

Abstract
Background:
Both gap junctional remodeling and interstitial fibrosis have been linked to impaired electrical conduction velocity (CV) and fatal ventricular arrhythmias in nonischemic heart failure (HF). However, the arrhythmogenic role of the ventricular gap junctional Cx43 in nonischemic HF remains in debate. Here, we assessed this in a newly developed arrhythmogenic canine model of nonischemic HF.
Methods and results:
Nonischemic HF was induced in canines by combined aortic valve insufficiency and aortic constriction. Left ventricular (LV) myocardium from HF dogs showed similar pathological changes to that of humans. HF dogs had reduced LV function, widened QRS complexes, and spontaneous nonsustained ventricular tachycardia. CV was measured in intact LV epicardium with high-density grid mapping. Total (Cx43-T) and nonphosphorylated Cx43 (Cx43-NP) and histological interstitial fibrosis were assessed from these mapped LV tissues. Longitudinal CV, which was slowed in HF (49 ± 1 vs. 65 ± 2 cm/s in Ctl), was positively correlated with reduced total junctional Cx43 and negatively correlated with markedly increased junctional Cx43-NP (2-fold) in HF. Cx43 dephosphorylation in HF was associated with enhanced colocalization of PP2A at the level of Cx43. Unchanged action potential upstroke and transverse CV were associated with unaltered Cx43 lateralization and interstitial fibrosis in the nonischemic HF canine LV.
Conclusion:
Our unique arrhythmogenic canine model of HF resembles human nonischemic HF (prior to the end stage). Cx43 remodeling occurs prior to the structural remodeling (with lack of fibrosis) in HF and it is crucial in slowed CV and ventricular arrhythmia development. Our findings suggest that altered Cx43 alone is arrhythmogenic and modulation of Cx43 has the anti-arrhythmic therapeutic potential for HF patients.
Keywords: Heart failure, connexin43 remodeling, Dephosphorylation, Conduction velocity, Fibrosis
1. Introduction
Five million people in the U.S. are affected by heart failure (HF), and sudden death occurs in nearly 50% of cases, primarily from ventricular tachycardia (VT) leading to ventricular fibrillation (VF) [1]. We and others have shown that the initiation of VT in nonischemic HF is primarily due to a non-reentrant mechanism (such as triggered activity) [2–4], but degeneration of VT to VF in HF is likely due to reentry from slow anisotropic conduction from altered gap junctions (GJ) and/or interstitial fibrosis [5].
Gap junctions, composed of connexins, are the principal membrane structures that conduct electrical impulses between adjacent cardiomyocytes of both atria and ventricles [6–10]. Gap junctional remodeling in the diseased heart have been linked to slow conduction and enhanced arrhythmogenicity. Connexin43 (Cx43) is a phosphoprotein and the major connexin expressed in ventricles [11–13]. We previously demonstrated Cx43 downregulation and increased Cx43 dephosphorylation in left ventricle (LV) from HF rabbits (arrhythmogenic model of nonischemic HF from pressure & volume overload) and from patients with nonischemic HF [14]. Interstitial fibrosis can form a physical separation to block this intercellular coupling [15–17]. There remains controversy as to the functional role of Cx43 downregulation and dephosphorylation on slow conduction in the intact failing heart [18–20] because increased interstitial fibrosis may mask the contribution of gap junctional remodeling in slowed conduction. While markedly increased interstitial fibrosis and patchy fibrotic scars are commonly seen in end-stage ischemic HF, studies in patients with nonischemic HF that is not end-stage show significant interstitial fibrosis in only ~30–50% of cases as assessed by cardiovascular magnetic resonance imaging [21–23]. With the other 50–70% of HF patients (not end-stage), the underlying electrophysiologic and molecular remodeling profiles remain unknown. Therefore, investigation is clearly needed to assess the impact of Cx43 remodeling (downregulation and dephosphorylation), in the absence of interstitial fibrosis, on conduction velocity (CV) in a large animal model of nonischemic HF that resembles human HF (prior to the end-stage of HF).
Here, we took advantage of our recently developed novel canine model of arrhythmogenic nonischemic HF (by combined aortic insufficiency and aortic constriction) [24] to quantitatively assess the relationship between altered CV and Cx43 remodeling as well as interstitial fibrosis profile on the same electrophysiologically mapped intact tissue. Electrophysiological assessment of CV was performed using a unique high-density grid mapping approach along with detailed immunohistological assessment of Cx43 expression and dephosphorylation in the same tissue being mapped. We found that markedly reduced intercalated disk located Cx43 proteins and increased dephosphorylated Cx43 lead to slow conduction in those mapped intact failing canine LV tissue with unchanged interstitial fibrosis. This loss of Cx43 in the setting of unchanged interstitial fibrosis is critically linked to impaired conduction velocity and enhanced spontaneous arrhythmogenicity in this non-ischemic HF canine model. Our findings suggest that Cx43 may be an anti-arrhythmic target for HF patients.
2. Methods
2.1. Animal model
All animal studies conformed to the Guide for the Care and Use of Laboratory Animals. This study was approved by the UAB Institutional Animal Use and Care Committee. Non-ischemic HF was induced in mixed breed dogs of either sex by creation of aortic valve insufficiency (AI, a Fogarty balloon catheter was used to perforate 1–2 aortic valve leaflets under fluoroscopic guidance to make holes ~4–5 mm) followed 6 ± 1 weeks later by constriction of the abdominal aorta (AC, a 2–0 silk ligature was tightened to reduce aortic diameter so that the systolic blood pressure gradient increased by ~50–60 mmHg compared to the baseline measurement). Aortic gradients were measured as the difference in systolic pressures between the femoral and carotid arteries. Both procedures were under isoflurane anesthesia. Prior to surgical intervention and at ~1–3 month intervals, dogs underwent echocardiographic examination and 24-h Holter monitoring in the conscious state. All data were obtained from 8 HF and 9 control dogs. HF dogs (N = 8) were used for terminal studies when LV fractional shortening (FS) decreased by ~25% compared to the baseline FS.
2.2. Holter monitoring
Prior to surgical intervention and at 240 days and 720 days post AC, digital Holter monitoring (GE SEER Light, GE Healthcare) was obtained from dogs in the conscious state. The chest and back were shaved and five ECG electrodes were applied to the chest secured by a specifically-designed nylon jacket. 24-h period Holter recording were made with the dogs’ movements being unrestricted. Holter data were analyzed to quantitate VT & PVCs and their time-of-day occurrence. VT episodes (3 or more consecutive beats), VT beats and PVCs were confirmed by an experienced cardiologist.
2.3. Electrocardiogram
Conscious dogs were shaved and had glue-on electrodes placed at the proximal portion of each limb, and a 6-lead electrocardiogram (ECG) was recorded using a Bard Electrophysiology system (C.R. Bard Inc.; Lowell, MA). ECG signals were recorded at 4 kHz sampling with a low frequency cutoff at 0.1 Hz, high frequency cutoff at 100 Hz, and a notch filter at 60 Hz for measurement of PR, QRS, and QT intervals. The corrected QT interval (QTc) was calculated using a regression algorithm specifically created for use in conscious dogs (van de Water’s QT correction); QTc = QT – 87(60/HR-1) [25,26].
2.4. Tissue harvest and grid mapping
Dog epicardial (Epi) heart tissue sections (~1.5 × 1.5 × 0.1 cm) were harvested from the posterior left ventricular (LV) free wall in a region devoid of coronary arteries and stored in cold cardioplegic solution for grid mapping followed by biochemical assays. High-density grid mapping on LV Epi was performed to measure longitudinal CV (CVL) and transverse CV (CVT) in vitro with a specially-designed 14 × 16 (224-site) high-density grid-electrode array (4.2 × 5.6 mm, inter-electrode distance of 350 μm; as described [27].) At the end of the grid mapping experiment, mapped epicardial tissue sections were labeled with sutures to mark orientation and then formalin-fixed, paraffin-embedded, and sectioned (5 μm per slide) for immunohistochemical (IHC) and histology studies. Activation time (AT) of each channel in the 224 site map array was assigned using a Matlab-based custom program with manual proofreading functions [9,27]. A vector field analysis method for calculating CV was developed based on a previously described [9,28] least-square algorithm with single value decomposition [9,28,29]. There were sufficient transverse vectors on the activation maps for accurately measuring transverse and longitudinal conduction velocities.
In each mapping procedure, one piece of tissue was superfused in a 37 °C tissue bath for a total of approximately 30 min. Tissue viability of grid mapped LV tissue pieces over the course of electrical pacing and recording were validated as evidenced by the consistent results of CVL and CVT in both Ctl and HF LV tissue pieces throughout the recording period (see more details in Online Supplement and Supplemental Fig. S1). Moreover, Western blot data demonstrated an unchanged expression and dephosphorylation of Cx43 with up to 60 min super-fusion of grid-mapped tissue pieces (Supplemental Fig. S2).
