

SUMMARY
Tissue-specific regulatory regions harbor substantial genetic risk for disease. Because brain development is a critical epoch for neuropsychiatric disease susceptibility, we characterized the genetic control of the transcriptome in 201 mid-gestational human brains, identifying 7,962 expression quantitative trait loci (eQTL) and 4,635 spliceQTL (sQTL), including several thousand prenatal-specific regulatory regions. We show that significant genetic liability for neuropsychiatric disease lies within prenatal eQTL and sQTL. Integration of eQTL and sQTL with genome-wide association studies (GWAS) via transcriptome-wide association identified dozens of novel candidate risk genes, highlighting shared and stage-specific mechanisms in schizophrenia (SCZ). Gene network analysis revealed that SCZ and autism spectrum disorder (ASD) affect distinct developmental gene co-expression modules. Yet, in each disorder, common and rare genetic variation converges within modules, which in ASD implicates superficial cortical neurons. More broadly, these data, available as a web browser and our analyses, demonstrate the genetic mechanisms by which developmental events have a widespread influence on adult anatomical and behavioral phenotypes.
In Brief
An atlas of expression and splice quantitative trait loci from mid-gestational human brain is integrated with genetic risk for schizophrenia, suggesting additional causal genes and highlighting the importance of QTL datasets derived from developmental stages most relevant to disease initiation.
Graphical Abstract
Graphical Abstract
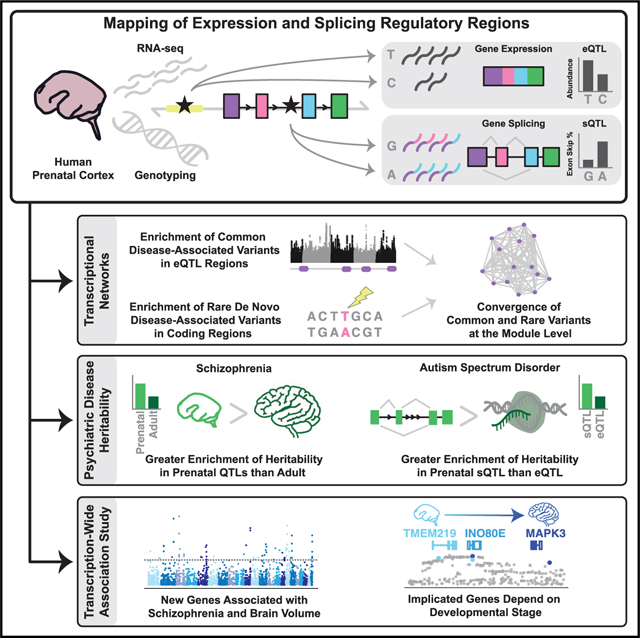
INTRODUCTION
Neurodevelopmental and neuropsychiatric disorders, such as autism spectrum disorder (ASD) and schizophrenia (SCZ), are highly heritable, complex conditions (Geschwind and Flint, 2015; Polderman et al., 2015), with hundreds of risk loci identified through large-scale genomic studies (Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium, 2017; Gratten et al., 2014; Pardiñas et al., 2018). However, the ability to interpret these variants has been hampered because many fall in non-coding regions of the genome or in regions of high linkage disequilibrium, making it challenging to identify causal mutations and their functional impact (Gandal et al., 2016; Nica and Dermitzakis, 2013; Schaid et al., 2018). Given the non-coding nature of the majority of these variants, as well as their enrichment in known regulatory regions (Cockerill, 2011; de la Torre-Ubieta et al., 2018), conserved regions (Siepel et al., 2005), and signature histone modifications (Schaub et al., 2012; Visel et al., 2009), many likely function through the regulation of gene expression and splicing (Li et al., 2016; Maurano et al., 2012; Ward and Kellis, 2012). Multiple large-scale projects, including Roadmap Epigenomics, GTEx, and Encode (Ernst et al., 2011; Roadmap Epigenomics Consortium et al., 2015; GTEx Consortium, 2015) have annotated regulatory regions across human tissues. However, little is known about how human allelic variation affects regulatory interactions during brain development, a crucial period for building the scaffold for human higher cognition and brain evolution (Geschwind and Rakic, 2013; Nord et al., 2015; Ward and Kellis, 2012).
Expression quantitative trait loci (eQTL) analysis identifies genetic loci that regulate gene expression. Critically, eQTL relationships are highly dependent on cell type and developmental stage (Dimas et al., 2009; Gerrits et al., 2009; Nica et al., 2011; GTEx Consortium, 2015), consistent with transcriptional surveys of brain development that show prominent temporal changes in gene expression (Colantuoni et al., 2011; Kang et al., 2011; Sunkin et al., 2013). Previous studies suggest that genetic disruption of these patterns, particularly during cortical development, increases risk for developmental and psychiatric disorders, highlighting the need to map regulatory variation within this critical time point (Gilman et al., 2012; Gulsuner et al., 2013; Parikshak et al., 2013; Willsey et al., 2013). However, prenatal brain eQTL analyses based on RNA sequencing (RNA-seq) are relatively small (Jaffe et al., 2018; O’Brien et al., 2018) and none include analysis of spliceQTL (sQTL) (Fromer et al., 2016; Ramasamy et al., 2014; Takata et al., 2017; GTEx Consortium, 2015).
Here, we performed a well-powered eQTL and sQTL analysis in developing human cortex to understand how functional genetic variation impacts phenotypes that likely originate in utero or early postnatal life (Hannon et al., 2016; Jaffe et al., 2016; Parikshak et al., 2015; Weinberger, 1987). We focused on mid-gestation, an epoch that captures neural progenitor proliferation, neurogenesis, and migration (Geschwind and Rakic, 2013; Johnson et al., 2009; Silbereis et al., 2016). We contrasted the genetic control of prenatal and adult expression, which show substantial differences. We found that both eQTL and sQTL contribute substantially to disease risk, but show mostly non-overlapping patterns. Integration of eQTL and sQTL with genome-wide association studies (GWAS) via transcriptome wide association identified new putative mechanisms undetected in adult brain datasets. These data provide insights into genes relevant to developmental disorders and into establishing which aspects of disease risk affect early corticogenesis, as compared to later processes.
RESULTS
We performed high-throughput RNA-seq and high-density genotyping in a set of 233 prenatal brains (Figure 1). After quality control and normalization of gene expression and genotypes (Figure S1; STAR Methods), we obtained a dataset of 15,925 genes and 6.6 million single nucleotide polymorphisms (SNPs) from 201 individuals. Analysis of ancestry indicate the donors in our study to be 40% Mexican, 25% African-American, 14% European-Mexican, 8% admixed of 3 or more ancestries, 6% Chinese, 5% African-American-Mexican, and 2% of European descent (Figure S2; STAR Methods).
Figure 1. Study Design and Overview of Analysis.

RNA-sequencing from 233 prenatal cortical samples was QCed resulting in 201 samples that was integrated with genotypes for eQTL and sQTL discovery. eQTL and sQTL were independently calculated using standard methods of covariate correction (Mostafavi et al., 2013). QTLs were characterized based on functional enrichment, cell type specificity, and compared to mature brain and non-brain tissues. We integrated prenatal brain QTLs with GWAS of neuropsychiatric disorders and other human brain phenotypes, performing LDSR, TWAS, and WGCNA to identify developmental disease risk and important biological processes under genetic control.
See also Figure S1.
Robust Identification of Prenatal Brain eQTLs
We identified cis-eQTLs using FastQTL (Ongen et al., 2016), adjusting for known and inferred covariates (Figure S3; STAR Methods) (Leek and Storey, 2007; Mostafavi et al., 2013). We identified 6,546 genes with a cis-eQTL (false discovery rate [FDR] <0.05), hereafter referred to as eGenes (Table S1). Conditioning on the most significant SNP (eSNP) permitted identification of an additional 1,416 independent, secondary eQTLs, for a total of 7,962 eQTLs (STAR Methods). We performed the analysis with two alternative methods to ensure that the eQTLs identified were not driven by ancestry differences within our dataset, which demonstrated high reproducibility of eQTLs identified by FastQTL; 94% of eGenes were also detected by EMMAX (Kang et al., 2008), and 84% of eQTLs were significant when a subpopulation meta-analysis was performed (Willer et al., 2010), despite substantially reduced power (Figure S4; STAR Methods).
eQTLs Mark Active Regulatory Regions
Positional enrichment of eSNPs shows that over 20% of significant eQTLs cluster within 10 kb of the transcription start site (TSS) of its eGene (Figures 2B and 2C), concordant with previous studies showing that promoter variants have a large influence on cognate gene expression (Kim et al., 2014; Stranger et al., 2007; Strunz et al., 2018). The overall distribution of eQTLs is consistent with previous work (GTEx Consortium, 2015; Veyrieras et al., 2008), with 70% of eSNPs located within 100 kb of the TSS, as well as a slight upstream bias (~56%) of eQTLs (Figure 2B). We reasoned that eQTL should overlap with regulatory regions defined by other methods (Figure 2A). Indeed, we find eQTLs are significantly enriched within regions of open chromatin identified in prenatal brain (odds ratio [OR] = 4.42, p < 2.2 × 10−16) (Figure 2D) (de la Torre-Ubieta et al., 2018). Distal eQTLs (>10 kb from TSS) are also enriched within 3D chromatin contacts detected at the same stage of brain development (OR = 2.96, p < 2.2 × 10−16) (Figure 2E) (Won et al., 2016) strengthening confidence that eSNPs are in accessible regions and distal eSNPs regulate their target gene (STAR Methods).
Figure 2. Characterization of eQTLs.

(A) A schematic showing that chromatin accessibility (ATAC-seq) and chromatin interactions (Hi-C) are overlapping and support eQTLs in linking genes to distal regulatory elements.
(B) Position of eSNPs in relation to the eGene TSS. Significant eQTLs are annotated based on support from prenatal brain Hi-C and distance from the TSS.
(C) Distribution of all primary and secondary eQTLs. eQTLs that lie within 10 kb of the TSS are colored green, as depicted in (B).
(D) Fraction of eQTLs with prenatal brain ATAC-seq support.
(E) Fraction of distal eQTLs (>10 kb from the TSS) with prenatal brain Hi-C support. eQTLs that show Hi-C support are colored purple, while those not supported by Hi-C are colored blue, as depicted in (B).
(F) Fold enrichment of eSNPs by their distribution within prenatal brain chromatin states, which are listed on the y axis. A red asterisk indicates significance at 5% FDR.
(G) eGenes are more tolerant to loss of function, as compared with those genes expressed in prenatal brain without significant eQTL (Wilcoxon rank-sum p < 2.2 × 10−16).
(H) Fold enrichments of eSNPs in TF binding sites (Arbiza et al., 2013). A red asterisk indicates significance at 5% FDR.
(I) Whole-gene view of SRR highlighting the association -log10(P) for all cis-SNPs with expression of SRR, the eSNP is denoted by the red line. Additionally, the eSNP falls within a CHD8 chromatin immunoprecipitation sequencing (CHIP-seq) binding site.
(J) The eQTL for SRR (p = 2.7 × 10−55), showing the distribution of expression values of SRR per genotype.
(K) The expression of SRR in control samples versus CHD8 knockdown (differential expression p = 0.042).
We next annotated eSNPs with chromatin state predictions from prenatal brain tissue (Roadmap Epigenomics Consortium et al., 2015) using GREGOR (Schmidt et al., 2015). As expected, we observed that eSNPs were most significantly enriched in TSSs, promoters, and transcribed regulatory promoter or enhancers (Figures 2F and S5A), providing further evidence that these eQTLs fall in regions functionally relevant to the regulation of gene expression (Ward and Kellis, 2012).
Functional Characterization of eQTLs Reveals Mutation Tolerance and Regulatory Drivers
Because changes in constrained genes are likely to have a higher impact on fecundity than those that are not highly constrained (Samocha et al., 2014), we reasoned that genes impacted by common regulatory variation would also be more tolerant to protein-disrupting variation (Lek et al., 2016). Indeed, comparing eGenes to genes that do not have a significant eQTL, we find that eGenes are more tolerant to loss of function mutations (Wilcoxon rank-sum test p < 2.2 × 10−16) (Figure 2G).
We next examined whether genomic regions tagged by eSNPs are enriched in transcription factor (TF) and DNA binding protein (DBP) binding sites to understand the mechanism by which eQTLs influence gene expression (STAR Methods) (Arbiza et al., 2013; Cotney et al., 2015). We found significant enrichment of eQTL in binding sites for 39 TFs and DBPs (Figure 2H), many with prominent known roles in brain development and patterning, including ELK4, NRF1, SMARCC1, SMARCC2, and CHD8 (Bestman et al., 2015; Durak et al., 2016; Eising et al., 2019; Lai et al., 2001; Ojeda et al., 1999; Preciados et al., 2016).
As a specific example, we chose CHD8 (fold change [FC] = 2.4, p = 7.8 × 10−23), because of the strong effect of CHD8 haploinsufficiency on ASD risk (Bernier et al., 2014; Neale et al., 2012; O’Roak et al., 2014; Sanders et al., 2012). We identified an eSNP falling directly within a CHD8 binding site for the gene serine-racemase (SRR) (Figures 2I and 2J (Cotney et al., 2015), which has been shown to play a role in glutamatergic neurotransmission and modulates neuropsychiatric phenotypes (Basu et al., 2009). CHD8 knockdown in neural progenitors led to significant differential expression of SRR (Figure 2K) (Sugathan et al., 2014). Thus, experimental data from prenatal brain and neural progenitors in vitro combined with these eQTL data supports the role of CHD8 and elucidates the mechanism by which CHD8 likely regulates this gene.
Robust Identification of Prenatal Brain sQTLs
Effects of genetic variation on RNA splicing are predicted to contribute to complex disease risk (Li et al., 2016). Because there has been no systematic analysis of splicing regulation in human prenatal brain, we reasoned that such analysis would enhance understanding of the link between splicing and disease. We identified 92,449 intronic excision clusters using an annotation free approach (Li et al., 2018), which allowed for discovery of alternative exons, an important advantage in the context of brain, for which isoform annotations are incomplete. We performed sQTL discovery (Ongen et al., 2016), identifying 4,635 significant sQTLs (5% FDR) in 2,132 genes (sGenes) (Figure 3A; Table S2). Of the 4,635 significant sQTLs, 3,295 were annotated introns (71%) and 1,255 (27%) were new cryptic introns (STAR Methods).
Figure 3. Characterization of sQTLs and Comparison to eQTLs.

(A) A schematic of sQTL detection and intron excision when genes are spliced into multiple isoforms.
(B) Position of sQTLs in relation to the splice junction. Significant sQTLs are annotated based on whether the sSNP lies within (green) or outside (blue) of the corresponding sGene.
(C) Fraction of sQTLs where the sSNP lies within versus outside its sGene.
(D) Fold enrichment of sSNPs by their distribution within prenatal brain chromatin states, which are depicted on they axis. A red asterisk indicates significance at 5% FDR.
(E) Fold enrichments of sSNPs in experimentally discovered RBP binding sites (Yang et al., 2015). A red asterisk indicates significance at 5% FDR.
(F) Whole-gene view of TRMT1 highlighting the association — log10(P) for all cis-SNPs with PSI values for a TRMT1 intron, the sSNP is denoted by the red line. Additionally, the sSNP falls within a SRRM4 CLIP-seq binding site.
(G) The sQTL for the TRMT1 intron, showing the distribution of PSI values of TRMT1 per genotype.
(H) The average PSI values of the TRMT1 intron in 3 control samples versus 3 SRRM4 overexpression samples.
