

Abstract
Alzheimer’s disease, one of the most important brain pathologies associated with neurodegenerative processes, is related to overactivation of calpain-mediated proteolysis. Previous data showed a compelling efficacy of calpain inhibition against abnormal synaptic plasticity and memory produced by the excess of amyloid-β, a distinctive marker of the disease. Moreover, a beneficial effect of calpain inhibitors in Alzheimer’s disease is predictable by the occurrence of calpain hyperactivation leading to impairment of memory-related pathways following abnormal calcium influxes that might ensue independently of amyloid-β elevation. However, molecules currently available as effective calpain inhibitors lack adequate selectivity. This work is aimed at characterizing the efficacy of a novel class of epoxide-based inhibitors, synthesized to display improved selectivity and potency towards calpain 1 compared to the prototype epoxide-based generic calpain inhibitor E64. Both functional and preliminary toxicological investigations proved the efficacy, potency, and safety of the novel and selective calpain inhibitors NYC438 and NYC488 as possible therapeutics against the disease.
Keywords: Alzheimer’s disease, amyloid-β, calpain, learning, long-term potentiation, memory
INTRODUCTION
Calcium-activated neutral cysteine proteases (calpains) are a variegated cluster of calcium-dependent proteases, able to modify the function of several target proteins by partial truncation. This limited non-digestive proteolysis is a particular form of post-translational modification that changes physiological activity and translocation of the target proteins [1], including calpains themselves [2]. Calpains regulate through proteolysis several cellular functions, including cytoskeleton assembly and disassembly.
In the central nervous system (CNS), where calpain I and calpain II are the main calpain isoforms, their activation is to synaptic plasticity and memory as well as to neurodegeneration [3, 4]. Events that have been proposed to participate in synaptic plasticity and memory, including cytoskeletal regulation, AMPA receptor trafficking, actin polymerization, and regulation of local protein synthesis are regulated by calpains [5] through a plethora of protein targets like CaMKIIα, protein kinase C and PP3alpha/calcineurin [6–10] and transcription factors such as the cAMP response element-binding protein (CREB) [11–16].
Abnormal calcium influxes intensify calpain activity at supra-physiological levels that are evident in a number of neurological disorders (i.e., Alzheimer’s disease (AD) [4, 17, 18]), generating a variety of detrimental effects in pathways related to synaptic plasticity and memory, including the decrease in CREB phosphorylation and activation [19, 20], and accompanied by synaptic dysfunction, which is a robust predictor of cognitive impairment in AD [21, 22].
Despite the intense effort in the field of AD directed to study and to provide a neuropathological substrate for an effective therapy toward AD, the current therapeutic arsenal (such as galantamine, rivastigmine, donepezil, and memantine) is at best symptomatic, and provides solely temporary relief without a real causative breakthrough [23, 24]. There are no FDA-approved drugs that can delay or halt the progression of the disease. Given the perspective epidemic of AD and other neurodegenerative diseases [25], it is rather urgent to develop a proficient line of therapeutics with high translational potential and optimal therapeutic index.
The vast majority of failing clinical efforts deal with the problem of amyloidogenic protein deposition, because proteinaceous aggregates consisting of deposition of extracellular amyloid plaques and intracellular neurofibrillary tangles are the major histopathological hallmarks of the disease. Our approach, instead, focuses on preserving synaptic functionality. This is justified by ample evidence suggesting that AD starts as a synaptic disorder [26]. It is likely that the very fine and variable amnesic symptoms, occurring at the beginning of the disease in the absence of any other clinical signs of brain injury, are caused by discrete changes in synaptic function, produced at least in part, by amyloid-β (Aβ) species (e.g., Aβ40 and Aβ42) [27–30], peptides derived from processing of amyloid-β protein precursor (AβPP). Previously, we validated the inhibition of calpains as a therapeutic target against their overactivation in AD toward the recovery of synaptic dysfunctions induced by Aβ [19, 31]. These findings led to an effort aimed to discovering novel calpain inhibitors that might be utilized against AD. Here we report findings from a phenotypic screening of three generations of peptidomimetic epoxide warhead containing molecules that have been previously proved to be unreactive toward reaction with free thiols while displaying irreversible active site calpain 1 inhibition with sub-micromolar potency [32]. We designed our drug screening for calpain inhibitors using a phenotypical modality combined with medicinal chemistry refined through target-based computational approach [32, 33], focusing on the capability of our candidate molecules to protect from the detrimental effect of oligomerized Aβ42 on hippocampal long-term potentiation (LTP), a type of synaptic plasticity thought to underlie learning and memory. Following this screening, the last generation of leads was further tested for pharmacokinetic and toxicological features, and then for the recovery of cognitive impairments in a mouse model of amyloid deposition, the AβPP/PS1 mouse [32].
MATERIAL AND METHODS
Animals
All experiments were performed with the approval of the Columbia University Animal Care and Use Committee in accordance with the guidelines for the humane treatment of animals (protocol #AC-AAAB9126). Hemizygous transgenic (HuAPP695SWE) 2576 mice expressing mutant human AβPP (K670N, M671L) [34] were crossed with hemizygous PS1 mice that express mutant human PS1 (M146V; line 6.2) [35]. The offspring, double-transgenic mice overexpressing AβPP/PS1, were compared with their wild type (WT) littermates so that age and background strain were comparable. To identify the genotype of the animals, we used DNA extracted from tail tissue as previously described [35, 36]. For pharmacokinetic testing, we used instead ICR mice.
Aβ peptide oligomerization
Recombinant Human Aβ42 peptide (American Peptides) was oligomerized as previously described [37]. Briefly, crude lyophilized A peptide was resuspended in cold 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP, Sigma) and aliquoted in polypropylene vials. After 24 h, the HFIP solution was allowed to evaporate in a fume hood until a thin film of monomeric peptide is formed on the bottom of the vials. Peptide films were dried under gentle vacuum and stored in sealed vials at –20°C. Prior to use, anhydrous DMSO (Sigma) was added to obtain a pure monomeric Aβ/DMSO solution and then sonicated for 10 min [37]. Low-order oligomer-enriched Aβ42 was obtained by incubating an aliquot of monomeric Aβ/DMSO solution in sterile artificial cerebrospinal fluid (ACSF) phosphate buffer at 4°C overnight. Oligomerized Aβ peptide was then further diluted up to 200 nM concentration with vehicle right before the experiments.
Drug administration
E64 from Sigma-Aldrich and all candidate compounds were solubilized in 100 μl Tween-80 + 100 μl dimethylsulfoxide (DMSO) and then diluted with vehicle solution to the appropriate experimental concentration for in vitro experiments or experimental dose for in vivo experiments. Oligomerized Aβ42 was either administered in vitro alone or co-administered in vitro with E64 or one of the candidate compounds. Drugs or vehicle were perfused for 20 min before plasticity induction in electrophysiological experiments or administered in vivo, as described [38], before the behavioral tests. Two month-old AβPP/PS1 and WT mice were evenly separated into 4 groups: AβPP/PS1 mice treated with vehicle, AβPP/PS1 mice treated with one of the candidate compounds, WT mice treated with vehicle, and WT mice treated with one of the candidate compounds. Separate groups of mice were administered with the compounds as described below for the pharmacokinetic assessment, spectrin western blotting, and histological analysis.
Electrophysiology
Mice were decapitated, and their hippocampi were removed. Transverse hippocampal slices of 400 μm thickness were cut on a tissue chopper and transferred to an interface chamber where they were maintained at 29°C. ACSF saline recording solution (124.0 mM NaCl, 4.4 mM KCl, 1.0 mM Na2HPO4, 25.0 mM NaHCO3, 2.0 CaCl2, 2.0 mM MgCl2, 10 mM glucose) was perfused at 1–2 ml/min and continuously bubbled with 95% O2 and 5% CO2. Slices were permitted to recover for at least 90 min before recording. A concentric bipolar platinum-iridium stimulation electrode was placed at the level of the Schaeffer collateral fibers, whereas the recording electrode, a low-resistance glass recording microelectrode filled with saline solution, was placed in CA1 stratum radiatum to record the extracellular field excitatory postsynaptic potential (fEPSP). An input–output curve was used to set the baseline fEPSP at ≈35% of the maximal slope. Baseline stimulation was delivered every minute (0.01-ms duration pulses) for 15 min before beginning the experiment to assure the stability of the response. LTP of evoked responses was induced by using θ-burst stimulation (4 pulses at 100 Hz, with the bursts repeated at 5 Hz and each tetanus including three 10-burst trains separated by 15 s). Responses were recorded for 120 min after tetanization. Responses are expressed as a percent of control values or normalized following the formula (values upon co-treatment Aβ42 + candidate compound)/(values upon treatment with sole Aβ42) *100.
Pharmacokinetic assessment
A total of 54 male ICR mice were divided into 3 dosing groups (18 mice in each group), which were administrated by intraperitoneal route at the same equimolar dose (NYC215, 7.57 mg/kg; NYC438, 7.86 mg/kg; NYC488, 7.83 mg/kg). Test article solutions were prepared by dissolving in 4% DMSO/4% Tween 80/92% deionized water to yield final concentrations as showed in the above table. Dose volume for each test animal was determined based on the most recent body weight. Blood (approximately 250 μl) was collected via retro-orbital puncture into tubes containing sodium heparin anticoagulant at 7.5, 15, 30, 60, 120, and 240 min post-dosing. Mice were sacrificed by cervical dislocation after blood harvest. The plasma were separated via centrifugation (11,000 rpm, 5 min) and stored in –80°C before analysis. Frozen unidentified plasma samples were thawed at room temperature and vortexed thoroughly. With a pipette, 25 μl of plasma was transferred into a 1.5 ml Eppendorf tube. To each sample, 25 μl of methanol methanol-water (1:1, v/v) and 25 μl of internal standard (IS) (100 ng/ml NYC488 for NYC215, 50 ng/ml YF2 for NYC438, and no IS was used for NYC488) were added, followed by the addition of 100 μl acetonitrile. The sample mixture was vortexed for approximately 1 min. After centrifugation at 11,000 rpm for 5 min, the upper layer was vaporized under nitrogen stream. The residue was dissolved with mobile phase and 20-μl aliquot was injected onto the LC/MS/MS system for analysis. Calibration standards were prepared by spiking 25 μl of the analyte standard solutions into 25 μl of heparinized blank mice plasma. The nominal standard concentrations in plasma were 3.00, 10.0, 30.0, 100, 300, 1,000, 3,000, and 10,000 ng/ml for each analyte. Quantification was achieved by the internal standard method using peak area ratios of the analyte to IS in plasma for NYC215 and NYC438. For NYC488, external standard method was used. Concentrations were calculated using a weighted least-squares linear regression (W = 1/x2). The assay was performed using an LC/MS/MS system consisting of the following components: HPLC system, G1379A vacuum degasser, G1311A quaternary pump, G1316A column oven (Agilent, Waldbronn, Germany) and NANOSPACE SI-2 HTS autosampler Z 3133 (Shisedo, Tokyo, Japan); MS/MS system, API 4000 triple quadrupole mass spectrometer, equipped with a TurboIonSpray (ESI) Interface (Applied Biosystems, Concord, Ontario, Canada). For the NYC215 and NYC438, we used a Capcell C18 column (100 mm × 4.6 mm I.D., 5 μm, Shiseido, Japan) while for the NYC488 we used a Synergi 4 μm Hydro-RP 80A (150 mm × 4.6 mm I.D., Phenomenex, Torrance, CA, USA). Mobile phase was acetonitrile versus 0.2% formic acid in 5 mM ammonium acetate at different mix depending on the candidate compound. The major pharmacokinetic parameters were calculated by non-compartmental analysis using WinNonlin 5.3 (Pharsight USA).
