

Abstract
Alpha-dystroglycan (αDG) is a highly glycosylated cell surface protein with a significant role in cell-to-extracellular matrix interactions in muscle. αDG interaction with extracellular ligands relies on the activity of the LARGE1 glycosyltransferase that synthesizes and extends the heteropolysaccharide matriglycan. Abnormalities in αDG glycosylation and formation of matriglycan are the pathogenic mechanisms for the dystroglycanopathies, a group of congenital muscular dystrophies. Muscle biopsies were evaluated from related 6-week-old Labrador retriever puppies with poor suckling, small stature compared to normal litter mates, bow-legged stance and markedly elevated creatine kinase activities. A dystrophic phenotype with marked degeneration and regeneration, multifocal mononuclear cell infiltration and endomysial fibrosis was identified on muscle cryosections. Single nucleotide polymorphism (SNP) array genotyping data on the family members identified three regions of homozygosity in 4 cases relative to 8 controls. Analysis of whole genome sequence data from one of the cases identified a stop codon mutation in the LARGE1 gene that truncates 40% of the protein. Immunofluorescent staining and western blotting demonstrated the absence of matriglycan in skeletal muscle and heart from affected dogs. Compared to control, LARGE enzyme activity was not detected. This is the first report of a dystroglycanopathy in dogs.
Keywords: dog, myopathy, α-dystroglycan, glycosylation
1. Introduction
Muscular dystrophies due to abnormal glycosylation of α-dystroglycan (αDG) are a common group of conditions referred to as dystroglycanopathies. Mutations in 18 genes have been identified in human patients with dystroglycanopathy [1]. A broad spectrum of clinical conditions ranging from a severe congenital onset with structural brain malformations (Walker Warburg syndrome, muscle-eye-brain disease, Fukuyama muscular dystrophy, CMD1D) to milder variants with no brain involvement (congenital muscular dystrophy type 1C and limb-girdle muscular dystrophy type 2 variants) have been reported [2–5]. Αlpha-dystroglycan is a cell surface protein that plays a significant role in cell-to-extracellular matrix interactions in muscle and undergoes complex glycosylation steps to regulate its ability to effectively interact with extracellular matrix proteins such as laminin, agrin and perlecan [2, 6–7].
Congenital muscular dystrophy 1D (CMD1D) is caused by mutations in the Like-acetyl-glucosaminyltransferase 1 (LARGE1) gene. The LARGE1 gene was identified as a possible candidate for human disease after the mutation in the equivalent gene was found in the spontaneously occurring Largemyd dystrophic mouse [8]. LARGE1 is ubiquitously expressed, with the highest levels in heart, brain and skeletal muscle [4]. Homozygous mice display a severe, progressive muscle weakness and myopathy with a maximum lifespan of 39 weeks. LARGE1 participates in the post-translational modification of αDG by synthesizing a heteropolysaccharide known as matriglycan, which is required for the binding of αDG and extracellular matrix ligands [9].
Several congenital myopathies and associated mutations have been described in recent years in the Labrador retriever breed, including two distinct DMD mutations in X-linked muscular dystrophy [10–12], a mild form of X-linked muscular dystrophy in which a mutation has not yet been published [13], a PTPLA1 mutation in centronuclear myopathy [14], an MTM1 mutation in X-linked myotubular myopathy [15–16], a COLQ mutation in congenital myasthenic syndrome [17], and two different COL6A3 mutations in collagen VI associated muscular dystrophy [18]. Here we describe for the first time a dystroglycanopathy in the Labrador retriever breed associated with a mutation in the LARGE1 gene.
2. Methods
2.1. Animals
Related Labrador retriever puppies were evaluated at 6 weeks of age as clinical cases by a practicing veterinarian. Owner approval for all diagnostic evaluations was obtained and IACUC approval was not required for evaluating diagnostic tissue specimens. Muscle or venous blood samples for DNA isolation from dogs in the original study pedigree, as well as other related dogs, were obtained under the University of Minnesota IACUC protocol 1903–36865A.
2.2. Histopathology and immunofluorescent staining
Samples from the biceps femoris, triceps and heart muscles of 3 affected Labrador retriever pups were collected immediately following euthanasia and were either wrapped in a saline-dampened gauze sponge and chilled or immersion-fixed in 10% neutral buffered formalin. Muscle samples were shipped by an overnight service under refrigeration to the Comparative Neuromuscular Laboratory at the University of California, San Diego, immediately flash frozen in isopentane pre-cooled in liquid nitrogen and stored frozen at −80°C until further processed. A full necropsy was performed on an additional related affected pup at Cornell University and DNA from this dog brought the number of cases for genomic studies to 4.
Skeletal muscle cross-sections (10 μm) were stained and reacted with a standard panel of histochemical stains and reactions [19]. Additional H&E staining on skeletal muscle and heart was carried out as previously described [2]. Immunofluorescent staining was performed on muscle cryosections from affected pups and archived control dogs (not age-matched) for detection of the carboxy terminus of dystrophin (1:250, Abcam, San Francisco, CA), for α-sarcoglycan (1:10, R98), β-sarcoglycan (1:100, 5B1), γ-sarcoglycan (1:100, 21B5), and δ-sarcoglycan (1:50, R214) all from the Campbell Laboratory, and laminin α2 chain (1:250, Enzo, Farmingdale, NY) by previously published methods [20]. Immunofluorescence was also performed to detect matriglycan and beta-dystroglycan (βDG) on skeletal muscle and heart cross-sections. For these studies, sections were blocked with Background Buster (Innovex Biosciences, Richmond, CA) prior to primary antibody incubation. A mouse monoclonal antibody IIH6 to matriglycan on alpha-dystroglycan (αDG; 1:25; Campbell Laboratory, University of Iowa; [21–22] was added to sections and allowed to incubate overnight at 4°C. The secondary antibody Alexa Fluor-conjugated goat anti-mouse IgM (1:500; Invitrogen, Carlsbad, CA) was added to the sections prior to adding mounting medium (ProLong Gold Antifade Mountant, Thermo Fisher Scientific, Waltham, MA) and coverslips. The sections were also stained with affinity purified βDG rabbit polyclonal AP83 (1:50; Campbell Laboratory, University of Iowa [23]) followed by Alexa-Fluor-conjugated 594 goat anti-rabbit IgG (1:500) and coverslip mounting. Phosphate buffered saline (PBS) was used for all immunofluorescence procedures. Digital images of stained sections were captured with a VS120-S5-FL Olympus slide scanner upright microscope (Olympus Cooperation, Tokyo, Japan).
Formalin fixed paraffin embedded sections from the brain frontal cortex of an affected pup and an archived control dog were routinely stained with H&E and Luxol fast blue (LFB). Additional brain sections were deparaffinized and rehydrated, followed by heat-induced antigen retrieval overnight at 50°C prior to staining for matriglycan localization (1:25, IIH6). Staining was visualized using ImmPACT DAB substrate, Peroxidase HRP (Vector laboratories, Burlingame, CA), and counterstained with hematoxylin.
