

Abstract
Niemann-Pick C is a rare neurodegenerative, lysosomal storage disease caused by accumulation of unesterified cholesterol. Diagnosis of the disease is often delayed due to its rarity, the heterogeneous presentation and the early non-specific symptoms. The discovery of disease-specific biomarkers – cholestane-3β,5α,6β-triol (C-triol), trihydroxycholanic acid glycinate (TCG) and N-palmitoyl-O-phosphocholineserine (PPCS, initially referred to as lysoSM-509) – has led to development of non-invasive, blood-based diagnostics. Dissemination of these rapid, sensitive, and specific clinical assays has accelerated diagnosis. Moreover, the superior receiver operating characteristic of the TCG bile acid biomarker and its detection in dried blood spots has also facilitated development of a newborn screen for NPC, which is currently being piloted in New York state. The C-triol, TCG and PPCS biomarkers have also proven useful for monitoring treatment response in peripheral tissues, but are uninformative with respect to treatment efficacy in the central nervous system (CNS). A major gap for the field is the lack of a validated, non-invasive biomarker to monitor the course of disease and CNS response to therapy.
Introduction
Niemann-Pick disease type C (NPC) is a rare neurovisceral lysosomal storage disorder caused by mutations in either the NPC1 gene (95% cases) [1] or the NPC2 gene (~5% cases) [2]. The disorder has a clinical incidence of 1/100,000–1/120,000 live births [3–5]. Analysis of whole exome sequence databases predict an incidence rate of ~1/90,000 in close agreement with the clinical data [4, 6]. These analyses also identified high frequency NPC1 variants with predicted incidence of 1/19,000–1/39,000, suggesting that there may be a significant under ascertainment of late onset phenotypes [6].
The NPC1 and NPC2 genes encode proteins that are central for regulation of cellular cholesterol homeostasis. NPC1 is a large transmembrane protein that resides in the limiting membrane of the lysosome [7], whereas NPC2 is a small soluble lysosomal protein that binds cholesterol within the lysosomal lumen [2]. Biochemical and structural studies support a hydrophobic handoff model in which NPC2 transfers cholesterol to NPC1 for lysosomal egress [8–10]. Recent structural characterization of the NPC1-NPC2 complex has elucidated the molecular basis of the cholesterol handoff and identified a central tunnel for cholesterol transit from the N-terminal domain to the sterol-sensing domain [11]. Deficiency in either NPC1 or NPC2 proteins impair cholesterol movement from the lysosome. The consequent lysosomal accumulation of unesterified cholesterol, as well as a broad range of sphingolipids, is the hallmark of the NPC cellular defect and is accompanied by dysregulation of cholesterol homeostatic pathways [12, 13]. The link between cholesterol dyshomeostasis and neurodegeneration is not fully understood, but appears to underlie the neuronal dysfunction and pathology that is central to NPC disease [14].
The clinical presentation of NPC is highly heterogeneous and can generally be classified into four categories based on age at onset of neurological manifestations and disease progression: early-infantile, late-infantile, juvenile, and adolescent/adult-onset [15, 16]. More than half of NPC patients present in the neonatal period with visceral signs such as hepatosplenomegaly and cholestatic jaundice. The liver disease typically resolves in the majority of patients. In a small subset, however, the liver disease is fatal during the neonatal period. Although patients usually present during childhood with neurological signs, in a subset of patients the disease is primarily systemic and neurological signs may not manifest for years or even decades. Therefore, NPC should be suspected not only in the neonates with cholestatic jaundice and organomegaly, but also in children of any age with isolated splenomegaly or hepatosplenomegaly. For patients that progress to neurological signs, many early signs are non-specific (e.g., delayed developmental milestones and clumsiness), and typically advance to more specific neurological manifestations, such as cerebellar ataxia, vertical supranuclear gaze palsy, cataplexy and dementia [15]. These manifestations are progressive and patients often succumb to the disease within the first two decades of life.
Diagnosis of NPC is challenging due to the rarity of the disorder, the heterogeneity of disease onset and presentation, its non-specific early symptoms, and the complexity of the laboratory testing [17]. Consequently, diagnosis is typically delayed 4–5 years for the late-infantile and juvenile forms of the disease, during which time opportunities for early intervention are lost [5, 18]. The discovery of blood-based diagnostics and dissemination of rapid, sensitive and specific clinical assays have lowered the barrier for testing and accelerated diagnosis [19–22]. In addition, the availability of molecular diagnostics (e.g., next generation sequencing) and phenotype-specific gene panels has further increased recognition of NPC disease in at-risk patient populations (e.g., neonates with cholestasis) [4, 23–25].
At present drug treatment options for NPC are limited. The only approved drug for treatment of NPC is the glycosphingolipid synthesis inhibitor miglustat (approved in the EU and >40 countries worldwide but not the US). The relatively modest disease-modifying effects of miglustat have prompted further drug development to address the unmet clinical needs. Two Phase 2b/3 trials in NPC have been completed over the past five years, though to date neither has led to drug approvals. These trials have underscored the significant challenges in developing and gaining regulatory approval for new treatments in a rare, slowly progressive neurodegenerative disease, as well as the urgent need for biomarkers that could serve as surrogate endpoints and accelerate drug approval.
Here, we review the development and current status of biomarkers for the diagnosis and treatment of NPC disease. This is not intended to be an exhaustive review of NPC biomarkers; rather, it is focused on the most widely accepted biomarkers that have the greatest clinical applicability. We discuss the application of these biomarkers to NPC diagnosis, newborn screening, and clinical development of new treatments, specifically highlighting our laboratory’s contribution to NPC biomarker discovery. For detailed discussion of the use of NPC biomarkers in diagnostic algorithms, the reader is referred to recent consensus statements and reviews [4, 17].
NPC diagnosis
Until recently, the standard for diagnosis of NPC disease was filipin staining of unesterified cholesterol in cultured skin fibroblasts obtained from patients. The filipin test identifies ~80–85% “classical” NPC patients; however, the test does not provide a clear result in patients with variant phenotypes [17]. The test is also susceptible to false positives among NPC carriers and other rare disorders that similarly store cholesterol – e.g., acid sphingomyelinase deficiency (ASMD), lysosomal acid lipase deficiency (LALD), mucolipidosis II/III, MEGDEL syndrome, Smith-Lemli-Opitz syndrome (SLOS), Tangier disease, and a rare congenital disorder of glycosylation due to Nogo-B receptor [17]. Other liabilities of the filipin assay are its invasiveness, high cost, and long turnaround times (> 7 weeks) [17, 22]. For these reasons filipin is no longer considered a first-line test for NPC diagnosis. However, it may still have utility in situations where biomarkers yield inconclusive results or to assess pathogenicity of variants of unknown significance identified through molecular diagnostics [26].
