

Abstract
Kinetic studies of a vinyl cyclopropane (VCP) cycloaddition, catalyzed by peptide-based thiyl radicals, are described. Reactions were analyzed using reaction progress kinetic analysis, revealing that ring-opening of the VCP is both rate- and enantio-determining. These conclusions are further corroborated by studies involving racemic and enantiopure VCP starting material. Noncovalent interactions play key roles throughout: both the peptide catalyst and VCP exhibit unproductive self-aggregation, which appears to be disrupted by binding between the catalyst and VCP. This in turn explains the requirement for the key catalyst feature, a substituent at the 4-position of the proline residue, which is required for both turnover/rate and selectivity.
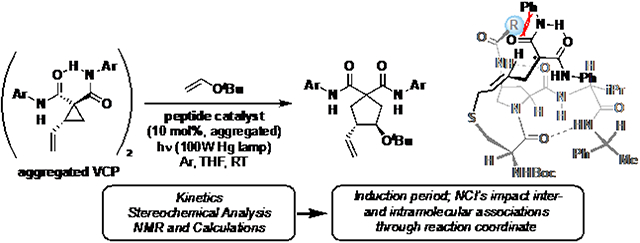
Graphical Abstract
Introduction
Inspired by the known biological roles for cysteine-based radicals,1-3 we recently developed a peptide catalyst that can mediate enantioselective radical transformations.4 Thiyl radicals, such as those generated from cysteine or cystine, are crucial reactive species in biological and synthetic contexts,5,6 including reports of chiral thiols as precursors for enantioselective H-atom donation.7,8 In particular, the addition of thiyl radicals to vinylcyclopropanes (VCPs)9-16 and related methylenecyclopropanes17-20 has garnered substantial interest over the past few decades. Early studies aspired to achieve asymmetric catalysis,21 but this was not realized until a relatively recent report of bulky aryl thiyl radicals that catalyze enantioselective ring-opening/cyclization of VCPs and related substrates.22, 23
This reaction offered a useful platform on which to develop a new peptide-based catalyst that could lead to further discoveries with radical-based reaction mechanisms (Scheme 1a). By leveraging the relatively weak bond dissociation energy of S–S bonds (~65 kcal/mol), and their vulnerability to UV light-promoted homolysis, two equivalents of active thiyl radical catalyst can be accessed from dimeric precatalysts (Scheme 1b). Strain-promoted ring-opening results from addition of the radical into the VCP olefin, affording malonyl-stabilized radical intermediate I. Addition of an electron-rich olefin (tert-butyl vinyl ether, TBVE) to I affords II, and the product is generated as a mixture of four stereoisomers via subsequent cyclization and fragmentation. The starting materials convert cleanly to products, and minimal byproducts are observed.
Scheme 1.

Enantioselective cysteine radical-mediated VCP cycloaddition.
We designed peptide 1, which features an amide substituent at the 4-proline position, to catalyze the VCP ring-opening/cycloaddition (Scheme 1c). This structural element was also new to our peptide scaffolds and predominately influences enantioselectivity in the thiyl radical-mediated cyclization.24, 25 In contrast, peptide 2, which lacks the 4-proline amide, is significantly less enantioselective. In all cases, the trans product is favored, as determined by x-ray crystallography.26 Despite extensive catalyst optimization, we were unable to improve the enantioselectivity beyond that obtained with 1. However, from these structure-function studies, we gained preliminary insight into how this catalyst may be operating. Our key observations were: (1) at the 4-proline position, amides provide superior enantiomeric excess relative to other functional groups; (2) enantioselectivity is sensitive to the steric or dispersive demands of this amide, as opposed to its H-bonding character; 27-29 (3) enhanced Lewis basicity of the proline backbone amide improves enantioselectivity; and (4) substrates bearing electron-deficient amides afford higher enantiomeric excess than substrates bearing electron-rich amides. We therefore proposed that the ring-opened intermediate is H-bonded to the peptide backbone (Scheme 1D).
However, despite the crucial effect of the 4-proline amide on enantioselectivity, an empirical approach to catalyst optimization did not lead to further enhancements in enantioselectivity. As many of the finer points of the mechanism remained unclear, we chose to carry out a detailed kinetic study of these reactions. In addition, we report an analysis of the overall reaction mechanism catalyzed by 1, the effect of VCP configuration on the reaction kinetics, and a comparison of catalysts 1 and 2. Several unexpected observations emerged from these investigations. These include issues related to catalyst aggregation under the reaction conditions; evidence that these reactions may not be fully stereoconvergent as previously assumed; and insights related to the rate-, enantiomer-, and diastereomer-determining steps.
The result is an enhanced understanding of this radical-mediated, peptide-catalyzed cycloaddition reaction, which emerges as a prototype of a high-complexity enantioselective reaction, far beyond the paradigm we anticipated at the outset of the work.30
Results and Discussion
Observation of induction period.
Our first goal was to identify the rate-determining step using reaction progress kinetic analysis.31,32 The concentration profile for this reaction reveals an induction period, with the maximum reaction rate attained at approximately 30% conversion (Figure 1A). Such an induction period is not unexpected and is consistent with the use of a precatalyst from which the active catalyst is generated via homolysis. The n-butyl disulfide-catalyzed reaction exhibits the same induction period and dependence on conversion, reinforcing our conclusion that the induction period is due to homolysis and not any specific features of the peptide catalyst.33
Figure 1.