2.3. Whole genome sequencing and genotyping
Genome wide association analysis–
Genomic DNA was isolated from whole blood or muscle using the Gentra PureGene blood kit (Qiagen). Four case and eight control dogs were genotyped on the Illumina CanineHD BeadChip 230 K array. After pruning the dataset as described previously [24], 113,057 SNPs remained. Assuming an autosomal recessive mode of inheritance, we utilized a homozygosity mapping approach with the GENO test in PLINK [25], in which SNPs were required to be homozygous in all four cases, and heterozygous or homozygous for the alternative allele in unaffected parents and siblings. All genome positions refer to the CanFam3.1 reference sequence assembly.
Whole genome sequence analysis–
A PCR-free library was prepared from one case and sequenced in one lane of an Illumina HiSeq 4000 sequencer by GeneWiz (South Plainfield, NJ 07080). Briefly, a fragment library with an average insert size of ~ 680 bp was prepared from which ~ 125 million 2 × 150 bp paired-end reads were generated, corresponding to roughly 17× genome-wide coverage. The reads were mapped against the dog reference genome assembly (CanFam3.1) as described [26–27] and are available in NCBI’s Short Read Archive https://www.ncbi.nlm.nih.gov/sra/PRJNA713494. Variants identified in the critical interval from the SNP genotype data in the case were compared to those of control genomes from the University of Minnesota’s private whole genome sequencing (WGS) database containing 523 dogs of 55 diverse breeds (including 16 Labrador retrievers from unrelated projects). Databases containing variants identified in WGS of more than 1300 dogs of 126 diverse breeds [28–29] were subsequently searched for the presence of an identified mutation. Forward (5’-AGACCAGCGATCCTGTCCTA-3’) and reverse (5’-CACCACCCTCACCAGAAGAC-3’) PCR primers were designed to produce a 470 bp amplicon containing the LARGE1 variant for genotyping by Sanger sequencing.
2.4. Western Blotting
Archived control and affected Labrador retriever skeletal and heart muscle was used for biochemical analysis. Thirty slices were cut at 30 μM thickness from frozen biceps muscle on a cryostat (Leica CM3050S Research Cryostat). Samples were solubilized in 1% Triton X-100 in 50 mM Tris (pH 7.6) and 150 mM NaCl with protease inhibitors. Samples were vortexed for 4 min and solubilized at 4°C with rotation. Muscle samples were then spun at 14,000 rpm for 30 min at 4°C. The supernatant was incubated with WGA-Agarose slurry (Vector BioLabs, Malvern, PA) overnight at 4°C with rotation. Samples were washed in 50mM Tris (pH 7.6) and 150 mM NaCl containing 0.1% TX-100. Laemmli sample buffer (5X LSB) was added, and samples were boiled for 10 min.
The mouse monoclonal antibody against matriglycan (IIH6) was characterized previously [22,30] and used at 1:100. The polyclonal antibody, AF6868 (R&D Systems, Minneapolis, MN), was used at a concentration of 1:100 for the detection of core αDG and βDG proteins. Blots were developed with infrared dye-conjugated secondary antibodies (1:13,000; LI-COR Biosciences, Lincoln, NE) and scanned using the Odyssey infrared imaging system (LI-COR Biosciences). Blot images were captured using the Odyssey image-analysis software.
Laminin overlay assays were performed as described previously [2]. PVDF-FL membranes were blocked in laminin-binding buffer containing 5% milk followed by incubation with mouse Engelbreth-Hom-Swarm (EHS) laminin (ThermoFisher) overnight at a concentration of 7.5 nM at 4°C in LBB containing 3% bovine serum albumin and 2 mM CaCl2. Membranes were washed and incubated with anti-laminin antibody (1:1000; Sigma-Aldrich) followed by IRDye 800 CW dye-conjugated donkey anti-rabbit IgG (1:13,000; LI-COR).
2.5. Analysis of Large enzymatic activity
Solubilization of canine skeletal and heart muscle–
Using a cryostat, 5 slices (30 μm each) were taken from both control and affected skeletal and heart muscle tissue. The tissue was solubilized in 250 μl of 50 mM Tris-HCl pH 7.4, 100mM NaCl, 1% TX-100, with protease inhibitors (0.6 μg/ml of Pepstatin A, 0.5 μg/ml of leupeptin and aprotinin, 0.75 mM benzamidine and 1 mM phenylmethanesulfonyl fluoride). After solubilization for one hour at 4°C, the samples were spun down at 30,000 × g for 30 minutes. The resulting supernatant was saved for the enzyme assays.
Enzymatic assays–
The HPLC-based LARGE enzymatic assay for 20 μl of the skeletal and heart muscle supernatants was performed using Xyl-1,3-GlcA -MU for GlcA-T activity as described previously [9,31]. For the assessment of endogenous LARGE GlcA-T activity, skeletal and heart muscle supernatants (20 μl) were incubated in a volume of 50 μl for 18 h at 37 °C, with 0.4 mM Xyl-1,3-GlcA-MU and 10 mM UDP-GlcA in 0.1 M MES buffer, pH 6.0, at 5 mM MnCl2, 5 mM MgCl2. The reaction was terminated by adding 25 μl of 0.2 M EDTA and boiling for 5 min, and the supernatants were analyzed using a LC18 column (4.6 × 250 mm Supelcosil LC-18 column (Supelco, Bellefonte, PA)) with Buffer A (50 mM ammonium formate pH 4.0) and Buffer B (80% acetonitrile in buffer A), using a 9% B isocratic run at 1 ml/min using HPLC (Shimadzu Scientific). The elution of MU derivatives was monitored by fluorescence detection (325 nm for excitation, and 380 nm for emission).
3. Results
3.1. Animals
Four affected Labrador retriever puppies (Figure 1A), produced by 2 pairs of parents each of which share relationships that go back several generations (Figure 1B) were evaluated for small stature, poor weight gain, bow legged stance, poor suckling, and weakness. Nine pups were born into litter one (4 yellow females, 4 black females, 1 yellow male) all with similar birth weights (0.3–0.5 kg). In the first week 4 pups required supplemental tube feeding due to poor weight gain, and difficulties in prehension of food and swallowing. One of these 4 pups died during the first week and another pup was euthanized at 2.5 weeks of age. At the time of euthanasia at 6 weeks of age, body weights of the two remaining affected pups were half that of the normal pups (1.3 and 1.4 kg compared to 2.7–2.9 kg). Four pups were born into litter two (2 yellow females, 1 black female,1 black male) all with similar birth weights (approximately 0.5 kg). Like the previous affected pups, difficulty feeding, and poor weight gain was found in 2 affected pups with weights at the time of euthanasia half that of normal littermates (2.7 and 2.9 kg vs 4.1 and 4.6 kg). Serum CK activities were markedly elevated in all affected pups ranging from 10,587 to 23,638 IU/L (reference 59–895 IU/L). Post-mortem muscle and heart specimens were collected on 3 affected pups and a full necropsy was performed on 1 affected pup at 6 weeks of age.