Oxysterols
Over the past decade the discovery of disease-specific, blood-based biomarkers have transformed the diagnosis of NPC, supplanting filipin as the first-line diagnostic. The first generation of these markers were non-enzymatic oxidation products of cholesterol known as oxysterols. We hypothesized that the intersection of lysosomal cholesterol storage and oxidative stress in NPC disease [27–29] would lead to an elevation in circulating oxysterols. Through targeted metabolomic screening of oxysterols first in npc1-deficient mice and then in the plasma of NPC1 patients, we discovered cholestane-3β,5α,6β-triol (C-triol) and 7-ketocholesterol (7-KC) as disease-specific biomarkers (Figure 1) [20]. This work led to the development of the first rapid, blood-based assay for diagnosis of NPC1 disease [19]. Although both C-triol and 7-KC have excellent receiver operating characteristics (ROC) with respect to diagnosing NPC1 patients (0.9958 for C-triol and 0.9907 for 7-KC) [19], the high correlation between the plasma oxysterol values (r2 = 0.86), indicating a common pathway for oxysterol generation, and the higher specificity of C-triol have made it the oxysterol biomarker of choice in the clinical laboratory [17, 19, 26, 30]. An early study showed that C-triol retrospectively identified 100% of NPC patients, whereas filipin staining only 65% of known NPC1 patients [31]. Clinical application of the C-triol biomarker for diagnosis of NPC1 patients has been demonstrated on different analytic platforms in multiple clinical laboratories [19, 21, 23, 30, 32–42]. The C-triol assay similarly detects NPC2 patients [38, 43]. The C-triol biomarker is also elevated in other rare genetic disorders including ASMD, cerebrotendinous xanthomathosis (CTX), and LALD [30, 36, 37, 40, 42, 44, 45], which may be relevant for the differential diagnosis depending upon the age of the patient.
Figure 1. Pathways leading to generation of disease biomarkers in NPC.
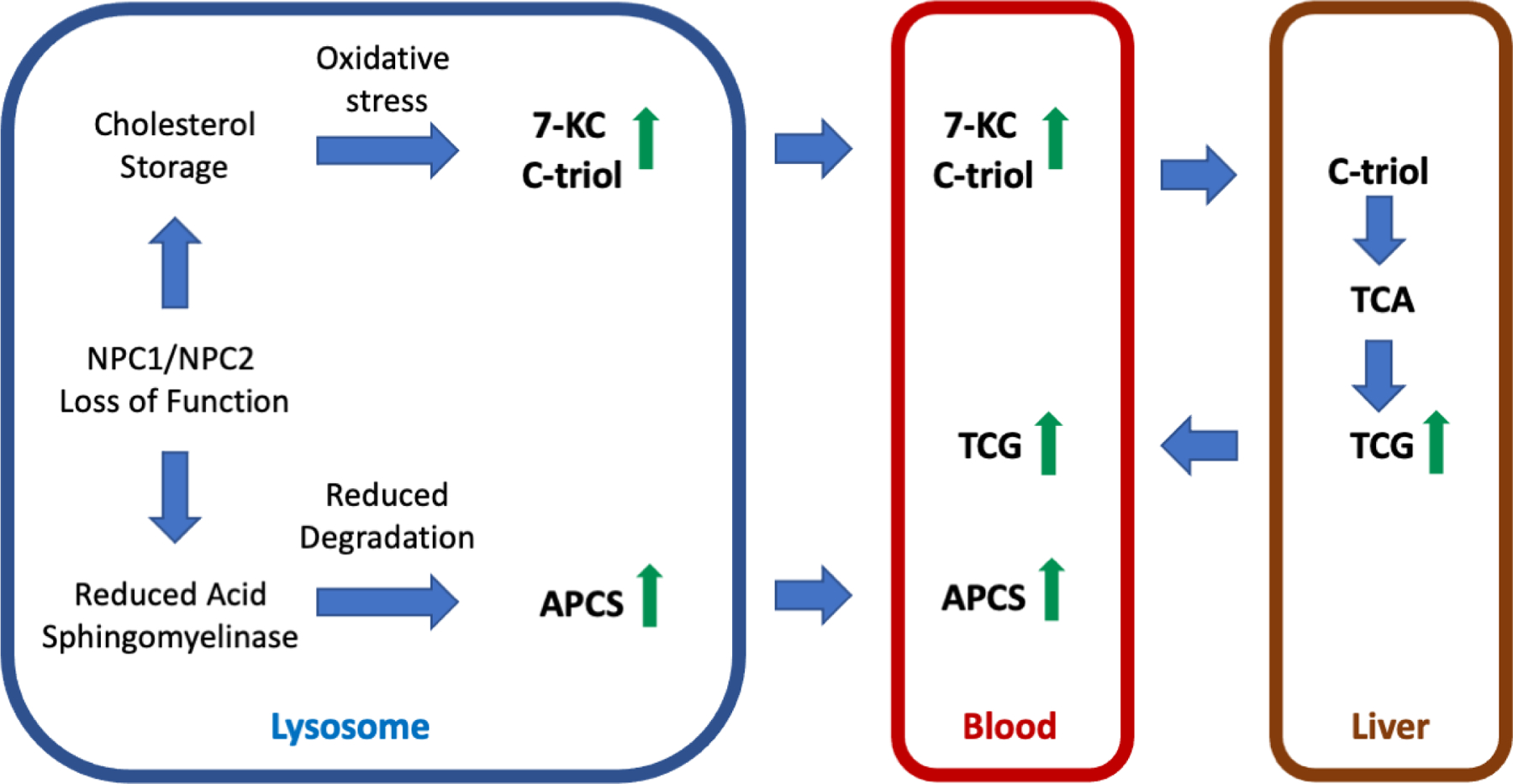
Either NPC1 or NPC2 loss of function leads to storage of unesterified cholesterol in lysosomes. The consequent oxidative stress promotes non-enzymatic oxidation of cholesterol leading to production of oxysterols (e.g., 7-KC and C-triol) that enter the circulation. C-triol is taken up by the liver and metabolized to TCA and to its glycinated derivative, TCG, which may enter the blood following bile acid secretion and uptake into the entero-hepatic circulation. The biosynthetic pathway for APCS species is unknown but catabolism may involve lysosomal acid sphingomyelinase. NPC1 or NPC2 deficiency creates an acid sphingomyelinase-deficient state allowing accumulation of APCS and eventual release into the circulation.
To date, the oxysterol assay has been implemented in >50 clinical laboratories worldwide and widely adopted in NPC diagnostic algorithms [4, 17]. The widespread application of the biomarker has also highlighted some of the assay’s limitations. To achieve sufficient sensitivity for mass spectrometric detection, C-triol requires derivatization that can result in artifactual generation of the oxysterol in situations where cholesterol and 5,6-epoxycholesterol have not been completely removed [40, 46]. C-triol may also show elevation due to autooxidation of cholesterol when the plasma sample is stored at room temperature for more than 24 hours [19, 21]. Perhaps the most significant liability of the biomarker is the overlap between NPC1 patients and the upper quartile of NPC1 carriers [19], which has precluded application of oxysterols for purposes of population screening.
