

Abstract
Motivation
Intrinsically disordered proteins (IDPs) are involved in numerous processes crucial for living organisms. Bias in amino acid composition of these proteins determines their unique biophysical and functional features. Distinct intrinsically disordered regions (IDRs) with compositional bias play different important roles in various biological processes. IDRs enriched in particular amino acids in human proteome have not been described consistently.
Results
We developed DisEnrich—the database of human proteome IDRs that are significantly enriched in particular amino acids. Each human protein is described using Gene Ontology (GO) function terms, disorder prediction for the full-length sequence using three methods, enriched IDR composition and ranks of human proteins with similar enriched IDRs. Distribution analysis of enriched IDRs among broad functional categories revealed significant overrepresentation of R- and Y-enriched IDRs in metabolic and enzymatic activities and F-enriched IDRs in transport. About 75% of functional categories contain IDPs with IDRs significantly enriched in hydrophobic residues that are important for protein–protein interactions.
Availability and implementation
The database is available at http://prodata.swmed.edu/DisEnrichDB/.
Supplementary information
Supplementary data are available at Bioinformatics Advances online.
1 Introduction
Intrinsically disordered proteins (IDPs) are macromolecules lacking distinct three-dimensional (3D) structure or containing a combination of ordered and intrinsically disordered regions (IDRs) under natural conditions. Two decades ago, the first hypothesis of proteins’ natural disorder was raised, and since that time it has been developed into a new, fast evolving field (Tompa, 2012). Today, we value the importance of these proteins that are involved in a large variety of crucial processes in a living cell (Uversky, 2021). Although estimations of IDP prevalence in different proteomes vary, it is commonly accepted that the abundance of disorder generally increases with an organism’s complexity (Peng et al., 2015; Xue et al., 2012a, b). Indeed, around 25% of eukaryotic proteins are predicted to be mostly disordered, and about 50% of them contain long IDRs (Peng et al., 2015). Human proteome is estimated to have up to 50% of disordered residues (Oldfield and Dunker, 2014). Human IDPs have been extensively studied due to their importance for a large variety of crucial processes (Dunker et al., 2008; Dyson and Wright, 2005; Iakoucheva et al., 2002). The abundance of IDPs in living organisms [and viruses (Xue et al., 2012a)] emerged due to a lack of rigid secondary structure, which allowed more promiscuous binding, faster evolution and larger functional variety than their structural protein counterparts (Uversky, 2021; Xie et al., 2007). For a long period of time, the structure–function paradigm with ‘lock and key’ explanation of enzymatic function has remained unquestioned due to the determination of static protein 3D structures that supported the concept of a rigid active site (lock) binding a single substrate (key; Uversky and Dunker, 2010). However, disordered proteins, which function outside of this ‘lock and key’ paradigm, have shifted our explanation of enzyme function toward Koshland’s induced fit theory where a flexible active site can adapt to substrate binding and allow the reaction to take place (Koshland, 1958). Thus, functioning of IDPs outside of the limits of ‘lock and key’ paradigm allows them to be multifunctional, non-specific binders, which are commonly involved in regulatory and signaling processes (Tompa, 2012; Wright and Dyson, 2015). Moreover, proteins with long disordered regions have been correlated with such functions as differentiation, transcription, cell cycle and RNA processing (Tompa, 2012; Xie et al., 2007). Many IDPs are involved in processes linked to various diseases and might contain disease-causing mutations, with up to 25% of disease-associated missense mutations located in IDRs (Vacic and Iakoucheva, 2012). Involvement of IDPs in such processes as carcinogenesis, protein aggregation and amyloid formation makes them important targets for the improvement of various diseases administration (Uversky et al., 2008).
IDP amino acid sequences are characterized by distinct compositional biases relative to ordered proteins, namely enrichment in polar amino acids and deficiency in hydrophobic amino acids (van der Lee et al., 2014). This feature is the key to the functional characteristics of these proteins. Based on the physico-chemical properties of amino acids, the concept of order- and disorder-promoting residues has been proposed (Williams et al., 2001). The majority of order-promoting residues are hydrophobic and usually reside within the hydrophobic core of the ordered protein structure, whereas disorder-promoting residues are polar and reside on the surface of ordered structures (Campen et al., 2008). However, proline represents an exception to this rule: it is a hydrophobic residue; however, it is the most disorder-promoting due to its unique chemical structure (Theillet et al., 2013). Distinct compositional biases in sequence have led to inability of IDPs to fold independently (Lise and Jones, 2004; van der Lee et al., 2014). However, due to distinct subsets of sequences (sequence archetypes), some structural characteristics can be identified within IDPs that are capable of collapsing and forming compact globules (Crick et al., 2006) or participating in liquid–liquid phase separation that help define proteinaceous membrane-less organelles such as stress granules (Uversky, 2021). The most important sequence archetypes for IDPs are polar tracts, polyampholytes and polyelectrolytes (Mao et al., 2013). Polar tracts are enriched in polar amino acids and deficient in charged and hydrophobic residues. They are capable to collapse and form globules that lack significant secondary structure preferences (Crick et al., 2006) and can be as compact as well-folded domains (Mao et al., 2013). Moreover, polar tracts have distinct effects on amyloid formation and are important for phase separation (Halfmann et al., 2011). Amino acid composition of polyelectrolytes is biased toward charge residues of one type. This sequence archetype can reverse the preference for collapsed structure of IDPs (Mao et al., 2013). And finally, polyampholytes are also enriched in charged residues but contain approximately equal portions of positively and negatively charged amino acids (Das and Pappu, 2013). Here, we studied IDRs in the human disordered proteome that are significantly enriched in different amino acids and calculated their over- and underrepresentation in biological processes (BPs). We developed the DisEnrich database, where this information is available for the whole human proteome along with scored comparison of enriched IDRs between proteins.