Figure 1.
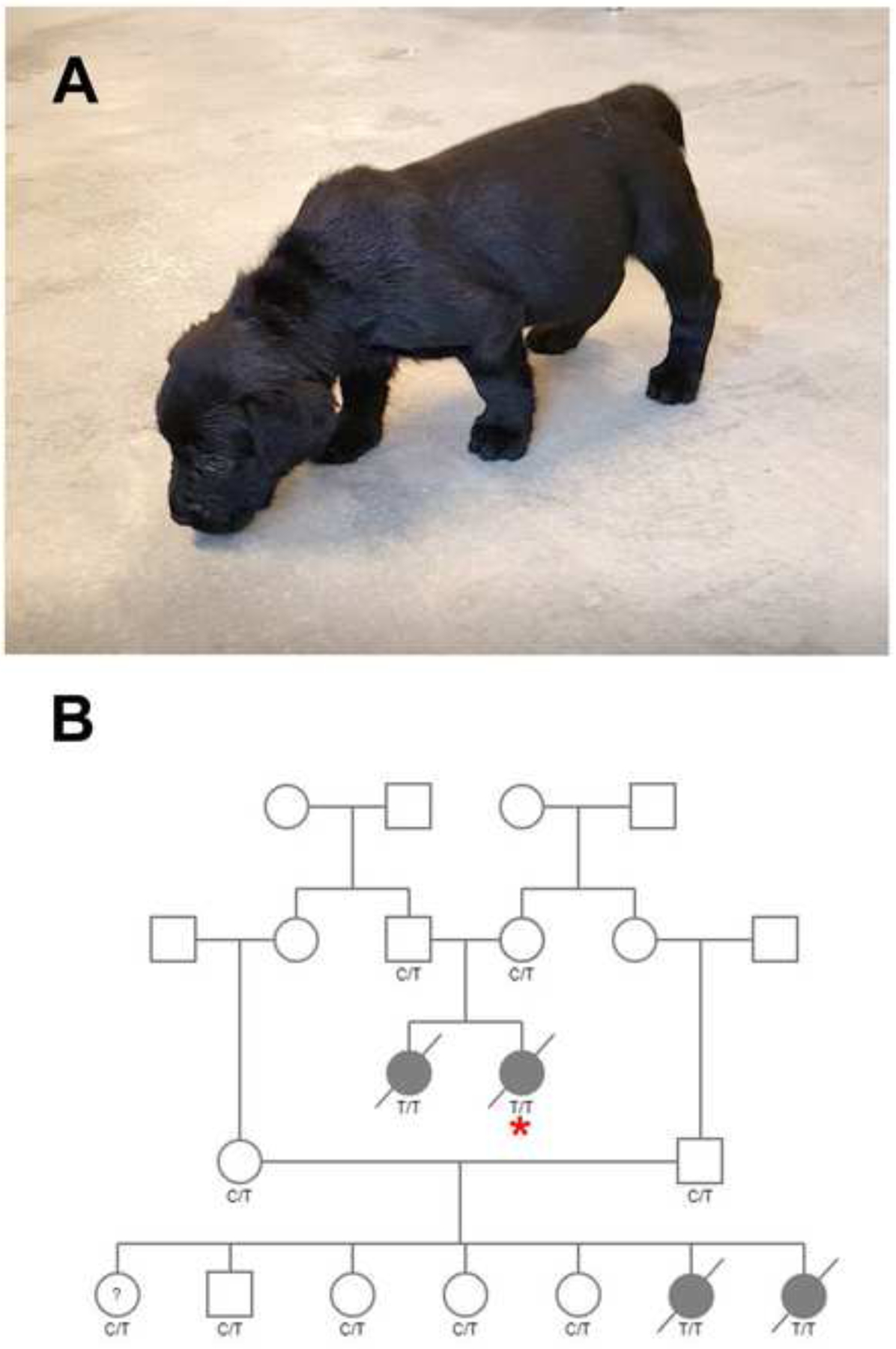
Affected Pup and Pedigree. A) 6-week-old Labrador retriever pup with poor suckling, small stature, and bow-legged stance. B) One family of Labrador retrievers formed the basis for this study. Males are squares and females’ circles. Cases are solid symbols, unaffected are open. ?=phenotype unknown. A diagonal strikethrough indicates deceased pups. Genotypes for the LARGE1 variant are provided for all dogs with available samples. TT = homozygous mutant, C/T = carrier, CC = clear. The case with WGS data is indicated with an asterisk.
3.2. Histopathology and Immunofluorescent Staining
Histopathology of skeletal muscle cryosections in three affected pups showed degenerative and regenerative changes consistent with a dystrophic phenotype (Figure 2A). Similar degenerative and regenerative changes were observed in the muscles of the fourth pup that underwent a complete necropsy. Immunofluorescent staining was performed with the IIH6 antibody to evaluate whether muscle from the three affected six-week-old pups lacked matriglycan. There was an absence of IIH6 staining in the affected muscle, whereas the control muscle displayed IIH6 staining along the sarcolemma. As detected by the AP83 antibody, βDG was observed along the sarcolemma in muscle sections from both control and affected pups (Figure 2B). Additional immunofluorescent stainings of skeletal muscle cryosections for the carboxy terminus of dystrophin, α, β, γ and δ- sarcoglycans, and laminin α−2 in affected muscles were like those of archived control muscle (Figure 3).
Figure 2.

Histopathology and Immunofluorescence Staining of Skeletal Muscle. Stainings are shown from a control dog and an affected Labrador retriever pup. A). With the H&E stain, a dystrophic phenotype was present in the muscle biopsy from the affected pup. B). Using the IIH6 monoclonal antibody to detect matriglycan on αDG, abnormal glycosylation consistent with a diagnosis of a dystroglycanopathy was identified. Positive staining was observed using the AP83 polyclonal antibody against βDG. Scale bars = 100 μM for all images.
Figure 3.