Spectrin western blotting
Hippocampal lysates for immunoblotting were prepared as previously described [19] with slight modifications. Hippocampal tissue was homogenized in lysis buffer (62.5 mM Tris-HCl, pH 6.8, 3% LDS, 1 mM DTT) and incubated at 4°C for 10 min, then sonicated before centrifugation at 20,000 rpm for 5 min. Whole cell extracts were electrophoresed on 3–8% gradient Tris-Acetate PAGE gel (Invitrogen) and then immunoblotted. Antibodies were used at a 1:1,000 concentration for immunoblotting. Spectrin antibody was from Millipore. β-III-Tubulin antibody was purchased from Promega.
Histologic analysis
Mouse organ (liver, heart, muscle, stomach, kidney, brain) samples collected at the end of the chronic treatment with either vehicle or candidate compounds were fixed in 10% buffered paraformaldehyde, processed through conventional histological techniques, and stained with hematoxylin and eosin. Microscopy was performed using an optical microscope (Olympus BX51) equipped with a camera (Olympus Q-Color-5), and the images were recorded in a computer using the Image Pro-Express software.
Behavioral assessment
A). Associative contextual memory
Associative memory was probed through fear conditioning in either vehicle or transgenic AβPP/PS1 mice, according to previously proposed method [39]. Our conditioning chamber was located inside a sound-attenuating box (72 × 51 × 48 cm). A clear Plexiglas window (2 × 12 × 20 cm) allowed the experimenter to film the mouse performance with a camera placed on a tripod and connected to FreezeFrame software (MED Associates Inc.). To provide background white noise (72 dB), a single computer fan was installed in one of the sides of the sound-attenuating chamber. The conditioning chamber (33 × 20 × 22 cm) was made of transparent Plexiglas on two sides and metal on the other two. One of the metal sides had a speaker and the other had a 24 V light. The chamber had a 36-bar insulated shock grid floor. The floor was removable and after each use we cleaned it with 75% ethanol and then with water. Only one animal at a time was present in the experimentation room. The other mice remained in their home cages. During the contextual conditioning experiment, mice were placed in the conditioning chamber for 2 min. In the last 2 s of the 2 min, mice were given a foot shock of 0.50 mA for 2 s through the bars of the floor, and left in the conditioning chamber for another 30 s before being placed back in their home cages. “Freezing" behavior, defined as the absence of all movements except for that necessitated by breathing, was assigned scores using FreezeView software (MED Associates Inc.). For evaluation of contextual fear learning, freezing at 24 h post-training was measured for 5 consecutive minutes in the chamber in which the mice were trained. Twenty-four hours after the contextual testing, cued fear conditioning was evaluated by placing the mice in a novel context (triangular cage with a smooth flat floor) for 2 min (pre-CS test), after which they were exposed to the CS for 3 min (CS test), and freezing was measured. In a separate set of experiments, we tested whether the four different experimental groups of mice had similar exploratory behavior and anxiety by carrying out the open field test. Animals were positioned in an open arena with a floor that was divided into compartments. The internal dimensions of the arena were 72 × 72 × 33 cm. An area measuring 36 × 36 cm in the center of the open field was defined as the “central compartment”. Behavioral scoring was evaluated by the percentage of time spent in the center compartment and the number of entries into the center compartment. No differences were found among the four groups of mice (data not shown).
B). Spatial memory
Spatial memory was assessed through the 2 day RAWM as previously described [40, 41]. Transgenic AβPP/PS1 mice were trained in fifteen daily sessions to identify a platform location by alternating between a visible and a hidden platform in the goal arm. The final three trials on day 1 and all fifteen trials on day 2 used a hidden escape platform to probe the ability of the mouse to find the goal arm location. The reaching of the learning criterion (max 1 arm error average in three consecutive trials) was obtained by WT vehicle-treated animals. The visible platform testing was used to exclude that visual, motor, and motivation deficits affect the performance of the mice. No difference in the time and the speed to reach the platform was observed among the different groups of mice indicating that visual, motor, and motivation skills were not affected by the experimental procedure (data not shown). Higher the scoring, less efficacy is associated to the experimental drug (by definition the vehicle-treated AβPP/PS1 display the worst score in criterion).
Amyloid-β assessment
Aβ content was assessed both in mouse hippocampi and plasma collected at the end of the chronic treatments, as described [42]. Hippocampi were homogenized in 880 μl of tissue lysate buffer (20 mM Tris-HCl (pH 7.4), 1 mM ethylenediaminetetraacetic acid, 1 mM ethyleneglycoltetraacetic acid, 250 mM sucrose) supplemented with 3X protease inhibitors (Roche). Blood samples were instead collected in EDTA-treated tubes, centrifuged to obtain the plasmatic fraction, and then mixed with an aliquot of the lysate buffer to maintain the matrix. ELISA assay (#EZBRAIN-SET, Millipore, USA) was performed according to the manufacturer’s protocol on a Costar-like 96-well plates were incubated overnight at 4°C with capture antibody (in 0.1 M sodium bicarbonate, pH 8.2) at a dilution of 4 μg/ml. Upon blocking, plate wells were incubated overnight with 50 μl of brain lysate and then washed with PBS and incubated with the antibody at a 1 μg/ml. Values were read at 620 nM wavelength 30 min after adding 100 μl of colorimetric buffer. The signal was normalized to the protein concentration for each sample.
Statistical analysis
For all experiments, mice were coded by “blind” investigators with respect to treatment and genotype. Data are expressed as mean ± SEM. Statistical analysis was performed with one-way ANOVA (for fear conditioning experiments), two-way ANOVA with repeated measures (for LTP and 2 day RAWM experiments) and Student’s t test (pairwise comparisons). The level of significance was set for p < 0.05.
RESULTS
Phenotypic screening of novel calpain 1 inhibitors using Aβ-induced reduction of LTP
For the present studies, we synthesized three generations of calpain inhibitors by modifying mostly the P2 and P3 domains of the inhibitor E64 backbone (Fig. 1). The P2 recognition group is important for the selectivity towards calpain 1 versus other proteases such as cathepsin B [32]. E64 was used as our benchmark compound for screening assay in synaptic plasticity. As a fundamental requirement, the screening of candidate compounds aimed to verify whether the new derivatives possessed at least equal capabilities as E64 to rescue the Aβ-induced defect in LTP of hippocampal slices [19].
Fig. 1.
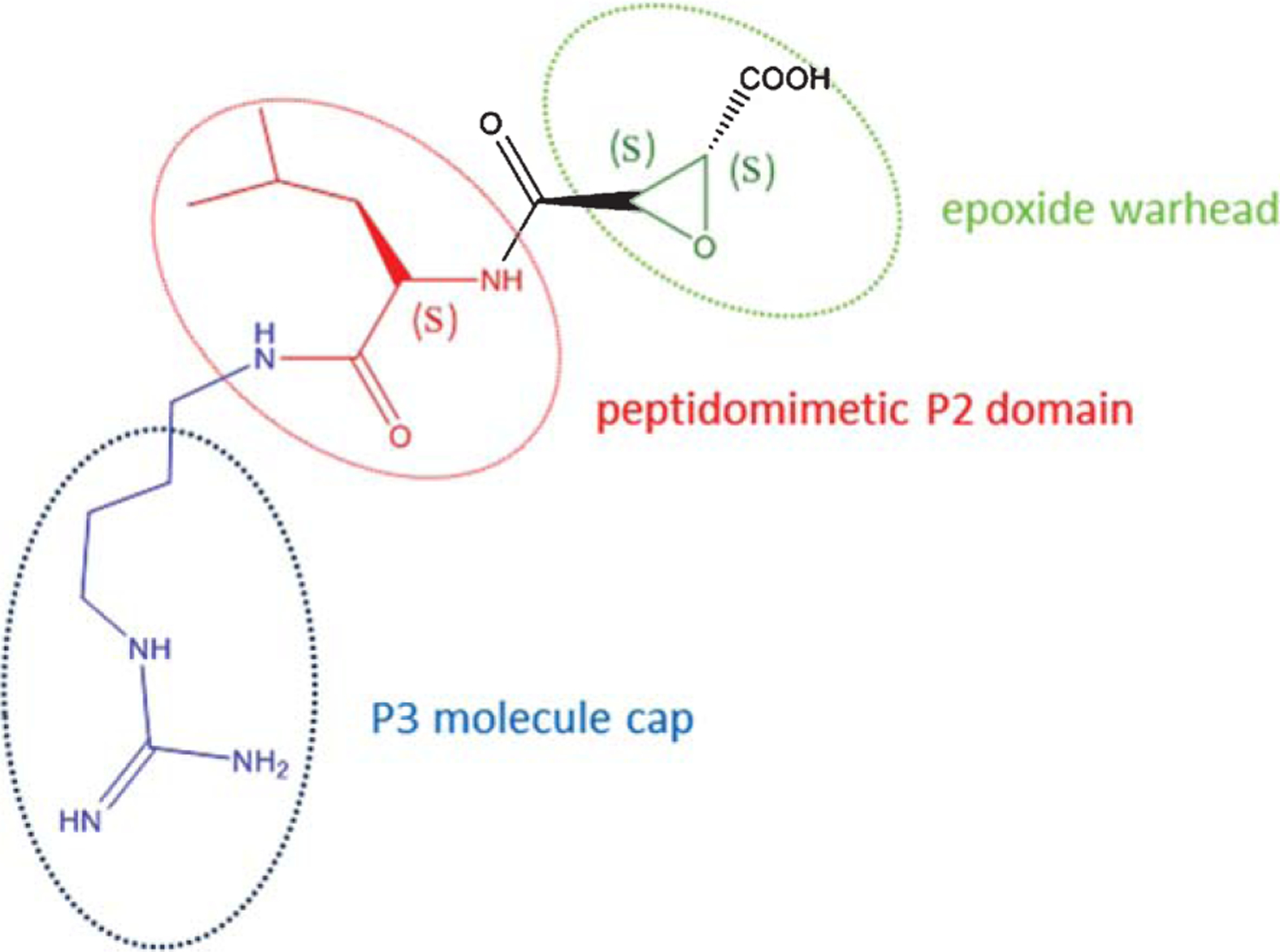
Chemical structure of the prototype epoxide compound E64. The structure of E64 can be functionally divided into three main domains: the epoxidic warhead that interacts with an enzymatic pocket of calpains producing the protease inhibition, a peptidomimetic leucin domain P2 that it is important to modulate the selectivity of any derivative compound for calpains, and finally a P3 compound cap that is useful for druggability development and for improving the pharmacological potency.
