

Abstract
Cisplatin has been a mainstay of cancer chemotherapy since the 1970s. Despite its broad anticancer potential, its clinical use has regularly been constrained by kidney toxicities. This review details those biochemical pathways and metabolic conversions that underlie the kidney toxicities. A wide range of redox events contribute to the eventual physiological consequences of drug activities.
1. Introduction
Cisplatin is a square–planar complex with two labile chloride (Cl) ion ligands and two relative inert ammonia (NH3) ligands coordinated to the central platinum (Pt) atom in a cis configuration (Fig. 1A). The drug was first approved as an antineoplastic agent in 1978 and remains an important and effective therapy today for the treatment of various cancers including bladder, breast, cervical, esophageal, head and neck, ovarian, prostate, small and non-small cell lung, stomach, testicular cancers, Hodgkin’s and non-Hodgkin’s lymphomas, melanoma, mesothelioma, multiple myeloma, neuroblastoma, and sarcomas (Koepsell, Lips, & Volk, 2007). When cisplatin enters the cells, the low intracellular chloride concentration can lead to the dissociation of the chlorides resulting in the formation of monoaquo and diaquo complexes, and the hydrolyzed product is a potent electrophile that can react with the purine bases on the DNA. The various adducts and crosslinks induced by cisplatin have a multitude of effects including DNA unwinding, DNA bending, replication inhibition, and transcription inhibition, which can further cause DNA strand breaks, impaired cell division and apoptotic cell death (Dasari & Tchounwou, 2014; Manohar & Leung, 2018).
Fig. 1.
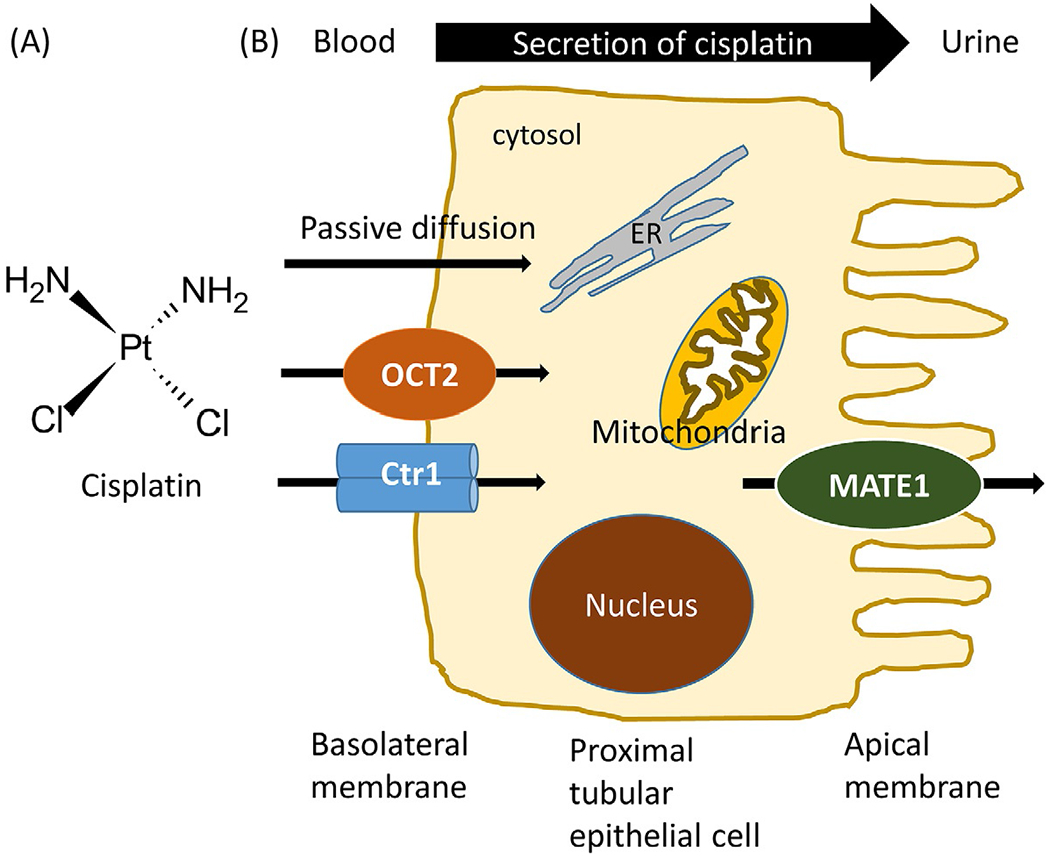
Schematic representation of cisplatin renal uptake and excretion. (A) Chemical structure of cisplatin. (B) Transporters involved in active tubular secretion of cisplatin into urine. Cisplatin is removed from the circulating blood and enters the proximal tubule cells either by passive diffusion or basolaterally expressed OCT2 and Ctr1. Cisplatin is then removed into the tubular lumen by apically expressed MATE1. OCT2, organic cation transporter 2; Ctr1, copper transporter 1; MATE1, multidrug and toxin extrusion transporter 1.
Although cisplatin has been on the market for more than 40 years, we are still struggling with its severe dose-limiting side effects, especially nephrotoxicity, which can be dose limiting in 30–40% of patients (Ridzuan et al., 2019). The clinical signs of kidney damage are a decrease in renal plasma flow and glomerular filtration rate (GFR) and an increase in blood urea nitrogen (BUN) and serum creatinine (Cr) concentrations (Volarevic et al., 2019). Cisplatin nephrotoxicity can present in a number of ways, e.g., acute kidney injury, hypomagnesemia, hypocalcemia, hyperuricemia, distal renal tubular acidosis, proximal tubular dysfunction, and chronic renal failure. Among these, the most serious and more common presentations is acute kidney injury, which occurs in 20–30% of patients (Gomez-Sierra, Eugenio-Perez, Sanchez-Chinchillas, & Pedraza-Chaverri, 2018). Primary approaches to protect from cisplatin-induced nephrotoxicity are based on avoiding extreme exposure of the kidneys to the drug. This is managed primarily by hydration with saline infusion and concomitant administration of mannitol, furosemide or magnesium, monitoring of renal function by serum Cr clearance, and decreasing cisplatin doses upon manifestation of renal dysfunction. However, even with aggressive hydration, renal toxicity occurs (Hakiminia, Goudarzi, & Moghaddas, 2019; Miller, Tadagavadi, Ramesh, & Reeves, 2010). Therefore, more effective preventative strategies without attenuation of tumoricidal activity need to be developed.
The development of effective tumor interventions with reduced nephrotoxicity relies heavily on understanding the molecular pathophysiology of cisplatin-induced nephrotoxicity. Cisplatin kills dividing cells by forming platinum-DNA crosslinks that prevent DNA synthesis and results in cell death. However, proximal tubular cells are not replicating, suggesting an alternative mechanism of toxicity (Tanase et al., 2019). The cellular process of nephrotoxicity can be attributed to local accumulation of cisplatin inside the renal proximal tubule by membrane transportation, and intracellular conversion of the drug into toxic metabolites (Hakiminia et al., 2019; Hanigan, Gallagher, & Taylor, 1996). Among the reported mechanisms of cisplatin nephrotoxicity are dysregulation of oxidative stress, mitochondrial dysfunction, endoplasmic reticulum (ER) stress, inflammatory responses (macrophage/monocyte infiltration into renal cortex and medulla), apoptosis, necrosis and autophagy (Hanigan & Devarajan, 2003). The roles of these factors seem to be closely related. Oxidative stress is one of the best characterized mechanisms for cisplatin nephrotoxicity and will be discussed later in this review.
2. Cisplatin renal uptake and excretion
During glomerular filtration and tubular secretion, cisplatin is markedly accumulated in the kidney, and the concentration in proximal tubules is about five times greater than that in blood. Accordingly, even non-toxic serum concentrations of cisplatin may reach toxic levels in the kidney, resulting in impaired renal function due to the severe injury of S3 segment of proximal tubules (Dasari & Tchounwou, 2014; Holditch, Brown, Lombardi, Nguyen, & Edelstein, 2019). Cisplatin can enter the renal tubular cells by either passive diffusion or active transport. Previous studies have indicated that movement of cisplatin through the renal tubular cells occurs in a basolateral to apical direction (Gomez-Sierra et al., 2018). Specifically, cisplatin is removed from the circulating blood and enters renal tubular cells by the actions of basolaterally expressed transporters such as organic cation transporter 2 (OCT2, the main OCT in human kidney (Yao, Panichpisal, Kurtzman, & Nugent, 2007)) and, to a lesser extent, copper transporter 1 (Ctr1). It is then removed into the tubular lumen by apically expressed multidrug and toxin extrusion transporter 1 (MATE1) (Ridzuan et al., 2019) (Fig. 1B). In line with these observations, cisplatin nephrotoxicity is positively correlated with OCT2 and Ctr1 expression, and negatively correlated with MATE1.
