

Abstract
Epigenetic mechanisms such as post-translational histone modifications are increasingly recognized for their contribution to gene activation and silencing in the brain. Histone acetylation in particular has been shown to be important both in hippocampal long-term potentiation (LTP) and memory formation in mice. The involvement of the epigenetic modulation of memory formation has also been proposed in neuropathological models, although up to now no clear-cut connection has been demonstrated between histone modifications and the etiology of Alzheimer’s disease (AD). Thus, we have undertaken preclinical studies in the APP/PS1 mouse model of AD to determine whether there are differences in histone acetylation levels during associative memory formation. After fear conditioning training, levels of hippocampal acetylated histone 4 (H4) in APP/PS1 mice were about 50% lower than in wild-type littermates. Interestingly, acute treatment with a histone deacetylase inhibitor, Trichostatin A (TSA), prior to training rescued both acetylated H4 levels and contextual freezing performance to wild-type values. Moreover, TSA rescued CA3-CA1 LTP in slices from APP/PS1 mice. Based on this evidence, we propose the hypothesis that epigenetic mechanisms are involved in the altered synaptic function and memory associated with AD. In this respect, histone deacetylase inhibitors represent a new therapeutic target to effectively counteract disease progression.
Keywords: Acetylation, Alzheimer’s disease, HDACs, histone, long term potentiation
INTRODUCTION
Evidence from several laboratories have shown that long-term memory and synaptic plasticity rely on gene expression after an early induction phase, which is characterized by the activation of a number of pathways both at the membrane and cytoplasmic level (for a review, see [1]). More recently, a fine regulation of memory-related genes and long-term synaptic plasticity have been discovered to involve epigenetic factors [2]. Indeed, epigenetic modifications, such as DNA methylation and histone post-translational modifications, profoundly affect the ability of polymerases to interact with the open reading frame of DNA without changing the DNA sequence itself. Hence, it would not be surprising that deregulation of epigenetic mechanisms might lead to the disruption of memory-associated gene expression and synaptic plasticity [2].
Memory loss is the main symptom of Alzheimer’s disease (AD). Interestingly, a large body of evidence supports the idea that AD begins as a synaptic disorder that progressively involves larger areas in the brain, leading to the deterioration of memory and learning [3]. Familial forms of AD [4,5] have been linked to mutations in the gene for amyloid-β protein precursor (AβPP) [4] and two other genes, presenilin 1 (PS1) and presenilin 2 (PS2) [6,7]. Proteolytic processing of AβPP through the presenilins leads to the formation of amyloid-β (Aβ) peptides. Indeed, one of the neuropathological hallmarks of AD is the progressive accumulation of Aβ inside the brain [8]. Such an accumulation can lead to synaptic disruption since high concentrations of Aβ have been found to inhibit long-term potentiation (LTP), a cellular model of memory [9-14]. In line with these observations, studies on human Aβ-producing transgenic mice have often revealed significant deficits in hippocampal basal synaptic transmission (BST), LTP, and memory (for a review see [15]).
Among the possible epigenetic factors, only DNA methylation has been previously hypothesized to play a possible role in AD [16,17]. Strikingly, there is no demonstration of defects in histone acetylation during memory formation in amyloid-depositing mice. We have now undertaken preclinical studies in a double transgenic mouse model (APP/PS1), carrying both the AβPP (K670N:M671L) and the PS1 mutation (M146L), to determine whether histone acetylation levels are different in comparison to WT mice during memory formation. These results identify AD as a disease with an epigenetic etiology at the level of histone acetylation.
MATERIALS AND METHODS
Animals
All experiments were performed with the approval of the Columbia University Animal Care and Use Committee in accordance with the guidelines for the humane treatment of animals (protocol #AC-AAAA8010). Hemizygous transgenic (HuAPP695SWE) 2576 mice expressing mutant human AβPP (K670N, M671L) [18] were crossed with hemizygous PS1 mice that express mutant human PS1 (M146V; line 6.2) [19]. The offspring, double-transgenic mice overexpressing APP/PS1, were compared with their wild type (WT) littermates so that age and background strain were comparable. To identify the genotype of the animals, we used DNA extracted from tail tissue as previously described [18-20].