Acute exposure to 200 nM Aβ42 blocks LTP induction [29] (Fig. 2A). A dose-response curve for E64, co-applied with oligomerized Aβ42 (200 nM) prior to the induction of LTP by tetanic stimulation showed that E64 restored LTP to the vehicle-treated level. The EC50 for E64 was 650 nM (Fig. 2B). Therefore, we evaluated all candidate lead compounds at a concentration corresponding to the EC50 for E64.
Fig. 2.
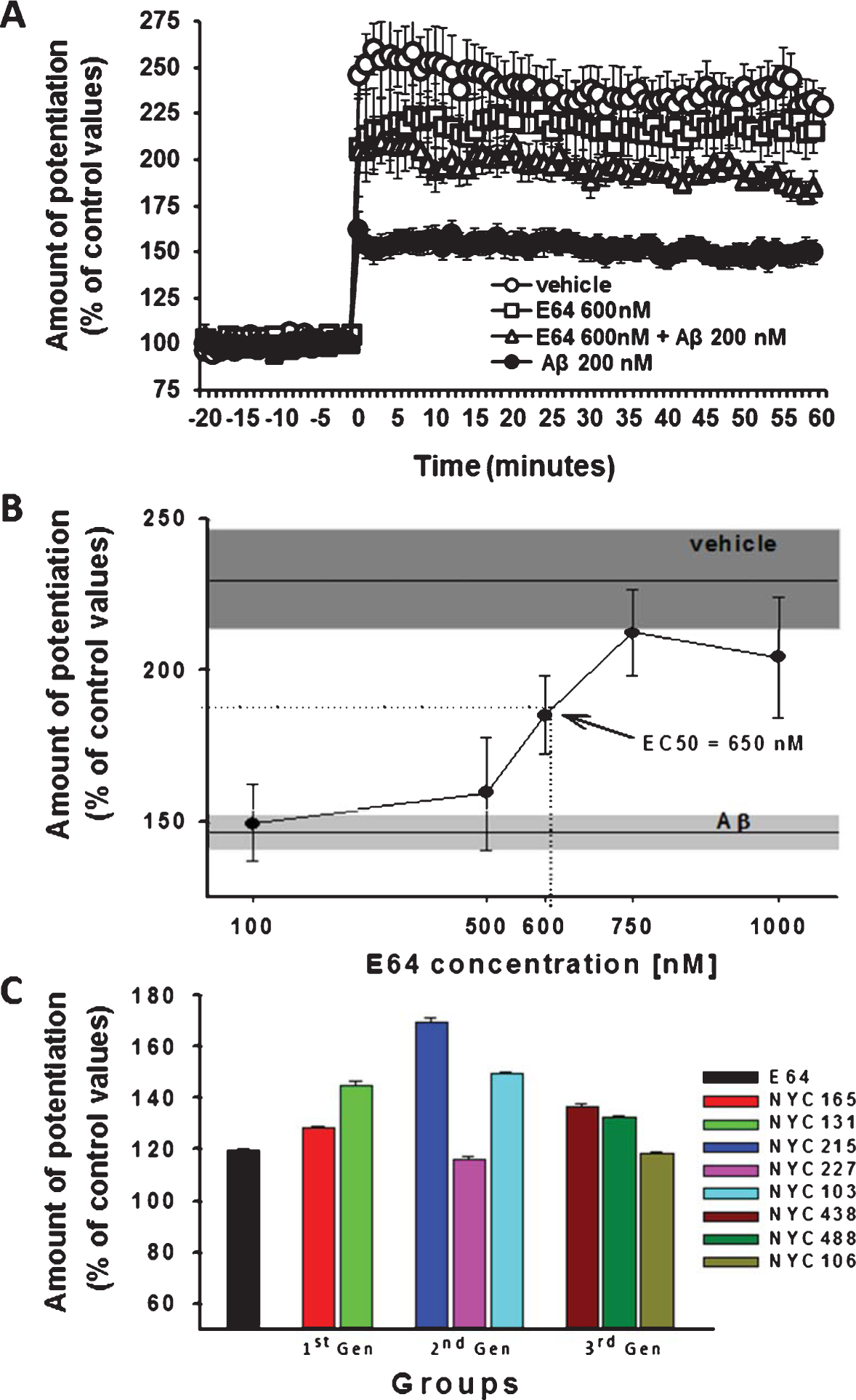
Electrophysiological screening of three generations of calpain inhibitors through evaluation of their ability to protect against the Aβ42-induced LTP impairment. A) Example of effect produced by the co-administration of E64, the prototype epoxide compound with calpain inhibitory activity, together with Aβ42 (200 nM, 20 min). E64 rescues the detrimental effect induced by Aβ42 onto LTP, recovering the potentiation values up to the levels observed upon perfusion with vehicle alone. B) Dose/response curve of LTP levels observed upon co-administration of different concentrations of E64 with Aβ42 (200 nM, 20 min). The ED50 for E64 is 650 nM. Boxes represent the S.E.M range. C) Phenotypical screening of three generations of new E64 derivatives as calpain inhibitors based on the ability to rescue the detrimental effect of Aβ42 (200 nM, 20 min) onto LTP in vitro when co-administered at 650 nM, the ED50 for E64. Compounds are grouped according to the progressive optimization throughout the drug discovery process: 1st generation compounds (1st Gen) are compounds with potency equal or slightly higher versus E64; 2nd generation compounds (2nd Gen) have higher potency than E64 in inhibiting calpain; and 3rd generation (3rd Gen) compounds, instead, are more potent than E64 displaying improved selectivity toward calpain 1 inhibition.
Novel calpain inhibitors developed throughout the three synthesis generation (1st generation: NYC165, NYC131; 2nd generation: NYC215, NYC227, NYC103; 3rd generation: NYC438, NYC488, and NYC106) provided robust results, except compounds NYC227 and NYC106. Slices perfused with our candidate compounds were able to preserve synaptic plasticity in the presence of 200 nM Aβ42, confirming the validity of the phenotypical drug screening [32] (Fig. 2C).
These results further validate the use of calpain inhibitors to recover synaptic plasticity in AD. All molecules were calpain inhibitors with equal or superior potency with respect to E64, while 2nd and 3rd generation molecules were developed to increase selectivity for calpain 1, as previously described [32].
Pharmacokinetic (PK) profile and brain drug activity of novel calpain 1 inhibitors
Next, we assessed the PK profile of the best 2nd and 3rd generation compounds obtained from the functional screening on synaptic plasticity (NYC215, NYC438, and NYC488) via LC-MS/MS determination of plasma concentrations. We treated i.p. three sets of mice for the PK assessment of different candidate compounds at equimolar concentrations: NYC215 (7.57 mg/kg), NYC438 (7.86 mg/kg), and NYC488 (7.83 mg/kg). The analysis of kinetics indicated that all three candidate compounds NYC215, NYC438, and NYC488 were rapidly absorbed. The peak plasma concentration occurred at 0.25, 0.5, and 1.25 h after dosing, respectively. Figure 3 shows the plasma concentrations at each sampling time. The absolute bioavailabilities of NYC215, NYC438, and NYC488 were 80.4%, 87.3%, and 41.3%, respectively. Their 1/2 lives were 0.6 h, 1.1 h, and 0.6 h.
Fig. 3.
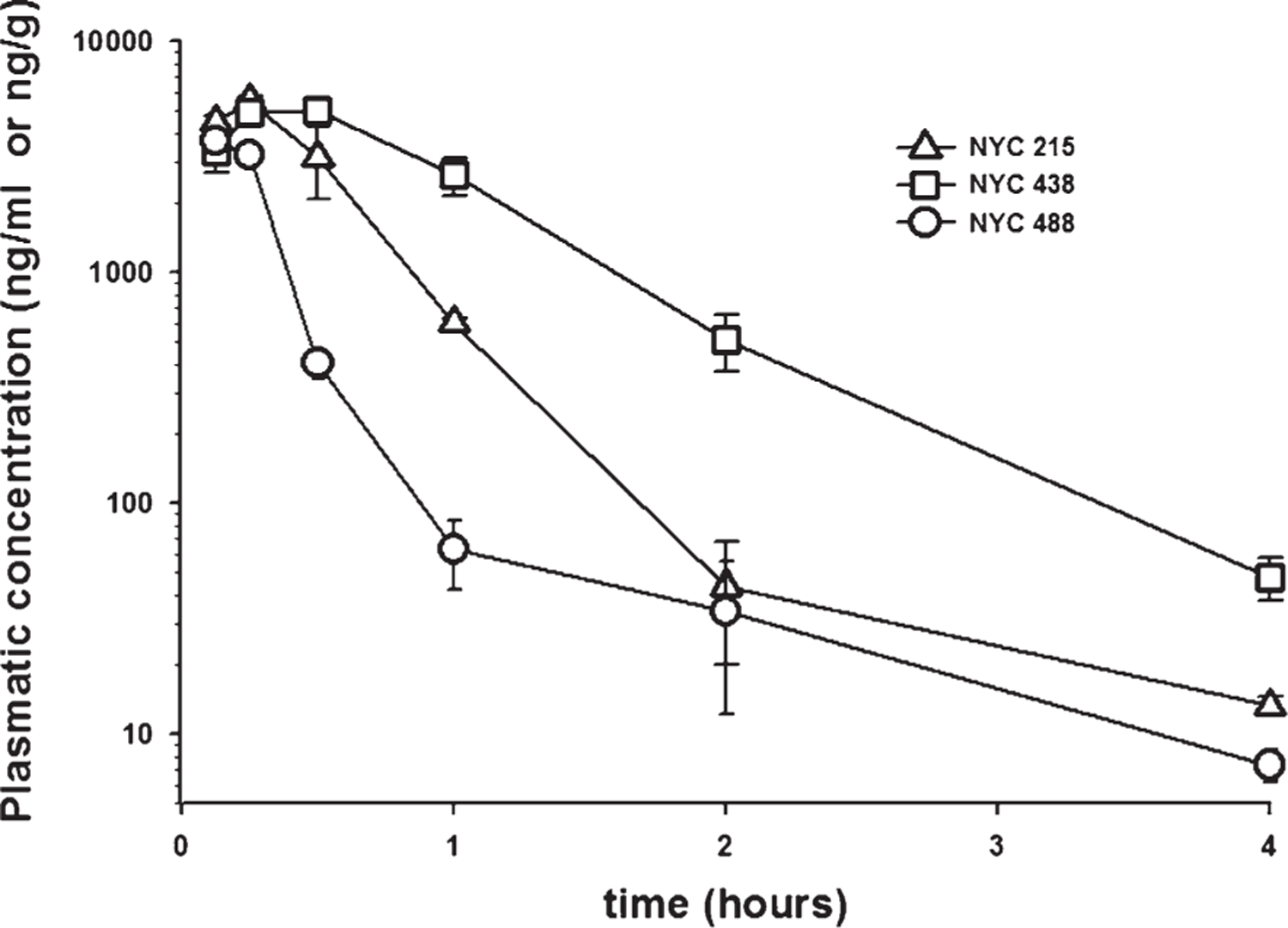
Analysis of drug kinetics in plasma. The analysis of kinetics indicates that all three candidate compounds NYC215 (7.57 mg/kg), NYC438 (7.86 mg/kg), and NYC488 (7.83 mg/kg) are rapidly absorbed upon i.p. injection. The peak plasma concentration occurred at 0.25, 0.5, and 0.125 h after dosing, respectively. Their half-life was ~0.6 h, ~1.1 h, and ~0.6 h, respectively.