Bile acids and newborn screening
The infeasibility of using C-triol or other oxysterol species for newborn screening in NPC disease prompted us to pursue further biomarker discovery. The known perturbations of sterol metabolism, coupled with reports of unusual urinary bile acids in NPC1 disease [47, 48] and the favorable chemical properties of bile acids for ionization and mass spectrometric detection, led us to pursue semi-targeted metabolomic profiling of bile acids in plasma from NPC1 subjects in order to identify a more suitable biomarker [49]. The profiling resulted in discovery of three unknown peaks specifically elevated in the plasma of NPC1 patients. Two of these structures were elucidated and identified as bile acid metabolites 3β,5α,6β-trihydroxycholan-24-oic acid (trihydroxycholanic acid, TCA) and its glycinated derivative, N-3β,5α,6β-trihydroxy-cholan-24-oyl)glycine (trihydroxycholanic acid glycinate, TCG). The low abundant third peak was not fully elucidated but provisionally assigned as the taurine derivative of TCA. The TCG bile acid was also reported by Clayton and colleagues who identified the biomarker through targeted mass spectrometric profiling of NPC1 patient plasma for presumptive bile acid metabolites of C-triol [50]. These bile acids are unusual in that they have a 3β-hydroxyl-5α-hydroxyl-cholanic acid rather than the 3α-hydroxyl-5β-hydroxyl-cholanic acid configuration typically found in circulating bile acids in human plasma. We further demonstrated that the bile acid biomarkers were derived from C-triol in stable isotopic labeling experiments (Figure 1), thus providing an explanation as to the origin of their unusual A and B steroid ring stereochemistry.
In comparison to oxysterols, the TCA and TCG bile acids exhibit robust ionization, do not require chemical derivatization, and are readily detected and stable at room temperature in plasma and dried blood spots. TCG can be separated from interferences within a total liquid chromatography run time of only 2.2 minutes. The latter property was crucial to enable development of a fully validated mass spectrometry-based method for detection of the glycine conjugated bile acid biomarker in dried blood spots with throughput of 500 samples per day [49], which is a prerequisite of newborn screening. In contrast to C-triol, the TCG biomarker measured in dried blood spots allowed complete separation between NPC1 carriers and patients, affording sensitivity and specificity of 100% (ROC 1.0) and provided the basis for a newborn screen for NPC1. An important limitation of the study, however, was that the dried blood spots were obtained from previously diagnosed NPC1 subjects. To address the key question as to whether the assay would have been able to detect NPC1 subjects at birth, we obtained the newborn dried blood spots from 15 NPC1 subjects residing in California, Michigan and New York – states that archive residual dried blood spots in biorepositories – and quantified TCA and TCG bile acids. Whereas TCG was undetectable in blood spots stored >13 years, suggesting reduced long-term stability for the glycinated bile acid, it was still able to identify all the NPC1 patients from dried blood spots that have been stored <10.5 years. The TCA precursor was detected in all the subjects – even those stored >20 years and discriminated NPC1 patients from carriers and controls completely [51]. These data conclusively demonstrated the feasibility of newborn screening for NPC1 using the bile acid biomarkers.
There is a compelling rationale for newborn screening in NPC disease. Implementation of universal screening has the potential to shift diagnosis to the neonatal period prior to the onset of neurological symptoms. This not only would greatly diminish the diagnostic delay but also allow for early pharmacological intervention that could delay disease progression and extend life. Newborn screening in the U.S. and decisions as to which genetic disorders to include in screening panels are determined at the state level. However, most states adopt Health and Human Services recommendations for disorders included in the Recommended Universal Screening Panel (RUSP). To support a nomination for NPC disease to the RUSP a requirement is to prospectively obtain data to demonstrate that the disorder can be detected using the screening assay. Working in close partnership with NPC patient advocacy groups (Firefly Fund and Ara Parseghian Medical Research Fund), we partnered with the NIH-funded ScreenPlus program to incorporate the TCG bile acid assay in a panel of 14 rare disorders that are being piloted at the New York State Department of Health [52]. Screening for NPC was initiated in May 2021 with the expectation that it may require up to five years to obtain sufficient experience to support a RUSP nomination.
N- palmitoyl-O-phosphocholineserine (LysoSM-509)
The strategy of differential liquid chromatography-mass spectrometry profiling of plasma samples from control and NPC1 patients led to discovery of another clinically useful biomarker, initially referred to as LysoSM-509 [53]. This biomarker was dramatically elevated in NPC1 plasma and showed even higher levels in ASMD [53–58]. Determination of its structure, however, proved elusive and in the original publication it was misassigned as an isomer of lysosphingomyelin. Following discovery of LysoSM-509, assays were developed for the biomarker using lysosphingomyelin in place of authentic compound for standard curves. Thus, assay accuracy was suspect and accurate reference ranges for control, heterozygotes, and NPC1 patients could not be obtained [53–58]. Nonetheless, application of these assays for relative quantification of the biomarker, despite concerns related to their accuracy, has been useful for discriminating NPC from ASMD [17].
To further explore the potential of lysoSM-509 as an NPC biomarker, we used accurate mass measurements, tandem mass spectra, and chemical derivatization to elucidate its structure. Unexpectedly, we found that lysoSM-509 was not a lysosphingomyelin species, as originally proposed by Rolfs and colleagues [53], but N-palmitoyl-O-phosphocholineserine (PPCS), the most abundant member of an entirely novel lipid class termed N-acyl-O-phosphocholineserines (APCS) [59]. The structure of PPCS was corroborated in a subsequent report by Maekawa and colleagues [60]. All APCS isoforms were dramatically elevated in NPC1 subjects, consistent with the earlier reports. The biosynthetic pathway for generation of the APCS metabolites is poorly understood. It is notable biomarkers are elevated in both ASMD and NPC subjects [53, 61], the former caused by genetic deficiency of acid sphingomyelinase and the latter a result of a secondary, post-translation defect in the activity of the lysosomal enzyme [62–65]. That these lipids contain a phosphorylcholine moiety implies a possible role for acid sphingomyelinase in the catabolism of these metabolites (Figure 1).
The discovery of the PPCS structure and synthesis of an authentic standard enabled development of a more reliable plasma assay [61]. The plasma PPCS test offers a sensitivity of 100.0% and specificity of 96.6% for identifying NPC1 subjects from control and NPC1 carriers, with 10% NPC1 carriers above the cutoff for NPC1. Only ASMD overlaps with NPC1 in the PPCS assay [53–56, 61]. PPCS is artifactually produced and elevated 8-fold when plasma, red blood cells, white blood cells, and whole blood are dried on newborn screening cards, suggesting that most PPCS generation may be triggered by the unknown substances in cotton fiber card [59]. Taken together, these findings indicate that the PPCS biomarker would appear to have limited utility for population screening.