2 Materials and methods
2.1 Prediction of disordered consensus for human proteome
We used protein sequences from the reference human proteome from UniProt KB (UniProt Consortium, 2019), proteome ID: UP000005640. Eighty-five collagen proteins were excluded from the dataset. Three methods were used to predict disorder regions: DISOPRED (cutoff = 0.5; Ward et al., 2004), IUPred2A (cutoff = 0.5; Mészáros et al., 2018) and SPOT-disorder (cutoff = 0.46; Hanson et al., 2017). Regions that can be characterized as: (i) signal peptide; (ii) transit peptide; (iii) transmembrane segment; (iv) intramembrane segment were excluded from the analysis. This information was retrieved for each protein from UniProt KB. Minimal disorder region length was set to 10 residues. We also tested a longer minimal IDR cutoff of 25 residues (as suggested by Mei et al. (2014)). Our analysis showed that increase of minimal IDR length does not significantly affect the functional distribution of human IDPs—top six overrepresented BPs remain the same (Supplementary Fig. S1). Predicted disordered consensus (DisEnrich consensus) was generated in the following way: if any of two methods, mentioned above, predicted a particular residue as disordered, it was considered as disordered. Additionally, if there are up to three ordered residues between two disorder regions and if any method predicted these residues as disordered, we consider them as disordered. Additionally, disordered consensus from MobiDB version 3.1.0 was used (Piovesan et al., 2018).
2.2 Identification of enriched IDRs
To identify IDRs that are enriched in a particular amino acid combination (amino acid category), two algorithms were used: ‘windows’ algorithm and fLPS (Harrison, 2017). ‘Windows’ algorithm was implemented as a script, which slides the window of a particular size along the protein sequence and finds IDR enriched in a particular amino acid category. IDR is considered enriched if the frequency of amino acid category inside this region is higher or equals to the frequency cutoff. For this algorithm, we used three window sizes: 10, 15 and 20 residues. Frequency cutoff was defined by cumulative binomial frequency for each amino acid category. Binominal frequency was calculated using the overall frequency of amino acid category in the whole disordered proteome. Overall frequency of any particular amino acid was calculated as the ratio of occurrences of this amino acid in all IDRs in the proteome over the overall length of all IDRs in the proteome. As amino acid categories, we considered single amino acids, as well as all their combinations as pairs and triplets. For each amino acid category, we used a cumulative binomial frequency cutoff which is less or equals 0.01. Additionally, if a category consists of more than one amino acid, we set cumulative binomial frequency cutoff to 0.05 for each single amino acid in this category.
fLPS algorithm requires minimal and maximal window size values, which cannot be equal. Similarly, we used three window sizes for fLPS: 10–11, 15–16 and 20–21 residues. As background frequencies, we used overall amino acids frequencies in disordered proteome for DisEnrich and MobiDB consensuses. Finally, enriched IDRs obtained by three window sizes for a particular protein and amino acid category were combined to generate total enriched IDRs. All intersected regions were merged together. Most of the enriched IDRs are around 20 residues (Supplementary Fig. S3). However, they originate from significantly longer IDRs and only a very small number of enriched IDRs (<1%) originate from IDRs shorter than 30 residues (Supplementary Fig. S4).
2.3 Analysis of IDP-involved BPs
Gene Ontology (GO; Ashburner et al., 2000) BP information was retrieved for each protein from UniProt KB (UniProt Consortium, 2019). BP GO terms were mapped to GO terms from a generic slim subset. Overall there are 69 top level BPs in GO generic slim subset. One protein can take part in several BPs. We limit our definition of IDPs to proteins with disordered content no <70% of the protein’s length. We tested three different cutoffs for disordered content: 60%, 70% and 80%. Our analysis did not reveal significant differences in functional distribution of human IDPs—top five overrepresented BPs remain the same (SupplementaryFigs S1 and S2). For 70% or more disordered proteins, we checked over and underrepresentation for each BP using the following algorithm. Over and underrepresentation of proteins with disordered content no <70% in BPs were calculated as ratio of observed and expected frequencies. The observed frequency in each BP was calculated as a ratio of the total number of the proteins with disordered content no <70% in a particular BP over the sum of all proteins with disordered content no <70% mapped to any BP. The expected frequency in each BP was calculated as ratio of total number of proteins with IDRs (with any length of disordered content) found for each particular BP to the total amount of proteins with IDRs mapped to any BP. Significance of overrepresentation was checked using chi-square test (P-value ≤ 0.0001 is considered significant). Statistical analysis was conducted using the R package, v3.6.0 (R Core Team, 2013). Amino acid categories, which showed significant overrepresentation in a particular BP in both disordered consensuses and obtained using both algorithms, are shown in Supplementary Table S1. All BPs from GO generic slim subset were grouped into five broad categories: metabolic and enzymatic, signaling, structural, transport and regulation. Over and underrepresentation of proteins with enriched IDRs in broad functional categories were calculated as ratio of observed and expected frequencies. The observed frequency in each broad functional category was calculated as a ratio of the total number of BPs with IDRs enriched in particular amino acid in this broad functional category over the sum of BPs with IDRs enriched in particular amino acid in all broad functional categories. The expected frequency in each broad functional category was calculated as ratio of total number of BPs with IDRs enriched in all amino acids to the total amount of BPs with IDRs mapped to any functional category. Sankey diagram was built using networkD3 library for R package v3.6.0. Significance of overrepresentation was calculated using the chi-square test (P-value ≤ 0.0001 is considered significant).