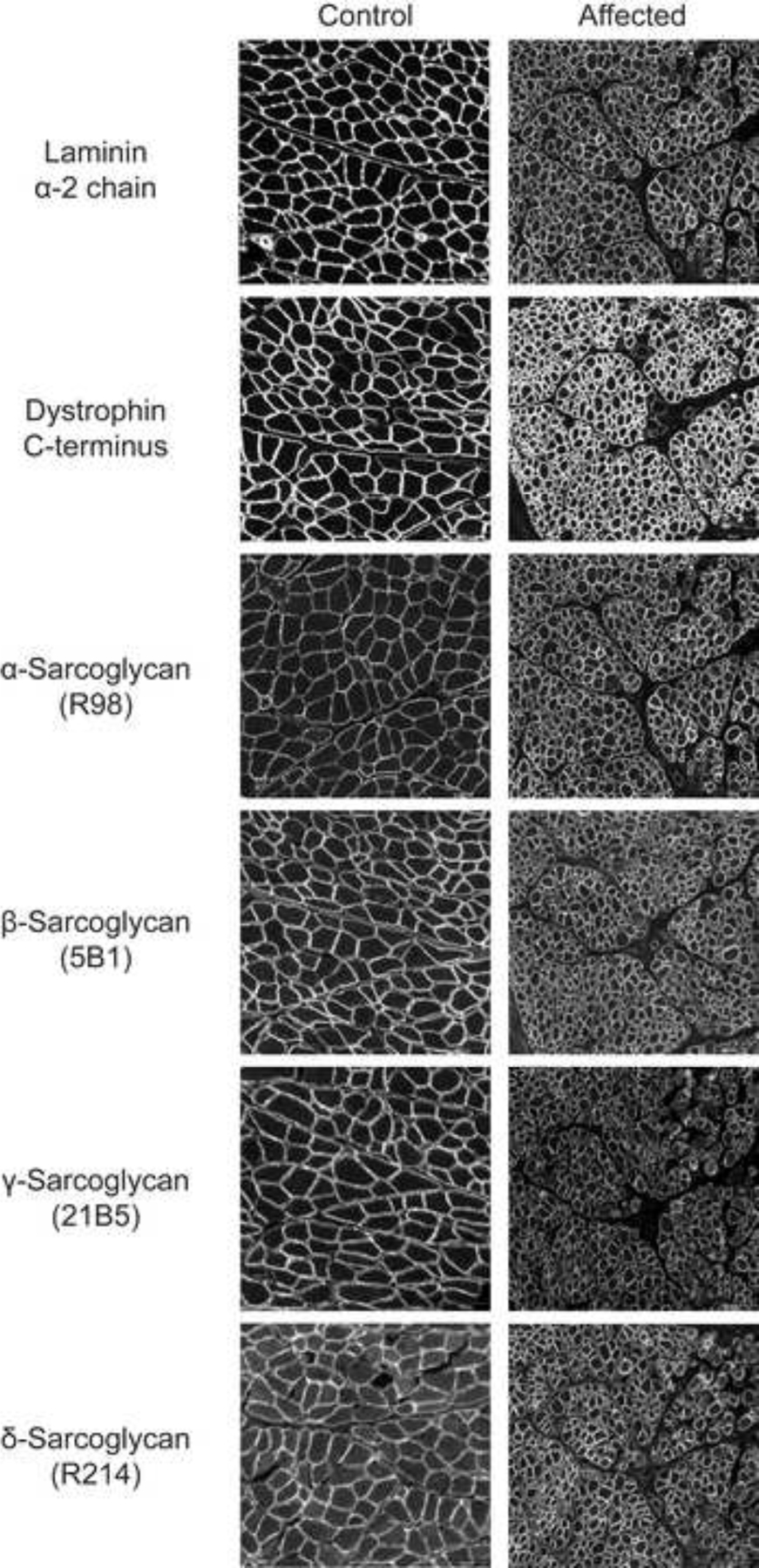
Additional Immunofluorescence Staining of Skeletal Muscle. Stainings with antibodies against laminin α−2 chain, dystrophin C-terminus, and α-, β-, γ-, and δ-sarcoglycans in the affected pup were like that of control muscle. Scale bars = 100 μM for all images.
Histopathology of the heart in affected pups showed no specific pathological changes (Figure 4). Like skeletal muscle, the heart muscle from affected pups showed an absence of IIH6 staining for matriglycan, whereas the control heart muscle displayed IIH6 staining along the sarcolemma. The brain frontal cortex of the pup submitted for necropsy did not show any specific pathological evidence of dysplasia or abnormal growth patterns (Figure 4). Using the IIH6 antibody, reddish brown staining localized matriglycan on neurons and associated axons in the control frontal cortex but was absent in the brain of the affected pup (Figure 5), indicating that brain dystroglycan is hypoglycosylated.
Figure 4.
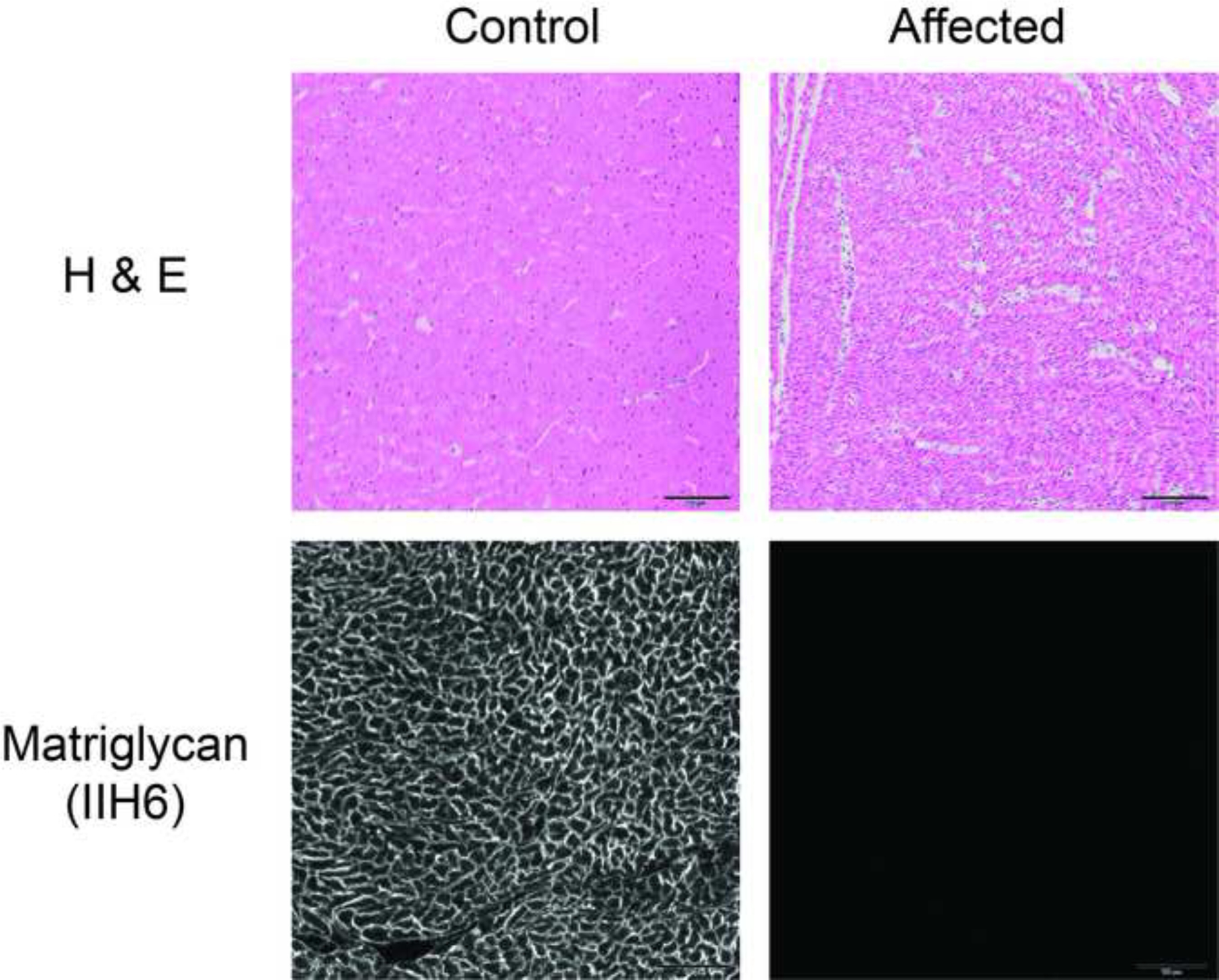
Histopathology and Immunofluorescence Staining of Heart Muscle. H&E stainings are shown for a control dog and an affected Labrador retriever pup. Except for the small muscle fiber size due to the young age of the affected pup, no pathological changes were identified compared to the control dog muscle. Using the IIH6 monoclonal antibody to detect matriglycan on αDG, abnormal glycosylation consistent with a diagnosis of a dystroglycanopathy was identified. Scale bars = 100 μm for all images.