In all graphs, duplicate data are simultaneously plotted and analyzed. (a) Different-excess experiment, [VCP]0 = 0.05 M, [1]T = 2.5 mM. (b) Different-excess experiment, [TBVE]0 = 0.1 M, [1]T = 2.5 mM. (c) Alternative Michaelis–Menten fit to experiment with [VCP]0 = 0.05 M, [TBVE]0 = 0.1 M, [1]T = 2.5 mM. (d) Determination of catalyst order in lower [1]T regime; overlay is attained when the x-axis is normalized assuming 0.5-order dependence on [1]T. (e) Determination of catalyst order in higher [1]T regime; overlay is attained when the x-axis is normalized assuming 0.2-order dependence on [1]T. (f) Same-excess experiment, [1]T = 2.5 mM. Experiment A = 0.05 M VCP, 0.1 M TBVE; Experiment B = 0.035 M VCP, 0.085 M TBVE; Experiment C = 0.035 M VCP, 0.085 M TBVE, 0.015 M P; truncated and time-adjusted to exclude induction period.
Determination of Kinetic Orders.
These kinetic studies reveal that the reaction rate is also independent of [TBVE], as evidenced by the concentration profiles for the different-excess experiment in which [TBVE]0 is doubled (Figure 1A). This conclusion was further supported using the variable time normalization analysis described by Burés,34 which provides overlaying plots when zeroth-order dependence on [TBVE] is assumed. To justify our subjective conclusions as to what constitutes sufficient overlay, plots representing other possibilities for kinetic orders are presented in the supporting information.
A second different-excess experiment, maintaining [TBVE]0 but changing [VCP]0, reveals a potential 0.5-order rate dependence on [VCP], indicating that the VCP may exist as a ground state dimer but is monomeric in the transition state (Figure 1B). This conclusion is also supported by the line-shape analysis described by Blackmond, as a plot of rate vs. [VCP]1/2 displays as a straight line in the post-induction period regime (Figure S4). The same kinetic orders are obtained from the n-butyl disulfide-catalyzed reaction, demonstrating that the peptide structure does not induce a change in mechanism or rate-determining step. However, this behavior is also consistent with simple Michaelis–Menten kinetics, in which the VCP is not aggregated but saturation of the catalyst is attained over the course of the reaction. Both scenarios appear to describe these data equally well, and cannot be distinguished on the basis of these kinetics experiments alone (Figure 1C).
Our experiments further reveal that the dependence of reaction rate on [1]T is complex. As [1]T increases from 1.25 to 3.5 mM, a corresponding increase in rate is observed. Beyond this, however, the reaction rate decreases with increasing [1]T. We quantified the order in [1]T at both lower and higher catalyst loadings,35 and found that at lower [1]T (1.25 mM), the reacton rate exhibits a 0.5-order dependence on [1]T, which is consistent with a resting state dimer (the disulfide precatalyst). The kinetic order shifts to a 0.2-order dependence in catalyst as [1]T increases to 4.85 mM (Figures 1D and 1E), suggesting that the disulfide is further aggregated intermolecularly at higher catalyst loadings.36-38 In contrast, n-butyl disulfide would not exhibit aggregation even at higher catalyst loadings; correspondingly, a neat 0.5-order rate dependence on [(nBuS)2]T is observed. Identification of the catalyst resting state as the dimer is further supported by the requirement for constant exposure of the reaction mixture to UV light. The reaction stops immediately when the light source is removed, likely due to rapid recombination of the thiyl radicals, further favored by close proximity of the radicals due to the aforementioned catalyst aggregation.39, 40
Identification of rate-determining step.
Taken together, these results reinforce that the ring-opening event is the rate-determining step for this reaction. The irreversibility of this step may derive from the relief of cyclopropyl ring strain as well as the highly stabilized nature of the malonyl radical. For these same reasons, we presume that addition of the radical and ring-opening occur in a single step, although a stepwise alternative cannot be ruled out. Given this strain relief, along with typically fast rates of ring-opening,41 we also considered that irreversible ring-opening may not be rate-determining but instead precedes rate-limiting (?) formation of catalyst-substrate adduct I (Scheme 1B); however, such a scenario is inconsistent with the data.
Effects of H-bonding on catalyst activation.
The H-bonding capability of the VCP is retained in the product cycloadduct (P), which still bears the two amide groups. We therefore investigated whether the product itself could engage in H-bonding with the catalyst using the same-excess experiment designed by Blackmond (Figure 1F). We chose to carry out the same-excess experiment using initial concentrations (0.035 M VCP, 0.085 M TBVE), that mimic 30% conversion of the standard reaction conditions (0.05 M VCP, 0.1 M TBVE), but exclude any product that would have been formed at 30% conversion. The experiments were compared only in the post-induction period regime, using a time-adjusted approach to directly compare the concentration profiles.42,43 The plots fail to overlay, but in a manner consistent with catalyst activation as opposed to product inhibition, as the reaction proceeds at a slower rate when product concentration is lower (Expt. A vs. B). These results could indicate that product formation facilitates catalysis by disrupting catalyst aggregation. Thus, we repeated the same-excess experiment, this time adding [P]0 equal to that obtained at 30% conversion (0.035 M VCP, 0.085 M TBVE, 0.015 M P). The resulting concentration profile overlays with that observed under the same conditions lacking product (Expt. B vs. C), indicating that in fact, the reaction rate is unaffected by [P]. Thus, the lack of overlay in the same-excess experiment results from the change in [VCP]0, and the observed catalyst activation results from interactions between the catalyst and the VCP.
