

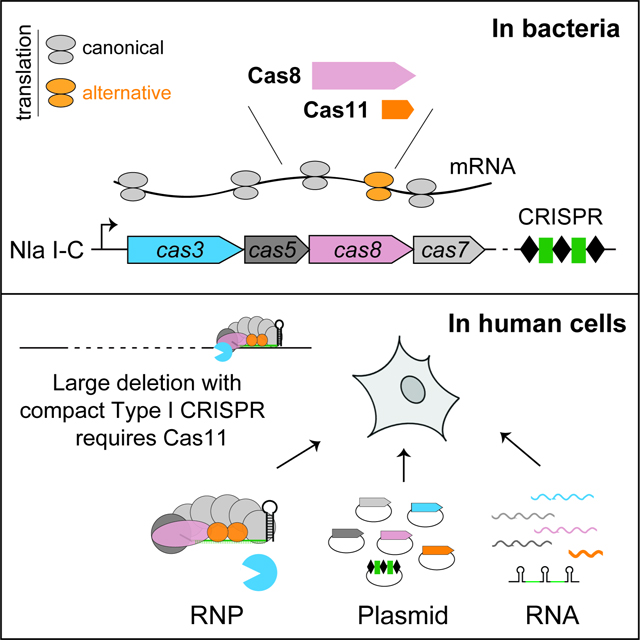
Summary
Leading CRISPR-Cas technologies employ Cas9 and Cas12 enzymes that generate RNA-guided dsDNA breaks. Yet, the most abundant microbial adaptive immune systems, Type I CRISPRs, are under-exploited for eukaryotic applications. Here we report the adoption of a minimal CRISPR-Cas3 from Neisseria lactamica (Nla) Type I-C system, to create targeted large deletions in the human genome. RNP delivery of its processive Cas3 nuclease and target recognition complex Cascade can confer ~95% editing efficiency. Unexpectedly, NlaCascade assembly in bacteria requires internal translation of a hidden component Cas11 from within the cas8 gene. Furthermore, expressing a separately encoded NlaCas11 is the key to enable plasmid- and mRNA- based editing in human cells. Finally, we demonstrate that supplying cas11 is a universal strategy to systematically implement divergent I-C, I-D, and I-B CRISPR-Cas3 editors with compact sizes, distinct PAM preferences and guide orthogonality. These findings greatly expand our ability to engineer long-range genome edits.
Keywords: CRISPR, gene editing, Cascade, Cas3, Cas11, DNA targeting, crRNA, large deletion, Neisseria, genome engineering
eTOC
Tan et al. discover a miniature CRISPR-Cas3 system that can efficiently create targeted large deletions in human genome. An inconspicuous subunit of its Cas machinery, Cas11, is encoded by a hidden ORF embedded in the cas operon. In human cells, Cas11 is the key enabler for compact CRISPR-Cas3 gene editors.
Graphical Abstract
INTRODUCTION
CRISPR-Cas systems employ diverse RNA-guided nucleases to help microbes fend off bacteriophages and other mobile genetic elements (Barrangou et al., 2007; Brouns et al., 2008; Makarova et al., 2015; Marraffini and Sontheimer, 2008). Current genome editing technologies primarily use single effector enzymes such as Cas9 or Cas12 from Class II CRISPR systems, for programmable DNA sequence alterations (Anzalone et al., 2020; Doudna, 2020). Cas9 or Cas12 is guided by its CRISPR RNA (crRNA) to the complementary target site flanked by a short protospacer-adjacent motif (PAM), and cleaves the target DNA at precise locations (Gasiunas et al., 2012; Jinek et al., 2012; Zetsche et al., 2015). Unlike these single-effector CRISPR systems, Type I CRISPR interference requires coordinated actions of a multi-subunit ribonucleoprotein (RNP) complex termed Cascade (CRISPR-associated complex for antiviral defense) that seeks out a PAM-flanked DNA target (Brouns et al., 2008; Wiedenheft et al., 2011), and a helicase-nuclease enzyme Cas3 that is recruited to the Cascade-induced R-loop and processively shreds the invader’s DNA genome (Hochstrasser et al., 2014; Huo et al., 2014; Liu and Doudna, 2020; Mulepati and Bailey, 2013; Sinkunas et al., 2011; Sinkunas et al., 2013; Westra et al., 2012; Xiao et al., 2018). Due to this unique targeting mechanism, CRISPR-Cas3 holds great potential for numerous eukaryotic applications, such as targeted deletion of large chromosomal regions, discovery of non-coding elements, removal of integrated viral genomes, and interrogation of structural variants impacting gene function or human disease (Cameron et al., 2019; Dolan et al., 2019; Hidalgo-Cantabrana and Barrangou, 2020; Morisaka et al., 2019).
Type I is the most abundant and diverse group of CRISPR-Cas systems and can be further classified into multiple subtypes (I-A through I-G) based on their cas gene composition (Koonin et al., 2017; Makarova et al., 2020). Despite their wide use for a broad range of tasks in microbial genome manipulation, including Cascade-based transcription regulation, Cascade-Cas3-based programmable antimicrobials, natural variant selection, homologous recombination (HR)-assisted gene editing, genome minimization, targeted DNA integration, etc. (Cameron et al., 2019; Csorgo et al., 2020; Hidalgo-Cantabrana and Barrangou, 2020; Klompe et al., 2019; Zheng et al., 2020), Type I CRISPRs have not been successfully adopted for eukaryotic application until recently.
Since 2019, Cascade-Cas3 is repurposed to effectively create targeted large chromosomal deletions of up to 30–100 kilobases (kb) in human cells (Cameron et al., 2019; Dolan et al., 2019; Morisaka et al., 2019). Tools based on Cascade alone without Cas3 were also developed. For example, Cascade fusions with FokI nuclease or other effector domains enabled programmable genome editing (Cameron et al., 2019) or transcription modulation in human cells (Chen et al., 2020; Pickar-Oliver et al., 2019), and gene activation in plants (Young et al., 2019). These applications mainly focused on four different Type I-E CRISPR-Cas systems from Thermobifida fusca (Tfu) (Dolan et al., 2019), Escherichia coli (Eco) (Morisaka et al., 2019), Pseudomonas aeruginosa (Pae) (Cameron et al., 2019) and Streptococcus thermophilus (Sth) (Young et al., 2019) that all prefer similar 5’-AAG or 5’-AA PAM sequence; although examples based on other subtypes also exist [e.g., Listeria monocytogenes (Lmo) I-B (Pickar-Oliver et al., 2019), Microcystis aeruginosa (Mae) I-D (Osakabe et al., 2020; Osakabe et al., 2021), and Pae I-F (Chen et al., 2020)]. Genetic engineering by Type I-E Cascade-Cas3 requires six cas genes and a CRISPR array, with a 7–8 kb total gene size that is 60–80% larger than the commonly used ~4.2 kb Streptococcus pyogenes cas9 (Cong et al., 2013; Gasiunas et al., 2012; Jinek et al., 2012; Mali et al., 2013). Such complexity and relatively larger payload size could hinder in vivo delivery using viral vectors that have cargo size constraints. To date, the most streamlined CRISPR-Cas3 system, subtype I-C, has never been adopted for eukaryotic use, despite the recent exploitation of P. aeruginosa I-C CRISPR for targeted bacterial genomic deletion of up to 424 kb (Csorgo et al., 2020). Indeed, most Type I CRISPRs remain untapped for eukaryotic development. Mining this treasure trove may lead to novel robust CRISPR-Cas3 editors with favorable attributes such as smaller size, guide orthogonality, and novel PAM specificity that expand the Cas3-targetable sequences.
Here, we present the first example of a Type I-C CRISPR repurposed for eukaryotic genome manipulation and further provide a framework to systematically implement divergent and compact CRISPR-Cas3 editors. We first defined a miniature Type I-C system from Neisseria lactamica (Nla) that elicits robust bacterial immunity with its preferred 5’-TTC PAM. Purified NlaCascade-Cas3 RNP can accomplish high-efficiency, multiplexed genome editing in human cells. Long-range PCR and sequencing revealed a spectrum of large, NlaCas3-induced unidirectional deletions, originating from the Cascade-programmed target site. Intriguingly, we uncovered a previously overlooked cas11 gene, which is encoded by internal prokaryotic translation from within cas8. The resulting Cas11 protein is an integral component of NlaCascade complex. Furthermore, we showed that this unconventional NlaCas11 must be supplied via a separate mammalian expression cassette to implement efficient editing with Nla CRISPR-Cas3 plasmids or mRNAs. Finally, we demonstrated that adding Cas11 is a broadly applicable approach to harness evolutionarily divergent and orthogonal CRISPR-Cas3 editors from the compact I-B, I-C, and I-D systems, paving the way for in vivo applications of Type I CRISPR technology.
RESULTS
A compact CRISPR-Cas3 from N. lactamica confers robust CRISPR interference in vivo.
In search for a previously uncharacterized compact Type I CRISPR, we examined the genomes of Neisseria spp. and identified a previously uncharacterized Type I-C CRISPR-Cas locus from N. lactamica strain ATCC 23970. It consists of a CRISPR array and seven annotated cas genes, including the spacer acquisition genes cas1, cas2, and cas4, the nuclease-helicase gene cas3, and the set of genes (cas5, cas8 and cas7) encoding protein subunits of Cascade (Figure 1A). The native CRISPR array contains thirty spacers 34–35 bp in length, sandwiched between 32 bp direct repeats. We then attempted to define the PAM sequence informatically by first looking for potential natural targets of all the natural spacers using CRISPRTarget (Biswas et al., 2013), allowing for up to 1 nt mismatch in spacer-target complementarity. A total of 28 unique targets (aka protospacers) were found, and upon alignment of these sequences with their 10 bp flanking regions on both the upstream and downstream sides, a strong 5’-TTC PAM was deduced (Figure 1B).
Figure 1. A compact CRISPR-Cas3 from N. lactamica confers bacterial immunity.
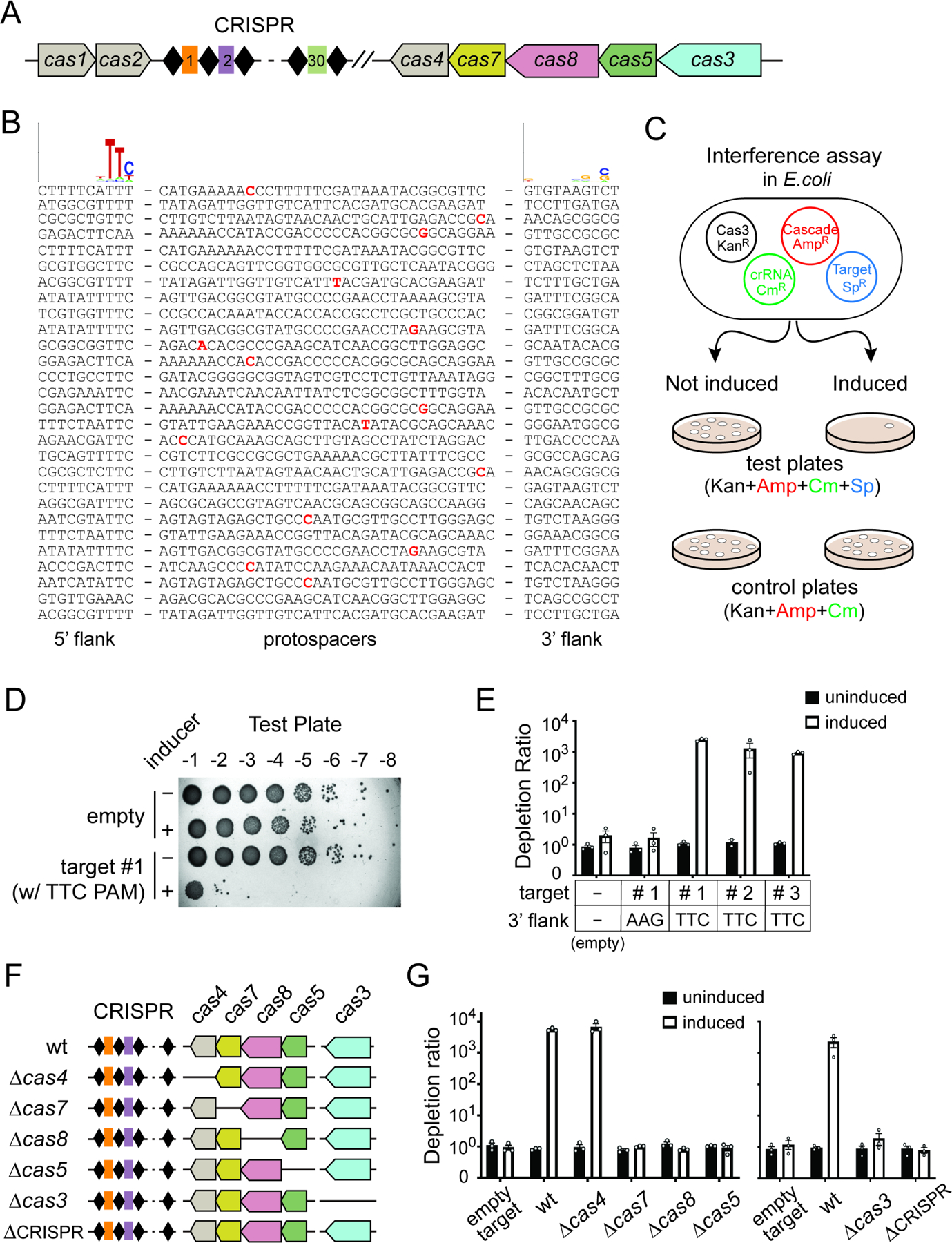
(A) Schematic of the miniature type I-C CRISPR-Cas locus from N. lactamica, with cas1, cas2 and cas4 in grey, cas7 in yellow, cas8 in pink, cas5 in green, and cas3 in blue. Black diamonds and colored rectangles, CRISPR repeats and spacers. Cas genes are drawn to scale, while CRISPR array is enlarged for clarity. (B) Informatic prediction defined a 5’- TTC PAM. Natural targets for native spacers of N. lactamica ATCC 23970 were defined using CRISPRTarget, with up to 1 nt mismatch in spacer-target complementarity allowed and denoted in bold and red. All target sequences and their 10-nt flanks on both 5’ and 3’ were aligned using Weblogo, and the resulting sequence logos are shown at the top. (C) Overview of the plasmid interference assay in E. coli. BL21-AI derivatives harboring four plasmids encoding crispr, cas3, cascade genes, and a target-PAM sequence were cultured with or without induction of crispr-cas expression, serially diluted, then plated on LB plates with triple or quadruple antibiotics to track cell survival. Reduced colony count on quadruple antibiotics plate for the induced culture indicates a CRISPR interference phenotype. (D) Representative image of an interference assay where isogenic E. coli strains were titered on a quadruple-antibiotics plate in 10-fold serial dilutions. Under induced condition, a matching target with a 5’-flanking TTC PAM led to drastic reduction in colony counts compared to the empty target control, indicative of robust CRISPR activity. (E) Induction of crispr-cas expression led to robust interference for three different targets flanked by a 5’-flanking TTC PAM, but not for the controls containing either no target or a target with a 5’-flanking AAG. Depletion ratios were calculated as the colony-forming units (CFUs) from triple antibiotic control plate divided by CFUs from quadruple-antibiotic test plate for the same sample. Data are displayed as log scale plots of the mean depletion ratio ± SEM, n=3. (F) Schematic of the crispr-cas loci in isogenic mutant strains used in (G). (G) CRISPR Interference mediated by Nla type I-C system genetically requires the cas7, cas8, cas5, cas3 and crispr genes but not cas4. Data are quantified and shown as in (D) and (E).