2.5. Immunohistochemical and histologic analysis
Fixed grid-mapped sections were probed with antibodies to total Cx43 (Cx43-T; BD Science), dephosphorylated Cx43 (Cx43-NP; Invitrogen), and N-cadherin (Chemicon) followed by confocal imaging (Zeiss 40× objective lens, Z-stack mode). Cell nuclei were stained with DAPI. Cx43 and Cx43-NP abundance and distribution based on IHC images were quantified using a previously developed Matlab-based algorithm [9,30]. Cx43 colocalized with N-cadherin was defined as end-to-end distributed Cx43 (Cx43E-E, junctional Cx43). Other Cx43 was defined as side-to-side Cx43 (Cx43S-S) [29].
Hematoxylin and eosin (H&E) and Masson’s trichrome staining were performed and analysis of interstitial collagen deposition (10–15 images with 40× magnification for each formalin-fixed tissue section) was performed as previously described [9,30].
2.6. Western blot and immunoprecipitation (IP)
Western blotting and IP were performed as described [14,31,32]. Primary antibodies were used for Cx43-T (BD Science), Cx43-NP (Invitrogen), PP1 (Abcam), PP2A (BD), and GAPDH (Chemicon), SCN5A (Abcam). IP was done with specific antibody to total Cx43 (Cx43-T) proteins as previously described [14,31,32] and Online Supplement.
2.7. Action potential (AP) recordings and cellular data
Intracellular recording of AP’s (CL = 600 ms) from the LV posterior wall epicardial surface of Krebs-perfused Ctl and HF hearts was performed using a conventional microelectrode as we have previously described [33]. Cardiac myocytes were isolated from Ctl and HF hearts by enzymatic digestion of cannulated LV wedges [14] for microscopic measurement of resting cell length.
2.8. Statistical analysis
All data were presented as Mean ± SEM. Statistical analyses were performed using GraphPad Prism 7 (GraphPad Software, CA). Linear regression was performed to find the best fit line without constraints. Statistically significant differences between two groups were performed using the nonparametric Mann-Whitney test. All data are presented as mean ± SEM. The criterion for statistical significance was a p value <0.05.
3. Results
3.1. LV contractile dysfunction in nonischemic HF dogs
With AI induction, pulse pressure increased 169% (from 22 ± 1 to 58 ± 4 mmHg) and severe AI was evident on aortography and color flow echo (Fig. 1A). Within 6 ± 1 weeks of AI induction, the LV dilated (LVEDD increased 21% from 3.60 ± 0.07 to 4.74 ± 0.13 cm;). Aortic constriction led to a 56 ± 4 mmHg increase in aortic gradient. Over the 9 ± 2 months following aortic constriction, LVEDD and LVESD increased 57% and 84%, respectively (from 3.61 ± 0.08 to 5.65 ± 0.18 cm and 2.03 ± 0.06 to 3.73 ± 0.17 cm; Fig. 1B&C). FS had decreased by 23% (43.9 ± 1.1 to 34.2 ± 1.3%; Fig. 1D). Dogs with induced HF showed lethargy, exercise intolerance, anorexia, tachypnea, and muscle wasting. These data suggest that this canine HF model exhibits typical non-ischemic HF phenotypes that are similar in those patients with a progressive nonischemic HF but not at the end-stage of HF.
Fig. 1.
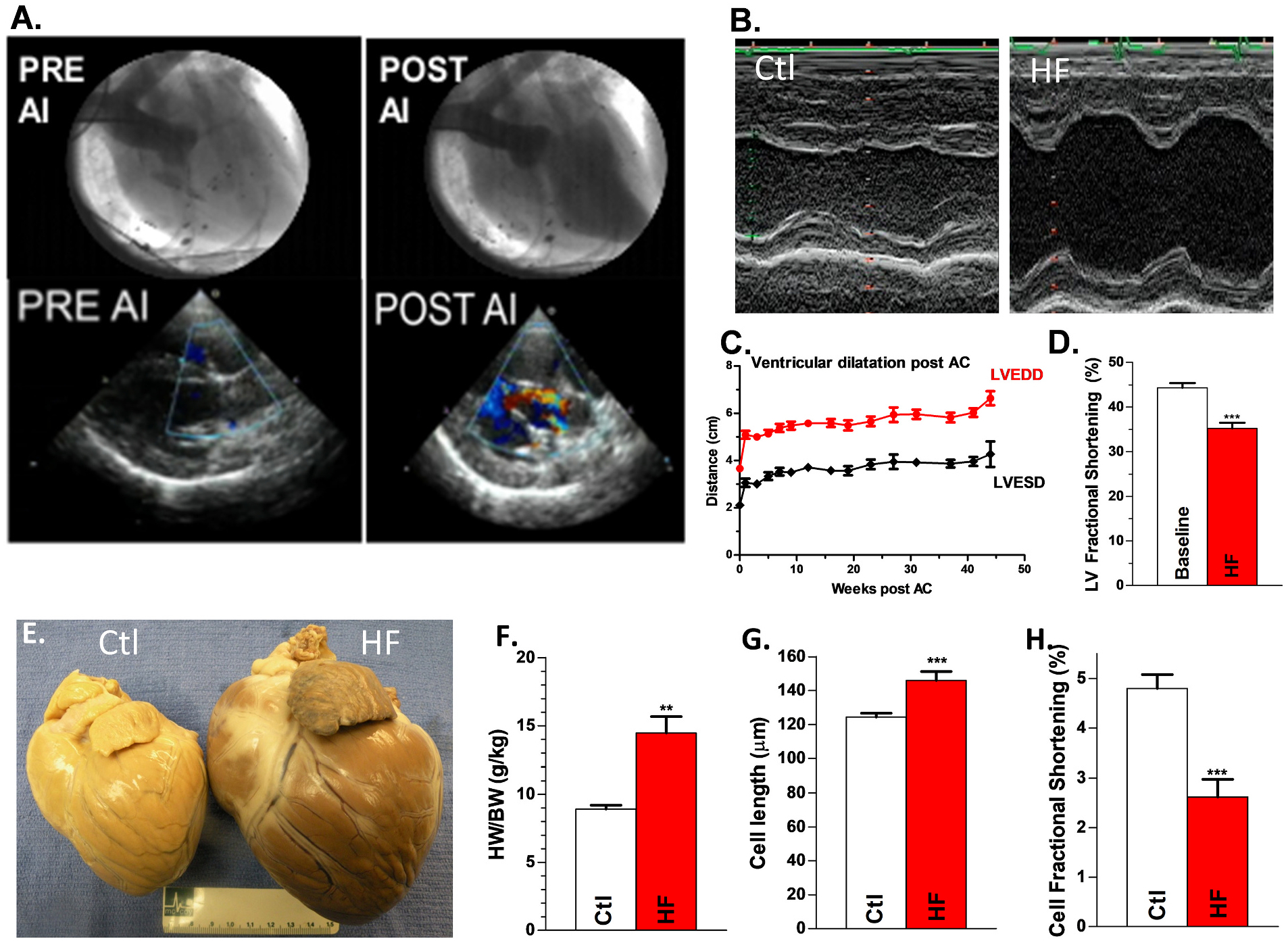
Canine HF model. A, Aortogram (top) and color-flow echocardiogram (bottom) demonstrating a competent aortic valve pre-AI and severe AI immediately post-AI induction. B, Representative M-mode echo images of LV at baseline and with induction of HF. Summary data for time course of change in LVEDD & LVESD (C) and LV fractional shortening (D) post aortic constriction (AC) Nanimal = 20. E, Representative hearts from control dog (left) and HF dog (right). Summary data for HW/BW, Nanimal = 9,8 (F), cell length, Nanimal = 9,8, ncell = 65,27 (G) and cell fractional shortening (FS) Nanimal = 9,8, ncell = 65,27 (H) for HF vs controls. **p < 0.01; ***p < 0.001. Each error bar indicates the standard error of the mean (SEM) of each experimental group. Mann-Whitney test was used for comparing difference between the two groups. Abbreviations: aortic insufficiency (AI), aortic constriction (AC), left ventricular end diastolic diameter (LVEDD), left ventricular end systolic diameter (LVESD).
Fig. 1E shows a formalin-fixed whole-heart specimen from a control and a HF dog demonstrating marked cardiomegaly with dilated ventricles. Heart weight normalized to bodyweight was increased in HF animals compared to Ctl (14.5 ± 1.2 g/kg vs. 8.9 ± 0.3 g/kg; Fig. 1F). Histologic analysis revealed that canine HF LV exhibited myocyte enlargement with myocytolysis and focal areas of inflammation with lymphocyte infiltration, characteristics of human HF [34]. Isolated HF LV myocytes showed enlarged longitudinal cell size (146 ± 5 vs. 124 ± 2 μm, N = 27, 65 cells; Fig. 1G) and reduced fractional shortening (FS, 2.6 ± 0.36 vs 4.8 ± 0.28%; N = 27, 65 cells; p < 0.001; Fig. 1H) compared to Ctl.
3.2. ECG features and spontaneous arrhythmogenicity in nonischemic HF dogs
ECG parameters including PR duration, QRS duration and corrected QT (QTc) were all significantly increased with HF compared to baseline (109 ± 4 vs 92 ± 4; 71 ± 4 vs 46 ± 3; and 230 ± 5 vs 245 ± 2 ms; N = 7,10; Fig. 2A, Supplemental Table 1). Holter monitoring was performed on 16 dogs at baseline and with development of HF. At baseline, none of the dogs had any ventricular ectopy. With HF, dogs exhibited up to 7953 PVCs/day, up to 227 runs of VT/day, and runs of VT up to 468 beats long. After a mean of 10.3 months, arrhythmia score increased from 0 ± 0 at baseline to 3.19 ± 0.34 for HF (p < 0.0001).
