

Abstract
Apolipoprotein E ε4 (APOE4) is the primary genetic risk factor for the late-onset form of Alzheimer’s disease (AD). Although the reason for this association is not completely understood, researchers have uncovered numerous effects of APOE4 expression on AD-relevant brain processes, including amyloid beta (Aβ) accumulation, lipid metabolism, endosomal-lysosomal trafficking, and bioenergetics. In this study, we aimed to determine the effect of APOE4 allelic dosage on regional brain lipid composition in aged mice, as well as in cultured neurons. We performed a targeted lipidomic analysis on an AD-vulnerable brain region (entorhinal cortex; EC) and an AD-resistant brain region (primary visual cortex; PVC) from 14–15 month-old APOE3/3, APOE3/4, and APOE4/4 targeted replacement mice, as well as on neurons cultured with conditioned media from APOE3/3 or APOE4/4 astrocytes. Our results reveal that the EC possesses increased susceptibility to APOE4-associated lipid alterations compared to the PVC. In the EC, APOE4 expression showed a dominant effect in decreasing diacylglycerol (DAG) levels, and a semi-dominant, additive effect in the upregulation of multiple ceramide, glycosylated sphingolipid, and bis(monoacylglycerol)phosphate (BMP) species, lipids known to accumulate as a result of endosomal-lysosomal dysfunction. Neurons treated with conditioned media from APOE4/4 vs. APOE3/3 astrocytes showed similar alterations of DAG and BMP species to those observed in the mouse EC. Our results suggest that APOE4 expression differentially modulates regional neuronal lipid signatures, which may underlie the increased susceptibility of EC-localized neurons to AD pathology.
Subject terms: Molecular neuroscience, Diseases
Introduction
Possession of the ε4 allele of apolipoprotein E (APOE) is the primary genetic risk factor for late-onset Alzheimer’s disease (AD). While early work investigating the association between APOE ε4 (APOE4) and AD focused on the ability of APOE4 to increase the aggregation and decrease the clearance of amyloid beta (Aβ) [1–7], more recent work has uncovered numerous effects of differential APOE isoform expression on other AD-relevant systems in the brain, including intracellular trafficking, energy metabolism, inflammation, and vascular integrity [see reviews by [8–11]]. It is important to note, however, that the primary role of the apoE protein, in both the brain and the periphery, is to mediate the transport of cholesterol and other lipids [12–14], a function that is also significantly altered by APOE4 expression [15–19]. Importantly, AD pathology and neuronal aging are linked to dysregulation of lipid metabolism [20], and specific lipid signatures provide diagnostic value in predicting phenoconversion to either amnestic mild cognitive impairment (aMCI) or AD [21]. Moreover, mitigation of cholesterol imbalances has been increasingly explored as a therapeutic strategy, with success in pre-clinical models of AD [22, 23].
While several groups have addressed APOE isoform-related lipid alterations in plasma and serum [24, 25], relatively few studies have reported the use of lipidomic analysis to investigate the lipid alterations that occur specifically in the brain [26–31]. The results from these studies include observations that APOE4 expression increased the levels of triacylglycerol (TAG) and decreased the levels of lysophosphatidylcholine (LPC), phosphatidylserine (PS), cholesterol, and 14-demethylanosterol (a cholesterol derivative) in the brains of APOE targeted replacement mice [28, 30]. In addition, APOE4 mice were more sensitive to lipid alterations caused by a high-fat and/or high-cholesterol diet, most notably showing increased levels of cholesterol-esters in the brain [28]. In humans, a post-mortem lipidomic analysis of the inferior parietal lobule from AD patients showed that APOE4 carriers had decreased levels of glycerophospholipids, most notably in phosphatidic acid (PA) [29].
Although this data is informative, there is still much to learn about the effects of APOE genotype on lipid signatures in the brain, especially in regards to the region- and cell-type-specific effects of APOE4 expression on individual lipid species. We have previously reported a multi-omic analysis on an AD-vulnerable brain region (entorhinal cortex; EC) and an AD-resistant brain region (primary visual cortex; PVC) of aged APOE targeted replacement mice, which has thus far revealed novel effects of APOE4 expression on several important biological pathways, including neuronal activity [32], endosomal-lysosomal trafficking [33], and bioenergetics [34]. Here, we present the results from our lipidomic analysis, which reveals specific effects of APOE4 expression on the regional lipid signatures of the EC and PVC from aged APOE mice, as well as results from a complementary lipidomic analysis on mouse primary neurons treated with conditioned media from APOE4/4 vs. APOE3/3 expressing astrocytes. Our findings reveal region- and cell-type-specific APOE4-associated lipid alterations in the brain, thus shedding light on novel features relevant for AD pathogenesis independent of Aβ.