Lysosphingolipids
Lysosphingolipids are sphingolipids lacking an N-acyl chain and have proven to be robust biomarkers for several lysosomal storage disorders, including Fabry disease (lyso-globotriaosylceramide, lyso-Gb3), Gaucher disease (glucosylsphingosine, lyso-GL-1) and Krabbe disease (galactosylsphingosine, psychosine). Their accumulation and tissue profile in sphingolipidoses led to the hypothesis that lysosphingolipids may be elevated in the plasma of NPC patients [17]. In two retrospective studies plasma lysosphingomyelin showed a modest (2.8-fold) elevation in NPC subjects and provided a sensitivity of 100% and specificity of 97–98% to distinguish NPC1 from controls [56, 66]. However, in subsequent studies no increase was seen in patients with NPC and there was a large overlap between NPC and controls [55, 67]. Whereas plasma lysosphingomyelin has little role in primary diagnosis of NPC, it may have greater utility in ASMD diagnostics, where it has shown to be elevated more than 10-fold and when used in combination with PPCS could discriminate between ASMD and NPC [54, 55, 67, 68]. Simultaneous assay of lysosphingolipids (hexosylsphingosine, lysosphingomyelin, lyso-Gb3) and PPCS was developed to diagnose Fabry disease (LysoGb3), Gaucher and Krabbe diseases (hexosylsphingosine), prosaposin deficiency (LysoGb3 and hexosylsphingosine), and ASMD and NPC (lysosphingomyelin and PPCS) [55, 56]. Lysosphingomyelin was also used for monitoring treatment with olipudase alfa in patients with ASMD [69].
Protein markers
Lysosomal lipid storage leads to downstream events in the pathogenic cascade, including neuroinflammation, neuroaxonal dystrophy, and neuronal cell death, in particular cerebellar Purkinje cells. Studies of these disease processes using proteomics and genomics have identified numerous plasma and cerebrospinal fluid (CSF) protein biomarkers, including galectin-3 [70, 71], cathepsin D [70], fatty acid binding protein 3 [72], heat shock protein 70 [73], chemokine ligand 3 [74], cathepsin S [75], lysozyme [75], calbindin D [76], and glycoprotein nonmetastatic melanoma protein B [77, 78]. At present none of these biomarkers has a role in primary NPC diagnostics, although some (e.g., calbindin D and fatty acid binding protein 3) have been examined in CSF as potential pharmacodynamic markers for monitoring treatment efficacy [79].
Comparison of plasma biomarkers for NPC diagnosis
We established in our laboratory fully-validated, FDA-compliant assays for quantification of plasma C-triol, TCG and PPCS, allowing us to directly compare the clinical performance the assays for NPC1 diagnosis in 301 consecutive, unknown patient samples submitted to the Washington University Metabolomics Core (Figure 2) [46]. As anticipated, the C-triol and TCG markers were more highly correlated than TCG and PPCS because C-triol and TCG are in the same metabolic pathway. All NPC1 patients were correctly identified by these biomarkers, but the C-triol and PPCS biomarkers led to more mis-diagnosed non-NPC1 patients. These results indicated that all three biomarkers were equally sensitive in detection of NPC1 patients, but that TCG was more specific [46]. As TCG is a metabolite of C-triol, it may also detect NPC2 patients similar to C-triol. The TCG assay does not require derivatization and is less prone to artefacts than oxysterol measurements; thus, the measurement of TCG is more accurate than C-triol, which may be of particular importance for diagnosing patients with milder phenotypes. TCG is stable in plasma at room temperature and even 37 °C for 5 days; therefore, the shipment of plasma sample for TCG assay with dry ice is not necessary and the handling of patient samples in a clinical lab is more forgiving. Based on these results, we expect that TCG will replace C-triol as first-line NPC diagnostic biomarker in the future.
Figure 2. Comparison biomarkers for diagnosis of NPC1 disease.
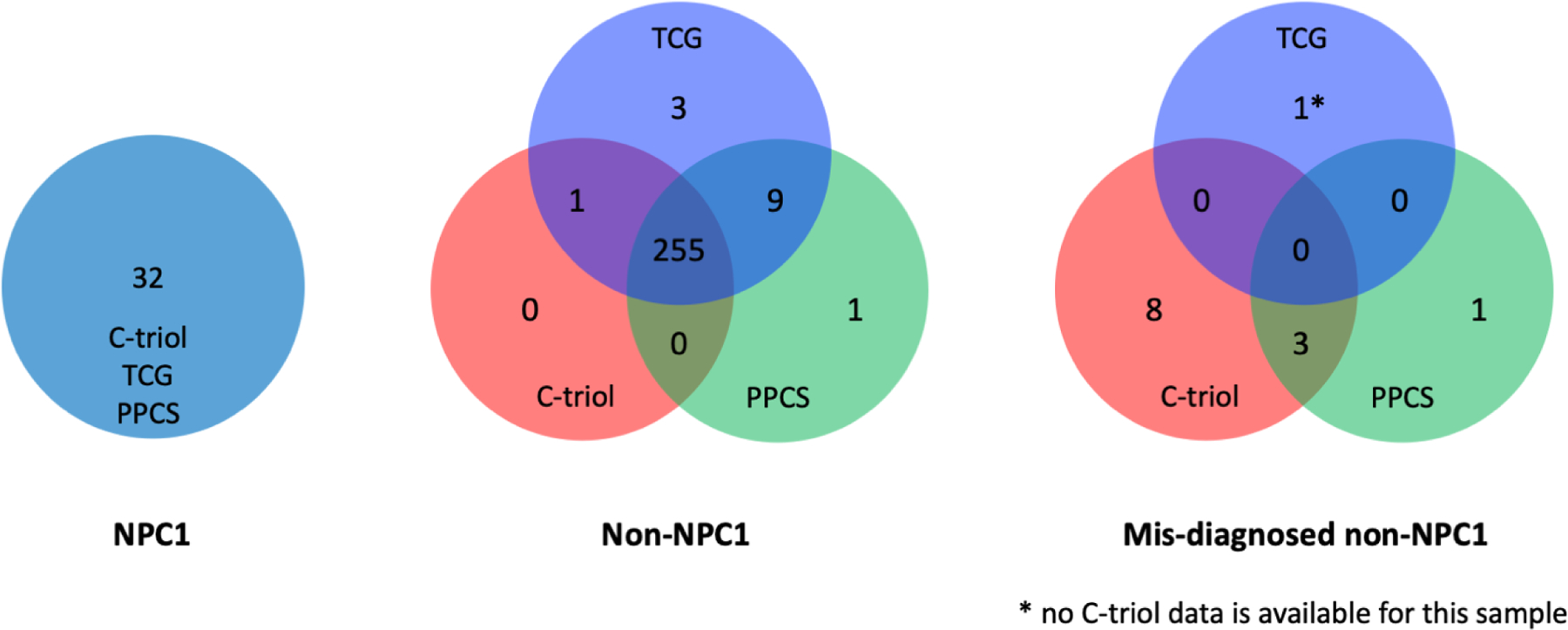
The performance of plasma C-triol, TCG and PPCS assays were directly compared in 301 consecutive, unknown patient samples submitted to the Washington University Metabolomics Core for NPC1 diagnosis. All three biomarkers correctly identified NPC1 patients (left). C-triol and PPCS assays were less specific, mis-identifying 11 and 4 normal subjects, respectively, as NPC1 patients, whereas TCG mis-diagnosed only 1 patient (right). Data reprinted form Sidhu et al. [46] with permission of the journal.