(I) Venn diagram showing the overlap of genes containing an eQTL versus sQTL.
(J) Distribution of the distance in base pairs between eSNP and sSNP for genes harboring both.
(K) Relative proportions of VEP-predicted SNP effects between eSNPs and sSNPs.
Genomic Features of sQTL Distinguish Them from eQTL
Positional enrichment of the most significant SNP per sQTL (sSNP) shows clustering around the splice junction, with 42% of sSNPs within 10 kb of the splice junction (Figure 3B), demonstrating that variants proximal to splicing junctions have a large effect. In contrast to eQTL, the majority of sSNPs (64%) lie within the gene body (Figure 3C), consistent with other tissues (Li et al., 2016). sQTLs were also most strongly enriched in promoters and transcribed regions (Figures 3D and S5B) (Lappalainen et al., 2013; Takataetal., 2017).
Identifying Drivers of Prenatal Brain RNA-Splicing
To validate that the identified sQTLs are tagging splicing regulatory regions, we evaluated sSNP enrichment in experimentally determined RNA binding protein (RBP) binding sites (Yang et al., 2015). We found significant enrichments for sSNPs in binding sites for 36 RBPs (Figure 3E; STAR Methods), many of which do not have well characterized roles in CNS function. Among the identified RBPs with known roles in neurodevelopment are HNRNPH, ATXN2, and SRRM4 (Almaguer-Mederos et al., 2018; Grammatikakiset al., 2016; Wang et al., 2007; Zhang et al., 2014a). SRRM4 was recently shown to regulate the splicing of microexons in neurons (Irimia et al., 2014), which is disrupted in ASD (Irimia et al., 2014) and srrm4 loss of function in mice leads to neurodevelopmental abnormalities (Quesnel-Vallières et al., 2015).
As a proof of principle, we focused on a single gene, TRMT1, which has been implicated in intellectual disability (ID) and whose sSNP falls within a SRRM4 cross-linking immunoprecipitation sequencing (CLIP-seq) peak (Yang et al., 2015). The strong sQTL signal in TRMT1 within this CLIP-seq peak (Figure 3G) suggested that SRRM4 would regulate this intron. Indeed, SRRM4 overexpression leads to significant differential splicing of the same intron for TRMT1 in human cells in vitro (Parikshak et al., 2016; Raj et al., 2014). Thus, we predict that splicing factors exhibiting significant enrichment with sSNPs provide functional links between allelic variation and the factors that modulate alternative splicing. The subsequent changes in expressed protein sequence are likely to have consequences for downstream neuronal functioning and provide high priority candidates for subsequent mechanistic investigation.
eQTL and sQTL Can Overlap but Are Mostly Independent
We next analyzed the overlap of genes harboring a significant eQTL compared to those with sQTL. Half of all genes with a sQTL were also an eGene (Figure 3I; STAR Methods), and 67% of sQTLs also affect total levels of expression. However, only 22% of eQTLs affect splice-junction usage (STAR Methods), suggesting independent regulation of most expression and splicing events, parallel with observations in other tissues (Li et al., 2016). Most of the identified regions for expression and splicing of the same gene are distinct, as evidenced by the large distances between eSNP and sSNPs influencing the same gene (Figure 3J), with 30% of the genes exhibiting a D′ <0.7 between the eSNP and sSNP (Figure S5F). For validation, we also used Ensembl’s Variant Effect Predictor (VEP) (McLaren et al., 2016) that annotates SNP function, and observed more sSNPs identified as an intronic variant than eSNPs and more eSNPs being identified as upstream gene variants (Figure 3K).
Tissue Specificity Corresponds to eQTL Effect Size
To examine eQTL sharing among tissue types, we next examined the correlation of effect sizes of prenatal brain eQTLs to those from 48 different tissue types from the Genotype-Tissue Expression Consortium (GTEx) (STAR Methods) (GTEx Consortium et al., 2017). Prenatal brain eQTLs found in one or more tissues showed significantly lower effect sizes (p < 2.2 × 10−16) than those that were prenatal brain-specific, consistent with previous studies that show eQTLs observed across tissues are more constrained, whereas tissue-specific eQTLs have a greater magnitude of effect (Mohammadi et al., 2017; GTEx Consortium et al., 2017). Prenatal brain-specific eGenes show lower tolerance to loss-of-function mutations as measured by pLI (Lek et al., 2016), compared with those eGenes shared between one or more tissue types (Figure 4B). This contrast between regulatory and coding variation highlights a model whereby tissue specific regulatory control provides a subtler means for evolution to vary protein expression levels, versus the often larger and disruptive effects of protein coding variation. To assess prenatal eQTL sharing between CNS and non-CNS tissues, we correlated the effect sizes of all significant prenatal eQTLs to the same eQTL across all GTEx tissues and PsychENCODE prefrontal cortex (STAR Methods). We observed the strongest correlations of prenatal brain eQTLs with adult brain and proliferative epithelial containing tissues (Figure 4C).
Figure 4. Age Specificity of Brain eQTLs.

(A) Distribution of effect size for prenatal-specific eQTLs versus prenatal eQTLs shared in any GTEx tissue (Wilcoxon rank-sum p < 2.2 × 10−16).
(B) Distribution of pLI scores for prenatal-specific eQTLs versus prenatal eQTLs shared in any GTEx tissue (Wilcoxon rank-sum p = 0.0163).
(C) Effect size correlations (Spearman’s ρ) between significant nominal prenatal eQTLs and corresponding eQTL across tissues grouped by similarity. Tissues from GTEx are denoted as circles, PsychENCODE prefrontal cortex is denoted by a diamond. The size of each point corresponds to the dataset’s sample size.
(D) Venn diagram comparing eGenes discovered in prenatal versus adult cortex from GTEx.
(E) Venn diagram comparing eGenes discovered in prenatal versus adult cortex from PsychENCODE.
(F) Venn diagram comparing sGenes discovered in prenatal versus adult cortex (Takata et al., 2017).
(G) The number of prenatal single cell marker genes containing an eQTL in prenatal-specific eQTLs, GTEx adult brain-specific eQTLs, and those overlapping both datasets. Prenatal, prenatal-specific; adult, adult-specific; both, overlapping and found in both prenatal and adult.
(H) LD score regression enrichments where annotations of prenatal eQTLs and adult eQTLs and sQTLs were added to the baseline annotations. A bold annotation and asterisk indicate significance (PBonferroni < 0.05) for all annotation categories tested, with the proportion of heritability explained in parenthesis and error bars representing the SE.
(I) LD score regression enrichment of the prenatal cortex and GTEx adult cortex annotations with varying window size around the eSNP.
See also Figure S6.
Comparison of Prenatal and Adult eQTL, which Differentially Enrich for SCZ Risk
Another important question is related to developmental stage: how do prenatal brain eQTLs compare with adult brain eQTLs? We first compared eGenes in our dataset to that of GTEx adult cortex (GTEx Consortium et al., 2017). We found 2,532 eGenes that overlapped between prenatal and adult stages, accounting for slightly more than one-third of both datasets (Figure 4D) and 68% of overlapping eQTLs tag the same regulatory region, even if the top SNP differed (Figure S6E; STAR Methods). Additionally, we compared our eGenes to those from PsychENCODE, the largest adult brain dataset to date (Wang et al., 2018a). Although most of the eGenes overlap in this comparison (STAR Methods), we still find more than 1,000 eGenes specific to the prenatal dataset (Figure 4E), which is likely an underestimate of the true number of prenatal specific eQTL, given the prenatal dataset’s nearly 10-fold smaller sample size.
We also compared genes that harbor a sQTL in our dataset to sQTLs identified in adult prefrontal cortex (Takata et al., 2017). We found that only 24% of the sGenes identified in prenatal brain were sGenes in the adult brain, which accounts for 39% of the adult brain sGenes (Figure 4F; STAR Methods), suggesting many splicing events may be stage-specific. Because the methods used to detect splicing differ between the studies, this comparison is not ideal, emphasizing the need for more comparable splicing comparisons at different stages of development.
To further examine the differences in eGenes at the different developmental time points, eGenes were annotated based on prenatal cell type markers (STAR Methods) (Polioudakis et al., 2019). We found that many more prenatal specific eGenes are prenatal cell type markers, compared with GTEx eGenes that are adult-specific (Figure 4G; STAR Methods), consistent with expectations based on the cellular composition of each epoch. One interesting exception are markers for microglia, which is a neural-immune cell present in prenatal and adult brain, but that showed enrichment for prenatal eGenes.
Previous work based on chromatin accessibility during cortical development had suggested that genetic liability for SCZ was significantly enriched in regions active in prenatal brain (de la Torre-Ubieta et al., 2018). Given the many differences between prenatal and adult brain eQTLs at both the level of SNPs and genes, we reasoned that partitioning disease risk imparted by common genetic variation could inform the question as to the timing of genetic contributions to disease risk. We found that SNP-based heritability for SCZ shows prenatal brain eQTLs, followed by sQTLs to be the highest enriched among all significant categories, with eQTLs accounting for 3.7% of SNP heritability (p= 9.1 × 10−4) (Figure 4H) and sQTLs accounting for 2.1% of SNP heritability (p = 6.6 × 10−3). In contrast, adult brain eQTLs from both GTEx and PsychENCODE do not reach significance (Figure 4H), suggesting that prenatal brain enriched regulatory regions harbor a greater proportion of SCZ risk variant than those enriched in adult. However, when the prenatal and adult annotations are combined, the proportion of SNP heritability explained for SCZ increases in an additive manner; the combined set of prenatal eQTLs and PsychENCODE eQTLs explain 6.6% SNP heritability for SCZ. This further supports the other analyses indicating that prenatal and adult regulatory regions are distinct and complementary.
In contrast to adult eQTL, adult sQTLs do contribute significant SNP heritability in SCZ, explaining 2.3% in SCZ, and when combined with the prenatal sQTLs, explain a total of 3.5% of the SNP-based heritability (Figure 4H). To test the robustness of this finding, we explored different window sizes around each eGene (STAR Methods). We observed consistent significant enrichment over almost an order of magnitude window sizes for prenatal annotations over adult for SCZ GWAS loci (Figure 4I).
eQTL within the Context of Transcriptional Networks
We applied robust weighted gene co-expression network analysis (WGCNA) (Zhang and Horvath, 2005) to construct transcriptional networks, identifying 19 modules (labeled by color) of co-expressed genes during mid-gestation cortical development (Figure 5A; Table S3). The modules identified represent genes that correspond to distinct biological functions defined through shared Gene Ontology (GO) enrichments and cell type markers (Figures 5B and 5C; STAR Methods). Six of these modules are enriched for specific brain cell types or brain-relevant GO terms: turquoise (mitotic progenitors and cell division), red (mitotic progenitors, outer radial glia, and splicing), yellow (superficial layer neurons and splicing), blue (developing neurons and axon guidance), green-yellow (adult neurons, synaptic transmission, and neuron projection development), and brown (adult neurons and CA2+ transport). These modules, by containing a full range of the major cell types in developing human brain, capture a substantial portion of the biological processes occurring during prenatal cerebral cortical development (Polioudakis et al., 2019; Pollen etal., 2015).
Figure 5. Prenatal Brain Co-expression Networks.

(A) Network analysis dendrogram based on hierarchical clustering of genes by their topological overlap, identifies 19 modules. Colored bars below the dendrogram show module membership and expression covariates. Importantly, covariates depicted below the dendrogram are not driving module clustering.
(B) Thetop 2 Gene Ontology biological processterms enriched for each module. The x axis depictsthe – log10(FDR), the dotted black line indicates significance –log10(0.05) with all listed categories significant.
(C) The top 2 cell types enriched for genes within each module. The × axis depicts the –log10(FDR), the dotted black line indicates significance –log10(0.05).
(D-F) Depiction of module GO term biological process summary and top hub genes along with edges supported by co-expression are shown forthe blue (D), red
(E), and yellow (F) module. Hub genes are defined by being the top 30 most connected genes based on kME intermodular connectivity.
(G) Per module QQ plot of SCZ GWAS SNP p values in regulatory regions defined by eQTLs. The red module and the blue module show the most inflation for enrichment of SCZ GWAS hits.
(H) Per module QQ plot ofASD GWAS SNP p values in regulatory regions defined by eQTLs. Theyellow module showsthe most inflation for enrichment ofASD GWAS hits.
(I) Per module rare variant enrichment for SCZ, ASD, ID, developmental disorder. Two asterisks indicate significance after Bonferroni correction, one asterisk indicates nominal significance.
(J) LD score regression partitioned heritabilityenrichmentsfor prenatal eQTL and sQTLannotations across multiple GWAS. Enrichmentsare colored by –log10(P) and error bars represent SE.
We next leveraged the identified eQTLs to link noncoding variants with target genes and asked whether there were any modules containing genes whose regulatory regions were enriched for ASD and SCZ GWAS signal (STAR Methods). We identified significant enrichments for SCZ (blue; p = 0.00099) and a marginal trend toward enrichment for ASD (yellow; p = 0.061)-associated common variants (Figures 5G, 5H, and S7). Interestingly, the blue module (Figure 5D), which showed eQTL GWAS enrichment for SCZ, is enriched for the GO category neurogenesis, including genes known to play major roles in brain development, such as DLX1, FGF2, and LHX6. The yellow module (Figure 5F), which shows suggestive eQTL GWAS enrichment for ASD, is enriched for the biological process of gene regulation in developing neurons and includes key genes such as MEF2, HNRNPH3, and FOXP4. Remarkably, single-cell sequencing data indicates that the genes within this module are also enriched in upper layer cortical projection neurons (Polioudakis et al., 2019), consistent with previous data suggesting that ASD-associated variation was enriched in superficial cortical layers (Parikshak et al., 2013). This analysis demonstrates the power of eQTLs, when integrated with co-expression modules to define where common genetic variation associated with a disease acts through regulation of genes with similar biological functions and potentially similar regulatory control.
Next, to determine whether there was overlap in pathways affected by common and rare variation, we examined if genes harboring rare mutations associated with risk for neuropsychiatric disease showed similar enrichment (Figure 5I). We find only the red module enriched for rare variation for SCZ (nominal p = 0.037), which is also trending toward enrichment in common variation from SCZ GWAS (Figures 5E and 5G). The yellow module exhibits the most significant enrichment for rare variation in ASD (FDR = 0.011) and is also the most enriched in common variation from ASD GWAS (Figure 5H). These analyses indicate that ASD and SCZ risk variants effect divergent gene sets during brain development. However, they also suggest that the pathways impacted by common and rare genetic variation may converge in each disorder.
eQTL and sQTL Differentially Enrich for GWAS Signal
Recent studies have shown heritability for complex disorders is disproportionately enriched in functional categories such as conserved regions and enhancers (Finucane et al., 2015), as well as regions regulating splicing (Li et al., 2016). However, little is known about how allelic variation affects splicing variation and disease risk in developing human brain, and how this compares to expression, so we compared GWAS enrichment patterns in sQTL and eQTL (Figure 5J; STAR Methods). SCZ (Pardiñas et al., 2018)-associated variants are significantly enriched for both prenatal eQTL and sQTL regions (p = 0.0004 and p = 0.002, respectively), whereas educational attainment (Okbay et al., 2016) is only significant for sQTL regions and not eQTL (p = 0.004) (Figure 5J). Genetic variants associated with risk for attention deficit hyperactivity disorder (ADHD) (Demontis et al., 2019) show a trend for enrichment in both eQTL and sQTL regions, whereas ASD GWAS (Grove et al., 2019) only shows a trend for enrichment in sQTLs (Figure 5J). As a negative control, we tested variants associated with risk for inflammatory bowel disease (Jostinset al., 2012) and observed no enrichment. Overall, these data are consistent with previous suggestions that sQTL harbor substantial disease risk, perhaps even more so than eQTL (Li et al., 2016), highlighting the relevance of these data to functional characterization of disease-associated risk variants, which will improve as sample sizes increase.