Figure 5.

Histopathology and Immunohistochemical Staining of Brain Frontal Cortex. H&E and Luxol fast blue stains of the frontal cortex are shown from a control dog and an affected Labrador retriever pup. No pathological evidence of dysplasia or abnormal growth patterns were identified in the affected pup compared to the control dog. Using the IIH6 monoclonal antibody to detect matriglycan on αDG, reddish brown staining identified matriglycan on neurons (arrowheads) and their associated axons (arrow) of control brain but not on similar structures from the affected pup. Unidentified cell processes are labeled around blood vessels. Scale bars = 100 μm for all images.
3.3. Genome wide association analysis, whole genome sequencing, and genotyping
As an autosomal recessive mode of inheritance was suspected, we employed a genotypic analysis model in which all four cases were required to be homozygous for an allele, while controls could be either heterozygous or homozygous for the alternate allele. In total, 115 SNPs met this criterion; 91 of these SNPs reside in an ~12.5 Mb segment on chromosome 10 between bp positions 22,958,819–35,483,256; 19 of the SNPs were on chromosome 11 in an ~5 Mb segment between 15,673,594–20,546,241; and the 5 remaining sporadic SNPs were on chromosome 36 (Supplemental Table 1).
We next obtained whole genome sequence from a case and searched for homozygous coding variants residing within the identified homozygous chromosomal regions that were unique to our case compared to 522 other dogs from 55 breeds within our laboratory’s private WGS database (including 16 Labrador retrievers from unrelated projects). After this filtering, a single variant remained; a premature stop codon within the LARGE1 gene on chromosome 10 (Chr10 g.30,357,716C>T; c.1363C>T; p.R455*, ENSCAFT00000074128.1, XM_038680042.1). The canine LARGE1 gene has 15 exons that encode 756 amino acids. The predicted premature stop codon truncates the protein, likely eliminating all enzymatic activity.
Dogs within the pedigree of Figure 1B were then genotyped for this c.1363C>T variant. All four cases were homozygous, all four parents were unaffected and heterozygous, and available littermates were unaffected and heterozygous; all consistent with an autosomal recessive trait. This LARGE1 variant was also not identified in external databases containing variants identified in whole genome sequence of more than 1300 dogs of 126 diverse breeds [28–29]. Further genotyping of 87 dogs closely related to those in Figure 1B, obtained from a breeding program developed for service dog training, identified 62 dogs who were homozygous wild type, 25 dogs who were heterozygous, and no dogs were homozygous mutant.
3.4. Western Blotting and Enzyme Activity
Wheat-germ agglutinin (WGA)-enriched extracts of skeletal muscle (biceps femoris, Figure 6A–C) and heart ventricular muscle (Figure 6D–F) from archived control dogs and affected pups were analyzed by western blotting for matriglycan and dystroglycan. Matriglycan, as detected by the IIH6 antibody, was absent in the skeletal muscles (Figure 6A) and heart (Figure 6D) from the affected pups, although αDG and βDG were present (Figures 6B and 6E). However, αDG protein was detected at a reduced molecular weight in the affected muscle, confirming that αDG glycosylation, not protein expression, was atypical. We then performed a laminin overlay assay to evaluate the ligand-binding capability in affected pup skeletal and heart muscle. Control skeletal and heart muscle showed broad bands of αDG-laminin-binding (Figure 6C and 6F). In contrast, αDG-laminin-binding was lacking in skeletal and heart muscle from affected pups, thus indicating a loss of αDG functional binding ability in affected pup muscle. Of note, the tissue-specific modification of αDG is observable by the increased molecular weight of cardiac matriglycan and laminin overlay (centered near 250 kDa) in controls, whereas the molecular weight in control skeletal muscle is centered near 150 kDa.
Figure 6.

Western blotting of Skeletal and Heart Muscle. Glycoproteins were enriched from the biceps femoris muscle using wheat-germ agglutinin (WGA)-agarose. Immunoblotting (three replicates) was performed on control and affected Labrador retriever skeletal and heart muscle with A,D, IIH6 antibody for matriglycan; B,E, antibody AF6868, which recognizes core αDG and βDG; and C,F, laminin overlay to detect laminin binding respectively.
To study how the LARGE1 stop codon mutation influenced enzymatic function, we assessed LARGE1 activity on control and affected muscle samples. LARGE1 acts to synthesize and extend matriglycan on αDG by adding xylose-glucuronic acid disaccharides; therefore, we examined LARGE1 enzymatic activity through the glucuronyltransferase (GlcA-T) activity assay. LARGE1 activity was not detected in either the affected skeletal (Figure 7A) or cardiac muscle samples (Figure 7B). Together, these data provide further evidence of the absence of LARGE1 in muscle from affected pups and are consistent with a diagnosis of a dystroglycanopathy.
Figure 7.