Next, we sought to test the functionality of this Nla I-C system by conducting a plasmid interference assay using E. coli as the surrogate host (Dillard et al., 2018) (Figure 1C). We cloned the Nla cas5-8-7-4 operon into pBAD vector under control of an arabinose inducible promoter, cas3 into pET28b under a T7 promoter, the native CRISPR into pACYC under a T7 promoter and the potential target sequences into pCDF1. Then, we built BL21-AI derivative strains harboring all four plasmids and plated the induced culture on quadruple antibiotics LB plates to track cell survival. Induction of crispr-cas expression led to ~1,000-fold reduction in colony counts for target plasmids that contain a 5’-TTC PAM followed by a target sequence complementary to any of the first three native CRISPR spacers, but not for the empty target plasmid control (Figures 1D–E). This indicates a potent plasmid interference phenotype in vivo. A control target for spacer 1 with a 5’-AAG motif failed to elicit interference, suggesting that a functional PAM is a prerequisite for Nla I-C system to mount successful CRISPR defense (Figure 1E).
To determine the other components genetically required for interference, we analyzed a series of deletion mutants each lacking a different crispr-cas gene (Figure 1F). Interference was completely abrogated by internal deletion of cas7, cas8, or cas5, but not the cas4 gene, from the pCascade plasmid, as well as in strains lacking cas3 or the CRISPR array (Figure 1G). Collectively, our results showed that DNA targeting by the Nla I-C system requires a matching spacer-target pair, a functional PAM, cas3 and all Cascade subunit genes, but not the putative spacer acquisition genes cas1, cas2 or cas4.
NlaCascade-Cas3 RNP achieves high-efficiency, multiplexed genome engineering in human cells
Having demonstrated the functionality of the compact Nla I-C CRISPR in bacterial immunity, we next asked if it can support genome engineering in human cells. We first aimed to test RNP-based genome editing by purifying recombinant Cas3 and Cascade separately from E. coli (Figure 2A), delivering them into various human cell lines via electroporation, and monitoring genome editing of a reporter gene using flow cytometry. Initial editing experiments were carried out in a human embryonic stem cell (hESC) dual reporter line (Dolan et al., 2019) with two CRISPR guides designed to target 5’-TTC-flanked sites in the integrated EGFP or tdTomato (tdTm) genes, respectively (Figure 2B). The corresponding Cascade complexes containing nuclear localization signal (NLS) sequences on the N-termini of all Cas7 subunits were purified via nickel affinity pulldown and size exclusion chromatography (SEC), and then tested with or without purified NLS-Cas3. We observed roughly 50% and 30% editing for EGFP and tdTm targeting respectively, when a cognate Cascade was used in conjunction with Cas3 (Figures 2C–D). Negative controls lacking Cas3 or with a Cascade targeting either the other non-corresponding reporter gene or an endogenous genomic locus (non-targeting, NT) all failed to produce a signal above the untreated background (Figures 2C–D). This 30–50% editing efficiency obtained in hESCs is quite impressive given that our prior work utilizing Tfu Type I-E RNP only gave up to 13% editing in the same reporter line (Dolan et al., 2019).
Figure 2. Nla CRISPR-Cas3 RNP achieves high-efficiency, multiplexed genome editing in human cells.
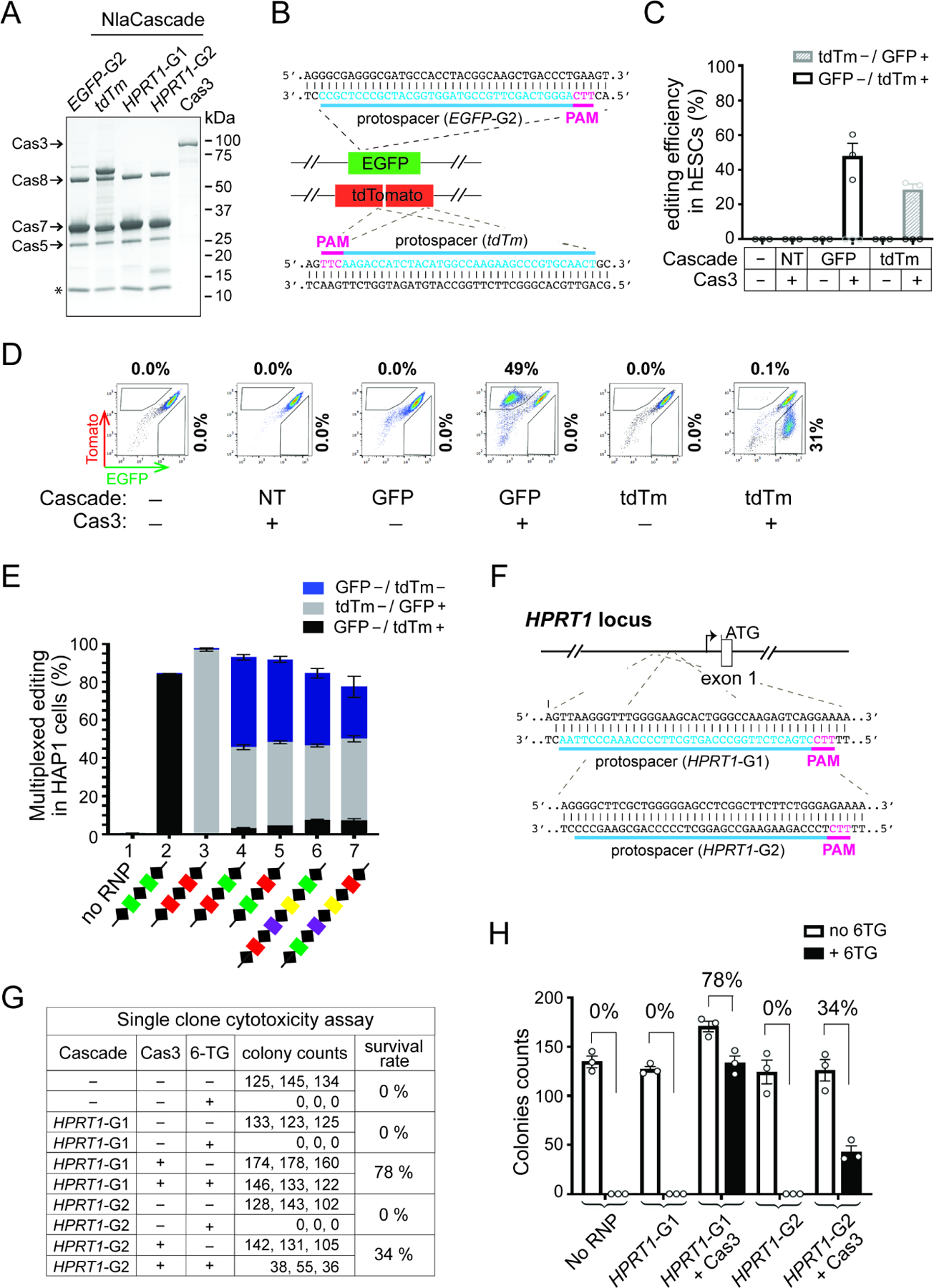
(A) SDS-PAGE of purified NlaCas3 protein and Cascade RNPs targeting different genes (EGFP-G2, tdTm, HPRT1-G1&G2). Star, an unexpected peptide consistently co-purified with NlaCascade and further examined in Figure 4. Cascade-tdTm was purified using a slightly different strategy and contains an extra band of 68 KDa corresponding to His-MBP-Cas5. (B) Schematic of the hESC dual-reporter cells used in (C-D), with protospacers for the EGFP- or tdTm- targeting Cascade indicated in blue and corresponding PAMs in magenta. (C) Cascade RNP targeting either EGFP, tdTm or a control locus (Non-targeting [NT], i.e., HPRT1) was electroporated into hESCs with or without Cas3. The gene editing efficiency was shown as the percentage of EGFP−/tdTm+ (white bar) or tdTm−/EGFP+ (shaded grey bar) cells in the total population. Data are shown as mean ± SEM, n=3. (D) Representative flow cytometry plots from an experiment in (C), with percentages of EGFP−/tdTm+ or EGFP+/tdTm− cells shown on the top or to the right, respectively. (E) Robust multiplexed editing in HAP1 dual reporter cells. Cascade RNP purified using each multi-spacer CRISPR array depicted at the bottom was electroporated together with Cas3. The gene editing efficiencies were shown as the percentage of EGFP−/tdTm+ (black bar), tdTm−/EGFP+ (grey bar), and EGFP−/tdTm− (blue bar) cells in the total population. Data are shown as mean ± SEM, n=3. The green, red, purple, and yellow spacers represent Nla guides targeting EGFP, tdTm, HPRT1, and CCR5 genes respectively. (F) Schematic of the HPRT1 locus, with protospacers for the two HPRT1-targeting Cascades shown in blue and corresponding PAMs in magenta. (G) Measurement of the HPRT1 targeting efficiency using a single-clone 6-TG survival assay. Survival rate is the ratio between the average colony counts from 6-TG+ vs. 6-TG− conditions. (H) Bar graph plotting the colony counts from (G). Data are shown as mean ± SEM, n=3.
We then performed parallel experiments in a HAP1 reporter cell line using the same EGFP-targeting Cascade and obtained dose-dependent editing of up to 83%. As the amount of Cascade went up from 4.5 to 35 pmol, editing efficiency gradually increased from 27% to 83% (Figures S1A and S1C). In contrast, for Cas3 titration the editing efficiency jumped to and plateaued at ~76% with as little as 3 pmol of Cas3 delivered (Figures S1B and S1D). These data imply that genome editing with our current Nla CRISPR-Cas3 RNP platform is limited by the assembly or target searching activity of Cascade, but not the DNA degradation activity afforded by Cas3.
To further leverage the high editing efficiency, we next sought to achieve simultaneous multiplexed editing of two distinct target sites in the same cell. Naturally, the CRISPR array of Type I system is transcribed into a multi-unit primary transcript which is then processed into individual mature crRNAs loaded inside Cascade (Brouns et al., 2008). The multi-spacer CRISPR cassette therefore offers an opportunity to co-express numerous guide RNAs and purify a collection of Cascade RNPs at once from E. coli. To explore this, we first created two CRISPR arrays in R-S-R-S-R configuration, each containing three repeats and two distinct intervening spacers at different relative positions (Figure 2E, samples 4–5). When each resulting Cascade preparation (Figure S2A) was electroporated with Cas3 into HAP1 cells, we saw concurrent disruption of both EGFP and tdTm fluorescence in 47% and 43% of the cells respectively, indicative of efficient multiplexed editing (Figure 2E). Importantly, flow cytometry showed that multiplexing indeed occurred in individual human cells, not just on a population level (Figure S2B). As controls, Cascade RNPs purified using CRISPR arrays with two identical spacers targeting the same reporter led to above 85% editing of just the cognate fluorescent target gene but not the non-cognate one (Figure 2E, samples 2–3).
We then further assayed longer CRISPR arrays containing five repeats and four different spacers for the ability to enable dual targeting of EGFP and tdTm. We observed 38% dual editing when the quadruple-spacer CRISPR has a tdTm-targeting spacer at the first position and an EGFP-targeting spacer at the last position (Figure 2E, samples 6). Conversely, 27% dual editing was achieved if the first spacer targets EGFP and the fourth spacer targets tdTm (Figure 2E, sample 7). These data showed the high potency of multiplexed editing using NlaCascade prepared from a multi-spacer CRISPR array.
Lastly, to further demonstrate facile programmability and broad applicability, we applied the NlaCRISPR-Cas3 RNP to target various endogenous genes in different human cell lines. We started with the HPRT1 locus of the near-haploid HAP1 cells, because its editing can be readily assessed using a single clone cytotoxicity assay measuring resistance to 6-thioguanine (6-TG) mediated cell killing. Cascade RNP targeting the promoter region 489 bp or 274 bp upstream of the ATG start codon of the HPRT1 gene was electroporated into wild-type (wt) HAP1 cells, leading to Cas3-dependent editing of 78% and 34%, respectively (Figures 2F–H, guides HPRT1-G1 & G2). The same HPRT1-G1 RNP also enabled DNA targeting in hESCs, HEK293T, and HeLa cells, as evidenced by the smaller-than-WT products in the long-range genomic PCRs (Figure S2C). Moreover, we also successfully programmed Cascade to edit another endogenous gene, CCR5, in HAP1 cells (Figure S2D). Altogether, we established Nla CRISPR-Cas3 as the first compact Type I-C editor for high-efficiency genome engineering in human cells.