Biomarkers for monitoring treatment efficacy
The slowly progressive nature and clinical heterogeneity in NPC is daunting with respect to development of new treatments. The disappointing regulatory outcomes for the recent Phase 2b/3 trials for arimoclomol (NCT02612129) and for 2-hydroxypropyl-β-cyclodextrin (HPβCD) (NCT02534844) underscore the challenges in conducting an interventional clinical trial for this disorder. The insistence by regulatory bodies on a well-controlled trial, the lack of a validated, clinical endpoint accepted by regulators, and the rarity of the disease all contribute to the infeasibility of conducting a trial of shorter than 4–5 years duration. For this reason, it would be highly desirable to use reliable, non-invasive biomarkers to assess response to interventions. Ideally, a panel of biomarkers that correspond to different aspects of the pathogenic cascade or relate to the biological pathway targeted by a therapeutic would be most useful for monitoring the course of disease and response to therapy.
Plasma C-triol, TCG, and PPCS biomarkers have been shown to be useful, but limited, tools for monitoring treatment responses in NPC1 patients. C-triol was significantly reduced in two siblings administered IV HPβCD through an expanded access protocol [80]. Likewise, plasma TCG and PPCS levels were rapidly reduced by IV HPβCD in three expanded access patients [46]. PPCS levels also decreased in a neonate receiving IV HPβCD [81]. However, in the Phase 1/2 trial of intrathecal HPβCD neither plasma nor CSF levels correlated with the demonstrated clinical benefit in slowing neurodegeneration [79]. This highlights a major caveat for use of these biomarkers, that they reflect almost exclusively metabolism in peripheral tissues and are uninformative with respect to treatment efficacy in the CNS. This is because the production of these metabolites is much lower in brain than liver tissue. As a result, biomarker levels in CSF primarily reflect transcytosis of plasma-derived metabolites down a steep concentration gradient across the blood-brain barrier rather than metabolites that are generated in situ in the brain [61].
By contrast, there is increasing evidence that proteomic rather than metabolomic biomarkers may be better suited to monitoring CNS treatment efficacy. Calbindin D is elevated in the CSF of NPC1 patients and is thought to be a marker for Purkinje cell loss [76]. Significant reduction was observed in CSF calbindin D in NPC1 patients following initiation of miglustat therapy [76] CSF calbindin D was also lowered in NPC1 patients treated with intrathecal HPβCD [79]. Similarly, levels of FABP3, which is considered as a marker of neuronal inflammation and cell damage, were significantly decreased in miglustat-treated patients relative to untreated patients and in patients receiving intrathecal HPβCD [72, 79]. More recently, neurofilament light chain, a non-specific marker of neurodegeneration, has been reported to be elevated in CSF and plasma of NPC1 patients and may prove useful as a pharmacodynamic marker in future clinical trials.
Summary
The discovery of NPC-specific blood-based biomarkers and their rapid dissemination over the past decade into clinical laboratories has transformed the approach to disease diagnosis. The circulating biomarkers most widely used in clinical laboratories – C-triol, TCG and PPCS – all have high sensitivity for identifying NPC disease. However, in contrast to C-triol and PPCS, TCG has significant advantages with respect to sample handling and stability and offers higher specificity for discrimination between carriers and NPC patients. For these reasons, the plasma TCG assay is recommended as the first-line diagnostic. TCG is also the only blood-based biomarker suitable for NPC newborn screening. A pilot screen based on quantification of TCG in dried blood spots is underway, the results of which should provide a more accurate assessment of the true disease incidence and facilitate therapeutic intervention in pre-symptomatic patients. Finally, the drug development landscape for NPC remains challenging. At present the field lacks a validated, non-invasive biomarker to monitor the course of disease and CNS response to therapy. Thus, there is an urgent need for new biomarkers that could serve as surrogate endpoints to accelerate drug approval.
References
- 1.Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, et al. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277:228–31. [DOI] [PubMed] [Google Scholar]
- 2.Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, et al. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 2000;290:2298–301. [DOI] [PubMed] [Google Scholar]
- 3.Jahnova H, Dvorakova L, Vlaskova H, Hulkova H, Poupetova H, Hrebicek M, et al. Observational, retrospective study of a large cohort of patients with Niemann-Pick disease type C in the Czech Republic: a surprisingly stable diagnostic rate spanning almost 40 years. Orphanet J Rare Dis. 2014;9:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patterson MC, Clayton P, Gissen P, Anheim M, Bauer P, Bonnot O, et al. Recommendations for the detection and diagnosis of Niemann-Pick disease type C: An update. Neurol Clin Pract. 2017;7:499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis. 2010;5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wassif CA, Cross JL, Iben J, Sanchez-Pulido L, Cougnoux A, Platt FM, et al. High incidence of unrecognized visceral/neurological late-onset Niemann-Pick disease, type C1, predicted by analysis of massively parallel sequencing data sets. Genet Med. 2016;18:41–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li X, Wang J, Coutavas E, Shi H, Hao Q, Blobel G. Structure of human Niemann-Pick C1 protein. Proc Natl Acad Sci U S A. 2016;113:8212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedland N, Liou HL, Lobel P, Stock AM. Structure of a cholesterol-binding protein deficient in Niemann-Pick type C2 disease. Proc Natl Acad Sci U S A. 2003;100:2512–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwon HJ, Abi-Mosleh L, Wang ML, Deisenhofer J, Goldstein JL, Brown MS, et al. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell. 2009;137:1213–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang ML, Motamed M, Infante RE, Abi-Mosleh L, Kwon HJ, Brown MS, et al. Identification of surface residues on Niemann-Pick C2 essential for hydrophobic handoff of cholesterol to NPC1 in lysosomes. Cell Metab. 2010;12:166–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qian H, Wu X, Du X, Yao X, Zhao X, Lee J, et al. Structural Basis of Low-pH-Dependent Lysosomal Cholesterol Egress by NPC1 and NPC2. Cell. 2020;182:98–111 e18. [DOI] [PubMed] [Google Scholar]
