

Abstract
Leber congenital amaurosis (LCA) is a severe congenital/early-onset retinal dystrophy. Given its monogenic nature and the immunological and anatomical privileges of the eye, LCA has been particularly targeted by cutting-edge research. In this review, we describe the current management of LCA, and highlight the clinical trials that are on-going and planned. RPE65-related LCA pivotal trials, which culminated in the first Food and Drug Administration-approved and European Medicines Agency-approved ocular gene therapy, have paved the way for a new era of genetic treatments in ophthalmology. At present, multiple clinical trials are available worldwide applying different techniques, aiming to achieve better outcomes and include more genes and variants. Genetic therapy is not only implementing gene supplementation by the use of adeno-associated viral vectors, but also clustered regularly interspaced short palindromic repeats (CRISPR)-mediated gene editing and post-transcriptional regulation through antisense oligonucleotides. Pharmacological approaches intending to decrease photoreceptor degeneration by supplementing 11-cis-retinal and cell therapy’s aim to replace the retinal pigment epithelium, providing a trophic and metabolic retinal structure, are also under investigation. Furthermore, optoelectric devices and optogenetics are also an option for patients with residual visual pathway. After more than 10 years since the first patient with LCA received gene therapy, we also discuss future challenges, such as the overlap between different techniques and the long-term durability of efficacy. The next 5 years are likely to be key to whether genetic therapies will achieve their full promise, and whether stem cell/cellular therapies will break through into clinical trial evaluation.
Keywords: genetics, retina, dystrophy, degeneration
Introduction
Leber congenital amaurosis (LCA) represents one of the severest diagnoses a family can receive regarding a child’s eyesight. It is characterised by early-onset visual impairment, nystagmus or roving eyes and severe photoreceptor dysfunction.1 LCA and early-onset severe retinal degeneration (EOSRD) belong to the same disease spectrum, with the latter being less severe; later onset of visual impairment, often better preserved visual acuity and decreased, but usually present electrophysiological responses.1 LCA/EOSRD affect approximately 1 in 80 000 children worldwide.2 Retinal examination can be unremarkable especially in the early stages or show mild signs such as retinal pigment epithelial mottling and vessel narrowing.3 Salt and pepper retinopathy, optic disc pallor and retinal pigment clumping (including nummular and bone spicule) usually appear in older individuals.
The diagnosis of LCA is clinical; by ophthalmological evaluation and electrophysiology. However, genetic testing is critical to confirm the specific diagnosis and is key towards the administration of genotype-specific treatment, inclusion in clinical trials, prognostication, as well as for family planning and tailored screening of systemic conditions.4 LCA can be an isolated eye condition or part of a syndrome, such as Senior Loken or Joubert syndrome.5 6 Moreover, it is genetically heterogeneous and can be caused by variants in at least 25 genes such as RPE65, GUCY2D, NMNAT1, CEP290, AIPL1, and RDH12.1 Some of these genotypes are associated with distinctive phenotypic features including, RDH12, CRX, CRB1 and NMNAT1 can present with early-onset macular atrophy,7 CEP290 may be associated with other ciliopathy manifestations,8 RPE65 may show absent or decreased autofluorescence.9 Furthermore, the pathophysiology and location where each faulty gene has its repercussion(s) is also different. AIPL1 is predominantly located in rod outer segments and reduces the intracellular concentration of cyclic guanosine monophosphate (cGMP).10 CRB1 is located in the cell membrane of Müller cells and has a role in protein scaffold.11 LRAT and RPE65 are located in retinal pigment epithelium (RPE) cells and are involved in the visual cycle.12 The retinal location of the proteins encoded by the genes that will be discussed in this review are depicted in figure 1.
Figure 1.
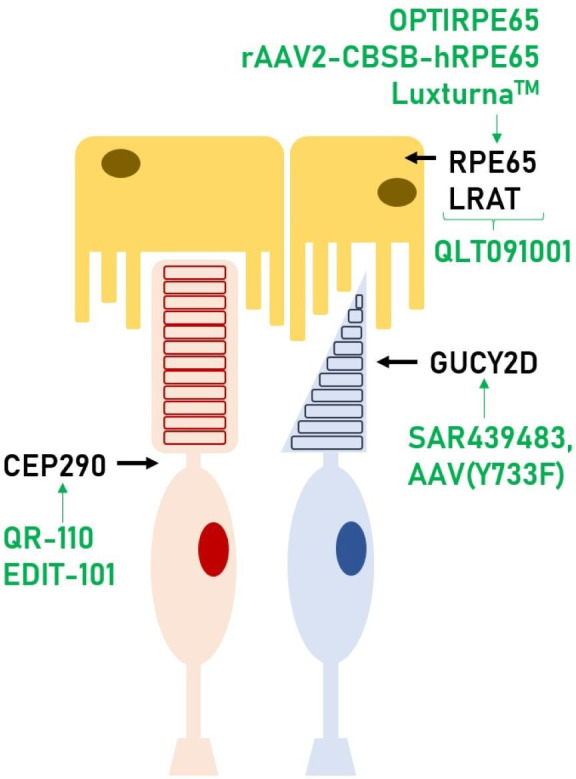
Photoreceptor (rod in red, cone in blue) and RPE cells. In this scheme, we can see where each gene discussed in this review has its function within the retina. These genes are present in both rods and cones, however, we depicted it showing the cell type where relative expression is highest. Also, in green, the therapies that are currently available or under investigation for those genes. RPE, retinal pigment epithelium.
Due to the severe visual impairment caused by LCA/EOSRD, the well-established genetic basis, availability of animal models and well-characterised clinical features, translational research has been particularly active. In this review, we aim to provide an update of the treatments that are currently available and those that are in clinical trial (table 1).
Table 1.