Fig. 2.
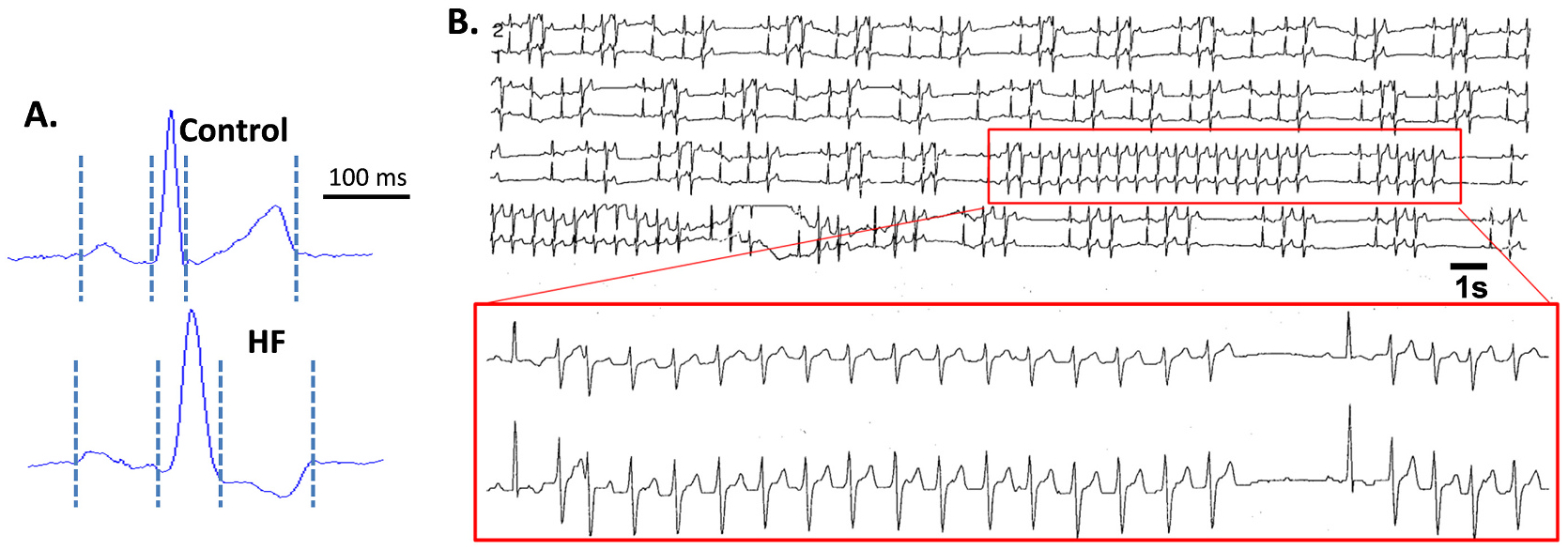
Spontaneous arrhythmias in canine HF model. A, Representative ECG from Ctl and HF dog hearts. B, Representative Holter monitor recording in conscious state for HF dog showing frequent PVCs, couplets, and runs of spontaneous nonsustained VT. Insert shows close up of a 16-beat and a 4-beat run of VT. Abbreviations: control (Ctl), heart failure (HF), premature ventricular complex (PVC), ventricular tachycardia (VT).
The 5 HF dogs whose tissue was studied biochemically and/or electrophysiologically (included in that group of 16) exhibited up to 1530 PVCs/day, up to 91 runs of VT/day, and runs of VT up to 28 beats long. Three exhibited non-sustained VT, one exhibited isolated premature ventricular complexes, and one exhibited no ventricular arrhythmias.
3.3. Slow longitudinal conduction but unchanged transverse conduction in nonischemic HF LV epicardium
Isochrones maps with visualized CV vectors show narrowed isochrones (reflecting slower conduction) in the longitudinal direction of HF LV Epi (compared to Ctl) during drive train stimuli (CVL of 48.6 ± 1.0 cm/s vs. 65.4 ± 1.5 cm/s; Figs.3A&B, left and Fig. 3C) and during premature stimulus (CL = 180 ms; 45.1 ± 1.9 cm/s vs. 64.2 ± 1.1; Fig. 3D). CVT was unaltered in HF LV vs Ctl LV during drive train stimuli (21.3 ± 1.5 cm/s vs. 24.1 ± 1.4 cm/s; Fig. 3E) and during premature stimulus (CL = 180 ms; 20.8 ± 1.8 cm/s vs. 23.1 ± 1.0 cm/s). With a decreased CVL but preserved CVT, HF dog Epi exhibited an altered anisotropic ratio (2.3 ± 0.2 vs 2.7 ± 0.1 in controls for paced beats and 2.2 ± 0.1 vs 2.8 ± 0.1 in controls for premature beats), which could contribute further to arrhythmogenesis.
Fig. 3.
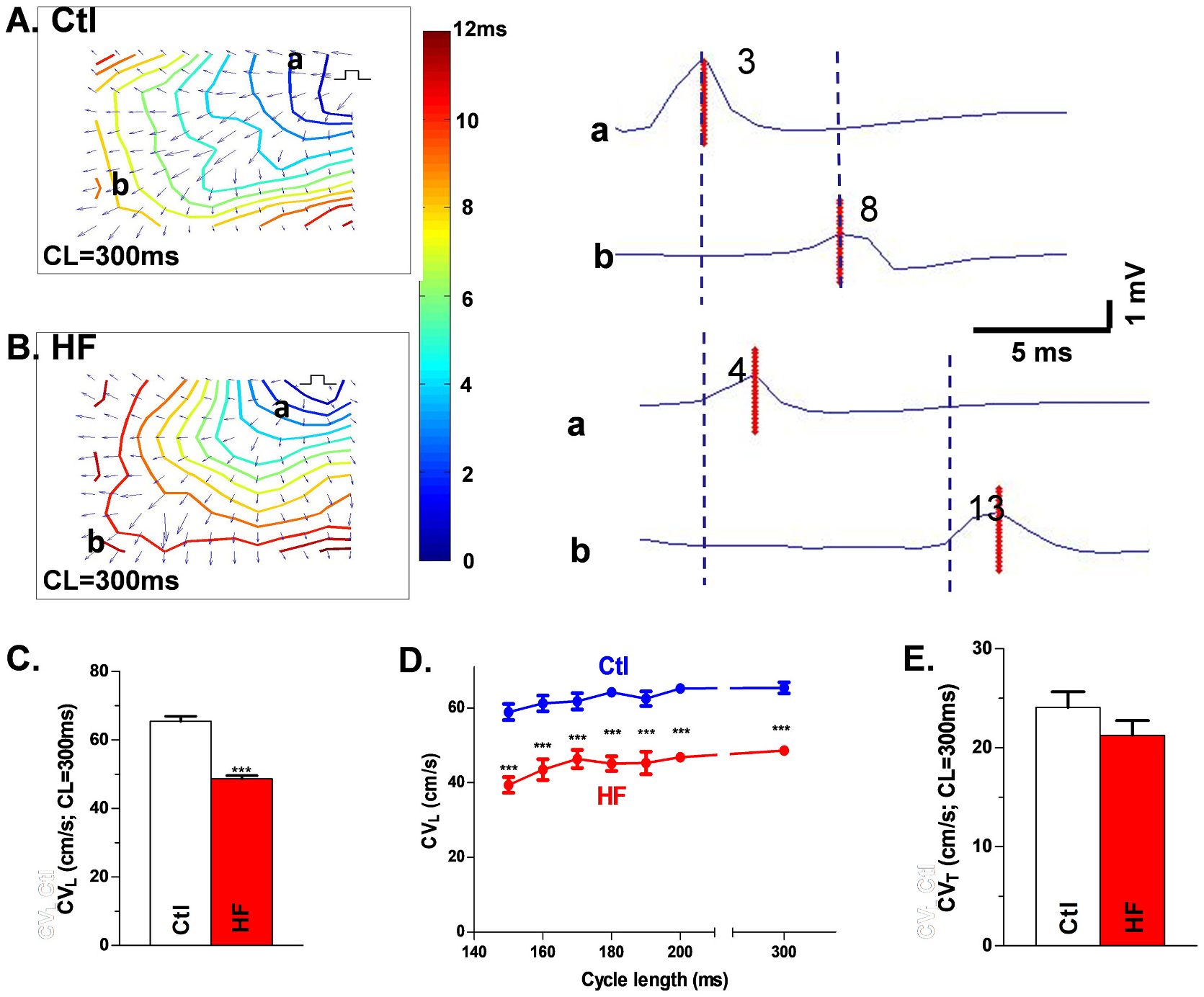
Slow conduction in HF LV. Isochronal maps with CV vector from each recording site for Ctl (A) and HF (B) tissue under drive train (A&B, left). Isochronal maps A and B show labeled recording sites a and b in the direction of CVL with electrograms from sites a and b (and the peak of the bipolar electrograms) at those respective sites (A&B, right), showing slow CVL in HF. Summary data for CVL with drive train stimuli (C) or with premature stimulation (D) and unchanged CVT (E). Nanimal = 9,8, ***p < 0.001. Each data point is the mean value from three technical repeats. Each error bar indicates the standard error of the mean (SEM) of each experimental group. Mann-Whitney test was used to compare the difference between the two groups. Abbreviations: control (Ctl), conduction velocity (CV), longitudinal conduction velocity (CVL), transverse conduction velocity (CVT), heart failure (HF).
