

Abstract
NMR has the resolution and specificity to determine atomic-level protein structures of isotopically-labeled proteins in complex environments and, with the sensitivity gains conferred by dynamic nuclear polarization (DNP), NMR has the sensitivity to detect proteins at their endogenous concentrations. However, DNP sensitivity enhancements are critically dependent on experimental conditions and sample composition. While some of these conditions are theoretically compatible with cellular viability, the effects of others on cellular sample integrity are unknown. Uncertainty about the integrity of cellular samples limits the utility of experimental outputs of in-cell experiments. Using several measures, we establish conditions that support DNP enhancements that can enable detection of micromolar concentrations of proteins in experimentally tractable times that are compatible with cellular viability. Taken together, we establish DNP assisted MAS NMR as a technique for structural investigations of biomolecules in intact viable cells that can be phenotyped both before and after NMR experiments.
Keywords: DNP solid-state NNR, in-cell NMR, cryo-preservation, HEK293, AMUPol
Introduction
Despite the importance of the environment, structural investigations of biomolecules are typically confined to highly purified in vitro systems. These investigations yield invaluable insights, but such investigations cannot fully recapitulate cellular environments. Capturing the effect of these complicated environments on biomolecular conformation is of particular importance for proteins that have more than one stable conformation, interact with cellular components or contain regions of intrinsic disorder.1
In-cell NMR allows us to obtain atomic resolution information of proteins in their native environments.2–8 Nuclear Magnetic Resonance (NMR) spectroscopy detects only NMR-active nuclei. These nuclei are non-perturbative, label-free probes that can be specifically incorporated into a protein of interest that is either delivered to or expressed inside the cell.5–9 In-cell NMR has been employed to investigate protein and nucleic acid structure, dynamics, interactions, and post-translational modifications like phosphorylation.4, 8–21 Solution state NMR spectroscopy can be used to study interactions in cells however, this technique is limited to small proteins that interact with small ligands or only transiently with larger biomolecules. This is because solution state NMR spectroscopy is limited by molecular tumbling times that depend upon molecular size and solvent viscosity and thus reports only on rapidly re-orienting molecules in the sample.
Magic angle spinning (MAS) solid-state NMR is not limited by molecular correlation times and is particularly useful to study large protein complexes, amyloid fibrils and membrane proteins.21–26 With the sensitivity gains conferred by dynamic nuclear polarization (DNP), MAS NMR has the sensitivity to detect proteins at their endogenous concentrations in complex biological environments.1, 21–22, 27–29 However, the effectiveness of DNP-enhanced MAS NMR is critically dependent on sample composition30–36 and experimental conditions23, 37–40. DNP increases the sensitivity of NMR spectroscopy through the transfer of the large spin polarization of an unpaired electron to nearby nuclei37 which are typically introduced into a sample by doping with millimolar concentrations of stable biological radicals34–36. In addition to millimolar concentrations of polarizing agents, a typical DNP sample of a hydrated biomolecule is cryoprotected by the addition of 60% d8-glycerol to aid in the formation of vitreous ice, deuterated to ~90% to aid spin diffusion, frozen to near liquid nitrogen temperatures and subjected to magic angle spinning. Pioneering work applying DNP MAS NMR to cultured mammalian cells suggested that measurement of low concentrations of proteins inside mammalian cells is possible21 but uncertainty about the biological integrity of the cellular sample limits the utility of the structural information for in-cell experiments.
Here, we establish an approach to homogenously introduce the polarization agent, AMUPol34, into cultured mammalian cells that results in high sensitivity gains and supports cellular viability throughout DNP NMR experimentation. After structural characterization via DNP MAS NMR, these cells can be cultured or imaged and their phenotype can be determined and compared with cells before structural characterization. Our work sets the stage to determine if and how cellular environments modulate protein structure, which is of particular interest for systems where protein conformation can affect cellular fate. Investigation of protein conformations inside viable cells using DNP MAS NMR creates an experimental system with the ability to tightly couple genotypes, phenotypes and environments (e.g. presence/absence of a drug) to specific structures or structural ensembles.
Results
HEK293 cells remain viable during DNP MAS NMR
Conditions that favor efficient DNP and HEK293 cell viability are compatible
To determine whether any of the components that are typically used to achieve high DNP-enhancements for biological systems compromise cellular viability, we used a trypan blue dye exclusion test to determine the percentage of cells with intact membranes present in a sample. Unsurprisingly, HEK293 viability was not compromised by replacement of media components with PBS, by addition of common working concentrations of cryoprotective agents (10% DMSO or 15% glycerol)41, or by either per-deuteration or complete deuteration of the buffer components (Figure S1A–C). Moreover, HEK293 viability was not compromised by addition of the polarization agent AMUPol at concentrations up to 50 mM (Figure S1E) even after 45 minutes of exposure at room temperature (Figure S1F). To ensure that AMUPol, or any of the other treatments, were not toxic to HEK293 cells, we assessed viability using an orthogonal method, a quantitative cell growth assay. HEK293 cells exposed to glycerol had a lag phase before exponential growth however the growth kinetics of the exponential phase were indistinguishable from untreated HEK293 cells once exponential growth began (Figure 1C). Neither per-deuteration nor exposure to AMUPol had any additional effects on cell growth (data not shown). Thus, HEK293 cell viability is not compromised by the components that support high DNP-enhancements. However, the amount of added cryoprotectants can affect viability. Suspension of cells in 60% glycerol, a percentage commonly used in biological DNP samples, resulted in the loss of membrane integrity of more than half of the cells (Figure S1C) and suspension of cells in 60% DMSO28 resulted in a larger loss of membrane integrity (Figure S1G) and a complete loss of propagative ability. To determine whether any of these components compromise cellular viability during or after cryopreservation, we used a trypan blue dye exclusion test to determine the percentage of viable cells after freezing in the presence of these compounds. Unsurprisingly, freezing cells that were cryoprotected in DNP-compatible media by direct immersion of the sample in liquid nitrogen (“flash freezing”) resulted in fewer than 20% of cells with trypan blue impermeable membranes, none of which were able to grow when the sample was plated (Figure S1D). The disconnect between trypan blue values and regrowth is not surprising. While under optimal handling conditions, trypan blue values are a good proxy for cultivability, they are grossly inaccurate and unpredictable following traumatic treatment of cell populations.42 Freezing cells that were cryoprotected in DNP-compatible media at the standard controlled rate of 1 °C/min had high post thaw viabilities (Figure 1E, Figure S1D) and had normal growth kinetics after being thawed (Figure 1C). Thus, HEK293 cells can be cryopreserved in matrices that support high DNP-enhancements in biological samples.
Figure 1:
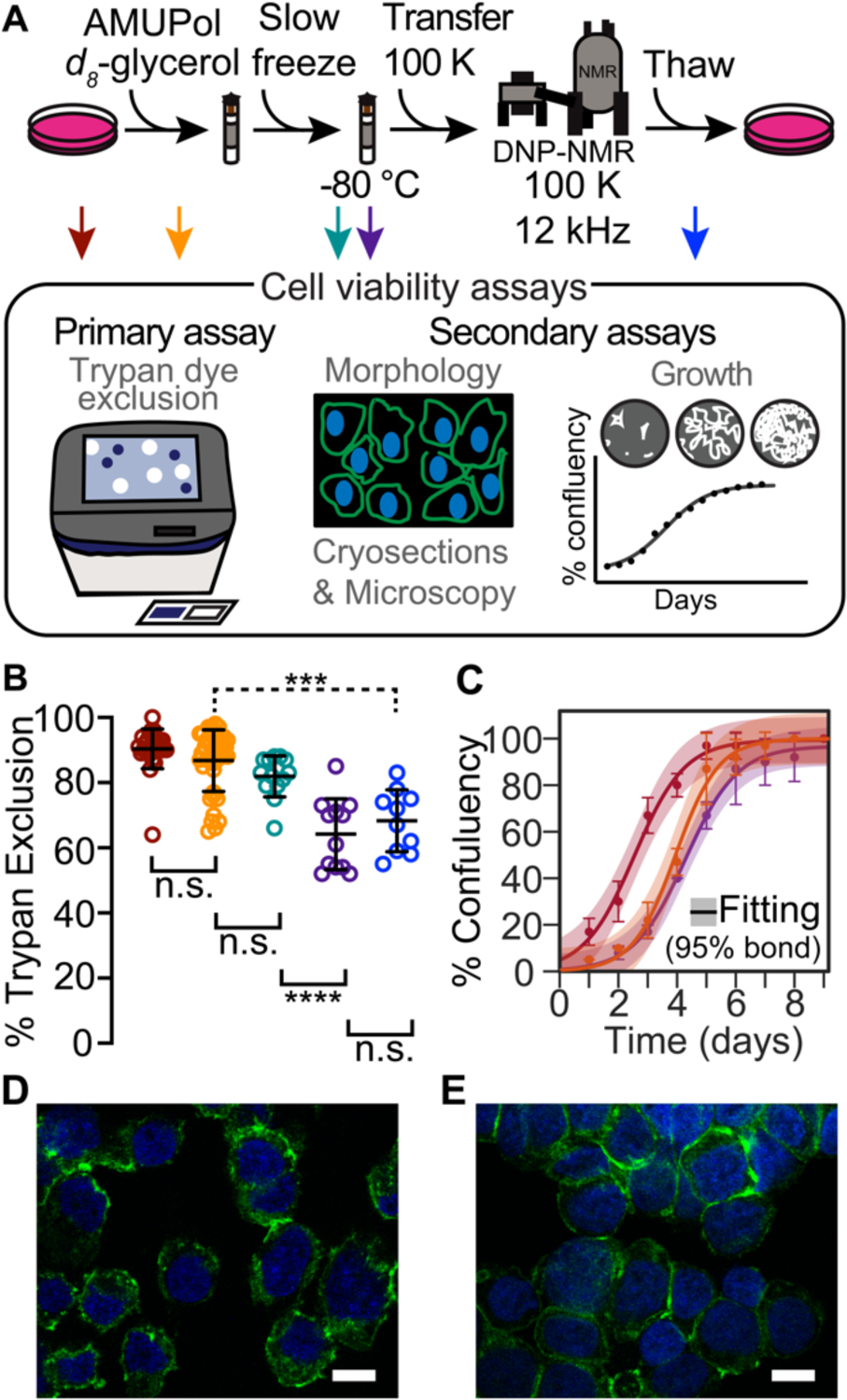
HEK293 cells are viable throughout the DNP NMR process. (A). Experimental scheme of the DNP NMR sample preparation procedure and cell viability assays. Colored arrows indicate points at which sample viability was assessed. Viability was assessed for cells after trypsinization and washing (dark red), after suspension in AMUPol and cryoprotectants (orange), after being frozen at 1 °C per min (green), after manipulation to remove the drive tip from the frozen rotor (purple) and after the entire DNP MAS NMR experiment (blue). (B) Frozen cells best represent the state of the sample during DNP NMR data collection. Percentage of cells with trypan impermeable membranes at each sample assessment point, colored as in A. Each point represents an independent sample. Black bars indicate average and standard deviation. Brackets indicate results of two-tailed homoscedastic student’s t-tests. Unpaired comparisons are indicated with solid brackets and paired comparisons with dashed brackets. (n.s. p > 0.05, * p < 0.05, *** p < 0.001, **** p < 0.0001). (C) Addition of glycerol affects growth kinetics, but further sample manipulations do not. Growth kinetics as assessed by confluency, colored as in A. The averages and standard deviations of three independent experiments are indicated by circles and error bars, respectively. The best fit of sigmoid is indicated in solid lines and the 95% confidence interval by the shaded area. (D and E) Fluorescence microscopy of HEK293 cells immediately before (D) and immediately after freezing (E) using Phalloidin (green) and Hoechst 33342 (blue) to stain actin cytoskeleton and nucleus (Scale bar 10 μm).