Materials and methods
Mice
Human APOE targeted replacement mice were first developed by Sullivan et al. [35, 36] and were acquired from Taconic Biosciences or directly from Dr. Patrick Sullivan. All mice used in this study were treated in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Columbia University Irving Medical Center Institutional Animal Care and Use Committee (IACUC). For this study, we utilized 14–15 month-old male APOE3/3, APOE3/4, and APOE4/4 mice, 8 mice per genotype. This sample size was chosen based on similar lipidomics studies in mice [37, 38], and our own metabolomics study in APOE mice [32].
Extraction of lipids from brain tissue
Mice were sacrificed by cervical dislocation to maintain the brain environment, and individual brain regions were immediately removed and snap-frozen on dry ice. Tissues were stored at −80 °C prior to extraction. Lipid and small-molecule metabolite extraction was performed using a methyl tert-butyl ether (MTBE)/methanol extraction protocol modified from previous reports [39, 40], as we have described previously [32]. Briefly, individual EC or PVC tissues were homogenized in 400 μl of ice-cold methanol using a bead mill homogenizer (TissueLyser II, Qiagen) at 25 beats/sec, 2x for 45 sec each. Following homogenization, samples were incubated in 1200 μl of MTBE for 1 hr at room temperature to separate organic-soluble lipids from aqueous-soluble lipids and other small-molecules. Finally, 360 μl of ultrapure water was added (for a final ratio of 3:1:0.9 MTBE:methanol:water) to resolve the two liquid phases, and each samples were centrifuged at 10,000 x g for 10 min. For this experiment, the upper organic phase was collected from each sample and stored in a separate tube, and the remaining protein pellets were resuspended in 25 mM ammonium bicarbonate, pH 8, with 2.5% SDS. A BCA protein assay was performed on each protein fraction, and the organic phase was normalized to their protein concentration equivalent with 100% methanol. All samples were then stored at −80 °C prior to analysis.
Extraction of lipids from astrocyte conditioned media-treated primary neurons
Astrocyte conditioned media (ACM) was obtained from immortalized astrocyte cell lines (a gift from Dr. David Holtzman) that were originally generated from primary astrocytes from P1-2 pups of APOE targeted replacement mice [41]. The immortalized astrocytes were conditioned with Neurobasal media supplemented with B27, Glutamax-I, Normocin and 1% penicillin/streptomycin for 24 h. This ACM was then collected, stored at −80 °C, and thawed prior to use. WT primary cortical neuronal cultures were obtained from embryonic day 17 (E17) C57Bl/6 (B6) embryos. Briefly, pregnant mice were euthanized by cervical dislocation and the embryo brains extracted. The meninges were removed and the cortices dissected out. The cortices were then enzymatically dissociated in 0.25% Trypsin-EDTA and resuspended in Neurobasal media supplemented with B27, Glutamax-I, Normocin and 1% penicillin/streptomycin. Dissociated cells were counted and 300 K neurons per well were plated directly into ACM in poly-D-lysine (PDL)-coated 6-well plates. ACM-treated neurons were then incubated at 37 °C for 7 days in a humidified chamber with 5% CO2, with 50% media exchange (with newly thawed ACM) once every 3 days. The neurons were then harvested, and the lipids were extracted using the MTBE/methanol extraction protocol described above. All samples were then stored at −80 °C prior to analysis.