- 12.Frolov A, Zielinski SE, Crowley JR, Dudley-Rucker N, Schaffer JE, Ory DS. NPC1 and NPC2 regulate cellular cholesterol homeostasis through generation of low density lipoprotein cholesterol-derived oxysterols. J Biol Chem. 2003;278:25517–25. [DOI] [PubMed] [Google Scholar]
- 13.Liscum L, Faust JR. Low density lipoprotein (LDL)-mediated suppression of cholesterol synthesis and LDL uptake is defective in Niemann-Pick type C fibroblasts. J Biol Chem. 1987;262:17002–8. [PubMed] [Google Scholar]
- 14.Walkley SU, Suzuki K. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim Biophys Acta. 2004;1685:48–62. [DOI] [PubMed] [Google Scholar]
- 15.Patterson MC, Hendriksz CJ, Walterfang M, Sedel F, Vanier MT, Wijburg F, et al. Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol Genet Metab. 2012;106:330–44. [DOI] [PubMed] [Google Scholar]
- 16.Wraith JE, Guffon N, Rohrbach M, Hwu WL, Korenke GC, Bembi B, et al. Natural history of Niemann-Pick disease type C in a multicentre observational retrospective cohort study. Mol Genet Metab. 2009;98:250–4. [DOI] [PubMed] [Google Scholar]
- 17.Vanier MT, Gissen P, Bauer P, Coll MJ, Burlina A, Hendriksz CJ, et al. Diagnostic tests for Niemann-Pick disease type C (NP-C): A critical review. Mol Genet Metab. 2016;118:244–54. [DOI] [PubMed] [Google Scholar]
- 18.Patterson MC, Mengel E, Wijburg FA, Muller A, Schwierin B, Drevon H, et al. Disease and patient characteristics in NP-C patients: findings from an international disease registry. Orphanet J Rare Dis. 2013;8:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang X, Sidhu R, Porter FD, Yanjanin NM, Speak AO, te Vruchte DT, et al. A sensitive and specific LC-MS/MS method for rapid diagnosis of Niemann-Pick C1 disease from human plasma. J Lipid Res. 2011;52:1435–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Porter FD, Scherrer DE, Lanier MH, Langmade SJ, Molugu V, Gale SE, et al. Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease. Sci Transl Med. 2010;2:56ra81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reunert J, Fobker M, Kannenberg F, Du Chesne I, Plate M, Wellhausen J, et al. Rapid Diagnosis of 83 Patients with Niemann Pick Type C Disease and Related Cholesterol Transport Disorders by Cholestantriol Screening. EBioMedicine. 2016;4:170–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang X, Ory DS. Towards a New Diagnostic Standard for Niemann-Pick C Disease. EBioMedicine. 2016;4:18–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bauer P, Balding DJ, Klunemann HH, Linden DE, Ory DS, Pineda M, et al. Genetic screening for Niemann-Pick disease type C in adults with neurological and psychiatric symptoms: findings from the ZOOM study. Hum Mol Genet. 2013;22:4349–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Synofzik M, Harmuth F, Stampfer M, Muller Vom Hagen J, Schols L, Bauer P. NPC1 is enriched in unexplained early onset ataxia: a targeted high-throughput screening. J Neurol. 2015;262:2557–63. [DOI] [PubMed] [Google Scholar]
- 25.Karpen SJ, Kamath BM, Alexander JJ, Ichetovkin I, Rosenthal P, Sokol RJ, et al. Use of a Comprehensive 66-Gene Cholestasis Sequencing Panel in 2171 Cholestatic Infants, Children, and Young Adults. J Pediatr Gastroenterol Nutr. 2021;72:654–60. [DOI] [PubMed] [Google Scholar]
- 26.Geberhiwot T, Moro A, Dardis A, Ramaswami U, Sirrs S, Marfa MP, et al. Consensus clinical management guidelines for Niemann-Pick disease type C. Orphanet J Rare Dis. 2018;13:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fu R, Yanjanin NM, Bianconi S, Pavan WJ, Porter FD. Oxidative stress in Niemann-Pick disease, type C. Mol Genet Metab. 2010;101:214–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vazquez MC, Balboa E, Alvarez AR, Zanlungo S. Oxidative stress: a pathogenic mechanism for Niemann-Pick type C disease. Oxid Med Cell Longev. 2012;2012:205713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zampieri S, Mellon SH, Butters TD, Nevyjel M, Covey DF, Bembi B, et al. Oxidative stress in NPC1 deficient cells: protective effect of allopregnanolone. J Cell Mol Med. 2009;13:3786–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boenzi S, Deodato F, Taurisano R, Goffredo BM, Rizzo C, Dionisi-Vici C. Evaluation of plasma cholestane-3beta,5alpha,6beta-triol and 7-ketocholesterol in inherited disorders related to cholesterol metabolism. J Lipid Res. 2016;57:361–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stampfer M, Theiss S, Amraoui Y, Jiang X, Keller S, Ory DS, et al. Niemann-Pick disease type C clinical database: cognitive and coordination deficits are early disease indicators. Orphanet J Rare Dis. 2013;8:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boenzi S, Deodato F, Taurisano R, Martinelli D, Verrigni D, Carrozzo R, et al. A new simple and rapid LC-ESI-MS/MS method for quantification of plasma oxysterols as dimethylaminobutyrate esters. Its successful use for the diagnosis of Niemann-Pick type C disease. Clin Chim Acta. 2014;437:93–100. [DOI] [PubMed] [Google Scholar]
- 33.Deodato F, Boenzi S, Taurisano R, Semeraro M, Sacchetti E, Carrozzo R, et al. The impact of biomarkers analysis in the diagnosis of Niemann-Pick C disease and acid sphingomyelinase deficiency. Clin Chim Acta. 2018;486:387–94. [DOI] [PubMed] [Google Scholar]
- 34.Hammerschmidt TG, de Oliveira Schmitt Ribas G, Saraiva-Pereira ML, Bonatto MP, Kessler RG, Souza FTS, et al. Molecular and biochemical biomarkers for diagnosis and therapy monitorization of Niemann-Pick type C patients. Int J Dev Neurosci. 2018;66:18–23. [DOI] [PubMed] [Google Scholar]
- 35.Kannenberg F, Nofer JR, Schulte E, Reunert J, Marquardt T, Fobker M. Determination of serum cholestane-3beta,5alpha,6beta-triol by gas chromatography-mass spectrometry for identification of Niemann-Pick type C (NPC) disease. J Steroid Biochem Mol Biol. 2017;169:54–60. [DOI] [PubMed] [Google Scholar]