Having established that the reaction rate is also independent of [TBVE], it seems that the variation in [VCP]0 is responsible for the observed rate difference in the same-excess experiment. Notably, in the same-excess experiment for the n-butyl disulfide-catalyzed reaction, the concentration profiles overlay. The myriad noncovalent interactions involving the peptide catalyst account for the divergent behavior between 1 and n-butyl disulfide. That is, the ring-opening reaction itself leads to an adduct that is unavailable for unproductive thiyl radical recombination. This, of course, is also true for the n-butyl disulfide catalyst. However, radical recombination is likely more favorable with the peptide catalysts than with n-butyl disulfide due to the aforementioned catalyst aggregation, and thus the disaggregating effect of VCP is more pronounced with the peptide catalyst than with n-butyl di-sulfide. Moreover, aggregation can be disrupted by intramolecular H-bonding within ring-opened VCP-catalyst adduct (I), as well as intermolecular H-bonding between 1 and VCP, thus leading to catalyst activation. These interactions are not possible between the VCP and n-butyl disulfide, and could explain the divergent behavior between 1 and n-butyl disulfide.
The same-excess experiment also reveals that regardless of [VCP]0 or [TBVE]0, the maximum rate occurs at ~32% conversion.44 These observations demand a more nuanced interpretation of the events defining the induction period. Classically, the induction period is generally a function of the reversible homolytic cleavage of the S–S bond in the precatalyst. However, the fact that the VCP:P ratio correlates with the induction period duration indicates that the VCP and product work in concert to influence homolysis and reaction progress, although the details of these noncovalent associations remain unclear. This scenario is consistent with the observation that changing [TBVE]0 does not influence the induction period, as it is not expected to compete in these noncovalent associations, and presumably is not competitive with the VCP in the initial radical addition.
Further investigation into induction period.
The origin of the induction period was further investigated in analogy to a previously reported scenario, in which irreversible pre-catalyst decomposition to form the active catalytic species resulted in a similar induction period and an apparent zeroth-order dependence on one of the starting materials.45 We considered the possibility that the same could be true of our system—that the consistent exposure of our disulfide precatalyst to light over the course of the reaction could render disulfide homolysis to be functionally irreversible. However, such a scenario does not describe our system, as multiple fits to the data obtained with different [VCP]0:[TBVE]0 ratios fail to provide similar parameters. Further details, including the fit attempts and the relevant rate equations, are provided in the supporting information.
From these reaction progress kinetics studies, we thus conclude that (1) ring-opening appears to be rate-determining; (2) the VCP may be aggregated as a dimer in the ground state; (3) the disulfide precatalyst is the catalyst resting state; (4) the precatalyst is also aggregated at higher catalyst loadings; (5) the induction period and associated cysteinyl thiyl radical recombination is strongly influenced by both intra- and intermolecular H-bonding interactions.
Hypotheses for stereoinduction.
The conclusion that ring-opening is rate-determining does not preclude later steps from being enantiodetermining. At the same time, while the stereo-centers are not constructed until the final cyclization event, enantiodetermination could occur in prior steps under certain circumstances. Considering the aforementioned effect of the 4-proline substituent, we advance two primary hypotheses for stereoinduction (Scheme 2). In Scheme 2A, we consider that the ring-opened intermediate is conformationally flexible; at the extreme, the conformers IIR and IIS orient different faces of the π-bond toward the 4-proline amide. After olefin addition, cyclization onto each π-face affords opposite enantiomers of product. If cyclization is rapid and the conformers cannot interconvert through bond rotation, then the ratio of conformers could directly relate to the enantiomeric ratio of products. The distribution of conformers could be influenced by the 4-substituent of the proline, which may define chiral space that better accommodates one conformation over the other. In Scheme 2B, we also consider that the 4-proline amide could affect the trajectory of olefin approach by disfavoring approach over one π-face of the ring-opened intermediate I. Common to Scheme 2A and 2B, if subsequent cyclization is rapid, then olefin approach and bond formation occur from the same π-face, leading to the two different enantiomers. We note that both of these scenarios assume that non-interconverting conformers are generated from ring-opening. While our intuition would suggest that these conformers would rapidly interconvert, if that is the case then no facial selectivity, and no enantioselectivity, governed by the 4-proline substituent would be possible. We discuss the precise structures of IR and IS in more detail, with attendant computational support, in Figure 5 below.
Scheme 2.

Stereochemical hypotheses
Figure 5.

Computed lowest-energy geometries of the post-ring-opening adducts IS and IR. Initial conformational searches were performed with metadynamics using CREST at the GFN2-xTB/ALPB(THF) level. DFT calculations were carried out at the B3LYP-D3BJ/6-31G(d)/PCM(THF) // B3LYP-D3BJ/6-311+G(d,p)/PCM(THF) level. Electronic energies are reported in parentheses.
The stereochemical picture is further complicated by the fact that this reaction also produces cis and trans diastereomers, each with different enantiomeric excess. This indicates that the enantio- and diastereodetermining steps are different. Cyclization is likely diastereodetermining, with C–C bond rotation leading to the cis and trans diastereomers (Scheme 2C). Correspondingly, either ring-opening or olefin addition is likely to be enantiodetermining. Our kinetic studies do not differentiate between these two scenarios.
Kinetics studies with enantiopure VCP.
We also studied the influence of the VCP chirality on the reaction. Our initial assumption was that the reaction was rigorously stereoablative, and therefore stereoconvergent. However, the kinetics experiments described below suggest otherwise. Preparatively, the VCP starting material is used as a racemic mixture. The determination that ring-opening is rate-determining led us to suspect that the enantiomers may in fact exhibit different reactivities. Indeed, in studies comparing (±)-VCP with enantiopure VCP, we noted several differences. Kinetics studies revealed that (R)-VCP and (S)-VCP react at different rates, and both react at faster rates than (±)-VCP (Figures 2A and 2B). We also observed a kinetic resolution of the racemic starting material, as the VCP becomes moderately enriched in the (S)-enantiomer over the course of the reaction, to 20% ee at 90% conversion. Consistent with irreversible ring-opening, no racemization of VCP is observed when enantiopure starting material is used. The reaction rate is still independent of [TBVE], indicating that the use of enantiopure VCP does not induce a change in rate-determining step.