Summary of ongoing clinical trials targeting LCA/EOSRD
| Gene | Drug | Clinical trial ID (NCT #) | Study phase | Route of delivery | Status | Sponsor | Location | Number of participants |
| RPE65 | rAAV 2/2.hRPE65p.hRPE65–OPTIRPE65 | NCT00643747– NCT02781480 | Phase I–II | Subretinal | Completed | MeiraGTx | UK | 12–15 |
| RPE65 | rAAV2-CBSB–hRPE65 | NCT00481546 | Phase I | Subretinal | Active, not recruiting | University of Pennsylvania | USA | 15 |
| RPE65 | voretigene neparvovec-rzyl (LuxturnaTM) | NCT00999609 | Phase III | Subretinal | Active, not recruiting | Spark Therapeutics | USA | 31 |
| RPE65 | rAAV2-CB–hRPE65 | NCT00749957 | Phase I-II | Subretinal | Completed | Applied Genetic Technologies Corp | USA | 12 |
| GUCY2D | SAR439483 | NCT03920007 | Phase I–II | Subretinal | Active, not recruiting | Sanofi | USA | 15 |
| CEP290 | QR-110 | NCT03140969 | Phase I/II | Intravitreal | Completed | ProQR Therapeutics | USA and Belgium | 11 |
| CEP290 | AGN-151587 | NCT03872479 | Phase I–II | Subretinal | Recruiting | Allergan | USA | 18 |
| RPE65 & LRAT | QLT091001 | NCT01521793 | Phase I | Oral | Completed | QLT Inc. | USA and Europe | 27 |
| RPE65 & LRAT | QLT091001 | NCT01014052 | Phase I | Oral | Completed | QLT Inc. | USA and Europe | 32 |
| – | Human RPE cells | NCT03566147 | Phase I | Subretinal | Unknown | Eyecure Therapeutics Inc. | China | 30 |
| – | Argus II retinal prosthesis | NCT01490827 | Observational | Subretinal | Terminated | Second Sight Medical Products | Europe | 52 |
| – | RST-001 | NCT02556736 | Phase I/II | Intravitreal | Active, not recruiting | Allergan | USA | 12 |
| – | GS030-DP and GS030-MD | NCT03326336 | Phase I/II | Intravitreal | Recruiting | GenSight Biologics | USA and Europe | 18 |
EOSRD, early-onset severe retinal degeneration; LCA, leber congenital amaurosis; RPE, retinal pigment epithelium.
General management of LCA/EOSRD
The current management for most patients with LCA/EOSRD remains symptomatic and supportive, with treatment of possible associated complications (cataract, vitreoretinal interphase abnormalities, cystoid macular oedema, keratoconus)13 14 and a recommendation for a balanced, healthy diet, rich in fruits and vegetables.15 Vitamin A, minerals and amino acids supplementation have failed to show a clear benefit for these patients, and are consequently not recommended.16 17 Ultraviolet (UV) light has been shown to cause oxidate stress in the retina, thus UV protection when outdoors is advised.18 Smoke and tobacco-free environments are also suggested, as there is evidence that smoking may further damage the retina.19 Patients benefit from correction of refractive error, use of low vision aids when possible and optimal access to educational and work-related opportunities. Infants with severe visual impairment may also have delays or difficulties with speech, social skills and behaviour, highlighting the importance of a multi-specialist approach.
Several clinical trials are currently available for individuals with inherited retinal degenerations (IRDs), including LCA/EOSRD, using different approaches. The optimal design of these studies is challenging. IRDs are rare, and therefore gathering large cohorts can be complicated; which is often addressed with multiple trial sites and consequently high costs. Also, determining the eligibility criteria and ideal therapeutic window may not be straightforward. Age is not always correlated with the severity of disease; the rate of degeneration differs significantly between individuals. Structural and functional retinal parameters are usually set as eligibility criteria, often based on natural history data. Robust prospective data take time to be gathered and there is rarely a consensus between different trials. The clinical trials currently available use different techniques as follows.
Genetic therapy
Given the relative immune privilege of the eye and its favourable anatomical characteristics, ophthalmology has been one of the areas in which genetic therapy has thrived,20 with IRD having been particularly targeted, partly due to its monogenic basis.
Gene supplementation
Gene supplementation is based on the delivery of DNA to the cells’ nuclei via a viral vector.21 This process leads to the transcription of a functional protein and is currently in trial in many autosomal recessive and X-linked conditions.22 Several LCA/EOSRD-associated genes have been targeted for this therapy.
RPE65-LCA/EOSRD
Retinal pigment epithelium-specific 65 kDa (RPE65, OMIM 180069) is an enzyme that catalyses a critical step in the visual cycle, within the RPE.23 Biallelic disease-causing variants in RPE65 cause 5%–10% of all LCA/EOSRD cases and these patients show severe, early-onset night blindness.1 Clinical trials targeting replacement of RPE65 have been in development since 2007.24 25
Bainbridge et al conducted a phase I/II trial in which twelve patients were treated with a recombinant adeno-associated virus 2 (rAAV2) containing RPE65 complementary DNA, at two dose levels (NCT00643747).26 The surgery involved the administration of up to 1 mL of viral vector through a subretinal bleb involving the fovea.25 After a 3-year follow-up, six (50%) patients showed an improvement in retinal sensitivity (assessed by microperimetry and photopic and scotopic static perimetry), which was evident after 1–2 months of surgery, maintained over 6–12 months, and declining after this point, although remaining improved when compared with preintervention. Three patients (3/12, 25%) had intraocular inflammation that led to macular thinning and decreased acuity in two of them (2/12, 16.6%). The authors suggested a possible dose–response effect but did not identify a correlation between better response and younger age. The fact that the sensitivity declined after the first year led to the conclusion that the effect of this drug was temporary, variable and incomplete; and that an optimised, more potent therapy was required.27 This led to the phase I/II study NCT02781480, which involved the AAV5 vector OPTIRPE65 and 15 patients divided into three dose escalation groups and a paediatric expansion cohort. This drug was found to be safe, with most adverse events being transient and mild/moderate. Nine patients received the optimal concentration (1.0×1011 vg/mL) and all of them showed a statistically significant improvement in vision-guided mobility (maze navigation at different lux levels), retinal sensitivity (Octopus static perimetry), visual acuity, contrast sensitivity and reading speed, 6 months after the injection.28
Concomitantly, Hawswirth et al conducted a phase I/II trial in which a subretinal injection of rAAV2-CBSB-hRPE65 was performed in 15 young adults (NCT00481546).29 The cohort was divided into five subcohorts of different doses and volume of the viral vector, delivered in one or two injections. There were no reported serious adverse events (SAEs) and visual function improved in all patients to different degrees in the treated areas (measured by full-field stimulus testing (FST)) and remained stable for up to 36 months postinjection; although there was continued evidence of retinal degeneration.30 31