3.4. Unaltered interstitial collagen deposition in nonischemic HF
Sample images taken from Masson’s trichrome-stained grid-mapped Epi LV are shown for Ctl and HF dogs (Fig. 4A). Collagen area percentage was calculated as the percentage of collagen area over the filled areas. No significant difference in mean collagen area percentage was demonstrated between grid mapped Ctl and HF LV (2.4 ± 0.2 vs.1.7 ± 0.3; Fig. 4B).
Fig. 4.
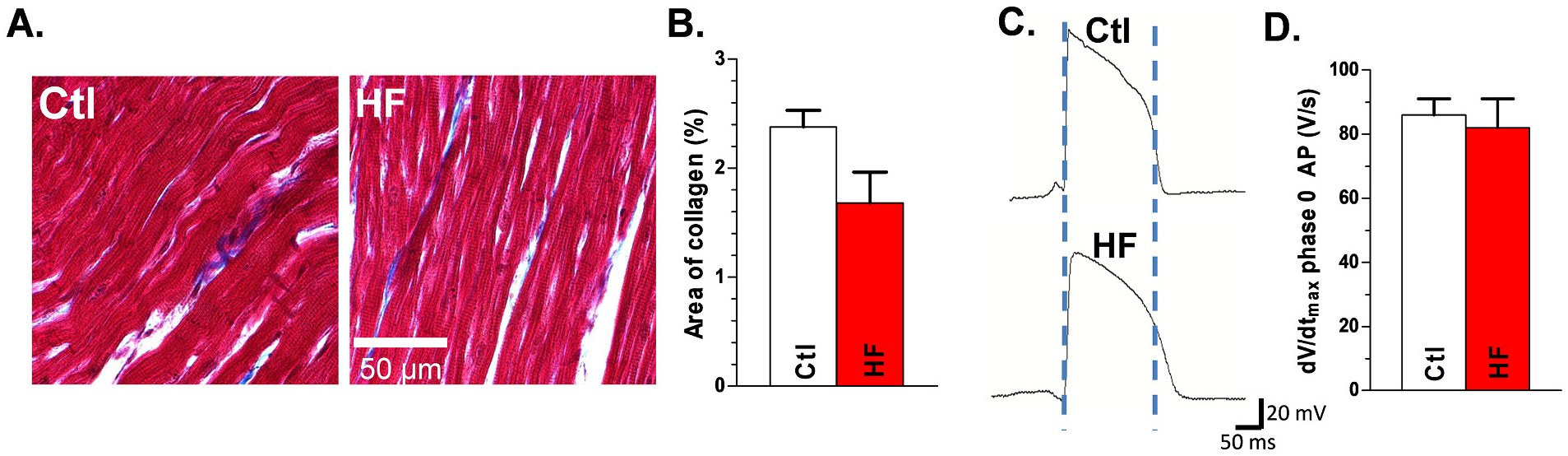
Unchanged interstitial collagen deposition and typical AP characteristics in HF dog hearts: A, Trichrome staining in mapped Ctl (left) and HF (right) dog LV Epi tissue. B, Summarized quantification of interstitial collagen in HF vs Ctl. Nanimal = 9,8, p = NS. Each quantitative interstitial collagen data point is the mean value from forty histology images of each tissue section. Representative AP’s from Ctl and HF LV epicardium (C) and summarized data for dV/dtVmax of phase 0 of AP (D). Nanimal = 9,8, p = NS. Each data point is the mean value from three technical repeats. Each error bar indicates the standard error of the mean (SEM) of each experimental group. Mann-Whitney test was used to compare the difference between the two groups. Abbreviations: action potential (AP), control (Ctl), heart failure (HF), left ventricle (LV), maximal upstroke velocity (Vmax).
3.5. No difference in dV/dtmax of AP, RMP in nonischemic HF LV
Action potentials from HF and Ctl Epi were assessed, and there were no differences in the dV/dtmax of phase 0 of the AP (82 ± 9 vs 86 ± 5 V/s for Ctl; p = NS; N = 5,5; Fig. 4C, D) or in the resting membrane potential (−76.7 ± 1.5 vs −75.4 ± 1.8 mV for Ctl; Fig. 4C). However, AP duration (APD90) was significantly increased with HF (222 ± 5 vs 182 ± 2 ms for Ctl) (Fig. 4C).
3.6. Reduced total Cx43 abundance in grid mapped nonischemic HF LV
Mapped HF LV Epi tissue sections showed reduced overall Cx43 fluorescence intensity compared to Ctl (Fig. 5). Cx43 colocalized with N-cadherin to a large extent in both Ctl and HF dogs (Figs.5A–5D). Summarized data showed a 33% decrease of Cx43E-E cumulative fluorescence intensity (p < 0.001, Fig. 5E), a 27% decrease of Cx43S-S cumulative fluorescent (p < 0.05, Fig. 5F), and unchanged Cx43 lateralization compared to controls (47.7% ± 4.0% vs. 44.3% ± 4.5%; Fig. 5G). Linear regression analysis showed a positive correlation between Cx43E-E and CVL (r2 = 0.5740, p < 0.001, Fig. 5H). Also, points representing CVL and level Cx43E-E in Ctl and HF dogs scarcely overlapped, further demonstrating significant differences in the relationship between Cx43E-E and CVL in HF vs Ctl dogs. In contrast, no significant correlation was demonstrated between Cx43S-S lateralization and CVT (r2 = 0.00091, p = 0.76, Fig. 5I), and the data points representing Ctl and HF animals largely overlapped, further suggesting that Cx43S-S does not play an active functional role in cardiac conduction.
Fig. 5.
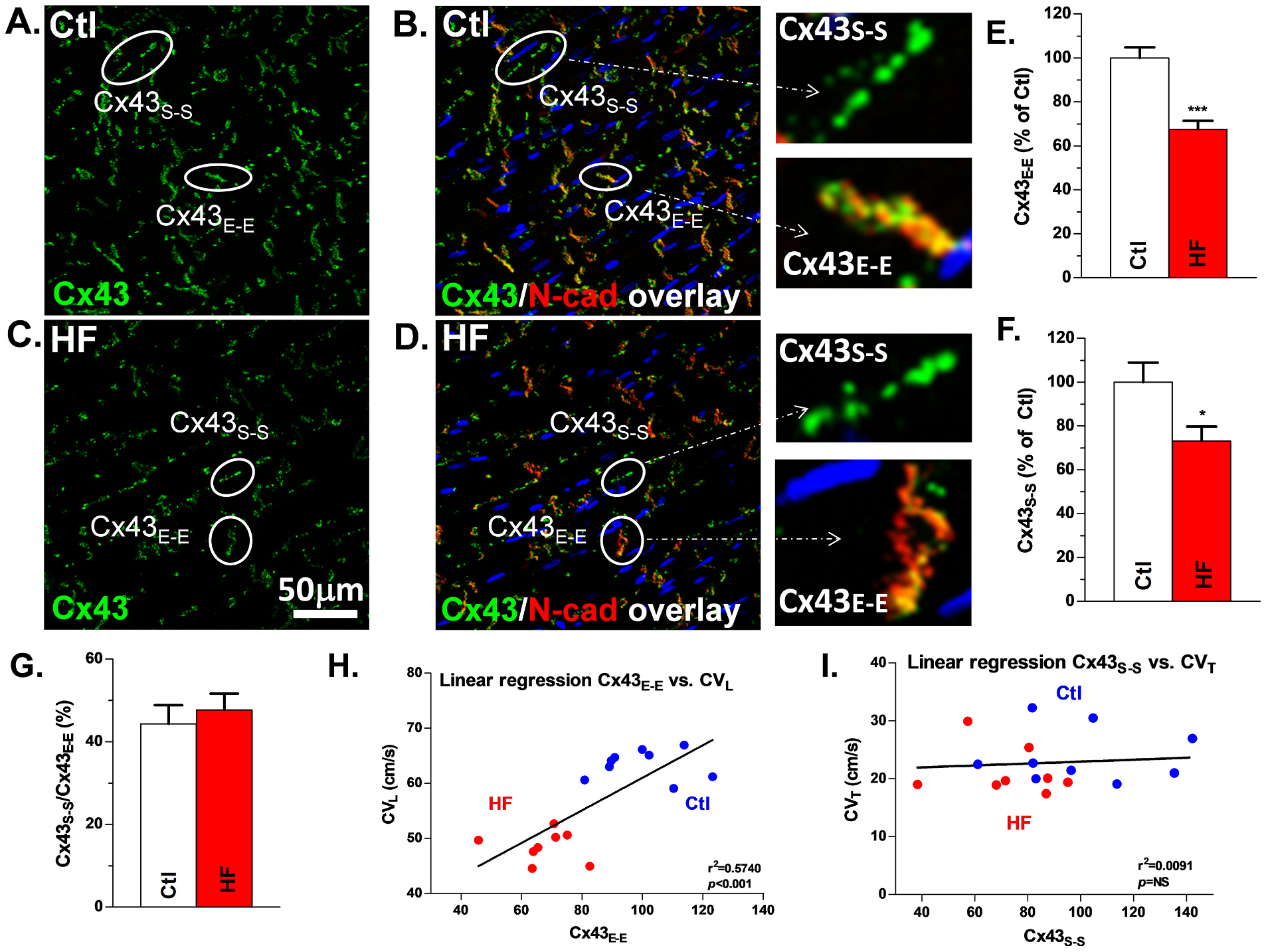
Reduced Cx43-T is positively correlated with slowed CVL in HF LV: Confocal images of Cx43 + N-cadherin double staining and Cx43 staining in Ctl (A, B) and HF (C, D) dog tissue used in grid mapping with Cx43 (green), N-cadherin (red) and overlap (yellow). Examples of Cx43E-E and Cx43S-S are encircled and labeled in each of the images as well as enlarged images of Cx43E-E and Cx43S-S. Quantification of Cx43E-E (E), Cx43S-S (F), and lateralization ratio (G) for HF vs Ctl, Nanimal = 9,8. Linear regression of Cx43E-E and CVL (H) and Cx43S-S and CVT (I), Nanimal = 9,8. *p < 0.05; ***p < 0.001. Each quantitative immunostaining Cx43 data point (Cx43E-E or Cx43S-S) is the mean value from forty confocal images of each tissue section. Each error bar indicates the standard error of the mean (SEM) of each experimental group. Mann-Whitney test was used to compare the difference between the two groups. Abbreviations: control (Ctl), longitudinal conduction velocity (CVL), heart failure (HF), junctional Cx43 (Cx43E-E), non-junctional Cx43 (Cx43S-S).