Drug administration
The class I/II HDAC inhibitor Trichostatin A (TSA) from Biomol was solubilized in 100% dimethylsulfoxide (DMSO) and then diluted with saline solution to a concentration of 0.5 μg/μl. TSA or vehicle was administered two hours before the behavioral tests by intraperitoneal injection (i.p.) of 2 mg/kg body weight. Three to four-month-old APP/PS1 and WT mice were evenly separated into 4 groups: APP/PS1 mice treated with vehicle, APP/PS1 mice treated with TSA, WT mice treated with vehicle, and WT mice treated with TSA. After the behavioral tests, animals were sacrificed for western blotting analysis. For electrophysiology experiments, TSA was perfused onto the slices for 30 min at a concentration of 1.65 μM.
Western blotting
Hippocampal lysates for immunoblotting were prepared as previously described [21] with slight modifications. Hippocampal tissue was homogenized in lysis buffer (62.5 mM Tris-HCl pH 6.8, 3% SDS, 1 mM DTT) and incubated at 4 °C for 10 min, then sonicated before centrifugation at 2,000 rpm for 5 min. Whole cell extracts were electrophoresed on 10–20% gradient PAGE gel (Invitrogen) and then immunoblotted. Antibodies were used at a 1:1,000 concentration for immunoblotting. All anti-histone antibodies were from Millipore. β-III-Tubulin antibody was purchased from Promega. Immunoblot data were quantified by measuring the band intensity using imaging software (NIH ImageJ). For quantitative immunoblot analysis, equal amounts of proteins were loaded into each lane. To confirm equal loading, blots were reprobed with corresponding pan-antibodies or antibodies for housekeeping proteins such as β-III-Tubulin. For quantification, we always used a signal in the linear range.
Contextual and cued fear conditioning
Our conditioning chamber was located inside a sound-attenuating box (72 × 51 × 48 cm). A clear Plexiglas window (2 × 12 × 20 cm) allowed the experimenter to film the mouse performance with a camera placed on a tripod and connected to Freezeframe software (MED Associates Inc.). To provide background white noise (72 dB), a single computer fan was installed in one of the sides of the sound-attenuating chamber. The conditioning chamber (33 × 20 × 22 cm) was made of transparent Plexiglas on two sides and metal on the other two. One of the metal sides had a speaker and the other had a 24 V light. The chamber had a 36-bar insulated shock grid floor. The floor was removable and after each use we cleaned it with 75% ethanol and then with water. Only one animal at a time was present in the experimentation room. The other mice remained in their home cages.
During the contextual conditioning experiment, mice were placed in the conditioning chamber for 2 min. In the last 2 seconds of the 2 min, mice were given a foot shock (US) of 0.50 mA for 2 seconds through the bars of the floor. After the US, the mice were left in the conditioning chamber for another 30 seconds and then were placed back in their home cages. “Freezing” behavior, defined as the absence of all movement except for that necessitated by breathing, was assigned scores using Freezeview software (MED Associates Inc.). For evaluation of contextual fear learning, freezing at 24 hours post-training was measured for 5 consecutive min in the chamber in which the mice were trained. Twenty-four hours after the contextual testing, cued fear conditioning was evaluated by placing the mice in a novel context (triangular cage with a smooth flat floor) for 2 min (pre-CS test), after which they were exposed to the CS for 3 min (CS test), and freezing was measured. In a separate set of experiments, we tested whether the four different experimental groups of mice had similar exploratory behavior and anxiety by carrying out the open field test. Animals were positioned in an open arena with a floor that was divided into compartments. The internal dimensions of the arena were 72 × 72 × 33 cm. An area measuring 36 × 36 cm in the center of the open field was defined as the “central compartment”. Behavioral scoring was evaluated by the percentage of time spent in the center compartment and the number of entries into the center compartment [22]. No differences were found among the four groups of mice (data not shown).
Measurement of LTP
Mice were decapitated, and their hippocampi were removed. Transverse hippocampal slices of 400 μm thickness were made on a tissue chopper and transferred to an interface chamber where they were maintained at 29°C. Saline recording solution (124.0 mM NaCl, 4.4 mM KCl, 1.0 mM Na2HPO4, 25.0 mM NaHCO3, 2.0 CaCL2, 2.0 mM MgSO4, 10 mM glucose) was perfused at 1–2 ml/minute and continuously bubbled with 95% O2 and 5% CO2. Slices were permitted to recover for at least 90 min before recording. A concentric bipolar platinum-iridium stimulation electrode was placed at the level of the Schaeffer collateral fibers, whereas the recording electrode, a low-resistance glass recording microelectrode filled with saline solution, was placed in CA1 stratum radiatum to record the extracellular field excitatory postsynaptic potential (fEPSP). An input – output curve was used to set the baseline fEPSP at ≈35% of the maximal slope. Baseline stimulation was delivered every minute (0.01-ms duration pulses) for 15 min before beginning the experiment to assure the stability of the response. TSA (1.65 μM) or vehicle (0.1%DMSO in recording solution) was added to perfused slices for 30 min in interleaved experiments. LTP was induced by using θ-burst stimulation (4 pulses at 100 Hz, with the bursts repeated at 5 Hz and each tetanus including three 10-burst trains separated by 15 s). Responses were recorded for 120 minutes after tetanization.