Our next goal was to determine if the inhibitor candidate is capable of lowering levels of spectrin proteolytic degradation products in hippocampi from adult animals upon in vivo administration. Spectrin is a cytoskeleton protein target for calpain cleavage. The presence of a specific calpain fragment around 145 kDa is an index of calpain activity [43] and a decreased immunoreactivity in western blotting assays would instead indicate low activity of calpain inhibitors [44]. This kind of investigation offers insights on specific calpain inhibition and brain penetration at the dose used in the efficacy studies in vivo. Using western blot analysis, we checked the prevention of calpain-generated spectrin fragments following i.p. treatment for 12 days with NYC215, the best 2nd generation compound, and NYC438 and NYC488, the two 3rd generation compounds that surpassed E64 benchmark in the LTP rescue assay, at the same concentrations used for PK assessment. The compound NYC215 was slightly less efficient at preventing the spectrin cleavage by calpains while the remaining two compounds NYC438 and NYC488 dramatically reduced the amount of fragments (Fig. 4). This result confirmed the ability of NYC215, NYC438, and NYC488 to cross the blood-brain barrier (BBB) and inhibit calpain in the brain. The brain penetration could result from either passive diffusion or active transport through a BBB transporter. Future experiments should involve in vitro studies with various BBB transporters to understand the transport modalities.
Fig. 4.
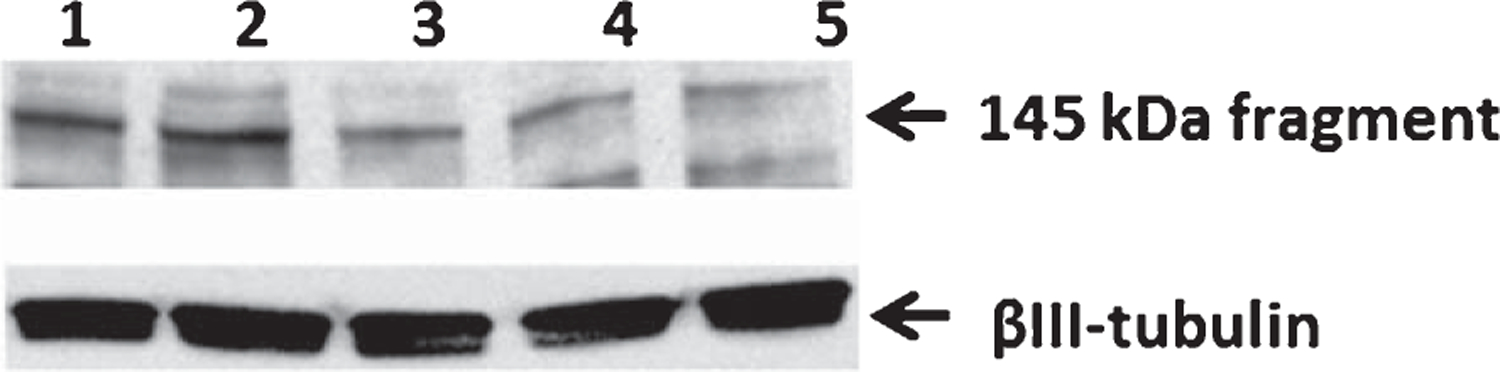
Phenotypic evaluation of brain activity of the new calpain 1 inhibitors through assessment of their proteolytic activity. Western blotting from hippocampi homogenates obtained from animals treated with vehicle, NYC215, NYC438, and NYC488. The lane 1 and 2 represents an array of vehicle samples loaded at 15 and 20 μg total protein/lane, respectively. Samples from compound-treated animals (NYC215, lane 3; NYC438, lane 4; NYC488, lane 5) were loaded at 15 μg/lane. The new calpain inhibitors were effective in decreasing the spectrin fragment at around 145 kDa that is generated specifically by calpain. The decrease of the specific calpain-generated fragment demonstrates that the drugs can reach the brain upon systemic administration, overcoming the problems related to first-pass metabolism and blood-brain barrier penetration.
Preliminary toxicity profile of optimized compounds
Our next goal was to have a preliminary assessment of the toxicity profile of NYC215, NYC438, and NYC488. Typically, toxicity studies would help in predicting possible side effects and deciding the safe dose of drugs to be administered during clinical studies [45, 46]. In a series of experiments, we determined the maximum tolerated dose (MTD) following NYC215, NYC438, and NYC488 administration in mice. MTD was computed as the maximum administered dose that does not produce any toxicity effect in terms of malaise (i.e., immobility, altered gait, hunched posture, spikey coat/stops, grooming, altered urination, and/or defecation, porphyrin staining around eyes and nose, vocalization, decrease access rate to food and water, PICA behavior) or death. This restrictive definition is actually superimposable with the one of “no observed adverse effect level” (NOAEL) dose [47]. From the NOAEL, it can be calculated the human equivalent dose (HED) and maximum recommending starting dose (MRSD) as prescribed by current FDA indications [48].
For assessment of the MTD, WT mice (3–5 month old) were acutely injected either with NYC215, NYC438, and NYC488 in an exploratory challenging dose-response treatment designed as sequential acute i.p. injections to establish the dose that produces a marked malaise. MTD was experimentally obtained as the dose immediately antecedent the one inducing malaise, and was found to be at 100 mg/kg i.p. for NYC215, at 150 mg/kg i.p. for NYC438, and at 200 mg/kg i.p. for NYC488. All these doses were >10 times higher than the concentration used in the efficacy study.
The evaluation of acute toxicity at MTD doses was then carried out in another set of animals (3–5 month old), acutely injected either with vehicle, NYC215, NYC438, or NYC488. No clinical signs of toxicity (as measured through food and liquid intake, weight change, locomotion and exploratory behavior, as well as mortality) were observed during the first 24 h with continuous monitoring given in the first 4 h, as well as for 14 days after acute, single dose administration. This observation was supported by the necropsy performed on the treated animals at 14 days after the acute treatment. Necropsy included weights and measurements of organs, appearance of organs (fat deposition, hemorrhage, pigment deposition or other changes, lesion, consistency), and examination of specific macro-lesions such as abnormal growths, fibrosis, and necrosis. We did not observe signs of anatomical modifications.
Next, we performed the evaluation of chronic toxicity. An additional set of experiments was performed with treatment for 15 days at the respective MTD for each drug or vehicle. Body weight, fluid and food intake, as well as any sign of behavioral distress, were continuously monitored during the treatment. No physical/behavioral distress or death was observed throughout the treatment. At the end of the chronic treatment, animals were sacrificed and necropsy was carried out in all the animals. Finally, in a separate set of experiments, treatment was perform for 15 days at the MTD and animals were monitored afterwards for additional 15 days to examine possible delayed signs of toxicity. Again, we did not observe any signs of toxicity.
In the absence of gross abnormalities, histopathologic evaluation after necropsy was limited to organs that have reported pathology linked to calpain inhibition or loss of function (for a review on the role of calpains in pathology see [49]). In particular, we focused on the condition of the liver (hepatotoxicity has been reported in association with protease inhibitors used in the treatment of HIV that inhibit calpain activity; hepatic steatosis and fibrosis, elevated free fatty acid levels and insulin resistance are associated with decreased activity of calpain 10), the kidney (looking for signs of diabetic nephropathy because of the possible inhibition of calpain 10, cellularity, inflammation, collagen deposition/fibrosis/sclerosis on glomeruli, vessels, tubules, collecting ducts and interstitium, common nephrotoxic effects as proximal tubular epithelial cell damage or renal papillary necrosis), the muscle (checking for myofiber size and fibrosis, necrotic fibers and dystrophy because the loss of function mutations in calpain 3 results in Limb Girdle Muscular Dystrophy Type 2A, regenerative fibers, fat deposition, inflammation), the stomach (assessing the possible presence of gastric cancer because calpain 9 has been proposed to act as a gastric cancer suppressor), and finally the brain (assessing cytoarchitecture, neuronal loss including both apoptosis and necrosis, inflammation, axonal degeneration, gliosis, myelination, body inclusions, neurotoxicity in neocortex, striatum, thalamus, hippocampus, brain stem, and cerebellum).
Overall, the histopathological evidence did not reflect any generalized toxicity induced by the chronic treatment at MTD for the three candidate inhibitors (Table 1). However, it is noteworthy that potential nephrotoxicity, induced by the 2nd generation compound NYC215, was suggested by the isometric vacuolization that is probably associated with osmolarity adjustment [50] (Fig. 5B). Nevertheless, no kidney toxicity was observed with the 3rd generation candidate compounds NYC438 (Fig. 5C) and NYC488 (Fig. 5D).
Table 1.
Assessment of the toxicity profile of new calpain inhibitors in vivo. Compounds NYC215, NYC438, and NYC488 were chronically administered in vivo at the respective MTDs (that are over 10 times higher than the expected therapeutic dose). Histological evaluation for probing gross modifications were carried out in different target organs known to be a possible source of concern in epoxide-based and/or calpain inhibition treatments (liver, kidney, muscle, stomach, and brain). The 3rd generation compounds NYC438 and NYC488 showed no sign of toxicity whereas the 2nd generation compound NYC215 induced a discrete isometric tubular epithelial vacuolization in the kidney, probably associated to osmolarity adjustment
| Organ | Pathology | Observation |
|---|---|---|
|
| ||
| Liver | fibrosis | no |
| necrosis | no | |
| steatosis | no | |
| Kidney | common nephrotoxic effects as proximal tubular epithelial cell damage or renal papillary necrosis | discrete isometric tubular epithelial vacuolization in the NYC215 group. No renal distress in all other groups |
| inflammation | no | |
| glomerular pathology | no | |
| signs of diabetic nephropathy | no | |
| Muscle | altered myofiber size and/or fibrosis | no |
| necrotic fibers and dystrophy | no | |
| regenerative fibers | no | |
| fat deposition | no | |
| inflammation | no | |
| Stomach | gastric cancer | no |
| Brain | neuronal loss (apoptosis and necrosis) | no |
| inflammation | no | |
| axonal degeneration | no | |
| gliosis | no | |
| demyelination | no | |
| abnormal inclusions | no | |
Fig. 5.