Lipidomic analysis
Lipid profiling was performed using an Agilent 1260 HPLC coupled to an Agilent 6490 triple quadrupole (QQQ) mass spectrometer [42]. Each sample was run through three separate chromatographic conditions (reverse-phase negative mode, reverse-phase positive mode and normal-phase positive mode) for the effective quantification of 337 distinct lipids from 28 lipid subclasses, as previously described [42]. From the mouse analysis, four samples (two APOE3/4 EC samples, one APOE3/3 PVC sample, and one APOE4/4 PVC sample) were removed prior to data processing due to chromatographic errors during the runs. No samples were removed prior to data processing from the cell analysis. Individual lipid species were measured by multiple reaction monitoring transitions and lipid concentration was calculated by referencing appropriate internal standards per lipid class added to each sample, using Agilent’s MassHunter Quantitative Analysis software (version 5). Lipid classes not internally standardized owing to commercial unavailability of respective standards were referenced to closely eluted standards. In mice, relative free cholesterol levels were calculated as relative mol% of all lipids detected for each sample; all other lipid species are expressed as relative mol% of all lipids measured except free cholesterol. In cultured neurons, all lipids are expressed as relative mol% of all lipids measured [43]. Blinding was performed during both the data acquisition and the data processing steps. Lipid nomenclature follows LIPID MAPS consortium guidelines. Lipid species are annotated as lipid class followed by total number of carbons and unsaturation degree of acyl chains. DAG and TAG species are annotated with the addition of acyl carbon and unsaturation of the product ion (e.g., DAG30:0/14:0). Sphingolipids contained d18:1 long-chain base except dhSM species, containing a d18:0 base. N-acyl-PS species contained C16:0 N-linked acyl chains.
Statistics
Statistical analysis was performed using SPSS Statistics 26 (IBM) and Prism 8.0 (Graphpad) software. Animals were randomly recruited to experimental groups based on genotype. Sample size was based on previous studies [44, 45], and is indicated in each legend. Samples were blinded during tandem biochemical analysis. Normality and homogeneity of variance were assessed with Shapiro–Wilk and Levene’s test, respectively. One-way ANOVA followed by Tukey’s post-hoc was performed for multiple comparisons and Kruskal–Wallis test was used for non-parametric testing, as referenced in each figure legend. A confidence interval of 95% was assumed for all statistical tests. No samples or animals were excluded from analysis.
Results
Regional effects of APOE4 expression on brain lipid composition
In order to understand the effects of differential APOE isoform expression on lipid signatures in the brain, we performed a targeted analysis of lipid metabolites extracted from the brains of aged APOE mice. Specifically, we utilized male 14–15 month-old APOE targeted replacement mice, which express the human APOE gene in place of the mouse Apoe gene. In order to understand the regional and dosage effects of APOE4 allelic expression, we studied APOE3/3, APOE3/4, and APOE4/4 mice (n = 8 mice per group) and extracted lipids from two brain regions: the EC, which is one of the first brain regions where neurofibrillary tangles (NFTs) accumulate in AD, and the PVC, which develops tangle pathology at a later stage [46]. The lipidomic analysis was performed using a previously described liquid chromatography-mass spectrometry (LC-MS) platform [42], allowing for the detection of 28 lipid classes and over 300 distinct lipid species (Extended Data File).
Our results confirm that cholesterol is the most abundant lipid subclass in the brain, followed by phosphatidylcholine (PC), phosphatidylethanolamine (PE)/plasmalogens, and sphingomyelin (SM), all of which play structural roles in the assembly of cellular membranes [47] (Extended Data File). While our findings corroborate previous observations reporting that differential APOE isoform expression does not profoundly alter lipid composition in the brain [26, 27], significant effects were observed in the levels of less abundant and bioactive lipid species in both brain regions. In the EC, 35 lipid species were differentially expressed in aged APOE mice, whereas only 9 lipid species were differentially expressed in the PVC between the different genotype groups (Fig. 1a).
Fig. 1. Comparative lipid profile of EC and PVC of aged APOE targeted replacement mice.

a Heatmap of lipids significantly altered by APOE3/4, APOE4/4 and APOE3/3 genotype in EC and PVC at 14–15 months of age. Each row represents a lipid species of a given category (see color legends); each column represents a sample. Results are expressed as Z-score [(average mol% of lipid species per genotype−average mol% of lipid species in all genotypes)/standard deviation of average mol% lipid species] represented in gradient color; blue and red indicate negative and positive Z-score, respectively. One-way ANOVA was performed and results thresholded by p < 0.05; n = 8, 6, and 8 for EC and n = 7, 8, and 7 for PVC in APOE3/3, APOE3/4, and APOE4/4 mice, respectively. b, c Volcano plots of differentially modulated lipid species in EC and PVC between APOE3/4 vs. APOE3/3, APOE4/4 vs. APOE3/3 and APOE4/4 vs. APOE3/4. Colored plots denote significantly affected species at Tukey’s post-hoc p < 0.05, black plots denote unaltered species (p > 0.05).