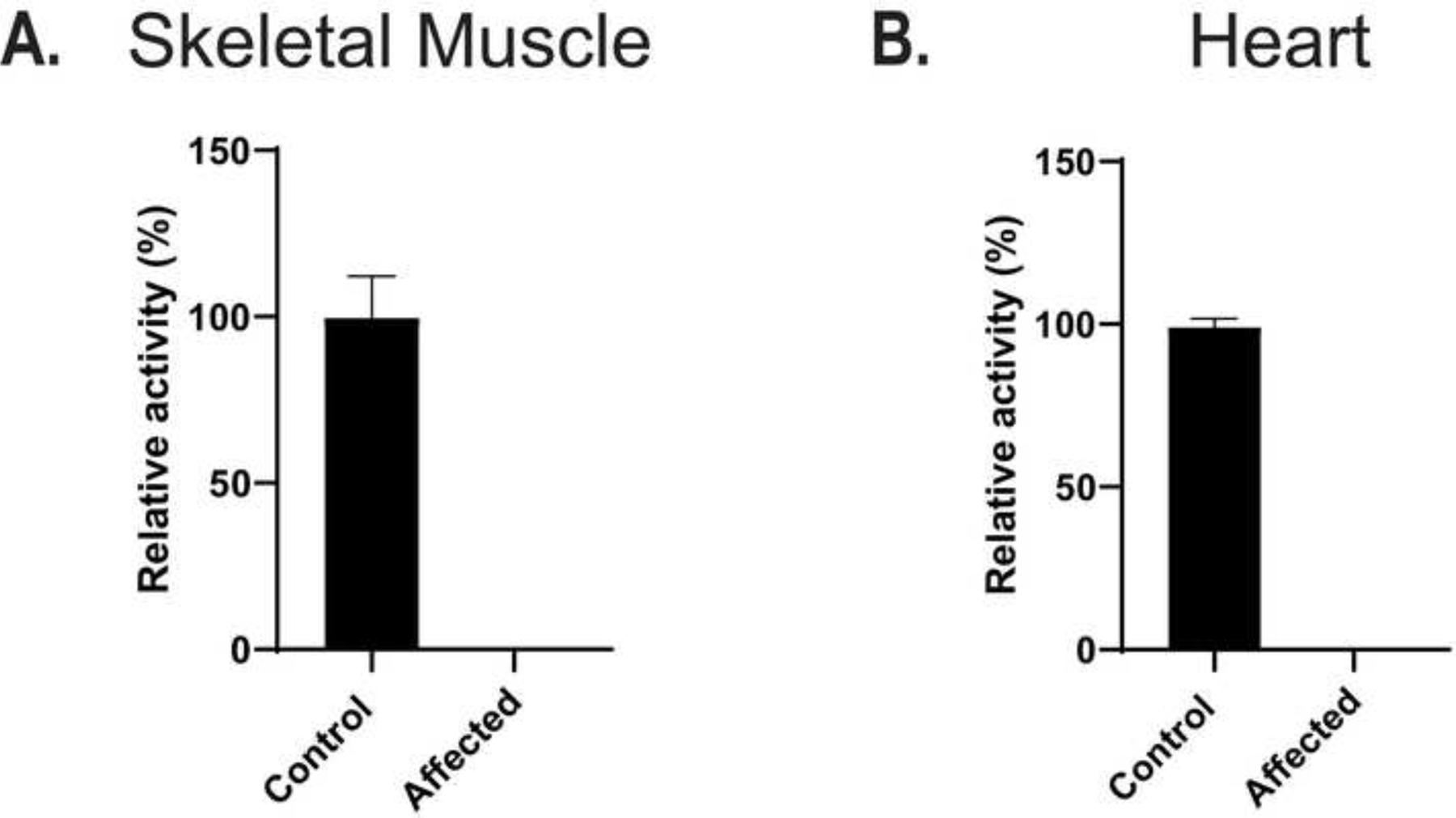
LARGE enzyme activity. Lysates (20 μL) from control dog skeletal (Panel A, n=3) and heart (Panel B, n=3) muscle and LARGE mutant skeletal (Panel A, n=3) and heart (Panel B, n=3) muscle (affected) were assayed for enzyme activity for LARGE. Relative activity (%) with respect to control and standard deviation is for triplicate experiments. The activity was normalized by amount of βDG signal from a blot of control and LARGE mutant lane using monoclonal 7D11 which has an epitope to the c-terminus of βDG.
4. Discussion
Over the past 10 years, several naturally occurring muscle diseases have been described in dogs with clinical and gene variant correlations to the human counterparts [10–18]. Spontaneously occurring canine models are now used for testing new cell, gene and gene editing therapies for the treatment of both human and canine muscle diseases, including XLMTM [32–33] and dystrophin-deficient muscular dystrophy [34–37]. Many of these myopathies occur in the Labrador retriever breed. Here we describe for the first time a LARGE1 gene mutation resulting in a dystroglycanopathy in a family of Labrador retriever dogs. The functional and causative nature of this mutation is strongly supported by the predicted significant truncation of the LARGE1 protein and the absence of detectable LARGE1 enzyme activity, coupled with the absence of matriglycan determined by immunofluorescent microscopy of affected skeletal muscles, heart and brain (frontal cortex).
As in people, the clinical phenotypes, pattern of tissue involvement and spectrum of severity in muscle diseases resulting from gene variants can vary widely in dogs. Mutations in LARGE1 have been identified in humans with congenital muscular dystrophy and central nervous system involvement [3, 38–41]. LARGE1 is expressed in skeletal muscle, heart and brain, but clinical signs may not be directed specifically to all three tissues. Heart irregularities were not detected from the necropsied pup’s histological evaluation. In dystroglycanopathies, heart abnormalities and dysfunction are often progressive and are not detectable early in life [42]. At only six-weeks of age, the necropsied pup may have been too young to display signs of cardiac disruption.
Further, in our family of Labrador retrievers, abnormal neurological signs or behavioral changes supportive of central nervous system disease were not clinically described despite the absence of matriglycan in the brain tissue examined (Figure 5). It is possible that these changes were subtle and not clinically obvious, or this may indicate a species variability. In the one dog that underwent a complete necropsy, no pathological evidence of dysplasia or abnormal growth patterns were identified in the brain. Another potential explanation for the apparent lack of neuropathology and clinical signs of brain disease is that LARGE2 may play a compensatory role in the central nervous system in the presence of a LARGE1 mutation; however, the expression and activity of LARGE2 in the central nervous system of dogs is not well understood. A limitation to the histological and immunohistochemical evaluation of the brain was the availability of only paraffin embedded tissue and only from limited areas of the brain. Despite this, we were able to document hypoglycosylation of dystroglycan in this dog brain.
LARGE1 mutations described in human dystroglycanopathies include missense and frameshift mutations as well as intragenic deletions/insertions [43]. In our family of dogs, a premature stop codon mutation was identified that truncates the protein. The position of the premature stop codon (p.R455*) disrupts the glucuronyltransferase catalytic domain (positions 414–756). Truncation of the protein presumably disrupts all enzymatic activity and increases the protein’s likelihood of being degraded. The absence of LARGE enzymatic activity was confirmed in a functional assay (Figure 7).
Although the mutation was not observed in whole genome sequence variant databases available to us that included many breeds including Labrador retrievers, it was detected within a Labrador retriever breeding program where surveillance for disease-causing mutations is rigorously performed. As with many other Mendelian disorders in dogs, owners, veterinarians, and breeders will now be able to use genetic testing to diagnose this condition, as well as reduce its incidence in pedigrees where it is being observed.
It was recently shown that overexpression of LARGE1 in cells from humans affected with different dystroglycanopathies restored α-DG laminin binding, indicating that the modulation of LARGE1 activity may represent a therapeutic strategy for the treatment of dystroglycanopathies [44–45]. As in X-linked myotubular myopathy and the X-linked dystrophinopathies, large animal models could play an important role in evaluating future therapies for this form of dystroglycanopathy.