Cryogenic transfer of slowly frozen cellular samples supports post-experiment viability
Because freezing rate affects the success of cryopreservation, we next assessed various methods of transferring cellular samples into the NMR spectrometer. We first assessed two common sample handling methods for low temperature MAS NMR; transfer of a room temperature sample into a pre-cooled instrument and cryogenic transfer of a flash frozen sample into a pre-cooled instrument43. Transfer of a room temperature rotor into an NMR probe that is pre-equilibrated to 100 K, as assessed by the temperature of the stator, reduces the sample temperature by ~200 K in ~2 minutes followed by a slow decrease to 100 K over 15 minutes. Freezing the sample inside of the spectrometer in this manner resulted in fewer than 5% viable cells at the end of the experiment. Flash freezing of the sample by immersion of the room temperature rotor into a liquid nitrogen bath followed by a cryogenic transfer into spectrometer39 resulted in fewer than 20% viable cells at the end of the experiment as assessed by trypan blue. As expected, freezing rates on these timescales resulted in substantial loss of cellular viability43–44. Thus, we froze cryoprotected cells at the controlled rate of 1 °C/min and then cryogenically transferred the sample from liquid nitrogen storage directly into a spectrometer that was pre-equilibrated to 100 K.43 Cryogenic transfer of slowly frozen sample resulted in high post DNP viability (Figure 1B).
Neither MAS nor DNP affect cellular viability
To determine whether the any of the manipulations required for DNP MAS NMR sample preparation compromise cellular viability, we assessed trypan blue membrane permeability at each step of our sample preparation workflow (Figure 1A, arrows). After harvesting adherent cells from tissue culture plates, the cells were rinsed with PBS and pelleted. At this point, cellular membrane integrity as assessed by trypan blue dye exclusion tests was high (92 ± 3%, Figure 1, dark red). Addition of cryoprotectants and AMUPol followed by transfer into 3.2 mm NMR rotors slightly reduced membrane integrity (87 ± 9%, Figure 1, orange; decrease of 5 ± 10%, p = 0.03). Freezing cryoprotected cells at the controlled rate of 1 °C/min did not significantly compromise membrane integrity as assessed by trypan blue dye exclusion test (82 ± 6%, Figure 1, green, p = 0.09). Post-NMR, trypan blue membrane integrity was lower than those for slow frozen samples (68 ± 9%, Figure 1, blue; decrease of 14 ± 11%, p = 4e-4). Interestingly, however, when membrane integrity was determined both pre-freezing (Figure 1A, orange arrow) and post-NMR experiment (Figure 1A, blue arrow) on the same sample – as opposed to comparing the averages - the decrease in viability was smaller (decrease of 11 ± 3%, p = 0.001, n = 10, rather than 19 ± 13%). This suggested that sample specific differences in rotor packing, DNP MAS NMR conditions, or unpacking may affect the membrane integrity. To determine if the membrane integrity decrease post-NMR experiment was a result of DNP MAS NMR or sample handling, we measured membrane integrity for samples that were frozen in NMR rotors with a cap and plug, but not subjected to NMR experimentation. We found that membrane integrities of capped samples that were not subjected to NMR were indistinguishable from samples that were subjected to NMR experimentation (64 ± 11%. Figure 1, purple, p = 0.35). Thus, the post-NMR decrease in membrane integrity was not a result of DNP MAS NMR but rather was a result of the sample handling required to remove the drive tip and silicon plug before the membrane integrity could be measured. Thus, cryoprotected frozen samples, rather than post-NMR samples, are most representative of the state of the sample when it is measured by NMR.
Cellular samples can be phenotyped before and after DNP MAS NMR analysis
Because membrane integrity only reports on one aspect of cellular sample integrity, we next assessed cellular integrity by two additional methods. To determine whether the any of the manipulations required for DNP MAS NMR sample preparation compromise cellular propagative ability, we next assessed cellular growth kinetics at each step in our workflow and again found that exposure to glycerol introduced a lag phase of 1.5 ± 0.5 days in growth, however no other perturbation significantly altered growth kinetics (Figure 1C). The disconnect between the membrane permeability and propagation indicated that cells with compromised membrane integrity are not necessarily dead. Cells can, and do, recover from transient insults the compromise membrane integrity.12 Finally, to determine if cells retained their morphology under these conditions, we used fluorescence microscopy to image cells immediately before (Figure 1D and S2) and immediately after (Figure 1E and S2) the freezing process. Because these cells did not contain an endogenous fluorophore, we fixed these cells and stained them with DAPI and Phalloidin to visualize both DNA and the actin network. We observed round and intact cells with well-defined nuclei for the stained cell suspensions of cells immediately before and after the freezing process. Taken together, this work indicated that majority of cells in cryopreserved samples for DNP were not only viable during DNP MAS NMR data collection but able to propagate after DNP MAS NMR data collection. Moreover, cellular samples can be imaged by fluorescence microscopy both before and after structural analysis by DNP MAS NMR.
Addition of AMUPol to HEK293 cells results in DNP enhancement of all biomass components.
Quantitative reporters of cellular biomass components
In order to assess whether the polarization agent, AMUPol, is able to increase the sensitivity of DNP experiments in intact cells, we first determined which peaks in the NMR spectra could be used as reporters of the different cellular biomass components. The majority of cellular biomass is comprised of proteins, which are found throughout the cell, lipids, about of quarter of which are found in the plasma membrane and the rest of which are found in the membrane of the ER and other internal organelles45–46, and nucleotides (RNA, most of which is found in the ribosomes in the cytoplasm and DNA, most of which is found in the nucleus). Each of these biomolecules has chemical moieties with characteristic chemical shifts (Figure 2A). Thus, as a first step, we determined whether the integrated intensity at the chemical shift of each characteristic moiety for the major biomass component could be used as a quantitative indicator of the major biomass constituents of HEK293 cells. To do so, we collected 13C direct polarization (DP) spectra and integrated the characteristic 13C peak intensity of each major biomass component to estimate their approximate quantities on samples that did not contain any polarization agent and were not subject to microwave irradiation. We found that the biomass compositions determined by 13C DP at 100 K for HEK293 cells were similar to the biomass composition for HEK293 cells determined by chemical means47 (Figure 2C and D). Thus, the integrated intensity of these characteristic chemical shifts reported quantitatively on the major biomass constituents of HEK293 cells.
Figure 2:
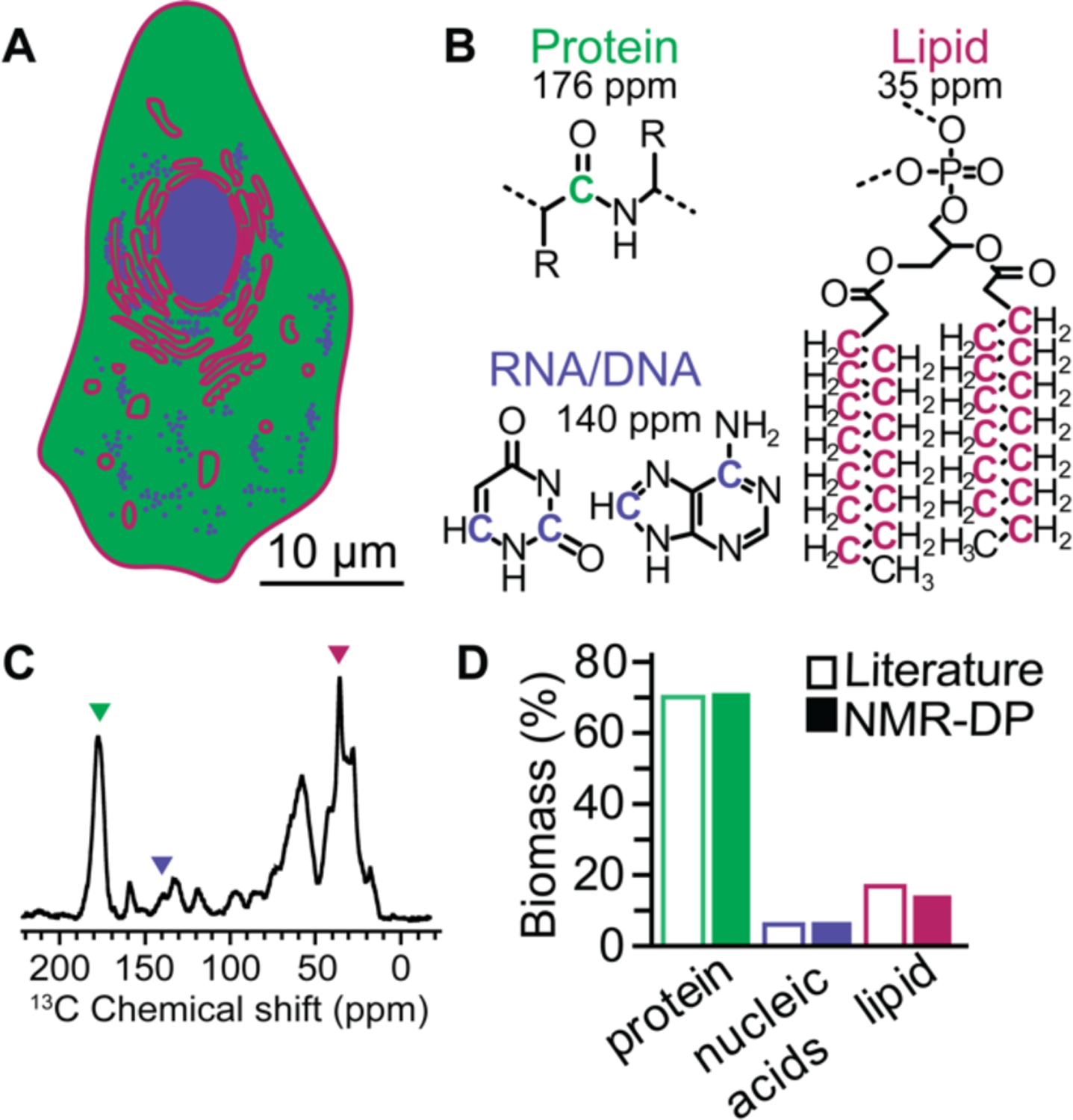
NMR report quantitively on cellular biomass composition. (A) Schematic of a mammalian cell depicting the heterogenous distribution of the three major components of the cellular biomass: proteins (green) which are dispersed throughout the cell, nucleotides (blue) which are found in the RNA component of ribosomes in the cytoplasm and as DNA in the nucleus and lipids (pink) which are found in the plasma membrane and enclose all of the membrane bound organelles. (B) Structures of the biomass moieties with the representative carbon for each biomass component highlighted. (C) 13C direct polarization spectra of cryopreserved HEK293 cells grown on isotopically enriched media at 100 K taken at 600 MHz with 12 kHz magic angle spinning and a recycle delay of 300 seconds. Colored arrowheads indicate integrated peaks. (D) Quantification of biomass by NMR (closed bars) is similar to quantification of biomass by orthogonal methods47 (open bars).
DNP is very efficient for all biomass components in cell lysates
After having confirmed that the integrated intensity of the selected peaks reported quantitatively on the major biomass constituents of HEK293 cells, we next determined the DNP performance, the DNP enhancement and DNP build-up times (TB,on)48, we could expect for complex mixtures of these biomolecules. We collected 13C cross-polarization (CP) spectra with and without microwave irradiation, to report DNP enhancement, for lysed cells prepared with a range of AMUPol concentrations. We found that the DNP enhancements for protein in lysed cells increased with increasing concentrations of AMUPol to a maximum value of ~93 at 5 mM AMUPol and then decreased with increasing AMUPol concentrations (Figure 3A). Taken together, these experiments on lysed cells demonstrate that AMUPol effectively enhanced all of the major biomass components.