OCT2 is most strongly expressed in the kidney and is localized to the basolateral membrane of renal proximal tubular cells (George, You, Joy, & Aleksunes, 2017; Yao et al., 2007). Notably, reduced cisplatin nephrotoxicity has been observed in vivo in cisplatin-treated (Koepsell et al., 2007) OCT2 knockout mice (Ciarimboli et al., 2010; Filipski, Mathijssen, Mikkelsen, Schinkel, & Sparreboom, 2009) or wild-type mice pretreated with cimetidine, a pharmacological inhibitor of OCT2 (Ciarimboli et al., 2010; Manohar & Leung, 2018) female rats whose OCT2 expression levels are much lower than those in males (Dasari & Tchounwou, 2014; Nematbakhsh et al., 2013; Urakami et al., 1999) cancer patients with a non-synonymous single-nucleotide polymorphism in the OCT2 gene SLC22A2 (rs316019) (Filipski et al., 2009; Ridzuan et al., 2019) cancer patients who received prehydration with magnesium which is assumed to down-regulate OCT2 levels (Muraki et al., 2012; Oka et al., 2014; Volarevic et al., 2019; Yamamoto et al., 2015; Yokoo et al., 2009) cancer patients who received cimetidine and verapamil (a calcium entry blocker) along with cisplatin (Sleijfer et al., 1987).
Ctr1 is highly expressed in adult kidney and the protein is localized on the basolateral side of both proximal and distal tubular cells (Pabla, Murphy, Liu, & Dong, 2009). In vitro downregulation of Ctr1 in kidney cells suppresses both cisplatin uptake and cytotoxicity, including cell death by both apoptosis and necrosis (Pabla et al., 2009). However, the role of Ctr1 in cisplatin nephrotoxicity has not yet been examined in vivo. Considering the crucial role of Ctr1 for cisplatin to enter the tumor cells (Holzer et al., 2004), concern has been raised that targeting this transporter for a renoprotective strategy may affect the antitumor effects of the drug. Thus, it is important to use tumor bearing in vivo models to determine the effects of therapies targeting cisplatin transporters on the cancers as well as renoprotection.
MATE1 is highly expressed in the brush-border membrane of renal proximal tubules and mediates the efflux of cisplatin from the renal epithelial cells into the urine. Higher renal accumulation of cisplatin, lower survival, and higher creatinine and BUN levels have been observed in cisplatin–treated MATE1 knockout mice or wild-type mice pretreated with pyrimethamine, a selective MATE inhibitor, suggesting the fundamental role of MATE1 in controlling the nephrotoxicity (Nakamura, Yonezawa, Hashimoto, Katsura, & Inui, 2010). Inversely, less accumulation of cisplatin in renal tissue has been found in cisplatin-treated kinin B1 receptor knockout mice or wild-type mice pretreated with a kinin B1 receptor antagonist R-715. Kinin B1 receptor deletion and blockage resulted in higher expression of MATE1 and consequently protected against cisplatin-induced acute kidney injury (Estrela et al., 2017). In humans the renal clearance of cisplatin exceeds the glomerular filtration rate, indicating that cisplatin is secreted across renal tubular cells (Nakamura et al., 2010). Additional studies are required to reveal the contribution of MATE family to the renal handling of cisplatin in humans.
3. Bioactivation of cisplatin in the kidney
Cisplatin undergoes metabolic activation in the kidney to a more potent toxin (Fig. 2). The first step of cisplatin metabolism is the formation of a glutathione (GSH)-conjugate, in which one of the chloride ions of cisplatin is replaced by the sulfur moiety of GSH (Townsend, Marto, Deng, Macdonald, & Hanigan, 2003). Buthionine sulfoximine, which depletes endogenous GSH by inhibiting GSH synthesis, has been shown to protect against cisplatin nephrotoxicity in rats (Mayer, Lee, & Cockett, 1987). GSH-conjugates are generally formed in the liver, which contains the highest level of glutathione-S-transferase (GST) (Hanigan, Gallagher, Taylor, & Large, 1994), and the conjugates are excreted from the cell into the serum or the bile via the multidrug-resistance-associated protein transporters (Homolya, Varadi, & Sarkadi, 2003; Zhang et al., 2014). Pharmacological inhibition of GST by ketoprofen has been shown to protect against cisplatin nephrotoxicity in rat (Sadzuka, Shimizu, Takino, & Hirota, 1994). However, the specific GST isoforms that may catalyze the formation of GSH-conjugate in the activation of cisplatin to a renal toxin have not been identified, nor has the location where this conjugate is formed in the body been determined. GstP1/P2 wild-type males had more extensive renal damage than either female wild-type or GstP1/P2 null mice (Townsend, Tew, He, King, & Hanigan, 2009). Wild-type males have ~10-fold higher levels of GSTP expression in the liver than females, and the liver is the only organ which shows gender difference of GSTP expression (Knight, Choudhuri, & Klaassen, 2007), suggesting that higher levels of GSTP expression in the liver may enhance the formation of cisplatin-GSH-conjugate that is metabolized to renal toxins. Contradictorily, a recent study indicated that cisplatin is not a substrate for GSTP as the GSH-adduct formation is not affected by human GSTP1–1. However, the conclusion was drawn based on the in vitro incubation of a high concentration of cisplatin (1 mM) with GSH (2mM) (De Luca et al., 2019). While the enzyme can catalyze GSH conjugation, other properties so far ascribed to GSTP include chaperone functions, regulation of nitric oxide pathways, regulation of a variety of kinase signaling pathways, participation in the forward reaction of protein S-glutathionylation and maintenance of cellular redox homeostasis (Tew et al., 2011; Zhang et al., 2014). Considering the pleiotropic functions of GSTP, its role in the cisplatin nephrotoxicity remains to be established.
Fig. 2.

Bioactivation of cisplatin to a nephrotoxin. The first step of cisplatin metabolism is the formation of a GSH-conjugate catalyzed by GST. As the cisplatin-GSH-conjugate passes through the kidney, it is cleaved by GGT to a cysteinyl-glycine-conjugate, which is further converted to a cysteine-conjugate by APN. GGT and APN are both plasma membrane-bound enzymes with the catalytic domain exposed to the extracellular surface. As the cisplatin-cysteine-conjugate enters the proximal tubular epithelial cell, it is further metabolized by a pyridoxal 5′-phosphate-dependent enzyme, CCBL, to a highly reactive and nephrotoxic thiol that would bind to proteins. The cytosolic glutamine transaminase K and especially the mitochondrial aspartate aminotransferase are the major CCBLs involved in the bioactiviation of cisplatin-cysteine-conjugate. GST, glutathione transferase; GGT, gamma glutamyl transpeptidase; APN, aminopeptidase N; CCBL, cysteine-S-conjugate beta-lyase.