3.7. Increased junctional Cx43-NP (Cx43-NPE-E) in nonischemic HF
While Cx43-NPE-E colocalized with N-cadherin to a large extent in both Ctl and HF dogs, HF Epi showed significantly enhanced Cx43-NPE-E fluorescence intensity compared to Ctl (Fig. 6A, B). Summarized data showed a 99% increase of Cx43-NPE-E cumulative fluorescence intensity in HF vs Ctl (Fig. 6C). Linear regression analysis was performed between Cx43-NPE-E and CVL, and a negative correlation was demonstrated between those two variables (r2 = 0.4623, p < 0.001, Fig. 6D) in Ctl and HF animals. There was no correlation between CVT and Cx43-NP (Supplemental Fig.S3).
Fig. 6.
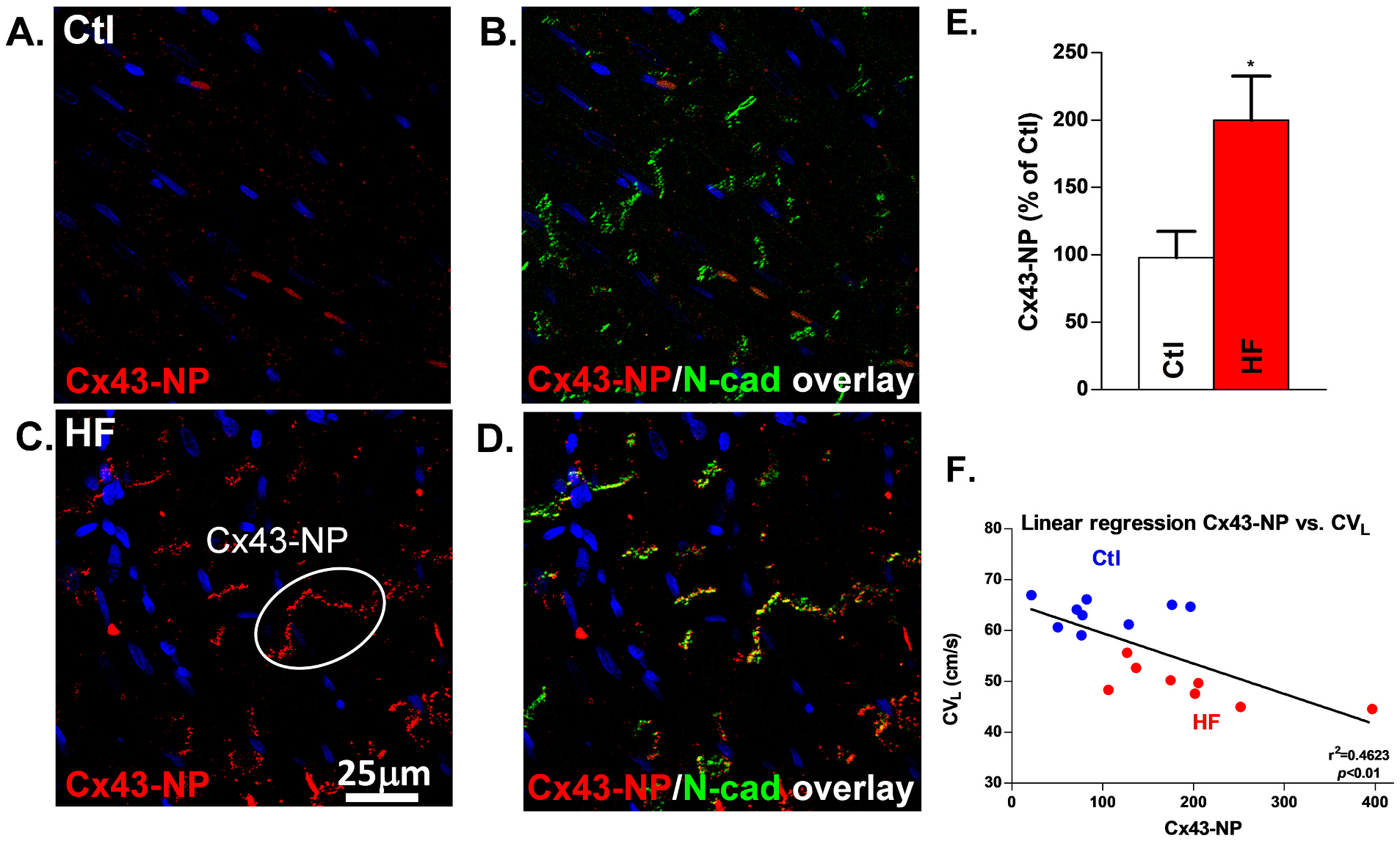
Enhanced Cx43-NP is negatively correlated with slowed CVL by IHC in HF: Confocal images of Cx43-NP + Cx43-T double staining and Cx43-NP staining in Ctl (A, B) and HF (C, D) dog tissue used in grid mapping with Cx43 (red), Cx43-NP (green) and overlap (yellow). An example of Cx43-NP is circled and labeled. E, Quantification of Cx43-NP. F, Linear regression of Cx43-NP and CVL. Nanimal = 9,8, *p < 0.05. Each quantitative Cx43-NP data point is the mean value from forty confocal images of each tissue section. Each error bar indicates the standard error of the mean (SEM) of each experimental group. Mann-Whitney test was used to compare the difference between the two groups. Abbreviations: longitudinal conduction velocity (CVL), non-phosphorylated Cx43 (Cx43-NP), HF (heart failure), IHC (immunohistochemistry).
3.8. Molecular basis for Cx43 dephosphorylation in nonischemic HF
To confirm the results obtained from IHC, we assessed total Cx43 (Cx43-T) and Cx43-NP using Western blot. We found that Cx43 protein was decreased 32% in canine HF LV, while the ratio of Cx43-NP to total Cx43 (Cx43-T) was increased ~80% (Figs.7A&B). These results were comparable to the IHC data in our canine HF LV as well as what we found in rabbit and human HF [14]. Moreover, we found that PP2A colocalized with Cx43 in Ctl canine LV, and with HF the amount of colocalized PP2A (with Cx43) is increased by 48% (Figs. 7C&D) although global expression of PP2A was decreased 20% (Figs.7E&F). The results of enhanced Cx43-NP associated with increased total Cx43 colocalized PP2A are consistent with what we reported in rabbit and human HF [14,31]. These results suggest that in HF enhanced PP2A-modulated Cx43 dephosphorylation along with downregulated Cx43 proteins leads to slowing of conduction that ultimately enhances arrhythmogenicity in LV of this nonischemic HF canine model.
Fig. 7.