Statistical analysis
For all experiments, mice were coded by “blind” investigators with respect to treatment and genotype. Data are expressed as mean ± SEM. Statistical analysis was performed with one-way ANOVA (for behavioral experiments), two-way ANOVA with repeated measures (for LTP) and Student’s t test (pairwise comparisons). The level of significance was set for p < 0.05.
RESULTS
APP/PS1 mice display a reduced endogenous level of histone 4 acetylation in response to a learning task
Similar to humans affected by AD [23], APP/PS1 mice show a deficit in associative memory [24]. In mice, associative learning can be assessed by contextual fear conditioning. This behavioral task, which is based on the association of a neutral stimulus with an aversive one, is dependent on hippocampal function [25]. To determine whether epigenetic changes at the level of histone acetylation might occur in AD, we asked whether the memory deficit observed in APP/PS1 mice are associated with altered chromatin structure in the hippocampus. To address this question, acetylation of histone 4 (H4) was measured in the hippocampi from 3 to 4 month old WT and APP/PS1 animals, after contextual fear conditioning. Vehicle control solution was administered via i.p. injection 2 hours before fear conditioning training and 1 hour after training APP/PS1 (n = 4) and WT (n = 4) mice were euthanized and hippocampi were extracted. Western blot analysis of hippocampal extracts demonstrated that, compared to WT mice, APP/PS1 mice showed an overall reduction of approximately 50% in acetylated H4 levels (t = 2.702, p = 0.0355) (Fig. 1). Interestingly, in interleaved experiments, it was found that HDAC inhibition rescued the H4 acetylation level defect observed in APP/PS1 mice (n = 4). Injection of the HDAC inhibitor, TSA (2 mg/kg body weight, i.p., 2 hours prior to training for contextual fear conditioning), enhanced H4 acetylation in APP/PS1 mice (t = 3.283, p = 0.0168 compared to vehicle treated APP/PS1 littermates), reaching the same levels of acetylation as TSA-treated WT mice (n = 5) (Fig. 1A). We next wanted to determine if there are any changes in the basal acetylation levels of H4 in APP/PS1 mice, compared to wild-type mice. We therefore measured acetylation of H4 in hippocampi from 3 to 4 month old WT (n = 3) and APP/PS1 (n = 3) animals which were exposed to the context without receiving an electrical shock. It was found that there is no difference between the two experimental groups in basal acetylated H4 (t = 0.076, p = 0.9427) (Fig. 1B). This demonstrates that the overexpression of mutated AβPP and PS1 transgenes affects epigenetic changes at the level of H4 acetylation after hippocampal-dependent learning, strongly supporting the hypothesis that AD is a disease with an epigenetic motif.
Fig. 1.