SCZ TWAS Prioritizes Dozens of Novel Risk Genes
To further leverage these data to characterize SCZ loci with developmental origins, we used a transcription-wide association study (TWAS) (Gusev et al., 2016), to integrate cis-eQTLs and GWAS to identify genes whose expression is correlated with SCZ. Previous TWAS studies in SCZ both relied on adult brain or non-brain tissues (Gandal et al., 2018b; Gusev et al., 2018). Given the evidence for a neurodevelopmental origin of SCZ (de la Torre-Ubieta et al., 2018; Gulsuner et al., 2013), we reasoned that these prenatal data would provide new perspectives on SCZ risk, especially given the sensitivity of TWAS to tissue source (Wainberg et al., 2017).
We used SCZ GWAS summary statistics (Pardiñas et al., 2018; Ripke et al., 2014) and our prenatal brain eQTL dataset to identify genes and splicing-events whose imputed cis-regulated expression is associated with SCZ. We identified 62 genes and 91 introns with significant transcriptome-wide SCZ associations (PBonferroni < 0.05) (Figure 6A; Table S4; STAR Methods). We also conducted a summary-data-based Mendelian randomization (SMR) and the associated heterogeneity in dependent instruments (HEIDI) test (Zhu et al., 2016) to test for pleiotropic association in the cis window of eQTL associations to distinguish pleiotropy from linkage. Eight genes and six introns are significant across both methods (PSMR < 0.05 and PHEIDI < 0.05; Table S5). With support from two different methods, these genes and introns provide high priority targets for further investigation.
Figure 6. SCZ TWAS.

(A) Manhattan plot ofTWAS resultsfrom genes and intron SCZ associations, highlighting the 62 genes and 91 introns significant at Pbonferroni < 0.05, overlaid with the SCZ GWAS significant loci. Key depicts significant GWAS loci (gray), TWAS genes (magenta), and introns (purple).
(B) Overlap of TWAS-significant genes from prenatal brain, adult brain GTEx, adult brain CMC, and adult brain PsychENCODE.
(C) Overlap ofTWAS genes from prenatal brain, high-confident adult brain, and whole-blood.
(D) Overlap ofTWAS introns from prenatal brain and adult brain.
(E and G) Illustration of two genomic regions on chromosome 16 (E) and on chromosome 3 (G) harboring at least one prenatal TWAS loci. The y axis shows Ensembl gene ID, LD, TWAS results overlapped with GWAS loci –log10(P); prenatal brain TWAS genes and introns are magenta dots, high-confident adult brain TWAS genes are blue dots, adult brain TWAS introns are green dots, and GWAS SNPs are gray dots) and SCZ GWAS loci (yellow line). Ensembl gene names and gene modelsare colored by significance in datasets; prenatal brain geneand intron associations(magenta), adult brain intron associations(green), high-confident adult brain gene associations(blue). Ifagene is significant in both prenatal and adult it is blackwith an asterisk colored bythedataset. SCZ GWAS loci are shown in yellow, with the LD block surrounding the SCZ locus in purple defined by r2 > 0.6.
(F and H) Summary data-based Mendelian randomization of prenatal brain genes with SCZ GWAS (F corresponds to the genomic region in E and H corresponds to the genomic region in G). Theyaxis shows – log10(P) ofSMR and GWAS loci and individual gene eQTL – log10(P). Genes are colored by passing the 5% FDR threshold for PSMR.
We next compared the overlap in SCZ candidate risk genes implicated by either prenatal or adult brain TWAS. Of 60 high-confidence adult brain SCZ TWAS implicated genes (Figure 6B; Table S4; STAR Methods), eleven overlapped with prenatal gene (binomial test p < 2.2 × 10−16; SNX19, VSP29, XRCC3, TSNAXIP1, DNAJA3, INO8OE, NAGA, SF3B1, TYW5, C2orf47, and DDHD2). These TWAS associations, significant across the multiple adult brain datasets and prenatal brain, implicate genes that are expressed throughout development and may impart risk across developmental stages.
We highlight three of these genes: IN080 complex subunit E (INO8OE), splicing factor 3b subunit 1 (SF3B1), and matrix AAA peptidase interacting protein 1 (MAIP1; C2orf47). INO8OE is a component of a chromatin remodeling complex involved nucleosome spacing and modulates transcriptional regulation during corticogenesis (Ayala et al., 2018; Sokpor et al., 2018). SF3B1 is within a SCZ GWAS locus and is supported by an animal model of psychosis (Ingason et al., 2015). MAIP1 (C2orf47) plays a role in mitochondrial Ca2+ handling and cell survival and was previously associated with SCZ in a large Swedish GWAS (Ripke et al., 2013). As a comparison, we performed a TWAS incorporating GTEx whole blood expression weights with the SCZ GWAS. We find that only 4 genes overlap between prenatal brain, high confidence adult brain, and whole blood SCZ transcription-level associations (binomial test p = 2.548 × 10−14) (Figure 6C).
At the level of splicing, we identify 13 genes with intron associations that overlapped between the 91 prenatal intron associations and the 120 adult intron associations (Figure 6D). We highlight two genes whose splicing is implicated by both prenatal and adult brain: adaptor related protein complex 3 subunit beta 2 (AP3B2) and M-phase phosphoprotein 9 (MPHOSPH9). AP3B2 has been linked to developmental disorders (Assoum et al., 2016) and is a component of the neuron-specific AP-3 complex that has been shown to interact with dysbindin-1, a SCZ-related protein, through direct binding to the AP-3 complex through though AP3B2 (Hashimoto et al., 2009; Oyama et al., 2009). MPHOSPH9 has been associated with SCZ through differential expression at the exon level in brain, associating splicing abnormalities of MPHOSPH9 with SCZ (Cohen et al., 2012).
To identify new risk regions for SCZ not identified by GWAS alone, we next examined the overlap between the 145-independent genome-wide significant SCZ GWAS loci (Pardiñas et al., 2018) and significant prenatal brain TWAS associations. We find 50 GWAS loci harbor prenatal TWAS associations and 23 additional SCZ risk loci where significant TWAS loci do not overlap GWAS significant loci (STAR Methods). Of these 23 newly identified loci, 18 regions were also identified by a high confident adult brain TWAS association and 5 were unique to prenatal brain (Table S6; STAR Methods).
We next conducted fine-mapping to refine TWAS associations by accounting for the correlation of linkage disequilibrium (LD) and SNP weights (Mancuso et al., 2019). We identified a credible set of 155 genes, with an average of 3 genes fine-mapped at each GWAS locus (Table S7), largely reflecting local patterns of LD, and indicative of the need for further experimental dissection at many of these complex loci.
A salient example of prenatal and adult TWAS overlap within a known GWAS region is at 16p11.2. This region is complex, containing 6 prenatal associations and 4 adult associations, with only one gene in common to both stages, INO8OE (Figure 6E). INO8OE shows both significant co-localization of its prenatal brain eQTL and SCZ GWAS in this region by SMR (PSRM INO8OE = 3.7 × 10−6) (Figure 6F) and is identified as a casual gene by fine-mapping (Table S7; STAR Methods), making it a high confidence locus identified in both tissues by both methods. TMEM219 is the only other gene in the credible set fine-mapped to this region and is specific to the prenatal period. Another SCZ GWAS locus on chromosome 3 harbors complex stage-specific splicing and expression associations (Figure 6G). Co-localization of prenatal brain eQTLs by SMR supports NT5DC2, GLN3, and PBRM1 at this locus (PSRM NT5DC2 = 1.4 × 10−6, PSRM GLN3 intron = 2.9 × 10−6, PSRM PBRM1 intron = 3.0 × 10−6) (Figure 6H), providing multiple lines of evidence linking expression changes in NT5DC2, and splicing of GLN3 and PBRM1 to SCZ risk. These analyses provide evidence across development supporting INO8OE, GLN3, and PBRM1 and prenatal specific associations, such as TMEM219 and NT5DC2.
Intracranial Volume TWAS
Given that cortical neurogenesis is a major driver of brain evolution and brain size (Baeet al., 2015; Geschwind and Rakic, 2013; Kostović and Jovanov-Milosević, 2006; Rakic, 1995, 2009), we reasoned that these developing cortex eQTL and sQTL would be particularly valuable in defining loci involved in intracranial volume (ICV), which is correlated with brain size in humans (Sgouros et al., 1999). We leveraged GWAS of intracranial volume (Adams et al., 2016) to perform TWAS, identifying 7 genes whose expression (NSF, LRRC37A, LRRC37A2, LRRC37A17P, RNF123, RP11–156P1.3, MAPT-AS1) and 8 introns whose splicing (NT5C2, CRHR1, LRRC37A, LRRC37A2, USMG5, KANSL1, USP4) were significantly associated with ICV (PBonferroni < 0.05) (Figure 7A; Table S4; STAR Methods). We also performed TWAS using adult brain (STAR Methods). LRRC37A2 is the only gene that overlaps between the prenatal implicated genes and the 16 significant adult cortex genes (Figure 7B; Table S4). Three genes (CRHR1, LRRC37A2, and USMG5) with splicing associations overlapped between the 8 prenatal intron associations and 10 adult intron associations (Figure 7C).
Figure 7. ICV TWAS.

(A) Manhattan plot ofTWAS results from genes and intron ICV associations, highlighting the 7 genes and 8 introns significant at Pbonferroni < 0.05, overlaid with ICV GWAS significant loci. Key depicts significant GWAS loci (gray), prenatal brain TWAS genes (magenta), and introns (purple).
(B) Overlap of TWAS genes from prenatal brain, adult brain from GTEx, and adult brain from CMC.
(C) Overlap of TWAS introns from prenatal brain and adult brain from CMC (Gusev et al., 2018).
(D and F) Illustration of two genomic regions on chromosome 17 (D) and on chromosome 10 (F) harboring at least one prenatal TWAS loci. The y axis shows Ensembl gene ID, LD, TWAS results overlapped with GWAS loci – log10(P); prenatal brain TWAS genes and introns are magenta dots, high-confident adult brain TWAS genes are blue dots, adult brain TWAS introns are green dots, and GWAS SNPs are gray dots) and ICV GWAS loci (yellow line). Ensembl gene names and gene models are colored by significance in datasets; prenatal brain gene and intron associations(magenta), adult brain intron associations(green), high-confident adult brain gene associations(blue). If the gene is implicated by more than one dataset there is an asterisk colored by the datasets. The ICV GWAS locus is shown in yellow with the LD block in purple defined by r2 > 0.8.
(E and G) Corresponding BrainSpan (BrainSpan, 2013) gene expression trajectories throughout human lifespan of highlighted TWAS genes (E corresponds to significant genes from D and G corresponds to significant genes from F).x axis corresponds to developmental periods defined in Kang et al. (2011), where the red line represents period 8, corresponding to birth.
See also Table S4.
We compared the overlap of significant GWAS regions with significant TWAS associations to identify any new regions harboring genetic modifiers of ICV (STAR Methods). Two prenatal associations (RNF123 and USP4) lie within close proximity to each other on chromosome 3, revealing a new locus associated with ICV. Of the adult loci identified, GPX1, overlaps the new locus identified from the prenatal brain dataset on chromosome 3, while IGFBP2 and CTD-2292M16.8 reveal adult-specific regions on chromosomes 2 and 14, respectively (Table S6).
One particularly interesting GWAS region lies at 17q21, where several significant TWAS associations are found (6 prenatal brain genes, 3 prenatal brain introns, 13 adult brain genes, and 6 adult brain introns) (Figure 7D). This region contains a common inversion polymorphism and has been associated with several brain-related diseases, including SCZ, dyslexia, and Parkinson’s disease (Chen et al., 2016; Latourelle et al., 2012; Veerappa et al., 2014). Fine-mapping of the TWAS associations at this region prioritizes LRRC37A, which is the only gene significant at both developmental time points (Figure 7E; STAR Methods) (BrainSpan, 2013). The only other ICV GWAS locus overlapping a prenatal brain TWAS association is on chromosome 10 (Figure 7F), which contains significant splicing associations in NT5C2 and USMG5, the latter also harboring an adult splicing association. USMG5 (ATP5MD) is a subunit of the mitochondrial ATP synthase (Ohsakaya et al., 2011), in which mutations cause Leigh syndrome (Barca et al., 2018). NT5C2 is a hydrolase that plays a role in cellular purine metabolism that has been linked to ID and spastic paraplegia (Darvish et al., 2017) and shows greater prenatal than postnatal expression (Figure 7G) (Brain-Span, 2013).
DISCUSSION
Our analysis provides the largest genome-wide map of human eQTL and the first map of sQTL during cerebral corticogenesis, a critical epoch in brain development, significantly expanding our understanding of gene and splicing regulation in the developing brain. We have comprehensively described the implicated regulatory regions defined by eSNPs and sSNPs, enabling the integration of developmental diversity to previous adult brain functional genomic analysis and genetic variant interpretation. This allows us to identify known major regulators of both expression (TFs) and splicing (RBPs) whose activity is predicted to be affected by common allelic variation. Several of these regulators are known to be disturbed in neurodevelopmental disease via rare variation (Carvill et al., 2013; Irimia et al., 2014; Lai et al., 2001; Suls et al., 2013; Zhang et al., 2014a), providing a link between the activities of common and rare variation on disease risk during brain development. We also show that genetic variation controlling regulation of expression and splicing in the human brain is sensitive to developmental stage. In the context of early onset neurological and psychiatric disorders, this provides a new window into genetic control of gene expression and splicing regulation during a critical time point for disease development. These data are available in the supplemental tables (Tables S1 and S2), while individual-level raw data has been deposited in dbGaP. To enable easier access to these prenatal brain eQTLs and sQTL, we have built the DevEloPing Cortex Transcriptome (DEPICT) viewer (https://labs.dgsom.ucla.edu/geschwind/pages/eqtl-browser), a web portal that allows browsing by gene or SNP summary-level data.
By way of comparison, a smaller prenatal brain eQTL dataset consisting of 120 different samples published while this work was under submission (O’Brien et al., 2018), shows high concordance among significant eQTLs found in both studies (effect size correlation, r = 0.67) (Figure S6F; STAR Methods). However, given our sample size, we identified nearly five times the number of eQTLs as that study (6,546 versus 1,329), consistent with the well-described relationship between eQTL discovery and sample size (Nica and Dermitzakis, 2013). Additionally, both studies identify a common inversion polymorphism at 17q21 (Stefansson et al., 2005) which harbors many eGenes. O’Brien et al. (2018) find associations between 7 eGenes in this region and variants associated with neuroticism, including LRRC37A, a gene we identify as a prenatal specific TWAS hit for ICV and have fine-mapped to this locus. Together, our studies emphasize the importance of understanding transcriptional regulation during prenatal periods.