2.4 Cosine similarity calculation
For each amino acid category ‘share of enriched regions in disordered part of the protein’ (S) was calculated as a ratio of length of enriched IDR to the overall length of IDRs in this protein. Each protein was represented as a vector of S values for all observed amino acid categories in all datasets. We studied four datasets, obtained by combination of two disordered consensus (DisEnrich and MobiDB) and two algorithms of defining enriched IDRs (‘windows’ and fLPS). Protein vectors were compared pairwise, all against all. Cosine similarity between vectors of proteins A and B was calculated using following equation:
| . | (1) |
2.5 Identification of enriched IDRs sequence archetypes and high-frequency repeats
Three sequence archetypes were considered for enriched IDRs: polar tracts, polyelectrolytes and polyampholytes (van der Lee et al., 2014). IDRs enriched in polar amino acids and deficient (frequency inside the IDR 0.1) in charged and hydrophobic ones were considered as polar tracts. IDRs enriched in charged residues of one type (positive or negative) were considered as polyelectrolytic. IDRs were considered as polyampholytic if it is enriched in charged residues, but the number of positive and negative charged is nearly equal (the ratio of positive/negative is no lesser than 0.8 and no higher than 1.2). Additionally, if the frequency of residues, in which a particular IDR is enriched, is higher than or equal to 0.9, this IDR is considered as a high-frequency repeat. Over and underrepresentation were calculated based on the length of IDRs. Significance of overrepresentation was checked using chi-square test (P-value ≤ 0.0001).
3 Results and discussion
3.1 Human proteome disordered consensus
We predicted IDRs for 20 150 proteins from the reference human proteome (UP000005640) retrieved from UniProt KB (UniProt Consortium, 2019) using an approach described in Section 2. To compare results obtained using our DisEnrich disordered consensus, we additionally retrieved disordered consensus for human proteome from MobiDB version 3.1.0 (Piovesan et al., 2018). In this study, we considered 10 residues as minimal IDR length. IDRs smaller than 10 residues were not considered. We tested a longer minimal IDR cutoff (25 residues as suggested by Mei et al. (2014)) and showed that increase of minimal IDR length did not change our main conclusions (see Section 2). Using this cutoff, we obtained 15 935 proteins with IDRs for DisEnrich consensus dataset (79%) and 11 719 proteins with IDRs for MobiDB consensus dataset (58%). Comparison of IDR length distribution between the two datasets revealed differences for short, disordered regions (Fig. 1A). MobiDB-lite, one of the main methods used for disorder prediction in MobiDB, utilizes 20 residues as the minimal IDR (Necci et al., 2017). However, overall MobiDB consensus might contain IDRs even shorter than 10 residues. Comparison of protein contents of both datasets revealed 11 476 common proteins that contain disordered regions (Fig. 1B). It constitutes 98% of the MobiDB dataset and 72% of our predicted dataset (DisEnrich).
Fig. 1.
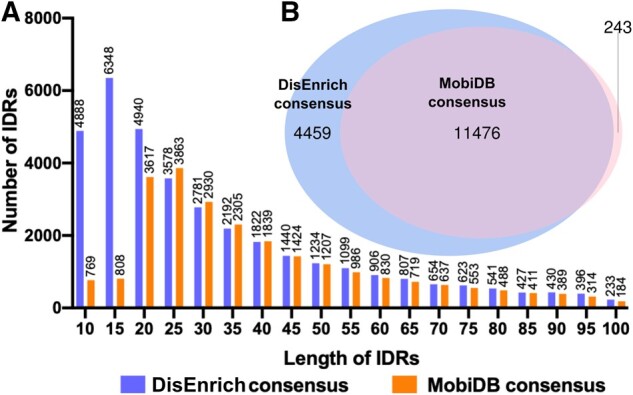
Comparison of DisEnrich and MobiDB disordered consensus datasets: (A) length distribution of IDRs in DisEnrich and MobiDB consensus datasets; (B) comparison of disordered proteins number in DisEnrich and MobiDB consensus datasets
3.2 DisEnrich—a database of enriched disordered regions in human proteins
We developed the DisEnrich database (http://prodata.swmed.edu/DisEnrichDB/) with web interfaces that display disordered consensus and enriched IDRs for any individual human protein, as well as a list of GO BP terms assigned to the protein. A web interface example is shown for the Mucin-4 (gene name: MUC4, UniProt accession: Q99102) in Figure 2. The top of the webpage lists the UniProt accession, UniProt entry name, gene name and protein name followed by the list of GO BPs (Fig. 2A). The second section of the webpage shows a table with the top 10 most similar proteins, based on cosine similarity of enriched IDRs (see Section 2) and up to three of their GO BPs (Fig. 2B). The Mucin-4 protein exhibits a high similarity of enriched IDRs with other mucins [cosine similarity is higher than 0.9 (Fig. 2B)]. The final section of the webpage illustrates the protein sequence (in sequence blocks of 100 amino acids), the STMI line [it denotes the location of signal peptide (S), transit peptide (T), transmembrane segment (M) and intramembrane regions (I) if they are present], disorder predictions by DISOPRED (Ward et al., 2004), IUPred2A (Mészáros et al., 2018) and SPOT-disorder (Hanson et al., 2017), DisEnrich and MobiDB (Piovesan et al., 2018) consensuses, and enriched IDRs (D stands for disordered, dot stands for ordered residue; Fig. 2C). The first 55 amino acids of Mucin-4 are shown in Figure 2C. There are four types of enriched IDRs: RICH–IDRs defined by the ‘windows’ algorithm using DisEnrich consensus (see Section 2); RICH_fLPS–IDRs defined by fLPS (Harrison, 2017) using DisEnrich consensus; RICH_MOBI–IDRs defined by the ‘windows’ algorithm using MobiDB consensus; RICH_fLPS_MOBI–IDRs defined by fLPS using MobiDB consensus. Additionally, using the top main menu one can access two lists of proteins in the human proteome: by UniProt accession and gene name. Full information about enriched IDRs in human proteins and full cosine similarity lists are also available for download in a plain text format.