Figure 3:
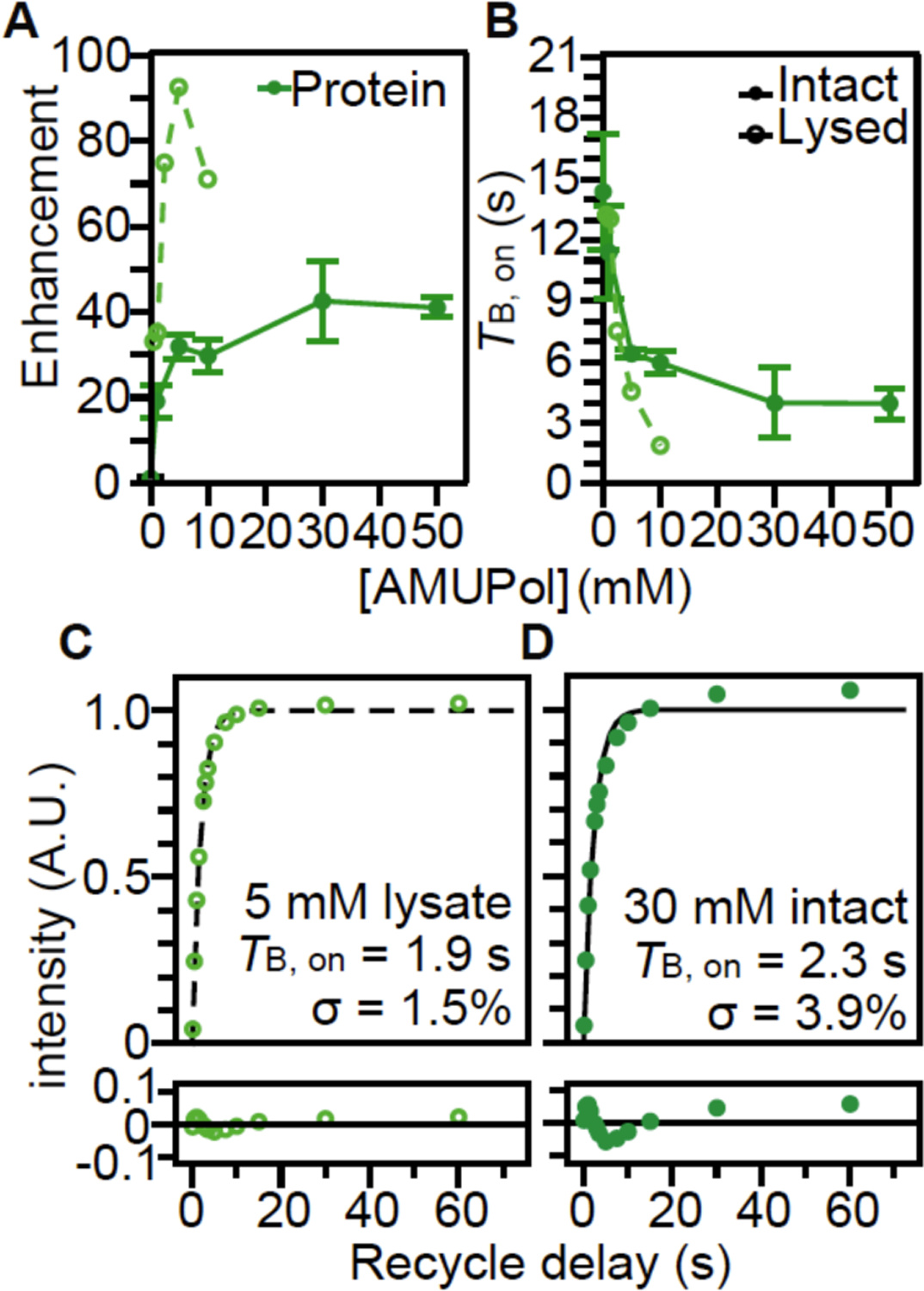
The polarization agent, AMUPol, effectively polarizes proteins in both lysed (dashed lines, light green open circles) and intact (solid lines, closed dark green circles) HEK293 cells. (A) DNP enhancement and (B) TB,on values from saturation recovery experiments are dependent upon the AMUPol concentration. Points indicate average and error bars indicate standard deviation of the measurements. (C) Fit of the TB,on data (open circles) for lysed cells containing 5 mM AMUPol to a mono-exponential equation (black dashed line) had a TB,on value of 1.9 seconds with a regression error (lower plot) of 1.5%. (D) Fit of the TB,on data (closed circles) for intact cells incubated with 30 mM AMUPol to a mono-exponential equation (black solid line) had a TB,on value of 2.3 seconds with a regression error (lower plot) of 3.9%. Analyses for additional biomass components are presented in Supplemental Figure 3.
Intact viable cells have moderate DNP efficiency when incubated the AMUPol
Mammalian cells are enclosed by selectively permeable membrane that regulates the movement of molecules across the cell and they exclude many exogenous molecules outside the plasma membrane. To determine if AMUPol, a small molecule with a molecular weight of 750 Da, could effectively enhance the signal for all the components of cellular biomass for intact HEK293 cells, we measured DNP enhancements and TB,on values of the major biomass components (Figure S3). DNP build-up time (TB,on) of NMR signal is shorter when radical concentrations increase. We collected DNP enhancements and TB,on build-up times for intact cells prepared by adding increasing concentrations of AMUPol to the sample. Interestingly, we found that the DNP enhancements for proteins in intact cells reached a maximum value of 43 ± 9, which is half of the maximal enhancement attained for proteins in lysates and that the addition of 30 mM AMUPol, rather than 5 mM AMUPol, was required to attain this enhancement. The lower enhancements and longer TB,on for the biomass components in intact cells relative to lysed cells indicate that the AMUPol concentration inside of the cell is lower than the concentration of AMUPol that was added to the sample, likely because the majority of the AMUPol is excluded by the semi-permeable plasma membrane. This suggested that AMUPol is heterogeneously distributed in samples of intact cells.
AMUPol is heterogeneously distributed in cells incubated with AMUPol
To assess the homogeneity of the AMUPol concentration throughout each biomass component (Figure 3C and D, Figure S3), we used the regression error of the fit of the TB,on data to a mono-exponential equation. If the concentration distribution of AMUPol is heterogenous, there will be a mixture of underlying TB,on values which will increase the regression error. The regression error of the fit of the TB,on data to a mono-exponential function of the amino acid proline suspended in a matrix of 60:30:10 (v/v) glycerol:D2O:H2O with 10 mM AMUPol was 0.5% and represents the error expected from experimental noise. For lysed biomass, the regression error for protein was 1.3 ± 0.2%, for nucleotide was 1.0 ± 0.2% (lower than protein, p < 0.01, n = 6), and for lipid was 1.1 ± 0.3% (indistinguishable from protein, p = 0.86, n = 6). The increase in regression error for the lysed cells relative to that for proline reflect the larger range of underlying TB,on values of the constituents in this complex mixture. For intact cells, the regression error for protein was 3.0 ± 0.8%, for nucleotide was 3.0 ± 0.8% (indistinguishable from protein, p = 0.62, n = 12), while the regression error for lipid was 2.5 ± 0.8% (somewhat lower than protein and nucleotide, p < 0.005, n = 12). The larger regression errors for the intact cells indicated that the concentration distribution of AMUPol is more heterogeneous in intact cells than in lysed cells. Taken together, this indicates that while AMUPol can polarize all of the biomass components in intact cells, possibly through a combination of both proximity-based and relayed DNP mechanisms48–50, the AMUPol concentration throughout the sample is heterogenous. This is likely because much of the AMUPol cannot efficiently access the interior of the cell.
Improved delivery of AMUPol to HEK293 cells via electroporation.
High viability of cells electroporated in the presence of AMUPol
Because only a small portion of the exogenously added AMUPol appears to be able to enter the cell, we sought to improve AMUPol delivery by transiently permeabilizing the plasma membrane via electroporation. We assessed trypan blue membrane permeability at each step in the protocol (arrows, Figure 4A) to determine whether the any of the steps during the DNP MAS NMR sample preparation of electroporated HEK293 cells compromises cellular viability. After harvesting adherent cells from tissue culture plates, the cells were rinsed with PBS, suspended in electroporation buffer and electroporated. Electroporation reduced trypan blue membrane integrity to 81 ± 9% (Figure 4A and B, red; a decrease of 9 ± 10%, p = 7e-5). Electroporation in the presence of AMUPol and the addition of cryoprotectants followed by transfer into the NMR rotors did not result in any further loss in trypan blue membrane integrity 79 ± 11% (Figure 4A and B, orange; p = 0.32). Freezing electroporated and cryoprotected cells at the controlled rate of 1 °C/min reduced trypan blue membrane integrity to 60 ± 15% (Figure 4A and B, green; a decrease of 19 ± 19%, p = 3e-7). However, the membrane integrity after rotor unpacking and DNP MAS NMR (Figure 4A and B, purple and blue, respectively) were indistinguishable from those for slow frozen samples. The trypan blue membrane permeability values have a wider range for these electroporated preparations than they do for cells incubated with AMUPol, likely due to differences in cell membrane re-sealing rates for different preparation of cells.12, 51–53
Figure 4:
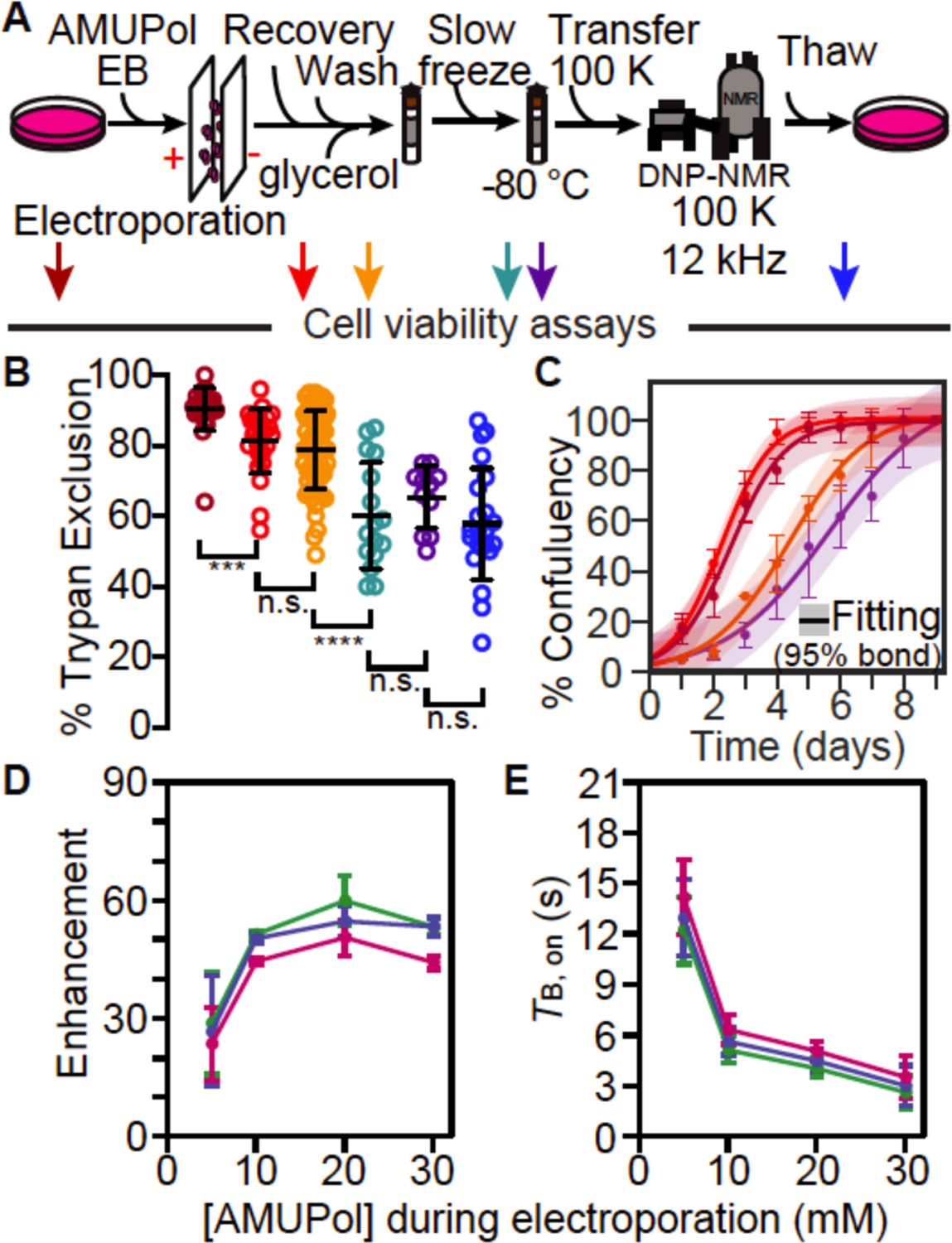
Electroporated HEK293 cells remain viable throughout the DNP NMR process. (A) Experimental scheme of the DNP NMR sample preparation procedure that includes an electroporation step to introduce AMUPol into the sample. Colored arrows indicate points at which sample viability were assessed. Viability was assessed for cells after trypsinization and washing (dark red), after electroporation (red), after suspension in AMUPol and cryoprotectants (orange), upon thaw (green), upon thaw after manipulation to remove the drive tip from the rotor (purple) and upon thaw after DNP MAS NMR (blue). (B) Frozen cells best represent the state of the sample during DNP NMR data collection. Percentage of cells with trypan blue impermeable membranes at each sample assessment point, colored as in A. Each point represents an independent experiment. Black bars indicate average value and standard deviations. Brackets indicate comparisons for unpaired two-tailed homoscedastic student’s t-tests (n.s. p > 0.05, **** p < 0.0001). (C) Addition of glycerol affects growth kinetics, but other sample manipulations, including electroporation, do not. Growth kinetics as assessed by confluency, colored as in A. The averages and standard deviations of three independent experiments are indicated by circles and error bars, respectively. The best fit to a sigmoid is indicated in solid lines and the 95% confidence interval by the shaded area. (D) DNP enhancement and (E) average TB,on values from saturation recovery experiments for all biomass components, protein (green), nucleotide (blue) and lipid (pink), are dependent upon the AMUPol concentration at the time of electroporation; the AMUPol concentration inside the rotor is estimated to be an order of magnitude lower than the AMUPol concentration at the time of electroporation. Error bars indicate standard deviations of the measurements.