Concurrently, Spark Therapeutics was developing Voretigene neparvovec (Luxturna, NCT00999609). Luxturna is the first and only gene therapy for an IRD approved by the Food and Drug Administration (FDA, 2017).32 It is also the only treatment trial for RPE65 that reached phase III. The latter involved 20 patients aged 3 and older, who had bilateral treatment with an AAV2 vector, with a 6 to 18 day interval between eyes.33 Primary endpoint was change in performance on a multiluminance mobility test, 1 year after the injection. A statistically significant difference was observed between the treated and control groups (p<0.05), with mild ocular adverse events related to the surgical procedure (transient ocular inflammation, elevated intraocular pressure and intraoperative retinal tears). After the approval from FDA, health insurances and healthcare systems are starting to cover this treatment for eligible patients.34 35 The National Health Service of England funds the use of Luxturna in patients who have two likely disease-causing variants in RPE65 and remaining viable retinal cells. An ophthalmologist has to evaluate the child to assess their fitness to participate and refer them to one of the four treatment centres in the UK (https://retinauk.org.uk/information-support/luxturna/). This will lead to more patients and longer follow-up, which will enable a better understanding of the durability of treatment response. The longest follow-up published so far has been 3 years, where the efficacy appeared relatively well maintained.36 Patients between 4 and 44 years old have been treated and followed for up to 10 years.37
Weleber et al conducted a phase I/II trial where they treated twelve patients with rAAV2-CB-hRPE65 (NCT00749957). The participants were divided into two groups and received two different doses.38 No SAEs were reported, and visual function improved in 9 of 12 patients, as per best-corrected visual acuity (BCVA), visual field testing or responses to a quality-of-life questionnaire. The greatest improvement in BCVA was observed in the younger patients with better baseline BCVA.38
GUCY2D-LCA/EOSRD
Biallelic variants in GUCY2D account for 10%–20% of cases worldwide.1 GUCY2D (OMIM 600179) encodes the enzyme guanylate cyclase 2D, which synthesises cGMP, the intracellular messenger of photoreceptor stimulation.39 While the majority of disease-causing variants are associated with LCA (88%), a minority cause autosomal dominant cone dystrophy/cone-rod dystrophy (CRD).40 A genotype–phenotype correlation has been postulated, including missense variants being more commonly associated with CRD and null alleles with LCA.40 GUCY2D-LCA is characterised by severe congenital onset, profoundly reduced visual acuity, nystagmus, light sensitivity and absent/markedly reduced electroretinogram responses. However, it has been noted that these patients tend to show relatively preserved central photoreceptor structure until late in the disease.41 42 This feature makes GUCY2D-LCA particularly suitable for interventional gene therapy trials.
Boye et al have developed SAR439483, using a modified AAV capsid (Y733F), a subretinal gene therapy aiming to restore expression of guanylate cyclase 2D and thereby preserve the structure of rods and cones.43 It was found to be effective in knock-out mice, with improvement in electroretinography (ERG) responses.44 45 A phase I/II trial (NCT03920007) is on-going to enrol 15 participants; 9 in a dose escalation phase, and 6 in the dose expansion phase.46
AIPL1-LCA
Aryl hydrocarbon receptor-interacting protein-like 1 (AIPL1, OMIM 604392)-LCA is a rare cause of LCA (1%–2% of cases).47 48 This gene is expressed in photoreceptors and has a role in the synthesis of cGMP phosphodiesterase, a key element in phototransduction. Individuals with a damaging variant in AIPL1 generally do not have residual outer retinal structure beyond the age of 4 years.49 A compassionate use gene therapy study is ongoing for AIPL1-LCA, in infants and young children with remaining outer retinal layers at the central macula. This consists of a unilateral subretinal injection of an AAV8 vector, and is taking place at Great Ormond Street Hospital and Moorfields Eye Hospital, London, UK.
Gene editing and post-transcriptional regulation
Clustered regularly interspaced short palindromic repeats (CRISPR) and their associated enzyme (Cas) play a role in prokaryotic adaptive immunity.50 This system works as a physiological genome editing tool that cuts and deletes exogenous DNA, such as from viruses. Their discovery has led to their implementation as a tool for gene editing, targeting cardiovascular, neurological and neoplastic disorders, with previously no available treatment.51
Another method to suppress the expression of a damaging variant is through the use of small molecules designed to interact with pre-mRNA, known as antisense oligonucleotides (AON). Through this bonding, these molecules can modulate the pre-mRNA splicing (eg, attaching to an exon, they can block its transcription), or cause the degradation of certain transcripts.52 This technique is particularly useful for frameshift variants that cause the inclusion of pseudoexons.
CEP290-LCA/EOSRD
This gene (OMIM 610142) encodes a centrosomal protein of 290 kDa, a key component of cilia.53 As with other ciliopathies, damaging variants in this gene can present as an isolated ophthalmic condition, or as a syndromic disorder with kidney, skeletal and multiorgan pathologies. Disease-causing variants in CEP290 have been associated with LCA, Joubert, Senior-Loken and Meckel syndromes.54 The intronic variant c.2991+1655A>G, p.(Cys998X) is the most frequent in LCA, with 60% to 90% of affected individuals carrying at least one c.2991+1655A>G allele.55 Patients with this variant rarely have extraocular manifestations. In vitro studies have shown that it creates a cryptic splice donor site resulting in the inclusion of an aberrant exon and a premature stop codon.56 This leads to a decreased amount of wildtype protein, which affects cilia function.
The first interventional phase I/II clinical trial treating individuals with CEP290-related LCA (NCT03140969) is targeting those carrying the variant c.2991+1655A>G in a homozygous or compound heterozygous state.57 The tested drug (QR-110) is an AON developed to block the aberrant splicing event and restore normal splicing. Ten patients were enroled and received a unilateral intravitreal injection of QR-110 every 3 months for up to 1 year.58 The study was deemed to be safe and no SAEs were reported. BCVA, macular optical coherence tomography (OCT) structure, mobility, nystagmus measurements and FST were selected as outcome measures.59 Improvement and/or stability was observed in all parameters in the treated eyes, after a follow-up period of at least 3 months after surgery. This study is now recruiting for phase III (NCT03913143).
