimm.2014.09.018 [DOI] [PubMed] [Google Scholar]
- 24. Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007;5:e38. DOI: 10.1371/journal.pbio.0050038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gouda HM, Kamel NM, Meshaal SS. Association of DNA methyltransferase 3B promotor polymorphism with childhood chronic immune thrombocytopenia. Lab Med. 2016;47:312‐317. DOI: 10.1093/labmed/lmw040 [DOI] [PubMed] [Google Scholar]
- 26. Pesmatzoglou M, Lourou M, Goulielmos GN, Stiakaki E. DNA methyltransferase 3B gene promoter and interleukin‐1 receptor antagonist polymorphisms in childhood immune thrombocytopenia. Clin Dev Immunol. 2012;2012:352059. DOI: 10.1155/2012/352059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Perrier S, Darakhshan F, Hajduch E. IL‐1 receptor antagonist in metabolic diseases: Dr Jekyll or Mr Hyde? FEBS Lett. 2006;580:6289‐6294. DOI: 10.1016/j.febslet.2006.10.061 [DOI] [PubMed] [Google Scholar]
- 28. Khorshied MM, El‐Ghamrawy MK. DNA methyltransferase 3B (DNMT3B −579G>T) promotor polymorphism and the susceptibility to pediatric immune thrombocytopenic purpura in Egypt. Gene. 2012;511:34‐37. DOI: 10.1016/j.gene.2012.09.024 [DOI] [PubMed] [Google Scholar]
- 29. Abd‐Elkader A, ElMelegy T, NasrEldin E, Abd‐Elhafez Z. DNA methyltransferases 3A −448 G/A and 3B −149C/T single‐nucleotide polymorphisms in primary immune thrombocytopenia. Egypt J Haematol. 2018;43:32. DOI: 10.4103/ejh.ejh_2_18 [DOI] [Google Scholar]
- 30. Zhao H, Xue F, Xu J, Fang Z. Aberrant histone methylation in the patients with immune thrombocytopenia. Platelets. 2014;25:207‐210. DOI: 10.3109/09537104.2013.859664 [DOI] [PubMed] [Google Scholar]
- 31. Ayoub SE, Hefzy EM, Abd El‐Hmid RG, Ahmed NA, Khalefa AA, Ali DY, et al. Analysis of the expression profile of long non‐coding RNAs MALAT1 and THRIL in children with immune thrombocytopenia. IUBMB Life. 2020;72:1941‐1950. DOI: 10.1002/iub.2310 [DOI] [PubMed] [Google Scholar]
- 32. Bay A, Coskun E, Oztuzcu S, Ergun S, Yilmaz F, Aktekin E. Plasma microRNA profiling of pediatric patients with immune thrombocytopenic purpura. Blood Coagul Fibrinolysis. 2014;25:379‐383. DOI: 10.1097/MBC.0000000000000069 [DOI] [PubMed] [Google Scholar]
- 33. Liu W, Zhou Z, Chen L, Wang X. Comparison of azacitidine and decitabine in myelodysplastic syndromes and acute myeloid leukemia: a network meta‐analysis. Clin Lymphoma Myeloma Leuk. 2021;21:e530‐e544. DOI: 10.1016/j.clml.2021.01.024 [DOI] [PubMed] [Google Scholar]
- 34. Zhou H, Hou Y, Liu X, Qiu J, Feng Q, Wang Y, et al. Low‐dose decitabine promotes megakaryocyte maturation and platelet production in healthy controls and immune thrombocytopenia. Thromb Haemost. 2015;113:1021‐1034. DOI: 10.1160/TH14-04-0342 [DOI] [PubMed] [Google Scholar]
- 35. Han P, Hou Y, Zhao Y, Liu Y, Yu T, Sun Y, et al. Low‐dose decitabine modulates T‐cell homeostasis and restores immune tolerance in immune thrombocytopenia. Blood. 2021;138:674‐688. DOI: 10.1182/blood.2020008477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Han P, Yu T, Hou Y, Zhao Y, Liu Y, Sun Y, et al. Low‐dose decitabine inhibits cytotoxic T lymphocytes‐mediated platelet destruction via modulating PD‐1 methylation in immune thrombocytopenia. Front Immunol. 2021;12:630693. DOI: 10.3389/fimmu.2021.630693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yurasov S, Wardemann H, Hammersen J, Tsuiji M, Meffre E, Pascual V, et al. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med. 2005;201:703‐711. DOI: 10.1084/jem.20042251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Suárez‐Fueyo A, Bradley SJ, Tsokos GC. T cells in systemic lupus erythematosus. Curr Opin Immunol. 2016;43:32‐38. DOI: 10.1016/j.coi.2016.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kleczynska W, Jakiela B, Plutecka H, Milewski M, Sanak M, Musial J. Imbalance between Th17 and regulatory T‐cells in systemic lupus erythematosus. Folia Histochem Cytobiol. 2011;49:646‐653. DOI: 10.5603/fhc.2011.0088 [DOI] [PubMed] [Google Scholar]
- 40. Quddus J, Johnson KJ, Gavalchin J, Amento EP, Chrisp CE, Yung RL, et al. Treating activated CD4+ T cells with either of two distinct DNA methyltransferase inhibitors, 5‐azacytidine or procainamide, is sufficient to cause a lupus‐like disease in syngeneic mice. J Clin Invest. 1993;92:38‐53. DOI: 10.1172/JCI116576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hanaei S, Sanati G, Zoghi S, Gharibzadeh S, Ziaee V, Rezaei N. The status of FOXP3 gene methylation in pediatric systemic lupus erythematosus. Allergol Immunopathol (Madr). 2020;48:332‐338. DOI: 10.1016/j.aller.2020.03.014 [DOI] [PubMed] [Google Scholar]
- 42. Yeung KS, Lee TL, Mok MY, Mak C, Yang W, Chong P, et al. Cell lineage‐specific genome‐wide DNA methylation analysis of patients with paediatric‐onset systemic lupus erythematosus. Epigenetics. 2019;14:341‐351. DOI: 10.1080/15592294.2019.1585176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Keshavarz‐Fathi M, Sanati G, Sadr M, Mohebbi B, Ziaee V, Rezaei N. Aberrant DNA methylation of the promoters of JAK2 and SOCS3 in juvenile systemic lupus erythematosus. Eur Cytokine Netw. 2021;32:48‐54. DOI: 10.1684/ecn.2021.0469 [DOI] [Google Scholar]
- 44. Li Y, Huang C, Zhao M, Liang G, Xiao R, Yung S, et al. A possible role of HMGB1 in DNA demethylation in CD4+ T cells from patients with systemic lupus erythematosus. Clin Dev Immunol. 2013;2013:206298. DOI: 10.1155/2013/206298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hu N, Qiu X, Luo Y, Yuan J, Li Y, Lei W, et al. Abnormal histone modification patterns in lupus CD4+ T cells. J Rheumatol. 2008;35:804‐810. [PubMed] [Google Scholar]
- 46. Zhu J, Huang X, Su G, Wang L, Wu F, Zhang T, et al. High expression levels of microRNA‐629, microRNA‐525‐5p and microRNA‐516a‐3p in paediatric systemic lupus erythematosus. Clin Rheumatol. 2014;33:807‐815. DOI: 10.1007/s10067-014-2583-5 [DOI] [PubMed] [Google Scholar]
- 47. Sourour SK, Aboelenein HR, Elemam NM, Abdelhamid AK, Salah S, Abdelaziz AI. Unraveling the expression of microRNA‐27a* & NKG2D in peripheral blood mononuclear cells and natural killer cells of pediatric systemic lupus erythematosus patients. Int J Rheum Dis. 2017;20:1237‐1246. DOI: 10.1111/1756-185X.13099 [DOI] [PubMed] [Google Scholar]
- 48. Li S, Li C, Zhang J, Tan X, Deng J, Jiang R, et al. Expression profile of long noncoding RNAs in children with systemic lupus erythematosus: a microarray analysis. Clin Exp Rheumatol. 2019;37:156‐163. [PubMed] [Google Scholar]
- 49. Guo Y, Sawalha AH, Lu Q. Epigenetics in the treatment of systemic lupus erythematosus: potential clinical application. Clin Immunol. 2014;155:79‐90. DOI: 10.1016/j.clim.2014.09.002 [DOI] [PubMed] [Google Scholar]