Nla CRISPR-Cas3 creates a spectrum of large, uni-directional genomic deletions.
To define the genomic lesions caused by Nla CRISPR-Cas3, we turned to comprehensive long-range PCR analysis. We and others have previously found that Type I-E CRISPR generates targeted uni-directional large deletions towards the PAM-proximal direction in human cells (Cameron et al., 2019; Dolan et al., 2019; Morisaka et al., 2019). Intriguingly, a recent report showed that Pae I-C CRISPR forms bi-directional large deletions in bacteria hosts (Csorgo et al., 2020). Without making any presumption about the directionality or size range for the NlaCas3-induced lesions, we performed three different sets of PCRs on genomic DNA extracted from HAP1 cells edited by Cascade-HPRT1-G1-Cas3 from Figure 2H. First, to specifically amplify regions downstream of CRISPR-programmed site, we used a fixed forward primer G annealing about 0.1 kb upstream of target site and paired it with tiling reverse primers B through F about 2.8–19 kb downstream of target (Figure 3A). Each PCR amplification gave rise to a collection of bands of varying sizes but all smaller than the corresponding full-length product (Figure 3B, lanes 6–10), indicative of a variety of large deletions firing downstream in the PAM-proximal direction. The control genomic DNA from untreated cells failed to yield any product (Figure 3B, lanes 1–5), likely due to a GC-rich region in exon 1 that prevents PCR amplification (Dolan et al., 2019).
Figure 3. NlaCRISPR-Cas3 creates targeted, large, and uni-directional DNA deletions.
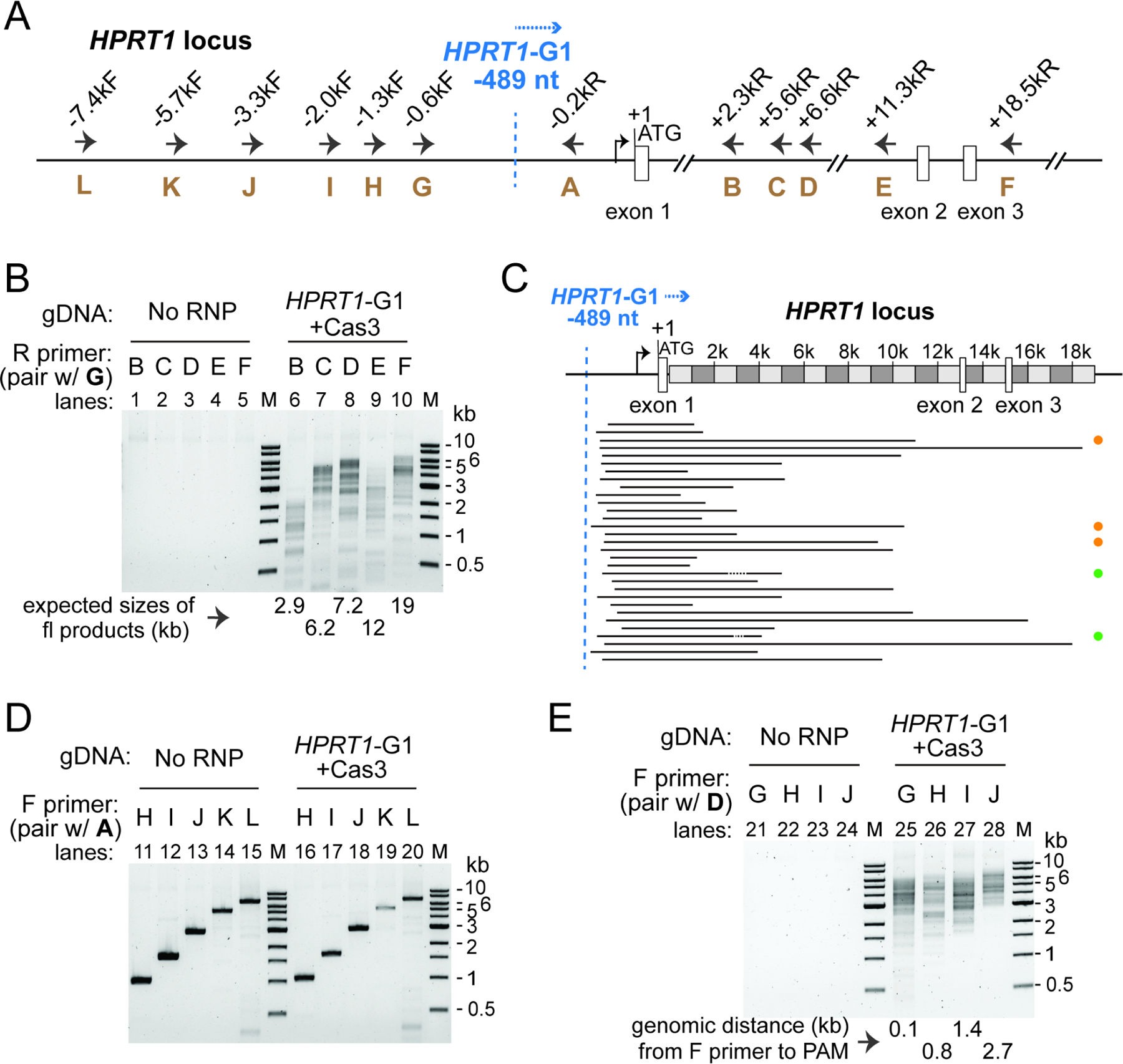
(A) Schematic of the HPRT1 locus and annealing sites for PCR primers used in (B), (D) and (E). All positions indicated are relative to HPRT translation start site (+1). Blue dashed line, recognition site (3rd nt of the TTC PAM) for guide HPRT1-G1; blue arrow, inferred direction of NlaCas3 translocation. (B)(D)(E) Characterization of genomic lesions by long-range PCRs, using primers amplifying regions downstream (B) or upstream (D) of the CRISPR-targeted site, or regions spanning both directions (E). A spectrum of large, uni-directional deletions was detected in the PAM-proximal genomic region, from cells treated with Cas3 and Cascade HPRT1-G1, but not the untreated control cells. PCR primers used are listed and their annealing sites depicted in (A). Smaller-than-full-length amplicons indicate large genomic deletions. M, DNA size markers. (C) Deletion locations at the HPRT1 locus, revealed by TOPO cloning of pooled tiling PCRs from lanes 6–10 in (B) and Sanger sequencing. Black lines, deleted genomic regions. Orange, green and the lack of dots on the right indicate Groups II, IV and I deletion junctions as described in Dolan et al., 2019.
To precisely define the boundaries of these NlaCas3-induced deletions, we pooled the PCR products from lanes 6–10 of Figure 3B, TOPO-cloned it and randomly selected 34 independent clones for Sanger sequencing. A total of 31 unique lesions were identified, and the overall pattern was similar to that exhibited by the Type I-E Tfu Cascade-Cas3 (Dolan et al., 2019). The onset of deletions was not uniformly at the presumed R-loop but instead was clustered in a window ~15–150 nt downstream, while the deletion endpoints were distributed across the ~20 kb PAM-proximal region analyzed (Figure 3C), highlighting the heterogenous nature of large deletions caused by NlaCas3. Furthermore, most of the resulting chromosomal junctions have the 5’ and 3’ sequences flanking the deletion rejoined seamlessly, presumably by end-joining DNA repair pathways in human cells (Figure 3C, supplemental Table 2).
Next, we conducted the converse PCR experiments to amplify regions upstream of the CRISPR-targeted site, using a fixed reverse primer A annealing 0.25 kb downstream of the target, in conjunction with serial forward primers H through L about 0.8–6.4 kb upstream of the target site (Figure 3A). No obvious large deletions were detected, as we only got the anticipated full-length bands from both edited and untreated cells (Figure 3D). This suggests that there are very few, if any, NlaCas3-induced deletions firing upstream in the PAM-distal direction. Collectively, the Nla I-C and Tfu I-E CRISPRs likely use similar mechanisms for processive DNA degradation by Cas3 and subsequent DNA repair by endogenous machinery of the human cell.
In the last set of long-range PCR, we paired serial forward primers G through J with a common reverse primer D annealing 7.1 kb downstream of the target (Figure 3A), and detected a spectrum of amplicons containing large deletions (Figure 3E, lanes 25–28). Of note, the size of the smallest amplicon in each reaction is larger than the genomic distance from the CRISPR target site to the annealing position of the forward primer used, implying that very few bi-directional large deletions exist that span both PAM- proximal and distal regions. In addition, similar editing and long-range PCR results were observed on the same HPRT1 target site in hESCs and HEK293T cells (Figure S3), as well as on the DNMT3b-EGFP target in hESCs (Figure S4), although in the latter experiment a few bi-directional lesions were detected (Figures S4D–E). Taken together, we concluded that Nla CRISPR-Cas3 creates a spectrum of large chromosomal deletions that are uni-directional relative to the target.
N. lactamica CRISPR-Cas3 encodes a “hidden” cas11 gene by alternative prokaryotic translation initiation
The expression and purification of Cascade-Cas3 could be laborious or technically challenging for certain Type I systems. So, to facilitate applications that involve screening a large number of individual guides, we tried to set up a plasmid-based editing platform. All four annotated Nla cas genes were human codon optimized, each fused with an NLS and separately cloned into a mammalian expression vector under control of an EF1a promoter and a bGH polyA signal (Figure 4A). A fifth plasmid expressing a mini-CRISPR targeting EGFP was co-transfected along with all four cas plasmids and gene editing efficiency was evaluated by flow cytometry. A total of four different Nla guides targeting 5’-TTC-flanked sequences in EGFP were tested (Figure S5A), but disappointingly none yielded any positive signal while the positive control SpyCas9-sgRNA gave 33% editing (Figures 4B–C). This failure to edit cannot be explained by lack of expression, as western blot confirmed that all plasmid-encoded Nla I-C Cas proteins were expressed in human cells (Figure S5B).
Figure 4. Cas11, a hidden product from internal translation, is essential for robust RNP editing with NlaCRISPR-Cas3.
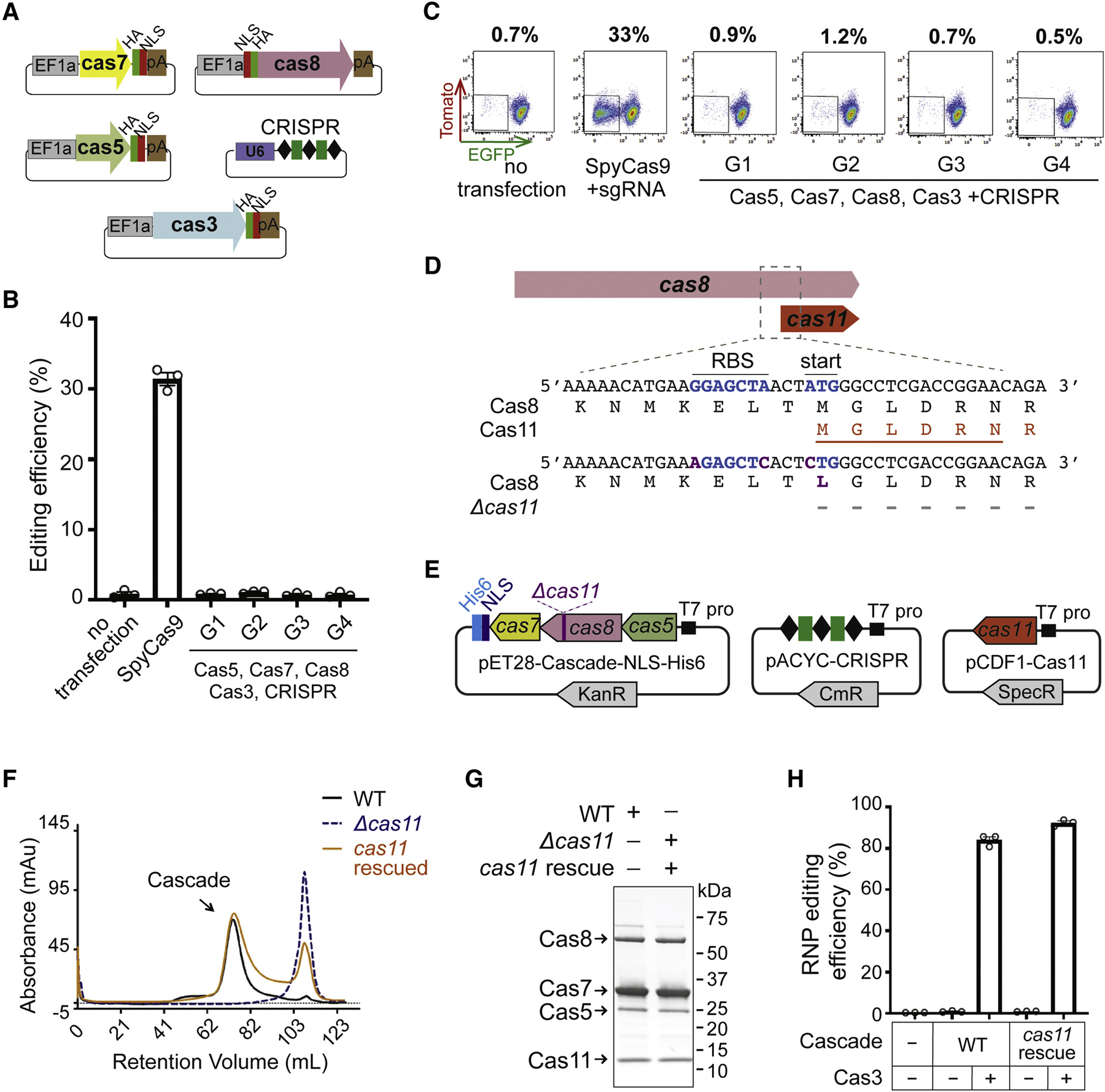
(A) Schematics of five plasmids used in (B) and (C), to express NlaCRISPR-Cas3 components in human cells. Grey, green, red, brown and purple rectangles indicate EF1a promoter, HA tag, NLS, bGH polyA signal, and U6 promoter, respectively. (B) The five crispr-cas plasmids from (A) were co-transfected into HAP1 reporter cells to evaluate genome editing efficiency. The editing efficiencies are shown as the percentage of EGFP- cells. G1 through G4, four different CRISPR guides targeting 5’-TTC flanked sites in EGFP; their sequences and locations are depicted in Figure S5A. A SpyCas9 plasmid targeting EGFP was the positive control. (C) Representative flow cytometry plots of an experiment in (B), with percentages of EGFP- cells in the population shown on the top. (D) Schematics of the Nla cas8 and cas11 genes. Blue, predicted RBS and internal translational start site for cas11; orange, the putative Cas11; purple, mutations introduced to create the Δcas11 construct. The first six amino acids of the ~13 kDa band obtained through Edman degradation were marked by orange line. (E) Plasmids used for purifying Δcas11 and cas11-rescued versions of NlaCascade in (F) and (G). (F) SEC chromatogram of NlaCascade RNPs purified via a C-terminal His tag on Cas7. Elution profiles of wt, Δcas11, and cas11-rescued NlaCascade RNPs are displayed as black, dashed gray, and orange lines. (G) SDS-PAGE of NlaCascades from (F). (H) Cas11-rescued Cascade RNP is as robust in mediating genome editing as the wt. Editing efficiencies were measured by flow cytometry and shown as the percentage of EGFP- cells in total population. Data in (B) and (H) are shown as mean ± SEM, n=3.