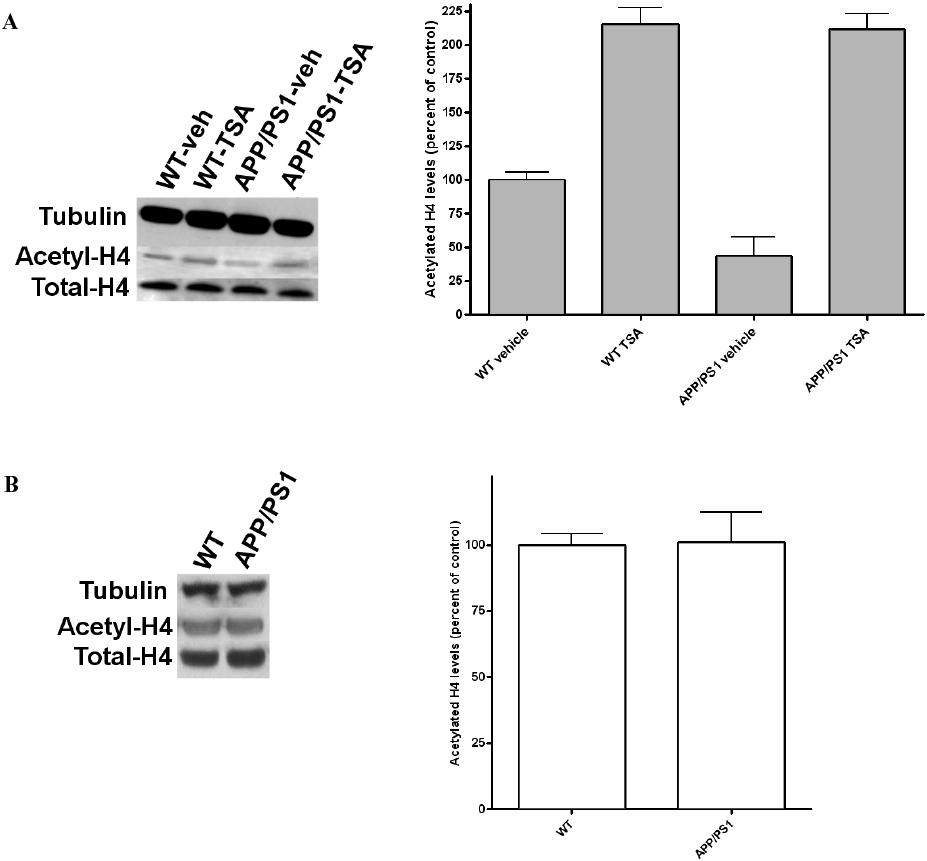
Histone acetylation reduction in APP/PS1 mice. (A) Western blotting of protein extracts from APP/PS1 and WT mice, which were injected with vehicle and TSA 2 hours before training, and euthanized 1 hour after contextual fear conditioning. Vehicle-treated APP/PS1 animals showed a decrease in acetylated H4 levels. However, TSA-treated APP/PS1 mice showed an enhancement of H4 acetylation, reaching the same levels of acetylation as TSA-treated WT mice. Results were normalized against vehicle-treated WT mice. (B) Basal acetylation levels of H4 in APP/PS1 mice and WT littermates, which were exposed to the context without receiving an electrical shock, were similar. The data shown in (A) and (B) are presented as a ratio of acetylated-H4 to total H4.
HDAC inhibition rescues associative memory in APP/PS1 mice
Next, we asked whether HDAC inhibition was also capable of ameliorating the associative memory deficit observed in the APP/PS1 mouse [24]. Mice were divided into four groups: vehicle-treated WT mice, vehicle-treated APP/PS1 mice, TSA-treated WT mice, and TSA-treated APP/PS1 mice. In a series of preliminary experiments, the four groups of mice were tested to determine if they had different perceptions of the electrical shock used in the behavioral task. We found no differences among the four experimental groups [24]. APP/PS1 and WT littermates of 3–4 months of age were subjected to a standard fear-conditioning paradigm [26]. Two hours after injection of TSA (2 mg/kg body weight; i.p), the animals were placed in a novel context (fear-conditioning box) and were exposed to a mild foot shock (training phase of the fear conditioning). Conditioning was assessed 24 hours later by measurement of “freezing” behavior–the absence of all movement except for that necessitated by breathing – in response to the context (contextual conditioning). During the training phase, no difference in the freezing behavior of the four experimental groups was seen (F3,47 = 0.02997, p = 0.9929, n = 12–13/group) (Fig. 2). Twenty-four hours later, when contextual conditioning was assessed, vehicle-treated APP/PS1 were unable to replicate the increase in the freezing time of vehicle-treated WT littermates (F3,47 = 0.0526, p = 0.0280, n = 12–13/group). Freezing in vehicle-treated APP/PS1 mice was about 46% that of vehicle-treated WT mice [vehicle-treated APP/PS1 mice: 14.88 ± 2.97% vs. vehicle-treated WT littermates: 31.68 ± 5.32%; n = 13 (11 females plus 2 males) and n = 13 (11 females plus 2 males), respectively; t = 2.76, p = 0.0109; Fig. 2]. However, the freezing time was increased in APP/PS1 mice after the injection of TSA to about 236.27% of vehicle-treated APP/PS1 mice [TSA-treated APP/PS1 mice: 35.15 ± 6.99%, n = 12 (10 females plus 2 males); t = 2.744, p = 0.0116; Fig. 2]. Statistical analysis revealed that freezing in APP/PS1 mice was not significantly different with respect to both TSA-treated (t = 0.1902, p = 0.8508) and vehicle-treated WT mice (t = 0.3982, p = 0.6941). TSA-treated WT mice showed a slight non-significant increase in freezing compared to vehicle-treated WT mice [TSA-treated WT mice: 36.95 ± 6.43%; n = 13 (11 females plus 2 males); t = 0.6316, p = 0.5336, Fig. 2]. We next tested cued fear conditioning, a hippocampus-independent task [24], and did not find a difference in freezing behavior among the 4 groups (both in the pre-CS group, F3,25 = 0.8763, p = 0.4666, and in the CS group, F3,24 = 0.6398, p = 0.5968, data not shown). This result suggests that the function of the amygdala, which is involved mainly in cued conditioning and is known to be normal in APP/PS1 mice [24], is not affected by the inhibition of HDACs by TSA, as previously demonstrated [27]. Taken together, these data show that the inhibition of histone deacetylation is able to re-establish normal associative memory in the APP/PS1 mouse while also restoring H4 acetylation levels.