Fig. 2.
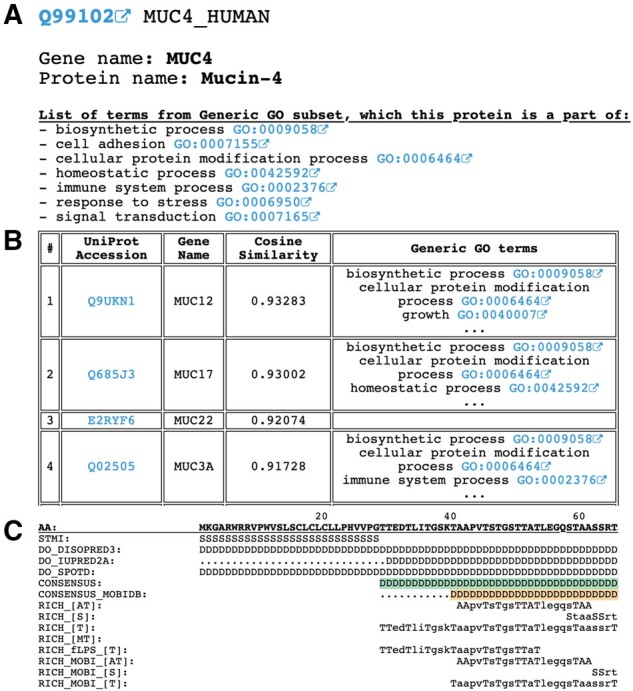
Example from the DisEnrich database: (A) basic information about protein with GO BPs; (B) cosine similarity of enriched IDRs with another proteins; (C) protein sequence with disorder predictions, consensuses and enriched IDRs
3.3 Functional distribution of human proteins with long IDRs
The IDR content of proteins can vary significantly, from containing a single short disordered region to the entire protein sequence being disordered (van der Lee et al., 2014). For example, almost 80% of the human proteome (15 935 out of 20 150 proteins defined by DisEnrich consensus) contain IDRs longer than 10 residues. We limit our definition of IDPs to proteins with disordered content no <70% of the protein’s length. Using this definition only 9% of human proteins (1955) are IDPs. Figure 3 shows the ratio of observed and expected frequencies of BPs for these IDPs based on DisEnrich consensus. All BPs were grouped into five broad categories: metabolic and enzymatic (e.g. mRNA processing), signaling (e.g. signal transduction), structural (e.g. chromosome organization), transport (e.g. nucleocytoplasmic transport) and regulation (e.g. homeostatic process). In general, our data confirm the observation that disorder is closely related to signaling and regulation, rather than metabolic and enzymatic activities (van der Lee et al., 2014). The top three BPs, in which IDPs are significantly overrepresented, are embryo development, mRNA processing and nucleocytoplasmic transport (Fig. 3). Moreover, our data showed that long disorder correlates with differentiation, cell cycle, mRNA processing and anticorrelates with transport in general, consistent with previous findings (Tompa, 2012; Xie et al., 2007).
Fig. 3.