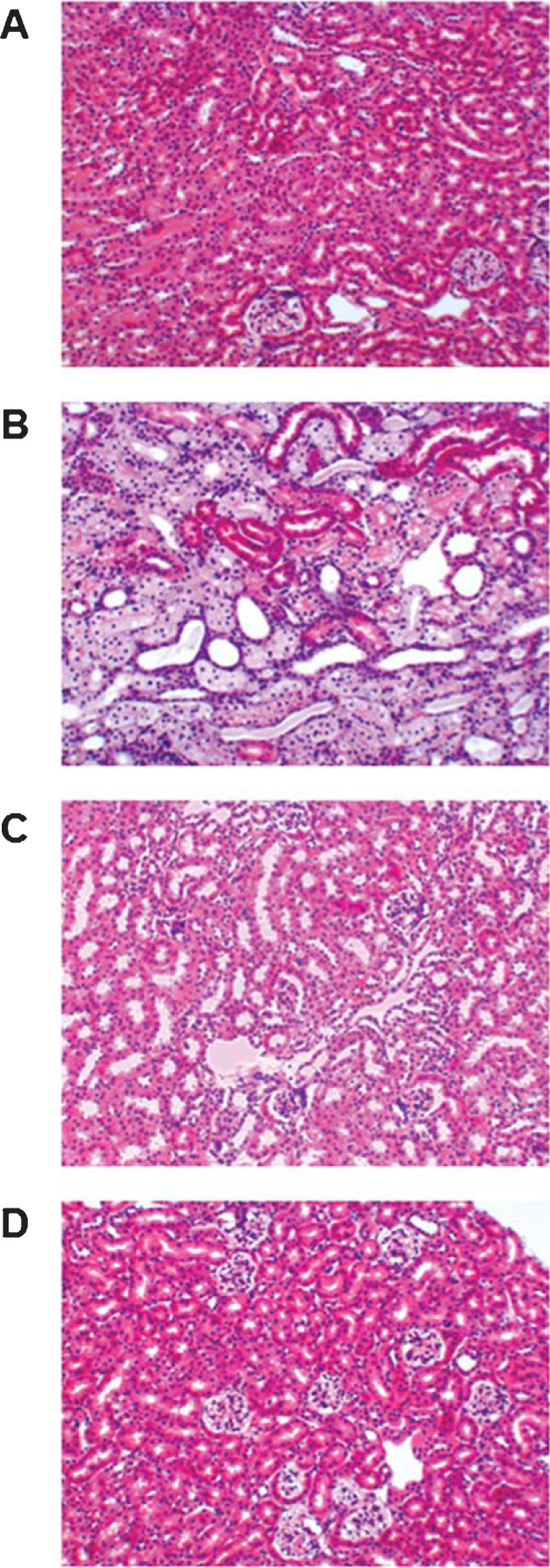
Light micrographs of sections from mouse kidneys stained with hematoxylin–eosin after treatment with novel calpain inhibitors. A) Normal histology of kidney tissue in mouse treated with vehicle. B) Staining of representative kidney slices obtained from the 2nd generation lead NYC215 group at the MTD (100 mg/kg i.p.). The renal distress, with vacuolization of the tubular epithelium, is quite evident throughout the examined section. Nevertheless, kidney sections of 3rd generation compounds NYC438 (150 mg/kg i.p.)- (C) and NYC488 (200 mg/kg i.p.)- (D) treated mice showed a normal microstructure of the kidney upon chronic treatment at respective MTDs.
Phenotypic screening of novel calpain 1 inhibitors with cognitive tests
Second and third generation lead compounds were then tested for the ability to ameliorate the defect in associative memory through fear conditioning assessment [39] and in short-term reference memory using the 2 day radial-arm water maze (RAWM) test [40, 41] in double transgenic AβPP/PS1 mice both at early stages of Aβ deposition (3 months) and late stages (7 months). Both associative memory and short-term spatial memory are early signs of cognitive decline in AD [51, 52].
We first examined contextual fear learning, a hippocampus-dependent task in both AβPP/PS1 and WT littermates treated with either vehicle or NYC215, NYC438, and NYC488 at equimolar doses (7.57 mg/kg, 7.86 mg/kg, and 7.83 mg/kg, respectively) from the age of 2 months until 3 months. According to the standard experimental paradigm, the animals must associate a neutral stimulus with an aversive one, so that when they are placed in a new context (fear-conditioning box), exposed to a conditional stimulus, i.e., a white noise cue, and they receive an unconditional stimulus, i.e., a mild foot electric shock, they display freezing behavior. Fear learning was then assessed 24 h later by measuring freezing behavior in response to presentation of the context (contextual conditioning) or of the auditory cue within a completely different context (cued conditioning). We found no difference in the freezing behavior of the vehicle and inhibitor-treated AβPP/PS1 mice compared with vehicle and inhibitor-treated WT littermates during the training phase of the fear-conditioning test (data not shown). Twenty-four hours later, we found decreased freezing behavior in vehicle-treated AβPP/PS1 mice compared with vehicle-treated WT littermates in the analysis of contextual learning. However, treatment with our lead molecules restored freezing in AβPP/PS1 mice and did not affect the performance of WT mice (Fig. 6A). Treatment with the compounds did not affect the performance of WT mice, further suggesting that they do not induce toxicity (Fig. 6A). Moreover, vehicle-treated WT mice showed similar freezing time as control untreated WT mice (data not shown). We also did not find a difference in freezing behavior during cued learning (data not shown). These results indicate that the impairment in contextual fear learning in AβPP/PS1 mice can be rescued by treatment with a calpain inhibitor. Testing of the effects of the novel inhibitors in older mice that were treated from the age of 2 months until 7 months confirmed the results of younger mice (Fig. 6B). The novel calpain inhibitors were able to prevent the cognitive disturbance in associative memory in double transgenic AβPP/PS1 mice at 7 month of age. AβPP/PS1 mice treated with the novel calpain inhibitors showed consistently more freezing in fear conditioning assessment than vehicle–treated mice, demonstrating the possibility to prevent the occurrence of cognitive disturbances in AβPP/PS1 mice even at ages when typically learning and memory is severely impaired.
Fig. 6.
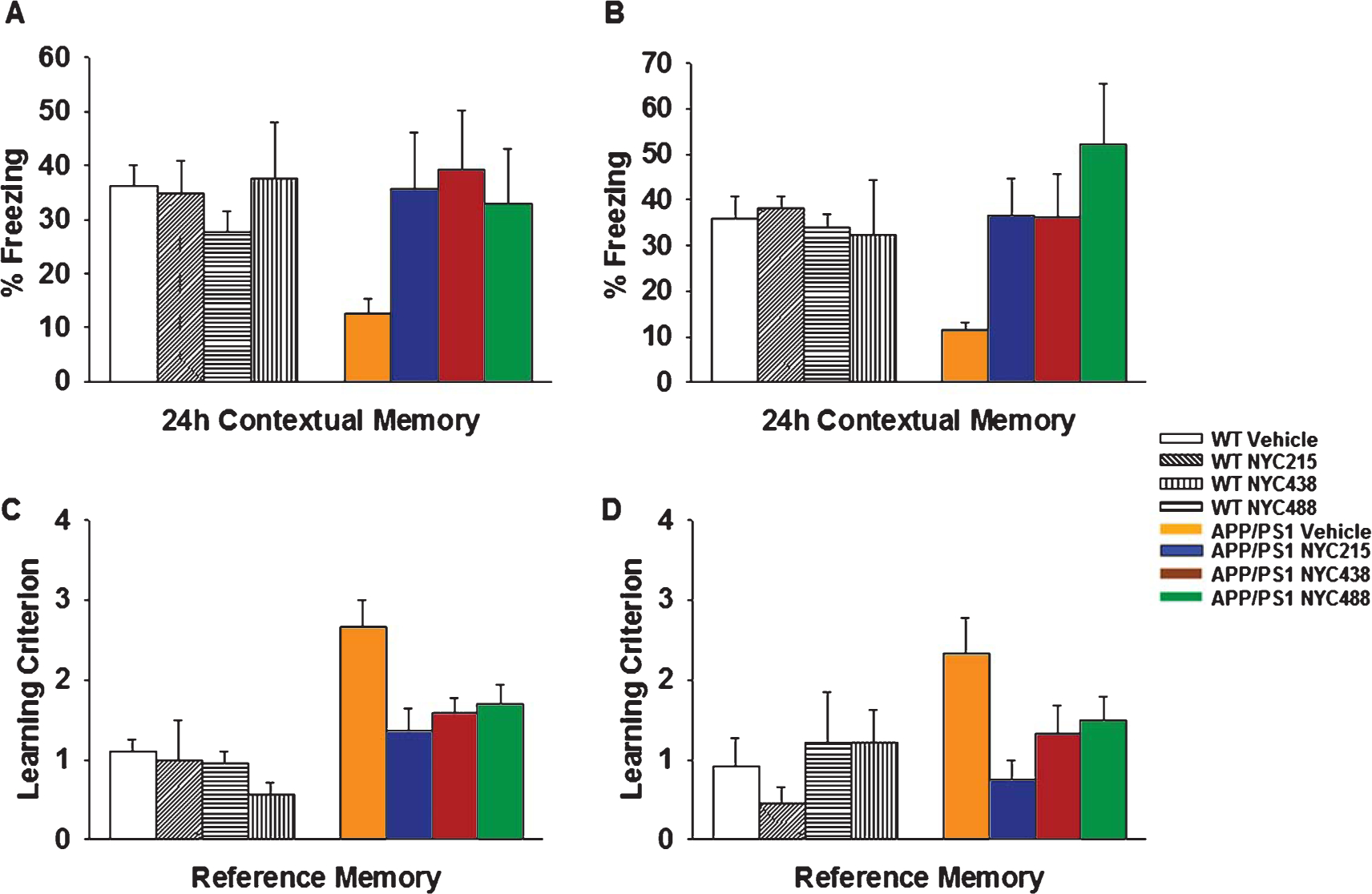
Behavioral evaluation of the ability of novel calpain inhibitor to rescue memory defects in AβPP/PS1 mice. A) Daily treatment with 2nd generation NYC215, and 3rd generation NYC438 and NYC488 from the age of 2 months until 3 months ameliorated the defect in contextual fear memory in AβPP/PS1 mice. WT-vehicle: n = 15, WT-NYC215: n = 8, WT-NYC438: n = 8, WT-NYC488: n = 8, AβPP/PS1-vehicle: n = 15, AβPP/PS1-NYC215: n = 10, AβPP/PS1-NYC438: n = 11, AβPP/PS1-NYC488: n = 10. p < 0.05 in all transgenic groups treated with compound compared to their respective vehicle-treated transgenics. B) Daily treatment with NYC215, NYC438 and NYC488 from the age of 2 months until 7 months ameliorated the defect in contextual fear memory. WT-vehicle: n = 18, WT-NYC215: n = 9, WT-NYC438: n = 9, WT-NYC488: n = 9, AβPP/PS1-vehicle: n = 17; AβPP/PS1-NYC215: n = 10; AβPP/PS1-NYC438: n = 10; AβPP/PS1-NYC488: n = 11. p < 0.05 in all transgenic groups treated with compound compared to their respective vehicle-treated transgenics. C) Daily treatment with NYC215, NYC438, and NYC488 from the age of 2 months until 3 months ameliorated the defect in spatial memory in AβPP/PS1 mice. WT-vehicle: n = 15, WT-NYC215: n = 8, WT-NYC438: n = 8, WT-NYC488: n = 8, AβPP/PS1-vehicle: n = 15, AβPP/PS1-NYC215: n = 10, AβPP/PS1-NYC438: n = 11, AβPP/PS1-NYC488: n = 10. p < 0.05 in all transgenic groups treated with compound compared to their respective vehicle-treated transgenics. D) Daily treatment with NYC215, NYC438, and NYC488 from the age of 2 months until 7 months ameliorated the defect in spatial memory in AβPP/PS1 mice. WT-vehicle: n = 18, WT-NYC215: n = 9, WT-NYC438: n = 9, WT-NYC488: n = 9, AβPP/PS1-vehicle: n = 17; AβPP/PS1-NYC215: n = 10; AβPP/PS1-NYC438: n = 10; AβPP/PS1-NYC488: n = 11. p < 0.05 in all transgenic groups treated with compound compared to their respective vehicle-treated transgenics.