To resolve the discrepancy in editing efficiencies between RNP- and plasmid- based experiments, we revisited the SDS-PAGE of NlaCasacde purification. An unexpected ~13 kDa protein band that consistently showed up in all preps drew our attention (Figure 2A, marked by star). We sequenced the N-terminus of this extra band through Edman degradation, to our surprise, it began with residue M485 of the NlaCas8 protein (Figure 4D, in orange). Further inspection of DNA sequence immediately upstream of M485 revealed a putative ribosome binding site (RBS) (Figure 4D); alternative translation from this predicted RBS and M485 would generate an expected in-frame product of 14.7 kDa, consistent with the ~13 kDa band on SDS-PAGE. Importantly, mutations disrupting the RBS and M485 abolished this extra peptide in E. coli culture (Figure S5C). Collectively, the above lines of evidence suggest that a previously overlooked internal small open reading frame (ORF) is encoded within Nlacas8. Our findings corroborate the recent report of independent translation of both the small and large Cascade subunits, Cas11 and Cas10, from a single gene in Type I-D CRISPR (McBride et al., 2020). Indeed, in silico prediction using the same algorithm employed by McBride et al. identified the translation start site at residue M485 of NlaCas8 with high-confidence (ΔG of −1.85 kcal/mol, where ΔG reflects the total Gibbs free energy change for the 16s rRNA to pair with the mRNA sequence) (Salis et al., 2009), supporting the idea that the ~13 kDa peptide in our Cascade prep is the Cas11 homolog for Nla I-C system. This is also consistent with the informatic prediction that the C-termini of large subunit protein Cas10 or Cas8 bear structural similarity with the separately encoded, conspicuous Cas11 homologs (Makarova et al., 2011).
Next, we aimed to understand the role of NlaCas11 in RNP-directed genome editing. Our attempts to purify a cas11 null version of NlaCascade failed, owning to the lack of stable Cascade formation during SEC (Δcas11, Figures 4D–F). Cas11 complementation from a separate E. coli expression plasmid restored Cascade assembly, resulting in a Cas11 rescue version of NlaCascade that is as effective in directing genome editing as WT (both with >85% efficiencies, Figures 4D–H). Taken together, NlaCas11 is essential for genome engineering because it is an integral part of the NlaCascade target recognition complex. This stands in contrast with the Synechocystis (Syn) Type I-D CRISPR where Δcas11 did not prevent Cas5, Cas7, Cas8 and crRNA from assembling into a stable complex that has severely impaired DNA binding capacity (McBride et al., 2020). Our findings also indicate that Cas11 homologs of different Type I subtypes may play distinct roles in Cascade assembly or functionality.
Cas11 is the key to implement plasmid- and mRNA- based genome editing with Nla CRISPR-Cas3
We hypothesized that the failure to enable high-efficiency, plasmid-based editing for Nla I-C CRISPR is due to the lack of Cas11 expression in human cells. This is because the prokaryotic and eukaryotic translation initiation mechanisms are distinct, and that the internal prokaryotic translation initiation site embedded within cas8 won’t be recognized by human ribosomes. Accordingly, to establish plasmid editing we may have to supply a separate mammalian expression cassette driving NlaCas11 from its own EF1a promoter and Kozak sequence which assists ribosome recruitment. To test this idea, we created a new vector expressing Nlacas11 transgene with an N-terminal NLS and a HA tag and transfected it into HAP1 reporter cells along with other crispr-cas plasmids (Figure 5A). Remarkably, a mixture of equal amounts of all plasmids exhibited 21% editing while the control mixture lacking pCas11 led to minimal editing (Figures 5B–C), unequivocally demonstrating that Cas11 was indeed the missing factor that precluded editing in Figure 4A–C. We then attempted to optimize editing efficiency by increasing the relative amount of Cas8 plasmid used, since Cas8 is the least expressed component in human cells (Figure S5B). The resulting optimized plasmid mixture enhanced editing efficiency to 33% (Figures 5B–C). The other three EGFP-targeting guides also gave robust editing, in a similar Cas11-dependent fashion (Figure S5D). Of note, the optimized control lacking pCas11 displayed low but noticeable levels of editing around 1–4%, implying that without NlaCas11 the processes of Cascade assembly, Cas3 recruitment and DNA targeting can still occur in human cells, although to a much lesser extent.
Figure 5. Cas11 enables efficient plasmid- and mRNA- based editing by NlaCRISPR-Cas3.
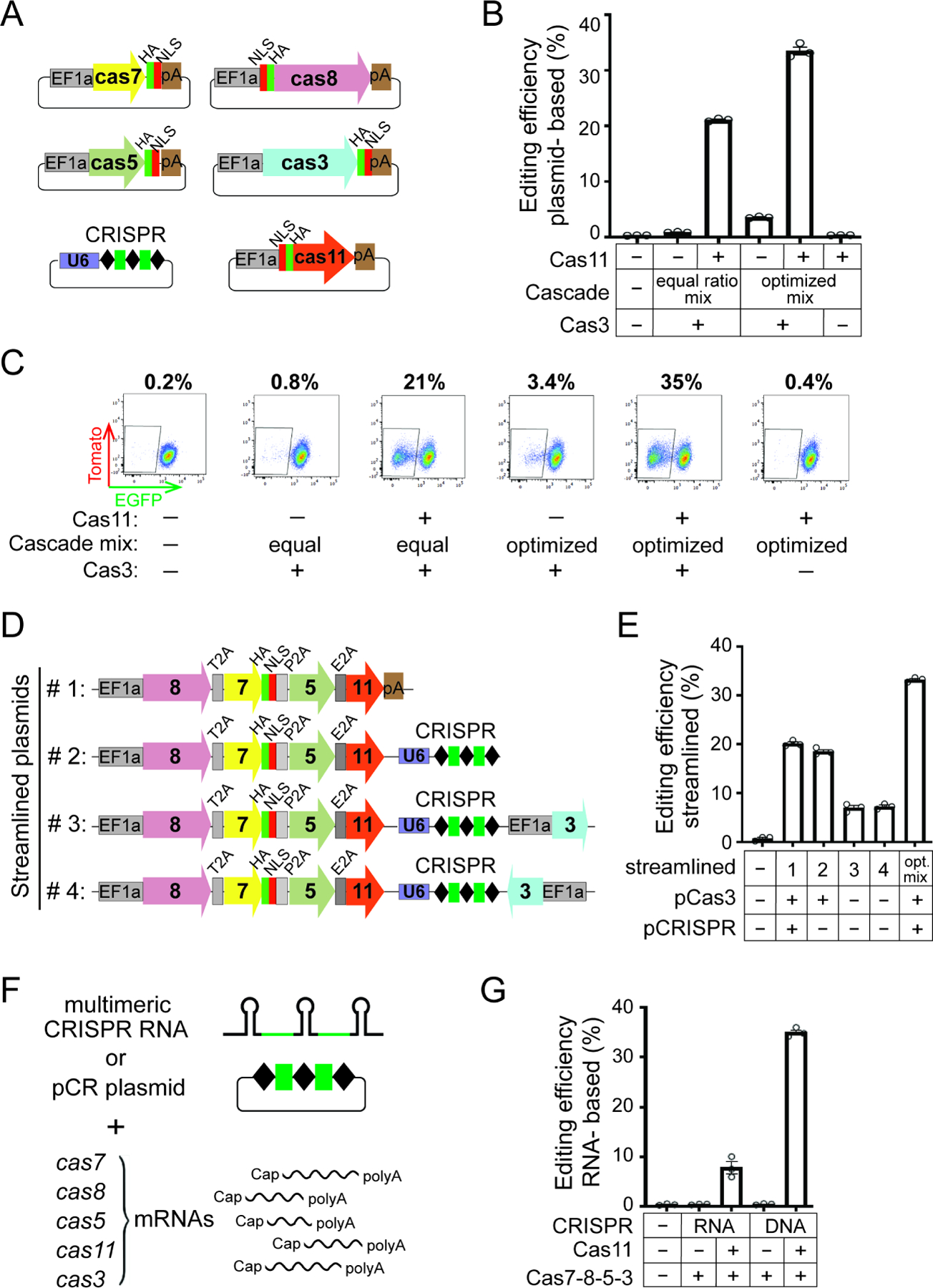
(A) Schematics of six plasmids used in (B) and (C). A separate Nlacas11 plasmid is included, and the rest are as in Figure 4A. (B) Crispr-cas plasmids from (A) were transfected into HAP1 cells and editing efficiencies evaluated and plotted as in Figure 4B. The equal ratio mix contains equal amounts of plasmids for each Cascade subunit, the optimized mix has more Cas8 and less Cas5. (C) Representative flow cytometry plots of an experiment in (B), with percentages of EGFP- cells in the population shown on the top. (D) Schematic of the pCascade and all-in-one plasmids used in (E). (E) Editing efficiencies for constructs in (D) were evaluated and plotted as in Figure 4B. (F) Schematics of cas mRNAs, pre-CRISPR RNA and pCR plasmid used in (G). Green, the EGFP-targeting CRISPR spacer. (G) mRNAs encoding NlaCascade subunits with or without Cas11 were electroporated into HAP1 cells, along with a EGFP-targeting CRISPR in the form of pre-CRISPR transcript (RNA) or plasmid (DNA). Editing efficiencies were plotted as in Figure 4B. Data in (B), (E) and (G) are shown as mean ± SEM, n=3.
To streamline applications that benefit from reduced number of plasmids used, we combined all Cascade subunit genes, including cas11, into a polycistronic cassette driven from a single promoter and connected them with 2A peptides (Liu et al., 2017); NLS sequence was also eliminated the from cas8, cas5, cas11 but not cas7. A panel of such constructs were created, varying the relative positions of each cas gene (Figure S5E). Their co-transfection with a pair of Cas3 and CRISPR plasmids resulted in 14–24% editing, with the 8-7-5-11 configuration being most efficient (Figure S5F). Then by combining in the crispr cassette, we arrived at a robust all-in-one Cascade plasmid that gave ~19% editing (Figures 5D–E). Further inclusion of cas3 in either orientation, however, reduced editing to 7%, possibly due the large plasmid size (Figures 5D–E). This set of experiments simplified the approach to reconstitute the Nla CRISPR-Cas3 activity in human cells with far fewer plasmids than originally used.
As we achieved RNP- and plasmid- mediated editing, a third format of delivery via electroporation of messenger RNA (mRNA) was explored. Using in vitro transcribed, 5’ capped and 3’ polyA-tailed mRNAs for cas5, cas7, cas8, cas11, and cas3, along with an in vitro transcribed multimeric pre-CRISPR transcript, we obtained 8% editing (Figures 5F–G). A switch from pre-crRNA to a plasmid-borne CRISPR array further boosted the efficiency substantially from 8% to 35% (Figures 5F–G), likely due to prolonged existence of plasmid versus RNA in cellular environment. Nonetheless, cas11 mRNA is essential to enable editing based on RNA delivery, regardless of the form of CRISPR provided.
Supplying Cas11 as a broadly applicable approach to adopt diverse miniature CRISPR-Cas3 editors
It was recently found that internal translation of a non-conventional Cas11 in microbes is a conserved phenomenon across Types I-B, I-C, and I-D systems, which together encompass nearly a quarter of all native CRISPRs (McBride et al., 2020; O’Brien et al., 2020). We hypothesized that not having a well-annotated hidden cas11 gene has broadly limited the utility of compact CRISPR-Cas3 and tested this idea with selective orthologs from other species (Figures 6A–B). In particular, we began with two I-C systems from Bacillus halodurans (Bha) and Desulfovibrio vulgaris (Dvu), both of which have been characterized biochemically and encode their own internal cas11 (Hochstrasser et al., 2016; McBride et al., 2020; Nam et al., 2012; O’Brien et al., 2020). For each system, six plasmids expressing a targeting CRISPR and all five cas genes including the internal cas11 were co-transfected into HAP1 cells, and displayed 17% and 2.5% editing for Bha and Dvu, respectively. Importantly, both systems strictly require Cas11, demonstrating the importance of the separately supplied Cas11 for effective genome editing with Type I-C CRISPR (Figures 6C–D).
Figure 6. Cas11 establishes diverse miniature CRISPR-Cas3 orthologs as gene editors.