- 50. Vanhaecke T, Papeleu P, Elaut G, Rogiers V. Trichostatin A‐like hydroxamate histone deacetylase inhibitors as therapeutic agents: toxicological point of view. Curr Med Chem. 2004;11:1629‐1143. DOI: 10.2174/0929867043365099 [DOI] [PubMed] [Google Scholar]
- 51. Salvi V, Bosisio D, Mitola S, Andreoli L, Tincani A, Sozzani S. Trichostatin A blocks type I interferon production by activated plasmacytoid dendritic cells. Immunobiology. 2010;215:756‐761. DOI: 10.1016/j.imbio.2010.05.023 [DOI] [PubMed] [Google Scholar]
- 52. Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007;13:1299‐1307. DOI: 10.1038/nm1652 [DOI] [PubMed] [Google Scholar]
- 53. Al‐Yacoub N, Fecker LF, Möbs M, Plötz M, Braun FK, Sterry W, et al. Apoptosis induction by SAHA in cutaneous T‐cell lymphoma cells is related to downregulation of c‐FLIP and enhanced TRAIL signaling. J Invest Dermatol. 2012;132:2263‐2274. DOI: 10.1038/jid.2012.125 [DOI] [PubMed] [Google Scholar]
- 54. Reilly CM, Mishra N, Miller JM, Joshi D, Ruiz P, Richon VM, et al. Modulation of renal disease in MRL/lpr mice by suberoylanilide hydroxamic acid. J Immunol. 2004;173:4171‐4178. DOI: 10.4049/jimmunol.173.6.4171 [DOI] [PubMed] [Google Scholar]
- 55. Thai TH, Patterson HC, Pham DH, Kis‐Toth K, Kaminski DA, Tsokos GC. Deletion of microRNA‐155 reduces autoantibody responses and alleviates lupus‐like disease in the Fas(lpr) mouse. Proc Natl Acad Sci U S A. 2013;110:20194‐20199. DOI: 10.1073/pnas.1317632110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ravelli A, Grom AA, Behrens EM, Cron RQ. Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment. Genes Immun. 2012;13:289‐298. DOI: 10.1038/gene.2012.3 [DOI] [PubMed] [Google Scholar]
- 57. Horton DB, Shenoi S. Review of environmental factors and juvenile idiopathic arthritis. Open Access Rheumatol. 2019;11:253‐267. DOI: 10.2147/OARRR.S165916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Duke O, Panayi GS, Janossy G, Poulter LW. An immunohistological analysis of lymphocyte subpopulations and their microenvironment in the synovial membranes of patients with rheumatoid arthritis using monoclonal antibodies. Clin Exp Immunol. 1982;49:22‐30. [PMC free article] [PubMed] [Google Scholar]
- 59. Wu SA, Yeh KW, Lee WI, Yao TC, Huang JL. Persistent improper upregulation of Th17 and TReg cells in patients with juvenile idiopathic arthritis. J Microbiol Immunol Infect. 2016;49:402‐408. DOI: 10.1016/j.jmii.2014.07.002 [DOI] [PubMed] [Google Scholar]
- 60. Cribbs AP, Kennedy A, Penn H, Amjadi P, Green P, Read JE, et al. Methotrexate restores regulatory T cell function through demethylation of the FoxP3 upstream enhancer in patients with rheumatoid arthritis. Arthritis Rheumatol. 2015;67:1182‐1192. DOI: 10.1002/art.39031 [DOI] [PubMed] [Google Scholar]
- 61. Chavez‐Valencia RA, Chiaroni‐Clarke RC, Martino DJ, Munro JE, Allen RC, Akikusa JD, et al. The DNA methylation landscape of CD4+ T cells in oligoarticular juvenile idiopathic arthritis. J Autoimmun. 2018;86:29‐38. DOI: 10.1016/j.jaut.2017.09.010 [DOI] [PubMed] [Google Scholar]
- 62. Ellis JA, Munro JE, Chavez RA, Gordon L, Joo JE, Akikusa JD, et al. Genome‐scale case‐control analysis of CD4+ T‐cell DNA methylation in juvenile idiopathic arthritis reveals potential targets involved in disease. Clin Epigenetics. 2012;4:20. DOI: 10.1186/1868-7083-4-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ghavidel AA, Shiari R, Hassan‐Zadeh V, Farivar S. The expression of DNMTs is dramatically decreased in peripheral blood mononuclear cells of male patients with juvenile idiopathic arthritis. Allergol Immunopathol (Madr). 2020;48:182‐186. DOI: 10.1016/j.aller.2019.08.003 [DOI] [PubMed] [Google Scholar]