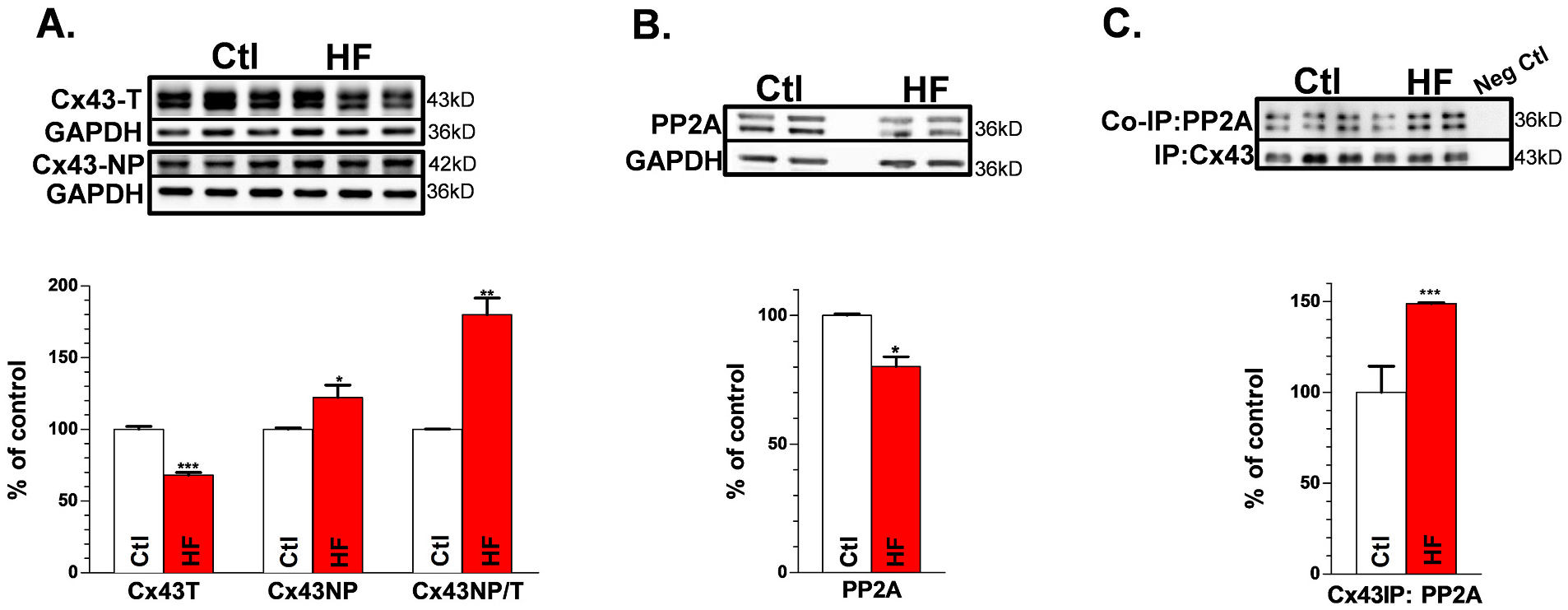
Reduced Cx43-T and enhanced Cx43-NP are associated with increased colocalized PP2A with Cx43 in HF LV: Representative Western blot images for Ctl and HF dog LV (top) and summarized data (bottom) for: Cx43-T and Cx43-NP (A & B), and PP1 (C & D) with GAPDH as controls. Co-immunoprecipitated PP2A with Cx43 for Ctl and HF dog LV (E & F), Nanimal = 9,8. *p < 0.05, **p < 0.01, ***p < 0.001. Each experiment had three technical repeats. The error bar indicates the standard error of the mean (SEM) of each experimental group. Mann-Whitney test was used to compare the between the two groups. Abbreviations: non-phosphorylated Cx43 (Cx43-NP), total Cx43 (Cx43-T), heart failure (HF), immuno-coprecipitation (IP), left ventricle (LV).
4. Discussion
Here we are the first to demonstrate the relationship of Cx43 expression and dephosphorylation to CV and arrhythmia development in the same exact tissue region mapped in a novel arrhythmogenic canine model of nonischemic HF. Another key finding in the current study is that this altered Cx43, in the absence of interstitial fibrosis, significantly contributes to increased spontaneous ventricular arrhythmias in vivo along with slow CV in intact LV tissue in this large animal model of nonischemic HF. Thus, our findings suggest that gap junctional remodeling occurs prior to the structural remodeling and it sufficiently impairs ventricular conduction and promotes arrhythmias at the early stage of HF.
4.1. The relationship between reduced Cx43, slow conduction, and ventricular arrhythmias in early stage of nonischemic HF
Extensive studies suggest that slow CV is linked to increased risk of cardiac reentrant arrhythmias [14,35–37]. Slowing of CV also presents in the failing heart has been linked to the development of fatal ventricular arrhythmias causing sudden cardiac death [14,35–38]. Cardiac conduction is determined by active membrane properties of each cell (largely a function of sodium channels) and tissue resistivity (as determined by gap junction channels and interstitial fibrosis) [39]. While Cx43 expression is decreased in both human HF and HF animal models, increased interstitial fibrosis has been found to also play a role in altered conduction velocity and even block of action potential propagation in the end-stage failing heart [14,31,38,40–43]. Thus, it remains unclear as to the relationship between Cx43 and interstitial fibrosis in slow conduction in nonischemic HF. In our novel canine model of nonischemic HF, we found slowing of CV along with significantly increased ventricular arrhythmogenicity that was associated with Cx43 remodeling (reduced abundance and increased dephosphorylation) but unchanged status of interstitial fibrosis. While we cannot rule out that this might occur in other regions of the failing heart that were not mapped or with development of more severe LV dysfunction, our new findings from the parallel functional assessment and in situ gap junction profiling suggest that gap junctional remodeling occurs prior to the structural remodeling, and it sufficiently impairs ventricular conduction to promote arrhythmias at an early stage of HF. We and others have previously showed reentry during ventricular tachycardia studies in nonischemic heart failure at late stage and the relationship between Cx43 remodeling and altered cellular coupling was further showed to lead to increased arrhythmias in HF at late stage. Moreover, the functional role of reduced Cx43 in slow conduction is strongly supported by our recent findings in either aged atria or young transgenic mouse atria with reduced Cx43 but absence of interstitial fibrosis [9,10].
It is notable that this lack of interstitial fibrosis has been found in up to 50–70% of hearts from patients with moderate to severe (but not end-stage) nonischemic HF [21–23]. While it is impossible to assess the link between molecular remodeling and functional impairment in those HF patients, our unique arrhythmogenic nonischemic HF model reflecting human HF prior to the development of end stage HF allows us explore this issue in a systematic fashion. First, our findings suggest that gap junctional remodeling occurs prior to the structural remodeling. Second, this gap junction remodeling sufficiently impairs conduction in the failing LV at the early stage, while progressively accumulated fibrosis may further aggravate the conduction impediment during the later stages of HF. Our unique model provides an ideal opportunity in which to assess the effects of altered Cx43 expression and phosphorylation on conduction velocity without the complication of concurrent interstitial fibrosis which itself could slow conduction (and limit the ability to assess the role of Cx43). Thus, our findings shed new light on targeting gap junction as an early intervention approach to prevent arrhythmias in HF patients.
4.2. Functinal impact of Cx43 remodeling in nonischemic HF
We have previously showed that downregulated Cx43 in either HF myocytes or cultured normal adult rabbit myocytes (with Cx43 siRNA knockdown) impaired cell-to-cell communication, while overexpressed wild-type Cx43 markedly improved the communication between adjacent HF myocytes [14,31]. On the other hand, the location or distribution of Cx43 proteins is also important in maintaining normal intercellular communication. Previous research suggested that lateralized Cx43, in which Cx43 is located at side-to-side surfaces of the cell borders distinct from intercalated disks, are not functional (i.e. such Cx43’s are internalized or form nonfunctional gap junctional channels due to lack of mechanical stability) [44]. However, the relationship between Cx43 remodeling (downregulation and/or lateralization) and conduction defects in intact cardiac tissue in various HF animal models remains controversial [40,43]. While under certain disease conditions, the ratio of Cx43S-S to Cx43E-E could be altered [40], unchanged lateralization of Cx43 has also been found in failing ventricles [43]. Several possibilities could be attributed to the discrepancy of the findings. It is known that cardiac muscle fibers are circumferentially and longitudinally arranged in three-dimensional space, and the abundance and distribution of GJs at different imaging focal planes and different regions could vary. Thus, this histological imaging limitation could cause inconsistent experimental results. In addition, pathologically enhanced fibrosis in structural heterogeneity [44,45] may also cause variations in quantified Cx43. We recently developed a unique layer-by-layer quantitative method using serial sequential z-stack confocal images [9,30] to utilize colocalization with N-cadherin as a hallmark for Cx43E-E [40], and as an internal control of expression level (based on the rationale that N-cadherin is a relatively stable housekeeping protein under both control and pathological conditions as we and others have previously shown) [9,30,46]. Using this novel method, we found that reduced Cx43E-E at intercalated disks showed a strong positive correlation with slowed CVL in LV from our nonischemic HF canine model (Fig. 5I). Reduced Cx43 has been shown to be critical in impaired cell-to-cell communication as we previously showed in isolated paired LV myocytes from a rabbit model of nonischemic HF and from Cx43 siRNA knockdown control myocytes [14]. In addition, we recently demonstrated the functional role of reduced Cx43 in impaired cell-to-cell communication and slowing of conduction in intact atrial myocardium, which significantly enhanced atrial arrhythmogenicity [9,10]. While our findings of reduced Cx43 are in agreement with findings in a canine rapid pacing HF model [43], different results were also reported by other groups [40,46] using the same pacing model. We speculate that these disparate findings could be due to different severity of the HF in their paced animals. Moreover, in humans enhanced Cx43 lateralization was found in RV biopsy samples of HF patients [47] which were restricted to regions of fibrosis and infarction [43]. Thus, the different progressive stages of HF could explain these differences and Cx43 lateralization could be a feature of later stages of cardiomyopathy [48,49] characterized by more severe LV dysfunction or at the regions containing a large amount of fibrosis. In those end-stage failing hearts, markedly increased fibrosis may also mask the functional contribution of remodeled Cx43 due to the physical separation by the fibrotic tissue.