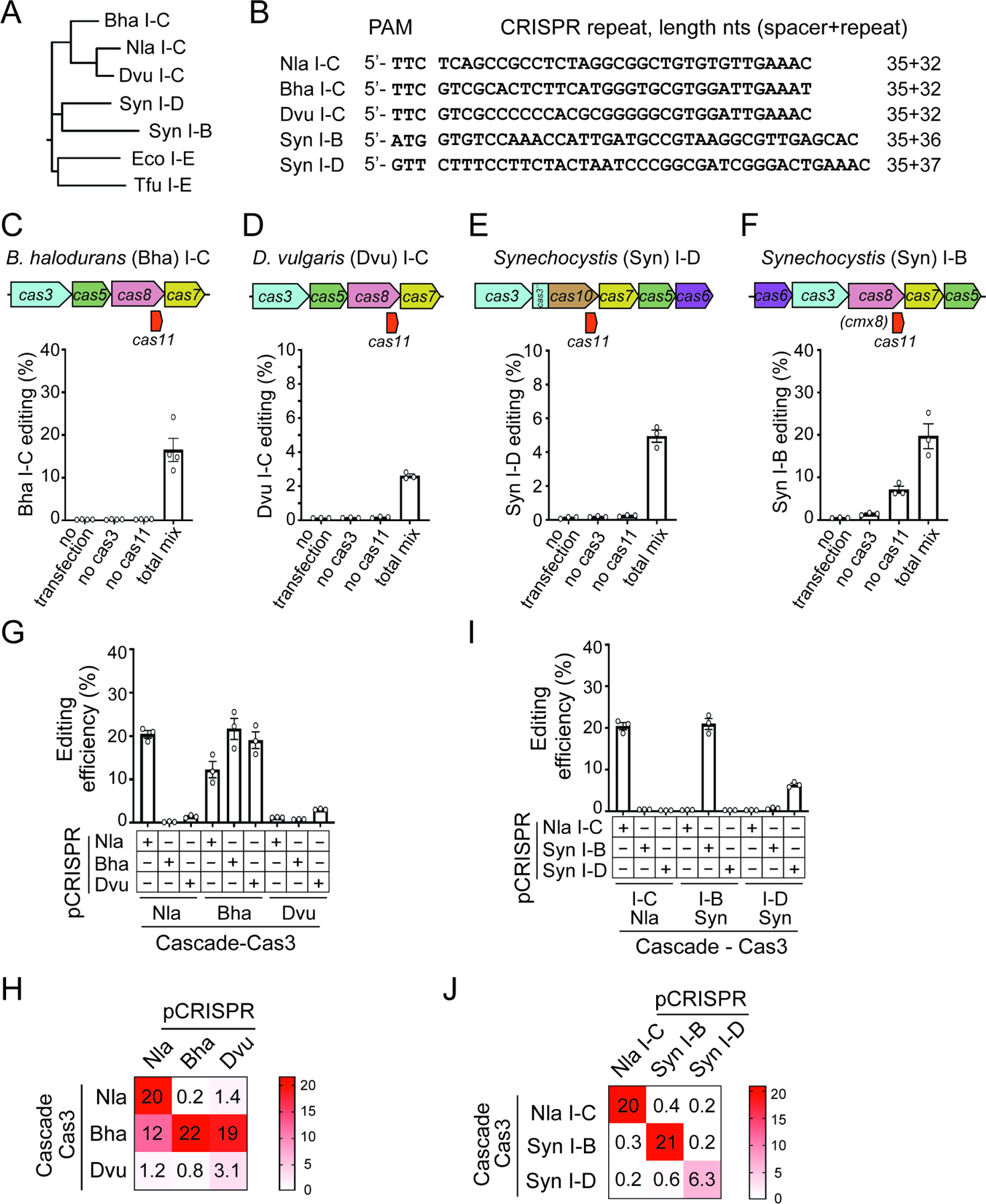
(A) Phylogenetic tree of the large subunit gene cas8 or cas10, from selective type I CRISPR systems analyzed for genome editing in human cells. The Tfu and Eco I-E systems are included at the bottom for comparison. (B) PAM and repeat sequence of CRISPR-Cas3 systems tested, with the lengths of native spacers and repeats indicated. (C)-(F). Schematics of the loci (top) and gene editing efficiencies (bottom) for Bha I-C, Dvu I-C, Syn I-D, and Syn I-B systems, respectively. (G)-(J) Mix-and-match experiments assaying Cas plasmids from different Type I systems paired with each other’s CRISPR constructs. Three distinct I-C editors are analyzed in (G), the Nla I-C, Syn I-D, and Syn I-B systems are tested in (I). Editing efficiencies were measured and plotted as in Figure 4B, except that for Syn I-B editing is measured as tdTm- cells in the population. Data in (C)-(F), (G) and (I) are shown as mean ± SEM, n=3 or 4. (H) (J) Heatmaps of gene editing efficiencies reported in (G) and (I).
Next, we extended our analysis to the I-D and I-B CRISPRs from cyanobacteria Synechocystis (Syn). The Syn I-D system contains five previously annotated cas genes, cas3, cas5, cas6, cas7 and cas10, plus the non-conventional cas11 embedded within cas10 (McBride et al., 2020; Scholz et al., 2013). After proper optimization of the amounts of plasmids used, we obtained ~5% editing in a Cas11-dependent manner (Figures 6E and S6B). In addition, we also analyzed a putative I-B system from Synechocystis sp. strain PCC 6714 that has not been characterized before. In-silico prediction identified a potential RBS and alternative start codon for its cas11 embedded within the large subunit gene cmx8 (Figure 6F). Co-expression of five well-annotated cas genes cas6, cas3, cmx8, cas7, and cas5 readily induced 7% editing, whereas further addition of a separate cas11 plasmid drastically boosted editing to 20% (Figure 6F). This suggests that although Cas11 is not a prerequisite for Syn I-B CRISPR to achieve Cascade assembly and DNA targeting, it can substantially elevate the overall editing efficiency. Altogether, supplying Cas11 offers a framework to exploit diverse and streamlined CRISPR-Cas3 systems for mammalian genome engineering. Moreover, the Syn I-D and I-B editors recognize 5’-GTT and 5’-ATG PAMs respectively, both motifs are different from the 5’-TTC PAM utilized by I-C systems (Csorgo et al., 2020; Leenay et al., 2016)(Figure 1) and the 5’-AAG PAM for I-E editors (Cameron et al., 2019; Dolan et al., 2019; Morisaka et al., 2019). Therefore, the compact editors established in this work greatly expanded the targeting scope of CRISPR-Cas3 in human genome.
CRISPR-Cas3 orthogonality in human cells
A myriad of CRISPR-Cas tools has been developed to achieve targeted activities including gene modification, transcription regulation, chromosomal loci imaging, and epigenetic control, etc. (Anzalone et al., 2020; Knott and Doudna, 2018; Komor et al., 2017). However, any individual tool can only mediate one activity at a time in any given cell. Multiple orthogonal Cas proteins can be used concurrently to mediate independent tasks, such as gene disruption, transcription control, genome imaging, DNA insertion, at different target sites (Esvelt et al., 2013; Fonfara et al., 2014; Kweon et al., 2017; Ma et al., 2015; McCarty et al., 2020). This type of application relies on the orthogonal nature of the CRISPR-Cas systems used, which means that each Cas machinery only functions with its own cognate crRNA. The set of CRISPR-Cas3 editors adopted in this work opens the possibility for orthogonal Type I applications. However, very little is known about the orthogonality barriers separating divergent CRISPR-Cas3 systems, prompting us to examine if their crRNAs are cross-functional in human genome engineering.
First, we conducted a mix-and-match experiment among all three I-C editors, by assaying each set of the I-C cas genes with every I-C CRISPR plasmid. The Nla and Dvu Cas proteins clearly prefer their own respective CRISPR from the same species; they also showed low but noticeable cross-reactivity with each other’s CRISPR, but no activity with the Bha CRISPR (Figures 6G–H). Strikingly, the Bha Cas proteins exhibited robust editing with all three I-C CRISPRs analyzed (12–22%, Figures 6G–H), revealing high degree of cross-species tolerance in crRNA usage. We reasoned that since these three I-C editors lack complete orthogonal barriers in crRNA usage and recognize identical 5’-TTC PAM, they are not the ideal choice to enable simultaneous and independent tasks.
Next, we performed a similar mix-and-match test among I-C, I-D, and I-B editors. We found that each set of the Cas proteins from the Nla I-C, Syn I-B, or Syn I-D system functions exclusively with their respective CRISPR but not those from other species, demonstrating true orthogonality (Figures 6I–J). For instance, the entire Syn I-B system displayed a robust editing efficiency of 21%; but when its CRISPR was replaced by the Nla I-C or Syn I-D CRISPR, we didn’t observe any editing above background, indicative of limited CRISPR interchangeability. The same trend held true for the Nla I-C and Syn I-D Cas proteins (Figures 6I–J). Such clear orthogonality barriers separating disparate compact CRISPR-Cas3 editors is instrumental for developing tools for simultaneous and independent Type I applications.
We then went one step further to disentangle the contributions of PAM and crRNA to this orthogonal barrier, by assaying chimeric CRISPR constructs in which the original guide sequence remains unchanged, but repeats are swapped among the I-C, I-B and I-D systems (Figure S6). For example, in the first set of tests the original guide would direct Nla I-C Cas proteins to recognize the same target sequence and 5’-TTC PAM, but yielded editing only when the respective repeat from the same species was utilized (Figures S6D–F). Similarly, the Syn I-B or I-D Cas proteins would be guided by their original spacer sequences to the matching target sites, but can only enable editing if the respective CRISPR repeat from the same system was used (Figures S6D–F). These results highlight that the specificity of each Cas protein set for its CRISPR repeat suffices for orthogonality in genome engineering.
DISCUSSION
A compact and high-efficiency Type I-C CRISPR for mammalian genome editing
We implemented the first minimal Cascade-Cas3, derived from a N. lactamica Type I-C CRISPR system, for eukaryotic genome engineering. This Nla I-C editor is superior to the existing Type I-E tools (Cameron et al., 2019; Dolan et al., 2019; Morisaka et al., 2019) in several regards. First, it requires a CRISPR and five cas genes including the 384 nt hidden Nlacas11 uncovered in this study, with a ~6.3 kb total gene size much reduced from the 7.2–8 kb I-E editors. It is therefore more favorable, among all Type I CRISPRs, for gene therapy and other in vivo applications especially those using DNA delivery methods with cargo size constraints (Lino et al., 2018). For adeno-associated virus (AAV) vector that has a 4.7 kb packing limit, Nla I-C system will not fit into a single AAV as the smaller Cas9s from Neisseria meningitidis, Campylobacter jejuni and Staphylococcus aureus do (Ibraheim et al., 2018; Kim et al., 2017; Ran et al., 2015), but may use a dual AAV strategy while allowing enough room for regulator elements and fusions with additional effector domains (Wang et al., 2020). Second, Nla I-C RNP is by far the most efficient Type I editor in human cells, achieving up to 95% targeting in HAP1 cells and 50% editing in hESCs (Figures 2E and 2C). These numbers substantially outperformed Tfu I-E RNP that mediates ~10% targeting in hESCs (Dolan et al., 2019), and are on par with what Cas9 RNP can achieve in multiple cell lines (Kim et al., 2014). Such high robustness may potentiate Cas3-based applications such as multiplexed targeting (Figure 2E), bi-allelic large deletions in diploid cells, long-range CRISPR screen, etc. Additionally, we optimized a procedure for rapid, simple, and high-yield production of NlaCascade-Cas3 RNP with decent purity and activity (Figure 2). Using a E. coli and T7-based expression system and a two-step purification protocol that involves nickel affinity resin and SEC, we routinely obtain 2 mg of recombinant NlaCascade or NlaCas3 from just one Liter of bacteria culture in two days. This is enough for hundreds of electroporation and human cell editing tests, representing a ten-fold improved yield compared to the production of Tfu I-E RNP (Dolan et al., 2019).
The genomic editing and repair outcome for Type I-C CRISPR
By analyzing NlaCas3-induced DNA lesions in four different human cell lines and on multiple target sites, we found that Nla I-C CRISPR creates a variety of targeted, large chromosomal deletions that are mostly uni-directional pointing to the PAM-proximal direction (Figures 3, S2–S4). This is consistent with the Type I-E generated lesions, profiled using long-range PCR and Next-Generation Sequencing (Cameron et al., 2019; Dolan et al., 2019; Morisaka et al., 2019). Although the boundaries of each deletion event cannot be precisely controlled to single nucleotide resolution, the overall pattern of Type I CRISPR induced deletions (i.e., location, direction, and sizes) in an edited human cell population is predictable.
Of note, it was recently shown that Pae Type I-C CRISPR led to bi-directional large deletions in various bacteria species including the native host (Csorgo et al., 2020). The apparent difference between PaeCas3 and NlaCas3 in deletion directionality warrants further studies. It may be caused by mechanistic differences in the repair of Cas3-induced breaks in bacteria versus human cells. Alternatively, PaeCas3 may operate differently from other Cas3 orthologs of I-E and Nla I-C systems by translocating bi-directionally from the CRISPR recognition site.