We find that integrating eQTLs with WGCNA-defined modules from prenatal brain expression shows distinct GWAS enrichment for ASD and SCZ within specific biological processes indicating disease-specific biology. This substantially advances previous work based on rare, gene disrupting de novo mutations (Iossifovet al., 2012; Ruzzo et al., 2019; Sanders et al., 2012) by showing convergence in genetic risk factors, even at the level of common variants that lie in regulatory regions. We show that genes involved in chromatin organization and splicing, as well as cell-type markers for superficial layer neurons are enriched for ASD GWAS loci in their eQTL-defined regulatory regions, similar to rare variants. For SCZ, we find enrichment of the biological pathways of neurogenesis and CNS development, parallel to previous observations based on analysis of chromatin confirmation in prenatal brain (Won et al., 2016), providing independent support for neurogenesis as a key process disrupted in SCZ risk. Furthermore, we show that SCZ GWAS enrichment from partitioned heritability significantly differs when using prenatal eQTL annotations versus adult eQTL annotations, consistently indicating the prenatal annotations to be the highest enriched among all functional annotations even when comparing to the PsychENCODE dataset with a sample size nearly 10 times larger than this cohort. Furthermore, the heritability explained by prenatal and adult eQTLs shows an additive effect, indicating the regulatory regions identified at these distinct time points indeed differ and are capturing different aspects of disease risk.
Similar significant enrichment of SCZ partitioned heritability has been shown for prenatal brain assay for transposase-accessible chromatin using sequencing (ATAC-seq) peaks (de la Torre-Ubieta et al., 2018), providing multiple lines of evidence that regulatory regions active in early cortical development harbor significant SCZ risk and are likely to be a crucial site of action for disease risk. Additionally, genetic risk for ADHD implicates prenatal eQTL and sQTL, while ASD risk shows a trending enrichment in sQTL. These trending enrichments are likely to gain significance as GWAS sample sizes increase. The identification of sQTL from prenatal brain will further allow mechanistic exploration of the effects of disease risk on gene isoform expression. Particularly interesting in this regard is the set of known splicing factors whose targets are enriched for disease-associated variation.
Finally, by integrating our map of prenatal gene expression and splicing regulation with the SCZ and ICV GWAS via TWAS, we are able to identify new candidate genes and candidate molecular mechanisms through which these disease-associated variants may be acting. Moreover, when comparing the implicated genes from prenatal brain expression with genes implicated via adult brain expression, there is little overlap. To some degree, this is expected and reflects TWAS’s known sensitivity to tissue (Wainberg et al., 2017), as well as the differences in eGenes and effect sizes between prenatal and adult eQTLs shown in our analysis. It does, however, highlight the importance of picking appropriate tissues and gene expression studies for a given phenotype. Here, the SCZ risk genes predicted by TWAS based on prenatal brain expression contributes to the growing list of candidate genes discovered from adult expression for a more complete view of potentially casual genes and splicing events. These results and others (de la Torre-Ubieta et al., 2018; Won et al., 2016) are consistent with the original framing of SCZ as a neurodevelopmental disorder (Weinberger, 1987). These data indicate that the developmental sensitivity for SCZ lies not only in adolescent development, but in the earliest periods of cortical neurogenesis and cortical patterning. This epoch is also particularly important for brain volume and patterning (de la Torre-Ubieta et al., 2018; Geschwind and Rakic, 2013), and the integration of eQTL data with the ICV GWAS identifies many new genes influencing human brain size.
GWAS regions that yield discordant TWAS gene associations depending on the developmental time period of reference panel are also interesting to consider. A salient example is the region containing the 16p11.2 copy number variant (CNV), which has been associated with multiple neurodevelopmental conditions, including SCZ, ASD, ID, and bipolar disorder (Bernier et al., 2017; Hanson et al., 2015; Shinawi et al., 2010; Weiss et al., 2008; Zhou et al., 2018). Fine mapping of this locus in SCZ implicates INO8OE and TMEM219 from prenatal brain, INO8OE being the only gene in this locus implicated by both prenatal and adult data. INO8OE is an interesting candidate, due to its similar role as the chromatin-helicase-DNA binding protein family, including CHD8, in chromatin remodeling during cortical neurogenesis (Clapier et al., 2017; Sokpor et al., 2018). Prenatal brain TWAS also implicated KCTD13, CTD-2574D22.2, PPP4C, and YPEL3, whereas MAPK3, DOC2A, and TAOK2, although implicated by adult brain expression data, are not supported by fine-mapping. This CNV contains 29 genes, so the relationship of this CNV to phenotype is unlikely to be simple or be driven by only one gene (Escamilla et al., 2017; Golzio et al., 2012). Additional fine mapping and functional analyses will be necessary for more conclusive genotype-phenotype associations.
These results demonstrate the importance of considering developmental stage of brain expression when using eQTL and sQTL data to interpret disease associated variants. They also highlight the value of using these data to further characterize developmental time points during which genetic variation acts to modulate risk for neurodevelopmental and early onset neuro-psychiatric diseases, suggesting that more detailed maps of the effects of common genetic variation effects throughout the lifespan will be of value.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Daniel H. Geschwind (dhg@mednet.ucla.edu). Summary statistics are available in the supplemental tables (Tables S1 and S2), while individual-level raw RNA-seq and genotype data has been deposited in dbGaP: phs001900. Also, to enable easier access to these prenatal brain eQTLs and sQTL, we have built the DevEloPing Cortex Transcriptome (DEPICT) viewer (https://labs.dgsom.ucla.edu/geschwind/pages/eqtl-browser), a web portal that allows browsing by gene or SNP summary-level data.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Developing Human Brain Samples
Prenatal tissue was obtained from the UCLA Gene and Cell Therapy core according to IRB guidelines from 233 donors (post-conception weeks: 14–21, final sample of 201 = 84 Female, 117 Male) following voluntary termination of pregnancy. This study was performed under the auspices of the UCLA Office of Human Research Protection, which determined that it was exempt because samples are anonymous pathological specimens. Full informed consent was obtained from all of the parent donors.
METHOD DETAILS
Library Preparation
Total RNA and genomic DNAfrom human prenatal brain tissue from PCW 14–21 that visually appeared to be cortical was extracted using miRNeasy-mini (QIAGEN) and DNeasy Blood and Tissue Kit (DNA) or were extracted using trizol with glycogen followed by column purification. Library preparation via Illumina Stranded TruSeq kit with Ribozero Gold ribosomal RNA depletion library prep was followed by sequencing on 233 brains and genotype array data on 212 brains was generated at the UCLA Neurogenomics Core. Pseudo-randomization was performed to decrease correlation between sequencing lane and biological variables such as sex and gestation week. RNA samples were pooled, randomized, and run across 4 lanes. Ribozero, ribosome depleted, 50 bp paired-end RNA sequencing was performed with mean sequencing depth of 60 million reads on an Illumina HiSeq2500.
QUANTIFICATION AND STATISTICAL ANALYSIS
Genotype pre-processing
Genotyping was performed at the UCLA Neurogenomics Core (UNGC) on either Illumina HumanOmni2.5 or HumanOmni2.5Exome platform in 8 batches. SNP genotypes were exported into PLINKformat. Batches were merged and markers that did not overlap genotyping platforms were removed. SNP marker names were converted from Illumina KGP IDs to rsIDs using the conversion file provided by Illumina. Quality control was performed in PLINK v1.9 (Chang et al., 2015). SNPs were filtered based on Hardy-Weinberg equilibrium (-hwe 1e6), minor allele frequency (-maf 0.01), individual missing genotype rate (-mind 0.10), variant missing genotype rate (-geno 0.05) resulting in 1,799,583 variants (Figure S1).
RNA-sequencing Data Processing Pipeline
All raw RNaseq fastq files, 4 per sample run on different lanes, were run through FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). FastQC output was visually inspected and sequencing lanes where the “per tile sequence quality” was red were removed, there was no sample with more than one sequencing lane removed. Fastq files were aligned to the GRCH37.p13 (hg19; Homo_sapiens.GRCh37.75.dna.primary_assembly.fa: ftp://ftp.ensembl.org/pub/release-75/fasta/homo_sapiens/dna/) reference genome using STAR v2.4 (Dobin et al., 2013). SAM files were sorted, indexed, converted to BAM files and merged across lanes from the same sample using Samtools v1.2 (Li et al., 2009). Gene quantifications were calculated using HTSeq-counts v0.6.0 (Anders et al., 2015) using an exon union model on the basis of Gencode v19 comprehensive gene annotations (Harrow et al., 2012). Quality control metrics were calculated using PicardTools v1.139 (http://broadinstitute.github.io/picard) and Samtools. A sex incompatibility check was also performed using XIST expression and Y chromosome non-pseudoautosomal expression which are known to show different patterns of expression in males and females. A scatterplot of XIST expression versus the sum of expression of genes in the non-pseudoautosomal region of the Y chromosome showed no gender mismatches. (Figure S1)
Sample Swap Identification
QC’d genotypes and sample BAM files were used to identify any sample identity swaps between the RNA and DNA experiments using VerifyBamID v1.1.2 (Jun et al., 2012). We identified 4 samples where [CHIPMIX] ~1 AND [FREEMIX] ~0, indicative of unmatching RNA and DNA, which were removed in the VCF file.
Genotyping Pipeline
PLINK genotype files were converted to vcf files using Plinkseq v0.10 (https://atgu.mgh.harvard.edu/plinkseq/). Genotypes were imputed into the 1000 Genomes Project phase 3 multi-ethnic reference panel (1000 Genomes Project Consortium et al., 2015) by chromosome using Beagle v4.1 (Browning and Browning, 2016) and subsequently merged. Multiallelic sites were removed using GATK v3.5 (Van der Auwera et al., 2013). Imputed genotypes were filtered for Hardy-Weinberg equilibrium p value <1 × 10–6 and minor allele frequency (MAF) 5%. Imputation quality was assessed filtering variants where allelic R-squared > 0.5 and dosage R-squared > 0.5 by GATK, resulting in ~6.6 million autosomal SNPs. We restricted to only autosomal due to sex chromosome dosage, as commonly done (GTEx Consortium, 2015).
RNA-seq Quality Control and Normalization
Gene counts were compiled from HTSeq Count (Anders etal., 2015) quantifications and imported into R version 3.2.1 for downstream analyses. Gene counts were put through quality control, removing genes that were not expressed in 80% of samples with 10 counts or more. Expression was then corrected for GC content, gene length, and quantile normalized to a standard normal distribution, as commonly done in QTL studies (Battle et al., 2015). Sample outliers were removed based on standardized sample network connectivity Z scores < 2 (Zhang and Horvath, 2005) Figure S2F). ComBat batch correction was performed (Johnson etal., 2007). After quality control and normalization, there remained 201 samples with 15,930 genes expressed (on the basis of Gencode v19 annotations) at sufficient levels (gene biotypes include 12,943 protein coding, 767 long noncoding RNAs, among others).
Covariate Selection
To evaluate and remove global effects of gene expression, we used PLINKv1.9 (Chang etal., 2015) to run multidimensional scaling on the QC’d imputed genotypes and to verify ancestral backgrounds of the samples. We aggregated the final 201 samples with HapMap3 (International HapMap Consortium, 2003) of 1397 samples across 11 populations (87 ASW, 165 CEU, 137 CHB, 109 CHD, 101 GIH, 113 JPT, 110 LWK, 86 MXL, 184 MKK, TSI 102, YRI 203). A plot of the first two MDs components of the merged data shows the genetic ancestry of our samples among a diverse reference population (Figure S2H). For eQTL analysis, the top 3 MDS components calculated only in the prenatal brain samples were used as covariates.
We also looked at the correlation of known measured biological covariates, measured technical covariates, as well as RNA quality control metrics from Picard tools (gestation week, RIN, sex, purification method, 260:230 ratio, 260:280 ratio, read depth, percent chimeras, 5′ bias, 3′ bias, AT dropout) with the top 10 principle components of the expression data and find the top principal component corresponds to the age of the sample (gestation week) and the second component corresponds to the RNA integrity number (RIN) (Figure S2G).
To measure hidden batch effects and confounders, hidden covariate analysis was performed using Hidden Covariates with a Prior (HCP) (Mostafavi et al., 2013). Hidden factors were calculated given the known measured factors. HCP was run separately for varying number of inferred hidden components: 5,10,15,20,25, 30. We included 20 HCPs in our eQTL model which we found to maximized eGene discovery along with gestation week, RIN, and sex (Figures S3A and S3C). We correlated the 20 HCPs along with gestation week, RIN, sex, and top 3 genotype PCs (all covariates used in the final model) with the measured factors and Picard metrics, as well as the top 20 PCs of expression to gain insight to meaning of the HCPs. We see each inferred hidden component’s relationship to the known variables is complex and distributed across variables.
cis-eQTL mapping
We performed cis-eQTL mapping using FastQTL (Ongen et al., 2016), a defined cis window of 1 megabase up- and down-stream of the gene start site regardless of strand for 15,930 expressed genes, and correction for the following covariates: gestation week, RIN, sex, 20 HCPs, 3 genotype PCs. FastQTL (Ongen et al., 2016) was run in the permutation pass mode (1000 permutations) to identify the best nominal associated SNP per phenotype and with a beta approximation to model the permutation outcome (Figure S3E) and correct for all SNPsin LD with the top SNP per phenotype. Beta approximated permutation p values were then multiple test corrected using the q-value Storey and Tibshirani FDR correction (Storey and Tibshirani, 2003). We define eQTL containing genes (eGenes) by having an FDR q-value ≤ 0.05. Secondary, independent eQTLs were identified by rerunning permutation tests in FastQTL for every eGene conditioning on the primary eSNP. To compare with studies that did not perform a permutation procedure, we tested all SNP-gene pairs and discover 893,813 significant eQTLs (5% FDR) corresponding to 11,625 eGenes.
To assess inflation in a Q-Q plot, we randomly chose 10 genes to run eQTL analysis in trans, testing all SNPs genome-wide with association with gene expression using MatrixEQTL (Shabalin, 2012). We corrected for the same covariates as in the cis-eQTL analysis. The Q-Q plot of the trans-eQTLs shows no inflation, an indication that our eQTL results are not confounded by population stratification. (Figure S3F)
qPCR
To validate a few eQTLs, we selected eGenes with the highest effect sizes in order to be able to detect a difference by qPCR and stratify by genotype, in which we find concordant significant eQTL signal. qPCR was performed on 5 samples of genotype 0 and 5 samples of genotype either 1 or 2 for each gene. (Figures S3G–S3I)
EMMAX
To further check that our eQTLs are not due to the population differences of our samples, we ran cis-eQTL analysis with EMMAX (Kang et al., 2008), which accounts for population structure using a genetic relationship matrix. We used the emmax-kin function (-v -h -s -d 10) to create the IBS kinship matrix. EMMAX was run for each gene with a cis-window of +−1MB around the TSS, correcting for the same covariates in the FastQTL analysis. Nominal EMMAXpvalues were corrected for multiple testing using the q-value Storey and Tibshirani FDR correction (Storey and Tibshirani, 2003).
To assess overlap between FastQTL and EMMAX, FastQTL was also run in the nominal pass mode to obtain nominal p values for all cis-SNPs tested per gene. FastQTL nominal p values were also corrected for multiple testing using the q-value Storey and Tibshirani FDR correction. We discover 920,356 nominal eQTLs at a 5% FDR threshold. To compare nominal results, we defined eGenes as a gene containing a significant SNP association at FDR ≤ 0.05. We found 93.8% of eGenes from the FastQTL analysis was an eGene in the EMMAX analysis. Additionally, we compared all SNP-gene pairs tested and found 92.8% of significant SNP-gene associations from nominal FastQTL to be significant in the EMMAX analysis (Figures S4D and S4E).
Meta-Analysis
We split up our sample into six groups based on hierarchical clustering of the top 3 PCs of the genotype data. (Figure S4B) The size of each group ranged from 12 samples to 47 samples and each group corresponded to distinct ancestries based on the MDS plots of samples merged with HapMap3. We performed association testing between the top SNP per gene, identified by FastQTL permutation pass eQTL analysis, within group using the lm() function in R, correcting for gestation week, RIN, age, and 20HCPs. A fixed effect meta-analysis was then run between groups using METAL v3.25.2011 (Willer et al., 2010), which implements a Cochran’s Q test for heterogeneity. We find significant heterogeneity at 10% of our eQTLs and find 87% of our eQTLs are significant in the meta-analysis at FDR 0.05% (q-value) strongly suggesting our results are not due to population stratification (Figure S4C).