Notabley, Cx43 is a phosphoprotein with numerous phosphorylation sites in the C-terminus. In addition to downregulated Cx43 proteins, we also discovered an increase in dephosphorylated Cx43 in HF that was associated with a marked elevation in the level of protein phosphatase PP2A that was colocalized with Cx43 proteins. Moreover, PP2A inhibition with okadaic acid improved intercellular coupling in isolated failing ventricular myocytes. Here we find similar Cx43 dephosphorylation in this HF canine model. Our IP studies support that enhanced colocalization (at the level of total Cx43 proteins) of PP2A. This increased dephosphorylated Cx43 was negatively correlated with CVL but had no correlation with CVT. Overall, we took advantage of our recently developed arrhythmogenic nonischemic HF canine model and demonstrated for the first time that Cx43 remodeling, in the absence of structural remodeling, plays a crucial role in impaired ventricular conduction and enhance arrhythmogenicity in nonischemic HF.
4.3. Other potential factors contributing to altered conduction in nonischemic HF
In addition to gap junctions and fibrosis, Na inward current and resting membrane potential (RMP) are major determinants of excitability of cardiac myocytes and can affect CV [50]. However by microelectrode recording we found no differences in dV/dtmax of the AP or in RMP with HF vs Ctl dogs, in line with findings in the canine rapid pacing HF model which showed unaltered kinetics of peak INa [51]. APD90 was prolonged consistent with the prolonged QTc and similar to that seen in animal and human HF [1]. Therefore, unchanged interstitial collagen content and cellular excitability are unlikely to be involved in slow CV and enhanced arrhythmogenicity in this nonischemic canine HF model. Although unchanged Cx43S-S and CVT along with the lack of correlation between Cx43S-S and CVT were found in our HF LV, the contribution of lateralized gap junction in impaired conduction in late-stage HF requires further investigation. In addition, several recently emerging theories such as gap junction hemichannels [12] and ephaptic coupling [52] have been proposed to contribute to slowed CV. As such, their possible roles in altered conduction in HF would also be worthy of future study.
4.4. Implications
HF is a deadly disease in large part because of lethal ventricular arrhythmias that underlie sudden death [1]. Conduction slowing is particularly critical in the transition of VT to VF. Here we show that our novel arrhythmogenic large animal model of nonischemic HF exhibited markedly enhanced arrhythmogenicity and reduced CVL but preserved CVT, a feature found in many other animal models of HF [53]. While this slowed CVL differs from findings from models with severely depressed LV function (where CVL and CVT are both decreased accompanied by significant structural remodeling) [40,54], gap junctional remodeling along with unchanged interstitial fibrosis could explain the differences in slowed conduction between moderate HF during the progress of the disease and end-stage HF with severely depressed LV function. While an arrhythmogenic substrate may be begun to develop during early stage of HF, increased interstitial fibrosis during the HF progression may contribute to further increase arrhythmogenicity at the end-stage of HF. The current findings along with our recently reported functional evidence [9,10] suggest that Cx43 remodeling occurred prior to the structural remodeling and is critical in slowed CV and ventricular arrhythmia development. Modulation of both Cx43 expression and phosphorylation state could be a novel approach to improve conduction and reduced arrhythmogenicity even at early stages of HF.
Supplementary Material
Acknowledgements
We acknowledge the excellent technical support from Dennis Rollins, Sharon Melnick, Weiwei Zhao, Joseph Barchue, Shannon Salter, and Frank Vance.
Funding sources
This study was supported by grants R01 HL-073966 (SMP), R21 HL-097268 (SMP), R01 HL-113640 (XA), R01-HL146744 (XA), and R01-AA024769 (XA) from the National Institute of Health.
Footnotes
Declaration of Competing Interest
None.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.yjmcc.2021.05.012.
References
- [1].Packer M, Sudden unexpected death in patients with congestive heart failure: a second frontier, Circulation 72 (1985) 681–685. [DOI] [PubMed] [Google Scholar]
- [2].Pogwizd SM, Nonreentrant mechanisms underlying spontaneous ventricular arrhythmias in a model of nonischemic heart failure in rabbits, Circulation 92 (1995) 1034–1048. [DOI] [PubMed] [Google Scholar]
- [3].Pogwizd SM, McKenzie JP, Cain ME, Mechanisms underlying spontaneous and induced ventricular arrhythmias in patients with idiopathic dilated cardiomyopathy, Circulation 98 (1998) 2404–2414. [DOI] [PubMed] [Google Scholar]
- [4].Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM, Arrhythmogenesis and contractile dysfunction in heart failure: roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness, Circ. Res 88 (2001) 1159–1167. [DOI] [PubMed] [Google Scholar]
- [5].Anderson KP, Walker R, Urie P, Ershler PR, Lux RL, Karwandee SV, Myocardial electrical propagation in patients with idiopathic dilated cardiomyopathy, J. Clin. Invest 92 (1993) 122–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Verheule S, van Kempen MJ, te Welscher PH, Kwak BR, Jongsma HJ, Characterization of gap junction channels in adult rabbit atrial and ventricular myocardium, Circ. Res 80 (1997) 673–681. [DOI] [PubMed] [Google Scholar]
- [7].Bruzzone R, White TW, Paul DL, Connections with connexins: the molecular basis of direct intercellular signaling, Eur. J. Biochem 238 (1996) 1–27. [DOI] [PubMed] [Google Scholar]
- [8].Saffitz JE, Davis LM, Darrow BJ, Kanter HL, Laing JG, Beyer EC, The molecular basis of anisotropy: role of gap junctions, J. Cardiovasc. Electrophysiol 6 (1995) 498–510. [DOI] [PubMed] [Google Scholar]
- [9].Yan J, Kong W, Zhang Q, Beyer EC, Walcott G, Fast VG, et al. , c-Jun N-terminal kinase activation contributes to reduced connexin43 and development of atrial arrhythmias, Cardiovascular research 97 (2013) 589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yan J, Thomson JK, Zhao W, Wu X, Gao X, DeMarco D, et al. , The stress kinase JNK regulates gap junction Cx43 gene expression and promotes atrial fibrillation in the aged heart, J. Mol. Cell. Cardiol 114 (2017) 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kanter HL, Saffitz JE, Beyer EC, Cardiac myocytes express multiple gap junction proteins, Circ. Res 70 (1992) 438–444. [DOI] [PubMed] [Google Scholar]
- [12].Leybaert L, Lampe PD, Dhein S, Kwak BR, Ferdinandy P, Beyer EC, et al. , Connexins in cardiovascular and neurovascular health and disease: pharmacological implications, Pharmacol. Rev 69 (2017) 396–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hood AR, Ai X, Pogwizd SM, Regulation of cardiac gap junctions by protein phosphatases, J. Mol. Cell. Cardiol 107 (2017) 52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ai X, Pogwizd SM, Connexin 43 downregulation and dephosphorylation in nonischemic heart failure is associated with enhanced colocalized protein phosphatase type 2A, Circ. Res 96 (2005) 54–63. [DOI] [PubMed] [Google Scholar]
- [15].Kostin S, Klein G, Szalay Z, Hein S, Bauer EP, Schaper J, Structural correlate of atrial fibrillation in human patients, Cardiovasc. Res 54 (2002) 361–379. [DOI] [PubMed] [Google Scholar]
- [16].Kanagaratnam P, Rothery S, Patel P, Severs NJ, Peters NS, Relative expression of immunolocalized connexins 40 and 43 correlates with human atrial conduction properties, J. Am. Coll. Cardiol 39 (2002) 116–123. [DOI] [PubMed] [Google Scholar]
- [17].Papageorgiou P, Monahan K, Boyle NG, Seifert MJ, Beswick P, Zebede J, et al. , Site-dependent intra-atrial conduction delay. Relationship to initiation of atrial fibrillation, Circulation 94 (1996) 384–389. [DOI] [PubMed] [Google Scholar]
- [18].Guerrero PA, Schuessler RB, Davis LM, Beyer EC, Johnson CM, Yamada KA, et al. , Slow ventricular conduction in mice heterozygous for a connexin43 null mutation, J. Clin. Invest 99 (1997) 1991–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Danik SB, Liu F, Zhang J, Suk HJ, Morley GE, Fishman GI, et al. , Modulation of cardiac gap junction expression and arrhythmic susceptibility, Circ. Res 95 (2004) 1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wiegerinck RF, van Veen TA, Belterman CN, Schumacher CA, Noorman M, de Bakker JM, et al. , Transmural dispersion of refractoriness and conduction velocity is associated with heterogeneously reduced connexin43 in a rabbit model of heart failure, Heart Rhythm 5 (2008) 1178–1185. [DOI] [PubMed] [Google Scholar]