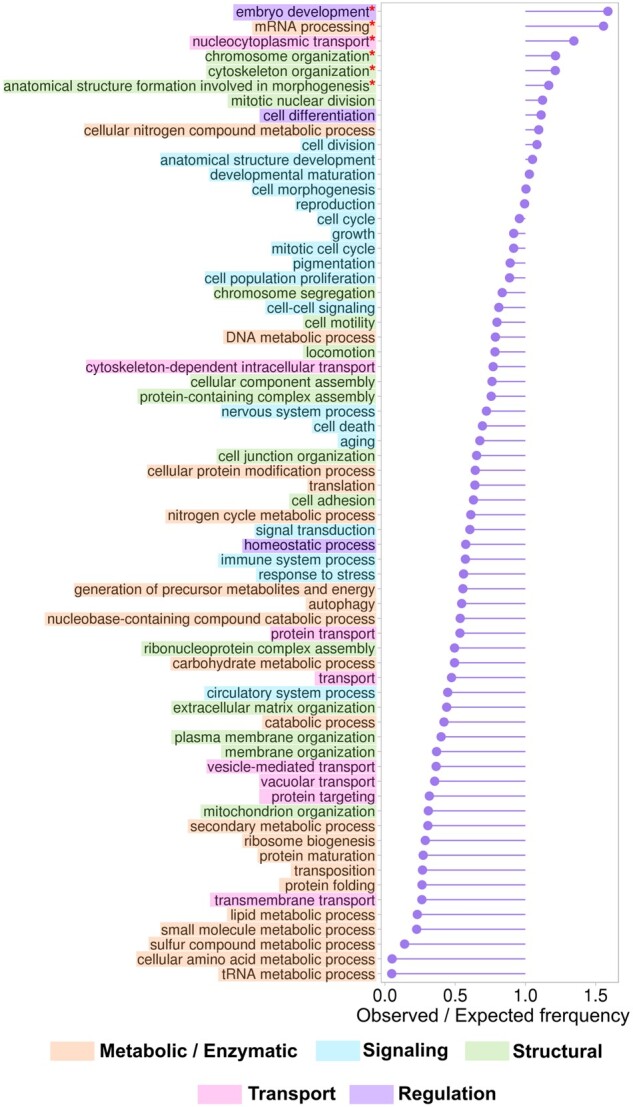
The ratio of observed and expected frequencies of BPs from GO generic subset defines over (ratio > 1) and under (ratio < 1) represented process categories for proteins with long disorder (with disordered content no <70%). Asterisks denote significant values according to chi-square test (P < 0.0001)
Embryogenesis is a crucial process for every multicellular organism involving several pathways where IDPs play important roles, including Wnt (Xue et al., 2012b) and Notch (Popovic et al., 2006), NF-κβ (Dyson and Komives, 2012). Moreover, these pathways are associated with embryonic stem cell development and cancer (Dreesen and Brivanlou, 2007; Dunker et al., 2015). Disordered regions were shown to play an important role in DNA demethylation during preimplantation embryonic development (Han et al., 2019). Indeed, 75% of proteins from our IDP dataset that function in embryo development are DNA-binding transcription factors, which also play important roles in embryogenesis (Rizzino and Wuebben, 2016). Abundance of disordered proteins in embryogenesis and development might be one of the reasons why IDPs are linked to the great variety of diseases.
As mentioned above disordered proteins play crucial role during mRNA processing (Tompa, 2012; Xie et al., 2007). Most of the proteins involved in mRNA processing in our dataset are linked to pre-mRNA splicing and alternative splicing, processes that are catalyzed by the spliceosome. The protein components of the spliceosome were studied in details and were shown to be highly enriched in intrinsic disorder (Korneta and Bujnicki, 2012; Wright and Dyson, 2015). Moreover, disordered proteins are capable to form membrane-less organelles by phase transition, which also contain RNA molecules and have been described as RNA granules (Frege and Uversky, 2015). Different types of these granules play important role in various processes including RNA metabolism (Weber and Brangwynne, 2012). IDPs involved in nucleocytoplasmic transport are disordered phenylalanine–glycine-rich nucleoporins. These proteins contain disordered FG-repeat regions, which play the role as the gate of the nuclear pore and for binding different proteins (Chug et al., 2015; Lemke, 2016).
3.4 Distribution of enriched IDRs in broad functional categories
We defined IDRs enriched in particular amino acid categories for DisEnrich and MobiDB disordered consensuses within proteins with long disorder (with disordered content no <70%, see Section 2). Proteins with these IDRs were mapped to GO generic slim BPs, which were grouped into broad categories (see Section 3.3). Figure 4 shows broad functional categories that contain proteins with IDRs significantly enriched (P-value < 0.0001) in particular amino acids. The thickness of the lines shows the number of BPs from GO generic slim subset. For this analysis, we selected amino acid categories, which IDRs are significantly overrepresented in a particular BP for both disordered consensuses: DisEnrich and MobiDB. Our analysis showed that GO generic slim BPs include proteins with IDRs significantly enriched in all amino acids except cysteine, leucine and tryptophan, which are considered order-promoting residues (Campen et al., 2008; Williams et al., 2001). However, at the same time, there are BPs that include proteins with IDRs significantly enriched in other order-promoting amino acids (e.g. aromatic residues tyrosine and phenylalanine), which are relatively rare in disordered proteins (Fig. 4). Overall, our results reveal that IDRs enriched in Tyr and Arg are significantly overrepresented in proteins linked to metabolic and enzymatic processes and Phe-enriched IDRs are significantly overrepresented in proteins linked to different types of transport (red lines in Fig. 4).
Fig. 4.