Figure 2.

Comparison of (±)-VCP, (S)-VCP, and (R)-VCP. In all graphs, duplicate data are simultaneously plotted and analyzed. (a) Relative rates and product enantiomeric excess. The ee values shown are the averages of multiple runs, and the standard deviation in these values is also shown. Ar = 4-CF3Ph. (b) Concentration profiles for enantiopure and racemic VCP. (c) Same-excess experiment with (R)-VCP, [1]T = 2.5 mM, truncated and time-adjusted to exclude induction period.46
Interpretation of the same-excess experiments, which were conducted under the same conditions as for (±)-VCP, is less straightforward (Figure 2C). Whereas the plots obtained with (±)-VCP did not overlay, the plots using both (R) - and (S)-VCP do appear to provide better, though imperfect, overlay, with a difference in line shape that suggests a corresponding difference in kinetic behavior. We also note that these enantiopure starting materials also exhibit behavior consistent with either dimerization or Michaelis–Menten kinetics.
In accord with the differential behavior of the enantioenriched starting materials, we observed that product ee is different depending on the absolute configuration of VCP starting material (Figure 2A). Relative to (±)-VCP, the product ee decreases when (S)-VCP is used and increases when (R)-VCP is used. These differences are quite small for the trans diastereomers, but more pronounced for the cis diastereomers. Indeed, this transmission of stereochemical information from starting material to product indicates that the reaction is not purely stereoablative, and is consistent with our conclusion that ring-opening is rate-determining.
The impact of the configuration of the VCP may be traced to its H-bonding capacity, both intra- and intermolecularly. The intermolecular H-bonding has been discussed above, in the context of VCP aggregation. There is also a H-bond within the VCP that is observable in the ground state. The amide N–H shifts in the VCP 1H NMR spectrum differ by 0.92 ppm; although these hydrogens are molecularly non-equivalent, this large difference could also be partially due to intramolecular H-bonding between the amides. Because each amide can act as either an H-bond donor or acceptor, the VCP can exist as four pseudo-diastereomers. Density functional theory (DFT) ground state optimizations paired with energies from local coupled-cluster theory (DLPNO-CCSD(T)) indicate that the configuration in which the vinyl group is trans to the H-bond donating amide is favored over the cis configuration (Figure 3A).47
Figure 3.

(a) Computational analysis of different configurations of the intramolecular H-bond. (b) Models for binding of (R)- and (S)-VCP to peptide catalyst. (c) Computational analysis of heterochiral and homochiral dimers. For all calculations, initial conformational searches were performed with metadynamics using CREST at the GFN2-xTB/ALPB(THF) level. Geometries were optimized at the B3LYP-D3BJ/6-31+G(d,p)/PCM(THF) level, followed by single-point energy calculations at the DLPNO-CCSD(T)/def2-TZVPP level, with M05-2X/6-31G(d)/SMD(THF) solvation free energies. Electronic energies are reported in parentheses.
These computational results aid in rationalizing the differences in rate and enantioselectivity for (R)- versus (S)-VCP. Based on the lower energy configuration of the intramolecular H-bond predominates, then only one amide is functionally available for binding to the catalyst. For (R)-VCP, the presumed mode of binding orients the vinyl group toward the cysteine radical; in contrast, for (S)-VCP, the vinyl group is oriented away from the radical (Figure 3B). This could account for the faster reaction rate of (R)-VCP. Although (S)-VCP still reacts rapidly, it may first have to rearrange to the less favorable trans configuration in order to bind to the catalyst and undergo ring-opening. It may also participate in a less selective pathway altogether, consistent with the decreased enantioselectivity when (S)-VCP is used.
We also performed conformational searches at the GFN2-xTB level, followed by DFT optimizations and high-level energy calculations to determine the most stable geometries for the heterochiral and homochiral dimers.48-50 Both geometries feature two H-bonding interactions between monomers, and the calculated dimerization free energies reflect that the heterochiral dimer is more stable relative to the homochiral dimer (Figure 3C). Based on these findings, we attribute the slower relative rate for (±)-VCP to differences in VCP dimer disaggregation, as described previously. Dissociation of the ground state VCP dimer will be less favorable by an order of magnitude for the more stable heterochiral dimer—which is inaccessible using enantiopure VCP—than for the homochiral dimer, leading to slower reaction rates for (±)-VCP. It should be noted that both computed dissociation equilibria favor the monomers overall (Kdiss > 1)—accordingly, we did not observe binding-related changes in 1H NMR chemical shifts of the amide protons in NMR titration experiments (see Supporting Information).
The key findings from the studies using enantiopure VCP are (1) the configuration of the VCP starting material influences the ee of the product, with modest changes observed depending on the enantiomer of VCP used; (2) (R)-VCP reacts at a faster rate than (S)-VCP, due to an intramolecular H-bond within the VCP that in turn influences VCP-catalyst interactions; (3) (±)-VCP reacts at a slower rate than either enantiopure starting material, as a result of different VCP dimerization energies.
From these observations, we also conclude that ring-opening seems likely to be the enantiodetermining step, given the effect of VCP configuration on enantioselectivity.
Kinetic analysis of catalyst lacking 4-proline substituent.