- [21].McCrohon JA, Moon JC, Prasad SK, McKenna WJ, Lorenz CH, Coats AJ, et al. , Differentiation of heart failure related to dilated cardiomyopathy and coronary artery disease using gadolinium-enhanced cardiovascular magnetic resonance, Circulation 108 (2003) 54–59. [DOI] [PubMed] [Google Scholar]
- [22].Hirakawa K, Yamamuro M, Uemura T, Takashio S, Kaikita K, Utsunomiya D, et al. , Correlation between microvascular dysfunction and B-type natriuretic peptide levels in non-ischemic heart failure patients with cardiac fibrosis, Int. J. Cardiol 228 (2017) 881–885. [DOI] [PubMed] [Google Scholar]
- [23].Takashio S, Yamamuro M, Uemura T, Utsunomiya D, Morita K, Izumiya Y, et al. , Correlation between extent of myocardial fibrosis assessed by cardiac magnetic resonance and cardiac troponin T release in patients with nonischemic heart failure, Am. J. Cardiol 113 (2014) 1697–1704. [DOI] [PubMed] [Google Scholar]
- [24].Zhu Y, Hanafy MA, Killingsworth CR, Walcott GP, Young ME, Pogwizd SM, Morning surge of ventricular arrhythmias in a new arrhythmogenic canine model of chronic heart failure is associated with attenuation of time-of-day dependence of heart rate and autonomic adaptation, and reduced cardiac chaos, PLoS One 9 (2014), e105379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Van de Water A, Verheyen J, Xhonneux R, Reneman RS, An improved method to correct the QT interval of the electrocardiogram for changes in heart rate, J Pharmacol Methods 22 (1989) 207–217. [DOI] [PubMed] [Google Scholar]
- [26].Tattersall ML, Dymond M, Hammond T, Valentin JP, Correction of QT values to allow for increases in heart rate in conscious beagle dogs in toxicology assessment, J. Pharmacol. Toxicol. Methods 53 (2006) 11–19. [DOI] [PubMed] [Google Scholar]
- [27].Cohen ML, Hoyt RH, Saffitz JE, Corr PB, A high density in vitro extracellular electrode array: description and implementation, Am. J. Phys 257 (1989) H681–H689. [DOI] [PubMed] [Google Scholar]
- [28].Bayly PV, KenKnight BH, Rogers JM, Hillsley RE, Ideker RE, Smith WM, Estimation of conduction velocity vector fields from epicardial mapping data, IEEE Trans. Biomed. Eng 45 (1998) 563–571. [DOI] [PubMed] [Google Scholar]
- [29].Yan J, Thomson JK, Zhao W, Fast VG, Ye T, Ai X, Voltage and calcium dual channel optical mapping of cultured HL-1 atrial myocyte monolayer, J. Vis. Exp 97 (2015) 563–52542, 10.3791/52542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yan J, Thomson JK, Wu X, Zhao W, Pollard AE, Ai X, Novel methods of automated quantification of gap junction distribution and interstitial collagen quantity from animal and human atrial tissue sections, PLoS One 9 (2014), e104357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ai X, Zhao W, Pogwizd SM, Connexin43 knockdown or overexpression modulates cell coupling in control and failing rabbit left ventricular myocytes, Cardiovasc. Res 85 (2010) 751–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yan J, Zhao W, Thomson JK, Gao X, DeMarco DM, Carrillo E, et al. , Stress signaling JNK2 crosstalk with CaMKII underlies enhanced atrial Arrhythmogenesis, Circ. Res 122 (6) (2018) 821–835, 10.1161/CIRCRESAHA.117.312536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Huang J, Zhou X, Smith WM, Ideker RE, Restitution properties during ventricular fibrillation in the in situ swine heart, Circulation 110 (2004) 3161–3167. [DOI] [PubMed] [Google Scholar]
- [34].Barison A, Grigoratos C, Todiere G, Aquaro GD, Myocardial interstitial remodelling in non-ischaemic dilated cardiomyopathy: insights from cardiovascular magnetic resonance, Heart Fail. Rev 20 (2015) 731–749. [DOI] [PubMed] [Google Scholar]
- [35].Greener ID, Sasano T, Wan X, Igarashi T, Strom M, Rosenbaum DS, et al. , Connexin43 gene transfer reduces ventricular tachycardia susceptibility after myocardial infarction, J. Am. Coll. Cardiol 60 (2012) 1103–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Baba S, Dun W, Cabo C, Boyden PA, Remodeling in cells from different regions of the reentrant circuit during ventricular tachycardia, Circulation 112 (2005) 2386–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Pogwizd SM, Corr B, The contribution of nonreentrant mechanisms to malignant ventricular arrhythmias, Basic Res. Cardiol 87 (Suppl. 2) (1992) 115–129. [DOI] [PubMed] [Google Scholar]
- [38].Glukhov AV, Fedorov VV, Kalish PW, Ravikumar VK, Lou Q, Janks D, et al. , Conduction remodeling in human end-stage nonischemic left ventricular cardiomyopathy, Circulation 125 (2012) 1835–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Walton MK, Fozzard HA, The conducted action potential. Models and comparison to experiments, Biophys. J 44 (1983) 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Akar FG, Spragg DD, Tunin RS, Kass DA, Tomaselli GF, Mechanisms underlying conduction slowing and arrhythmogenesis in nonischemic dilated cardiomyopathy, Circ. Res 95 (2004) 717–725. [DOI] [PubMed] [Google Scholar]
- [41].Ai X, Jiang A, Ke Y, Solaro RJ, Pogwizd SM, Enhanced activation of p21-activated kinase 1 in heart failure contributes to dephosphorylation of connexin 43, Cardiovasc. Res 92 (2011) 106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Dupont E, Matsushita T, Kaba RA, Vozzi C, Coppen SR, Khan N, et al. , Altered connexin expression in human congestive heart failure, J. Mol. Cell. Cardiol 33 (2001) 359–371. [DOI] [PubMed] [Google Scholar]
- [43].Poelzing S, Rosenbaum DS, Altered connexin43 expression produces arrhythmia substrate in heart failure, Am. J. Physiol. Heart Circ. Physiol 287 (2004) H1762–H1770. [DOI] [PubMed] [Google Scholar]
- [44].Solan JL, Lampe PD, Spatio-temporal regulation of connexin43 phosphorylation and gap junction dynamics, Biochim. Biophys. Acta 2018 (1860) 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Boulaksil M, Bierhuizen MF, Engelen MA, Stein M, Kok BJ, van Amersfoorth SC, et al. , Spatial heterogeneity of Cx43 is an Arrhythmogenic substrate of polymorphic ventricular Tachycardias during compensated cardiac hypertrophy in rats, Front Cardiovasc Med 3 (2016) 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Hesketh GG, Shah MH, Halperin VL, Cooke CA, Akar FG, Yen TE, et al. , Ultrastructure and regulation of lateralized connexin43 in the failing heart, Circ. Res 106 (2010) 1153–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kitamura H, Ohnishi Y, Yoshida A, Okajima K, Azumi H, Ishida A, et al. , Heterogeneous loss of connexin43 protein in nonischemic dilated cardiomyopathy with ventricular tachycardia, J. Cardiovasc. Electrophysiol 13 (2002) 865–870. [DOI] [PubMed] [Google Scholar]
- [48].Akar FG, Nass RD, Hahn S, Cingolani E, Shah M, Hesketh GG, et al. , Dynamic changes in conduction velocity and gap junction properties during development of pacing-induced heart failure, Am. J. Physiol. Heart Circ. Physiol 293 (2007) H1223–H1230. [DOI] [PubMed] [Google Scholar]
- [49].Fontes MS, van Veen TA, de Bakker JM, van Rijen HV, Functional consequences of abnormal Cx43 expression in the heart, Biochim. Biophys. Acta 2012 (1818) 2020–2029. [DOI] [PubMed] [Google Scholar]
- [50].Shaw RM, Rudy Y, Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling, Circ. Res 81 (1997) 727–741. [DOI] [PubMed] [Google Scholar]
- [51].Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, et al. , Increased late sodium current in myocytes from a canine heart failure model and from failing human heart, J. Mol. Cell. Cardiol 38 (2005) 475–483. [DOI] [PubMed] [Google Scholar]
- [52].Hoagland DT, Santos W, Poelzing S, Gourdie RG, The role of the gap junction perinexus in cardiac conduction: potential as a novel anti-arrhythmic drug target, Prog. Biophys. Mol. Biol 144 (2019) 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Boulaksil M, Noorman M, Engelen MA, van Veen TA, Vos MA, de Bakker JM, et al. , Longitudinal arrhythmogenic remodelling in a mouse model of longstanding pressure overload, Neth Heart J 18 (2010) 509–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sato T, Ohkusa T, Honjo H, Suzuki S, Yoshida MA, Ishiguro YS, et al. , Altered expression of connexin43 contributes to the arrhythmogenic substrate during the development of heart failure in cardiomyopathic hamster, Am. J. Physiol. Heart Circ. Physiol 294 (2008) H1164–H1173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.