Electroporated cellular samples can be phenotyped before and after DNP MAS NMR analysis
We next assessed growth kinetics and again found that exposure to glycerol introduced a lag phase of 2.2 ± 0.5 days (p = 0.02, n = 3) but did not affect growth rate (Figure 4C). Freezing cells increased the lag phase by 1.2 ± 0.9 days although this change not significant (p = 0.13, n = 3). However, no perturbation altered the growth rate, including electroporation (p = 0.7, n = 3) and freezing (p = 0.23, n = 3). Given that we found that membrane integrity and propagative ability were not tightly coupled for cells incubated with AMUPol, this is perhaps not particularly surprising. Taken together, this work indicated that majority of cells in a sample that is cryopreserved after delivery of AMUPol via electroporation were not only viable during DNP MAS NMR data collection but also able to propagate after DNP MAS NMR data collection.
Improved DNP efficiency and homogeneity when AMUPol is delivered via electroporation
To determine if introduction of AMUPol into HEK293 cells via electroporation can enhance all of the major biomass components of intact mammalian cells, we collected DNP enhancements and TB,on build-up times for intact cells electroporated with increasing concentrations of AMUPol (Figure 4D and E). Relative to lysed cells (Figure 3A and B, dotted lines), electroporated cells had lower enhancements and longer TB,on values, while relative to cells incubated with AMUPol (Figure 3A and B, solid lines), electroporated cells had higher enhancements and shorter TB,on values; DNP enhancements for proteins in electroporated cells reached a maximum value of 60 ± 6 (Figure 4D, green). However, as observed earlier, the TB,on values for the AMUPol concentration (at the time of electroporation) that resulted in the highest DNP enhancements for electroporated cells were similar to those of the lysed samples (Figure 4E, green; 20 mM AMUPol, TB,on = 4.0 ± 0.6 s, n = 4). Taken together, this indicated that the AMUPol concentration inside of electroporated cells is lower than the concentration of AMUPol that was added to the sample which reflects the difference in sample preparation. For electroporated samples, only a fraction of AMUPol that is present in the electroporation buffer enters the cells and the extracellular AMUPol is washed out of the sample after electroporation and before sample packing. The concentration of AMUPol in the sample is an order of magnitude lower than the concentration of AMUPol in the buffer at the time of electroporation12, 51, 54.
To assess the homogeneity of the AMUPol concentration throughout each biomass component in these electroporated samples, we used the regression error of the fit of the TB,on data to a mono-exponential equation (Figure S4). For electroporated cells, the regression error for protein was 1.7 ± 0.4%, for nucleotide was 1.4 ± 0.4% (lower than protein, p = 2e-7, n = 12) and lipid was 1.1 ± 0.3% (lower than protein, p = 6e-5, n = 12). When compared with lysed samples, the regression error was indistinguishable for lipids (p = 0.38, unpaired), and the regression errors were slightly higher for protein and nucleotide (p < 0.05, unpaired). This indicated that the concentration distribution of AMUPol is more heterogeneous in electroporated cells than in lysed cells. When compared with the incubated but not electroporated samples, the regression errors were significantly smaller for all of the components (p < 0.003, unpaired). This showed that the concentration of AMUPol throughout the biomass is more homogenous in electroporated samples relative to samples that were incubated with AMUPol. Taken together, this indicates that delivery of AMUPol to mammalian cells by electroporation followed by removal of extracellular AMUPol results in polarization of all the biomass components with increased homogeneity of the AMUPol concentration throughout the sample relative to delivery of AMUPol by incubation.
AMUPol is slowly deactivated by cellular environments at room temperature.
Samples of lysed cells are flash frozen immediately upon addition of AMUPol. However, AMUPol is added to intact cells 8 minutes (for cells incubated with AMUPol) and 15 minutes (for cells electroporated with AMUPol) before starting to reduce the sample temperature at a rate of 1 °C per minute. Because sample manipulations can impact cellular integrity but exposure to cellular environments can reduce nitroxide radicals,22 we characterized the effect of sample preparation time on the DNP performance of AMUPol. First, we confirmed that AMUPol was not deactivated by cellular environments once samples were frozen. To do so, we measured DNP enhancements and TB,on values for electroporated samples that were stored for various times after freezing and before sample measurement. These times ranged from several hours to one month. As expected, the DNP enhancements and TB,on values were not sensitive to the length of time of frozen storage (data not shown). Next, to determine if AMUPol is deactivated by cellular environments at room temperature, we measured DNP enhancements and TB,on values for samples prepared with different room temperature delay intervals before freezing. For lysed cells in the presence of 10 mM AMUPol, the enhancements decreased by half and the TB,on values doubled after an hour at room temperature (Figure 5 and S5). When cells are electroporated in the presence of 10 mM AMUPol, allowed to recover, and then washed to remove extracellular AMUPol, the DNP enhancements decreased by more than half and TB,on values more than doubled when the sample was allowed to sit for an additional 45 minutes at room temperature before freezing (Figure 5 and S5). The decreases in DNP enhancement for the lysed and electroporated cells indicate that exposure to cellular constituents at room temperature decreases the polarization ability of AMUPol and the increases in TB,on values suggest that this deactivation process is via destruction of the radical. In contrast, when intact cells were incubated with 10 mM AMUPol, the enhancements and TB,on values were unchanged with increasing room-temperature incubation periods. Interestingly, we know that cellular constituents deactivate AMUPol. Yet, there was no net change in DNP enhancement or TB,on value with increasing incubation times in these samples. This observation could be explained if AMUPol both continuously enters and is also continuously reduced by the cell during the room temperature incubation time. Taken together, the effect of room temperature incubation times DNP enhancements demonstrates that a short time interval between exposure to AMUPol and freezing results in the best DNP performance. However, the rate of DNP performance loss was slow relative to most sample manipulation steps. Therefore, sample preparation times after addition of AMUPol should be minimized, but not at the expense of sample handling steps that can compromise sample integrity.
Figure 5:
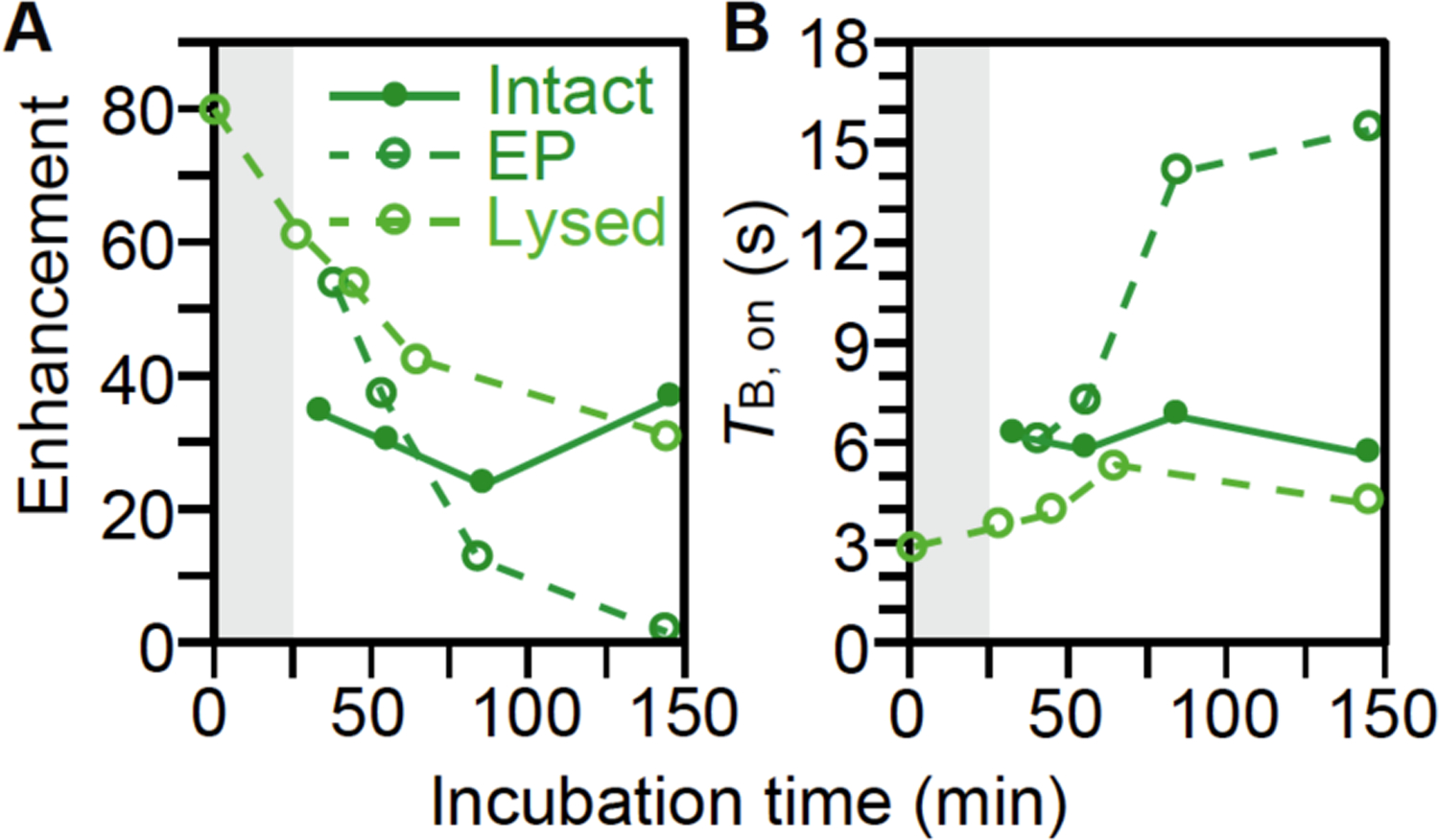
AMUPol is deactivated by cellular biomass. (A) DNP enhancements decrease and (B) TB,on values from saturation recovery experiments for protein increase with longer room temperature exposure to AMUPol to cellular biomass before freezing. Lysed cells with 10 mM AMUPol (light green open circles) were flash frozen after indicated room temperature incubation times. Intact cells where the 10 mM AMUPol was introduced by incubation (dark green closed circles) and where the 10 mM AMUPol was introduced by electroporation (dark green open circles) require room temperature manipulation before reducing the temperature at a rate of 1 °C/min. An additional 25 minutes (gray bar) was added to the room temperature incubation time for intact cells to visually represent this difference in sample preparation. Analyses for additional biomass components are presented in Supplemental Figure S5.