Intron Cluster Quantifications
We used Leafcutter (Li et al., 2018) to leverage information from reads that span introns to quantify clusters of variably spliced introns. From the already aligned FASTQ files by STAR, output bam files were converted into junction files. Intron clustering was performed using default settings of 50 reads per cluster and a maximum intron length of 500kb. The Leafcutter prepare_genotype_table script was then used to calculate intron excision ratios and to filter out introns used in less than 40% of individuals with almost no variation. Intron excision ratios were then standardized and quantile normalized. Leafcutter’s leafviz annotation code were used to annotate detected introns as annotated or cryptic. New cryptic introns were annotated by being cryptic 5′, cryptic_3′, cryptic unanchored, or novel annotated pair based on Gencode hg19 gene annotations.
sQTL mapping
Standardized and normalized intron excision ratios calculated by leafcutter was used as the phenotype for sQTL mapping. FastQTL (Ongen et al., 2016) was used to test for association between SNPs within a cis-region of +−100kb of the intron cluster and intron ratios within cluster. Hidden covariate analysis was performed using Hidden Covariates with a Prior (HCP) (Mostafavi et al., 2013) on intron excision ratios given the same known covariates used for eQTL HCP calculations. We included 5 HCPs in our spliceQTL model which we found to maximized intron QTL (Figure S3D) discovery along with gestation week, RIN, and sex. FastQTL was run in the permutation pass mode (1000 permutations). Beta approximated permutation p values were then multiple test corrected using the q-value Storey and Tibshirani FDR correction. We define sQTL as an intron having an FDR q-value ≤ 0.05, and an sGene as a gene containing a significant sQTL at any intron.
ATAC-seq Overlap of eQTLs
Prenatal brain ATAC-seq peaks were obtained from (de la Torre-Ubieta et al., 2018). We annotated eQTLs as being supported by ATAC if the LD block (r2 > 0.8 PLINK) around its eSNP overlapped an open chromatin region. To test for significance, we created a null set of eQTLs (q-value > 0.2), annotated overlap with ATAC peaks, and then ran a Fisher’s exact test.
Hi-C Overlap of eQTLs
Prenatal brain CP and GZ Hi-C topological association domain bed files were obtained from Won et al. (2016). eSNPs located within 10kb of the eGene TSS were removed, as Hi-C cannot detect any chromosomal interaction less than 10kb apart. We defined any remaining eQTL as overlapping Hi-C if the LD block (r2 > 0.8 PLINK) around its eSNP fell in one 10kbTAD bins and the corresponding eGene +−2kb overlapped with the other 10kb TAD bin in either CP or GZ. To test for significance, we created a null set of eQTLs (q-value > 0.2), annotated overlap with Hi-C, and then ran a Fisher’s exact test.
Functional enrichment of QTLs in epigenetic marks, transcription factor, and splicing factor binding sites
We performed functional enrichment of both eQTLs and sQTLs using GREGOR (Genomic Regulatory Elements and Gwas Overlap algoRithm) (Schmidt et al., 2015) to evaluate enrichment of variants in genome wide annotations. We downloaded the 25 state ChromHMM model BED files from the Roadmap Epigenetics Project (Ernst and Kellis, 2015; Roadmap Epigenomics Consortium et al., 2015), generated from a set of 5 core chromatin marks assayed in prenatal brain (Figure S5). We downloaded consensus transcription factor and DNA-binding protein binding site BED files (Arbiza et al., 2013), which called consensus binding sites from multiple cell types from Encode CHIP-seq data which was used to computationally annotate all possible genome-wide sites for 78 binding proteins. We filtered to 62 binding proteins that showed cortical brain expression in BrainSpan (BrainSpan, 2013). Lastly, we obtained human RNA binding protein (RBP) binding site BED files from CLIPdb (Yang et al., 2015) database of publicly available cross-linking immunoprecipitation (CLIP)-seq datasets from 51 RBPs.
GREGOR evaluates the enrichment of QTL variants in these genomic annotations by estimating the significance of observed overlap of the eSNP or sSNP relative to the expected overlap using a set of matched control variants. GREGOR creates a list of possible causal SNPs by extending the list of eSNPs or sSNPs (index SNPs) to all SNPs in high linkage disequilibrium (r2 > 0.7). A set of matched control SNPs (SNPs are selected based on matching the index SNP for number of variants in LD, minor allele frequency, and distance to nearest gene/intron) is then created, and enrichments are calculated based on the observed and expected overlap within each annotation.
We downloaded CHD8 ChIP-seq peaks from ChIP experiments performed in human mid-gestation prenatal brain (Cotney et al., 2015) and expression counts for CHD8 knockdown in neural progenitors (Sugathan et al., 2014). ChIP-seq peaks were overlapped with prenatal eQTLs using the GenomicRanges package in R. eGenes were identified in the knockdown and control expression data and plotted using ggplot.
We downloaded SRRM4 CLIP-seq peaks (Yang et al., 2015) and PSI values from SRRM4 overexpression in human 293T cells from averaged from 3 overexpression samples and 3 control camples (Parikshaket al., 2016; Raj et al., 2014). CLIP-seq peaks were overlapped with prenatal sQTLs using the GenomicRanges package in R. The leafcutter identified intron of the sGene ws identified in the overexpression and control PSI data and plotted using ggplot.
eQTL sQTL Overlap
We used the Storey’s π1 statistic described in Nicaet al. (2011), to assess the proportion of true associations among sQTLs that were also detected by the eQTL analysis and eQTLs that were also detected by the sQTL analysis. The overlap was assessed by taking all significant SNP-gene associations from the eQTLs and estimating the proportion of true associations (π1) on the distribution of corresponding p values of the overlapping SNP-gene pairs in the sQTL dataset and vice versa. This is done by first estimating π0, the proportion of true null associations based on their distribution. Then π1 = 1-π0 estimates the lower bound of true positive associations.
Estimation of Variant Effect of eQTL and sQTL
Ensembl’s Variant Effect Predictor (VEP) version 90 (McLaren et al., 2016) was used to annotate the effects of variants of significant QTLs on genes, transcripts, protein sequence, and regulatory regions. VEP annotations are based off of a wide range of reference data including Ensembl database verion 92, GRCH37.p13 genome assembly, Gencode 19 gene annotations, RefSeq 2015–01, Poly-Phen 2.2.2, SOFT 5.2.2, dbSNP 150, COSMIC 81, ClinVar 2017–06, and gnomAD r2.0.
Prenatal Cell Type Markers
Cell type enriched genes were obtained from Polioudakis et al. (2019), a single-cell RNA-seq dataset from GW17–18 human prenatal cortex. Briefly, Drop-seq was run on single cells isolated from human prenatal neocortex according to the online Drop-seq protocol v.3.1 (http://mccarrolllab.com/download/905/) and the methods published in Macosko et al. (2015). The raw Drop-seq data was processed using the Drop-seq tools v1.12 pipeline from the McCarroll Laboratory (http://mccarrolllab.com/wp-content/uploads/2016/03/Drop-seqAlignmentCookbookv1.2Jan2016.pdf). Normalization was performed using Seurat v2.0.1 (Butler et al., 2018). Raw counts were read depth normalized by dividing by the total number of UMIs per cell, then multiplying by 10,000, adding a value of 1, and log transforming (ln (transcripts-per-10,000 + 1)) using the Seurat function ‘CreateSeuratObject’. To identify cell-type enriched genes, differential expression analysis was performed for each cluster individually versus all other cells in the dataset for genes detected in at least 10% of cells in the cluster. Differential expression analysis was performed using a linear model implemented in R as follows: lm(expression ñumber_of_UMI + donor + lab_batch). P values were then Benjamini-Hochberg corrected (Benjamini and Hochberg, 1995). Genes were considered enriched if they were detected in at least 10% of cells in the cluster, > 0.2 log2 fold enriched, and Benjamini-Hochberg corrected p value < 0.05. Cell type enriched genes were annotated based on the gene harboring an eQTL in either the prenatal brain dataset or GTEx adult cortex dataset (GTEx Consortium et al., 2017).
Cross age-tissue comparison
We downloaded GTEx v7 eQTL summary statistics for all 48 tissue types (GTEx Consortium et al., 2017). To compare effect sizes consistently between studies, we calculated effect sizes by running a linear model with scaled log tpm expression values for significant prenatal eQTLs and calculated in the same way for corresponding SNP-gene pairs in the GTEx data to obtain a beta value from non-standard normalized expression. Significant prenatal eQTLs were identified as prenatal specific if the corresponding SNP-gene pair was not found in any GTEx tissue or as shared if it was found in at least one GTEx tissue.
Effect size correlations between all GTEx tissues and PsychENCODE (GTEx Consortium et al., 2017; Wang et al., 2018a) were calculated by first obtaining all FDR ≤ 0.05 (q-value) nominal prenatal eQTLs. Nominal eQTL analysis was run in FastQTL, using the same input for the permutation pass, to obtain all SNP-gene pairs tested. Spearman’s p correlations were calculated per tissue on the absolute value of the slope from FastQTL output of all FDR ≤ 0.05 prenatal eQTLs and corresponding absolute value of slope from SNP-gene pairs in GTEx nominal associations. The absolute value of the slope was used for all correlations to control for strandflips. For each tissue compared in this analysis, we indicate how many of the prenatal eGenes were found in the tissue of comparison by the sample size of that tissue (Figure S6C). A scatterplot of the prenatal brain versus PsychEncode adult brain effect sizes shows corresponding eQTL regression beta values from each dataset (Figure S6D).
Additionally, we used Storey’s Qvalue software (Storey and Tibshirani, 2003) to assess overlap between prenatal brain eQTLs and the eQTLs from the GTEx v7 tissues (GTEx Consortium et al., 2017). The proportion of true associations(π1) was estimated by looking up significant prenatal brain eQTLs in each of the GTEx tissues, creating a distribution of corresponding p values of the overlapping SNP-gene pairs used to calculate π0, the proportion of true null associations based on their distribution. Then π1 = 1 -π0 estimates the lower bound of true positive associations. We performed the reciprocal overlap by looking up GTEx significant eQTLs per tissue in the prenatal brain dataset (Figures S6A and S6B)
Overlap between prenatal brain eGenes and GTEx Cortex eGenes (N = 136 individuals, eGenes = 6,146) was performed by intersecting all significant eGenes detected by permutation test followed by FDR correction. Overlap between prenatal brain eGenes (11,625; 5% FDR all SNP-gene pairs) and PsychENCODE eGenes (N = 1,866, eGenes = 32,944), was performed by intersection all significant eGenes detected by all nominal eQTLs with an FDR ≤ 0.05. Overlap between prenatal brain sGenes and adult sGenes was performed by intersecting the genes of significant sQTLs by permutation test followed by FDR correction.
Overlap between our prenatal brain eQTL dataset and a prenatal brain eQTL dataset consisting of 120 samples (O’Brien et al., 2018), were performed by intersecting all nominal eQTLs with an FDR ≤ 0.05. Spearman’s p correlations were calculated for eQTL effect sizes (slope) for overlapping FDR-significant eQTLs (Figure S6F).
Partitioned Heritability
Partitioned heritability was measured using LD Score Regression v1.0.0 (Finucane et al., 2015) to identify enrichment of GWAS summary statistics among functional genomic annotations by accounting for LD, specifically eQTL and sQTL regulatory regions. The full baseline model of 53 functional categories was downloaded from Finucane et al. (2015) (https://github.com/bulik/ldsc/wiki/Partitioned-Heritability). Prenatal and adult eQTL annotations from both GTEx Cortex and PsychENCODE, and prenatal sQTL and adult sQTL annotations (Takata et al., 2017), were created by taking a 500bp window (+−250) around each eSNP or sSNP. This resulted in 6,163 prenatal eQTL annotations, 5,690 adult eQTL GTEx annotations, 32,944 adult PsychENCODE eQTL annotations, 4,635 prenatal sQTL annotations, and 8,966 adult CMC sQTL annotations. An annotation file was then created by marking all HapMap3 (International HapMap Consortium, 2003) SNPs that fell within the QTL annotations. LD scores were calculated for the QTL annotation SNPs using an LD window of 1cM using LD reference panel 1000 Genomes European Phase 3 (1000 Genomes Project Consortium et al., 2015). This LD reference panel was chosen due to the CLOZUK+PGC SCZ GWAS (Pardiñas et al., 2018; Ripke et al., 2014) being comprised of mainly European ancestry samples (Ripke et al., 2014). Baseline LD-scores and QTL LD-scores were simultaneously included in computation of partitioned heritability. Enrichment for each annotation was calculated by the proportion of heritability explained by each annotation divided by the proportion on SNPs in the genome falling in that annotation category. Enrichment p values were then Bonferroni corrected. To test robustness of partitioned heritability with 500bp window annotation, we explored different window sizes (500bp, 1000bp, 5kb, 10kb, 20kb, 50kb, 100kb, 250kb) around each eGene.
All prenatal eQTLs and sQTLs were tested separately for enrichment in genetic variants associated with SCZ (Pardiñas et al., 2018; Ripke et al., 2014), major depressive disorder (CONVERGE Consortium, 2015), intracranial volume (Adams etal., 2016), inflammatory bowel disease (Jostins et al., 2012), head circumference (Taal et al., 2012), epilepsy (International League Against Epilepsy Consortium on Complex Epilepsies, 2014), educational attainment (Rietveld et al., 2013), cognitive performance (Benyamin et al., 2014), ASD (Grove et al., 2019), Alzheimer’s disease (Lambert et al., 2013), and attention deficit hyperactivity disorder (Demontis etal., 2019).
WGCNA
After gene counts were put through quality control removing genes that were not expressed at a level of 10 counts or more in 80% of samples, expression was conditional quantile normalized, adjusting for gene length and GC content. Sample outliers were removed based on standardized sample network connectivity Z scores < 2. ComBat batch correction was performed (Johnson et al., 2007). Gestation week, RIN, and the top 4 Picard PCs were regressed from the expression dataset.
Network analysis was performed with robust consensus WGCNA (rWGCNA) (Zhang and Horvath, 2005) assigning genes to specific modules based on biweight midcorrelations among genes. Soft threshold power of 11 was chosen to achieve scale-free topology (r2 > 0.8) (Figures S7A and S7B). Then, 50 signed co-expression networks were generated on 50 independent bootstraps of the samples; each co-expression network uses the same estimated power parameter. The 50 topological overlap matrices were combined edge-wise by taking the median of each edge across all bootstraps. The topological overlap matrices were clustered hierarchically using average linkage hierarchical clustering (using ‘1 - TOM’ as a dis-similarity measure). The topological overlap dendrogram was used to define modules using minimum module size of 100, deep split of 4, merge threshold of 0.2, and negative pamStage.
Module preservation was run in an independent RNA-seq dataset of cortical development from 8 post conception weeks to 12 months after birth (Parikshak et al., 2013; Sunkin et al., 2013), showing that these modules are all highly preserved (Figure S7).
GO enrichment
GO definitions were downloaded from Ensembl release 86. GO terms with a small (< 35) or large (> 100) number of genes were removed. Logistic regression was performed using the model: is.go ~is.module + gene covariates (GC content and gene length) for an indicator-based enrichment, and p values were Bonferroni FDR corrected. The top two significant terms are reported.