Given the significant effect of the 4-substituent of the proline on enantioselectivity, we compared the kinetics of reactions catalyzed by 1 and 2, which lacks this substituent (Figure 4A). The data obtained with 2 display some enigmatic, but reproducible, features. The reactions catalyzed by peptides with unsubstituted proline residues appear to occur in two distinct phases, with a period of reduced rate in between (Figure 4B). This suggests that the dependence of rate on [VCP] with catalyst 2 is more complicated than the simple 0.5-order scenario observed with catalyst 1. Because 2 is less sterically congested, it may be able to bind aggregated VCP, which then reacts directly in ring-opening without prior dissociation. However, because catalyst 2 is also simply a less stereoselective catalyst, we did not investigate this behavior further.
Figure 4.

Comparison of catalysts 1 and 2. In all graphs, duplicate data are simultaneously plotted and analyzed. (a) Relative rates and product enantiomeric excess. Ar = 4-CF3Ph. Values in parenthesis indicate the relative rate when compared with the 1-catalyzed reaction. (b) Concen-tration profiles. (c) Different-excess experiment, [VCP]0 = 0.05 M, [2]T = 2.5 mM. (d) Same-excess experiment, [2]T = 2.5 mM, truncated and time-adjusted to exclude induction period. (e) Comparison of (±)-VCP and enantiopure VCP using 2.
Catalyst 2 is also less active than catalyst 1. That is, the overall rate of the 2-catalyzed reaction is reduced relative to the 1-catalyzed reaction (maximum rate, krel = 1 for 1; krel = 0.6 for 2). The different-excess experiment reveals a change in the two-phase nature of the reaction, but the overall reaction rate appears unaffected by the changes in [TBVE] (Figure 4C). Thus, we conclude that the absence of the 4-proline amide does not induce a change in rate-determining step. The same-excess experiment indicates that 2 experiences catalyst deactivation (Figure 4D) in contrast to the catalyst activation behavior observed with 1. Deactivation of 2 could result from unproductive H-bonding between the product and catalyst, which is perhaps less likely with 1 due to repulsive nonbonding interactions involving the 4-proline substituent.
As in the 1-catalyzed reaction, differences in reaction rates are observed when (±)-, (R)-, and (S)-VCP are used in the 2-catalyzed reaction (Figure 4E). We attribute the reactivity differences between (R)- and (S)-VCP to different orientations of the vinyl group in the VCP-catalyst complex. Because the 4-proline amide may not play a specific role in binding VCP, we expect that the same rate differences between the enantiomers of starting material would be observed with both catalysts 1 and 2. The reduced reaction rate of (±)-VCP relative to enantiopure VCP can still be attributed to the lower reactivity of the heterochiral dimer as opposed to the homochiral dimer. Furthermore, the stalled reactivity observed with (±)-VCP and catalyst 2 is absent when enantiopure VCP is used, which suggests that the reduced reactivity is also related to aggregation.
These data support a scenario wherein the VCP remains aggregated to a greater extent when catalyst 2 is employed. This hypothesis is also consistent with the overall decreased rate of reaction with catalyst 2 relative to catalyst 1; the more complex rate dependence on [VCP] when 2 is used, which may reflect that the VCP does not readily dissociate under these conditions; and the lack of kinetic resolution of (±)-VCP in the 2-catalyzed reaction. The same-excess experiments also suggest that the deaggregation phenomena are mutually associated with the interaction between catalyst 2 and the VCP.
Our comparison of catalysts 1 and 2 thus show that (1) the 2-catalyzed reaction proceeds at a slower rate relative to the 1-catalyzed reaction; (2) the relative rates of reaction using (R)-, (S)-, and (±)-VCP follow the same trends in both the 1- and 2-catalyzed reaction, indicating that H-bonding between the VCP and peptide backbone is responsible for these reactivity differences; (3) no kinetic resolution is observed in the 2-catalyzed reaction, suggesting that catalyst-assisted deaggregation of the VCP dimer is facilitated by the 4-proline amide, and proceeds in a configuration-dependent manner.
Updated catalytic cycle.
Our kinetic evaluation of the peptide-catalyzed VCP cycloaddition reveal several previously undocumented facts and unappreciated insights: (1) ring-opening is likely both the rate- and enantio-determining step; (2) cyclization is diastereodetermining; and (3) the VCP and catalyst engage in intermolecular associations that impact overall rate, stereoselectivity, and even the degree of stereoconvergence (Scheme 3). Accordingly, the kinetics also support a complicated scenario wherein aggregated species exist on the productive, photochemical path leading to S–S bond homolysis. Similarly, as shown with dashed lines in Scheme 3, the mechanism of thiyl recombination to reform the disulfide precatalyst is also likely subject to analogous aggregation effects, but our data do not resolve the details of this process.
Scheme 3.

Updated catalytic cycle.
One key point of stereochemical significance —that ring-opening, which we and others generally assumed to be stereoablative, is the enantiodetermining step—is particularly notable. Ring-opening does not merely lead cleanly to a common intermediate I. Rather, ring-opening likely leads to a distribution of intermediates IR and IS—each of which also presents multiple conformers of varying reactivity (Scheme 2A). The populations of the non-interconverting conformers, generated directly from the ring-opening step, and their relative reactivities thus directly influence the stereochemical outcomes in the usual manner. Notably, the different binding modes for (R)- and (S)-VCP also translate to different distributions of these conformers, resulting in the observed differences in enantiomeric excess. Rapid olefin addition to these conformers also does not lead to a common intermediate II. Instead, different intermediates (IIR and IIS) are likely formed, which each cyclize with different diastereoselectivity. Rotation about the C–C bond (IIR vs IIS) leads to the different diastereomers, and it is also conceivable that this rotation is influenced by the conformation and operation of other noncovalent interactions within these intermediates as well. This hypothesis also accounts for the fact that the cis and trans diastereomers are formed with different enantiomeric excesses.