Implementing diverse compact CRISPR-Cas3 editors
Given the robustness of Nla I-C RNP (Figures 2 and S1), we were initially puzzled by the complete lack of editing when all previously annotated Nla I-C cas genes were expressed via mammalian vectors (Figures 4A–C). Further investigation led to the unexpected discovery of NlaCas11 as an internal translation product (Figures 4D and S5C). Most importantly, by providing a separately encoded Nlacas11 with its own Kozak sequence for human ribosomal entry, we set up a framework to systematically harness divergent I-B, I-C, and I-D CRISPRs as effective gene editors (Figures 5–6). Our findings could help explain the limited number of streamlined CRISPR-Cas3 editors developed so far. Most Type I eukaryotic tools available now are based on the more complicated I-E and I-F systems (Cameron et al., 2019; Chen et al., 2020; Dolan et al., 2019; Hidalgo-Cantabrana and Barrangou, 2020; Morisaka et al., 2019; Pickar-Oliver et al., 2019; Young et al., 2019), and this may not be a mere coincidence. I-E CRISPRs contain their cas11 as a well-annotated, independent ORF; whereas I-F neither encodes nor requires any Cas11 homolog for functionality (Chowdhury et al., 2017; Makarova et al., 2011; McBride et al., 2020). In contrast, the streamlined I-B, I-C, I-D systems demand Cas11 as part of functional Cascade but encode it as an unannotated hidden gene, hindering their wide adoption for eukaryotic genome manipulation. In cryo-EM structures of Syn I-D and Dvu I-C Cascade, two copies of the “hidden” Cas11 proteins form a filament continuous with the large subunit protein Cas10 or Cas8, together constituting the inner belly of the overall helical Cascade architecture (McBride et al., 2020; O’Brien et al., 2020). This position is analogous to that occupied by conventional Cas11 (aka Cse2) in I-E Cascade (Jackson et al., 2014; Mulepati et al., 2014; Xiao et al., 2018; Xiao et al., 2017; Zhao et al., 2014).
Of note, Syn I-B CRISPR can achieve moderate editing in the absence of its cas11 vector (~8%, Figure 6E). This is in line with the successful exploitation of Lmo I-B Cascade-P300 fusion for targeted gene activation in human cells without expressing the hidden Lmocas11 (Pickar-Oliver et al., 2019), implying that Type I-B in general depends on Cas11 to a lesser extent than I-C or I-D systems.
We devised three highly accessible avenues to conduct Cas3-mediated genome manipulation via RNP, plasmid and mRNA delivery. Each method has its own advantages and drawbacks. CRISPR RNP is an attractive approach to edit hard-to-transfect primary, cancer and stem cells, due to its high efficacy, minimal off-targets, low cytotoxicity, and bypass of optimization for expression, codon usage, etc. (Lino et al., 2018). However, for every new guide, a separate Cascade complex must be purified. A robust method to reconstitute Cascade in vitro using individual proteins and synthetic crRNA is needed to alleviate this limitation. Plasmid vectors are easier to produce and would benefit applications that require sustained Cascade-Cas3 expression, while RNA delivery offers high efficiency with little risk of insertional mutagenesis.
New additions to the CRISPR-Cas toolkit
The repertoire of compact Type I editors established in this work greatly expanded the CRISPR toolbox, due to features such as long-range editing, high efficiency, smaller size, distinct PAMs and guide orthogonality. Our cas11 strategy breaks down the barrier to allow further mining of the prevalent I-B, I-C and I-D CRISPRs for novel editors with special properties in Cas3 processivity, PAM stringency, guide-target mismatch tolerance, etc. Repurposing CRISPR-guided Tn7-like transposons that have domesticated I-B CRISPR variants (Makarova et al., 2020; Peters et al., 2017; Saito et al., 2021) for eukaryotic applications may also hinge on providing their hidden Cas11. Moreover, Cas11’s existence within gene fusion is noteworthy (Makarova et al., 2020). Single protein effector of the recently identified Type III-E CRISPR, Cas7-11, is the natural fusion of a Cas11 and multiple Cas7 subunits, and functions a CRISPR-guided RNase with great potential for programmable RNA knockdown and editing (Makarova et al., 2020; Ozcan et al., 2021; van Beljouw et al., 2021).
LIMITATIONS OF THE STUDY
Using a Cas11-centric strategy, we established a set of CRISPR-Cas3 gene editors. Our long-range PCR-Sanger sequencing in Figures 3 and S3–S4 showed a uni-directional large deletion pattern for Nla I-C system. While this pattern is consistent with previous NGS results for I-E editors (Dolan et al., 2019; Morisaka et al., 2019), future deep sequencing work is needed to unbiasedly profile the deletions for diverse Type I orthologs. Furthermore, additional work is also required to carefully predict and evaluate potential off-target effects for the compact Type I editors; and to define the principles for designing effective guides at different chromatin regions.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yan Zhang (yzhangbc@med.umich.edu)
Materials availability
The following plasmids generated in this study have been deposited to Addgene: pSmart-EF1a-Nla-cas5 (Addgene ID 178878), pSmart-EF1a-Nla-cas7 (Addgene ID 178879), pSmart-EF1a-Nla-cas8 (Addgene ID 178880), pSmart-EF1a-Nla-cas3 (Addgene ID 178881), pSmart-EF1a-Nla-cas11 (Addgene ID 178882), pSmart HC Kan-U6-Nla-repeat-BbsI-Nla-repeat (Addgene ID 178883), pET-NlaCascade-NLS-6xHis (Addgene ID 180213), pET28-T7-NlaCas3-NLS-6xHis (Addgene ID 180214), pACYC-T7-Nla-repeat-BbsI (Addgene ID 180215).
Data and Code Availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Escherichia coli BL21 (DE3)
E. coli BL21 (DE3) cells were used for protein production. Cells were grown in Lysogeny Broth (LB) supplemented with appropriate antibiotics.
Escherichia coli DH5 alpha
This strain was used for cloning. Cells were grown at 37°C in LB supplemented with appropriate antibiotics.
Human embryonic stem cell (hESC) culture
Human ESCs were cultured in E8 medium on matrigel (Corning) coated tissue culture plates at 37°C and 5% CO2 in a humidified incubator, with daily media change. Cells were split every 4–5 days with 0.5 mM EDTA in 1x PBS.
HAP1 cell culture
Human HAP1 cells (Horizon Discovery) were cultured in IMDM (Gibco) supplemented with 10% FBS (Corning) at 37°C and 5% CO2 in a humidified incubator, with daily media change. Cells were split every 2 to 3 days using TrypLE Express (Gibco).
HEK293T cell culture
HEK293T cells were cultured in DMEM/F12 (Gibco) supplemented with 10% FBS (Corning) at 37°C and 5% CO2 in a humidified incubator, with media change every two days. Cells were split every 4 to 5 days using TrypLE Express (Gibco).
Hela cell culture
Hela cells were cultured in DMEM/F12 (Gibco) supplemented with 10% FBS (Corning) at 37°C and 5% CO2 in a humidified incubator, with media change every three days. Cells were split every 5 to 6 days using TrypLE Express (Gibco).
METHOD DETAILS
Informatic prediction of the protospacer adjacent motif (PAM)
Genomic sequence of N. lactamica strain ATCC 23970 was analyzed using CRISPRCasFinder (Couvin et al., 2018), and a putative type I-C system was predicted. Its native CRISPR array contains 30 spacers. We first bioinformatically searched for natural targets for all spacers using CRISPRTarget (Biswas et al., 2013). A total of 28 unique potential targets were identified when allowing for 1 nt mismatch in spacer-target base pairing. We then extracted the spacer-matched (i.e., protospacer) sequences together with their 5’ and 3’ 10 nt flanks, aligned them using WebLogo (Crooks et al., 2004), and deduced a consensus 5’- TTC PAM.
CRISPR interference assay in E. coli
The Nla I-C cas3-, cascade-, and CRISPR- encoding plasmids were co-transformed into BL21-AI (ThermoFisher). The resulting intermediate strain was made competent using the Mix & Go E. coli Transformation Kit (Zymo), transformed with a target-containing pCDF1 plasmid, leading to a final BL21-AI derivative strain harboring four compatible plasmids. For CRISPR interference assay, a single colony of this final strain was used to inoculate an overnight culture of 3 mL LB with four antibiotics (50 μg/mL kanamycin, 50 μg/mL carbenicillin, 25 μg/mL chloramphenicol, and 100 μg/mL spectinomycin). On the next morning, 30 μL was 1:100 back diluted into 3 mL LB broth with quadruple antibiotics, grown at 37°C 220 rpm until OD600 reaches 0.3–0.8. 1.5 mL of this starter culture was pelleted, resuspended in 1mL LB with 50 μg/mL kanamycin, μg/mL carbenicillin, and 25 μg/mL chloramphenicol (w/o spectinomycin), and then split in two 0.5 mL halves. One half was induced with 0.2% L-arabinose and 1 mM IPTG. Both the induced and un-induced half cultures were grown for three more hours at 37°C 220rpm, their 10-fold serial dilutions were plated onto LB plates with quadruple vs. triple antibiotics (w/o spectinomycin). CRISPR interference efficiency (i.e., depletion ratio) is calculated as the CFUs of triple antibiotic control plate divided by CFUs from quadruple-antibiotic test plate.
Construction of HAP1-AAVS1-EGFP and HAP1-AAVS1-EGFP-tdTomato reporter cell lines
The HAP1-EGFP reporter line was created by knocking-in an EF1a promoter-driven EGFP cassette into AAVS1 safe harbor locus. Briefly, wt HAP1 cells were individualized with TrypLE Express (Gibco), washed once with IMDM supplemented with 10% FBS and resuspended in Neon buffer R to a concentration of 2×107 cells/mL. 3 μg SpyCas9 protein was assembled with 0.6 μg of AAVS1-targeting sgRNA, the resulting Cas9 RNP was then mixed with 1 μg pSmart-AAVS1-EF1alpha-EGFP plasmid and approximately 105 cells in buffer R in a total volume of 10 μL. The mixture was electroporated with a 10 μL Neon tip (Invitrogen, 1575V, 10ms, 3 pulses), and plated in one well of a 24-well tissue culture plate containing 500 μL IMDM supplemented with 10% FBS. The cells were cultured for 2 weeks after electroporation, with daily medium change. EGFP positive cells were then sorted into 96 well plates at 1 cell per well via FACS. The sorted single cells were expanded, correctly targeted clones were identified by genomic junction PCRs. The final clone we picked happen to have a diploid genome with just one AAVS1 allele targeted by EGFP reporter; the other allele remained wt. We used this single clone to construct HAP1-AAVS1-EGFP-tdTomato dual reporter line, by knocking-in an EF1a driven tdTomato cassette into the untargeted wt AAVS1 allele using the same procedure described above, except that pSmart-AAVS1-EF1alpha-tdTomato plasmid was used and that EGFP+/ tdTm+ double positive cells were sorted for single clone expansion.
Plasmid transfection
CRISPR-Cas3 plasmid transfection was conducted using Lipofectamine 3000 Transfection Reagent (ThermoFisher) per manufacturer’s instructions. HAP1 reporter cells were seeded one day before transfection at 1×105 cells per well of a 24-well plate. For each transfection, we used 1 μL P3000 Enhancer Reagent, 1.5 μL Lipofectamine 3000 reagent, and a total of 500 ng crispr-cas3 plasmids. To monitor genome editing efficiency, cells were analyzed by flow cytometry 4–5 days post transfection. For Nla I-C system, we used 45, 22.5, 67.5, 270, 45 and 50 ng of Cas3, Cas5, Cas7, Cas8, Cas11 and CRISPR plasmids, respectively. For Bha I-C system, we used 45, 22.5, 67.5, 270, 45 and 50 ng of Cas3, Cas5, Cas7, Cas8, Cas11 and CRISPR plasmids, respectively. For Dvu I-C system, we used 90 ng each for Cas3, Cas5, Cas7, Cas8, and Cas11, and 50 ng for CRISPR plasmid. For Syn I-D system, we used 76.5, 180, 76.5, 15, 76.5, 22.5, and 50 ng of Cas3, Cas5, Cas6, Cas7, Cas10, Cas11 and CRISPR plasmids, respectively. For Syn I-B system, we used 75 ng each for Cas3, Cas5, Cas6, of Cas7, Cmx8 (Cas8), Cas11 and 50 ng of CRISPR plasmid.
Purification of NlaCascade RNP
NlaCascade is recombinantly expressed in E. coli BL21(DE3) cells in LB using two plasmids. All NlaCascade RNPs used in this study (except for the tdTm-G1 in Figure 2) were purified via Ni affinity purification followed by SEC. Briefly, we first co-transformed two plasmids encoding Cascade-His (e.g., pYZ1668 pET28-NlaCascade-NLS-6xHis) and CRISPR (e.g., pYZ288 pACYC-T7-Nla-repeat-EGFP-guide-G2–2x) into BL21(DE3). A single colony of the strain was inoculated into 10 mL LB broth with 50 μg/mL of kanamycin and 20 μg/ml of chloramphenicol, grown O/N at 37°C, 220rpm. This overnight culture was inoculated into 1 L of LB broth containing 50 μg/mL kanamycin and 20 μg/mL chloramphenicol, grown at 37°C 220rpm until OD600 reaches 0.6, cooled down to 18°C and induced with 1mM IPTG at 18°C O/N. Cells were pelleted and resuspended in 30 mM HEPES pH 7.5, 500 mM NaCl and 0.5 mM TCEP, and lysed with sonication. His-tagged protein was bound to Ni-NTA resin (Qiagen) and eluted with 30 mM HEPES pH 7.5, 500 mM NaCl, and 300 mM imidazole. The eluate was concentrated and loaded onto a sephacryl S300 column, using 30 mM HEPES pH 7.5, 150 mM NaCl and 0.5 mM DTT as elution buffer. The NlaCascade-containing fractions were pooled, concentrated, filter sterilized, aliquoted and frozen in liquid nitrogen. To purify the cas11-rescued version of NlaCascade in Figures 4E–H, we used a BL21 (DE3) derivative strain harboring three plasmids to express NlaCascadeΔcas11 (pYZ1670), CRISPR (pYZ288), and NlaCas11 (pYZ1673).
NlaCascade-tdTm was purified using MBP affinity purification followed by SEC. Briefly, we co-transformed two plasmids encoding MBP-Cascade (e.g. pYZ1664 pET-His-MBP-NlaCascade-NLS) and CRISPR (e.g., pYZ320, pACYC-T7-Nla-repeat-tdTomato-guide-G1–2x) into BL21 (DE3). Protein expression was done as described earlier except that 0.2% glucose was added to the 1 L LB large culture. Cell pellet was resuspended in 20 mM HEPES pH 7.5 and 500 mM NaCl and lysed by sonication. MBP-tagged protein was bound to amylose resin (NEB) and eluted with 20 mM HEPES pH 7.5, 500 mM NaCl, and 10 mM maltose. The eluate was incubated with TEV protease (homemade) O/N to cleave off His-MBP tag, concentrated, and loaded onto sephacryl S300 column. Cascade-containing fractions were pooled, dialyzed into 20 mM HEPES pH 7.5 and 150 mM NaCl, concentrated, filter sterilized, aliquoted and frozen.