AMUPol transverses the nuclear envelope of intact cells.
While RNA is largely localized to the cytoplasm, DNA is mostly localized to the nucleus. RNA and DNA can be distinguished using 13C-15N correlation spectroscopy. The 13C chemical shift of the C1 carbon of the ribose in RNA molecules is 8 ppm greater than that of the C1 carbon of the deoxyribose in DNA molecules and the 15N chemical shift of the N9 nitrogen of the pyrimidine is ~20 ppm greater than that of the N1 nitrogen of the purine (Figure 6). Thus, to determine if AMUPol can enter membrane bound organelles, we collected DNP-enhanced 2D 13C-15N correlation spectra (TEDOR)55 on lysed and intact HEK293 cells that were either incubated or electroporated with AMUPol and examined the deoxyribose-purine and pyrimidine regions. We observed strong DNA signals in all the samples (Figure 6), indicating that AMUPol was able to enter the nucleus in intact viable cells.
Figure 6:
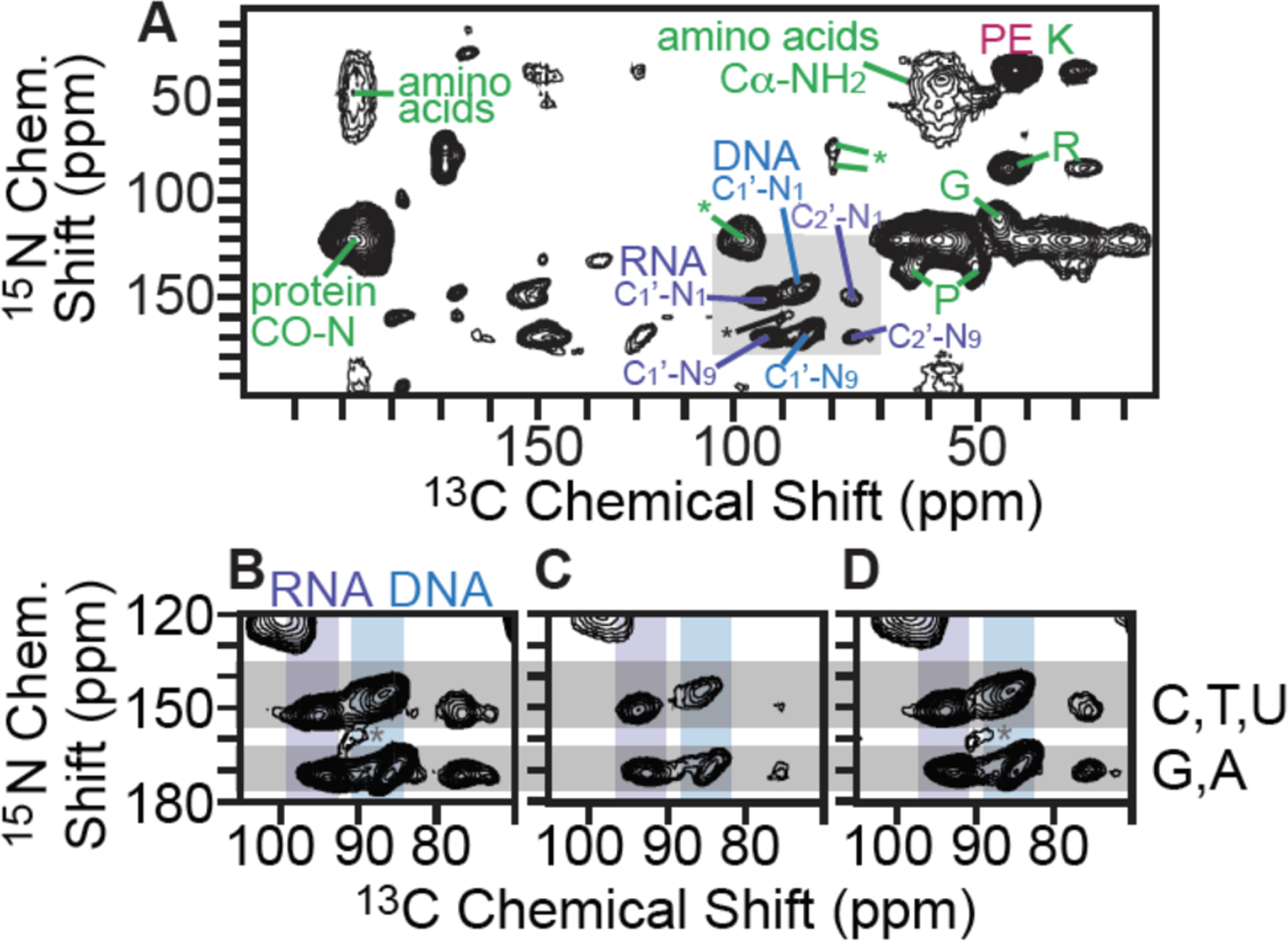
AMUPol freely enters the nucleus. A) 2D heteronuclear correlation spectra (TEDOR) of intact cells electroporation in the presence of 20 mM AMUPol. Selected 13C-15N correlations from the protein back bone and sidechains (green), from RNA (purple), from DNA (blue) and from lipid (pink) are annotated and the signal to noise ratios are reported in Table S1. Signals from RNA (purple bar) and DNA (blue bar) are resolved in the 2D TEDOR spectra (gray box, figure A) and are expanded to show details for B) lysed cells with 10 mM AMUPol, C) intact cells incubated with 30 mM AMUPol and D) intact cells electroporated in the presence of 20 mM AMUPol. Chemical shifts for pyrimidines and purines are indicated by the upper and lower gray bars, respectively. Spinning side bands are annotated by *. Full views and additional comparison of the data in B, C and D are available in Figure S6, Figure S7, Figure S8 and Table S1.
To further distinguish the cytosolic and nucleic locations of AMUPol, we compared the normalized peak intensities of the cytosolic and nucleic components, for the lysed sample to intact samples that were either incubated or electroporated with AMUPol. The TEDOR peak intensities were normalized to either the ribose-purine peak of RNA or DNA for each sample to control for differences in DNP-enhancements and cross-polarization efficiencies. TEDOR spectra have distinct peaks for DNA, RNA, protein backbone sites, protein side chain moieties, and free amino acids (Figure S6 and S7). DNA is located only in the nucleus, while RNA, proteins and free amino acids are entirely or largely cytoplasmically localized (i.e. more than 80% of the protein content of a cell is non-nuclearly localized).56 In addition to the RNA and DNA ribose purine and pyrimidine peaks, we determined peak intensities for the amide-carbonyl and amide-Cα sites for both proteins and free amino acids as well as the carbon-nitrogen bonds in the protein side chains of arginine and glycine (Table S1). When the intensity of the amino acids peaks are compared to the ribose-purine peak of RNA all samples had similar ratios (p > 0.08). This indicated that the cytoplasmic distribution of AMUPol is similar for all of these samples. When the intensity of the amino acid and RNA peaks are compared to the deoxyribose-purine peak of DNA, we found that lysed and electroporated samples have indistinguishable peak intensity ratios (p = 0.22). However, the DNA peaks for intact cells incubated with AMUPol were less intense than expected; the ratios of peak intensities for cytoplasmic to nuclear components were larger by 60% ± 50% (p < 0.05); (Table S1). This suggested that the AMUPol concentration in the nucleus of cells incubated with AMUPol is lower than the concentration of AMUPol in the cytoplasm. Because DNP enhancements for cells incubated with AMUPol are independent of room temperature incubation times despite the time-dependent reduction of AMUPol by cellular biomass and these samples have heterogenous AMUPol distribution, this suggests that there is an AMUPol concentration gradient inside intact cells incubated with AMUPol. Despite the heterogeneity of the AMUPol distribution for incubated cells, however, AMUPol is able to enter the nucleus. Taken together, examination of the peak intensity ratios for the different biomass components indicates that AMUPol is able to transverse the nuclear membrane of intact cells and effectively polarizes biomolecules localized in both the cytoplasm and the nucleus.
Sensitivity enhancements from DNP enable detection of a minority component in experimentally tractable acquisition times.
To explore the sensitivity of DNP MAS NMR, we examined the signal to noise ratio (SNR) for different biomass peaks from DNP-enhanced 2D heteronuclear (TEDOR) and homonuclear (DARR) correlation spectra for lysed and intact cells that had been either incubated or electroporated with AMUPol. While RNA and DNA together comprise ~6% of the cellular biomass, DNA alone makes up a fifth of the total nucleotide content. DNA comprises ~1.3% of the biomass of HEK293 cells, half of which is pyrimidines.47 When cells were prepared without AMUPol, the peak that corresponds to the deoxyribose-pyrimidine site in DNA had an SNR of 5 after 18 hours of signal averaging. In contrast, after only two hours of signal averaging, we found that this peak for lysed HEK cells with 10 mM AMUPol had an SNR of 200, while that of intact cells incubated with 30 mM AMUPol had an SNR of 40 and that of intact cells electroporated with 20 mM AMUPol had an SNR of 110 (Figure 6 and S7). In 2D homonuclear correlation spectra, without DNP, the peak for the deoxyribose C4-C5 site had an SNR of 10 after 25 hours of signal averaging. In contrast, after only 8 hours of signal averaging, we found that this peak for lysed HEK cells with 10 mM AMUPol had an SNR of 27, while that of intact cells incubated with 30 mM AMUPol had an SNR of 18 and that of intact cells electroporated with 20 mM AMUPol had an SNR of 24. As expected, the SNRs were larger for samples with higher DNP enhancements. Comparison of the SNR of these two-dimensional spectra collected without DNP to those collect with DNP reveal a time savings of three to five orders of magnitude. Overall, DNP-enhanced MAS NMR enabled detection of a component by both 2D homo and heteronuclear spectroscopy that comprises under 0.6% of the cellular biomass in a few hours with high signal to noise.
DISCUSSION
NMR has the resolution and specificity to determine atomic-level protein structures of isotopically-labeled proteins in complex environments7 and, with the sensitivity gains conferred by dynamic nuclear polarization (DNP), NMR has the sensitivity to detect proteins at their endogenous concentrations1. However, just as for other structural biology techniques, DNP sensitivity enhancements are critically dependent on experimental conditions and sample composition.37 While some of these conditions are at least theoretically compatible with cellular viability, the effects of others on cellular sample integrity, like magic angle spinning and exposure to polarization agents, are unknown. Uncertainty about the integrity of cellular samples complicates interpretation of the data of in-cell experiments, limiting the utility of that information. We establish that sample conditions that favor efficient DNP enhancements are compatible with cellular viability. Using several measures of sample viability, we find that the rate of temperature change during freezing has the largest effect. With this in mind, we established methods that maintain cellular viability throughout the DNP NMR experiments. This allows for sample validation both before and after structural analysis. We also find that the polarization agent, AMUPol, does not affect cellular viability. Using electroporation to deliver AMUPol to cells, we find that AMUPol is distributed homogenously throughout the cytoplasm and the membrane-bound nucleus. Finally, we find that the magnitude of the sensitivity enhancements is high enough to enable detection of a protein at micromolar concentrations in experimentally tractable experimental times. We establish an approach for DNP NMR on viable cells that can be phenotyped before and after experiments to eliminate uncertainty about sample integrity.