The first CRISPR-mediated retinal gene therapy clinical trial (NCT03872479) using EDIT-101 is ongoing. This phase I/II study started in 2019 and aims to enrol 18 participants harbouring the c.2991+1655A>G variant. The study consists of a subretinal injection of an AAV5 vector containing Staphylococcus aureus Cas9 and CEP290-specific guide RNAs.60 61 This was tested in mice and non-human primates, with no SAEs reported. Furthermore, the Cas9 expression was noted to be restricted to photoreceptors and the CEP290 gene-editing rates correlated with the levels of both Cas9 mRNA and gRNA.61
Pharmacotherapy
Visual cycle modulators have been under development for the last 10 years, with the aim of either decreasing the accumulation of various retinoid derivatives or to supply deficient compounds.62 In RPE65-related and LRAT (OMIM 604863)-related LCA/EOSRD, all-trans-retinal does not get converted back to 11-cis-retinal, interfering with the visual cycle, and ultimately leading to retinal degeneration and visual loss.63 It has been suggested that exogenous supply of 11-cis-retinal may prevent (or slow) photoreceptor degeneration.
A few studies (NCT01014052, NCT01521793) have tested the effect of supplementing 9-cis-retinyl acetate orally (QLT091001), in patients with LRAT and RPE65-related retinal degeneration. The first study involved a daily dose of 40 mg/m2 for 7 days, was completed in 2013 and included 18 participants.64 No SAEs were reported, and mild AEs were transient. An improvement in visual function was noted in 44% of participants with respect to visual field (20% expansion in kinetic perimetry documented at two consecutive study visits starting within 2 months of treatment) and in 67% for BCVA (≥5 Early Treatment Diabetic Retinopathy Study (ETDRS) letter score increase). A correlation between the thickness of the outer segments and the responsiveness to treatment was reported, suggesting that this structural parameter may predict the efficacy of the intervention.65 After a 2-year follow-up, 11 patients had returned to their baseline visual field and 10 to their baseline BCVA letter scores.66 A second trial tested the effect of repeated courses of this drug, in patients who had already received the first dose. Subjects received up to three more courses of once-daily oral QLT091001 (40 or 60 mg/m2) for 7 days, with a minimum of 3 weeks in between treatment courses.67 This study included 27 participants from various locations worldwide, with results pending. The effects of QLT091001 have also been tested in adults with delayed dark adaptation secondary to age-related macular dystrophy (NCT01999764) and in patients with RPE65-related autosomal dominant retinitis pigmentosa (RP, NCT01543906). The latter study showed an improvement in visual field in three out of five patients and in BCVA in one patient.68
Photoreceptor and RPE transplantation
Patient-derived somatic cells can be reprogrammed to induced pluripotent stem cells (iPSCs), retinal precursor cells and specialised cells such as photoreceptors or RPE.69 70 This process aims to replace dystrophic photoreceptors and provide a trophic and metabolic structure to prevent their degeneration.71 72 Given this an autologous transplant, it has a relatively low risk of rejection and would likely not require long-term immunosuppression. Although the first animal studies suggested successful integration of the transplant into the host retina, more recent studies have identified that a cytoplasmic transfer of RNA and proteins can occur between donor and host tissue, instead of integration.73 74 Nevertheless, work is on-going with hopes of human clinical trials in the near future.
Subretinal transplantation of human embryonic stem cell derived RPE cells has been undertaken in individuals with age-related macular degeneration (AMD) and Stargardt macular dystrophy.75 76 No adverse proliferation, rejection or other safety issue related to the transplant were reported. After a 1 year follow-up, the investigators mentioned an improvement in BCVA in more than half of treated eyes. Increased subretinal pigmentation was also noted in 13 of 18 patients, although this was not correlated with improved visual function. iPSC–RPE cells have also been transplanted into the subretinal space as patches on biodegradable scaffolds, in animal models resembling AMD.77 Sharma et al reported improved integration and functionality of the RPE in patches, when compared with RPE cells in suspension. These cell therapies are now being attempted for the first time in patients with RP and LCA; including plans for MERTK-associated LCA.
Optoelectronic devices
An optoelectronic device is able to transform an electrical signal to an optical one, and vice versa.78 There are different types regarding where they are located; epiretinal, subretinal, suprachoroidal and cortical implants are currently available or under evaluation.79 In the case of the retinal prostheses, their concept is to stimulate the preserved inner retinal layers by the implantation of an electronic device that would compensate for the outer retina not functioning properly.80 These are all composed of an external camera that capture light stimuli, a video processor that converts them into electric currents and a stimulator chip that directs them onto the retinal neuronal network. Some devices have been studied mainly for AMD (such as IRIS), whereas others target retinal and/or optic nerve degenerations.79
Argus II (Second Sight Medical Products) is the first retinal prosthetic device that became FDA approved, in 2013. It remains the only FDA approved retinal prosthesis and has been widely used, with over 350 implanted patients worldwide.81 Its production has been currently suspended, due to the company focusing on the development of new devices.82 A 5-year follow-up of 30 patients showed 24 SAEs among 12 patients (conjunctival erosion and dehiscence, hypotony, presumed endophthalmitis and retack of the device were the most common; corneal melt and retinal detachment were among the severest), treated with standard ophthalmic approaches. Three patients had the device completely or partially explanted due to associated complications, and two experienced device failure but were left implanted.83 This group also described an improvement in visual function, assessed by visual tasks such as following a high contrast line or spotting a door. This device is intended for individuals with advanced RP with a history of useful vision.84 However, in one of their clinical trials (NCT01490827), participants with severe to profound outer retinal degeneration were included; thereby patients with LCA may also have been included. Despite the improvements in visual function, as well as in quality of life assessments, limitations such as a field of view dependent on head position and limited electrodes translating into limited field remain to be addressed.85
Optogenetics
The field of optogenetics aims to repurpose second-order and third-order neurons of the visual pathway (bipolar and ganglion cells),86 to replace the degenerated or misfunctioning photoreceptors.87 To do so, a gene encoding a light-sensitive opsin-based protein is introduced in these cells, photosensitising them.88 This approach is particularly useful due to being independent of the causative gene, and therefore suitable for a broad range of patients with IRDs. It also takes advantage of the usually preserved and functioning inner retinal layers. The feasibility of this strategy has been tested in rats and mice, where they showed a positive impact measured by restored visual evoked potentials and optomotor responses.89–91
Due to favourable preclinical data, there are currently two trials involving optogenetics’ techniques in patients with advanced RP. We believe that patients with LCA may be eligible for these trials as well, given that they meet the requirement of severely decreased visual acuity and retinal degeneration. NCT02556736 is a phase I/IIa, open label, dose-escalation study launched in 2015 at multiple locations in the USA. It has fully recruited 12 adults who have received an intravitreal injection of the drug RST-001, a gene therapy encoding the protein channelrhodopsin-2.92 The second trial, NCT03326336, launched in 2017, is still recruiting in the USA, Europe and UK. It is a phase I/II study that combines a gene therapy called GS030-DP (rAAV2 vector containing the optimised channelrhodopsin ChrimsonR-tdTomato, delivered intravitreally) and stimulating glasses named GS030-MD. It will involve 18 participants divided into three dose-escalating cohorts and one extension cohort of three more individuals. Results are still pending for both studies.