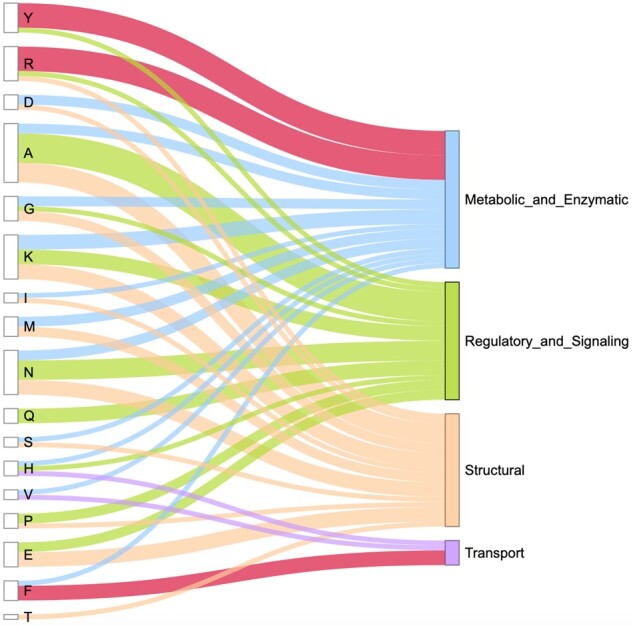
Broad functional categories and amino acid categories enriched in IDRs, which are significantly overrepresented in BPs included in broad categories. Left column shows amino acids, right column broad functional categories. Each functional category and lines pointed toward it are denoted by separate color. The thickness of the lines shows the number of BPs from GO generic slim subset. Red lines denote significant overrepresentation (P < 0.0001) of particular enriched IDRs among broad functional categories
Proteins with Tyr-enriched IDRs are mostly involved in metabolic and enzymatic processes, such as mRNA processing and mRNA stabilization. For example, TATA-binding protein-associated factor 2N (gene name: TAF15, UniProt accession: Q92804) is a transcription factor that plays an important role during transcription initiation (Jobert et al., 2009). C-terminal part of this protein contains a tri-RGG motif (IDRs enriched in Arg and Gly) that is required for RNA binding (Thandapani et al., 2013). RGG motif in TAF15 (and some other proteins with similar enriched IDRs, e.g. RNA-binding protein FUS; cosine similarity between FUS and TAF15 is 0.923) is intertwined with Tyr-enriched IDRs, which also play a significant role in RNA recognition (Kondo et al., 2018). Phe-enriched IDRs are mostly linked to transport processes and were discussed previously.
There are two amino acid categories that are exclusively significantly enriched in IDRs of only one broad functional category: Thr-enriched IDRs are observed only for structural processes and Gln-enriched IDRs are observed only for regulatory and signaling processes. The only BP which includes proteins with Thr-enriched IDRs and belongs to structural functional category is cell adhesion. Mucins are known to contain long IDRs enriched in serine and threonine that anchor the O-glycans (Ambort et al., 2012). Involvement of mucins in modulation of cell adhesion has been proposed (Wesseling et al., 1995). Moreover, mucin-4 (Fig. 2) was shown to represses cell aggregation in cancer cells (Singh et al., 2004). Gln-enriched IDRs are significantly overrepresented in proteins involved in cell differentiation, cell death and anatomical structure development (Supplementary Table S1). One example is myocyte-specific enhancer factor 2D, which is a transcription factor involved in all processes mentioned above and contains signature Gln- and Pro-enriched IDRs. These enriched IDRs play important role in activation of transcription (Aude-Garcia et al., 2010; Wang et al., 2016).
Interestingly, 75% of BPs from GO generic slim subset contain proteins with IDRs significantly enriched in hydrophobic residues that are order-promoting (Campen et al., 2008) (Supplementary Table S1). In spite of being order-promoting, IDRs enriched in hydrophobic residues are crucial for protein–protein interaction. It was revealed that IDRs involved in protein binding tend to be enriched in hydrophobic residues (Mészáros et al., 2007; Wong et al., 2013).
3.5 Functional distribution of major sequence archetypes of IDRs
We studied the distribution of IDPs’ three broad sequence archetypes (van der Lee et al., 2014) and high-frequency repeats in GO BPs. Figure 5 shows over and underrepresentation of polar tracts, polyelectrolytic IDRs and polyampholytes IDRs in BPs. Distribution of high-frequency repeats is shown in Supplementary Figure S5. Overall, sequence archetypes are more overrepresented in metabolic and enzymatic BPs than IDRs in general (Fig. 3). All three sequence archetypes and high-frequency repeats are significantly overrepresented mostly in groups linked to metabolic and enzymatic and structural BPs (exception—nucleocytoplasmic transport). Involvement in these BPs is not common for disordered proteins (van der Lee et al., 2014).
Fig. 5.

The ratio of observed and expected frequencies of BPs from GO generic subset defines overrepresented (ratio > 1) and underrepresented (ratio < 1) process categories for (A) polar tracts, (B) polyelectrolytic IDRs, (C) polyampholytic IDRs. Asterisks denote significant values according to chi-square test (P < 0.0001)
Nucleocytoplasmic transport stands out of the rest BPs with significant overrepresentation of sequence archetypes, being the only top group linked to transport activity. Polar tracts and polyelectrolytic regions are significantly overrepresented in nucleocytoplasmic transport in human IDPs in comparison to the whole proteome (Fig. 5A and B). Nucleocytoplasmic transport group contains 55 proteins, most of them bind DNA or RNA, and polar tracts take part in these processes as well. For example, RNA-binding protein with serine-rich domain 1 (RNPS1, UniProt accession: Q15287), which is the component of the splicing-dependent exon-junction complex (EJC) involved in pre-mRNA splicing and mRNA export from nucleus, contains serine-enriched polar tract at the N-terminal region (McCracken et al., 2003). Serine-enriched polar tract of RNPS1 is crucial for RNA recognition and binding (Sakashita et al., 2004). In fact, most of the proteins from nucleocytoplasmic transport group, which bind DNA or RNA, are also linked to metabolic and enzymatic activities (e.g. RNA splicing).