As the cisplatin-GSH-conjugate passes through the kidney by the multidrug resistance-associated protein 2, it is cleaved by gamma glutamyl transpeptidase (GGT) to a cysteinyl-glycine-conjugate, which is further converted to a cysteine-conjugate by aminopeptidase N (APN). Both GGT (Hanigan & Pitot, 1985) and APN (Jardinaud et al., 2004) are abundantly expressed on the luminal surface of the renal proximal tubule epithelial cells, and both reactions happen extracellularly. GGT is a plasma membrane-bound enzyme with the active side directed into the extracellular space, and it is the first enzyme in the metabolic pathway for the activation of cisplatin-GSH-conjugate. Kidney has the highest GGT activity (Hanigan & Pitot, 1985), and the activity of GGT has been shown to be essential for the cisplatin nephrotoxicity. Pretreatment with GGT inhibitor acivicin in rats and mice, or knockout of Ggt genes in mice significantly protected the animals from the cisplatin-induced renal toxicity without reducing platinum concentration in the kidney. The protective effect was confirmed by BUN, and Cr levels, and quantitative histologic analysis (Hanigan et al., 1994; Hanigan et al., 2001; Tanase et al., 2019; Townsend & Hanigan, 2002). In line with these results, in vitro data showed that pharmacological inhibition of GGT by OU749 or acivicin in renal proximal tubular epithelial cells (RPTEC-SV40 or LLC-PK1) eliminated the toxicities of cisplatin and cisplatin-GSH-conjugate, but had no significant effect on the toxicity of cisplatin-cysteinyl-glycine-conjugate (Fliedl et al., 2014; Townsend, Deng, Zhang, Lapus, & Hanigan, 2003). In addition, high doses of GSH have been found to reduce nephrotoxicity In vivo (Hanigan et al., 1994), but the biochemical mechanism of this protective action is not fully understood. GSH is the major physiological substrate for GGT (Hanigan & Ricketts, 1993). High concentration of GSH may protect against cisplatin nephrotoxicity by serving as a competitive inhibitor of GGT activity (Hanigan et al., 1994). Consistently, NOV-002, a mimetic of glutathione disulfide (GSSG), has been shown to be an effective protector against cisplatin-induced kidney toxicity in mice possibly through its ability to act as a competitive substrate for GGT ( Jenderny, Lin, Garrett, Tew, & Townsend, 2010). Intriguingly, GGT-negative prostate cancer xenografts grew at less than twice the rate of the GGT-positive tumors, and were significantly more sensitive to cisplatin toxicity than GGT-positive tumors, suggesting that GGT inhibition may reduce the nephrotoxicity of cisplatin, yet enhance its antitumor effect (Hanigan, Gallagher, Townsend, & Gabarra, 1999). Taken together, GGT was considered as a good target for the attenuation of cisplatin nephrotoxicity. GSH is the most abundant cellular antioxidant preventing protein thiols from oxidation either directly or indirectly through catalytic systems such as glutathione peroxidases (Ye, Zhang, Townsend, & Tew, 2015; Zhang, Ye, Singh, Townsend, & Tew, 2018). NOV-002 was shown to decrease GSH/GSSG ratio, increase oxidative stress and increase S-glutathionylation of protein thiols, thus influences multiple redox pathways critical to the maintenance of cell viability (Townsend et al., 2008; Townsend, Pazoles, & Tew, 2008; Townsend & Tew, 2009; Uys et al., 2010). The specific role of thiol homeostasis in the cisplatin nephrotoxicity remains to be established.
APN, like GGT, is a membrane associated enzyme with the catalytic domain exposed to the extracellular surface, and is the key enzyme immediately downstream in the GGT toxification pathway (Hausheer et al., 2011). Many peptides and small molecules, including GSH and cysteinyl-glycine dipeptides, have been shown to inhibit APN (Bauvois & Dauzonne, 2006; Hausheer et al., 2011). Accordingly, in human proximal tubular epithelial HK-2 cells, both GSH and cysteinyl-glycine have been shown to be able to ameliorate cisplatin toxicity (Paolicchi et al., 2003). In addition, Hausheer and colleagues proposed that the cisplatin nephroprotection effect of the disulfide BNP7787 (disodium 2,2′-dithio-bis ethane sulfonate, dimesna, Tavocept™) could result from the inhibition of GGT activity, mediated by BNP7787-dervied mesna-disulfide heteroconjugates that contain a terminal gamma-glutamate moiety (e.g., mesna-GSH, and mesna-cyteinyl-glutamate) (Hausheer et al., 2010), and/or the inhibition of APN activity by mesnacysteine, mesna-GSH, and mesna-cysteinyl-glycine (Hausheer et al., 2011). BNP7787 can act as a pharmacological surrogate/modulator of physiological thiols and disulfides and its active pharmacophore, mesna, was able to attach to cisplatin rendering it non-toxic since the mesna-cisplatin conjugate is not the substrate for GGT toxification (Hausheer et al., 2010). Due to the capacity to prevent the formation of a GSH-conjugates, thiol-generating agents have been tested as nephroprotective drugs in cisplatin-treated patients. However, their clinical values have been limited by the unwanted tumor protecting effect as well as their own intrinsic toxicities. The only agent that is approved for prevention of cisplatin nephrotoxicity in patients with non-small cell lung cancer and advanced ovarian cancer is amifostine (ethanethiol, 2-[(3-aminopropyl) amino] dihydrogen phosphate ester). However, its administration is associated with several potentially serious side effects, including ototoxicity and hypotension (Hausheer et al., 2011; Ridzuan et al., 2019).
As the cisplatin-cysteine-conjugate enters the proximal tubular epithelial cells, it is further metabolized by a pyridoxal 5′-phosphate (PLP)-dependent enzyme, cysteine-S-conjugate beta-lyase (CCBL), to a highly reactive and nephrotoxic thiol that would bind to proteins. Mammalian tissues contain at least eleven PLP-dependent enzymes with CCBL activity (Cooper et al., 2011; Cooper & Pinto, 2006). Among them, overexpression of the cytosolic glutamine transaminase K and the mitochondrial aspartate aminotransferase (mitAAT) has been shown to significantly increase the cisplatin toxicity in confluent LLC-PK1 cells (Zhang et al., 2006; Zhang & Hanigan, 2003). Consistently, inhibition of the CCBL activities by aminooxyacetic acid (AOAA), a general inhibitor of PLP-containing enzymes, led to amelioration of cisplatin nephrotoxicity both in vitro and in vivo (Townsend, Deng, et al., 2003; Townsend & Hanigan, 2002; Zhang et al., 2006). In the kidney, the proximal tubules are targeted by cisplatin and mitochondria are especially vulnerable. In mice pretreated with AOAA, platination of renal proteins was decreased in mitochondria, but not in the cytosol, suggesting that mitAAT is a major CCBL involved in the bioactiviation of cisplatin-cysteine-conjugate. The highly reactive cisplatin-thiol compound rapidly binds to the sulfhydryl moieties of the proteins in proximity in mitochondria, such as alpha-ketoglutarate dehydrogenase and to a lesser extent aconitase, and the deactivation of these proteins may contribute to the cisplatin-induced mitochondrial dysfunction of energy metabolism in kidney cells (Cooper et al., 2011; Zhang et al., 2006). Unlike GGT and APN, mitAAT is widely distributed among various organs. Liver, heart, brain, kidney and skeletal muscles exhibit high levels of mitAspAT activity (Parli, Godfrey, & Ross, 1987). Therefore, the nephrotoxicity of cisplatin cannot be explained on the basis of the tissue expression of this enzyme.
4. Oxidative stress in cisplatin-induced nephrotoxicity
Oxidative stress is a hallmark of cisplatin-induced nephrotoxicity, and it is characterized as an imbalance between the production and destruction of reactive oxygen species (ROS), the latter is regulated by antioxidant defenses. The term ROS refers to a number of chemically reactive molecules derived from O2, and the most common and biologically important ROS are O2− (superoxide anions), H2O2 (hydrogen peroxide) and OH• (hydroxyl radicals). Of these, OH• is invariably toxic, O2− is a byproduct of mitochondrial oxidation reactions and H2O2 has evolved into an important intermediary signaling molecule. Primary sites of intracellular ROS include the mitochondrial electron transport chain (ETC), ER and NADPH oxidase (NOX) complex. Antioxidant enzymes include superoxide dismutase (SOD), catalase, glutathione peroxidase (GPx), glutathione reductase (GR), peroxiredoxin (Prx), GST, glutaredoxin (Grx), thioredoxin (Trx), and thioredoxin reductase (TrxR). Low molecular weight cofactor, such as GSH and NADPH also play an important role in the antioxidant defense. In addition, selenium has a critical role in maintaining antioxidant defenses (Ye et al., 2015).
A substantial source of ROS production is from mitochondria, the organelle responsible for producing ATP. Cisplatin accumulation in mitochondria is favored as the positively charged hydrolysis product of cisplatin is preferentially attracted by the negatively charged mitochondria, and this phenomenon may partly explain the particular sensitivity of the renal proximal tubule to cisplatin toxicity, as this segment exhibits one of the highest densities of mitochondria in the kidney (Gomez-Sierra et al., 2018). Following cisplatin administration, mitochondrial ROS levels significantly increase through different mechanisms which contribute to nephrotoxicity (Hanigan & Devarajan, 2003). First of all, cisplatin, as an electrophile, can directly disrupt the action of mitochondrial complexes, leading to increased production of ROS. In addition, recent studies have focused on the indirect effects of cisplatin on mitochondrial ROS generation. Mitochondria require narrow parameters to maintain their function, and reduction of mitochondrial function would result in increased ROS production and decreased ATP production. For example, cisplatin binds to mitochondrial DNA which blocks transcription, thus leading to decreased ETC protein expression, impaired respiration, and enhanced ROS generation (Marullo et al., 2013). Increased ROS formation promotes cell death and further damages respiratory complexes, thus enhancing cellular damage (Volarevic et al., 2019; Waseem &Parvez, 2013). In addition, cisplatin disrupts mitochondrial bioenergetics through inhibition of PPARα-mediated expression of genes involved in fatty acid metabolism. Fatty acid oxidation is the major source of energy for the renal proximal tubule, and treatment of PPARα agonists has been reported to reduce cisplatin nephrotoxicity in vivo (Gomez-Sierra et al., 2018). Another important effect of cisplatin on mitochondrial bioenergetics is the blockage of mitochondrial glutamate oxaloacetate transaminase (Ozaki et al., 2013), which catalyzes the conversion of glutamate and oxaloacetate with aspartate and α-ketoglutarate, and plays an important role in NADH (the main electron donor for oxidative phosphorylation) entry into the mitochondria. Cisplatin is able to inhibit mitochondrial transcription factor A by upregulating miR-709 (Guo et al., 2018), and inhibit the nuclear translocation of estrogen receptor alpha, an orphan nuclear receptor that acts as a transcription factor to regulate the transcription of genes required for mitochondrial biogenesis and oxidative phosphorylation (Tsushida et al., 2018). Furthermore, cisplatin increases mitochondrial ROS accumulation by decreasing mitochondrial GSH levels, which may result from GSH oxidation or decreased GR activity (Volarevic et al., 2019).