Next, we aimed to verify whether the same treatment could reverse the spatial learning impairment in AβPP/PS1 mice. The 2-day RAWM task was performed as previously described [41]. The mouse had to swim in a 6 arm maze filled with milky water until it was able to find a hidden platform at the end of one of the arms (submerged platform) using the visual orientation cues placed within sight above the maze. During the first day (training), mice were trained to identify the platform location by alternating between a visible and a hidden platform in a specific maze arm (goal arm) during several consecutive training trials. We evaluated the number of mouse entries in an arm with no platform (incorrect arm entries). Failure to select an arm after 15 s was counted as an error. Each trial lasted up to 1 min. After 1 min, if the platform had not been located, the mouse was then directed towards the platform while swimming.
NYC215 or NYC438 or NYC488 improved the performance of AβPP/PS1 mice with the 2-day RAWM task without affecting the performance of WT littermates (Fig. 6C). Indeed, vehicle-treated AβPP/PS1 failed to reach the learning criterion ( 1 error) in the 2-day RAWM task by block 9 and 10 of day 2 whereas vehicle-injected WT littermate mice succeeded. NYC215, NYC438, and NYC488 ameliorated the deficit in RAWM performance in transgenic mice on the last block. NYC215, NYC438, and NYC488 did not affect the WT littermate performance with the RAWM. Vehicle-treated WT mice had similar performance as control untreated WT mice (data not shown). To test for visual, motor, and motivational deficits, all mice also underwent visible platform task after performing the RAWM test. We found no difference in speed and latency period to the platform for the various groups of mice (data not shown).
Testing of the effects of the novel inhibitors in older mice that were treated from the age of 2 months until 7 months confirmed the results in younger mice (Fig. 6D). Our novel calpain inhibitors prevented deficits in spatial memory in double transgenic AβPP/PS1 mice at 7 months of age.
Overall, AβPP/PS1 mice treated with the novel calpain inhibitors showed consistently more freezing in fear conditioning assessment and fewer errors in 2-days RAWM than vehicle–treated mice, demonstrating the possibility to prevent the occurrence of cognitive disturbances in AβPP/PS1 mice even at ages when typically learning and memory is severely impaired. Taken together, these data suggest that the novel calpain inhibitors are capable and quite effective in restoring the cognitive abilities in the AβPP/PS1 AD mouse model. This implies that early treatments with calpain inhibitors may protect from the progressive cognitive sequelae already established in the early stage of the disease. These results have significant translational value in further reinforcing the need for clinical trials of calpain inhibitors in AD. Nevertheless, in the current study we verified whether the 3rd generation compound NYC438, which has longer half-life and better bioavailability, altered Aβ40 and Aβ42 levels in hippocampal tissue and blood. ELISA analysis of Aβ40 and Aβ42 revealed readily quantifiable levels of these peptides in the hippocampus and blood following 5-month treatment with NYC438 or vehicle in double transgenic AβPP/PS1 mice. Daily treatment with NYC438 did not reduce hippocampal or blood levels of Aβ40 and Aβ42 in AβPP/PS1 mice (Fig. 7). Overall, these data support the hypothesis that the beneficial effects of calpain inhibition are produced by mechanisms downstream of Aβ production, while still counteracting the Aβ-induced detrimental effects.
Fig. 7.
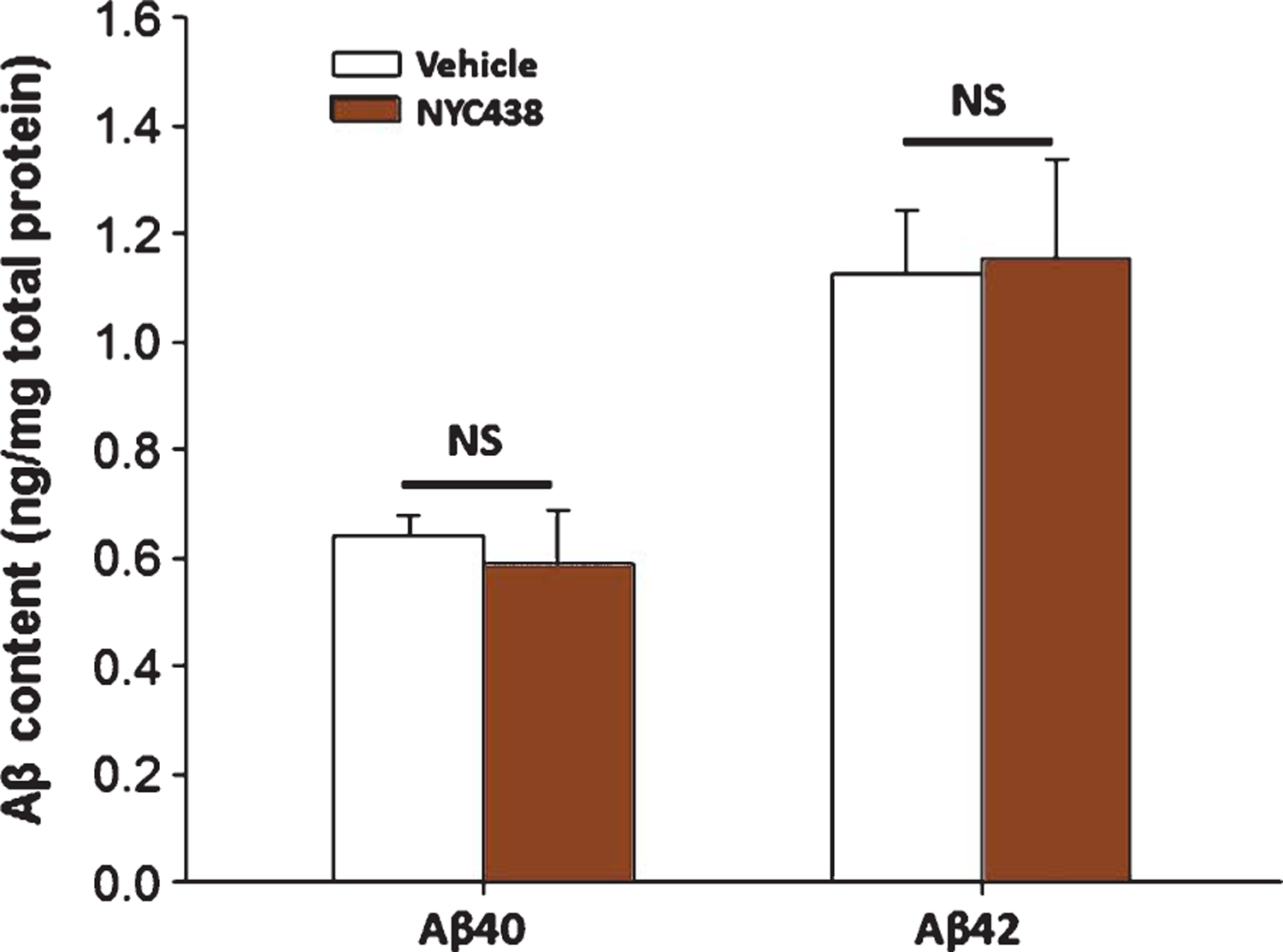
ELISA analyses of Aβ40 and Aβ42 levels after treatment with the novel calpain inhibitor NYC438. 3rd generation compound NYC438 did not affect Aβ40 and Aβ42 levels in hippocampi of 7–8 month old AβPP/PS1 mice (n = 5 per group).
Novel calpain 1 inhibitors do not alter cerebral and plasma Aβ content
We previously demonstrated that both E64 and BDA-410 do not have any effect on Aβ levels [19].
DISCUSSION
AD is a neurological, multifactorial illness, of epidemic proportions, which is characterized by a number of neuropathological features, including the steady presence of diffuse proteinaceous aggregates in the brain diffused along brain areas, neuronal death, and synaptic changes leading to dementia [53, 54]. AD incidence in the population is growing together with progressively increasing lifespans [25, 55]. Thus, there is an undisputable need for robust and safe therapeutics to treat the disease. One of the putative causes of AD is attributable to the increased presence or circulation of soluble oligomers of Aβ providing both a substrate for synaptic degeneration and diffusion of the pathology along brain areas [30]. Substantial evidence supports the ability of Aβ oligomers to reduce plasticity and memory both in translational experimental models of the disease (for a review see: [56]) and in humans [57–59]. It is therefore conceivable to counteract the detrimental effects of oligomeric Aβ exposure as a possible target for the development of a causal therapy in AD. Indeed, the potential therapeutic value in AD of our novel calpain inhibitors was demonstrated in their ability to ameliorate deficits in LTP and memory induced by soluble Aβ-induced, overcoming the possible criticism of drugs developed solely to block protein aggregation and deposition [48].