4 Conclusion
We developed DisEnrich database that contain all IDRs in human proteome significantly enriched in particular amino acids. Analysis of IDP distribution in broad functional categories based on DisEnrich disordered consensus revealed that disorder is closely related to regulation and signaling, rather than metabolic and enzymatic activities. Among GO BPs IDPs are significantly overrepresented in embryogenesis and take part in pathways that have been implicated in embryogenesis, embryonic stem cell development and cancer. In general, our results reveal that IDRs enriched in Tyr and Arg are significantly overrepresented in proteins linked to metabolic and enzymatic processes and Phe-enriched IDRs are significantly overrepresented in proteins linked to different types of transport. Moreover, IDPs involved in 75% of BPs contain IDRs significantly enriched in hydrophobic residues, that are known to be important for protein–protein interactions. Analysis of distinct sequence biases of IDRs revealed that polar tracts, polyelectrolytic and polyampholytic disordered regions are significantly overrepresented in metabolic and enzymatic and structural BPs.
Funding
This work was supported by the National Institutes of Health [GM127390 to N.V.G.] and the Welch Foundation [I-1505 to N.V.G.].
Conflict of Interest: none declared.
Supplementary Material
Contributor Information
Kirill E Medvedev, Department of Biophysics, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Jimin Pei, McDermott Center for Human Growth and Development, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Nick V Grishin, Department of Biophysics, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA; Department of Biochemistry, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA; Howard Hughes Medical Institute, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
References
- Ambort D. et al. (2012) Perspectives on mucus properties and formation—lessons from the biochemical world. Cold Spring Harb. Perspect. Med., 2, a014159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M. et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet., 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aude-Garcia C. et al. (2010) Dual roles for MEF2A and MEF2D during human macrophage terminal differentiation and c-Jun expression. Biochem. J., 430, 237–244. [DOI] [PubMed] [Google Scholar]
- Campen A. et al. (2008) TOP-IDP-scale: a new amino acid scale measuring propensity for intrinsic disorder. Protein Pept. Lett., 15, 956–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chug H. et al. (2015) Crystal structure of the metazoan Nup62•Nup58•Nup54 nucleoporin complex. Science, 350, 106–110. [DOI] [PubMed] [Google Scholar]
- Crick S.L. et al. (2006) Fluorescence correlation spectroscopy shows that monomeric polyglutamine molecules form collapsed structures in aqueous solutions. Proc. Natl. Acad. Sci. USA, 103, 16764–16769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R.K., Pappu R.V. (2013) Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. USA, 110, 13392–13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreesen O., Brivanlou A.H. (2007) Signaling pathways in cancer and embryonic stem cells. Stem Cell Rev., 3, 7–17. [DOI] [PubMed] [Google Scholar]
- Dunker A.K. et al. (2008) Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol., 18, 756–764. [DOI] [PubMed] [Google Scholar]
- Dunker A.K. et al. (2015) Intrinsically disordered proteins and multicellular organisms. Semin. Cell Dev. Biol., 37, 44–55. [DOI] [PubMed] [Google Scholar]
- Dyson H.J., Komives E.A. (2012) Role of disorder in IκB-NFκB interaction. IUBMB Life, 64, 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson H.J., Wright P.E. (2005) Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol., 6, 197–208. [DOI] [PubMed] [Google Scholar]
- Frege T., Uversky V.N. (2015) Intrinsically disordered proteins in the nucleus of human cells. Biochem. Biophys. Rep., 1, 33–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfmann R. et al. (2011) Opposing effects of glutamine and asparagine govern prion formation by intrinsically disordered proteins. Mol. Cell, 43, 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C. et al. (2019) Functions of intrinsic disorder in proteins involved in DNA demethylation during pre-implantation embryonic development. Int. J. Biol. Macromol., 136, 962–979. [DOI] [PubMed] [Google Scholar]
- Hanson J. et al. (2017) Improving protein disorder prediction by deep bidirectional long short-term memory recurrent neural networks. Bioinformatics, 33, 685–692. [DOI] [PubMed] [Google Scholar]
- Harrison P.M. (2017) fLPS: fast discovery of compositional biases for the protein universe. BMC Bioinform., 18, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iakoucheva L.M. et al. (2002) Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol., 323, 573–584. [DOI] [PubMed] [Google Scholar]
- Jobert L. et al. (2009) PRMT1 mediated methylation of TAF15 is required for its positive gene regulatory function. Exp. Cell Res., 315, 1273–1286. [DOI] [PubMed] [Google Scholar]
- Kondo K. et al. (2018) Plastic roles of phenylalanine and tyrosine residues of TLS/FUS in complex formation with the G-quadruplexes of telomeric DNA and TERRA. Sci. Rep., 8, 2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korneta I., Bujnicki J.M. (2012) Intrinsic disorder in the human spliceosomal proteome. PLoS Comput. Biol., 8, e1002641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshland D.E. (1958) Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. USA, 44, 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke E.A. (2016) The multiple faces of disordered nucleoporins. J. Mol. Biol., 428, 2011–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lise S., Jones D.T. (2004) Sequence patterns associated with disordered regions in proteins. Proteins, 58, 144–150. [DOI] [PubMed] [Google Scholar]