Cisplatin also stimulates ROS formation mediated by NOX enzymes. O2− production significantly increased in glomeruli and proximal tubules after cisplatin treatment, and the enhancement has been shown to be associated with the increased expression of the NOX subunits gp91 (phox) and p47 (phox). In agreement with this phenomenon, the enhanced O2− production has been shown to be prevented by the NOX2 selective inhibitor diphenylene iodonium (Trujillo et al., 2015). Among NOX1—5, NOX1 was found to have a nuclear location with plasma membrane and cytoplasmic distribution, NOX2, 3, and 5 are detected at the plasma membrane, and NOX4 has been identified in nucleus and ER (Brown & Griendling, 2009). NOX4 is highly expressed in the kidney and its expression is significantly upregulated in cisplatin-induced acute kidney injury model. NOX4 aggravates cisplatin-induced nephrotoxicity by promoting ROS-mediated programmed cell death and inflammation, and disruption of NOX4 has been shown to contribute to renal function recovery and kidney damage relief (Meng et al., 2018).
Cytochrome P450 (CYP) is an ER-anchored family of heme-containing monooxygenases that can produce O2− and H2O2, and can cause the release of catalytic iron, and through Fenton reactions to the generation of potent oxidants. Among the isoforms, CYP2E1 has been identified and localized to the kidney proximal tubules. CYP2E1 null mice demonstrated marked functional and histological protection against cisplatin-induced renal injury as compared to wild-type mice (Liu & Baliga, 2003). In addition, renal proximal tubular epithelial cells that were transfected with anti-caspase 12 (an ER-specific caspase) antibody significantly attenuated cisplatin-induced apoptosis. It is proposed that the interaction of cisplatin with CYP2E1 promotes the generation of ROS leading to activation of caspase 12 and subsequent apoptosis. (Liu & Baliga, 2005; Quintanilha et al., 2017). Oxidative stress is widely known to be related to ER stress as ROS may disrupt proper protein folding (Cao & Kaufman, 2014). Indeed, cisplatin has been shown to induce ER stress and unfolded protein response which triggers cell death (Peyrou, Hanna, & Cribb, 2007). ER stress in turn further increases ROS production in ER, mitochondria, and NOX system. Oxidative protein folding is another important resource of ROS production in ER. After accepting electrons from protein disulfide isomerases, ER oxidoreductase 1 transfers the electrons to O2 and produces H2O2 in the ER lumen (Ye et al., 2015). In addition, ER stress promotes Ca2+ release from ER into cytosol. Ca2+ is taken by mitochondria, resulting in mitochondrial transition pore opening, cytochrome c release, improper respiration and enhanced ROS production (Cao & Kaufman, 2014). Moreover, cytosolic Ca2+ is able to activate calcium/calmodulin-dependent kinase II, which stimulates NOX-mediated ROS production (Pandey, Gratton, Rafikov, Black, & Fulton, 2011).
As evidenced above, cisplatin, through multiple pathways, promotes ROS production, and the main downstream consequence of uncontrolled ROS accumulation is cell death. ROS play a central role in apoptosis induction mediated by intrinsic mitochondrial dysfunction, ER stress, and extrinsic death receptor activation. In addition to the processes described above, cisplatin-induced ROS has been reported to be involved in the activation of p53, the master regulator of apoptosis, leading to enhanced expression of Bax and PUMA (p53 upregulated modulator of apoptosis, also known as Bcl-2-binding component 3), which participate in mitochondrial pore formation and subsequent cytochrome c release, as well as upregulation of genes that encode for the extrinsic apoptosis protein Fas, FasL and TNFα (tumor necrosis factor-alpha). The latter is also the main driver for the development of renal inflammation after cisplatin treatment (Volarevic et al., 2019). ROS have also been proposed to promote cytochrome c release by direct oxidation of cardiolipin, a mitochondrial phospholipid that binds to cytochrome c (Santos et al., 2008). Therefore, oxidative stress is one of the best characterized mechanisms for cisplatin nephrotoxicity and is closely related with other mechanisms of cisplatin nephrotoxicity, such as mitochondrial dysfunction, ER stress, inflammatory responses, and apoptosis (Fig. 3).
Fig. 3.

Summary of the major pathways involved in cisplatin nephrotoxicity. Cisplatin, through multiple pathways, promotes ROS production in mitochondria, ER and NOX system, and the main downstream consequence of uncontrolled ROS accumulation (oxidative stress) is cell death. Oxidative stress is one of the best characterized mechanisms for cisplatin nephrotoxicity and is closely related with other mechanisms of cisplatin nephrotoxicity. For example, oxidative stress plays a central role in apoptosis induction mediated by intrinsic mitochondrial dysfunction, and ER stress. In addition, cisplatin-induced oxidative stress has been reported to be involved in the activation of p53, leading to enhanced expression of Bax and PUMA, which participate in mitochondrial pore formation and subsequent cytochrome c release, as well as upregulation of genes that encode for the extrinsic apoptosis protein Fas, FasL and TNFα, the latter is also the main driver for the development of renal inflammation after cisplatin treatment. AIF, apoptosis inducing factor; DAMPs, danger-associated molecular pattern molecules; ER, endoplasmic reticulum; PUMA, p53 upregulated modulator of apoptosis, also known as Bcl-2-binding component 3; TLR, toll-like receptor; TNFα, tumor necrosis factor-alpha.
5. Antioxidants against cisplatin-induced nephrotoxicity
Unchecked production of ROS is a major contributor to cisplatin-induced nephrotoxicity, making antioxidant-based therapies a popular choice for therapeutic intervention. Several natural antioxidant compounds have been shown to protect against cisplatin-induced kidney toxicity (Dasari & Tchounwou, 2014). Both in vivo and in vitro studies demonstrated that extracts from different types of ginseng (American ginseng (Ma et al., 2017; Ma et al., 2017), Chinese ginseng (Liu et al., 2014), Korean black ginseng (Han et al., 2016; Jung, An, Eom, Kang, & Kim, 2017), and Korean red ginseng (Kim et al., 2014)) and active constituents of ginseng (ginsenosides (Baek, Shin, Kim, Chang, & Park, 2017; Li et al., 2016), pseudoginsenoside (Wang et al., 2014) and anthocyanin (Qi et al., 2017)) are able to protect against cisplatin-induced nephrotoxicity by restoring redox balance and exerting anti-apoptotic and anti-inflammatory effect, as evidenced by decreased ROS generation and lipid peroxidation, increased GSH and antioxidant enzyme (catalase and SOD) levels, restored Bcl2: Bax ratio, and suppressed activation of p53, caspase 3 and 9, NFκB, IL1β, COX2 and iNOS. Similarly, licorice extract and its active components (glycyrrhizic acid, 18β-glycyrrhizic acid, and isoliquiritigenin) (Arjumand & Sultana, 2011; Ju et al., 2017; Patricia Moreno-Londono, Bello-Alvarez, & Pedraza-Chaverri, 2017; Wu, Chen, & Yen, 2015), curcumin (the most active and major phytochemical found in the rhizome of turmeric) (Kumar, Barua, Sulakhiya, & Sharma, 2017; Sahin et al., 2014; Topcu-Tarladacalisir, Sapmaz-Metin, & Karaca, 2016; Ueki, Ueno, Morishita, & Maekawa, 2013; Waseem & Parvez, 2013), grape seed proanthocyanidin extract (Hassan, Edrees, El-Gamel, & El-Sayed, 2014; Saad, Youssef, & El-Shennawy, 2009), honey (Hamad et al., 2015; Huang et al., 2017; Ibrahim, Eldaim, & Abdel-Daim, 2016), and pomegranate (flower, rind and seed) extract (Bakir et al., 2015; Boroushaki et al., 2015; Karwasra et al., 2016; Motamedi et al., 2014) have the potential to attenuate the nephrotoxic side effects of cisplatin through reduction of oxidative stress, mitochondrial apoptosis and inflammation responses. Among these natural products, the renal protective effect of grape seed proanthocyanidin has also been linked to inhibition of ER stress-induced apoptosis (caspase 12 activation) (Gao et al., 2014). In addition, curcumin is able to prevent cisplatin-induced renal alterations in transporter expression in mitochondrial bioenergetics, ultrastructure and dynamic (Ortega-Dominguez et al., 2017). Curcumin has also been reported to exert protection when used as a pre-treatment or co-treatment with cisplatin therapy, but not as a post-treatment (Kumar et al. ,2017). Amelioration of cisplatin-induced nephrotoxicity by pomegranate flower extract is gender-related, although reasons for this are not clear (Jilanchi et al., 2014; Motamedi et al., 2014).