5. Conclusions
This study expands our knowledge of congenital myopathies affecting dogs with direct correlations to the corresponding human conditions. Here we describe a mutation in the LARGE1 gene in a family of Labrador retriever dogs in which histopathology, immunostaining, western blotting and enzyme activity confirm a diagnosis of a dystroglycanopathy.
Supplementary Material
Highlights.
Whole genome sequencing revealed a dystroglycanopathy in dogs with a LARGE1 mutation
Matriglycan was absent in skeletal muscle and heart from affected dogs
LARGE enzyme activity was not detected
This is the first report of a dystroglycanopathy in dogs
Acknowledgements
KPC is an investigator of the Howard Hughes Medical Institute.
This work was supported in part by a Paul D. Wellstone Muscular Dystrophy Specialized Research Center Grant (1U54NS053672 to KPC).
This work was also supported by the Cardiovascular Institutional Research Fellowship (5T32HL007121–45 to JMH).
SGF is supported in part by an NIH Special Emphasis Research Career Award (1 K01 OD027058) in Pathology and Comparative Medicine sponsored by the Division of Comparative Medicine, Office of Research Infrastructure Programs.
The authors thank Andrew David Miller and Kathleen Kelly for pathologic evaluation of the brain and heart on the affected dog necropsied at Cornell University.
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Nickolls AR, Bonnemann CG. The roles of dystroglycan in the nervous system: insights from animal models of muscular dystrophy. Dis Model Mech 2018;11:(12):dmm035931. doi: 10.1242/dmm.035931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Michele DE, Barresi R, Kanagawa M, Saito F, Cohn RD, Satz JS et al. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature 2002;418:417–22. doi: 10.1038/nature00837. [DOI] [PubMed] [Google Scholar]
- [3].Longman C, Brockington M, Torelli S, Jimenez-Mallebrera C, Kennedy C, Khalil N et al. Mutations in the human LARGE gene cause CMD1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum Mol Genet 2003;12:2853–61. doi: 10.1093/hmg/ddg307. [DOI] [PubMed] [Google Scholar]
- [4].Grewal PK, McLaughlan JM, Moore CJ, Browning CA, Hewitt JE. Characterization of the LARGE family of putative glycosyltransferases associated with dystroglycanopathies. Glycobiology 2005;15:912–23. doi: 10.1093/glycob/cwi094. [DOI] [PubMed] [Google Scholar]
- [5].Muntoni F, Torelli S, Brockington M. Muscular dystrophies due to glycosylation defects. Neurotherapeutics 2008;5:627–32. doi: 10.1016/j.nurt.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Endo T Glycobiology of alpha-dystroglycan and muscular dystrophy. J Biochem 2015;157:1–12. doi: 10.1093/jb/mvu066. [DOI] [PubMed] [Google Scholar]
- [7].Yoshida-Moriguchi T, Campbell KP. Matriglycan: A novel polysaccharide that links dystroglycan to the basement membrane. Glycobiology 2015;25:702–13. doi: 10.1093/glycob/cwv021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Grewal PK, Holzfeind PJ, Bittner RE, Hewitt JE. Hewitt JE. Mutant glycosyltransferase and altered glycoslyation of alpha-dystroglycan in the myodystrophy mouse. Nat Genet 2001; 28:151–4. doi: 10.1038/88865. [DOI] [PubMed] [Google Scholar]
- [9].Inamori K, Yoshida-Moriguchi T, Hara Y, Anderson ME, Yu L, Campbell KP. Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science 2012;335:93–6. doi: 10.1126/science.1214115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bergman RL, Inzana KD, Monroe WE, Shell LG, Liu LA, Engvall E et al. Dystrophin-deficient muscular dystrophy in a Labrador retriever. J Am Anim Hosp Assoc 2002; 38:255–61. doi: 10.5326/0380255. [DOI] [PubMed] [Google Scholar]
- [11].Kornegay JN, Bogan JR, Bogan DJ, Childers MK, Li J, Nghiem P, Detwiler DA et al. Canine models of Duchenne muscular dystrophy and their use in therapeutic strategies. Mamm Genome. 2012;23:85–108. doi: 10.1007/s00335-011-9382-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Barthélémy I, Calmels N, Weiss RB, Tiret L, Vulin A, Wein N et al. X-linked muscular dystrophy in a Labrador Retriever strain: phenotypic and molecular characterization. Skelet Muscle. 2020;10:23. doi: 10.1186/s13395-020-00239-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Vieira NM, Guo LT, Estrela E, Kunkel LM, Zatz M, Shelton GD. Muscular dystrophy in a family of Labrador Retrievers with no muscle dystrophin and a mild phenotype. Neuromuscul Disord. 2015. May;25(5):363–70. doi: 10.1016/j.nmd.2015.02.012. [DOI] [PubMed] [Google Scholar]
- [14].Pelé M, Tiret L, Kessler JL, Blot S, Panthier JJ. SINE exonic insertion in the PTPLA gene leads to multiple splicing defects and segregates with the autosomal recessive centronuclear myopathy in dogs. Hum Mol Genet 2005;14:1417–27. doi: 10.1093/hmg/ddi151. [DOI] [PubMed] [Google Scholar]
- [15].Beggs AH, Böhm J, Snead E, Kozlowski M, Maurer M, Minor K et al. MTM1 mutation associated with X-linked myotubular myopathy in Labrador Retrievers. Proc Natl Acad Sci U S A. 2010;107:14697–702. doi: 10.1073/pnas.1003677107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Snead EC, Taylor SM, van der Kooij M, Cosford K, Beggs AH, Shelton GD. Clinical phenotype of X-linked myonuclear myopathy in Labrador Retriever puppies J Vet Intern Med. 2015;29:254–60. doi: 10.1111/jvim.12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rinz CJ, Levine J, Minor KM, Humphries HD, Lara R, Starr-Moss AN et al. A COLQ missense mutation in Labrador Retrievers with congenital myasthenic syndrome. PLoS One 2014;9:e106425. doi: 10.1371/journal.pone.0106425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bolduc V, Minor KM, Hu Y, Kaur R, Friedenberg SG, Van Buren S et al. Pathogenic variants in COL6A3 cause Ullrich-like congenital muscular dystrophy in young Labrador Retriever dogs. Neuromuscul Disord. 2020;30:360–7. doi: 10.1016/j.nmd.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dubowitz V, Sewry CA, Oldfors A (2013). Histological and histochemical stains and reactions. In Dubowitz V, Sewry CA, Oldfors A eds. Muscle Biopsy: a practical approach. 4th ed. Oxford: Saunders Elsevier, 16–27. [Google Scholar]
- [20].Mickelson JR, Minor KM,Guo LT, Friedenberg SG, Cullen JN, Ciavarella A et al. Sarcoglycan A mutation in miniature dachshund dogs causes limb-girdle muscular dystrophy 2D. Skelet Muscle 2021;11:2. doi: 10.1186/s13395-020-00257-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ervasti JM, Ohlendieck K, Kahl SD, Gaver MG, Campbell KP. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 1990;345:315–9. doi: 10.1038/345315a0. [DOI] [PubMed] [Google Scholar]