To gain additional atomic-level resolution insight into the structures and energetics of species on the catalytic cycle, we identified intermediates IR and IS as candidates for computational analysis, on account of their key enantiodetermining role in our hypothesis. These intermediates also exhibit relatively tractable structural complexity and flexibility, compared to the ensuing open-shell transition states and intermediates IIR and IIS. The results of these calculations are shown in Figure 5. Notably, in addition to their common β-turn geometry, both structures feature an intramolecular H-bond between the prolyl carbonyl O and the 4-proline amide N–H. The ring-opened VCP in IS is held over the β-turn by a H-bond between the C-terminal amide carbonyl O and a VCP amide N–H (Figure 5, left). In this arrangement, one face of the formal radical center is sterically obstructed by the 4-proline substituent. This arrangement may well bias the approach of TBVE to favor the unobstructed face of the radical center—reinforcing our previous experimental findings that suggested a steric role of the 4-proline substituent in enantioinduction.4 However, in IR the ring-opened VCP component extends away from the peptide backbone, anchored only by a C–H–π interaction between the amide phenyl group and the t-Bu of the N-terminal Boc group (Figure 5, right). Consequently, both faces of the radical center are accessible to TBVE approach. At the same time, IR exhibits a higher energy, likely as a consequence of fewer intramolecular noncovalent interactions and its extended structure.
These calculated structures provide atomistic pictures that support the plausibility of IR and IS as intermediates. A comprehensive computational assessment of various contingencies for these flexible systems, for example combining molecular dynamics simulations with DFT-level refinement,51-54 could well reveal more insight. Nevertheless, the structures of IR and IS ground each as reasonable catalyst-substrate complexes of differing energy along the complex reaction coordinate surface.
Conclusions
The detailed kinetic analysis of many enantioselective reactions unveils phenomena that are often not apparent at the outset of a project. In the present case, the design principles involving highly functionalized and flexible catalysts, and similarly functionalized substrates, created complex scenarios with a multitude of noncovalent interactions that simultaneously operate to deliver the observed results. These studies, which shed light on the current VCP-based cycloaddition, are almost certainly harbingers of similarly complicated phenomena underlying many other enantioselective catalytic reactions where noncovalent interactions operate.55-57 Thus, in depth kinetic studies of this type, while often time-intensive and difficult to interpret, are an indispensable activity for not only supporting hypotheses but also unveiling blind spots in an experimentally grounded manner.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Professor Donna Blackmond (Scripps Research), Professor Nilay Hazari (Yale University) and Dr. Jonathan M. Ryss (Yale University) for helpful discussions, as well as the Yale Center for Research Computing for computational resources.
Funding Sources
This research was supported by the National Institutes of Health (NIHGMS R35 132092).
Footnotes
The authors report no competing financial interests.
Supporting Information. Full experimental details, kinetics plots, raw data, and details of 1H NMR binding studies, as well as full computational methods, energies, and coordinates. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Knappe J; Elbert S; Frey M; Wagner AFV Pyruvate formase-lyase mechanism involving the protein-based glycyl radical. Biochem. Soc. Trans 1993, 21, 731–734. [DOI] [PubMed] [Google Scholar]
- (2).Licht S; Gerfen GJ; Stubbe J Thiyl Radicals in Ribonucleotide Reductase. Science 1996, 271, 477–481. [DOI] [PubMed] [Google Scholar]
- (3).Stubbe J; van der Donk WA Protein Radicals in Enzyme Catalysis. Chem. Rev 1998, 98, 705–762. [DOI] [PubMed] [Google Scholar]
- (4).Ryss JM; Turek AK; Miller SJ Disulfide-Bridged Peptides That Mediate Enantioselective Cycloadditions through Thiyl Radical Catalysis. Org. Lett 2018, 20, 1621–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Dénès F; Pichowicz M; Povie G; Renaud P Thiyl Radicals in Organic Synthesis. Chem. Rev 2014, 114, 2587–2693, and references therein. [DOI] [PubMed] [Google Scholar]
- (6).McLean JT; Benny A; Nolan MD; Swinand G; Scanlan EM, Cysteinyl radicals in chemical synthesis and in nature. Chem. Soc. Rev 2021, Advance Article. doi: 10.1039/D1CS00254F. [DOI] [PubMed] [Google Scholar]
- (7).Cai Y; Roberts BP Tocher DA Carbohydrate-derived thiols as protic polarity-reversal catalysts for enantioselective radical-chain reactions. J. Chem. Soc., Perkin Trans 1 2002, 1376–1386. [Google Scholar]