Conclusions
Undoubtfully, tremendous progress has been made since the first description of LCA by Theodore Leber 150 years ago. In depth multimodal evaluation and genotypic characterisation have made this condition attractive for pioneering research that could improve patients’ (and their families) quality of life, and advance the frontiers of medicine. Outstanding research has led to this condition being the only IRD with an FDA-approved gene therapy, and with 29 registered clinical trials, 19 of which are interventional (www.clinicaltrials.gov, accessed on October 2020).
In this review, we have focused on recent and ongoing clinical trials with the greatest likelihood of successfully treating LCA/EOSRD. A wide range of different approaches and interventions are in clinical evaluation, with many more in late preclinical stages, hoping to treat conditions caused by variants in other genes, as well as applying different techniques to hopefully obtain better outcomes.93–96
There is great optimism (that we should share with our patients) that over the next 5 years, clinical trials for LCA/EOSRD will continue to increase in number, with more treatments being approved. More genes and more treatment approaches are in the pipeline and will continue their development to a clinical trial and, hopefully, an approved drug in the next few years. There are many challenges, including establishing these therapies to be cost effective and scalable—including if more genetic therapies could be delivered intravitreally compared with subretinally, with all the inherent benefits. In addition, should durability wane, it will need to be determined if and when to retreat individuals that have already undergone gene therapy. A further future area of investigation will be the pros and cons of a patient receiving different treatments both in the same or in the contralateral eye, at the same or subsequent timepoints. As more therapies are developed, research exploring their synergic (or antagonistic) effect(s) will need to be undertaken.
Footnotes
Contributors: All authors contributed to the design of this review article, literature review, manuscript preparation and review. The authors were responsible for all content and editorial decision. MDV illustrated the figure.
Funding: This work has been supported by grants from The Wellcome Trust [099173/Z/12/Z], the National Institute forHealth Research Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology, Moorfields Eye Charity, Retina UK and the Foundation Fighting Blindness (no specific grant/award number for the latter).
Disclaimer: The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Competing interests: The authors alone are responsible for the content and writing of this article. MM consults for MeiraGTx Ltd.
Provenance and peer review: Not commissioned; externally peer reviewed.
Ethics statements
Patient consent for publication
Not required.
References
- 1. Kumaran N, Moore AT, Weleber RG, et al. Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions [published correction appears in Br J Ophthalmol. 2019 Jun;103(6):862]. Br J Ophthalmol 2017;101:1147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tsang SH, Sharma T. Leber congenital amaurosis. Adv Exp Med Biol 2018;1085:131–7. 10.1007/978-3-319-95046-4_26 [DOI] [PubMed] [Google Scholar]
- 3. Chacon-Camacho OF, Zenteno JC. Review and update on the molecular basis of Leber congenital amaurosis. World J Clin Cases 2015;3:112–24. 10.12998/wjcc.v3.i2.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pontikos N, Arno G, Jurkute N, et al. Genetic basis of inherited retinal disease in a molecularly characterized cohort of more than 3000 families from the United Kingdom. Ophthalmology 2020;127:1384–94. 10.1016/j.ophtha.2020.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tsang SH, Aycinena ARP, Sharma T. Ciliopathy: Senior-Løken syndrome. Adv Exp Med Biol 2018;1085:175–8. 10.1007/978-3-319-95046-4_34 [DOI] [PubMed] [Google Scholar]
- 6. Radha RamA Devi a, Naushad Sm, Lingappa L. clinical and molecular diagnosis of Joubert syndrome and related disorders. Pediatr Neurol 2020;106:43–9. [DOI] [PubMed] [Google Scholar]
- 7. Fahim AT, Bouzia Z, Branham KH, et al. unique features and natural history of autosomal recessive RDH12-associated retinal degeneration. Br J Ophthalmol 2019;103:1789–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Coppieters F, Lefever S, Leroy BP, et al. Cep290, a gene with many faces: mutation overview and presentation of CEP290base. Hum Mutat 2010;31:1097–108. 10.1002/humu.21337 [DOI] [PubMed] [Google Scholar]
- 9. Kumaran N, Georgiou M, Bainbridge JWB, et al. Retinal structure in RPE65-Associated retinal dystrophy. Invest Ophthalmol Vis Sci 2020;61:47. 10.1167/iovs.61.4.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yadav RP, Artemyev NO. Aipl1: a specialized chaperone for the phototransduction effector. Cell Signal 2017;40:183–9. 10.1016/j.cellsig.2017.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ray TA, Cochran KJ, Kay JN. The enigma of CRB1 and CRB1 retinopathies. Adv Exp Med Biol 2019;1185:251–5. 10.1007/978-3-030-27378-1_41 [DOI] [PubMed] [Google Scholar]
- 12. den Hollander AI, Roepman R, Koenekoop RK, et al. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res 2008;27:391–419. 10.1016/j.preteyeres.2008.05.003 [DOI] [PubMed] [Google Scholar]
- 13. Davies EC, Pineda R. Cataract surgery outcomes and complications in retinal dystrophy patients. Can J Ophthalmol 2017;52:543–7. 10.1016/j.jcjo.2017.04.002 [DOI] [PubMed] [Google Scholar]
- 14. Hogden MC, Tsang S. Clinical and Pathological Features of Selected Human Retinal Degenerative Diseases. In: Zarbin M, Singh M, Casaroli-Marano R, eds. Cell-Based therapy for degenerative retinal disease. stem cell biology and regenerative medicine. Cham: Humana Press, 2019. [Google Scholar]