Fig. 2.
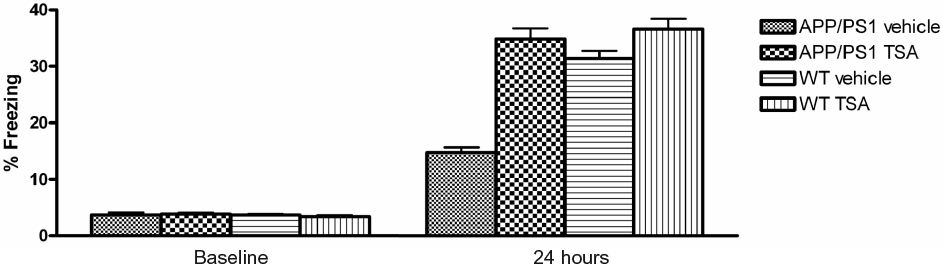
TSA injection improves contextual fear conditioning performance in APP/PS1 mice. 3 to 4 month-old APP/PS1 and WT littermates treated with TSA or vehicle 2 hours prior to training show no difference in immediate freezing in the training chamber. However, vehicle-treated APP/PS1 mice show reduced freezing responses compared to vehicle-treated littermates when tested for contextual fear conditioning 24 hours after training. Injection of TSA two hours prior to training ameliorates the deficit in freezing responses in APP/PS1 mice after 24 hours but does not further improve freezing in WT mice.
HDAC inhibition ameliorates deficits in hippocampal long-term potentiation in APP/PS1 mice
Given that learning and memory are thought to be associated with the long-term strengthening of synaptic connections, we next decided to test if TSA ameliorates hippocampal synaptic function in APP/PS1 mice. These animals show an impairment of LTP by 3 months of age [20]. Therefore, we tested whether inhibition of histone de-acetylation can re-establish normal synaptic function in 3-4 month old transgenic mice. As previously shown [20], vehicle-treated APP/PS1 mice had a reduction in LTP compared to vehicle-treated WT littermates. A two-way ANOVA revealed a significant difference in the LTP of APP/PS1 and WT mice (F1,10 = 5.536, p = 0.04). Treatment of hippocampal slices with TSA (1.65 μM) for 30 min before inducing LTP through tetanic stimulation was able to ameliorate the potentiation deficit in APP/PS1 slices (Fig. 3A). Two-way ANOVA revealed a significant difference between the two groups (F1,11 = 13.166, p = 0.004). On the other hand, TSA did not change the amplitude of LTP in hippocampal slices from WT mice compared to WT slices treated with vehicle alone (F1,12 = 0.512, p = 0.488, Fig. 3B). Moreover, TSA had no effect on the basal synaptic responses in experiments in which no tetanic stimulation was applied, in both slices from APP/PS1 mice and WT littermates either during TSA application or 120 min after the end of the application (n = 3; Fig. 3A and B). Thus, consistent with our findings on H4 acetylation and contextual fear memory, inhibition of histone deacetylation is able to improve hippocampal synaptic function in the APP/PS1 mouse.
Fig. 3.