- (8).Shin NY; Ryss JM; Zhang X; Miller SJ; Knowles RR Light-driven deracemization enabled by excited-state electron transfer. Science 2019, 366, 364–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Feldman KS; Romanelli AL; Ruckle RE Jr.; Miller RF Cyclopentane Synthesis via Free-Radical-Mediated Addition of Functionalized Alkenes to Substituted Vinylcyclopropanes. J. Am. Chem. Soc 1988, 110, 3300–3302. [Google Scholar]
- (10).Miura K; Fugami K; Oshima K; Utimoto K Synthesis of vinylcyclopentanes from vinylcyclopropanes and alkenes promoted by benzenethiyl radical. Tetrahedron Lett. 1988, 29, 5135–5138. [Google Scholar]
- (11).Feldman KS; Simpson RE Oxygenation of Substituted Vinylcyclopropanes: Preparative and Mechanistic Studies. J. Am. Chem. Soc 1989, 777, 4878–4886. [Google Scholar]
- (12).Feldman KS; Ruckle RE Jr.; Romanelli AL Vinylcyclopentene synthesis via phenylthio radical catalyzed addition of electron deficient alkynes to substituted vinylcyclopropanes. Tetrahedron Lett. 1989, 30, 5845–5848. [Google Scholar]
- (13).Feldman KS; Romanelli AL; Ruckle RE Jr.; Jean G Vinylcyclopenate Synthesis via Phenylthio Radical Catalyzed Alkenylation of Vinylcyclopropanes: Preparative and Mechanistic Studies. J. Org. Chem 1992, 57; 100–110. [Google Scholar]
- (14).Feldman KS; Berven HM; Weinreb PH 2,2-Dihalovinylcyclopropanes as Highly Diastereoselective Three-Atom Addends in Phenylthio Radical Mediated Vinylcyclopentane Synthesis. J. Am. Chem. Soc 1993, 115, 11364–11369. [Google Scholar]
- (15).Andrey O; Camuzat-Dedenis B; Chabaud L; Julienne K; Landais Y; Parra-Rapado L; Renaud P Free-radical functionalisation of vinylcyclopropanes. Tetrahedronh 2003, 59, 8543–8550. [Google Scholar]
- (16).Rahaman H; Ueda M; Miyata O; Naito T Two Novel Domino Re-actions Triggered by Thiyl-Radical Addition to Vinylcyclopropyl Oxime Ether. Org. Lett 2009, 77, 2651–2654. [DOI] [PubMed] [Google Scholar]
- (17).Singleton DA; Church KM [3+2] Methylenecyclopentane annulations of unactivated and electron-rich olefins with 2-(phenylsulfonyl)-1-methylenecyclopropanes. J. Org. Chem 1990, 55, 4780–4782 [Google Scholar]
- (18).Singleton DA; Huval CC; Church KM; Priestley ES Methylenecyclopropanecarboxylates and -dicarboxylates, efficient reagents for the [3+2] methylenecyclopentane annulation of unactivated and electron-rich alkenes. Tetrahedron Lett. 1991, 32, 5765–5768 [Google Scholar]
- (19).Huval CC; Church KM; Singleton DA Free-Radical Mediated [3+2] Methylenecyclopentane Annulations of Electron-Poor Alkenes. Synlett 1994, 1994, 273–274. [Google Scholar]
- (20).Huval CC; Singleton DA Versatile [3+2] methylenecyclopentane annulations of unactivated and electron-rich olefins with [(trimethylsilyl)methylene]cyclopropanedicarboxylates. J. Org. Chem 1994, 59, 2020–2024. [Google Scholar]
- (21).Feldman KS; Ruckle RE Jr.; Ensel SM; Weinreb PH Synthesis of a Chiral Binaphthyldisulfide: A Potentially Useful Reagent for Catalytic Asymmetric Synthesis. Tetrahedron Lett. 1992, 33, 7101–7102. [Google Scholar]
- (22).Hashimoto T; Kawamata Y; Maruoka K An organic thiyl radical catalyst for enantioselective cyclization. Nat. Chem 2014, 6, 702–705. [DOI] [PubMed] [Google Scholar]
- (23).Hashimoto T; Takino K; Hato K; Maruoka K A Bulky Thiyl-Radical Catalyst for the [3+2] Cyclization of N-Tosyl Vinylaziridines and Alkenes. Angew. Chem. Int. Ed 2016, 55, 8081–8085. [DOI] [PubMed] [Google Scholar]
- (24).Sonntag L-S; Schweizer S; Ochsenfeld C; Wennemers H The “Azido Gauche Effect” – Implications for the Conformation of Azidoprolines. J. Am. Chem. Soc 2006, 128, 14697–14703. [DOI] [PubMed] [Google Scholar]
- (25).Schnitzer T; Wennemers H Effect of γ-Substituted Proline Derivatives on the Performance of the Peptidic Catalyst H-DPro-Pro-Glu-NH2. Synthesis 2018, 50, 4377–4382. [Google Scholar]
- (26).Based on the literature, the cis product is predicted to be favored. Beck-with ALJ; Easton CJ Serelis AK Some Guidelines for Radical Reactions. J. Chem. Soc., Chem. Commun 1980, 482–483. [Google Scholar]
- (27).Wagner JP; Schreiner PR, London Dispersion in Molecular Chemistry—Reconsidering Steric Effects. Angew. Chem. Int. Ed 2015, 54 (42), 12274–12296. [DOI] [PubMed] [Google Scholar]
- (28).Yepes D; Neese F; List B; Bistoni G, Unveiling the Delicate Balance of Steric and Dispersion Interactions in Organocatalysis Using High-Level Computational Methods. J. Am. Chem. Soc 2020, 142(7), 3613–3625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Eschmann C; Song L; Schreiner PR, London Dispersion Interactions Rather than Steric Hindrance Determine the Enantioselectivity of the Corey–Bakshi–Shibata Reduction. Angew. Chem. Int. Ed 2021, 60 (9), 4823–4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Whitesides GM; Ismagilov RF Complexity in Chemistry. Science 1999, 284, 89–92. [DOI] [PubMed] [Google Scholar]
- (31).Blackmond DG Reaction Progress Kinetic Analysis: A Powerful Methodology for Mechanistic Studies of Complex Catalytic Reactions. Angew. Chem. Int. Ed 2005, 44, 4302–4320. [DOI] [PubMed] [Google Scholar]
- (32).Blackmond DG Kinetic Profiling of Catalytic Organic Reactions as a Mechanistic Tool. Am. Chem. Soc 2015, 137, 10852–10866. [DOI] [PubMed] [Google Scholar]
- (33).This plot and all other plots not shown in the main text are presented in the supporting information.