- 15. Chao DL, Burr A, Pennesi M. RPE65-Related Leber Congenital Amaurosis / Early-Onset Severe Retinal Dystrophy. In: Adam MP, Ardinger HH, Pagon RA, eds. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993-2020, 2019. https://www.ncbi.nlm.nih.gov/books/NBK549574/ [PubMed] [Google Scholar]
- 16. Zhao Y, Feng K, Liu R, et al. Vitamins and mineral supplements for retinitis pigmentosa. J Ophthalmol.; 2019;2019:8524607. 10.1155/2019/8524607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schwartz SG, Wang X, Chavis P, et al. Vitamin A and fish oils for preventing the progression of retinitis pigmentosa. Cochrane Database Syst Rev 2020;6:CD008428. 10.1002/14651858.CD008428.pub3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vaz VVA, Jardim da Silva L, Geihs MA, et al. Single and repeated low-dose UVB radiation exposures affect the visual system. J Photochem Photobiol B 2020;209:111941. 10.1016/j.jphotobiol.2020.111941 [DOI] [PubMed] [Google Scholar]
- 19. Hammond BR, Wooten BR, Snodderly DM. Cigarette smoking and retinal carotenoids: implications for age-related macular degeneration. Vision Res 1996;36:3003–9. 10.1016/0042-6989(96)00008-9 [DOI] [PubMed] [Google Scholar]
- 20. Streilein JW. Ocular immune privilege: therapeutic opportunities from an experiment of nature. Nat Rev Immunol 2003;3:879–89. 10.1038/nri1224 [DOI] [PubMed] [Google Scholar]
- 21. Smith AJ, Bainbridge JWB, Ali RR. Gene supplementation therapy for recessive forms of inherited retinal dystrophies. Gene Ther 2012;19:154–61. 10.1038/gt.2011.161 [DOI] [PubMed] [Google Scholar]
- 22. Sinha D, Steyer B, Shahi PK, et al. Human iPSC modeling reveals mutation-specific responses to gene therapy in a genotypically diverse dominant maculopathy. Am J Hum Genet 2020;107:278–92. 10.1016/j.ajhg.2020.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jin M, Yuan Q, Li S, et al. Role of LRAT on the retinoid isomerase activity and membrane association of RPE65. J Biol Chem 2007;282:20915–24. 10.1074/jbc.M701432200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miraldi Utz V, Coussa RG, Antaki F, et al. Gene therapy for RPE65-related retinal disease. Ophthalmic Genet 2018;39:671–7. 10.1080/13816810.2018.1533027 [DOI] [PubMed] [Google Scholar]
- 25. Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med 2008;358:2240–8. 10.1056/NEJMoa0802315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bainbridge JWB, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber's congenital amaurosis. N Engl J Med 2008;358:2231–9. 10.1056/NEJMoa0802268 [DOI] [PubMed] [Google Scholar]
- 27. Bainbridge JWB, Mehat MS, Sundaram V, et al. Long-Term effect of gene therapy on Leber's congenital amaurosis. N Engl J Med 2015;372:1887–97. 10.1056/NEJMoa1414221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. MeiraGTx . AAO 2019 data review. Available: https://investors.meiragtx.com/static-files/13f8f6eb-3ba5-4509-985a-2b7a830a13f4
- 29. Hauswirth WW, Aleman TS, Kaushal S, et al. Treatment of Leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther 2008;19:979–90. 10.1089/hum.2008.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cideciyan AV, Hauswirth WW, Aleman TS, et al. Human RPE65 gene therapy for Leber congenital amaurosis: persistence of early visual improvements and safety at 1 year. Hum Gene Ther 2009;20:999–1004. 10.1089/hum.2009.086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jacobson SG, Cideciyan AV, Ratnakaram R, et al. Gene therapy for Leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol 2012;130:9–24. 10.1001/archophthalmol.2011.298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. U.S. Food & Drug Administration . FDA approves novel gene therapy to treat patients with a rare form of inherited vision loss. Available: https://www.fda.gov/news-events/press-announcements/fda-approves-novel-gene-therapy-treat-patients-rare-form-inherited-vision-loss
- 33. Russell S, Bennett J, Wellman JA, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 2017;390:849–60. 10.1016/S0140-6736(17)31868-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. NHS . NHS to fund revolutionary treatment for blindness in children. Available: https://www.england.nhs.uk/2019/09/nhs-to-fund-revolutionary-treatment-for-blindness-in-children/
- 35. The Oregonian/Oregon Live . 4-Year-Old sees a brighter future after brand-new vision treatment. Available: https://www.oregonlive.com/news/erry-2018/10/095a0fdf409726/4yearold-sees-a-brighter-futur.html
- 36. Bennett J, Wellman J, Marshall KA, et al. Safety and durability of effect of contralateral-eye administration of AAV2 gene therapy in patients with childhood-onset blindness caused by RPE65 mutations: a follow-on phase 1 trial. Lancet 2016;388:661–72. 10.1016/S0140-6736(16)30371-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Could LUXTURNA® be right for you? Available: https://luxturna.com/about-luxturna/
- 38. Weleber RG, Pennesi ME, Wilson DJ, et al. Results at 2 years after gene therapy for Rpe65-deficient Leber congenital amaurosis and severe Early-Childhood-Onset retinal dystrophy. Ophthalmology 2016;123:1606–20. 10.1016/j.ophtha.2016.03.003 [DOI] [PubMed] [Google Scholar]
- 39. Bouzia Z, Georgiou M, Hull S, et al. GUCY2D-Associated Leber congenital amaurosis: a retrospective natural history study in preparation for trials of novel therapies. Am J Ophthalmol 2020;210:59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sharon D, Wimberg H, Kinarty Y, et al. Genotype-functional-phenotype correlations in photoreceptor guanylate cyclase (GC-E) encoded by GUCY2D. Prog Retin Eye Res 2018;63:69–91. 10.1016/j.preteyeres.2017.10.003 [DOI] [PubMed] [Google Scholar]