Several food-derived antioxidants are known to have bifunctionality, since they not only scavenge ROS, but also increase GSH content, as well as enhance the expression and activities of antioxidant enzymes (catalase, GPx, GR, GST, HO-1, NQO1, SOD, and TrxR) through Nrf2. Among these dietary antioxidants, capsaicin, ellagic acid, epigallocatechin-3-O-gallate, α-lipoic acid, lycopene, quercetin, resveratrol, sulforaphane, tannins, and vitamin B2, C, and E, have been reported to not only ameliorate cisplatin-induced renal damage through their antioxidant and anti-inflammatory effects, but also potentiate the antineoplastic effects of cisplatin by mechanisms such as downregulation of drug efflux transporters, and upregulation of p53 protein in cancer cells, as well as reduction of cancer stem cell population, generally accepted to be important for drug resistance and rumor relapse (Dasari & Tchounwou, 2014; Miller et al., 2010; Volarevic et al., 2019).
Based on all these preclinical studies, dietary intervention with antioxidants from natural products or food seems to be a favorable approach to prevent cisplatin-induced kidney damage without compromising its antitumor activities (Kumar et al., 2017; Patricia Moreno-Londono et al., 2017; Volarevic et al., 2019; Wang et al., 2014). However, current animal models used in cisplatin-induced renal damage research have several limitations, e.g., the use of animals that do not present cancer or are young, as well as models that induce acute not chronic kidney disease, which has greater clinical relevance (Volarevic et al., 2019). Some effects have been made to demonstrate the beneficial effect of antioxidants as an adjuvant therapy in humans but to date, these have not been comprehensive. Daily honey consumption by cancer patients has been shown to significantly reduce BUN and Cr levels during cisplatin treatment (Osama et al., 2017). Lycopene has been reported to improve renal functions (GFR) in cancer patients under cisplatin therapy (Mahmoodnia, Mohammadi, & Masumi, 2017). In addition, in patients with reduced: oxidized vitamin C ratios >7.5, vitamin supplementation (vitamin C, vitamin E and selenium) attenuated cisplatin-induced loss of renal function (Weijl et al., 2004). At this stage, more clinical approaches are still needed to support these types of “natural antioxidant” interventions as a way of reducing cisplatin toxicities.
6. Conclusion and future direction
Cisplatin combines an unusual clinical pharmacology, with bifunctional alkylating functions sharing heavy metal characteristics. With the halogens acting as leaving groups, electrophilic intermediates are free to interact with an extensive range of cellular nucleophiles. Given that there is more than 40 years of clinical experience dealing with the side effects of cisplatin, there is a certain degree of familiarity with how patients under treatment should be managed. Unusually, detoxification of the drug with GSH systems contributes to kidney toxicity and the obvious importance of redox pathways in its metabolism do provide a fascinating background to view pharmacology of other metal containing alkylating agents, including analogues.
Abbreviation
- AOAA
aminooxyacetic acid
- APN
aminopeptidase N
- BUN
blood urea nitrogen
- CCBL
cysteine-S-conjugate beta-lyase
- Cr
creatinine
- Ctr1
copper transporter 1
- CYP
cytochrome P450
- ER
endoplasmic reticulum
- ETC
electron transport chain
- GFR
glomerular filtration rate
- GGT
gamma glutamyl transpeptidase
- GPx
glutathione peroxidase
- GR
glutathione reductase
- Grx
glutaredoxin
- GSH
glutathione
- GSSG
glutathione disulfide
- GST
glutathione S-transferase
- MATE1
multidrug and toxin extrusion transporter 1
- mitAAT
mitochondrial aspartate aminotransferase
- NOX
NADPH oxidase
- OCT2
organic cation transporter 2
- PLP
pyridoxal 5′-phosphate
- Prx
peroxiredoxin
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- Trx
thioredoxin
- TrxR
thioredoxin reductase
References
- Arjumand W, & Sultana S (2011). Glycyrrhizic acid: A phytochemical with a protective role against cisplatin-induced genotoxicity and nephrotoxicity. Life Sciences, 89, 422–429. [DOI] [PubMed] [Google Scholar]
- Baek SH, Shin BK, Kim NJ, Chang SY, & Park JH (2017). Protective effect of ginsenosides Rk3 and Rh4 on cisplatin-induced acute kidney injury in vitro and in vivo. Journal of Ginseng Research, 41, 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakir S, Yazgan UC, Ibiloglu I, Elbey B, Kizil M, & Kelle M (2015). The protective effect of pomegranate extract against cisplatin toxicity in rat liver and kidney tissue. Archives of Physiology and Biochemistry, 121, 152–156. [DOI] [PubMed] [Google Scholar]
- Bauvois B, & Dauzonne D (2006). Aminopeptidase-N/CD13 (EC 3.4.11.2) inhibitors: Chemistry, biological evaluations, and therapeutic prospects. Medicinal Research Reviews, 26, 88–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroushaki MT, Rajabian A, Farzadnia M, Hoseini A, Poorlashkari M, Taghavi A, et al. (2015). Protective effect of pomegranate seed oil against cisplatin-induced nephrotoxicity in rat. Renal Failure, 37, 1338–1343. [DOI] [PubMed] [Google Scholar]
- Brown DI, & Griendling KK (2009). Nox proteins in signal transduction. Free Radical Biology & Medicine, 47, 1239–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao SS, & Kaufman RJ (2014). Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxidants & Redox Signaling, 21, 396–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciarimboli G, Deuster D, Knief A, Sperling M, Holtkamp M, Edemir B, et al. (2010). Organic cation transporter 2 mediates cisplatin-induced oto- and nephrotoxicity and is a target for protective interventions. The American Journal of Pathology, 176, 1169–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AJ, Krasnikov BF, Niatsetskaya ZV, Pinto JT, Callery PS, Villar MT, et al. (2011). Cysteine S-conjugate beta-lyases: Important roles in the metabolism of naturally occurring sulfur and selenium-containing compounds, xenobiotics and anticancer agents. Amino Acids, 41, 7–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AJ, & Pinto JT (2006). Cysteine S-conjugate beta-lyases. Amino Acids, 30, 1–15. [DOI] [PubMed] [Google Scholar]
- Dasari S, & Tchounwou PB (2014). Cisplatin in cancer therapy: Molecular mechanisms of action. European Journal of Pharmacology, 740, 364–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca A, Parker LJ, Ang WH, Rodolfo C, Gabbarini V, Hancock NC, et al. (2019). A structure-based mechanism of cisplatin resistance mediated by glutathione transferase P1–1. Proceedings of the National Academy of Sciences of the United States of America, 116, 13943–13951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrela GR, Wasinski F, Felizardo RJF, Souza LL, Camara NOS, Bader M, et al. (2017). MATE-1 modulation by kinin B1 receptor enhances cisplatin efflux from renal cells. Molecular and Cellular Biochemistry, 428, 101–108. [DOI] [PubMed] [Google Scholar]
- Filipski KK, Mathijssen RH, Mikkelsen TS, Schinkel AH, & Sparreboom A (2009). Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clinical Pharmacology and Therapeutics, 86, 396–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliedl L, Wieser M, Manhart G, Gerstl MP, Khan A, Grillari J, et al. (2014). Controversial role of gamma-glutamyl transferase activity in cisplatin nephrotoxicity. ALTEX, 31, 269–278. [DOI] [PubMed] [Google Scholar]
- Gao Z, Liu G, Hu Z, Li X, Yang X, Jiang B, et al. (2014). Grape seed pro-anthocyanidin extract protects from cisplatin-induced nephrotoxicity by inhibiting endoplasmic reticulum stress-induced apoptosis. Molecular Medicine Reports, 9, 801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George B, You D, Joy MS, & Aleksunes LM (2017). Xenobiotic transporters and kidney injury. Advanced Drug Delivery Reviews, 116, 73–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Sierra T, Eugenio-Perez D, Sanchez-Chinchillas A, & Pedraza-Chaverri J (2018). Role of food-derived antioxidants against cisplatin induced-nephrotoxicity. Food and Chemical Toxicology, 120, 230–242. [DOI] [PubMed] [Google Scholar]