The approach to sample preparation described in this work is likely to be easily generalized to other cultured mammalian cell lines. The freezing rate had the largest influence on post-experiment sample viability. Indeed, while protocols for cryopreservation of mammalian cells differ in the composition of the freezing media57, most use a standard freezing rate of 1 °C per minute44. Here, we eliminated carbon and nitrogen-containing components of the freezing media because doing so will facilitate spectral interpretation for samples where the target molecule is at concentrations that are low enough that signals from natural abundance components make a significant contribution to the spectra.27 However, DNP efficiency is unlikely to be compromised by nutrient-rich components of the freezing media since cell lysates and other complex mixtures of biomolecules support efficient DNP.1, 27, 39 Thus, as long as samples are frozen slowly and cryogenically transferred into a pre-cooled spectrometer43, the freezing media can be tailored to the cell line.
The method used to introduce the polarization agent affects the experimental output and therefore must be chosen to address the structural question under consideration. In-cell structural biology enables the study of protein conformation in environments that maintain the identity, stoichiometry, concentrations and organization of the myriad of biomolecules that can interact with a protein of interest.1, 7, 18, 58 NMR is uniquely suited to study proteins in these complicated contexts with atomic level resolution. However, choice of approach and interpretation of the results requires consideration of the potential sources of bias. Solution state NMR experiments are biased towards the observation of mobile components, although interactions that transiently modify rotational correlation times can be inferred.58 MAS NMR of frozen samples are not biased by rotational correlation times but lack experimental sensitivity necessary to observe molecules at their endogenous concentrations. The sensitivity enhancements from DNP rely upon proximity to a polarization agent.27 Thus, DNP-enhanced MAS NMR experiments are biased towards observation of molecules that are accessible to polarization agents. In this work, we characterized samples prepared in three different ways; intact cells where AMUPol was introduced by incubation, lysed cells, and intact cells where AMUPol was introduced by electroporation. For cells incubated with AMUPol, a minority of the AMUPol enters the cell; the peak intensities for DNA are lower than expected and the TB,on fits indicate that the AMUPol concentration is heterogenous. While the identity, stoichiometry, concentrations and organization of the cellular components for cells incubated with AMUPol are all maintained, the experimental read-out is spatially biased. Thus, data from experiments performed on intact cells incubated with AMUPol are qualitative rather than quantitative. While any observed conformation inside cells incubated with AMUPol exists, the relative populations of different conformations cannot be inferred from peak intensities. Lysing cells results in a homogenous distribution of the polarization agent throughout the sample. For lysed cells, the identity and stoichiometry of the sample components are maintained, however lysing results in a small decrease in concentration of cellular components and loss of organization. Because the distribution of AMUPol is homogenous, the data from experiments on lysed cells are quantitative but the decrease of the concentrations and loss of organization of the environment upon lysis may alter the conformational ensemble. Introduction of the polarization agent into cells by electroporation may best address questions that require quantitative information about the structural ensemble for proteins that are potentially sensitive to not only stoichiometry and identity but also to the concentration and organization of the cellular components21, 59–62 Introduction of AMUPol via electroporation results in a homogenous distribution of the polarization agent throughout the cell. Using the electroporation approach for AMUPol delivery, the cellular organization is maintained and cellular propagation is unaltered. While transient permeabilization of the plasma membrane could potentially alter the identity, stoichiometry and/or concentration of cellular components, phenotyping before and after experiments eliminates uncertainty about sample integrity. Thus, structural information from experiments on electroporated cells will report quantitatively about the ensemble with minimal or no perturbation to the identity, concentration, stoichiometry or organization of the cellular constituents. For mixed populations of cells, either as a result of an unavoidable decrease in cellular integrity or because of standing biological heterogeneity in the system, the structural ensemble will provide quantitative information about the ensemble in samples where the polarization is homogenously dispersed. On the other hand, in the case of heterogenous distribution of the polarization agent, the structural information about the structural ensemble will provide only qualitative rather than quantitative information. Here we described two, of many potential approaches, to deliver polarization agents to intact cells. Indeed, preliminary work in our group using compounds that permeate cellular membranes, like DMSO, indicates that these agents result in homogenous dispersion. Ultimately, choice of methodology to introduce polarization agents therefore depends upon the structural question to be addressed.
DNP efficiencies for intact, viable samples are sufficient to collect data for proteins at their endogenous levels inside cells. Robust protocols describe successful delivery of endogenous concentrations isotopically enriched proteins into mammalian cells via electroporation.6, 21, 58 Although in cases where this technique is not suited, there are other delivery options.8, 17, 63 Because the workflow described here does not destroy the sample, the cellular localization of the isotopically-enriched proteins can be assessed immediately before and after the NMR experiment in either live cells, if the molecule carries a fluorophore, or fixed, stained cells. The quality and quantity of the target (or any other) protein can be assessed by orthogonal methods, like western blotting6, 12 and any perturbation that result from protein delivery can be controlled for in quantitative regrowth assays. In this work, because DNA is spectrally resolved from other biomass components, we could specifically detect DNA, a component that comprises less than 1% of the biomass, with high signal to noise ratios in short acquisition time. The endogenous concentration of most proteins is two to three orders of magnitude lower than DNA. However, using DNA as a benchmark suggests that a protein at a micromolar concentrations could be detected by two-dimensional spectroscopy with a SNR of ~10 in experimental acquisition times of a few days to a week. These experimental times and SNR are similar to prior reports of proteins at micromolar concentrations in complicated biological mixtures using DNP-enhanced MAS NMR.1, 21, 27 Thus, the magnitude of the DNP sensitivity enhancements in preparations of intact viable cells is sufficient to enable detection of a protein at micromolar concentrations in experimentally tractable experimental times.
A concern that plagues structural biology is that the manipulations that permit structural characterization may also alter molecular structure. Such alternations are of particular concern in cases for proteins whose conformations are environmentally sensitive, like those that can adopt more than one stable structure or contain regions that are intrinsically disordered in some settings. If cells survive structural characterization, then biological phenotypes can be assessed both before and after DNP MAS NMR. If the manipulations do not affect phenotype, then the observed structural ensembles will report on the biologically relevant conformations. Here we describe a generalizable approach for in-cell structural biology that allows both pre- and post-experimental sample validation. Such studies could significantly expand our understanding of protein conformation changes that influence cellular fate.
Materials and Methods
Sample preparation
To reduce experimental acquisition times, we uniformly isotopically labeled HEK293 cells by culturing them in isotopically-enriched media. Human embryonic kidney 293 (HEK293) cells were cultured in 13C, 15N labelled media (BioExpress 6000 Mammalian U-13C, 98%; U-15N, 98%, Cambridge Isotope Laboratories, USA) with 10% (v/v) fetal bovine serum (FBS, qualified, Gibco) and 1% (v/v) PenStrep (Gibco) at 37 °C and 5% CO2. Confluent plates were harvested using Tryp-LE Express (Gibco) and BioExpress 6000 media, transferred to 15 mL conical tube and centrifuged at 233 × g for 5 min at 22 °C using a swinging bucket rotor (Beckman Coulter). Pelleted cells were resuspended and washed once with 1x PBS (−CaCl2, −MgCl2, pH 7.4, Gibco). AMUPol was delivered to cells in two ways: incubation and electroporation. For delivery by incubation, a 50 μL cell pellet was mixed with 50 μL perdeuterated 1x PBS (85% D2O + 10% H2O, pH 7.4) containing AMUPol (Cortecnet, USA) and 18 μL of d8-glycerol. The 118 μL cell suspension had a final composition of 15% (v/v) d8-glycerol, 75% (v/v) D2O and 10% (v/v) H2O. For delivery by electroporation, a 50 μL cell pellet was mixed with 100 μL electroporation buffer (SF cell line solution, Lonza) containing AMUPol and electroporated (HEK293 pulse sequence, Lonza 4D-nucleofactor) using manufacturer’s instructions. Post electroporation, cells were allowed to recover for 10 minutes in electroporation buffer containing AMUPol inside the tissue culture hood. Next, cells were washed twice with 50–100 μL (depending on cell pellet volume) of 1x PBS to eliminate the electroporation buffer and any extracellular AMUPol from the sample and the 50 μL cell pellet was resuspended in perdeuterated 1x PBS and d8-glycerol for a final composition of 15% (v/v) d8-glycerol, 75% (v/v) D2O and 10% (v/v) H2O.
In case of intact cells, after delivery of AMUPol, cells were transferred into 3.2 mm sapphire rotor (Bruker) by centrifugation in a swinging bucket rotor at 100 × g for 2 minutes at 22 °C. The supernatant was removed, and rotors were frozen at a controlled rate (–1°C/min) in ‘Cool Cell LX’ (Corning) in the −80 °C freezer for 12–16 hours. Finally, frozen rotors were transferred to liquid nitrogen storage until measurement by DNP NMR. In case of lysates, after AMUPol delivery by incubation, cells were transferred into 3.2 mm sapphire rotor (Bruker) by centrifugation in a swinging bucket rotor at 100 × g for 2 minutes at 22 °C. The rotors were then sealed and the intact cells were lysed inside the rotor by flash freezing in liquid nitrogen and stored till DNP NMR. During sample insert, rotors were taken out of liquid nitrogen, thawed to room temperature and then inserted into pre-cooled spectrometer at 100 K (adding one additional freeze thaw cycle to the sample handling protocol).
Trypan Blue exclusion assay
Pelleted cells (10 μL) were diluted into 100 μL unlabeled DMEM and 10 μL of this cell suspension were mixed with 10 μL of Trypan Blue (0.4% solution). 10 μL of the Trypan Blue cell suspension was loaded onto Countess Chamber. Trypan blue membrane permeability was assessed using Countess automated cell counter (Life technologies) using the manufacturer’s protocol.
Growth assay
Equal number of cells (0.1 million cells) were plated in 10 cm dish containing complete media (DMEM) and grown for 9–14 days (as indicated before). After cells have settled down (post 8–10 hours), media was removed to get rid of floating dead cells. 10–12 mL of DMEM is added to the 10 cm culture dish and cell growth is monitored using inverted light microscope till 100% confluency. Fitting of sigmoidal curves was performed with an equation of , where y(t) denotes the cell culture time t, a and k are fitting parameters, and t0 defines a lag time of tL as tL = t0 −2/k.64 The error range for the fitting was estimated at the 95% confidence level.
Microscopy
HEK293 cells were grown to confluency in 10 cm dish and harvested. Cells were washed with 1x PBS twice and centrifuged at 233 × g for 5 minutes. 50 μL of cell pellet was resuspended in 50 μL of 1x perdeuterated PBS containing 10 mM AMUPol with 15% (v/v) d8-glycerol. Next, the cells were centrifuged as mentioned above and the supernatant was discarded. The cell pellet was then either prepared for imaging or slow frozen (1 °C/min) and then prepared for imaging. Before freezing sample: Cell pellet was mixed with warm complete media (DMEM with 10% FBS and 1% Pen-Strep), centrifuged (233 × g, 5 min) and supernatant was discarded. After freezing samples: Frozen cell pellets were thawed for 2 min in a 37 °C water bath, resuspended in warm complete media (DMEM with 10% FBS and 1% Pen-Strep), centrifuged (233 × g, 5 min) and supernatant was discarded. The cell pellets were resuspended in 250 μL of PBS containing 4% paraformaldehyde for 30 min to fix the cells. Excess paraformaldehyde was quenched with 0.1 M glycine in PBS for 5 min. 300 μL of 0.1% Triton X-100 (in PBS) was added to the cell pellet and resuspended for 3–5 min to increase permeability followed by 1x PBS wash (3 times). Phalloidin solution [1x Phalloidin (ab176753, Abcam) + 1% BSA in PBS] with Hoechst 33342 (Invitrogen, R37605) was added to the cells and they were incubated at room temperature for 60 minutes. Cells were pelleted and rinsed 2–3 times with 1x PBS, 5 min per wash. Cell pellets were resuspended in 100 μL of PBS. 5 μL of the resuspension solution was dropped on glass slide and imaged using confocal microscopy (Zeiss LSM780 inverted). Images were analyzed with Zen Black and Fiji software (NIH).