- 36.Klinke G, Rohrbach M, Giugliani R, Burda P, Baumgartner MR, Tran C, et al. LC-MS/MS based assay and reference intervals in children and adolescents for oxysterols elevated in Niemann-Pick diseases. Clin Biochem. 2015;48:596–602. [DOI] [PubMed] [Google Scholar]
- 37.Pajares S, Arias A, Garcia-Villoria J, Macias-Vidal J, Ros E, de las Heras J, et al. Cholestane-3beta,5alpha,6beta-triol: high levels in Niemann-Pick type C, cerebrotendinous xanthomatosis, and lysosomal acid lipase deficiency. J Lipid Res. 2015;56:1926–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Polo G, Burlina A, Furlan F, Kolamunnage T, Cananzi M, Giordano L, et al. High level of oxysterols in neonatal cholestasis: a pitfall in analysis of biochemical markers for Niemann-Pick type C disease. Clin Chem Lab Med. 2016;54:1221–9. [DOI] [PubMed] [Google Scholar]
- 39.Ribas GS, Souza HM, de Mari J, Deon M, Mescka C, Saraiva-Pereira ML, et al. Selective screening of Niemann-Pick type C Brazilian patients by cholestane-3beta,5alpha,6beta-triol and chitotriosidase measurements followed by filipin staining and NPC1/NPC2 gene analysis. Clin Chim Acta. 2016;459:57–62. [DOI] [PubMed] [Google Scholar]
- 40.Cooper JA, Church HJ, Wu HY. Cholestane-3beta, 5alpha, 6beta-triol: Further insights into the performance of this oxysterol in diagnosis of Niemann-Pick disease type C. Mol Genet Metab. 2020;130:77–86. [DOI] [PubMed] [Google Scholar]
- 41.Mandia D, Plaze M, Le Ber I, Ewenczyk C, Morin A, Carle G, et al. High diagnostic value of plasma Niemann-Pick type C biomarkers in adults with selected neurological and/or psychiatric disorders. J Neurol. 2020;267:3371–7. [DOI] [PubMed] [Google Scholar]
- 42.Romanello M, Zampieri S, Bortolotti N, Deroma L, Sechi A, Fiumara A, et al. Comprehensive Evaluation of Plasma 7-Ketocholesterol and Cholestan-3beta,5alpha,6beta-Triol in an Italian Cohort of Patients Affected by Niemann-Pick Disease due to NPC1 and SMPD1 Mutations. Clin Chim Acta. 2016;455:39–45. [DOI] [PubMed] [Google Scholar]
- 43.Reunert J, Lotz-Havla AS, Polo G, Kannenberg F, Fobker M, Griese M, et al. Niemann-Pick Type C-2 Disease: Identification by Analysis of Plasma Cholestane-3beta,5alpha,6beta-Triol and Further Insight into the Clinical Phenotype. JIMD Rep. 2015;23:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bjorkhem I, Diczfalusy U, Lovgren-Sandblom A, Starck L, Jonsson M, Tallman K, et al. On the formation of 7-ketocholesterol from 7-dehydrocholesterol in patients with CTX and SLO. J Lipid Res. 2014;55:1165–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin N, Zhang H, Qiu W, Ye J, Han L, Wang Y, et al. Determination of 7-ketocholesterol in plasma by LC-MS for rapid diagnosis of acid SMase-deficient Niemann-Pick disease. J Lipid Res. 2014;55:338–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sidhu R, Kell P, Dietzen DJ, Farhat NY, Do AND, Porter FD, et al. Application of a glycinated bile acid biomarker for diagnosis and assessment of response to treatment in Niemann-pick disease type C1. Mol Genet Metab. 2020;131:405–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alvelius G, Hjalmarson O, Griffiths WJ, Bjorkhem I, Sjovall J. Identification of unusual 7-oxygenated bile acid sulfates in a patient with Niemann-Pick disease, type C. J Lipid Res. 2001;42:1571–7. [PubMed] [Google Scholar]
- 48.Maekawa M, Misawa Y, Sotoura A, Yamaguchi H, Togawa M, Ohno K, et al. LC/ESI-MS/MS analysis of urinary 3beta-sulfooxy-7beta-N-acetylglucosaminyl-5-cholen-24-oic acid and its amides: new biomarkers for the detection of Niemann-Pick type C disease. Steroids. 2013;78:967–72. [DOI] [PubMed] [Google Scholar]
- 49.Jiang X, Sidhu R, Mydock-McGrane L, Hsu FF, Covey DF, Scherrer DE, et al. Development of a bile acid-based newborn screen for Niemann-Pick disease type C. Sci Transl Med. 2016;8:337ra63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mazzacuva F, Mills P, Mills K, Camuzeaux S, Gissen P, Nicoli ER, et al. Identification of novel bile acids as biomarkers for the early diagnosis of Niemann-Pick C disease. FEBS Lett. 2016;590:1651–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang XT, Sidhu R, Orsini JJ, Farhat NY, Porter FD, Berry-Kravis E, et al. Diagnosis of niemann-pick C1 by measurement of bile acid biomarkers in archived newborn dried blood spots. Molecular Genetics and Metabolism. 2019;126:183–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.ScreenPlus. Available from: https://einsteinmed.org/research/screenplus/.
- 53.Giese AK, Mascher H, Grittner U, Eichler S, Kramp G, Lukas J, et al. A novel, highly sensitive and specific biomarker for Niemann-Pick type C1 disease. Orphanet J Rare Dis. 2015;10:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuchar L, Sikora J, Gulinello ME, Poupetova H, Lugowska A, Malinova V, et al. Quantitation of plasmatic lysosphingomyelin and lysosphingomyelin-509 for differential screening of Niemann-Pick A/B and C diseases. Anal Biochem. 2017;525:73–7. [DOI] [PubMed] [Google Scholar]
- 55.Pettazzoni M, Froissart R, Pagan C, Vanier MT, Ruet S, Latour P, et al. LC-MS/MS multiplex analysis of lysosphingolipids in plasma and amniotic fluid: A novel tool for the screening of sphingolipidoses and Niemann-Pick type C disease. PLoS One. 2017;12:e0181700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Polo G, Burlina AP, Kolamunnage TB, Zampieri M, Dionisi-Vici C, Strisciuglio P, et al. Diagnosis of sphingolipidoses: a new simultaneous measurement of lysosphingolipids by LC-MS/MS. Clin Chem Lab Med. 2017;55:403–14. [DOI] [PubMed] [Google Scholar]
- 57.Polo G, Burlina AP, Ranieri E, Colucci F, Rubert L, Pascarella A, et al. Plasma and dried blood spot lysosphingolipids for the diagnosis of different sphingolipidoses: a comparative study. Clin Chem Lab Med. 2019;57:1863–74. [DOI] [PubMed] [Google Scholar]
- 58.Voorink-Moret M, Goorden SMI, van Kuilenburg ABP, Wijburg FA, Ghauharali-van der Vlugt JMM, Beers-Stet FS, et al. Rapid screening for lipid storage disorders using biochemical markers. Expert center data and review of the literature. Mol Genet Metab. 2018;123:76–84. [DOI] [PubMed] [Google Scholar]