- 41. Bouzia Z, Georgiou M, Hull S, et al. GUCY2D-Associated Leber congenital amaurosis: a retrospective natural history study in preparation for trials of novel therapies. Am J Ophthalmol 2020;210:59–70. 10.1016/j.ajo.2019.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Boye SE. Leber congenital amaurosis caused by mutations in GUCY2D. Cold Spring Harb Perspect Med 2014;5:a017350. 10.1101/cshperspect.a017350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Boye SL, Peshenko IV, Huang WC, et al. Aav-Mediated gene therapy in the guanylate cyclase (RetGC1/RetGC2) double knockout mouse model of Leber congenital amaurosis. Hum Gene Ther 2013;24:189–202. 10.1089/hum.2012.193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Boye SL, Peterson JJ, Choudhury S, et al. Gene Therapy Fully Restores Vision to the All-Cone Nrl(-/-) Gucy2e(-/-) Mouse Model of Leber Congenital Amaurosis-1. Hum Gene Ther 2015;26:575–92. 10.1089/hum.2015.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gene therapy for GUCY2D-associated retinal disease. Available: http://ittsd2018.org/wp-content/uploads/2019/01/Boye_Monell-conference-11-14-18-1.pdf
- 46. Upenn education. Available: https://clinicalresearch.itmat.upenn.edu/clinicaltrial/6129/tbd-phase-12-dose-escalation/
- 47. Sacristan-Reviriego A, Bellingham J, Prodromou C, et al. The integrity and organization of the human AIPL1 functional domains is critical for its role as a Hsp90-dependent co-chaperone for rod PDE6. Hum Mol Genet 2018;27:1309. 10.1093/hmg/ddy024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sacristan-Reviriego A, Le HM, Georgiou M, et al. Clinical and functional analyses of AIPL1 variants reveal mechanisms of pathogenicity linked to different forms of retinal degeneration. Sci Rep 2020;10:17520. 10.1038/s41598-020-74516-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Aboshiha J, Dubis AM, van der Spuy J, et al. Preserved outer retina in AIPL1 Leber's congenital amaurosis: implications for gene therapy. Ophthalmology 2015;122:862–4. 10.1016/j.ophtha.2014.11.019 [DOI] [PubMed] [Google Scholar]
- 50. Makarova KS, Wolf YI, Iranzo J, et al. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol 2020;18:67–83. 10.1038/s41579-019-0299-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang B. Crispr/Cas gene therapy. J Cell Physiol. 2020. [Google Scholar]
- 52. Collin RWJ, Garanto A. Applications of antisense oligonucleotides for the treatment of inherited retinal diseases. Curr Opin Ophthalmol 2017;28:260–6. 10.1097/ICU.0000000000000363 [DOI] [PubMed] [Google Scholar]
- 53. Kim J, Krishnaswami SR, Gleeson JG. CEP290 interacts with the centriolar satellite component PCM-1 and is required for Rab8 localization to the primary cilium. Hum Mol Genet 2008;17:3796–805. 10.1093/hmg/ddn277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen HY, Kelley RA, Li T, et al. Primary cilia biogenesis and associated retinal ciliopathies. Semin Cell Dev Biol 2021;110:70–88. 10.1016/j.semcdb.2020.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sheck L, Davies WIL, Moradi P, et al. Leber congenital amaurosis associated with mutations in CEP290, clinical phenotype, and natural history in preparation for trials of novel therapies. Ophthalmology 2018;125:894–903. 10.1016/j.ophtha.2017.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. den Hollander AI, Koenekoop RK, Yzer S, et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet 2006;79:556–61. 10.1086/507318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Collin RW, den Hollander AI, van der Velde-Visser SD, et al. Antisense Oligonucleotide (AON)-based Therapy for Leber Congenital Amaurosis Caused by a Frequent Mutation in CEP290. Mol Ther Nucleic Acids 2012;1:e14. 10.1038/mtna.2012.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cideciyan AV, Jacobson SG, Drack AV, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat Med 2019;25:225–8. 10.1038/s41591-018-0295-0 [DOI] [PubMed] [Google Scholar]
- 59. Jacobson SG, Cideciyan AV, Sumaroka A, et al. Outcome measures for clinical trials of Leber congenital amaurosis caused by the intronic mutation in the CEP290 gene. Invest Ophthalmol Vis Sci 2017;58:2609–22. 10.1167/iovs.17-21560 [DOI] [PubMed] [Google Scholar]
- 60. Quinn J, Musa A, Kantor A, et al. Genome-Editing strategies for treating human retinal degenerations. Hum Gene Ther 2020. 10.1089/hum.2020.231. [Epub ahead of print: 19 Nov 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Maeder ML, Stefanidakis M, Wilson CJ, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med 2019;25:229–33. 10.1038/s41591-018-0327-9 [DOI] [PubMed] [Google Scholar]
- 62. Hussain RM, Gregori NZ, Ciulla TA, et al. Pharmacotherapy of retinal disease with visual cycle modulators. Expert Opin Pharmacother 2018;19:471–81. 10.1080/14656566.2018.1448060 [DOI] [PubMed] [Google Scholar]
- 63. Redmond TM, Yu S, Lee E, et al. Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat Genet 1998;20:344–51. 10.1038/3813 [DOI] [PubMed] [Google Scholar]
- 64. Scholl HPN, Moore AT, Koenekoop RK, et al. Safety and Proof-of-Concept Study of Oral QLT091001 in Retinitis Pigmentosa Due to Inherited Deficiencies of Retinal Pigment Epithelial 65 Protein (RPE65) or Lecithin:Retinol Acyltransferase (LRAT). PLoS One 2015;10:e0143846. 10.1371/journal.pone.0143846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wen Y, Birch DG. Outer Segment Thickness Predicts Visual Field Response to QLT091001 in Patients with RPE65 or LRAT Mutations. Transl Vis Sci Technol 2015;4:8. 10.1167/tvst.4.5.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Koenekoop RK, Sui R, Sallum J, et al. Oral 9-cis retinoid for childhood blindness due to Leber congenital amaurosis caused by RPE65 or LRAT mutations: an open-label phase 1B trial. Lancet 2014;384:1513–20. 10.1016/S0140-6736(14)60153-7 [DOI] [PubMed] [Google Scholar]
- 67. U.S. National Institutes of Health . Repeated treatments of QLT091001 in subjects with Leber congenital amaurosis or retinitis pigmentosa (extension of study RET IRD 01). Available: https://clinicaltrials.gov/ct2/show/NCT01521793?term=QLT091001&draw=2&rank=3