Cold insert
Rotors were transferred in liquid nitrogen directly into the NMR probe that was pre-equilibrated at 100 K43.
DNP NMR spectroscopy
All dynamic nuclear polarization magic angle spinning nuclear magnetic resonance (DNP MAS NMR) experiments were performed on a 600 MHz Bruker Ascend DNP NMR spectrometer/7.2 T Cryogen-free gyrotron magnet (Bruker), equipped with a 1H, 13C, 15N triple-resonance, 3.2 mm low temperature (LT) DNP MAS NMR Bruker probe (600 MHz). The sample temperature was 104 K and the MAS frequency was 12 kHz. The DNP enhancement for the instrumentation setup for a standard sample of 1.5 mg of uniformly 13C, 15N labeled proline (Isotech) suspended in 25 mg of 60:30:10 d8-glycerol:D2O:H2O containing 10 mM AMUPol was between 130 and 140 and a TB,on of 4.6 s. For 13C cross-polarization (CP) MAS experiments, the 13C radio frequency (RF) amplitude was linearly swept from 75 kHz to 37.5 kHz with an average of 56.25 kHz. 1H RF amplitude was 68~72 kHz for CP, 83 kHz for 90 degree pulse, and 85 kHz for 1H TPPM decoupling with phase alternation of ± 15° during acquisition of 13C signal. The DNP enhancements were determined by comparing 1D 13C CP spectra collected with and without microwaves irradiation. For TB,on measurements, recycle delays ranged from 0.1 s to 300 s. To determine the TB,on, the dependence of the recycle delay using saturation recovery on both 13C peak intensity or volume was fit to the mono-exponential equation and the stretched-exponential equation , respectively.
13C-13C 2D correlations were measured using 20 ms DARR mixing with the 1H amplitude at the MAS frequency. A total of 280 complex t1 points with increment of 25 μs were recorded. For 13C-15N 1D and 2D correlations, a 24 rotor periods (2 ms) TEDOR sequence was applied with 13C and 15N pulse trains at 55.5 kHz and 41.7 kHz, respectively. A total of 64 complex t1 points with an increment of 80 μs were recorded. The recycle delay was 3.9 s and the same 1H decoupling was applied. The experimental time required to collect a 2D TEDOR spectra with 32 scans was 2 h and to collect a 2D DARR of 16 scans was 5 h.
DNP NMR Data analysis\
For 1D experiments, the data were processed using NMRPipe65. The real part of the processed spectrum was exported using pipe2txt.tcl command. Peaks were integrated, and the time constants were obtained by least-squares fitting with a single-exponential function. To obtain the error estimates for DNP enhancements, the DNP enhancements were measured on three independent sample preparations for each time point. The reported errors were calculated by pooling variances across all incubation time points. DNP enhancements were determined by peak intensity. For 2D experiments, the TEDOR and DARR data were both apodized with a Lorenz-to-Gauss window function with IEN of 15 Hz and GB of 75 Hz in the t1 and t2 time domains. The noise level and peak height from the 2D NMR spectrum was detected by the NMRDraw software for S/N estimation. Experimental data are available upon request.
Quantification of the biomass components
Each biomass component is represented by a characteristic chemical moiety. We quantified the biomass components by integrating the peaks for specific chemical moieties in the quantitative 13C-direct polarization (DP) spectra (Figure 2B, colored carbon atoms). However, the peak integration reports only the quantity of carbons in those specific moieties, not the quantity of the entire molecule. To convert the peak intensity to biomass quantity, we determined the average molecular weight for each biomass component and divided it by the weight of the number of carbons with the quantitated moiety present in the biomass component. We calculated conversion factors for protein, nucleotides and lipids. The identity, molecular weights, and number of moiety carbons for individual amino acids, nucleotides and lipids66 that we used to determine the conversion factor, as well as the exact integration windows, are listed in Table S3. The conversion factor for protein was 10.8, for nucleotides was 11.7 and for lipids was 3.12. The integrated peak intensities were multiplied by their respective conversion factor to calculate the biomass quantity. Some chemical moieties, like carbonyls, are found in both in proteins as well as in lipid head group. Using the literature abundance of the different biomass component we estimated the contribution of lipid head groups could be up to 7% of the “protein” peak and the contribution of aliphatic side chains could be up to 22% of the “lipid” peak47.
Supplementary Material
Acknowledgements:
This work was supported by grants from the National Institute of Health (NS-111236), the Welch Foundation (I-1923_20170325, the Lupe Murchison Foundation and the Kinship Foundation (Searle Scholars Program) to K.K.F. The authors would like to acknowledge the assistance of the UT Southwestern Live Cell Imaging Facility, a Shared Resource of the Harold C. Simmons Cancer Center, supported in part by an NCI Cancer Center Support Grant, 1P30 CA142543-01
Footnotes
Competing Financial interests:
The authors declare no competing financial interests.
Supporting Information
The Supporting Information is available free of charge at “link”.
Cellular viability results for different DNP matrix compositions, Fluorescence microscopy images of cells before and after freezing. Results for protein, nucleotides and lipids for; the dependence of DNP enhancement on [AMUPol], the TB,on dependance on [AMUPol], the TB,on fitting error analysis, the AMUPol deactivation by cellular components. Two dimensional spectra for lysed cells and for cells where AMUPol was introduced by incubation or electroporation, 1D slices from these spectra plotted to compare SNR across preparations.
Supplementary tables summarize SNR of selected peaks from the 2D spectra, DNP enhancement, buildup times and fitting parameters for all samples, and values used for biomass quantification calculations.
Classification
Biophysics and Structural Biology
Reference
- 1.Frederick KK; Michaelis VK; Corzilius B; Ong T-C; Jacavone AC; Griffin RG; Lindquist S, Sensitivity-enhanced NMR reveals alterations in protein structure by cellular milieus. Cell 2015, 163 (3), 620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banci L; Barbieri L; Bertini I; Luchinat E; Secci E; Zhao Y; Aricescu AR, Atomic-resolution monitoring of protein maturation in live human cells by NMR. Nature chemical biology 2013, 9 (5), 297–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Freedberg DI; Selenko P, Live cell NMR. Annual Review of Biophysics 2014, 43, 171–192. [DOI] [PubMed] [Google Scholar]
- 4.Sakakibara D; Sasaki A; Ikeya T; Hamatsu J; Hanashima T; Mishima M; Yoshimasu M; Hayashi N; Mikawa T; Wälchli M, Protein structure determination in living cells by in-cell NMR spectroscopy. Nature 2009, 458 (7234), 102–105. [DOI] [PubMed] [Google Scholar]
- 5.Selenko P; Serber Z; Gadea B; Ruderman J; Wagner G, Quantitative NMR analysis of the protein G B1 domain in Xenopus laevis egg extracts and intact oocytes. Proceedings of the National Academy of Sciences 2006, 103 (32), 11904–11909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Theillet F-X; Rose HM; Liokatis S; Binolfi A; Thongwichian R; Stuiver M; Selenko P, Site-specific NMR mapping and time-resolved monitoring of serine and threonine phosphorylation in reconstituted kinase reactions and mammalian cell extracts. Nature protocols 2013, 8 (7), 1416. [DOI] [PubMed] [Google Scholar]
- 7.Burmann BM; Gerez JA; Matečko-Burmann I; Campioni S; Kumari P; Ghosh D; Mazur A; Aspholm EE; Šulskis D; Wawrzyniuk M, Regulation of α-synuclein by chaperones in mammalian cells. Nature 2020, 577 (7788), 127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inomata K; Ohno A; Tochio H; Isogai S; Tenno T; Nakase I; Takeuchi T; Futaki S; Ito Y; Hiroaki H, High-resolution multi-dimensional NMR spectroscopy of proteins in human cells. Nature 2009, 458 (7234), 106–109. [DOI] [PubMed] [Google Scholar]
- 9.Majumder S; Xue J; DeMott CM; Reverdatto S; Burz DS; Shekhtman A, Probing protein quinary interactions by in-cell nuclear magnetic resonance spectroscopy. Biochemistry 2015, 54 (17), 2727–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waudby CA; Camilloni C; Fitzpatrick AW; Cabrita LD; Dobson CM; Vendruscolo M; Christodoulou J, In-cell NMR characterization of the secondary structure populations of a disordered conformation of α-synuclein within E. coli cells. PloS one 2013, 8 (8), e72286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Monteith WB; Pielak GJ, Residue level quantification of protein stability in living cells. Proceedings of the National Academy of Sciences 2014, 111 (31), 11335–11340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Theillet F-X; Binolfi A; Bekei B; Martorana A; Rose HM; Stuiver M; Verzini S; Lorenz D; van Rossum M; Goldfarb D; Selenko P, Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 2016, 530 (7588), 45–50. [DOI] [PubMed] [Google Scholar]
- 13.Pielak GJ; Li C; Miklos AC; Schlesinger AP; Slade KM; Wang G-F; Zigoneanu IG, Protein nuclear magnetic resonance under physiological conditions. Biochemistry 2009, 48 (2), 226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bertrand K; Reverdatto S; Burz DS; Zitomer R; Shekhtman A, Structure of proteins in eukaryotic compartments. Journal of the American Chemical Society 2012, 134 (30), 12798–12806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamatsu J; O’Donovan D; Tanaka T; Shirai T; Hourai Y; Mikawa T; Ikeya T; Mishima M; Boucher W; Smith BO, High-resolution heteronuclear multidimensional NMR of proteins in living insect cells using a baculovirus protein expression system. Journal of the American Chemical Society 2013, 135 (5), 1688–1691. [DOI] [PubMed] [Google Scholar]
- 16.Banci L; Barbieri L; Luchinat E; Secci E, Visualization of redox-controlled protein fold in living cells. Chemistry & biology 2013, 20 (6), 747–752. [DOI] [PubMed] [Google Scholar]
- 17.Luchinat E; Banci L, In-cell NMR in human cells: Direct protein expression allows structural studies of protein folding and maturation. Accounts of chemical research 2018, 51 (6), 1550–1557. [DOI] [PubMed] [Google Scholar]
- 18.Luchinat E; Barbieri L; Campbell TF; Banci L, Real-Time Quantitative In-Cell NMR: Ligand Binding and Protein Oxidation Monitored in Human Cells Using Multivariate Curve Resolution. Analytical chemistry 2020, 92 (14), 9997–10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hänsel R; Luh LM; Corbeski I; Trantirek L; Dötsch V, In-cell NMR and EPR spectroscopy of biomacromolecules. Angewandte Chemie International Edition 2014, 53 (39), 10300–10314. [DOI] [PubMed] [Google Scholar]
- 20.Bodart J-F; Wieruszeski J-M; Amniai L; Leroy A; Landrieu I; Rousseau-Lescuyer A; Vilain J-P; Lippens G, NMR observation of Tau in Xenopus oocytes. Journal of magnetic resonance 2008, 192 (2), 252–257. [DOI] [PubMed] [Google Scholar]