Another important feature of our approach is targeting calpain inhibition. Over-activation of calpains, whether as a direct consequence of Aβ activity or due to other factors affecting calcium signaling (for a review, see [60, 61]), is one of the culprits of AD [62]. Moreover, calpain hyperactivation has been shown in other proteinopathies [63–65]. Therefore, the identification of calpain inhibitors might serve to attenuate elevated calpain activity while preserving physiological levels of calpain activation [66, 67]. Among the known calpain inhibitors which have been studied in AD is A-705253, developed by Abbvie Pharma, a notable ketoamide-based calpain inhibitor that is active in the 3xTgAD mouse model rescuing memory defects, and reducing levels of BACE enzyme, Aβ deposits, and overall neuroinflammation [68, 69]. We designed a novel series of epoxide-based calpain inhibitors using E64 as a lead. Two 3rd generation inhibitors, NYC438 and NYC488, were potent inhibitors of calpain 1 (IC50 <100 nM) with improved selectivity and easy synthetic scalability [32], Inhibition of spectrin cleavage provided evidence of functional brain bioavailability, and both inhibitors were able to recover both the plasticity and the memory impairment associated with the exposure to Aβ42.
Traditionally, medicinal chemistry addresses the need to optimize toward more potent and more selective compounds during in vitro screening. These are extremely important drug characteristics yet still void of interest if not associated with translational significance in vivo. In our phenotypical screening effort, we parallelized the discovery of the most active compound towards maintenance and recovery of LTP in hippocampus, a key cognitive brain area, in a transgenic model of amyloid deposition regardless of the severity of the plaque load, with the identification of the most selective and potent compound for calpain inhibition. These results are translationally important because cognitive disturbances are the most compelling debilitating symptoms of AD. Therapy should sustain the ability of synaptic terminals to undergo plasticity throughout the different stages of the disease, and to recover and maintain the cognitive reserve, in order to greatly reduce the risk of dementia [70].
The development of a class of molecules that are more selective toward calpains than previous inhibitors has proved to be difficult. Novel synthesis has also been challenged by the potential large number of toxic effects due to the high number of physiological effectors of calpains [71]. Interestingly, our studies in which computation guided medicinal chemistry efforts led to a new library of very promising molecules displaying marked inhibition of calpain 1 and selectivity over cathepsin B [32]. These molecules had an epoxide warhead which, differently than other potent and well-known inhibitors including compounds BDA-410 and E64 that have been previously tested in AD [19], did not show reactivity toward non-specific targets [32]. BDA-410 and E64 bear cyclopropenone and epoxide warheads, respectively [19]. Both cyclopropenone and epoxide warheads are electrophilic, with the potential to covalently modify the cysteine active site of calpain 1, or to non-selectively modify other protein-thiols, or even to form glutathione conjugates and therefore be rapidly cleared [72]. In our drug discovery design, therefore, we considered the knowledge of reactivity toward proteins and free thiols as a specific requirement early in the drug development project, in addition to the simple structure-based, “rational” drug design strategy towards selective calpain 1 inhibitors using phenotypical screening.
Both of our 3rd generation compounds, NYC438 and NYC488, had very good PK characteristics with a slightly longer half-life for NYC438 versus NYC488 (~1.1 h and ~0.6 h). The inhibitors, moreover, showed no overall toxicity, despite the general idea that epoxides are liable because of non-specific interaction with thiol groups in off-targets [71]. This is important as our toxicological tests were performed at the MTD, which is at least ten-times higher than specific therapeutic doses [48]. This signifies an intrinsic safety of our lead 3rd generation compounds [73]. Epoxide inhibitors might display a number of advantages over other types of cysteine protease inhibitors. In fact, epoxides are less reactive than other classes of inhibitors, such as halomethylketones or aldehydes, showing to be more directed to a single target (whether calpains or cathepsins) and therefore they could cause lower toxicity in vivo [72]. Since epoxide inhibitors are irreversible, therapeutic doses can be administered less frequently than those of reversible inhibitors, which are effective only when not yet cleared from blood circulation.
While our new drugs display an optimal efficacy toward translational AD models involving the activity of Aβ both in terms of behavioral and plasticity outcomes, they do not modify the levels of Aβ40 and Aβ42 in the AβPP/PS1 mouse model.
This signifies a potent action due to blocking AD-associated calpain overactivation, which is either downstream or independent of Aβ, and suggests a convergence of AD pathophysiology on calpains [19]. This feature potentially broadens the therapeutic value of our compounds from AD to other neurodegenerative diseases displaying calpain overactivation [63–65]. This is particularly important in light of the fact that clinical trials attempting to cure AD through a strategy aimed mostly at the reduction of Aβ levels, have failed, a negative outcome that has prompted FDA to put a warning on the use of this strategy [74].
As recently remarked by the Alzheimer’s Association [25], the slow, insidious nature of AD progression, the staggering impoverishment of the life quality for AD patients, and the destructive public health outcomes such as family disintegration/impoverishments, and cost of caregiving supports the quest for new symptom- and disease-modifying treatments. The availability of a new series of effective, non-toxic therapeutics, exerting their activity on the molecular mechanisms of the disease, is a positive step toward a better cure for AD and any other disease associated with calpain overactivation.
ACKNOWLEDGMENTS
We would like to thank Dr. Gregory Hook (American Life Science Pharmaceuticals, La Jolla, CA, USA) and Dr. Virginia Hook (UCSD, La Jolla, CA, USA) for their generous and helpful insights on experimental data and manuscript; Dr. Dafang Zhong (Center for Drug Metabolism Research, Shanghai Institute of Materia Medica, China) for the pharmacokinetic testing.
This work was supported by grants from the National Institutes of Health, # U01-AG028713. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/15-0618r1).
REFERENCES
- [1].Wu HY, Lynch DR (2006) Calpain and synaptic function. Mol Neurobiol 33, 215–236. [DOI] [PubMed] [Google Scholar]
- [2].Cong J, Goll DE, Peterson AM, Kapprell HP (1989) The role of autolysis in activity of the Ca2+-dependent proteinases (mu-calpain and m-calpain). J Biol Chem 264, 10096–10103. [PubMed] [Google Scholar]
- [3].Liu F, Grundke-Iqbal I, Iqbal K, Oda Y, Tomizawa K, Gong CX (2005) Truncation and activation of calcineurin A by calpain I in Alzheimer disease brain. J Biol Chem 280, 37755–37762. [DOI] [PubMed] [Google Scholar]
- [4].Saito K, Elce JS, Hamos JE, Nixon RA (1993) Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: A potential molecular basis for neuronal degeneration. Proc Natl Acad Sci U S A 90, 2628–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Baudry M, Zhu G, Liu Y, Wang Y, Briz V, Bi X (2015) Multiple cellular cascades participate in long-term potentiation and in hippocampus-dependent learning. Brain Res 1621, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hu GY, Hvalby O, Walaas SI, Albert KA, Skjeflo P, Andersen P, Greengard P (1987) Protein kinase C injection into hippocampal pyramidal cells elicits features of long term potentiation. Nature 328, 426–429. [DOI] [PubMed] [Google Scholar]
- [7].Lledo PM, Hjelmstad GO, Mukherji S, Soderling TR, Malenka RC, Nicoll RA (1995) Calcium/calmodulin-dependent kinase II and long-term potentiation enhance synaptic transmission by the same mechanism. Proc Natl Acad Sci U S A 92, 11175–11179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Malinow R, Schulman H, Tsien RW (1989) Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science 245, 862–866. [DOI] [PubMed] [Google Scholar]
- [9].Otmakhov N, Griffith LC, Lisman JE (1997) Postsynaptic inhibitors of calcium/calmodulin-dependent protein kinase type II block induction but not maintenance of pairing-induced long-term potentiation. J Neurosci 17, 5357–5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wu HY, Tomizawa K, Oda Y, Wei FY, Lu YF, Matsushita M, Li ST, Moriwaki A, Matsui H (2004) Critical role of calpain-mediated cleavage of calcineurin in excitotoxic neurodegeneration. J Biol Chem 279, 4929–4940. [DOI] [PubMed] [Google Scholar]
- [11].Watt F, Molloy PL (1993) Specific cleavage of transcription factors by the thiol protease, m-calpain. Nucleic Acids Res 21, 5092–5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jin N, Qian W, Yin X, Zhang L, Iqbal K, Grundke-Iqbal I, Gong CX, Liu F (2013) CREB regulates the expression of neuronal glucose transporter 3: A possible mechanism related to impaired brain glucose uptake in Alzheimer’s disease. Nucleic Acids Res 41, 3240–3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kandel ER (2012) The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain 5, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Silva AJ, Kogan JH, Frankland PW, Kida S (1998) CREB and memory. Annu Rev Neurosci 21, 127–148. [DOI] [PubMed] [Google Scholar]
- [15].Barco A, Alarcon JM, Kandel ER (2002) Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell 108, 689–703. [DOI] [PubMed] [Google Scholar]
- [16].Benito E, Barco A (2010) CREB’s control of intrinsic and synaptic plasticity: Implications for CREB-dependent memory models. Trends Neurosci 33, 230–240. [DOI] [PubMed] [Google Scholar]
- [17].Ferreira A, Bigio EH (2011) Calpain-mediated tau cleavage: A mechanism leading to neurodegeneration shared by multiple tauopathies. Mol Med 17, 676–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Higuchi M, Tomioka M, Takano J, Shirotani K, Iwata N, Masumoto H, Maki M, Itohara S, Saido TC (2005) Distinct mechanistic roles of calpain and caspase activation in neurodegeneration as revealed in mice overexpressing their specific inhibitors. J Biol Chem 280, 15229–15237. [DOI] [PubMed] [Google Scholar]