- Mao A.H. et al. (2013) Describing sequence-ensemble relationships for intrinsically disordered proteins. Biochem. J., 449, 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken S. et al. (2003) An evolutionarily conserved role for SRm160 in 3ʹ-end processing that functions independently of exon junction complex formation. J. Biol. Chem., 278, 44153–44160. [DOI] [PubMed] [Google Scholar]
- Mei Y. et al. (2014) Intrinsically disordered regions in autophagy proteins. Proteins, 82, 565–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mészáros B. et al. (2007) Molecular principles of the interactions of disordered proteins. J. Mol. Biol., 372, 549–561. [DOI] [PubMed] [Google Scholar]
- Mészáros B. et al. (2018) IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res., 46, W329–W337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Necci M. et al. (2017) MobiDB-lite: fast and highly specific consensus prediction of intrinsic disorder in proteins. Bioinformatics, 33, 1402–1404. [DOI] [PubMed] [Google Scholar]
- Oldfield C.J., Dunker A.K. (2014) Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem., 83, 553–584. [DOI] [PubMed] [Google Scholar]
- Peng Z. et al. (2015) Exceptionally abundant exceptions: comprehensive characterization of intrinsic disorder in all domains of life. Cell. Mol. Life Sci., 72, 137–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piovesan D. et al. (2018) MobiDB 3.0: more annotations for intrinsic disorder, conformational diversity and interactions in proteins. Nucleic Acids Res., 46, D471–D476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic M. et al. (2006) Gene synthesis, expression, purification, and characterization of human Jagged-1 intracellular region. Protein Expr. Purif., 47, 398–404. [DOI] [PubMed] [Google Scholar]
- R Core Team (2013) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/. [Google Scholar]
- Rizzino A., Wuebben E.L. (2016) Sox2/Oct4: a delicately balanced partnership in pluripotent stem cells and embryogenesis. Biochim. Biophys. Acta, 1859, 780–791. [DOI] [PubMed] [Google Scholar]
- Sakashita E. et al. (2004) Human RNPS1 and its associated factors: a versatile alternative pre-mRNA splicing regulator in vivo. Mol. Cell. Biol., 24, 1174–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A.P. et al. (2004) Inhibition of MUC4 expression suppresses pancreatic tumor cell growth and metastasis. Cancer Res., 64, 622–630. [DOI] [PubMed] [Google Scholar]
- Thandapani P. et al. (2013) Defining the RGG/RG motif. Mol. Cell, 50, 613–623. [DOI] [PubMed] [Google Scholar]
- Theillet F.X. et al. (2013) The alphabet of intrinsic disorder: I. Act like a Pro: on the abundance and roles of proline residues in intrinsically disordered proteins. Intrinsically Disord. Proteins, 1, e24360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P. (2012) Intrinsically disordered proteins: a 10-year recap. Trends Biochem. Sci., 37, 509–516. [DOI] [PubMed] [Google Scholar]
- UniProt Consortium (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res., 47, D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V.N. (2021) Recent developments in the field of intrinsically disordered proteins: intrinsic disorder-based emergence in cellular biology in light of the physiological and pathological liquid-liquid phase transitions. Annu. Rev. Biophys., 50, 135–156. [DOI] [PubMed] [Google Scholar]
- Uversky V.N., Dunker A.K. (2010) Understanding protein non-folding. Biochim. Biophys. Acta, 1804, 1231–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V.N. et al. (2008) Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu. Rev. Biophys., 37, 215–246. [DOI] [PubMed] [Google Scholar]
- Vacic V., Iakoucheva L.M. (2012) Disease mutations in disordered regions—exception to the rule? Mol. Biosyst., 8, 27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lee R. et al. (2014) Classification of intrinsically disordered regions and proteins. Chem. Rev., 114, 6589–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. et al. (2016) Discovery, characterization, and functional study of a novel MEF2D CAG repeat in duck (Anas platyrhynchos). DNA Cell Biol., 35, 398–409. [DOI] [PubMed] [Google Scholar]
- Ward J.J. et al. (2004) The DISOPRED server for the prediction of protein disorder. Bioinformatics, 20, 2138–2139. [DOI] [PubMed] [Google Scholar]
- Weber S.C., Brangwynne C.P. (2012) Getting RNA and protein in phase. Cell, 149, 1188–1191. [DOI] [PubMed] [Google Scholar]
- Wesseling J. et al. (1995) Episialin (MUC1) overexpression inhibits integrin-mediated cell adhesion to extracellular matrix components. J. Cell Biol., 129, 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R.M. et al. (2001) The protein non-folding problem: amino acid determinants of intrinsic order and disorder. Pac. Symp. Biocomput., 89–100. [DOI] [PubMed] [Google Scholar]
- Wong E.T. et al. (2013) On the importance of polar interactions for complexes containing intrinsically disordered proteins. PLoS Comput. Biol., 9, e1003192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright P.E., Dyson H.J. (2015) Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol., 16, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H. et al. (2007) Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J. Proteome Res., 6, 1882–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B. et al. (2012a) Orderly order in protein intrinsic disorder distribution: disorder in 3500 proteomes from viruses and the three domains of life. J. Biomol. Struct. Dyn., 30, 137–149. [DOI] [PubMed] [Google Scholar]
- Xue B. et al. (2012b) The roles of intrinsic disorder in orchestrating the Wnt-pathway. J. Biomol. Struct. Dyn., 29, 843–861. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.