- 21.Narasimhan S; Scherpe S; Lucini Paioni A; Van Der Zwan J; Folkers GE; Ovaa H; Baldus M, DNP-Supported Solid-State NMR Spectroscopy of Proteins Inside Mammalian Cells. Angewandte Chemie 2019, 131 (37), 13103–13107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Albert BJ; Gao C; Sesti EL; Saliba EP; Alaniva N; Scott FJ; Sigurdsson ST; Barnes AB, Dynamic nuclear polarization nuclear magnetic resonance in human cells using fluorescent polarizing agents. Biochemistry 2018, 57 (31), 4741–4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta R; Lu M; Hou G; Caporini MA; Rosay M; Maas W; Struppe J; Suiter C; Ahn J; Byeon I-JL, Dynamic nuclear polarization enhanced MAS NMR spectroscopy for structural analysis of HIV-1 protein assemblies. The Journal of Physical Chemistry B 2016, 120 (2), 329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaplan M; Narasimhan S; de Heus C; Mance D; van Doorn S; Houben K; Popov-Čeleketić D; Damman R; Katrukha EA; Jain P, EGFR dynamics change during activation in native membranes as revealed by NMR. Cell 2016, 167 (5), 1241–1251. e11. [DOI] [PubMed] [Google Scholar]
- 25.Qiang W; Yau W-M; Lu J-X; Collinge J; Tycko R, Structural variation in amyloid-β fibrils from Alzheimer’s disease clinical subtypes. Nature 2017, 541 (7636), 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scherpelz KP; Wang S; Pytel P; Madhurapantula RS; Srivastava AK; Sachleben JR; Orgel J; Ishii Y; Meredith SC, Atomic-level differences between brain parenchymal- and cerebrovascular-seeded Aβ fibrils. Scientific Reports 2021, 11 (1), 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Costello WN; Xiao Y; Frederick KK, DNP-Assisted NMR Investigation of Proteins at Endogenous Levels in Cellular Milieu. Methods in enzymology 2019, 615, 373–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schlagnitweit J; Friebe Sandoz S; Jaworski A; Guzzetti I; Aussenac F; Carbajo RJ; Chiarparin E; Pell AJ; Petzold K, Observing an Antisense Drug Complex in Intact Human Cells by in-Cell NMR Spectroscopy. ChemBioChem 2019, 20 (19), 2474–2478. [DOI] [PubMed] [Google Scholar]
- 29.Renault M; Tommassen-van Boxtel R; Bos MP; Post JA; Tommassen J; Baldus M, Cellular solid-state nuclear magnetic resonance spectroscopy. Proceedings of the National Academy of Sciences 2012, 109 (13), 4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Akbey Ü; Franks WT; Linden A; Lange S; Griffin RG; van Rossum BJ; Oschkinat H, Dynamic nuclear polarization of deuterated proteins. Angewandte Chemie International Edition 2010, 49 (42), 7803–7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takahashi H; Fernández-de-Alba C; Lee D; Maurel V; Gambarelli S; Bardet M; Hediger S; Barra A-L; De Paëpe G, Optimization of an absolute sensitivity in a glassy matrix during DNP-enhanced multidimensional solid-state NMR experiments. Journal of Magnetic Resonance 2014, 239, 91–99. [DOI] [PubMed] [Google Scholar]
- 32.Lee M; Hong M, Cryoprotection of lipid membranes for high-resolution solid-state NMR studies of membrane peptides and proteins at low temperature. Journal of biomolecular NMR 2014, 59 (4), 263–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tran NT; Mentink-Vigier F; Long JR, Dynamic Nuclear Polarization of Biomembrane Assemblies. Biomolecules 2020, 10 (9), 1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sauvée C; Rosay M; Casano G; Aussenac F; Weber RT; Ouari O; Tordo P, Highly efficient, water-soluble polarizing agents for dynamic nuclear polarization at high frequency. Angewandte Chemie 2013, 125 (41), 11058–11061. [DOI] [PubMed] [Google Scholar]
- 35.Lund A; Casano G; Menzildjian G; Kaushik M; Stevanato G; Yulikov M; Jabbour R; Wisser D; Renom-Carrasco M; Thieuleux C, TinyPols: a family of water-soluble binitroxides tailored for dynamic nuclear polarization enhanced NMR spectroscopy at 18.8 and 21.1 T. Chemical Science 2020, 11 (10), 2810–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stevanato G; Casano G; Kubicki DJ; Rao Y; Esteban Hofer L; Menzildjian G; Karoui H; Siri D; Cordova M; Yulikov M, Open and closed radicals: local geometry around unpaired electrons governs magic-angle spinning dynamic nuclear polarization performance. Journal of the American Chemical Society 2020, 142 (39), 16587–16599. [DOI] [PubMed] [Google Scholar]
- 37.Ni QZ; Daviso E; Can TV; Markhasin E; Jawla SK; Swager TM; Temkin RJ;Herzfeld J; Griffin RG, High frequency dynamic nuclear polarization. Accounts of chemical research 2013, 46 (9), 1933–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Akbey Ü; Oschkinat H, Structural biology applications of solid state MAS DNP NMR. journal of magnetic resonance 2016, 269, 213–224. [DOI] [PubMed] [Google Scholar]
- 39.Narasimhan S; Pinto C; Paioni AL; van der Zwan J; Folkers GE; Baldus M, Characterizing proteins in a native bacterial environment using solid-state NMR spectroscopy. Nature Protocols 2021, 16 (2), 893–918. [DOI] [PubMed] [Google Scholar]
- 40.Jaudzems K; Bertarello A; Chaudhari SR; Pica A; Cala-De Paepe D; Barbet-Massin E; Pell AJ; Akopjana I; Kotelovica S; Gajan D, Dynamic Nuclear Polarization-Enhanced Biomolecular NMR Spectroscopy at High Magnetic Field with Fast Magic-Angle Spinning. Angewandte Chemie International Edition 2018, 57 (25), 7458–7462. [DOI] [PubMed] [Google Scholar]
- 41.Miyamoto Y; Ikeuchi M; Noguchi H; Hayashi S, Long-term Cryopreservation of Human and other Mammalian Cells at −80 °C for 8 Years. Cell Med 2018, 10, 2155179017733148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tennant JR, EVALUATION OF THE TRYPAN BLUE TECHNIQUE FOR DETERMINATION OF CELL VIABILITY. Transplantation 1964, 2 (6), 685–694. [DOI] [PubMed] [Google Scholar]
- 43.Ghosh R; Kragelj J; Xiao Y; Frederick KK, Cryogenic sample loading into a magic angle spinning nuclear magnetic resonance spectrometer that preserves cellular viability. Journal of Visualized Experiments 2020, (163), e61733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mazur P, Kinetics of water loss from cells at subzero temperatures and the likelihood of intracellular freezing. The Journal of general physiology 1963, 47 (2), 347–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang Y; Lee M; Fairn GD, Phospholipid subcellular localization and dynamics. Journal of Biological Chemistry 2018, 293 (17), 6230–6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Meer G; de Kroon AIPM, Lipid map of the mammalian cell. Journal of Cell Science 2011, 124 (1), 5–8. [DOI] [PubMed] [Google Scholar]
- 47.Dietmair S; Hodson MP; Quek L-E; Timmins NE; Gray P; Nielsen LK, A Multi-Omics Analysis of Recombinant Protein Production in Hek293 Cells. PLOS ONE 2012, 7 (8), e43394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pinon AC; Schlagnitweit J; Berruyer P; Rossini AJ; Lelli M; Socie E; Tang M; Pham T; Lesage A; Schantz S; Emsley L, Measuring Nano- to Microstructures from Relayed Dynamic Nuclear Polarization NMR. The Journal of Physical Chemistry C 2017, 121 (29), 15993–16005. [Google Scholar]
- 49.Zhao L; Pinon AC; Emsley L; Rossini AJ, DNP-enhanced solid-state NMR spectroscopy of active pharmaceutical ingredients. Magnetic Resonance in Chemistry 2018, 56 (7), 583–609. [DOI] [PubMed] [Google Scholar]
- 50.Berruyer P; Björgvinsdóttir S; Bertarello A; Stevanato G; Rao Y; Karthikeyan G; Casano G; Ouari O; Lelli M; Reiter C; Engelke F; Emsley L, Dynamic Nuclear Polarization Enhancement of 200 at 21.15 T Enabled by 65 kHz Magic Angle Spinning. The Journal of Physical Chemistry Letters 2020, 11 (19), 8386–8391. [DOI] [PubMed] [Google Scholar]
- 51.Sözer EB; Pocetti CF; Vernier PT, Transport of charged small molecules after electropermeabilization — drift and diffusion. BMC Biophysics 2018, 11 (1), 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Binolfi A; Limatola A; Verzini S; Kosten J; Theillet F-X; May Rose H; Bekei B; Stuiver M; van Rossum M; Selenko P, Intracellular repair of oxidation-damaged α-synuclein fails to target C-terminal modification sites. Nature Communications 2016, 7 (1), 10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zu Y; Huang S; Lu Y; Liu X; Wang S, Size Specific Transfection to Mammalian Cells by Micropillar Array Electroporation. Scientific Reports 2016, 6 (1), 38661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang S; Wang C; Lu J; Ma X; Liu Z; Li D; Liu Z; Liu C, In-Cell NMR Study of Tau and MARK2 Phosphorylated Tau. International Journal of Molecular Sciences 2019, 20 (1), 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jaroniec CP; Filip C; Griffin RG, 3D TEDOR NMR experiments for the simultaneous measurement of multiple carbon− nitrogen distances in uniformly 13C, 15N-labeled solids. Journal of the American Chemical Society 2002, 124 (36), 10728–10742. [DOI] [PubMed] [Google Scholar]
- 56.Shaiken TE; Opekun AR, Dissecting the cell to nucleus, perinucleus and cytosol. Scientific Reports 2014, 4 (1), 4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chaytor JL; Tokarew JM; Wu LK; Leclère M; Tam RY; Capicciotti CJ; Guolla L; von Moos E; Findlay CS; Allan DS; Ben RN, Inhibiting ice recrystallization and optimization of cell viability after cryopreservation. Glycobiology 2011, 22 (1), 123–133. [DOI] [PubMed] [Google Scholar]
- 58.Theillet F-X; Binolfi A; Bekei B; Martorana A; Rose HM; Stuiver M; Verzini S; Lorenz D; Van Rossum M; Goldfarb D, Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 2016, 530 (7588), 45–50. [DOI] [PubMed] [Google Scholar]
- 59.Laustsen A; Bak RO, Electroporation-Based CRISPR/Cas9 Gene Editing Using Cas9 Protein and Chemically Modified sgRNAs. In CRISPR Gene Editing: Methods and Protocols, Luo Y, Ed. Springer New York: New York, NY, 2019; pp 127–134. [DOI] [PubMed] [Google Scholar]
- 60.Pasquet L; Chabot S; Bellard E; Markelc B; Rols M-P; Reynes J-P; Tiraby G;Couillaud F; Teissie J; Golzio M, Safe and efficient novel approach for non-invasive gene electrotransfer to skin. Scientific Reports 2018, 8 (1), 16833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grys M; Madeja Z; Korohoda W, Avoiding the side effects of electric current pulse application to electroporated cells in disposable small volume cuvettes assures good cell survival. Cellular & Molecular Biology Letters 2017, 22 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.DiFranco M; Quinonez M; Capote J; Vergara J, DNA Transfection of Mammalian Skeletal Muscles using In Vivo Electroporation. JoVE 2009, (32), e1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ogino S; Kubo S; Umemoto R; Huang S; Nishida N; Shimada I, Observation of NMR Signals from Proteins Introduced into Living Mammalian Cells by Reversible Membrane Permeabilization Using a Pore-Forming Toxin, Streptolysin O. Journal of the American Chemical Society 2009, 131 (31), 10834–10835. [DOI] [PubMed] [Google Scholar]
- 64.Nielsen L; Khurana R; Coats A; Frokjaer S; Brange J; Vyas S; Uversky VN; Fink AL, Effect of environmental factors on the kinetics of insulin fibril formation: Elucidation of the molecular mechanism. Biochemistry 2001, 40 (20), 6036–6046. [DOI] [PubMed] [Google Scholar]
- 65.Delaglio F; Grzesiek S; Vuister GW; Zhu G; Pfeifer J; Bax A, Nmrpipe - a Multidimensional Spectral Processing System Based on Unix Pipes. Journal of Biomolecular Nmr 1995, 6 (3), 277–293. [DOI] [PubMed] [Google Scholar]
- 66.Dawaliby R; Trubbia C; Delporte C; Noyon C; Ruysschaert J-M; Van Antwerpen P; Govaerts C, Phosphatidylethanolamine is a key regulator of membrane fluidity in eukaryotic cells. Journal of Biological Chemistry 2016, 291 (7), 3658–3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.