- [22].Goddeeris MM, Wu B, Venzke D, Yoshida-Moriguchi T, Saito F, Matsumura K et al. LARGE glycans on dystroglycan function as a tunable matrix scaffold to prevent dystrophy. Nature 2013:136–40. doi: 10.1038/nature12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Williamson RA, Henry MD, Daniels KJ, Hrstka RF, Lee JC, Sunada Y, et al. Dystroglycan is essential for early embryonic development: Disruption of Reichert’s membrane in dag1-null mice. Hum Mol Genet 1997;6:831–41. doi: 10.1093/hmg/6.6.831. [DOI] [PubMed] [Google Scholar]
- [24].Becker D, Minor KM, Letko A, Ekenstedt KJ, Jagannathan V, Leeb T et al. A GJA9 frameshift variant is associated with polyneuropathy in Leonberger dogs. BMC Genomics. 2017;18:662. doi: 10.1186/s12864-017-4081-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D et al. PLINK, a tool set for whole-genome association and population-based linkage analysis. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Meurs KM, Friedenberg SG, Kolb J, Saripalli C, Tonino P, Woodruff K et al. A missense variant in the titin gene in Doberman pinscher dogs with familial dilated cardiomyopathy and sudden cardiac death. Hum Genet 2019;138:515–24. doi: 10.1007/s00439-019-01973-2 [DOI] [PubMed] [Google Scholar]
- [27].Shelton GD, Minor KM, Li K, Naviaux JC, Monk J, Guzik E et al. A mutation in the mitochondrial aspartate/glutamate carrier leads to intramitochondrial oxidation and an inflammatory myopathy in Dutch Shepherd Dogs. J Neuromusc Dis 2019;6:485–501. doi: 10.3233/JND-190421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jagannathan V, Drögemüller C, Leeb T, et al. Dog Biomedical Variant Database Consortium (DBVDC). A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and eight wolves. Anim Genet. 2019;50:695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Plassais J, Kim J, Davis BW, Karyadi DM, Hogan AN, Harris AC et al. Whole genome sequencing of canids reveals genomic regions 494 under selection and variants influencing morphology. Nat Commun 2019;10:1489. doi: 10.1038/s41467-019-09373-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell 1991;66:1121–31. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- [31].Inamori K, Hara Y, Willer T, Anderson ME, Zhu Z, Yoshida-Moriguchi T et al. Xylosyl- and glucuronyltransferase functions of LARGE in alpha-dystroglycan modification are conserved in LARGE2. Glycobiology 2013; 23:295–302. doi: 10.1093/glycob/cws152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Elverman M, Goddard MA, Mack D, Snyder JM, Lawlor MW, Meng H et al. Long-term effects of systemic gene therapy in a canine model of myotubular myopathy. Muscle Nerve. 2017;56:943–53. doi: 10.1002/mus.25658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mack DL, Poulard K, Goddard MA, Latournerie V, Snyder JM, Grange RW et al. Systemic AAV8-mediated gene therapy drives whole-body correction of myotubular myopathy in dogs. Mol Ther. 2017;25:839–54. doi: 10.1016/j.ymthe.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nghiem PP, Kornegay JN. Gene therapies in canine models for Duchenne Muscular Dystrophy. Hum Genet 2019;138:483–89. doi: 10.1007/s00439-019-01976-z. [DOI] [PubMed] [Google Scholar]
- [35].Barraza-Flores P, Fontelonga TM, Wuebbles RD, Hermann HJ, Nunes AM, Kornegay JN et al. Laminin-111 protein therapy enhances muscle regeneration and repair in the GRMD dog model of Duchenne muscular dystrophy. Hum Mol Genet 2019;28:2686–95 doi: 10.1093/hmg/ddz086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Song Y, Morales L, Malik AS, Mead AF, Greer CD, Mitchell MA et al. Non-immunogenic utrophin gene therapy for the treatment of muscular dystrophy animal models. Nat Med 2019;25:1505–11. doi: 10.1038/s41591-019-0594-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mata López S, Balog-Alvarez C, Vitha S, Bettis AK, Canessa EH, Kornegay JN et al. Challenges associated with homologous directed repair using CRISPR-Cas 9 and TALEN to edit the DMD genetic mutation in canine Duchenne muscular dystrophy. PLoS One 2020;15:e0228072. doi: 10.1371/journal.pone.0228072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Godfrey C, Clement E, Mein R, Brockington M, Smith J, Talim B et al. Refining genotype-phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 2007;130:2725–35. doi: 10.1093/brain/awm212. [DOI] [PubMed] [Google Scholar]
- [39].Clement E, Mercuri E, Godfrey C, Smith J, Robb S, Kinali M et al. Brain involvement in muscular dystrophies with defective dystroglycan glycosylation. Ann Neurol 2008; 64: 573–82. doi: 10.1002/ana.21482. [DOI] [PubMed] [Google Scholar]
- [40].Mercuri E, Messina S, Bruno C, Mora M, Pegoraro E, Comi GP et al. Congenital muscular dystrophies with defective glycosylation of dystroglycan: A population study. Neurology 2009;72:1802–9. doi: 10.1212/01.wnl.0000346518.68110.60. [DOI] [PubMed] [Google Scholar]
- [41].Clarke NF, Maugenre S, Vandebrouck A, Urtizberea JA, Willer T, Peat RA et al. Congenital muscular dystrophy type 1D (CMD1D due to a large intragenic insertion/deletion, involving intron10 of the LARGE gene. Eur J Hum Genet 2011;19:452–7. doi: 10.1038/ejhg.2010.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Libell EM, Richardson JA, Lutz KL, Ng BY, Mockler SRH, Laubscher KM et al. Cardiomyopathy in limb girdle muscular dystrophy. Muscle Nerve 2020; 62:626–632. doi: 10.1002/mus.27052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Righino B, Bozzi M, Pirolli D, Sciandra F, Biogotti MG, Brancaccio A et al. , Identification and modeling of a GT-A fold in the α-dystroglycan glycosylating enzyme LARGE1. J Chem Inf Model 2020;60: 3145–56. doi: 10.1021/acs.jcim.0c00281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Barresi R, Michele DE, Kanagawa M, Harper HA, Dovico SA, Satz JS et al. LARGE can functionally bypass α-dystroglycan glycosylation defects in distinct congenital muscular dystrophies. Nat Med 2004;10:696–703. doi: 10.1038/nm1059. [DOI] [PubMed] [Google Scholar]
- [45].Beltran D, Anderson ME, Bharathy N, Settelmeyer TP, Svalina MN, Bajwa Z et al. Exogenous expression of the glycosyltransferase LARGE1 restores α-dystroglycan matriglycan and laminin binding in rhabdomyosarcoma. Skelet Muscle 2019,9:11. doi: 10.1186/s13395-019-0195-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.