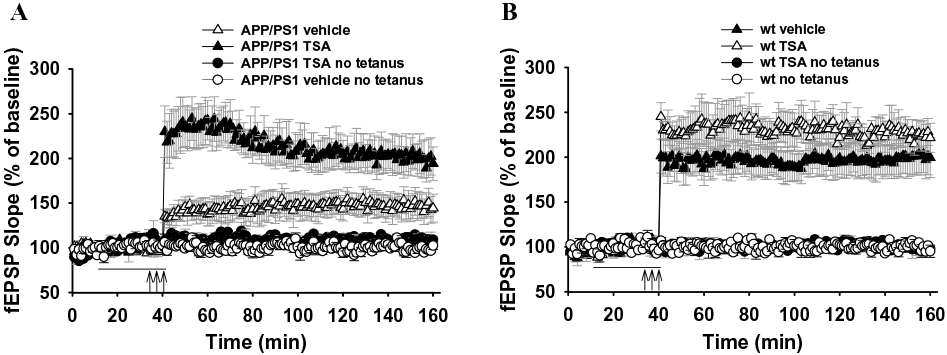
TSA reverses CA1-LTP impairment in slices from APP/PS1 mice. (A) Summary graph showing that 30 min perfusion with TSA abrogates LTP impairment in 3–4 month old APP/PS1 mice without affecting the baseline transmission. (B) Summary graph showing that 30 min perfusion with TSA does not affect LTP and baseline transmission in WT mice. These experiments were interleaved with those of APP/PS1 mice. The horizontal bar represents TSA application. The three arrows correspond to the theta-burst stimulation.
DISCUSSION
The use of HDAC inhibitors in rodents has demonstrated the role of chromatin modification in the transcriptional regulation of processes underlying memory. A cursory review of literature reveals that HDAC inhibition can increase both learning and hippocampal LTP in association with a selective increase of histone acetylation [27-30]. Also, in neuropathological models, a role for the epigenetic modulation of memory and learning has been proposed [31]. However, no clear-cut connection has been demonstrated between histone epigenetic modifications and AD etiology [31]. We have shown that a brief HDAC inhibition enables the recovery of impaired memory in the APP/PS1 amyloid-depositing mouse through histone epigenetic modification. Specifically, we observed that the APP/PS1 mice display a reduced endogenous level of H4 acetylation after a learning task. Nevertheless, the acute exposure to a class I/II HDAC inhibitor, TSA, rescued contextual spatial learning and hippocampal CA3-CA1 LTP, as well as H4 acetylation in APP/PS1 mice. Our results suggest that these effects from TSA are likely to be mediated, at least in part, by the re-elevation in H4 acetylation (hyperacetylation), which might be low as a consequence of either reduced endogenous HAT activity or increased endogenous HDAC activity following overexpression of the AβPP and PS1 transgenes. Thus, our findings unravel a possible epigenetic component in AD.
Notably, HDAC inhibitors could affect neuronal function through a variety of mechanisms including epigenetic and non-epigenetic changes [21,32]. Thus, it is possible that the block of HDACs class I/II may increase the acetylation of non-histone substrates that, in turn, can contribute to the amplification of cellular processes associated with memory. In fact, Green and colleagues [33] showed that inhibition of class III NAD+-dependent HDACs using vitamin B3 restored cognitive deficits in the triple transgenic AD mice, via a mechanism involving the reduction of Thr231-phosphotau. In addition, Fisher et al. [21] showed that sodium butyrate, a known HDAC inhibitor, induced sprouting of dendrites, increased the number of synapses, and reinstated learning and access to long-term memories in CK-p25 transgenic mice with neuronal loss. However, both groups did not investigate levels of histone acetylation in their transgenic mouse models. In contrast, we were able to show, for the first time, that LTP and memory defects in APP/PS1 mice are likely to be mediated at least in part by decreased H4 acetylation. We were also able to show that promoting histone acetylation restores learning after synaptic dysfunction has already ensued. Together, these findings provide compelling evidence that increased histone acetylation can overcome the decrease of memory function seen in an amyloid-depositing mouse model.
Previous studies investigating the mechanisms underlying LTP in acute hippocampal slices suggested that transcription is not necessary for potentiation occurring prior to 2 hours [34]. However, we observed an effect of TSA immediately after induction of LTP (Fig. 3). This finding is consistent with the result of Levenson and collaborators [30]. The LTP induction paradigm they applied induces a form of LTP that is dependent upon transcription for either induction or expression. This was proved by using 5,6-dichlorobenzimidazole riboside (DRB), a transcription inhibitor, in wild-type mice. DRB was able to block both the early and late phase of a TSA-induced LTP increase, suggesting that both the early and late enhancement of LTP by TSA is dependent upon transcriptional modulation [30].