- Guo Y, Ni J, Chen S, Bai M, Lin J, Ding G, et al. (2018). MicroRNA-709 mediates acute tubular injury through effects on mitochondrial function. Journal of the American Society of Nephrology, 29, 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakiminia B, Goudarzi A, & Moghaddas A (2019). Has vitamin E any shreds of evidence in cisplatin-induced toxicity. Journal of Biochemical and Molecular Toxicology, 33, e22349. [DOI] [PubMed] [Google Scholar]
- Hamad R, Jayakumar C, Ranganathan P, Mohamed R, El-Hamamy MM, Dessouki AA, et al. (2015). Honey feeding protects kidney against cisplatin nephrotoxicity through suppression of inflammation. Clinical and Experimental Pharmacology & Physiology, 42, 843–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MS, Han IH, Lee D, An JM, Kim SN, Shin MS, et al. (2016). Beneficial effects of fermented black ginseng and its ginsenoside 20(S)-Rg3 against cisplatin-induced nephrotoxicity in LLC-PK1 cells. Journal of Ginseng Research, 40, 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanigan MH, & Devarajan P (2003). Cisplatin nephrotoxicity: Molecular mechanisms. Cancer Therapy, 1, 47–61. [PMC free article] [PubMed] [Google Scholar]
- Hanigan MH, Gallagher BC, & Taylor PT Jr. (1996). Cisplatin nephrotoxicity: Inhibition of gamma-glutamyl transpeptidase blocks the nephrotoxicity of cisplatin without reducing platinum concentrations in the kidney. American Journal of Obstetrics and Gynecology, 175, 270–273, (discussion 273–274). [DOI] [PubMed] [Google Scholar]
- Hanigan MH, Gallagher BC, Taylor PT Jr., & Large MK (1994). Inhibition of gamma-glutamyl transpeptidase activity by acivicin in vivo protects the kidney from cisplatin-induced toxicity. Cancer Research, 54, 5925–5929. [PubMed] [Google Scholar]
- Hanigan MH, Gallagher BC, Townsend DM, & Gabarra V (1999). Gamma-glutamyl transpeptidase accelerates tumor growth and increases the resistance of tumors to cisplatin in vivo. Carcinogenesis, 20, 553–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanigan MH, Lykissa ED, Townsend DM, Ou CN, Barrios R, & Lieberman MW (2001). Gamma-glutamyl transpeptidase-deficient mice are resistant to the nephrotoxic effects of cisplatin. The American Journal of Pathology, 159, 1889–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanigan MH, & Pitot HC (1985). Gamma-glutamyl transpeptidase—Its role in hepatocarcinogenesis. Carcinogenesis, 6, 165–172. [DOI] [PubMed] [Google Scholar]
- Hanigan MH, & Ricketts WA (1993). Extracellular glutathione is a source of cysteine for cells that express gamma-glutamyl transpeptidase. Biochemistry, 32, 6302–6306. [DOI] [PubMed] [Google Scholar]
- Hassan HA, Edrees GM, El-Gamel EM, & El-Sayed EA (2014). Amelioration of cisplatin-induced nephrotoxicity by grape seed extract and fish oil is mediated by lowering oxidative stress and DNA damage. Cytotechnology, 66, 419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausheer FH, Parker AR, Petluru PN, Jair KW, Chen S, Huang Q, et al. (2011). Mechanistic study of BNP7787-mediated cisplatin nephroprotection: Modulation of human aminopeptidase N. Cancer Chemotherapy and Pharmacology, 67, 381–391. [DOI] [PubMed] [Google Scholar]
- Hausheer FH, Shanmugarajah D, Leverett BD, Chen X, Huang Q, Kochat H, et al. (2010). Mechanistic study of BNP7787-mediated cisplatin nephroprotection: Modulation of gamma-glutamyl transpeptidase. Cancer Chemotherapy and Pharmacology, 65, 941–951. [DOI] [PubMed] [Google Scholar]
- Holditch SJ, Brown CN, Lombardi AM, Nguyen KN, & Edelstein CL (2019). Recent advances in models, mechanisms, biomarkers, and interventions in cisplatin-induced acute kidney injury. International Journal of Molecular Sciences, 20, 3011–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer AK, Samimi G, Katano K, Naerdemann W, Lin X, Safaei R, et al. (2004). The copper influx transporter human copper transport protein 1 regulates the uptake of cisplatin in human ovarian carcinoma cells. Molecular Pharmacology, 66, 817–823. [DOI] [PubMed] [Google Scholar]
- Homolya L, Varadi A, & Sarkadi B (2003). Multidrug resistance-associated proteins: Export pumps for conjugates with glutathione, glucuronate or sulfate. BioFactors, 17, 103–114. [DOI] [PubMed] [Google Scholar]
- Huang H, Shen Z, Geng Q, Wu Z, Shi P, & Miao X (2017). Protective effect of Schisandra chinensis bee pollen extract on liver and kidney injury induced by cisplatin in rats. Biomedicine & Pharmacotherapy, 95, 1765–1776. [DOI] [PubMed] [Google Scholar]
- Ibrahim A, Eldaim MA, & Abdel-Daim MM (2016). Nephroprotective effect of bee honey and royal jelly against subchronic cisplatin toxicity in rats. Cytotechnology, 68, 1039–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardinaud F, Banisadr G, Noble F, Melik-Parsadaniantz S, Chen H, Dugave C, et al. (2004). Ontogenic and adult whole body distribution of aminopeptidase N in rat investigated by in vitro autoradiography. Biochimie, 86, 105–113. [DOI] [PubMed] [Google Scholar]
- Jenderny S, Lin H, Garrett T, Tew KD, & Townsend DM (2010). Protective effects of a glutathione disulfide mimetic (NOV-002) against cisplatin induced kidney toxicity. Biomedicine & Pharmacotherapy, 64, 73–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jilanchi S, Nematbakhsh M, Mazaheri S, Talebi A, Zolfaghari B, Pezeshki Z, et al. (2014). Pomegranate flower extract does not prevent cisplatin-induced nephrotoxicity in female rats. International Journal of Preventive Medicine, 5, 1621–1625. [PMC free article] [PubMed] [Google Scholar]
- Ju SM, Kim MS, Jo YS, Jeon YM, Bae JS, Pae HO, et al. (2017). Licorice and its active compound glycyrrhizic acid ameliorates cisplatin-induced nephrotoxicity through inactivation of p53 by scavenging ROS and overexpression of p21 in human renal proximal tubular epithelial cells. European Review for Medical and Pharmacological Sciences, 21, 890–899. [PubMed] [Google Scholar]
- Jung K, An JM, Eom DW, Kang KS, & Kim SN (2017). Preventive effect of fermented black ginseng against cisplatin-induced nephrotoxicity in rats. Journal of Ginseng Research, 41, 188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karwasra R, Kalra P, Gupta YK, Saini D, Kumar A, & Singh S (2016). Antioxidant and anti-inflammatory potential of pomegranate rind extract to ameliorate cisplatin-induced acute kidney injury. Food & Function, 7, 3091–3101. [DOI] [PubMed] [Google Scholar]
- Kim YJ, Lee MY, Son HY, Park BK, Ryu SY, & Jung JY (2014). Red ginseng ameliorates acute cisplatin-induced nephropathy. Planta Medica, 80, 645–654. [DOI] [PubMed] [Google Scholar]
- Knight TR, Choudhuri S, & Klaassen CD (2007). Constitutive mRNA expression of various glutathione S-transferase isoforms in different tissues of mice. Toxicological Sciences, 100, 513–524. [DOI] [PubMed] [Google Scholar]
- Koepsell H, Lips K, & Volk C (2007). Polyspecific organic cation transporters: Structure, function, physiological roles, and biopharmaceutical implications. Pharmaceutical Research, 24, 1227–1251. [DOI] [PubMed] [Google Scholar]
- Kumar P, Barua CC, Sulakhiya K, & Sharma RK (2017). Curcumin ameliorates cisplatin-induced nephrotoxicity and potentiates its anticancer activity in SD rats: Potential role of curcumin in breast cancer chemotherapy. Frontiers in Pharmacology, 8, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Yan MH, Liu Y, Liu Z, Wang Z, Chen C, et al. (2016). Ginsenoside Rg5 ameliorates cisplatin-induced nephrotoxicity in mice through inhibition of inflammation, oxidative stress, and apoptosis. Nutrients, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, & Baliga R (2003). Cytochrome P450 2E1 null mice provide novel protection against cisplatin-induced nephrotoxicity and apoptosis. Kidney International, 63, 1687–1696. [DOI] [PubMed] [Google Scholar]