Cell Type enrichment
Cell type markers from human and mouse brain were downloaded from Hawrylycz et al. (2015), Lein et al. (2007), Mancarci et al. (2017), Miller et al. (2014), Tasic et al. (2016), Winden et al. (2009), Zhang et al. (2014b, 2016) as well as the prenatal types from Polioudakis et al. (2019). Logistic regression was performed using the model: is.cell type ~is.module + gene covariates (GC content and gene length) for an indicator-based enrichment, and p values were Bonfefrroni FDR corrected. The top two significant cell types are reported.
Rare variant enrichment
Genes containing disease associated de novo variants were downloaded from denovo-db v1.5, which is a collection of germline de novo single nucleotide variants and small indels consolidated across many studies for many disorders including for ASD, Developmental Disorder, ID, and SCZ (http://denovo-db.gs.washington.edu) De Rubeiset al., 2014; Fromeretal., 2014; Girirajan et al., 2013; Glessner et al., 2009; Gulsuner et al., 2013; International Schizophrenia Consortium, 2008; Iossifov et al., 2014; Krumm et al., 2015; Levinson et al., 2011; Malhotra and Sebat, 2012; Marshall et al., 2008; McCarthy et al., 2009, 2014; Michaelson et al., 2012; Moreno-De-Luca et al., 2010, 2013; O’Roak et al., 2012, 2014; Rujescu et al., 2009; Sanders et al., 2011; Stefansson et al., 2005; Tavassoli et al., 2014; Turner etal., 2016,2017; Vacicetal., 2011; Weiss etal., 2008; Yuen etal., 2015). We compiled a list of all genes containing at least one de novo mutation of annotation functional class: frameshift, frameshift near splice, splice acceptor, splice donor, start lost, stop gained, stop gained near splice, and stop lost. We then analyzed the union of these genes with genes implicated by CNVs in Gandal et al. (2018a). Gandal et al. (2018a) included CNVs that were in at least two studies with a p value < 0.01 or in one study with a p value < 10−6, from well powered studies of over 500 subjects per group.(Girirajan et al., 2013; Glessner et al., 2009; International Schizophrenia Consortium, 2008; Levinson et al., 2011; Malhotra and Sebat, 2012; Marshall et al., 2008; McCarthy et al., 2009; Moreno-De-Luca et al., 2010, 2013; Rujescu et al., 2009; Sanders et al., 2011; Stefansson et al., 2005; Vacic et al., 2011; Weiss et al., 2008). Logistic regression was performed using the model: is.disease ~is.module + gene covariates (GC content and gene length) for an indicator-based enrichment, and p values were Bonferroni FDR corrected.
| Disorder | # Genes with de novo variants | # Genes with CNVs | Total # unique genes |
|---|---|---|---|
|
| |||
| ASD | 688 | 288 | 966 |
| Developmental Disorder | 516 | 0 | 516 |
| ID | 167 | 0 | 167 |
| SCZ | 81 | 289 | 365 |
GWAS Module Enrichment
To assess module enrichment of GWAS by gene regulatory regions, we first created a map of gene specific regulatory regions. Every eGene was mapped to its eSNP (1st and 2nd if it had one) and extended to the LD block (r2 > 0.8 PLINK) calculated in our sample. GWAS p values for ASD (Grove et al., 2019) and SCZ (Ripke et al., 2014) were assigned to modules based on overlapping positions with eSNP LD block annotations. To assess significance, 1000 permutations were performed for each module, randomly selecting the number of annotations corresponding to the number of genes in each module from all eSNP LD block annotations (Figures S7E–S7G). Significance was calculated by the proportion of permuted observed p values at the expected p value of 0.001 that are larger than the actual module’s observed p value at the expected p value of 0.001.
TWAS
We performed SCZ and intercranial volume TWASs using the FUSION package (http://gusevlab.org/projects/fusion/) with gene and splice expression measured in prenatal brain tissue. To identify genes with evidence of genetic control, we used GCTA (Yang et al., 2011) software to estimate cis-SNP heritability h2g (+−1MB window around gene TSS). We identified 3,784 genes and 5,738 splicing-events with significant cis-h2g (nominal p value < 0.05), which were used to calculate the SNP-based predictive weights per gene/intron.
Using the FUSION package, five-fold cross-validation of five models of expression prediction (best cis-eQTL, best linear unbiased predictor, Bayesian sparse linear mixed model (BSLMM) (Zhou et al., 2013), Elastic-net regression, LASSO regression) were calculated and evaluated for accuracy. The model with largest cross-validation R2 was chosen for downstream association analyses.
TWAS statistics were calculated using prenatal weights, as well as published adult expression weights (Gusev et al., 2018) calculated from the CMC eQTL dataset for gene and intron level weights (Fromer et al., 2016) and GTEx Cortex expression gene weights (http://gusevlab.org/projects/fusion/), LD SNP correlations from the 1000 Genomes European Phase 3 reference panel (as the GWAS used are from European populations) (1000 Genomes Project Consortium et al., 2015), and GWAS summary statistics from CLOZUK +PGC SCZ GWAS (105,318 individuals) (Pardiñas et al., 2018; Ripke et al., 2014) and CHARGE ENIGMA Meta-Analysis ICV GWAS (26,577 individuals) (Adams et al., 2016). TWAS association statistics were Bonferroni corrected per GWAS, gene and intron separately. Additional Fusion parameters include running colocalization by COLOC (-coloc_P 0.05) and increasing the maximum fraction of missing SNPs to be imputed due to ancestry differences between the prenatal eQTLs and the GWAS (-max_impute 0.06).
We created a set of high confident adult brain TWAS gene associations, designated by being significant in 2 of 3 adult brain datasets (CMC, GTEx Cortex, and PsychENCODE). The CMC, GTEx Cortex weights were run with the CLOZUK+ PCG SCZ GWAS and PsychENCODE SCZ TWAS results were downloaded from Gandal et al. (2018b)
Overlap of TWAS hits (+−500kb) with GWAS significant loci (LD block reported in paper) was assessed for both prenatal TWAS hits and CMC, GTEx cortex, and PsychENCODE adult prefrontal cortex TWAS hits. Novel regions were identified by genes and introns (+−500k) that did not overlap a GWAS significant loci, and then regions were merged if the gene/intron +−500kb overlapped.
Summary-data Bases Mendelian Randomization (SMR)
We performed summary based mendelian randomization (SMR) (Zhu et al., 2016) to identify gene-trait associations by integrating eQTL and GWAS data, as a complementary approach to TWAS. All nominal snp-gene pair prenatal eQTLs and CLOZUK+ PGC SCZ GWAS (Pardiñas et al., 2018) were used as input to SMP. The set based SMR test was run (-smr-multi) using SNPs in the cis window with a Peql < 10−4 (-peqtl-smr 1e-4) and the associated HEIDI test. For replication of significant TWAS hits, we require genes to have a significant SMR association (PSMR < 0.05) and HEIDI test (PHEIDI < 0.05).
FOCUS fine mapping
TWAS association statistics at genomic risk regions tend to be correlated as a function of linkage and eQTL overlap between predictive models (Mancuso et al., 2019; Wainberg et al., 2017). To prioritize candidate susceptibility genes in our TWAS we performed statistical fine-mapping using FOCUS (Mancuso et al., 2019). FOCUS models the correlation structure induced by LD and overlapping eQTL weights across predictive models and computes posterior probabilities for a gene/intron to explain all observed TWAS association signal at a region. To account for missing gene predictions models, we include gene expression prediction models for genes for genes not predictable from the prenatal brain dataset across 45 tissues measured in 47 reference panels and include the model with the best accuracy among the other tissues. We next computed 90% credible gene-sets by taking genes with largest posterior probability until 90% density was explained.
DATA AND CODE AVAILABILITY
The RNaseq and genotype dataset used for eQTL and sQTL discovery generated in this study are available at dbGaP with accession number: phs001900.
Additional Resources
We have created DevEloPing Cortex Transcriptome (DEPICT) viewer (https://labs.dgsom.ucla.edu/geschwind/pages/eqtl-browser), a web resource for viewing summary level eQTL and sQTL.
Supplementary Material
KEY RESOURCES TABLE
Highlights.
We identify eQTLs and sQTLs specific to human prenatal brain development
Regulation of expression and splicing differs substantially across development
Distinct relationships exist between expression, splicing, and disease risk
Variation in splicing carries as much, if not more, disease risk than expression
ACKNOWLEDGMENTS
Tissue was collected from the UCLA CFAR (5P30 AI028697). RNA-seq libraries were sequenced by the UCLA Neuroscience Genomics Core. We specifically thank Sandeep Deverasetty for creating the web browser. This work was supported by the NIH (5R37 MH060233, 5R01 MH094714, and 1R01 MH110927 to D.H.G.; R00MH102357 to J.L.S.; and 1F32MH114620 toG.R.) and the National Institute of Neurological Disorders and Stroke of the NIH Training Grant (T32NS048004 to R.L.W.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2019.09.021.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams HH, Hibar DP, Chouraki V, Stein JL, Nyquist PA, Rentería ME, Trompet S, Arias-Vasquez A, Seshadri S, Desrivières S, et al. (2016). Novel genetic loci underlying human intracranial volume identified through genome-wide association. Nat. Neurosci 19, 1569–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almaguer-Mederos LE, Mesa JML, González-Zaldívar Y, Almaguer-Go-tay D, Cuello-Almarales D, Aguilera-Rodríguez R, Falcón NS, Gispert S, Auburger G, and Velázquez-Pérez L. (2018). Factors associated with ATXN2 CAG/CAA repeat intergenerational instability in Spinocerebellar ataxia type 2. Clin.Genet 94, 346–350. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W. (2015). HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbiza L, Gronau I, Aksoy BA, Hubisz MJ, Gulko B, Keinan A, and Siepel A. (2013). Genome-wide inference of natural selection on human transcription factor binding sites. Nat. Genet 45, 723–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assoum M, Philippe C, Isidor B, Perrin L, Makrythanasis P, Sondheimer N, Paris C, Douglas J, Lesca G, Antonarakis S, et al. (2016). Autosomal-Recessive Mutations in AP3B2, Adaptor-Related Protein Complex 3 Beta 2 Subunit, Cause an Early-Onset Epileptic Encephalopathy with Optic Atrophy. Am. J. Hum. Genet 99, 1368–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium (2017). Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol. Autism 8, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala R, Willhoft O, Aramayo RJ, Wilkinson M, McCormack EA, Ocloo L, Wigley DB, and Zhang X. (2018). Structure and regulation of the human IN080-nucleosome complex. Nature 556, 391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae BI, Jayaraman D, and Walsh CA (2015). Genetic changes shaping the human brain. Dev. Cell 32, 423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barca E, Ganetzky RD, Potluri P, Juanola-Falgarona M, Gai X, Li D, Jalas C, Hirsch Y, Emmanuele V, Tadesse S, et al. (2018). USMG5 Ashkenazi Jewish founder mutation impairs mitochondrial complex V dimerization and ATP synthesis. Hum. Mol. Genet 27, 3305–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu AC, Tsai GE, Ma CL, Ehmsen JT, Mustafa AK, Han L, Jiang ZI, Benneyworth MA, Froimowitz MP, Lange N, et al. (2009). Targeted disruption of serine racemase affects glutamatergic neurotransmission and behavior. Mol. Psychiatry 14, 719–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle A, Khan Z, Wang SH, Mitrano A, Ford MJ, Pritchard JK, and Gilad Y. (2015). Genomic variation. Impact of regulatory variation from RNA to protein. Science 347, 664–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B Met 57, 289–300. [Google Scholar]
- Benyamin B, Pourcain B, Davis OS, Davies G, Hansell NK, Brion MJ, Kirkpatrick RM, Cents RA, Franić S, Miller MB, et al. ; Wellcome Trust Case Control Consortium 2 (WTCCC2) (2014). Childhood intelligence is heritable, highly polygenic and associated with FNBP1L. Mol. Psychiatry 19, 253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier R, Golzio C, Xiong B, Stessman HA, Coe BP, Penn O, Witherspoon K, Gerdts J, Baker C, Vulto-van Silfhout AT, et al. (2014). Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158, 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier R, Hudac CM, Chen Q, Zeng C, Wallace AS, Gerdts J, Earl R, Peterson J, Wolken A, Peters A, et al. ; Simons VIP consortium (2017). Developmental trajectories for young children with 16p11.2 copy numbervariation. Am. J. Med. Genet. B. Neuropsychiatr. Genet 174, 367–380. [DOI] [PubMed] [Google Scholar]
- Bestman JE, Huang LC, Lee-Osbourne J, Cheung P, and Cline HT (2015). An in vivo screen to identify candidate neurogenic genes in the developing Xenopus visual system. Dev. Biol 408, 269–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BrainSpan(2013). BrainSpan: Atlas of the Developing Human Brain(Allen Institute for Brain Science). [Google Scholar]
- Browning BL, and Browning SR (2016). Genotype Imputation with Millions of Reference Samples. Am. J. Hum. Genet 98, 116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryois J, Garrett ME, Song L, Safi A, Giusti-Rodriguez P, Johnson GD, Shieh AW, Buil A, Fullard JF, Roussos P, et al. (2018). Evaluation of chromatin accessibility in prefrontal cortex of individuals with schizophrenia. Nat. Commun 9, 3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R. (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvill GL, Heavin SB, Yendle SC, McMahon JM, O’Roak BJ, Cook J, Khan A, Dorschner MO, Weaver M, Calvert S, et al. (2013). Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat. Genet 45, 825–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, and Lee JJ (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Calhoun VD, Perrone-Bizzozero NI, Pearlson GD, Sui J, Du Y, and Liu J. (2016). A pilot study on commonality and specificity of copy number variants in schizophrenia and bipolar disorder. Transl. Psychiatry 6, e824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR, Iwasa J, Cairns BR, and Peterson CL (2017). Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol 18, 407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockerill PN (2011). Structure and function of active chromatin and DNase I hypersensitive sites. FEBS J. 278, 2182–2210. [DOI] [PubMed] [Google Scholar]
- Cohen OS, Mccoy SY, Middleton FA, Bialosuknia S, Zhang-James Y, Liu L, Tsuang MT, Faraone SV, and Glatt SJ (2012). Transcriptomic analysis of postmortem brain identifies dysregulated splicing events in novel candidate genes for schizophrenia. Schizophr. Res 142, 188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT, Colantuoni EA, Elkahloun AG, Herman MM, Weinberger DR, and Kleinman JE (2011). Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 478, 519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium CONVERGE (2015). Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 523, 588–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotney J, Muhle RA, Sanders SJ, Liu L, Willsey AJ, Niu W, Liu W, Klei L, Lei J, Yin J, et al. (2015). The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat. Commun 6, 6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darvish H, Azcona LJ, Tafakhori A, Ahmadi M, Ahmadifard A, and Paisán-Ruiz C. (2017). Whole genome sequencing identifies a novel homozygous exon deletion in the NT5C2 gene in a family with intellectual disability and spastic paraplegia. NPJ Genom. Med 2, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre-Ubieta L, Stein JL, Won H, Opland CK, Liang D, Lu D, and Geschwind DH (2018). The Dynamic Landscape of Open Chromatin during Human Cortical Neurogenesis. Cell 172, 289–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. ; DDD Study; Homozygosity Mapping Collaborative for Autism; UK10K Consortium (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demontis D, Walters RK, Martin J, Mattheisen M, Als TD, Agerbo E, Baldursson G, Belliveau R, Bybjerg-Grauholm J, Bækvad-Hansen M, et al. ; ADHD Working Group of the Psychiatric Genomics Consortium (PGC); Early Lifecourse & Genetic Epidemiology (EAGLE) Consortium; 23andMe Research Team (2019). Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet 51, 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimas AS, Deutsch S, Stranger BE, Montgomery SB, Borel C, Attar-Cohen H, Ingle C, Beazley C, Gutierrez Arcelus M, Sekowska M, et al. (2009). Common regulatory variation impacts gene expression in a cell type-dependent manner. Science 325, 1246–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durak O, Gao F, Kaeser-Woo YJ, Rueda R, Martorell AJ, Nott A, Liu CY, Watson LA, and Tsai LH (2016). Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat. Neurosci 19, 1477–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, and Huber W. (2009). Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc 4, 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eising E, Carrion-Castillo A, Vino A, Strand EA, Jakielski KJ, Scerri TS, Hildebrand MS, Webster R, Ma A, Mazoyer B, et al. (2019). A set of regulatory genes co-expressed in embryonic human brain is implicated in disrupted speech development. Mol. Psychiatry 24, 1065–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, and Kellis M. (2015). Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nat. Biotechnol 33, 364–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. (2011). Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escamilla CO, Filonova I, Walker AK, Xuan ZX, Holehonnur R, Espinosa F, Liu S, Thyme SB, López-García IA, Mendoza DB, et al. (2017). Kctd13 deletion reduces synaptic transmission via increased RhoA. Nature 551, 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane HK, Bulik-Sullivan B, Gusev A,Trynka G, Reshef Y, Loh PR, Anttila V, Xu H, Zang C, Farh K, et al. ; ReproGen Consortium; Schizophrenia Working Group of the Psychiatric Genomics Consortium; RACI Consortium (2015). Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet 47, 1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E, Palta P, Ruderfer DM, et al. (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, Ruderfer DM, Oh EC, Topol A, Shah HR, et al. (2016). Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci 19, 1442–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal MJ, Leppa V, Won H, Parikshak NN, and Geschwind DH (2016). The road to precision psychiatry: translating genetics into disease mechanisms. Nat. Neurosci 19, 1397–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal MJ, Haney JR, Parikshak NN, Leppa V, Ramaswami G, Hartl C, Schork AJ, Appadurai V, Buil A, Werge TM, et al. ; CommonMind Consortium; PsychENCODE Consortium; iPSYCH-BROAD Working Group (2018a). Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 359, 693–697.29439242 [Google Scholar]
- Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, Won H, van Bakel H, Varghese M, Wang Y, et al. (2018b). Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362, eaat8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrits A, Li Y, Tesson BM, Bystrykh LV, Weersing E, Ausema A, Dontje B, Wang X, Breitling R, Jansen RC, and de Haan G. (2009). Expression quantitative trait loci are highly sensitive to cellular differentiation state. PLoS Genet. 5, e1000692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, and Flint J. (2015). Genetics and genomics of psychiatric disease. Science 349, 1489–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, and Rakic P. (2013). Cortical evolution: judgethe brain by its cover. Neuron 80, 633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman SR, Chang J, Xu B, Bawa TS, Gogos JA, Karayiorgou M, and Vitkup D. (2012). Diverse types of genetic variation converge on functional gene networks involved in schizophrenia. Nat. Neurosci 15, 1723–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Dennis MY, Baker C, Malig M, Coe BP, Campbell CD, Mark K, Vu TH, Alkan C, Cheng Z, et al. (2013). Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. Am. J. Hum. Genet 92, 221–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, et al. (2009). Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golzio C, Willer J, Talkowski ME, Oh EC, Taniguchi Y, Jacquemont S, Reymond A, Sun M, Sawa A, Gusella JF, et al. (2012). KCTD13is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature 485, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammatikakis I, Zhang P, Panda AC, Kim J, Maudsley S, Abdelmohsen K, Yang X, Martindale JL, Motifno O, Hutchison ER, et al. (2016). Alternative Splicing of Neuronal Differentiation Factor TRF2 Regulated by HNRNPH1/H2. Cell Rep. 15, 926–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratten J, Wray NR, Keller MC, and Visscher PM (2014). Large-scale genomics unveils the genetic architecture of psychiatric disorders. Nat. Neurosci 17, 782–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, Pallesen J, Agerbo E, Andreassen OA, Anney R, et al. ; Autism Spectrum Disorder Working Group of the Psychiatric Genomics Consortium; BUPGEN; Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium; 23andMe Research Team (2019). Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet 51, 431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium (2015). Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium, Aguet F, Brown AA, Castel SE, Davis JR, He Y, Jo B, Mohammadi P, Park Y, Parsana P, et al. (2017). Genetic effects on gene expression across human tissues. Nature 550, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulsuner S, Walsh T, Watts AC, Lee MK, Thornton AM, Casadei S, Rippey C, Shahin H, et al. ; Consortium on the Genetics of Schizophrenia (COGS) PAARTNERS Study Group (2013). Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 154,518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BW, Jansen R, de Geus EJ, Boomsma DI, Wright FA, et al. (2016). Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet 48, 245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev A, Mancuso N, Won H, Kousi M, Finucane HK, Reshef Y, Song L, Safi A, Schizophrenia Working Group of the Psychiatric Genomics Consortium, and McCarroll S, et al. (2018). Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nat. Genet 50, 538–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne F, and Ivanek R. (2016). Visualizing Genomic Data Using Gviz and Bioconductor. Methods Mol. Biol 1418, 335–351. [DOI] [PubMed] [Google Scholar]
- Hannon E, Spiers H, Viana J, Pidsley R, Burrage J, Murphy TM, Troakes C, Turecki G, O’Donovan MC, Schalkwyk LC, et al. (2016). Methylation QTLs in the developing brain and their enrichment in schizophrenia risk loci. Nat. Neurosci 19, 48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Irizarry RA, and Wu Z. (2012). Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics 13, 204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson E, Bernier R, Porche K, Jackson FI, Goin-Kochel RP, Snyder LG, Snow AV, Wallace AS, Campe KL, Zhang Y, et al. ; Simons Variation in Individuals Project Consortium (2015). The cognitive and behavioral phenotype of the 16p11.2 deletion in a clinically ascertained population. Biol. Psychiatry 77, 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. (2012). GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto R, Ohi K, Okada T, Yasuda Y, Yamamori H, Hori H, Hikita T, Taya S, Saitoh O, Kosuga A, et al. (2009). Association analysis between schizophrenia and the AP-3 complex genes. Neurosci. Res 65, 113–115. [DOI] [PubMed] [Google Scholar]
- Hawrylycz M, Miller JA, Menon V, Feng D, Dolbeare T, Guillozet-Bongaarts AL, Jegga AG, Aronow BJ, Lee CK, Bernard A, et al. (2015). Canonical genetic signatures of the adult human brain. Nat. Neurosci 18, 1832–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingason A, Giegling I, Hartmann AM, Genius J, Konte B, Friedl M, Schizophrenia Working Group of the Psychiatric Genomics Consortium (PGC), Ripke S, Sullivan PF, St Clair D, et al. (2015). Expression analysis in a rat psychosis model identifies novel candidate genes validated in a large case-control sample of schizophrenia. Transl. Psychiatry 5, e656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap Consortium (2003). The International HapMap Project. Nature 426, 789–796. [DOI] [PubMed] [Google Scholar]
- International League Against Epilepsy Consortium on Complex Epilepsies (2014). Genetic determinants of common epilepsies: a meta-analysis of genome-wide association studies. Lancet Neurol. 13, 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Schizophrenia Consortium (2008). Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, et al. (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515,216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia M, Weatheritt RJ, Ellis JD, Parikshak NN, Gonatopoulos-Pournatzis T, Babor M, Quesnel-Vallieres M, Tapial J, Raj B, O’Hanlon D, et al. (2014). A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell 159, 1511–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AE, Gao Y, Deep-Soboslay A, Tao R, Hyde TM, Weinberger DR, and Kleinman JE (2016). Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat. Neurosci 19, 40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AE, Straub RE, Shin JH, Tao R, Gao Y, Collado-Torres L, Kam-Thong T, Xi HS, Quan J, Chen Q, et al. ; BrainSeq Consortium (2018). Developmental and genetic regulation of the human cortex transcriptome illuminate schizophrenia pathogenesis. Nat. Neurosci 21, 1117–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C, and Rabinovic A. (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- Johnson MB, Kawasawa YI, Mason CE, Krsnik Z, Coppola G, Bogdanović D, Geschwind DH, Mane SM, State MW, and Sestan N. (2009). Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron 62, 494–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. ; International IBD Genetics Consortium (IIBDGC) (2012). Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491,119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun G, Flickinger M, Hetrick KN, Romm JM, Doheny KF, Abecasis GR, Boehnke M, and Kang HM (2012). Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. Am. J. Hum. Genet 91, 839–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, and Eskin E. (2008). Efficient control of population structure in model organism association mapping. Genetics 178, 1709–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, Sousa AM, Pletikos M, Meyer KA, Sedmak G, et al. (2011). Spatio-temporal transcriptome of the human brain. Nature 478, 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Xia K, Tao R, Giusti-Rodriguez P, Vladimirov V, van den Oord E, and Sullivan PF (2014). A meta-analysis of gene expression quantitative trait loci in brain. Transl. Psychiatry 4, e459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostović I, and Jovanov-Milosević N. (2006). The development of cerebral connections during the first 20–45 weeks’ gestation. Semin. Fetal Neonatal Med 11,415–422. [DOI] [PubMed] [Google Scholar]
- Krumm N, Turner TN, Baker C, Vives L, Mohajeri K, Witherspoon K, Raja A, Coe BP, Stessman HA, He ZX, et al. (2015). Excess of rare, inherited truncating mutations in autism. Nat. Genet 47, 582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CS, Fisher SE, Hurst JA, Vargha-Khadem F, and Monaco AP (2001). A forkhead-domain gene is mutated in a severe speech and language disorder. Nature 413, 519–523. [DOI] [PubMed] [Google Scholar]
- Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, et al. ; European Alzheimer’s Disease Initiative (EADI); Genetic and Environmental Risk in Alzheimer’s Disease; Alzheimer’s Disease Genetic Consortium; Cohorts for Heart and Aging Research in Genomic Epidemiology (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet 45, 1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, and Horvath S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, and Horvath S. (2012). Fast R Functions for Robust Correlations and Hierarchical Clustering. J. Stat. Softw 46, i11. [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T, Sammeth M, Friedländer MR, ‘t Hoen PA, Monlong J, Rivas MA, Gonzàlez-Porta M, Kurbatova N, Griebel T, Ferreira PG, et al. (2013). Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latourelle JC, Dumitriu A, Hadzi TC, Beach TG, and Myers RH (2012). Evaluation of Parkinson disease risk variants as expression-QTLs. PLoS ONE 7, e46199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, and Carey VJ (2013). Software for computing and annotating genomic ranges. PLoS Comput. Biol 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, and Storey JD (2007). Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 3, 1724–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. ; Exome Aggregation Consortium (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinson DF, Duan J, Oh S, Wang K, Sanders AR, Shi J, Zhang N, Mowry BJ, Olincy A, Amin F, et al. (2011). Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am. J. Psychiatry 168, 302–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YI, van de Geijn B, Raj A, Knowles DA, Petti AA, Golan D, Gilad Y, and Pritchard JK (2016). RNA splicing is a primary link between genetic variation and disease. Science 352, 600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YI, Knowles DA, Humphrey J, Barbeira AN, Dickinson SP, Im HK, and Pritchard JK (2018). Annotation-free quantification of RNA splicing using LeafCutter. Nat. Genet 50, 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra D, and Sebat J. (2012). CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell 148, 1223–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancarci BO, Toker L, Tripathy SJ, Li B, Rocco B, Sibille E, and Pavlidis P. (2017). Cross-Laboratory Analysis of Brain Cell Type Transcriptomes with Applications to Interpretation of Bulk Tissue Data. eNeuro 4, ENEUR0.0212–17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso N, Freund MK, Johnson R, Shi H, Kichaev G, Gusev A, and Pasaniuc B. (2019). Probabilistic fine-mapping of transcriptome-wide association studies. Nat. Genet 51, 675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, et al. (2008). Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet 82, 477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. (2012). Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy SE, Makarov V, Kirov G, Addington AM, McClellan J, Yoon S, Perkins DO, Dickel DE, Kusenda M, Krastoshevsky O, et al. ; Wellcome Trust Case Control Consortium (2009). Microduplications of 16p11.2 are associated with schizophrenia. Nat. Genet 41, 1223–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy SE, Gillis J, Kramer M, Lihm J, Yoon S, Berstein Y, Mistry M, Pavlidis P, Solomon R, Ghiban E, et al. (2014). De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol. Psychiatry 19, 652–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, and Cunningham F. (2016). The Ensembl Variant Effect Predictor. Genome Biol. 17, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson JJ, Shi Y, Gujral M, Zheng H, Malhotra D, Jin X, Jian M, Liu G, Greer D, Bhandari A, et al. (2012). Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell 151, 1431–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Ding SL, Sunkin SM, Smith KA, Ng L, Szafer A, Ebbert A, Riley ZL, Royall JJ, Aiona K, et al. (2014). Transcriptional landscape of the prenatal human brain. Nature 508, 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi P, Castel SE, Brown AA, and Lappalainen T. (2017). Quantifying the regulatory effect size of cis-acting genetic variation using allelic fold change. Genome Res. 27, 1872–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-De-Luca D, SGENE Consortium, Mulle JG, Simons Simplex Collection Genetics Consortium, Kaminsky EB, Sanders SJ, GeneSTAR, Myers SM, Adam MP, Pakula AT, et al. (2010). Deletion 17q12 is a recurrent copy number variant that confers high risk of autism and schizophrenia. Am. J. Hum. Genet 87, 618–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-De-Luca D, Sanders SJ, Willsey AJ, Mulle JG, Lowe JK, Geschwind DH, State MW, Martin CL, and Ledbetter DH (2013). Using large clinical data sets to infer pathogenicity for rare copy number variants in autism cohorts. Mol. Psychiatry 18, 1090–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafavi S, Battle A, Zhu X, Urban AE, Levinson D, Montgomery SB, and Koller D. (2013). Normalizing RNA-sequencing data by modeling hidden covariates with prior knowledge. PLoS ONE 8, e68141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepusz G, and Csárdi G. (2006). The igraph software package for complex network research. Complex Syst. 1695, 1–9. [Google Scholar]
- Nica AC, and Dermitzakis ET (2013). Expression quantitative trait loci: present and future. Philos. Trans. R. Soc. Lond. B Biol. Sci 368, 20120362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nica AC, Parts L, Glass D, Nisbet J, Barrett A, Sekowska M, Travers M, Potter S, Grundberg E, Small K, et al. ; MuTHER Consortium (2011).The architecture of gene regulatory variation across multiple human tissues: the MuTHER study. PLoS Genet. 7, e1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nord AS, Pattabiraman K, Visel A, and Rubenstein JLR (2015). Genomic perspectives of transcriptional regulation in forebrain development. Neuron 85, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien HE, Hannon E, Hill MJ, Toste CC, Robertson MJ, Morgan JE, McLaughlin G, Lewis CM, Schalkwyk LC, Hall LS, et al. (2018). Expression quantitative trait loci in the developing human brain and their enrichment in neuropsychiatric disorders. Genome Biol. 19, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, et al. (2012). Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Stessman HA, Boyle EA, Witherspoon KT, Martin B, Lee C, Vives L, Baker C, Hiatt JB, Nickerson DA, et al. (2014). Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat. Commun 5, 5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsakaya S, Fujikawa M, Hisabori T, and Yoshida M. (2011). Knockdown of DAPIT (diabetes-associated protein in insulin-sensitive tissue) results in loss of ATP synthase in mitochondria. J. Biol. Chem 286, 20292–20296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojeda SR, Hill J, Hill DF, Costa ME, Tapia V, Cornea A, and Ma YJ (1999). The Oct-2 POU domain gene in the neuroendocrine brain: a transcriptional regulator of mammalian puberty. Endocrinology 140, 3774–3789. [DOI] [PubMed] [Google Scholar]