- 68. Kenna PF, Humphries MM, Kiang A-S, et al. Advanced late-onset retinitis pigmentosa with dominant-acting D477G RPE65 mutation is responsive to oral synthetic retinoid therapy. BMJ Open Ophthalmol 2020;5:e000462. 10.1136/bmjophth-2020-000462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chen FK, McLenachan S, Edel M, et al. iPS cells for modelling and treatment of retinal diseases. J Clin Med 2014;3:1511–41. 10.3390/jcm3041511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861–72. 10.1016/j.cell.2007.11.019 [DOI] [PubMed] [Google Scholar]
- 71. Pearson RA, Barber AC, Rizzi M, et al. Restoration of vision after transplantation of photoreceptors. Nature 2012;485:99–103. 10.1038/nature10997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Liu Y, Xu HW, Wang L, et al. Human embryonic stem cell-derived retinal pigment epithelium transplants as a potential treatment for wet age-related macular degeneration. Cell Discov 2018;4:50. 10.1038/s41421-018-0053-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. West EL, Gonzalez-Cordero A, Hippert C, et al. Defining the integration capacity of embryonic stem cell-derived photoreceptor precursors. Stem Cells 2012;30:1424–35. 10.1002/stem.1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Pearson RA, Gonzalez-Cordero A, West EL, et al. Donor and host photoreceptors engage in material transfer following transplantation of post-mitotic photoreceptor precursors. Nat Commun 2016;7:13029. 10.1038/ncomms13029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Schwartz SD, Regillo CD, Lam BL, et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt's macular dystrophy: follow-up of two open-label phase 1/2 studies. Lancet 2015;385:509–16. 10.1016/S0140-6736(14)61376-3 [DOI] [PubMed] [Google Scholar]
- 76. Mehat MS, Sundaram V, Ripamonti C, et al. Transplantation of human embryonic stem cell-derived retinal pigment epithelial cells in macular degeneration. Ophthalmology 2018;125:1765–75. 10.1016/j.ophtha.2018.04.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sharma R, Khristov V, Rising A, et al. Clinical-Grade stem cell-derived retinal pigment epithelium patch rescues retinal degeneration in rodents and pigs. Sci Transl Med 2019;11:eaat5580. 10.1126/scitranslmed.aat5580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dowling J. Current and future prospects for optoelectronic retinal prostheses. Eye 2009;23:1999–2005. 10.1038/eye.2008.385 [DOI] [PubMed] [Google Scholar]
- 79. Wang V, Kuriyan AE. Optoelectronic devices for vision restoration. Curr Ophthalmol Rep 2020;8:69–77. 10.1007/s40135-020-00232-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yue L, Weiland JD, Roska B, et al. Retinal stimulation strategies to restore vision: fundamentals and systems. Prog Retin Eye Res 2016;53:21–47. [DOI] [PubMed] [Google Scholar]
- 81. Bloch E, Luo Y, da Cruz L. Advances in retinal prosthesis systems. Ther Adv Ophthalmol 2019;11:2515841418817501. 10.1177/2515841418817501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. MarketScope. Available: https://www.market-scope.com/pages/news/3418/second-sight-to-focus-on-orion-suspend-production-of-argus-ii
- 83. da Cruz L, Dorn JD, Humayun MS, et al. Five-Year safety and performance results from the argus II retinal prosthesis system clinical trial. Ophthalmology 2016;123:2248–54. 10.1016/j.ophtha.2016.06.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. SecondSight. Available: https://secondsight.com/faq/
- 85. Sommerhalder J, Pérez FA. Prospects and Limitations of Spatial Resolution. In: Gabel VP, ed. Artif. Vis. Clin. Guide. Cham: Springer International Publishing, 2017: 29–45. [Google Scholar]
- 86. Schiefer U, Hart W. Functional anatomy of the human visual pathway. In: Clinical neuro-ophthalmology. Berlin, Heidelberg: Springer, 2007: 19–28. [Google Scholar]
- 87. Yue L, Weiland JD, Roska B, et al. Retinal stimulation strategies to restore vision: fundamentals and systems. Prog Retin Eye Res 2016;53:21–47. 10.1016/j.preteyeres.2016.05.002 [DOI] [PubMed] [Google Scholar]
- 88. Simunovic MP, Shen W, Lin JY, et al. Optogenetic approaches to vision restoration. Exp Eye Res 2019;178:15–26. 10.1016/j.exer.2018.09.003 [DOI] [PubMed] [Google Scholar]
- 89. Bi A, Cui J, Ma Y-P, et al. Ectopic expression of a microbial-type rhodopsin restores visual responses in mice with photoreceptor degeneration. Neuron 2006;50:23–33. 10.1016/j.neuron.2006.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Tomita H, Sugano E, Isago H, et al. Channelrhodopsin-2 gene transduced into retinal ganglion cells restores functional vision in genetically blind rats. Exp Eye Res 2010;90:429–36. 10.1016/j.exer.2009.12.006 [DOI] [PubMed] [Google Scholar]
- 91. Pan Z-H, Lu Q, Bi A, et al. Optogenetic approaches to restoring vision. Annu Rev Vis Sci 2015;1:185–210. 10.1146/annurev-vision-082114-035532 [DOI] [PubMed] [Google Scholar]
- 92. Eyewire News . Allergan acquires gene therapy company RetroSense therapeutics, 2021. Available: https://eyewire.news/articles/allergan-acquires-gene-therapy-company-retrosense-therapeutics/
- 93. Feathers KL, Jia L, Perera ND, et al. Development of a Gene Therapy Vector for RDH12-Associated Retinal Dystrophy. Hum Gene Ther 2019;30:1325–35. 10.1089/hum.2019.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Jo DH, Song DW, Cho CS, et al. CRISPR-Cas9-mediated therapeutic editing of Rpe65 ameliorates the disease phenotypes in a mouse model of Leber congenital amaurosis. Sci Adv 2019;5:eaax1210. 10.1126/sciadv.aax1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Murray SF, Jazayeri A, Matthes MT, et al. Allele-Specific inhibition of rhodopsin with an antisense oligonucleotide slows photoreceptor cell degeneration. Invest Ophthalmol Vis Sci 2015;56:6362–75. 10.1167/iovs.15-16400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. LaVail MM, Yasumura D, Matthes MT, et al. Gene therapy for MERTK-Associated retinal degenerations. Adv Exp Med Biol 2016;854:487–93. 10.1007/978-3-319-17121-0_65 [DOI] [PMC free article] [PubMed] [Google Scholar]