- 64. Wong L, Jiang K, Chen Y, Jarvis JN. Genetic insights into juvenile idiopathic arthritis derived from deep whole genome sequencing. Sci Rep. 2017;7:2657. DOI: 10.1038/s41598-017-02966-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hu Z, Jiang K, Frank MB, Chen Y, Jarvis JN. Complexity and specificity of the neutrophil transcriptomes in juvenile idiopathic arthritis. Sci Rep. 2016;6:27453. DOI: 10.1038/srep27453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Demir F, Çebi AH, Kalyoncu M. Evaluation of plasma microRNA expressions in patients with juvenile idiopathic arthritis. Clin Rheumatol. 2018;37:3255‐3262. DOI: 10.1007/s10067-018-4277-x [DOI] [PubMed] [Google Scholar]
- 67. Schulert GS, Fall N, Harley JB, Shen N, Lovell DJ, Thornton S, et al. Monocyte microRNA expression in active systemic juvenile idiopathic arthritis implicates microRNA‐125a‐5p in polarized monocyte phenotypes. Arthritis Rheumatol. 2016;68:2300‐2313. DOI: 10.1002/art.39694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li HW, Xie Y, Li F, Sun GC, Chen Z, Zeng HS. Effect of miR‐19a and miR‐21 on the JAK/STAT signaling pathway in the peripheral blood mononuclear cells of patients with systemic juvenile idiopathic arthritis. Exp Ther Med. 2016;11:2531‐2536. DOI: 10.3892/etm.2016.3188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Huang N, Fan Z, Ma L, Ma H, Huang H, Yu H, et al. Long non‑coding RNA RP11‑340F14.6 promotes a shift in the Th17/Treg ratio by binding with P2X7R in juvenile idiopathic arthritis. Int J Mol Med. 2020;46:859‐868. DOI: 10.3892/ijmm.2020.4618 [DOI] [PubMed] [Google Scholar]
- 70. Kamiya Y, Kawada J, Kawano Y, Torii Y, Kawabe S, Iwata N, et al. Serum microRNAs as potential biomarkers of juvenile idiopathic arthritis. Clin Rheumatol. 2015;34:1705‐1712. DOI: 10.1007/s10067-015-2922-1 [DOI] [PubMed] [Google Scholar]
- 71. Chifotides HT, Bose P, Verstovsek S. Givinostat: an emerging treatment for polycythemia vera. Expert Opin Investig Drugs. 2020;29:525‐536. DOI: 10.1080/13543784.2020.1761323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Leoni F, Fossati G, Lewis EC, Lee JK, Porro G, Pagani P, et al. The histone deacetylase inhibitor ITF2357 reduces production of pro‐inflammatory cytokines in vitro and systemic inflammation in vivo . Mol Med. 2005;11:1‐15. DOI: 10.2119/2006-00005.Dinarello [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Vojinovic J, Damjanov N, D'Urzo C, Furlan A, Susic G, Pasic S, et al. Safety and efficacy of an oral histone deacetylase inhibitor in systemic‐onset juvenile idiopathic arthritis. Arthritis Rheum. 2011;63:1452‐1458. DOI: 10.1002/art.30238 [DOI] [PubMed] [Google Scholar]
- 74. Hartmann M, Hakobyan M, Langstein J, Schönung M, Stäble S, Touzart A, et al. Epigenetic regulation of normal hematopoiesis and its dysregulation in hematopoietic malignancies. Epigenetics Immun Syst. 2020;16:285‐313. [Google Scholar]
- 75. Stajic D, Jansen L. Empirical evidence for epigenetic inheritance driving evolutionary adaptation. Philos Trans R Soc Lond B Biol Sci. 2021;376:20200121. DOI: 10.1098/rstb.2020.0121 [DOI] [PMC free article] [PubMed] [Google Scholar]