Purification of NlaCas3
Plasmid pYZ1669 pET28-T7-NlaCas3-NLS-6xHis was transformed into BL21(DE3). NlaCas3 expression was carried out as described earlier for His-tagged NlaCacade except that only 50 μg/mL kanamycin was included in all LB cultures. The cell pellet was resuspended in 30 mM HEPES pH 7.5, 500 mM NaCl and 0.5 mM TCEP, lysed with sonication. His-tagged Cas3 was bound to Ni-NTA resin (Qiagen) and eluted with 30 mM HEPES pH 7.5, 500 mM NaCl, and 300 mM imidazole. The eluate was concentrated and loaded onto a sephacryl S300 column, using 30 mM HEPES pH 7.5, 150 mM NaCl and 0.5 mM DTT as elution buffer. Fractions containing Cas3 were pooled, concentrated, filter sterilized, aliquoted and frozen in liquid nitrogen.
RNP electroporation
NlaCascade-Cas3 RNPs were electroporated into human cells using Neon Transfection system (ThermoFisher) per manufacturer’s instructions. Briefly, cells were individualized with TrypLE Express (Gibco), washed once with culturing media and resuspended in Neon buffer R to a concentration of 2×107 cells/mL. 36 pmol of NLS-NlaCascade with or without 50 pmol of NLS-NlaCas3 were mixed with approximately 105 cells in buffer R in a total volume of 10 μL. This mixture was electroporated with a 10 μL Neon tip (1575V 10ms 3 pulses for HAP1 cells, 1100V 20ms 2 pulses for hESCs, 1150V 20ms 2 pulses for HEK293T; 1005V 35ms 2 pulses for Hela cells), and plated in a 24-well tissue culture plate containing 500 μL culturing media.
In vitro transcription of mRNAs
5’ capped and 3’ polyadenylated cas mRNAs were synthesized by in vitro transcription using mMessage mMachine T7 Ultra kit (ThermoFisher) per manufacture’s protocols. DNA templates used were purified PCR amplifications generated from plasmids pYZ147–150 and pYZ586, with the T7 promoter and polyT sequences incorporated via the primers. The pre-CRISPR transcript was synthesized with AmpliScribe T7-Flash Transcription Kit (Lucigen) per manufacturer’s instructions, using PCR amplicons generated from for pYZ288 as transcription template.
mRNA delivery into HAP1 cells by electroporation
CRISPR-Cas3 mRNAs were electroporated into HAP1 cells as described for RNP delivery with minor modifications. Individualized cells were washed once with IMDM supplemented with 10% FBS and resuspended in Neon buffer R to a concentration of 4×107 cells/mL. Approximately 2×105 cells were mixed with 50, 120, 120, 140, 120 ng of cas3, cas5, cas7, cas8, cas11 mRNAs, along with either 200 ng CRISPR plasmid or 2 μg pre-CRISPR RNA, in buffer R in a total volume of 10 μL. Each mixture was electroporated with a 10 μL Neon tip (1575V, 10ms, 3 pulses) and plated in 24-well tissue culture plates containing 500 μL IMDM with 10% FBS.
DNA lesion analysis by long-range PCR, cloning and Sanger sequencing
Genomic DNA of the edited cells was isolated using Gentra Puregene Cell Kit (Qiagen) per manufacturer’s instructions and used as template for long-range PCR with Q5 DNA Polymerase (NEB). PCR products were resolved on 1% agarose gel, stained with SYBR Safe (Invitrogen) and visualized using a ChemiDoc MP imager (BioRad). To precisely define Cas3-induced deletions, the entire PCR reaction was purified using QIAquick PCR Purification Kit (Qiagen), before cloning into pCR-BluntII-TOPO vector (Invitrogen). The resulting single colonies were randomly picked for colony PCR analysis using M13 forward and reverse primers. Clones with lesion-containing inserts were sent for Sanger sequencing (Eurofins). Deletion junctions were identified by aligning sequencing results to the reference WT sequence using Snapgene.
Flow cytometry
About 4–5 days post RNP electroporation or plasmid transfection, cells were individualized with TrypLE Express (Gibco), resuspended in IMDM supplemented with 10% FBS (for HAP1 cells) or DMEM/F12 supplemented with 10% FBS (for hESCs), and kept on ice until analysis. Flow cytometry was done using LSR Fortessa (BD) with a 488 nm laser for EGFP and a 561 nm laser for TdTomato; the data were analyzed with FlowJo v10.4.1.
6-TG cytotoxicity assay
HAP1 cells were individualized with TrypLE Express (Gibco) two days after electroporation and seeded in a 6-well plate at 200 cells/well density. Two days post seeding, 6-TG (Sigma) was added to each well to final concentration of 15 μM. Medium containing 6-TG was changed every two days until six days post 6-TG treatment, when cells were fixed with ice-cold 90% methanol for 30 min, washed with 1x PBS, stained with 0.5% crystal violet at RT for 5 min and destained with water. The plates are then air-dried at RT O/N and imaged with ChemiDoc MP imaging system (BioRad). Surviving colonies were counted using OpenCFU (Geissmann, 2013).
Western blot
Human cells were lysed directly on plate using 100 μL lysis buffer (1x PBS, 1x Laemmli sample buffer, 50 mM DTT, 100 units benzonase [Sigma-Aldrich]) per well of a 24-well plate two days after transfection. The lysed sample was denatured at 95°C for 5 min, immediately placed on ice or stored at −20°C until use. 10 μL of each denatured sample and 5 μL of Precision Plus Protein Dual Color Standard (BioRad) were separated by 12% SDS-PAGE and transferred to a 0.2 μm PVDF membrane (GE Amersham) using TranBlot SD Semi-Dry transfer (BioRad), in blotting buffer (25 mM Tris, 192 mM glycine, 0.03% SDS, 20% methanol, pH 8.3) for 30 min at 15 V at RT. After transfer, the membrane was blocked in blocking buffer (3% non-fat milk in 1x TBST) at RT for 40 min, and then incubated with mouse anti-HA (Sigma-Aldrich, H9658, 1:20,000) in blocking buffer at RT for 2 hrs. After washing three times with 1x TBST at RT for 10 min, the membrane was incubated with an HRP conjugated anti-mouse IgG secondary antibody (Promega, W4021, 1:10,000) in blocking buffer at RT for 1 hr. Then, the membrane was washed three times with 1x TBST at RT for 10 min, treated with Clarity Western ECL Substrate (BioRad) and imaged using ChemiDoc MP Imaging system (BioRad). For loading control, the same membrane was rinsed once with 1x TBST and stripped with 5 mL One-Minute Plus Western Blot Stripping Buffer (GM Biosciences) per manufacturer’s instructions. Stripped membrane was re-probed with mouse anti-GAPDH (Santa Cruz, sc-32233, 1:2,000) as described above.
E. coli strains sYZ276 and sYZ277 were grown and induced as described in the earlier CRISPR interference assay section. Cells equivalent to 150 μL of OD600 3.75 culture were pelleted and resuspended in 150 μL 1x PBS. 12 μL of resuspension was mixed with 3 μL 1M DTT and 15 μL of 2x Laemmli sample buffer (BioRad), denatured at 95°C for 5 min. 10 μL of each sample was separated on 15% SDS-PAGE. Protein transfer and detection were done as described above, except that mouse anti-Flag (Sigma-Aldrich, F1804, 1:5000) was used as the primary antibody.
N-Terminal protein sequencing with Edman degradation
5 μL NlaCascade-EGFP-G2 RNP (8 μM) was mixed with 1 μL 1 M DTT and 6 μL 2x Laemmli sample buffer, denatured at 95°C for 5 min, separated on a 4–15% gradient SDS-PAGE (BioRad), and transferred to 0.2 μm PVDF membrane as described above. After transfer, the membrane was stained with 0.1% Ponceau S solution. The ~14 kDa mysterious protein band was excised and sequenced with Edman degradation (Tufts University Core Facility).
Phylogenetic analysis
Cas8 protein sequences from Bha I-C, Nla I-C, Dvu I-C, Eco I-E, Tfu I-E and Syn I-B systems and the Syn I-D Cas10 protein sequence were aligned by MUSCLE (Edgar, 2004) with default parameters. A phylogenetic tree was then constructed using FastTree version 2.1.10 (Price et al., 2010) with default parameters. Tree visualization was rendered using Drawgram 3.696.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analysis were performed using Graphpad Prism version 9. Parameters of statistical analysis is provided in figure legends for respective figures.
Supplementary Material
Supplemental Table 1. Oligonucleotides, plasmids, strains and DNA fragments used in this study. Related to Star Methods
Supplemental Table 2. Sanger sequencing results of DNA deletions introduced by NlaCascade-Cas3 in human cells. Related to Figure 3, Figure S3 and S4
KEY RESOURCES TABLE
Tan* and Krueger* et al.
| Reagent or Resource | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal anti-HA antibody, mouse | Sigma-Aldrich | H9658 |
| Monoclonal anti-GAPDH antibody, mouse | Santa Cruz Biotechnology | sc-32233 |
| Monoclonal anti-Flag M2 antibody, mouse | Sigma-Aldrich | F1804 |
| Anti-Mouse IgG (H+L) HPR Conjugate | Promega | W4021 |
| Bacterial Strains | ||
| Escherichia coli JM109 | Promega | L2005 |
| Escherichia coli 5-alpha | NEB | C2987H |
| Escherichia coli BL21 (DE3) | Novagen | 70235 |
| Escherichia coli BL21-AI | ThermoFisher | C607003 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| L-arabinose | Sigma-Aldrich | A3256 |
| Isopropyl-β-D-thiogalactopyranoside (IPTG) | Fisher Scientific | BP1755–10 |
| FastBreak Cell Lysis Reagent, 10x | Promega | V8573 |
| Bond-Breaker TCEP Solution, neutral pH | ThermoScientific | 77720 |
| Ni-NTA agarose | Qiagen | 30210 |
| Amylose Resin | New England Biolabs | E8021S |
| Matrigel, GFR | Corning | 354230 |
| TGFbeta1 | PeproTech | 100–21C |
| Albumin human | Sigma-Aldrich | A9731 |
| L-Ascorbic acid 2-phosphate sesquimagnesium salt hydrate | Sigma-Aldrich | A8960 |
| Insulin solution | Sigma-Aldrich | I9278 |
| Human Holo-Transferrin | R&D systems | 2914HT100MG |
| Y-27632 | Cayman Chemical | 100055831 |
| TrypLE Express Enzyme | Gibco | 12605010 |
| Fetal Bovine Serum | Corning | 35010CV |
| 6-Thioguanine (6-TG) | Sigma-Aldrich | A4660 |
| Q5 hot start high-fidelity DNA Polymerase | New England Biolabs | M0493L |
| 2x GoTaq Green Master Mix | Promega | M7122 |
| Invitrogen SYBR Safe DNA Gel Stain | ThermoFisher | S33102 |
| Critical Commercial Assays | ||
| Lipofectamine 3000 Transfection Reagent | ThermoFisher | L3000015 |
| HiPrep Sephacryl S-300 HR Column | Cytiva | 17116701 |
| HiPrep Sephacryl S-400 HR Column | Cytiva | 28935605 |
| Gentra Puregene Cell Kit | Qiagen | 158767 |
| Zero Blunt TOPO PCR Cloning Kit | ThermoFisher | 450245 |
| Neon Transfection System 10 μL Kit | ThermoFisher | MPK1096 |
| mMESSAGE mMACHINE T7 Ultra Transcription Kit | ThermoFisher | AM1345 |
| AmpliScribe T7-Flash Transcription Kit | Lucigen | ASF3507 |
| Clarity Western ECL Substrate | BioRad | 1705060 |
| Experimental Models: Cell Line | ||
| HAP1 cells | Horizon Discovery | C859 |
| HAP1-AAVS1-EGFP | This paper | N/A |
| HAP1-AAVS1-EGFP-tdTomato | This paper | N/A |
| Human embryonic stem cell H9 | WiCell | WA09 |
| Human embryonic stem cell H9-DNMT3b-EGFP-tdTomato | Dolan et al., 2019 | N/A |
| HEK293T | ATCC | CRL-11268 |
| HeLa | ATCC | CCL-2 |
| Experimental Models: Organisms/strains | ||
| See Supplemental Table 1 for E. coli BL21-AI derivative strains used in this study. | ||
| Oligonucleotides | ||
| See Supplemental Table 1 for sequences of oligonucleotides and gBlocks used in this work. | ||
| Recombinant DNA | ||
| See Supplemental Table 1 for recombinant DNA used in this study. | ||
| Software and Algorithms | ||
| CRISPRCasFinder | https://crisprcas.i2bc.paris-saclay.fr | |
| MUSCLE | Edgar 2004 | |
| FastTree | Price et al., 2010 | |
| Drawgram | https://evolution.gs.washington.edu/phylip/doc/drawgram.html | |
Highlights.
A miniature CRISPR-Cas3 from N. lactamica confers bacterial immunity
NlaCascade-Cas3 RNP enables high-efficiency targeted large deletions in human cells
Cas11, a hidden internal translation product, is essential for genome editing
Supplying Cas11 as a framework to harness divergent compact CRISPR-Cas3 editors
ACKNOWLEDGEMENTS
We thank Erik Sontheimer for critical reading of the manuscript; Phyllis Hanson, Mats Ljungman, Alice Telesnitsky, Ruma Banerjee, Matthew Chapman, and Daniel Goldman for helpful discussions; Ying Liu for technical support; Univ. of Michigan flow cytometry core facility for equipment support; and all Zhang lab members and Chunyi Hu for thoughtful discussions. This work was supported by National Institutes of Health grants GM117268 and GM137883 to Y.Z., GM118174 to A.K., Univ. of Michigan institutional fund, Biological Scholar Award, Endowment for the Basic Sciences Accelerator Award, and MICHR Translational Research Diamond grant to Y.Z.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
A patent application has been filed describing the invention reported herein.