The therapies for AD that are currently in use include augmentation of the cholinergic system by inhibition of acetylcholinesterases or, more recently, the use of NMDA antagonists that may act by blocking glutamate neurotoxicity. These agents have a limited efficacy and only the latter appears to have an even modest effect on the course of the disease. A major effort is underway to decrease the Aβ load in the brain either by agents that block the AβPP-processing secretases or by the use of treatments that appear to augment the removal of Aβ from the brain, such as immunization with Aβ peptides [35]. Animal studies have demonstrated that the neuritic dystrophy that develops in animals that overproduce Aβ can be reversed by immunization [36]. Unfortunately, human trials of an Aβ vaccine had to be terminated because of encephalitic complications in some of the patients [37]. TSA and other HDAC inhibitors represent a new approach to AD treatment that appears to make the synapse more robust and resistant to the effects of Aβ. With regards to the use of HDAC inhibitors, it has been criticized that inhibition of HDACs might alter gene expression globally and thus affect memory processes in a nonspecific manner. However, Vecsey and colleagues [27] showed that TSA does not globally alter gene expression but instead increases the expression of specific genes during memory consolidation. They were able to show that HDAC inhibitors, including TSA, enhance memory and synaptic plasticity mainly by the activation of key genes that are dependent on CREB transcriptional activation [27]. Thus, it is likely that TSA may be capable of stopping memory degradation in the presence of Aβ accumulation as well as improving brain functions that have already deteriorated, as in the case of the 3-month-old double-transgenic mouse. Interestingly, ‘rewiring’ of the brain and recovery of memory using HDAC inhibitors was recently reported in CK-p25 transgenic mice [21]. Therefore, it is possible that HDAC inhibitors could be capable of re-establishing neural networks in the AD brain. This suggests that using small molecules to target HDACs in AD patients could facilitate access to long-term memories. Novel HDAC inhibitors with minimized side-effects are currently being developed by the pharmaceutical industry. It remains to be seen if these newer inhibitors can readily enter the brain and if they are as effective as TSA.
ACKNOWLEDGMENTS
This work was supported in part by Alzheimer Disease Research Zenith Award ZEN-07-58977, National Institutes of Health Grant R01 NS049442 (to O.A.), and by United Kingdom Alzheimer’s Research Trust Pilot Grant, The International Sephardic Educational Foundation (ISEF) Scholarship, The Lewis Family Trust Scholarship, The Sidney & Elizabeth Corob Charitable Trust Scholarship, the Charlotte and Yule Bogue Research Fellowships (to Y.I.F). We wish to thank Vivian and Norman Belmonte, Michal Luzac and Linda Lee for their support.
Footnotes
Authors’ disclosures available online (http://www.jalz.com/disclosures/view.php?id=14).
REFERENCES
- [1].Pittenger C, Kandel ER (2003) In search of general mechanisms for long-lasting plasticity: Aplysia and the hippocampus. Philos Trans R Soc Lond B Biol Sci 358, 757–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Levenson JM, Sweatt JD (2005) Epigenetic mechanisms in memory formation. Nat Rev Neurosci 6, 108–118. [DOI] [PubMed] [Google Scholar]
- [3].Masliah E (1995) Mechanisms of synaptic dysfunction in Alzheimer’s disease. Histol Histopathol 10, 509–519. [PubMed] [Google Scholar]
- [4].Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B (1987) The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 325, 733–736. [DOI] [PubMed] [Google Scholar]
- [5].Czech C, Tremp G, Pradier L (2000) Presenilins and Alzheimer’s disease: biological functions and pathogenic mechanisms. Prog Neurobiol 60, 363–384. [DOI] [PubMed] [Google Scholar]
- [6].Sherrington R, Rogaev EI, Liang Y,Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, et al. (1995) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375, 754–760. [DOI] [PubMed] [Google Scholar]
- [7].Levy-Lahad E, Wijsman EM, Nemens E, Anderson L, Goddard KA, Weber JL, Bird TD, Schellenberg GD (1995) A familial Alzheimer’s disease locus on chromosome 1. Science 269, 970–973. [DOI] [PubMed] [Google Scholar]
- [8].Takahashi H, Mercken M, Honda T, Saito Y, Murayama M, Song S, Takashima A (1999) Impaired proteolytic processing of presenilin-1 in chromosome 14-linked familial Alzheimer’s disease patient lymphocytes. Neurosci Lett 260, 121–124. [DOI] [PubMed] [Google Scholar]
- [9].Cullen WK, Suh YH, Anwyl R, Rowan MJ (1997) Block of LTP in rat hippocampus in vivo by beta-amyloid precursor protein fragments. Neuroreport 8, 3213–3217. [DOI] [PubMed] [Google Scholar]
- [10].Itoh A, Akaike T, Sokabe M, Nitta A, Iida R, Olariu A, Yamada K, Nabeshima T (1999) Impairments of long-term potentiation in hippocampal slices of beta-amyloid-infused rats. Eur J Pharmacol 382, 167–175. [DOI] [PubMed] [Google Scholar]