- 59.Sidhu R, Mondjinou Y, Qian M, Song H, Kumar AB, Hong X, et al. N-acyl-O-phosphocholineserines: structures of a novel class of lipids that are biomarkers for Niemann-Pick C1 disease. J Lipid Res. 2019;60:1410–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maekawa M, Jinnoh I, Matsumoto Y, Narita A, Mashima R, Takahashi H, et al. Structural Determination of Lysosphingomyelin-509 and Discovery of Novel Class Lipids from Patients with Niemann-Pick Disease Type C. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sidhu R, Kell P, Dietzen DJ, Farhat NY, Do AND, Porter FD, et al. Application of N-palmitoyl-O-phosphocholineserine for diagnosis and assessment of response to treatment in Niemann-Pick type C disease. Mol Genet Metab. 2020;129:292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pentchev PG, Gal AE, Booth AD, Omodeo-Sale F, Fouks J, Neumeyer BA, et al. A lysosomal storage disorder in mice characterized by a dual deficiency of sphingomyelinase and glucocerebrosidase. Biochim Biophys Acta. 1980;619:669–79. [DOI] [PubMed] [Google Scholar]
- 63.Reagan JW, Jr., Hubbert ML, Shelness GS. Posttranslational regulation of acid sphingomyelinase in niemann-pick type C1 fibroblasts and free cholesterol-enriched chinese hamster ovary cells. J Biol Chem. 2000;275:38104–10. [DOI] [PubMed] [Google Scholar]
- 64.Thomas GH, Tuck-Muller CM, Miller CS, Reynolds LW. Correction of sphingomyelinase deficiency in Niemann-Pick type C fibroblasts by removal of lipoprotein fraction from culture media. J Inherit Metab Dis. 1989;12:139–51. [DOI] [PubMed] [Google Scholar]
- 65.Vanier MT, Rodriguez-Lafrasse C, Rousson R, Gazzah N, Juge MC, Pentchev PG, et al. Type C Niemann-Pick disease: spectrum of phenotypic variation in disruption of intracellular LDL-derived cholesterol processing. Biochim Biophys Acta. 1991;1096:328–37. [DOI] [PubMed] [Google Scholar]
- 66.Welford RW, Garzotti M, Marques Lourenco C, Mengel E, Marquardt T, Reunert J, et al. Plasma lysosphingomyelin demonstrates great potential as a diagnostic biomarker for Niemann-Pick disease type C in a retrospective study. PLoS One. 2014;9:e114669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu C, Iwamoto T, Hossain MA, Akiyama K, Igarashi J, Miyajima T, et al. A combination of 7-ketocholesterol, lysosphingomyelin and bile acid-408 to diagnose Niemann-Pick disease type C using LC-MS/MS. Plos One. 2020;15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Iwahori A, Maekawa M, Narita A, Kato A, Sato T, Ogura J, et al. Development of a Diagnostic Screening Strategy for Niemann-Pick Diseases Based on Simultaneous Liquid Chromatography-Tandem Mass Spectrometry Analyses of N-Palmitoyl-O-phosphocholine-serine and Sphingosylphosphorylcholine. Biol Pharm Bull. 2020;43:1398–406. [DOI] [PubMed] [Google Scholar]
- 69.Diaz GA, Jones SA, Scarpa M, Mengel KE, Giugliani R, Guffon N, et al. One-year results of a clinical trial of olipudase alfa enzyme replacement therapy in pediatric patients with acid sphingomyelinase deficiency. Genet Med. 2021;23:1543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cluzeau CV, Watkins-Chow DE, Fu R, Borate B, Yanjanin N, Dail MK, et al. Microarray expression analysis and identification of serum biomarkers for Niemann-Pick disease, type C1. Hum Mol Genet. 2012;21:3632–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rauniyar N, Subramanian K, Lavallee-Adam M, Martinez-Bartolome S, Balch WE, Yates JR 3rd. Quantitative Proteomics of Human Fibroblasts with I1061T Mutation in Niemann-Pick C1 (NPC1) Protein Provides Insights into the Disease Pathogenesis. Mol Cell Proteomics. 2015;14:1734–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cologna SM, Jiang XS, Backlund PS, Cluzeau CV, Dail MK, Yanjanin NM, et al. Quantitative proteomic analysis of Niemann-Pick disease, type C1 cerebellum identifies protein biomarkers and provides pathological insight. PLoS One. 2012;7:e47845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mengel E, Bembi B, Del Toro M, Deodato F, Gautschi M, Grunewald S, et al. Clinical disease progression and biomarkers in Niemann-Pick disease type C: a prospective cohort study. Orphanet J Rare Dis. 2020;15:328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cologna SM, Cluzeau CV, Yanjanin NM, Blank PS, Dail MK, Siebel S, et al. Human and mouse neuroinflammation markers in Niemann-Pick disease, type C1. J Inherit Metab Dis. 2014;37:83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Alam MS, Getz M, Yi S, Kurkewich J, Safeukui I, Haldar K. Plasma markers to distinguish brain and liver inflammation in a lysosomal disorder. Molecular Genetics and Metabolism. 2014;111:S18–S. [Google Scholar]
- 76.Bradbury A, Bagel J, Sampson M, Farhat N, Ding WG, Swain G, et al. Cerebrospinal Fluid Calbindin D Concentration as a Biomarker of Cerebellar Disease Progression in Niemann-Pick Type C1 Disease. Journal of Pharmacology and Experimental Therapeutics. 2016;358:254–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fukaura M, Ishitsuka Y, Shirakawa S, Ushihama N, Yamada Y, Kondo Y, et al. Intracerebroventricular Treatment with 2-Hydroxypropyl-beta-Cyclodextrin Decreased Cerebellar and Hepatic Glycoprotein Nonmetastatic Melanoma Protein B (GPNMB) Expression in Niemann-Pick Disease Type C Model Mice. Int J Mol Sci. 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marques ARA, Gabriel TL, Aten J, van Roomen CPAA, Ottenhoff R, Claessen N, et al. Gpnmb Is a Potential Marker for the Visceral Pathology in Niemann-Pick Type C Disease. Plos One. 2016;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ory DS, Ottinger EA, Farhat NY, King KA, Jiang X, Weissfeld L, et al. Intrathecal 2-hydroxypropyl-beta-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: a non-randomised, open-label, phase 1–2 trial. Lancet. 2017;390:1758–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tortelli B, Fujiwara H, Bagel JH, Zhang J, Sidhu R, Jiang X, et al. Cholesterol homeostatic responses provide biomarkers for monitoring treatment for the neurodegenerative disease Niemann-Pick C1 (NPC1). Hum Mol Genet. 2014;23:6022–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Reynolds M, Linneman LA, Luna S, Warner BB, Turmelle YP, Kulkarni SS, et al. A Phase 1/Phase 2 open label nonrandomized clinical trial of intravenous 2-hydroxypropyl-β-cyclodextrin for acute liver disease in infants with Niemann-Pick C. Molecular Genetics and Metabolism Reports. 2021;28:100772. [DOI] [PMC free article] [PubMed] [Google Scholar]