- Liu H, & Baliga R (2005). Endoplasmic reticulum stress-associated caspase 12 mediates cisplatin-induced LLC-PK1 cell apoptosis. Journal of the American Society of Nephrology, 16, 1985–1992. [DOI] [PubMed] [Google Scholar]
- Liu X, Huang Z, Zou X, Yang Y, Qiu Y, & Wen Y (2014). Panax notoginseng saponins attenuates cisplatin-induced nephrotoxicity via inhibiting the mitochondrial pathway of apoptosis. International Journal of Clinical and Experimental Pathology, 7, 8391–8400. [PMC free article] [PubMed] [Google Scholar]
- Ma ZN, Li YZ, Li W, Yan XT, Yang G, Zhang J, et al. (2017). Nephroprotective effects of saponins from leaves of Panax quinquefolius against cisplatin-induced acute kidney injury. International Journal of Molecular Sciences, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma ZN, Liu Z, Wang Z, Ren S, Tang S, Wang YP, et al. (2017). Supplementation of American ginseng berry extract mitigated cisplatin-evoked nephrotoxicity by suppressing ROS-mediated activation of MAPK and NF-kappaB signaling pathways. Food and Chemical Toxicology, 110, 62–73. [DOI] [PubMed] [Google Scholar]
- Mahmoodnia L, Mohammadi K, & Masumi R (2017). Ameliorative effect of lycopene effect on cisplatin-induced nephropathy in patient. Journal of Nephropathology, 6, 144–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manohar S, & Leung N (2018). Cisplatin nephrotoxicity: A review of the literature. Journal of Nephrology, 31, 15–25. [DOI] [PubMed] [Google Scholar]
- Marullo R, Werner E, Degtyareva N, Moore B, Altavilla G, Ramalingam SS, et al. (2013). Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS One, 8, e81162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer RD, Lee KE, & Cockett AT (1987). Inhibition of cisplatin-induced nephrotoxicity in rats by buthionine sulfoximine, a glutathione synthesis inhibitor. Cancer Chemotherapy and Pharmacology, 20, 207–210. [DOI] [PubMed] [Google Scholar]
- Meng XM, Ren GL, Gao L, Yang Q, Li HD, Wu WF, et al. (2018). NADPH oxidase 4 promotes cisplatin-induced acute kidney injury via ROS-mediated programmed cell death and inflammation. Laboratory Investigation, 98, 63–78. [DOI] [PubMed] [Google Scholar]
- Miller RP, Tadagavadi RK, Ramesh G, & Reeves WB (2010). Mechanisms of cisplatin nephrotoxicity. Toxins (Basel), 2, 2490–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motamedi F, Nematbakhsh M, Monajemi R, Pezeshki Z, Talebi A, Zolfaghari B, et al. (2014). Effect of pomegranate flower extract on cisplatin-induced nephrotoxicity in rats. Journal of Nephropathology, 3, 133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraki K, Koyama R, Honma Y, Yagishita S, Shukuya T, Ohashi R, et al. (2012). Hydration with magnesium and mannitol without furosemide prevents the nephrotoxicity induced by cisplatin and pemetrexed in patients with advanced non-small cell lung cancer. Journal of Thoracic Disease, 4, 562–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Yonezawa A, Hashimoto S, Katsura T, & Inui K (2010). Disruption of multidrug and toxin extrusion MATE1 potentiates cisplatin-induced nephrotoxicity. Biochemical Pharmacology, 80, 1762–1767. [DOI] [PubMed] [Google Scholar]
- Nematbakhsh M, Ebrahimian S, Tooyserkani M, Eshraghi-Jazi F, Talebi A, & Ashrafi F (2013). Gender difference in csplatin-induced nephrotoxicity in a rat model: Greater intensity of damage in male than female. Nephro-Urology Monthly, 5, 818–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T, Kimura T, Suzumura T, Yoshimoto N, Nakai T, Yamamoto N, et al. (2014). Magnesium supplementation and high volume hydration reduce the renal toxicity caused by cisplatin-based chemotherapy in patients with lung cancer: A toxicity study. BMC Pharmacology and Toxicology, 15, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Dominguez B, Aparicio-Trejo OE, Garcia-Arroyo FE, Leon-Contreras JC, Tapia E, Molina-Jijon E, et al. (2017). Curcumin prevents cisplatin-induced renal alterations in mitochondrial bioenergetics and dynamic. Food and Chemical Toxicology, 107, 373–385. [DOI] [PubMed] [Google Scholar]
- Osama H, Abdullah A, Gamal B, Emad D, Sayed D, Hussein E, et al. (2017). Effect of honey and royal jelly against cisplatin-induced nephrotoxicity in patients with cancer. Journal of the American College of Nutrition, 36, 342–346. [DOI] [PubMed] [Google Scholar]
- Ozaki T, Ishiguro S, Itoh H, Furuhama K, Nakazawa M, & Yamashita T (2013). Cisplatin binding and inactivation of mitochondrial glutamate oxaloacetate transaminase in cisplatin-induced rat nephrotoxicity. Bioscience, Biotechnology, and Biochemistry, 77, 1645–1649. [DOI] [PubMed] [Google Scholar]
- Pabla N, Murphy RF, Liu K, & Dong Z (2009). The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. American Journal of Physiology. Renal Physiology, 296, F505–F511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey D, Gratton JP, Rafikov R, Black SM, & Fulton DJ (2011). Calcium/calmodulin-dependent kinase II mediates the phosphorylation and activation of NADPH oxidase 5. Molecular Pharmacology, 80, 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicchi A, Sotiropuolou M, Perego P, Daubeuf S, Visvikis A, Lorenzini E, et al. (2003). gamma-Glutamyl transpeptidase catalyses the extracellular detoxification of cisplatin in a human cell line derived from the proximal convoluted tubule of the kidney. European Journal of Cancer, 39, 996–1003. [DOI] [PubMed] [Google Scholar]
- Parli JA, Godfrey DA, & Ross CD (1987). Separate enzymatic microassays for aspartate aminotransferase isoenzymes. Biochimica et Biophysica Acta, 925, 175–184. [DOI] [PubMed] [Google Scholar]
- Patricia Moreno-Londono A, Bello-Alvarez C, & Pedraza-Chaverri J (2017). Isoliquiritigenin pretreatment attenuates cisplatin induced proximal tubular cells (LLC-PK1) death and enhances the toxicity induced by this drug in bladder cancer T24 cell line. Food and Chemical Toxicology, 109, 143–154. [DOI] [PubMed] [Google Scholar]
- Peyrou M, Hanna PE, & Cribb AE (2007). Cisplatin, gentamicin, and p-aminophenol induce markers of endoplasmic reticulum stress in the rat kidneys. Toxicological Sciences, 99, 346–353. [DOI] [PubMed] [Google Scholar]
- Qi ZL, Wang Z, Li W, Hou JG, Liu Y, Li XD, et al. (2017). nephroprotective effects of anthocyanin from the fruits of Panax ginseng (GFA) on cisplatin-induced acute kidney injury in mice. Phytotherapy Research, 31, 1400–1409. [DOI] [PubMed] [Google Scholar]
- Quintanilha JCF, de Sousa VM, Visacri MB, Amaral LS, Santos RMM, Zambrano T, et al. (2017). Involvement of cytochrome P450 in cisplatin treatment: Implications for toxicity. Cancer Chemotherapy and Pharmacology, 80, 223–233. [DOI] [PubMed] [Google Scholar]
- Ridzuan NRA, Rashid NA, Othman F, Budin SB, Hussan F, & Teoh SL (2019). Protective role of natural products in cisplatin-induced nephrotoxicity. Mini Reviews in Medicinal Chemistry, 19, 1134–1143. [DOI] [PubMed] [Google Scholar]
- Saad AA, Youssef MI, & El-Shennawy LK (2009). Cisplatin induced damage in kidney genomic DNA and nephrotoxicity in male rats: The protective effect of grape seed proanthocyanidin extract. Food and Chemical Toxicology, 47, 1499–1506. [DOI] [PubMed] [Google Scholar]
- Sadzuka Y, Shimizu Y, Takino Y, & Hirota S (1994). Protection against cisplatin-induced nephrotoxicity in the rat by inducers and an inhibitor of glutathione S-transferase. Biochemical Pharmacology, 48, 453–459. [DOI] [PubMed] [Google Scholar]