REFERENCES
- Anzalone AV, Koblan LW, and Liu DR (2020). Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol 38, 824–844. [DOI] [PubMed] [Google Scholar]
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, and Horvath P (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. [DOI] [PubMed] [Google Scholar]
- Biswas A, Gagnon JN, Brouns SJ, Fineran PC, and Brown CM (2013). CRISPRTarget: bioinformatic prediction and analysis of crRNA targets. RNA Biol 10, 817827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, and van der Oost J (2008). Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron P, Coons MM, Klompe SE, Lied AM, Smith SC, Vidal B, Donohoue PD, Rotstein T, Kohrs BW, Nyer DB, et al. (2019). Harnessing type I CRISPR-Cas systems for genome engineering in human cells. Nat Biotechnol 37, 1471–1477. [DOI] [PubMed] [Google Scholar]
- Chen Y, Liu J, Zhi S, Zheng Q, Ma W, Huang J, Liu Y, Liu D, Liang P, and Songyang Z (2020). Repurposing type I-F CRISPR-Cas system as a transcriptional activation tool in human cells. Nat Commun 11, 3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury S, Carter J, Rollins MF, Golden SM, Jackson RN, Hoffmann C, Nosaka L, Bondy-Denomy J, Maxwell KL, Davidson AR, et al. (2017). Structure Reveals Mechanisms of Viral Suppressors that Intercept a CRISPR RNA-Guided Surveillance Complex. Cell 169, 47–57 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couvin D, Bernheim A, Toffano-Nioche C, Touchon M, Michalik J, Neron B, Rocha EPC, Vergnaud G, Gautheret D, and Pourcel C (2018). CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic acids research 46, W246–W251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, and Brenner SE (2004). WebLogo: a sequence logo generator. Genome Res 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csorgo B, Leon LM, Chau-Ly IJ, Vasquez-Rifo A, Berry JD, Mahendra C, Crawford ED, Lewis JD, and Bondy-Denomy J (2020). A compact Cascade-Cas3 system for targeted genome engineering. Nature methods 17, 1183–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillard KE, Brown MW, Johnson NV, Xiao Y, Dolan A, Hernandez E, Dahlhauser SD, Kim Y, Myler LR, Anslyn EV, et al. (2018). Assembly and Translocation of a CRISPR-Cas Primed Acquisition Complex. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan AE, Hou Z, Xiao Y, Gramelspacher MJ, Heo J, Howden SE, Freddolino PL, Ke A, and Zhang Y (2019). Introducing a Spectrum of Long-Range Genomic Deletions in Human Embryonic Stem Cells Using Type I CRISPR-Cas. Molecular cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA (2020). The promise and challenge of therapeutic genome editing. Nature 578, 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ, and Church GM (2013). Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nature methods 10, 1116–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfara I, Le Rhun A, Chylinski K, Makarova KS, Lecrivain AL, Bzdrenga J, Koonin EV, and Charpentier E (2014). Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic acids research 42, 2577–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasiunas G, Barrangou R, Horvath P, and Siksnys V (2012). Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proceedings of the National Academy of Sciences of the United States of America 109, E2579–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann Q (2013). OpenCFU, a new free and open-source software to count cell colonies and other circular objects. PLoS One 8, e54072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo-Cantabrana C, and Barrangou R (2020). Characterization and applications of Type I CRISPR-Cas systems. Biochem Soc Trans 48, 15–23. [DOI] [PubMed] [Google Scholar]
- Hochstrasser ML, Taylor DW, Bhat P, Guegler CK, Sternberg SH, Nogales E, and Doudna JA (2014). CasA mediates Cas3-catalyzed target degradation during CRISPR RNA-guided interference. Proc Natl Acad Sci U S A 111, 6618–6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser ML, Taylor DW, Kornfeld JE, Nogales E, and Doudna JA (2016). DNA Targeting by a Minimal CRISPR RNA-Guided Cascade. Molecular cell 63, 840–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo Y, Nam KH, Ding F, Lee H, Wu L, Xiao Y, Farchione MD Jr., Zhou S, Rajashankar K, Kurinov I, et al. (2014). Structures of CRISPR Cas3 offer mechanistic insights into Cascade-activated DNA unwinding and degradation. Nature structural & molecular biology 21, 771–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibraheim R, Song CQ, Mir A, Amrani N, Xue W, and Sontheimer EJ (2018). All-inone adeno-associated virus delivery and genome editing by Neisseria meningitidis Cas9 in vivo. Genome biology 19, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RN, Golden SM, van Erp PB, Carter J, Westra ER, Brouns SJ, van der Oost J, Terwilliger TC, Read RJ, and Wiedenheft B (2014). Structural biology. Crystal structure of the CRISPR RNA-guided surveillance complex from Escherichia coli. Science 345, 1473–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, and Charpentier E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Koo T, Park SW, Kim D, Kim K, Cho HY, Song DW, Lee KJ, Jung MH, Kim S, et al. (2017). In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nature communications 8, 14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim D, Cho SW, Kim J, and Kim JS (2014). Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24, 1012–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klompe SE, Vo PLH, Halpin-Healy TS, and Sternberg SH (2019). Transposonencoded CRISPR-Cas systems direct RNA-guided DNA integration. Nature 571, 219–225. [DOI] [PubMed] [Google Scholar]
- Knott GJ, and Doudna JA (2018). CRISPR-Cas guides the future of genetic engineering. Science 361, 866–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor AC, Badran AH, and Liu DR (2017). CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 168, 20–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, and Zhang F (2017). Diversity, classification and evolution of CRISPR-Cas systems. Current opinion in microbiology 37, 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kweon J, Jang AH, Kim DE, Yang JW, Yoon M, Rim Shin H, Kim JS, and Kim Y (2017). Fusion guide RNAs for orthogonal gene manipulation with Cas9 and Cpf1. Nature communications 8, 1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenay RT, Maksimchuk KR, Slotkowski RA, Agrawal RN, Gomaa AA, Briner AE, Barrangou R, and Beisel CL (2016). Identifying and Visualizing Functional PAM Diversity across CRISPR-Cas Systems. Molecular cell 62, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lino CA, Harper JC, Carney JP, and Timlin JA (2018). Delivering CRISPR: a review of the challenges and approaches. Drug Deliv 25, 1234–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TY, and Doudna JA (2020). Chemistry of Class 1 CRISPR-Cas effectors: Binding, editing, and regulation. The Journal of biological chemistry 295, 14473–14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Chen O, Wall JBJ, Zheng M, Zhou Y, Wang L, Vaseghi HR, Qian L, and Liu J (2017). Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci Rep 7, 2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Naseri A, Reyes-Gutierrez P, Wolfe SA, Zhang S, and Pederson T (2015). Multicolor CRISPR labeling of chromosomal loci in human cells. Proceedings of the National Academy of Sciences of the United States of America 112, 3002–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Aravind L, Wolf YI, and Koonin EV (2011). Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol Direct 6, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJ, Charpentier E, Haft DH, et al. (2015). An updated evolutionary classification of CRISPR-Cas systems. Nature reviews Microbiology 13, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns SJJ, Charpentier E, Cheng D, Haft DH, Horvath P, et al. (2020). Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nature reviews Microbiology 18, 67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, and Church GM (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, and Sontheimer EJ (2008). CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322, 1843–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride TM, Schwartz EA, Kumar A, Taylor DW, Fineran PC, and Fagerlund RD (2020). Diverse CRISPR-Cas Complexes Require Independent Translation of Small and Large Subunits from a Single Gene. Molecular cell 80, 971–979 e977. [DOI] [PubMed] [Google Scholar]
- McCarty NS, Graham AE, Studena L, and Ledesma-Amaro R (2020). Multiplexed CRISPR technologies for gene editing and transcriptional regulation. Nature communications 11, 1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisaka H, Yoshimi K, Okuzaki Y, Gee P, Kunihiro Y, Sonpho E, Xu H, Sasakawa N, Naito Y, Nakada S, et al. (2019). CRISPR-Cas3 induces broad and unidirectional genome editing in human cells. Nat Commun 10, 5302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulepati S, and Bailey S (2013). In vitro reconstitution of an Escherichia coli RNA-guided immune system reveals unidirectional, ATP-dependent degradation of DNA target. J Biol Chem 288, 22184–22192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulepati S, Heroux A, and Bailey S (2014). Structural biology. Crystal structure of a CRISPR RNA-guided surveillance complex bound to a ssDNA target. Science 345, 1479–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam KH, Haitjema C, Liu X, Ding F, Wang H, DeLisa MP, and Ke A (2012). Cas5d protein processes pre-crRNA and assembles into a cascade-like interference complex in subtype I-C/Dvulg CRISPR-Cas system. Structure 20, 1574–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien RE, Santos IC, Wrapp D, Bravo JPK, Schwartz EA, Brodbelt JS, and Taylor DW (2020). Structural basis for assembly of non-canonical small subunits into type I-C Cascade. Nature communications 11, 5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osakabe K, Wada N, Miyaji T, Murakami E, Marui K, Ueta R, Hashimoto R, AbeHara C, Kong B, Yano K, et al. (2020). Genome editing in plants using CRISPR type I-D nuclease. Commun Biol 3, 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osakabe K, Wada N, Murakami E, Miyashita N, and Osakabe Y (2021). Genome editing in mammalian cells using the CRISPR type I-D nuclease. Nucleic acids research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan A, Krajeski R, Ioannidi E, Lee B, Gardner A, Makarova KS, Koonin EV, Abudayyeh OO, and Gootenberg JS (2021). Programmable RNA targeting with the single-protein CRISPR effector Cas7-11. Nature 597, 720–725. [DOI] [PubMed] [Google Scholar]
- Peters JE, Makarova KS, Shmakov S, and Koonin EV (2017). Recruitment of CRISPR-Cas systems by Tn7-like transposons. Proceedings of the National Academy of Sciences of the United States of America 114, E7358–E7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickar-Oliver A, Black JB, Lewis MM, Mutchnick KJ, Klann TS, Gilcrest KA, Sitton MJ, Nelson CE, Barrera A, Bartelt LC, et al. (2019). Targeted transcriptional modulation with type I CRISPR-Cas systems in human cells. Nat Biotechnol 37, 1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, and Arkin AP (2010). FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, et al. (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Ladha A, Strecker J, Faure G, Neumann E, Altae-Tran H, Macrae RK, and Zhang F (2021). Dual modes of CRISPR-associated transposon homing. Cell 184, 2441–2453 e2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salis HM, Mirsky EA, and Voigt CA (2009). Automated design of synthetic ribosome binding sites to control protein expression. Nature biotechnology 27, 946–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz I, Lange SJ, Hein S, Hess WR, and Backofen R (2013). CRISPR-Cas systems in the cyanobacterium Synechocystis sp. PCC6803 exhibit distinct processing pathways involving at least two Cas6 and a Cmr2 protein. PloS one 8, e56470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkunas T, Gasiunas G, Fremaux C, Barrangou R, Horvath P, and Siksnys V (2011). Cas3 is a single-stranded DNA nuclease and ATP-dependent helicase in the CRISPR/Cas immune system. EMBO J 30, 1335–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkunas T, Gasiunas G, Waghmare SP, Dickman MJ, Barrangou R, Horvath P, and Siksnys V (2013). In vitro reconstitution of Cascade-mediated CRISPR immunity in Streptococcus thermophilus. EMBO J 32, 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Beljouw SPB, Haagsma AC, Rodriguez-Molina A, van den Berg DF, Vink JNA, and Brouns SJJ (2021). The gRAMP CRISPR-Cas effector is an RNA endonuclease complexed with a caspase-like peptidase. Science 373, 1349–1353. [DOI] [PubMed] [Google Scholar]
- Wang D, Zhang F, and Gao G (2020). CRISPR-Based Therapeutic Genome Editing: Strategies and In Vivo Delivery by AAV Vectors. Cell 181, 136–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra ER, van Erp PB, Kunne T, Wong SP, Staals RH, Seegers CL, Bollen S, Jore MM, Semenova E, Severinov K, et al. (2012). CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Mol Cell 46, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedenheft B, Lander GC, Zhou K, Jore MM, Brouns SJJ, van der Oost J, Doudna JA, and Nogales E (2011). Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature 477, 486–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Luo M, Dolan AE, Liao M, and Ke A (2018). Structure basis for RNA-guided DNA degradation by Cascade and Cas3. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Luo M, Hayes RP, Kim J, Ng S, Ding F, Liao M, and Ke A (2017). Structure Basis for Directional R-loop Formation and Substrate Handover Mechanisms in Type I CRISPR-Cas System. Cell 170, 48–60 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JK, Gasior SL, Jones S, Wang L, Navarro P, Vickroy B, and Barrangou R (2019). The repurposing of type I-E CRISPR-Cascade for gene activation in plants. Commun Biol 2, 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, et al. (2015). Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Sheng G, Wang J, Wang M, Bunkoczi G, Gong W, Wei Z, and Wang Y (2014). Crystal structure of the RNA-guided immune surveillance Cascade complex in Escherichia coli. Nature 515, 147–150. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Li J, Wang B, Han J, Hao Y, Wang S, Ma X, Yang S, Ma L, Yi L, et al. (2020). Endogenous Type I CRISPR-Cas: From Foreign DNA Defense to Prokaryotic Engineering. Front Bioeng Biotechnol 8, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. Oligonucleotides, plasmids, strains and DNA fragments used in this study. Related to Star Methods
Supplemental Table 2. Sanger sequencing results of DNA deletions introduced by NlaCascade-Cas3 in human cells. Related to Figure 3, Figure S3 and S4
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.