- [11].Vitolo OV, Sant’Angelo A, Costanzo V, Battaglia F, Arancio O, Shelanski M (2002) Amyloid beta -peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci U S A 99, 13217–13221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. [DOI] [PubMed] [Google Scholar]
- [13].Chen QS, Kagan BL, Hirakura Y, Xie CW (2000) Impairment of hippocampal long-term potentiation by Alzheimer amyloid beta-peptides. J Neurosci Res 60, 65–72. [DOI] [PubMed] [Google Scholar]
- [14].Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL (1998) Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A 95, 6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sant’Angelo A, Trinchese F, Arancio O (2003) Usefulness of behavioral and electrophysiological studies in transgenic models of Alzheimer’s disease. Neurochem Res 28, 1009–1015. [DOI] [PubMed] [Google Scholar]
- [16].Fuso A, Seminara L, Cavallaro RA, D’Anselmi F, Scarpa S (2005) S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Mol Cell Neurosci 28, 195–204. [DOI] [PubMed] [Google Scholar]
- [17].Rogaev EI, Lukiw WJ, Lavrushina O, Rogaeva EA, St George-Hyslop PH (1994) The upstream promoter of the beta-amyloid precursor protein gene (APP) shows differential patterns of methylation in human brain. Genomics 22, 340–347. [DOI] [PubMed] [Google Scholar]
- [18].Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274, 99–102. [DOI] [PubMed] [Google Scholar]
- [19].Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S (1996) Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature 383, 710–713. [DOI] [PubMed] [Google Scholar]
- [20].Trinchese F, Liu S, Battaglia F, Walter S, Mathews PM, Arancio O (2004) Progressive age-related development of Alzheimer-like pathology in APP/PS1 mice. Ann Neurol 55, 801–814. [DOI] [PubMed] [Google Scholar]
- [21].Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH(2007) Recovery of learning and memory is associated with chromatin remodelling. Nature 447, 178–182. [DOI] [PubMed] [Google Scholar]
- [22].Crawley JN (1999) Behavioral phenotyping of transgenic and knockout mice: experimental design and evaluation of general health, sensory functions, motor abilities, and specific behavioral tests. Brain Res 835, 18–26. [DOI] [PubMed] [Google Scholar]
- [23].Sperling RA, Bates JF, Chua EF, Cocchiarella AJ, Rentz DM, Rosen BR, Schacter DL, Albert MS (2003) fMRI studies of associative encoding in young and elderly controls and mild Alzheimer’s disease. J Neurol Neurosurg Psychiatry 74, 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gong B, Vitolo OV, Trinchese F, Liu S, Shelanski M, Arancio O (2004) Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J Clin Invest 114, 1624–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Phillips RG, LeDoux JE (1992) Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci 106, 274–285. [DOI] [PubMed] [Google Scholar]
- [26].Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ (1994) Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 79, 59–68. [DOI] [PubMed] [Google Scholar]
- [27].Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA (2007) Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci 27, 6128–6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Alarcon JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A (2004) Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron 42, 947–959. [DOI] [PubMed] [Google Scholar]
- [29].Korzus E, Rosenfeld MG, Mayford M (2004) CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron 42, 961–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Levenson JM, O’Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD (2004) Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem 279, 40545–40559. [DOI] [PubMed] [Google Scholar]
- [31].Abel T, Zukin RS (2008) Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr Opin Pharmacol 8, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, Puigserver P, Sinclair DA, Tsai LH (2007) SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J 26, 3169–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Green KN, Steffan JS, Martinez-Coria H, Sun X, Schreiber SS, Thompson LM, LaFerla FM (2008) Nicotinamide restores cognition in Alzheimer’s disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J Neurosci 28, 11500–11510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Frey U, Krug M, Brodemann R, Reymann K, Matthies H (1989) Long-term potentiation induced in dendrites separated from rat’s CA1 pyramidal somata does not establish a late phase. Neurosci Lett 97, 135–139. [DOI] [PubMed] [Google Scholar]
- [35].Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P (1999) Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400, 173–177. [DOI] [PubMed] [Google Scholar]
- [36].Lombardo JA, Stern EA, McLellan ME, Kajdasz ST, Hickey GA, Bacskai BJ, Hyman BT (2003) Amyloid-beta antibody treatment leads to rapid normalization of plaque-induced neuritic alterations. J Neurosci 23, 10879–10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO (2003) Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med 9, 448–452. [DOI] [PubMed] [Google Scholar]