- [19].Trinchese F, Fa M, Liu S, Zhang H, Hidalgo A, Schmidt SD, Yamaguchi H, Yoshii N, Mathews PM, Nixon RA, Arancio O (2008) Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J Clin Invest 118, 2796–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Liang Z, Liu F, Grundke-Iqbal I, Iqbal K, Gong CX (2007) Down-regulation of cAMP-dependent protein kinase by over-activated calpain in Alzheimer disease brain. J Neurochem 103, 2462–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Brewer GJ (2000) Neuronal plasticity and stressor toxicity during aging. Exp Gerontol 35, 1165–1183. [DOI] [PubMed] [Google Scholar]
- [22].Arendt T (2009) Synaptic degeneration in Alzheimer’s disease. Acta Neuropathol 118, 167–179. [DOI] [PubMed] [Google Scholar]
- [23].Galimberti D, Scarpini E (2012) Progress in Alzheimer’s disease. J Neurol 259, 201–211. [DOI] [PubMed] [Google Scholar]
- [24].Hua DH (2013) Design, synthesis, and evaluation of bioactive small molecules. Chem Rec 13, 60–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Alzheimer’s Association (2014) 2014 Alzheimer’s disease facts and figures. Alzheimers Dement 10, e47–e92. [DOI] [PubMed] [Google Scholar]
- [26].Masliah E (1995) Mechanisms of synaptic dysfunction in Alzheimer’s disease. Histol Histopathol 10, 509–519. [PubMed] [Google Scholar]
- [27].Vitolo OV, Sant’Angelo A, Costanzo V, Battaglia F, Arancio O, Shelanski M (2002) Amyloid beta -peptide inhibition of the PKA/CREB pathway and long-term potentiation: Reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci U S A 99, 13217–13221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cullen WK, Suh YH, Anwyl R, Rowan MJ (1997) Block of LTP in rat hippocampus in vivo by beta-amyloid precursor protein fragments. Neuroreport 8, 3213–3217. [DOI] [PubMed] [Google Scholar]
- [29].Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. [DOI] [PubMed] [Google Scholar]
- [30].Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297, 353–356. [DOI] [PubMed] [Google Scholar]
- [31].Di Rosa G, Odrijin T, Nixon RA, Arancio O (2002) Calpain inhibitors: A treatment for Alzheimer’s disease. J Mol Neurosci 19, 135–141. [DOI] [PubMed] [Google Scholar]
- [32].Schiefer IT, Tapadar S, Litosh V, Siklos M, Scism R, Wijewickrama GT, Chandrasena EP, Sinha V, Tavassoli E, Brunsteiner M, Fà M, Arancio O, Petukhov P, Thatcher GRJ (2013) Design, synthesis, and optimization of novel epoxide incorporating peptidomimetics as selective calpain inhibitors. J Med Chem 56, 6054–6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Priest BT, Erdemli G (2014) Phenotypic screening in the 21st century. Front Pharmacol 5, 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274, 99–102. [DOI] [PubMed] [Google Scholar]
- [35].Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S (1996) Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature 383, 710–713. [DOI] [PubMed] [Google Scholar]
- [36].Trinchese F, Liu S, Battaglia F, Walter S, Mathews PM, Arancio O (2004) Progressive age-related development of Alzheimer-like pathology in APP/PS1 mice. Ann Neurol 55, 801–814. [DOI] [PubMed] [Google Scholar]
- [37].Fa M, Orozco IJ, Francis YI, Saeed F, Gong Y, Arancio O (2010) Preparation of oligomeric beta-amyloid 1–42 and induction of synaptic plasticity impairment on hippocampal slices. J Vis Exp (41), e1884, doi:10.3791-1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fa M, Staniszewski A, Saeed F, Francis YI, Arancio O (2014) Dynamin 1 is required for memory formation. PLoS One 9, e91954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Phillips RG, LeDoux JE (1992) Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci 106, 274–285. [DOI] [PubMed] [Google Scholar]
- [40].Fiorito J, Saeed F, Zhang H, Staniszewski A, Feng Y, Francis YI, Rao S, Thakkar DM, Deng SX, Landry DW, Arancio O (2013) Synthesis of quinoline derivatives: Discovery of a potent and selective phosphodiesterase 5 inhibitor for the treatment of Alzheimer’s disease. Eur J Med Chem 60, 285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Alamed J, Wilcock DM, Diamond DM, Gordon MN, Morgan D (2006) Two-day radial-arm water maze learning and memory task; robust resolution of amyloid-related memory deficits in transgenic mice. Nat Protoc 1, 1671–1679. [DOI] [PubMed] [Google Scholar]
- [42].Teich AF, Patel M, Arancio O (2013) A reliable way to detect endogenous murine beta-amyloid. PLoS One 8, e55647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wang KK (2000) Calpain and caspase: Can you tell the difference? Trends Neurosci 23, 20–26. [DOI] [PubMed] [Google Scholar]
- [44].Hu RJ, Bennett V (1991) In vitro proteolysis of brain spectrin by calpain I inhibits association of spectrin with ankyrin-independent membrane binding site(s). J Biol Chem 266, 18200–18205. [PubMed] [Google Scholar]
- [45].Yoon M, Campbell JL, Andersen ME, Clewell HJ (2012) Quantitative in vitro to in vivo extrapolation of cell-based toxicity assay results. Crit Rev Toxicol 42, 633–652. [DOI] [PubMed] [Google Scholar]
- [46].Bae J-W, Kim D-H, Lee W-W, Kim H-Y, Son C-G (2015) Characterizing the human equivalent dose of herbal medicines in animal toxicity studies. J Ethnopharmacol 162, 1–6. [DOI] [PubMed] [Google Scholar]
- [47].Dorato MA, Engelhardt JA (2005) The no-observed-adverse-effect-level in drug safety evaluations: Use, issues, and definition(s). Regul Toxicol Pharmacol 42, 265–274. [DOI] [PubMed] [Google Scholar]
- [48].Food US and Administration Drug (1995) Guidance for Industry, Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. US Food and Drug Administration, Silver Spring, MD. [Google Scholar]
- [49].Bertipaglia I, Carafoli E (2007) Calpains and human disease. Subcell Biochem 45, 29–53. [DOI] [PubMed] [Google Scholar]
- [50].Choudhury D, Ahmed Z (2006) Drug-associated renal dysfunction and injury. Nat Clin Pract Nephrol 2, 80–91. [DOI] [PubMed] [Google Scholar]
- [51].Swainson R, Hodges JR, Galton CJ, Semple J, Michael A, Dunn BD, Iddon JL, Robbins TW, Sahakian BJ (2001) Early detection and differential diagnosis of Alzheimer’s disease and depression with neuropsychological tasks. Dement Geriatr Cogn Disord 12, 265–280. [DOI] [PubMed] [Google Scholar]
- [52].Cushman LA, Stein K, Duffy CJ (2008) Detecting navigational deficits in cognitive aging and Alzheimer disease using virtual reality. Neurology 71, 888–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Blennow K, de Leon MJ, Zetterberg H (2006) Alzheimer’s disease. Lancet 368, 387–403. [DOI] [PubMed] [Google Scholar]
- [54].Forlenza OV, Diniz BS, Gattaz WF (2010) Diagnosis and biomarkers of predementia in Alzheimer’s disease. BMC Med 8, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM (2007) Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement 3, 186–191. [DOI] [PubMed] [Google Scholar]
- [56].Puzzo D, Lee L, Palmeri A, Calabrese G, Arancio O (2014) Behavioral assays with mouse models of Alzheimer’s disease: Practical considerations and guidelines. Biochem Pharmacol 88, 450–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kar S, Quirion R (2004) Amyloid beta peptides and central cholinergic neurons: Functional interrelationship and relevance to Alzheimer’s disease pathology. Prog Brain Res 145, 261–274. [DOI] [PubMed] [Google Scholar]
- [58].Kar S, Slowikowski SP, Westaway D, Mount HT (2004) Interactions between beta-amyloid and central cholinergic neurons: Implications for Alzheimer’s disease. J Psychiatry Neurosci 29, 427–441. [PMC free article] [PubMed] [Google Scholar]
- [59].Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD (2000) Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA 283, 1571–1577. [DOI] [PubMed] [Google Scholar]
- [60].Egorova P, Popugaeva E, Bezprozvanny I (2015) Disturbed calcium signaling in spinocerebellar ataxias and Alzheimer’s disease. Semin Cell Dev Biol 40, 127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Chakroborty S, Stutzmann GE (2014) Calcium channelopathies and Alzheimer’s disease: Insight into therapeutic success and failures. Eur J Pharmacol 739, 83–95. [DOI] [PubMed] [Google Scholar]
- [62].Ferreira A (2012) Calpain dysregulation in Alzheimer’s disease. ISRN Biochemistry 2012, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Menzies FM, Garcia-Arencibia M, Imarisio S, O’Sullivan NC, Ricketts T, Kent BA, Rao MV, Lam W, Green-Thompson ZW, Nixon RA, Saksida LM, Bussey TJ, O’Kane CJ, Rubinsztein DC (2015) Calpain inhibition mediates autophagy-dependent protection against polyglutamine toxicity. Cell Death Differ 22, 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Diepenbroek M, Casadei N, Esmer H, Saido TC, Takano J, Kahle PJ, Nixon RA, Rao MV, Melki R, Pieri L, Helling S, Marcus K, Krueger R, Masliah E, Riess O, Nuber S (2014) Overexpression of the calpain-specific inhibitor calpastatin reduces human alpha-Synuclein processing, aggregation and synaptic impairment in [A30P]alphaSyn transgenic mice. Hum Mol Genet 23, 3975–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Rao MV, McBrayer MK, Campbell J, Kumar A, Hashim A, Sershen H, Stavrides PH, Ohno M, Hutton M, Nixon RA (2014) Specific calpain inhibition by calpastatin prevents tauopathy and neurodegeneration and restores normal lifespan in tau P301L mice. J Neurosci 34, 9222–9234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Donkor IO (2011) Calpain inhibitors: A survey of compounds reported in the patent and scientific literature. Expert Opin Ther Pat 21, 601–636. [DOI] [PubMed] [Google Scholar]
- [67].Pietsch M, Chua KC, Abell AD (2010) Calpains: Attractive targets for the development of synthetic inhibitors. Curr Top Med Chem 10, 270–293. [DOI] [PubMed] [Google Scholar]
- [68].Nikkel AL, Martino B, Markosyan S, Brederson JD, Medeiros R, Moeller A, Bitner RS (2012) The novel calpain inhibitor A-705253 prevents stress-induced tau hyperphosphorylation in vitro and in vivo. Neuropharmacology 63, 606–612. [DOI] [PubMed] [Google Scholar]
- [69].Medeiros R, Kitazawa M, Chabrier MA, Cheng D, Baglietto-Vargas D, Kling A, Moeller A, Green KN, LaFerla FM (2012) Calpain inhibitor A-705253 mitigates Alzheimer’s disease-like pathology and cognitive decline in aged 3xTgAD mice. Am J Pathol 181, 616–625. [DOI] [PubMed] [Google Scholar]
- [70].Stern Y (2012) Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol 11, 1006–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Baudry M, Chou MM, Bi X (2013) Targeting calpain in synaptic plasticity. Expert Opin Ther Targets 17, 579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Powers JC, Asgian JL, Ekici OD, James KE (2002) Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem Rev 102, 4639–4750. [DOI] [PubMed] [Google Scholar]
- [73].Ishiura S, Hanada K, Tamai M, Kashiwagi K, Sugita H (1981) The effect of an in vivo-injected thiol protease inhibitor, E-64-c, on the calcium-induced degeneration of myofilaments. J Biochem 90, 1557–1560. [DOI] [PubMed] [Google Scholar]
- [74].Kozauer N, Katz R (2013) Regulatory innovation and drug development for early-stage Alzheimer’s disease. N Engl J Med 368, 1169–1171. [DOI] [PubMed] [Google Scholar]