- (34).Burés J Variable Time Normalization Analysis: General Graphical Elucidation of Reaction Orders from Concentration Profiles. Angew. Chem. Int. Ed 2016, 55, 16084–16087. [DOI] [PubMed] [Google Scholar]
- (35).Burés J A Simple Graphical Method to Determine the Order in Catalyst. Angew. Chem. Int. Ed 2016, 55, 2028–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kagan HB Practical Consequences of Non-Linear Effects in Asymmetric Synthesis. Adv. Synth. Catal 2001, 343, 227–233. [Google Scholar]
- (37).Satyanarayana T; Abraham S; Kagan HB Nonlinear Effects in Asymmetric Catalysis. Angew. Chem. Int. Ed 2009, 48, 456–494. [DOI] [PubMed] [Google Scholar]
- (38).Ford DD; Lehnherr D; Kennedy CR; Jacobsen EN On- and Off-Cycle Catalyst Cooperativity in Anion-Binding Catalysis. J. Am. Chem. Soc 2016, 138, 7860–7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).This is true for reactions catalyzed both by 1 and by n-butyl disulfide.
- (40).Wongkongkathep P; Li H; Zhang X; Orgorzalek Loo RR; Julian RR; Loo JA Enhancing protein disulfide bond cleavage by UV excitation and electron capture dissociation for top-down mass spectrometry. Int. J. Mass. Spectrom 2015, 390, 137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Bowry VW; Lusztyk J; Ingold KU Calibration of a New Horologery of Fast Radical “Clocks.” Ring-Opening Rates for Ring- and α-Alkyl-Substituted Cyclopropylcarbinyl Radicals and for the Bicyc lo[2.1.0]pent-2-yl Radical. J Am. Chem. Soc 1991, 131, 5687–5698. [Google Scholar]
- (42).Baxter RD; Sale D; Engle KM; Yu J-Q; Blackmond DG Mechanistic Rationalization of Unusual Kinetics in Pd-Catalyzed C–H Olefination . J. Am. Chem. Soc 2012, 134, 4600–4606. [DOI] [PubMed] [Google Scholar]
- (43).The post-induction period was considered to be t = 0, and the other experiments were shifted accordingly.
- (44).See supporting information for a graphical representation of these data.
- (45).Rosner T; Pfaltz A; Blackmond DG “Observation of Unusual Kinetics in Heck Reactions of Aryl Halides: The Role of Non-Steady-State Catalyst Concentration. J. Am. Chem. Soc 2001, 123, 4621–4622. [DOI] [PubMed] [Google Scholar]
- (46).Same-excess experiment using (S)-VCP can be found in the supporting information.
- (47).Frisch MJ et al. Gaussian 16, Revision B.01; Gaussian Inc.: Wallingford, CT, 2016; see Supporting Information for full citation. [Google Scholar]
- (48).Bannwarth C; Ehlert S; Grimme S GFN2-xTB-An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theoiy Comput 2019, 15, 1652–1671. [DOI] [PubMed] [Google Scholar]
- (49).Grimme S Exploration of Chemical Compound, Conformer, and Reaction Space with Meta-Dynamics Simulations Based on Tight-Binding Quantum Chemical Calculations. J. Chem. Theory Comput 2019, 15, 2847–2862. [DOI] [PubMed] [Google Scholar]
- (50).Pracht P; Bohle F; Grimme S Automated exploration of the low-energy chemical space with fast quantum chemical methods. Phys. Chem. Chem. Phys 2020, 22, 7169–7192. [DOI] [PubMed] [Google Scholar]
- (51).Thiel W, Computational Catalysis—Past, Present, and Future. Angew. Chem. Int. Ed 2014, 53, 8605–8613. [DOI] [PubMed] [Google Scholar]
- (52).Harvey JN; Himo F; Maseras F; Perrin L, Scope and Challenge of Computational Methods for Studying Mechanism and Reactivity in Homogeneous Catalysis. ACS Catal. 2019, 9, 6803–6813. [Google Scholar]
- (53).Yan XC; Metrano AJ; Robertson MJ; Abascal NC; Tirado-Rives J; Miller SJ; Jorgensen WL Molecular Dynamics Simulations of a Conformationally Mobile Peptide-Based Catalyst for Atroposelective Bromination. ACS Catal. 2018, 8, 9968–9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Stone EA; Hosseinzadeh P; Craven TW; Robertson MJ; Han Y; Hsieh S-Y; Metrano AJ; Baker D; Miller SJ Isolating Conformers to Assess Dynamics of Peptidic Catalysts Using Computationally Designed Macrocyclic Peptides. ACS Catal. 2021, 11, 4395–4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Toste FD; Sigman MS; Miller SJ “Pursuit of Noncovalent Interactions for Strategic Site-Selective Catalysis.” Acc. Chem. Res 2017, 58, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Knowles RR; Jacobsen EN “Attractive Noncovalent Interactions in Asymmetric Catalysis” PNAS 2010, 107, 20678–20685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Metrano AJ; Chinn AJ; Shugrue CR; Stone EA; Kim B; Miller SJ “Asymmetric Catalysis Mediated by Synthetic Peptides, Version 2.0: Expansion of Scope and Mechanisms” Chem. Rev 2020, 120, 11479–11615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.