- Sahin K, Orhan C, Tuzcu M, Muqbil I, Sahin N, Gencoglu H, et al. (2014). Comparative in vivo evaluations of curcumin and its analog difluorinated curcumin against cisplatin-induced nephrotoxicity. Biological Trace Element Research, 157, 156–163. [DOI] [PubMed] [Google Scholar]
- Santos NA, Bezerra CS, Martins NM, Curti C, Bianchi ML, & Santos AC (2008). Hydroxyl radical scavenger ameliorates cisplatin-induced nephrotoxicity by preventing oxidative stress, redox state unbalance, impairment of energetic metabolism and apoptosis in rat kidney mitochondria. Cancer Chemotherapy and Pharmacology, 61, 145–155. [DOI] [PubMed] [Google Scholar]
- Sleijfer DT, Offerman JJ, Mulder NH, Verweij M, van der Hem GK, Schraffordt Koops HS, et al. (1987). The protective potential of the combination of verapamil and cimetidine on cisplatin-induced nephrotoxicity in man. Cancer, 60, 2823–2828. [DOI] [PubMed] [Google Scholar]
- Tanase DM, Gosav EM, Radu S, Costea CF, Ciocoiu M, Carauleanu A, et al. (2019). The predictive role ofthe biomarker kidney molecule-1 (KIM-1) in acute kidney injury (AKI) cisplatin-induced nephrotoxicity. International Journal of Molecular Sciences, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tew KD, Manevich Y, Grek C, Xiong Y, Uys J, & Townsend DM (2011). The role of glutathione S-transferase P in signaling pathways and S-glutathionylation in cancer. Free Radical Biology & Medicine, 51, 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topcu-Tarladacalisir Y, Sapmaz-Metin M, & Karaca T (2016). Curcumin counteracts cisplatin-induced nephrotoxicity by preventing renal tubular cell apoptosis. Renal Failure, 38, 1741–1748. [DOI] [PubMed] [Google Scholar]
- Townsend DM, Deng M, Zhang L, Lapus MG, & Hanigan MH (2003). Metabolism of cisplatin to a nephrotoxin in proximal tubule cells. Journal of the American Society of Nephrology, 14, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DM, & Hanigan MH (2002). Inhibition of gamma-glutamyl transpeptidase or cysteine S-conjugate beta-lyase activity blocks the nephrotoxicity of cisplatin in mice. The Journal of Pharmacology and Experimental Therapeutics, 300, 142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DM, He L, Hutchens S, Garrett TE, Pazoles CJ, & Tew KD (2008). NOV-002, a glutathione disulfide mimetic, as a modulator of cellular redox balance. Cancer Research, 68, 2870–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DM, Marto JA, Deng M, Macdonald TJ, & Hanigan MH (2003). High pressure liquid chromatography and mass spectrometry characterization of the nephrotoxic biotransformation products of cisplatin. Drug Metabolism and Disposition, 31, 705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DM, Pazoles CJ, & Tew KD (2008). NOV-002, a mimetic of glutathione disulfide. Expert Opinion on Investigational Drugs, 17, 1075–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DM, & Tew KD (2009). Pharmacology of a mimetic of glutathione disulfide, NOV-002. Biomedicine & Pharmacotherapy, 63, 75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DM, Tew KD, He L, King JB, & Hanigan MH (2009).Role of glutathione S-transferase Pi in cisplatin-induced nephrotoxicity. Biomedicine & Pharmacotherapy, 63, 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo J, Molina-Jijon E, Medina-Campos ON, Rodriguez-Munoz R, Reyes JL, Barrera D, et al. (2015). Superoxide anion production and expression of gp91(phox) and p47(phox) are increased in glomeruli and proximal tubules of cisplatin-treated rats. Journal of Biochemical and Molecular Toxicology, 29, 149–156. [DOI] [PubMed] [Google Scholar]
- Tsushida K, Tanabe K, Masuda K, Tanimura S, Miyake H, Arata Y, et al. (2018). Estrogen-related receptor alpha is essential for maintaining mitochondrial integrity in cisplatin-induced acute kidney injury. Biochemical and Biophysical Research Communications, 498, 918–924. [DOI] [PubMed] [Google Scholar]
- Ueki M, Ueno M, Morishita J, & Maekawa N (2013). Curcumin ameliorates cisplatin-induced nephrotoxicity by inhibiting renal inflammation in mice. Journal of Bioscience and Bioengineering, 115, 547–551. [DOI] [PubMed] [Google Scholar]
- Urakami Y, Nakamura N, Takahashi K, Okuda M, Saito H, Hashimoto Y, et al. (1999). Gender differences in expression of organic cation transporter OCT2 in rat kidney. FEBS Letters, 461, 339–342. [DOI] [PubMed] [Google Scholar]
- Uys JD, Manevich Y, Devane LC, He L, Garret TE, Pazoles CJ, et al. (2010). Preclinical pharmacokinetic analysis of NOV-002, a glutathione disulfide mimetic. Biomedicine & Pharmacotherapy, 64, 493–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volarevic V, Djokovic B, Jankovic MG, Harrell CR, Fellabaum C, Djonov V, et al. (2019). Molecular mechanisms of cisplatin-induced nephrotoxicity: A balance on the knife edge between renoprotection and tumor toxicity. Journal of Biomedical Science, 26, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Kong L, Zhang J, Yu G, Lv G, Zhang F, et al. (2014). The pseudoginsenoside F11 ameliorates cisplatin-induced nephrotoxicity without compromising its anti-tumor activity in vivo. Scientific Reports, 4, 4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waseem M, & Parvez S (2013). Mitochondrial dysfunction mediated cisplatin induced toxicity: Modulatory role of curcumin. Food and Chemical Toxicology, 53, 334–342. [DOI] [PubMed] [Google Scholar]
- Weijl NI, Elsendoorn TJ, Lentjes EG, Hopman GD, Wipkink-Bakker A, Zwinderman AH, et al. (2004). Supplementation with antioxidant micronutrients and chemotherapy-induced toxicity in cancer patients treated with cisplatin-based chemotherapy: A randomised, double-blind, placebo-controlled study. European Journal of Cancer, 40, 1713–1723. [DOI] [PubMed] [Google Scholar]
- Wu CH, Chen AZ, & Yen GC (2015). Protective effects of glycyrrhizic acid and 18beta-glycyrrhetinic acid against cisplatin-induced nephrotoxicity in BALB/c Mice. Journal of Agricultural and Food Chemistry, 63, 1200–1209. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Watanabe K, Tsukiyama I, Matsushita H, Yabushita H, Matsuura K, et al. (2015). Nephroprotective effects of hydration with magnesiumin patients with cervical cancer receiving cisplatin. Anticancer Research, 35, 2199–2204. [PubMed] [Google Scholar]
- Yao X, Panichpisal K, Kurtzman N, & Nugent K (2007). Cisplatin nephrotoxicity: A review. The American Journal of the Medical Sciences, 334, 115–124. [DOI] [PubMed] [Google Scholar]
- Ye ZW, Zhang J, Townsend DM, & Tew KD (2015). Oxidative stress, redox regulation and diseases of cellular differentiation. Biochimica et Biophysica Acta, 1850, 1607–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoo K, Murakami R, Matsuzaki T, Yoshitome K, Hamada A, & Saito H (2009). Enhanced renal accumulation of cisplatin via renal organic cation transporter deteriorates acute kidney injury in hypomagnesemic rats. Clinical and Experimental Nephrology, 13, 578–584. [DOI] [PubMed] [Google Scholar]
- Zhang L, Cooper AJ, Krasnikov BF, Xu H, Bubber P, Pinto JT, et al. (2006). Cisplatin-induced toxicity is associated with platinum deposition in mouse kidney mitochondria in vivo and with selective inactivation of the alpha-ketoglutarate dehydrogenase complex in LLC-PK1 cells. Biochemistry, 45, 8959–8971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Grek C, Ye ZW, Manevich Y, Tew KD, & Townsend DM (2014). Pleiotropic functions of glutathione S-transferase P. Advances in Cancer Research, 122, 143–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, & Hanigan MH (2003). Role of cysteine S-conjugate beta-lyase in the metabolism of cisplatin. The Journal of Pharmacology and Experimental Therapeutics, 306, 988–994. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ye ZW, Singh S, Townsend DM, & Tew KD (2018). An evolving understanding of the S-glutathionylation cycle in pathways of redox regulation. Free Radical Biology & Medicine, 120, 204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]