- Okbay A, Beauchamp JP, Fontana MA, Lee JJ, Pers TH, Rietveld CA, Turley P, Chen GB, Emilsson V, Meddens SF, et al. ; LifeLines Cohort Study (2016). Genome-wide association study identifies 74 loci associated with educational attainment. Nature 533, 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ongen H, Buil A, Brown AA, Dermitzakis ET, and Delaneau O. (2016). Fast and efficient QTL mapper for thousands of molecular phenotypes. Bioinformatics 32, 1479–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyama S, Yamakawa H, Sasagawa N, Hosoi Y, Futai E, and Ishiura S. (2009). Dysbindin-1, a schizophrenia-related protein, functionally interacts with the DNA-dependent protein kinase complex in an isoform-dependent manner. PLoS ONE 4, e4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagès H, Carlson M, Falcon S, and Li N. (2018). AnnotationDbi: Annotation Database Interface. R package version 1421. [Google Scholar]
- Pardiñas AF, Holmans P, Pocklington AJ, Escott-Price V, Ripke S, Carrera N, Legge SE, Bishop S,Cameron D, Hamshere ML,et al. ;GERAD1 Consortium; CRESTAR Consortium (2018). Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet 50, 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, and Geschwind DH (2013). Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Gandal MJ, and Geschwind DH (2015). Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat. Rev. Genet 16, 441–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Swarup V, Belgard TG, Irimia M, Ramaswami G, Gandal MJ, Hartl C, Leppa V, Ubieta LT, Huang J, et al. (2016). Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 540, 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polderman TJ, Benyamin B, de Leeuw CA, Sullivan PF, van Bochoven A, Visscher PM, and Posthuma D. (2015). Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat. Genet 47, 702–709. [DOI] [PubMed] [Google Scholar]
- Polioudakis D, de la Torre-Ubieta L, Langerman J, Elkins AG, Shi X, Stein JL, Vuong CK, Nichterwitz S, Gevorgian M, Opland CK, et al. (2019). A Single-Cell Transcriptomic Atlas of Human Neocortical Development during Mid-gestation. Neuron 103, 785–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollen AA, Nowakowski TJ, Chen J, Retallack H, Sandoval-Espinosa C, Nicholas CR, Shuga J, Liu SJ, Oldham MC, Diaz A, et al. (2015). Molecular identity of human outer radial glia during cortical development. Cell 163, 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preciados M, Yoo C, and Roy D. (2016). Estrogenic Endocrine Disrupting Chemicals Influencing NRF1 Regulated Gene Networks in the Development of Complex Human Brain Diseases. Int. J. Mol. Sci 17, 2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesnel-Vallieres M, Irimia M, Cordes SP, and Blencowe BJ (2015). Essential roles for the splicing regulator nSR100/SRRM4 during nervous system development. Genes Dev. 29, 746–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj B, Irimia M, Braunschweig U, Sterne-Weiler T, O’Hanlon D, Lin ZY, Chen GI, Easton LE, Ule J, Gingras AC, et al. (2014).Aglobal regulatory mechanism for activating an exon network required for neurogenesis. Mol. Cell 56, 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P. (1995). A small step for the cell, a giant leap for mankind: a hypothesis of neocortical expansion during evolution. Trends Neurosci. 18, 383–388. [DOI] [PubMed] [Google Scholar]
- Rakic P. (2009). Evolution ofthe neocortex: a perspective from developmental biology. Nat. Rev. Neurosci 10, 724–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramasamy A, Trabzuni D, Guelfi S, Varghese V, Smith C, Walker R, De T, Coin L, de Silva R, Cookson MR, et al. ; UK Brain Expression Consortium; North American Brain Expression Consortium (2014). Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat. Neurosci 17, 1418–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rietveld CA, Medland SE, Derringer J, Yang J, Esko T, Martin NW, Westra HJ, Shakhbazov K, Abdellaoui A, Agrawal A, et al. ; LifeLines Cohort Study (2013). GWAS of 126,559 individuals identifies genetic variants associated with educational attainment. Science 340, 1467–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, O’Dushlaine C, Chambert K, Moran JL, Kahler AK, Akterin S, Bergen SE, Collins AL, Crowley JJ, Fromer M, et al. ; Multicenter Genetic Studies ofSchizophrenia Consortium; Psychosis Endophenotypes International Consortium; Wellcome Trust Case Control Consortium 2 (2013). Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet 45, 1150–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, Neale BM, Corvin A, Walters JTR, Farh K-H, Holmans PA, Lee P, Bulik-Sullivan B, Collier DA, Huang H, et al. ; Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rujescu D, Ingason A, Cichon S, Pietilainen OP, Barnes MR, Toulopoulou T, Picchioni M, Vassos E, Ettinger U, Bramon E, et al. ; GROUP Investigators (2009). Disruption of the neurexin 1 gene is associated with schizophrenia. Hum. Mol. Genet 18, 988–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzzo EK, Perez-Cano L, Jung JY, Wang LK, Kashef-Haghighi D, Hartl C, Singh C, Xu J, Hoekstra JN, Leventhal O, et al. (2019). Inherited and De Novo Genetic Risk for Autism Impacts Shared Networks. Cell 178, 850–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM, Kosmicki JA, Rehnström K, Mallick S, Kirby A, et al. (2014). A framework for the interpretation of de novo mutation in human disease. Nat. Genet 46, 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, et al. (2011). Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70, 863–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaid DJ, Chen W, and Larson NB (2018). From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet 19, 491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub MA, Boyle AP, Kundaje A, Batzoglou S, and Snyder M. (2012). Linking disease associations with regulatory information in the human genome. Genome Res. 22, 1748–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EM, Zhang J, Zhou W, Chen J, Mohlke KL, Chen YE, and Willer CJ (2015). GREGOR: evaluating global enrichment of trait-associated variants in epigenomic features using a systematic, data-driven approach. Bioinformatics 31, 2601–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgouros S, Goldin JH, Hockley AD, Wake MJ, and Natarajan K. (1999). Intracranial volume change in childhood. J. Neurosurg 91, 610–616. [DOI] [PubMed] [Google Scholar]
- Shabalin AA (2012). Matrix eQTL: ultrafast eQTL analysis via large matrix operations. Bioinformatics 28, 1353–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinawi M, Liu P, Kang SH, Shen J, Belmont JW, Scott DA, Probst FJ, Craigen WJ, Graham BH, Pursley A, et al. (2010). Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J. Med. Genet 47, 332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. (2005). Evolutionary conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silbereis JC, Pochareddy S, Zhu Y, Li M, and Sestan N. (2016). The Cellular and Molecular Landscapes ofthe Developing Human Central Nervous System. Neuron 89, 248–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokpor G, Castro-Hernandez R, Rosenbusch J, Staiger JF, and Tuoc T. (2018). ATP-Dependent Chromatin Remodeling During Cortical Neurogenesis. Front. Neurosci 12, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Helgason A, Thorleifsson G, Steinthorsdottir V, Masson G, Barnard J, Baker A, Jonasdottir A, Ingason A, Gudnadottir VG, et al. (2005). A common inversion under selection in Europeans. Nat. Genet 37, 129–137. [DOI] [PubMed] [Google Scholar]
- Storey JD, and Tibshirani R. (2003). Statistical significancefor genomewide studies. Proc. Natl. Acad. Sci. USA 1OO, 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, Beazley C, Ingle CE, Dunning M, Flicek P, Koller D, et al. (2007). Population genomics of human gene expression. Nat. Genet 39, 1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strunz T, Grassmann F, Gayán J, Nahkuri S, Souza-Costa D, Maugeais C, Fauser S, Nogoceke E, and Weber BHF (2018). A mega-analysis of expression quantitative trait loci (eQTL) provides insight into the regulatory architecture of gene expression variation in liver. Sci. Rep 8, 5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugathan A, Biagioli M, Golzio C, Erdin S, Blumenthal I, Manavalan P, Ragavendran A, Brand H, Lucente D, Miles J, et al. (2014). CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc. Natl. Acad. Sci. USA 111, E4468–E4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suls A, Jaehn JA, Kecskés A, Weber Y, Weckhuysen S, Craiu DC, Siekierska A, Djémié T, Afrikanova T, Gormley P, et al. ; EuroEPINOMICS RES Consortium (2013). De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am. J. Hum. Genet 93, 967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunkin SM, Ng L, Lau C, Dolbeare T, Gilbert TL, Thompson CL, Hawrylycz M, and Dang C. (2013). Allen Brain Atlas: an integrated spatio-temporal portal for exploring the central nervous system. Nucleic Acids Res. 41, D996–D1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taal HR, Pourcain BS, Thiering E, Das S, Mook-Kanamori DO, Warrington NM, Kaakinen M, Kreiner-M0ller E, Bradfield JP, Freathy RM, et al. ; Cohorts for Heart and Aging Research in Genetic Epidemiology (CHARGE) Consortium; Early Genetics & Lifecourse Epidemiology (EAGLE) consortium; Early Growth Genetics (EGG) Consortium (2012). Common variants at 12q15 and 12q24 are associated with infant head circumference. Nat. Genet 44, 532–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata A, Matsumoto N, and Kato T. (2017). Genome-wide identification of splicing QTLs in the human brain and their enrichment among schizophrenia-associated loci. Nat. Commun 8, 14519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Menon V, Nguyen TN, Kim TK, Jarsky T, Yao Z, Levi B, Gray LT, Sorensen SA, Dolbeare T, et al. (2016). Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci 19, 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavassoli T, Kolevzon A, Wang AT, Curchack-Lichtin J, Halpern D, Schwartz L, Soffes S, Bush L, Grodberg D, Cai G, and Buxbaum JD (2014). De novo SCN2Asplicesite mutation in a boywith Autism spectrum disorder. BMC Med. Genet 15, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner SD (2014). qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots. bioRxiv. 10.1101/005165. [DOI] [Google Scholar]
- Turner TN, Hormozdiari F, Duyzend MH, McClymont SA, Hook PW, Iossifov I, Raja A, Baker C, Hoekzema K, Stessman HA, et al. (2016). Genome Sequencing of Autism-Affected Families Reveals Disruption of Putative Noncoding Regulatory DNA. Am. J. Hum. Genet 98, 58–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TN, Yi Q, Krumm N, Huddleston J, Hoekzema K, F Stessman HA, Doebley AL, Bernier RA, Nickerson DA, and Eichler EE (2017). denovo-db: a compendium of human de novo variants. Nucleic Acids Res. 45, D804–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacic V, McCarthy S, Malhotra D, Murray F, Chou HH, Peoples A, Makarov V, Yoon S, Bhandari A, Corominas R, et al. (2011). Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia. Nature 471, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van derAuwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. (2013). From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10.1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veerappa AM, Saldanha M, Padakannaya P, and Ramachandra NB (2014). Family based genome-wide copy number scan identifies complex rearrangements at 17q21.31 in dyslexics. Am. J. Med. Genet. B. Neuropsychiatr. Genet 165B, 572–580. [DOI] [PubMed] [Google Scholar]
- Veyrieras JB, Kudaravalli S, Kim SY, Dermitzakis ET, Gilad Y, Stephens M, and Pritchard JK (2008). High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet. 4, e1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Rubin EM, and Pennacchio LA (2009). Genomic views of distant-acting enhancers. Nature 461, 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainberg M, Sinnott-Armstrong N, Knowles D, Golan D, Ermel R, Ruusalepp A, Quertermous T, Hao K, Bjorkegren JL, Rivas MA, et al. (2017). Vulnerabilities of transcriptome-wide association studies. bioRxiv. 10.1101/206961. [DOI] [Google Scholar]
- Wang E, Dimova N, and Cambi F. (2007). PLP/DM20 ratio is regulated by hnRNPH and F and a novel G-rich enhancer in oligodendrocytes. Nucleic Acids Res. 35, 4164–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Liu S, Warrell J, Won H, Shi X, Navarro FCP, Clarke D, Gu M, Emani P, Yang YT, et al. ; PsychENCODE Consortium (2018a). Comprehensive functional genomic resource and integrative model for the human brain. Science 362, eaat8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward LD, and Kellis M. (2012). Interpreting noncoding genetic variation in complex traits and human disease. Nat. Biotechnol 30, 1095–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei T, and Simko V. (2017). corrplot: Visualization of a Correlation Matrix (Version 0.84). R package version 1421. [Google Scholar]
- Weinberger DR (1987). Implications of normal brain development for the pathogenesis of schizophrenia. Arch. Gen. Psychiatry 44, 660–669. [DOI] [PubMed] [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T, et al. ; Autism Consortium (2008). Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med 358, 667–675. [DOI] [PubMed] [Google Scholar]
- Willer CJ, Li Y, and Abecasis GR (2010). METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, Reilly SK, Lin L, Fertuzinhos S, Miller JA, et al. (2013). Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winden KD, Oldham MC, Mirnics K, Ebert PJ, Swan CH, Levitt P, Rubenstein JL, Horvath S, and Geschwind DH (2009). The organization of the transcriptional network in specific neuronal classes. Mol. Syst. Biol 5, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won H, de laTorre-Ubieta L, Stein JL, Parikshak NN, Huang J, Opland CK, Gandal MJ, Sutton GJ, Hormozdiari F, Lu D, et al. (2016). Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 538, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Lee SH, Goddard ME, and Visscher PM (2011). GCTA:atool for genome-wide complex trait analysis. Am. J. Hum. Genet 88, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YC, Di C, Hu B, Zhou M, Liu Y, Song N, Li Y, Umetsu J, and Lu ZJ (2015). CLIPdb: a CLIP-seq database for protein-RNA interactions. BMC Genomics 16, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen RK, Thiruvahindrapuram B, Merico D, Walker S, Tammimies K, Hoang N, Chrysler C, Nalpathamkalam T, Pellecchia G, Liu Y, et al. (2015). Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med 21, 185–191. [DOI] [PubMed] [Google Scholar]
- Zhang B, and Horvath S. (2005).Ageneral frame work for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol 4, Article17. [DOI] [PubMed] [Google Scholar]
- Zhang F, Wang G, Shugart YY, Xu Y, Liu C, Wang L, Lu T, Yan H, Ruan Y, Cheng Z, et al. (2014a). Association analysis of a functional variant in ATXN2 with schizophrenia. Neurosci. Lett 562, 24–27. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014b). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MS, Li G, et al. (2016). Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 89, 37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Carbonetto P, and Stephens M. (2013). Polygenic modeling with bayesian sparse linear mixed models. PLoS Genet. 9, e1003264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Shi Y, Li F, Wu X, Huai C, Shen L, Yi Z, He L, Liu C, and Qin S. (2018). Study of the association between Schizophrenia and microduplication at the 16p11.2 locus in the Han Chinese population. Psychiatry Res. 265, 198–199. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, Montgomery GW, Goddard ME, Wray NR, Visscher PM, and Yang J. (2016). Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet 48, 481–487. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNaseq and genotype dataset used for eQTL and sQTL discovery generated in this study are available at dbGaP with accession number: phs001900.
Additional Resources
We have created DevEloPing Cortex Transcriptome (DEPICT) viewer (https://labs.dgsom.ucla.edu/geschwind/pages/eqtl-browser), a web resource for viewing summary level eQTL and sQTL.