Specifically, we observe that in the EC, several neutral lipid species were affected by APOE4 expression (Fig. 1a), including numerous short-length diacylglycerol species (DAG 28-34 C), which showed decreasing levels with APOE4 expression, as did several longer DAG species (38-40 C). Cholesterol-ester (CE) 18:0 was also reduced in the EC with increasing number of APOE4 alleles. We detected differential effects in sphingolipid species, namely APOE4-associated increases in ceramides (Cer) 16:0 and 18:1; hexosylceramides (HexCer) 16:0, 16:1, 18:0, and 26:0; lactosylceramide (LacCer) 16:0; and monosialodihexosylganglioside (GM3) 22:0, while sphingomyelin (SM) 18:0 and dihydrosphingomyelin (dhSM) 16:1 were decreased with APOE4 expression. For glycerophospholipids, phosphatidylcholines (PC) 36:0 and 42:7; plasmalogen phosphatidylethanolamine (PEp) 34:2; and PS 42:4 and 42:5 were increased with APOE4 expression, while phosphatidylglycerol (PG) 32:0 and phosphatidylinositols (PI) 38:3 and 38:4 were decreased. In addition, lysoglycerophospholipids, such as lysophosphatidylinositol (LPI) 16:0 and lysoetherphosphatidylcholine (LPCe) 18:0, were also decreased. Interestingly, multiple species (32:0, 34:0, 34:1, 36:0, 36:1, and 38:4) of the atypical phospholipid bis(monoacylglycerol)phosphate (BMP) [which is also commonly referred to as lysobisphosphatidic acid (LBPA)] were elevated with increasing APOE4 alleles. Importantly, while BMP is exclusively found in the intraluminal vesicles (ILVs) of late endocytic compartments and is considered a bona fide marker of these structures [48], we have also previously observed increased levels of BMP in the EC of late-onset AD patients [42].
In the PVC, we found decreased levels of DAG 38:3, Cer 18:1, SM 16:0, SM 20:0, PE 36:1, and LPCe 16:0 with APOE4 expression, while BMP 34:2 was significantly increased (Fig. 1a, right panel). These observations support a reduced effect of APOE4 on the lipid composition of the PVC as compared to the EC, which may be due to the fact that APOE displays increased gene expression in the medial temporal lobe compared to other brain regions [49, 50]. We also note that most of the differentially expressed lipid subclasses we observed are affected in both regions, namely DAG, Cer, SM, and BMP, albeit to a lesser degree in the PVC. Remarkably, these lipids are all associated with the endosomal-lysosomal pathway, as either bioactive signaling molecules (DAG) [51], substrates of lysosomal degradation (Cer, SM) [52], or lysosomal resident lipids (BMP) [48]. This suggests that while some effects of APOE4 expression may be regionally restricted, others are common throughout the brain, including previously described effects of APOE4 expression on endosomal-lysosomal trafficking [33, 53].
Co-dominant and additive effects of APOE4 alleles in the EC
In terms of AD susceptibility, one copy of APOE4 confers a ~3-fold increased risk, while two copies of APOE4 confers a ~12-fold increased risk of developing AD [14]. For this reason, it is important to understand which biological effects of APOE4 expression are in agreement with this additive model (i.e., a second APOE4 allele has an additional impact), and which features are associated with a dominant APOE4 effect (i.e., it is common to either one or two alleles of APOE4). To better understand the effect magnitude of each APOE4 allele on lipid signatures, we performed paired comparisons between the three APOE genotypes (APOE3/3, APOE3/4, and APOE4/4; Fig. 1b, c). Using post-hoc multiple comparison analysis, we detected 7 and 31 significantly altered lipid species in the EC when APOE3/4 or APOE4/4 mice, respectively, were compared to APOE3/3 mice (Fig. 1b). Notably, 71% (5 of 7) of the lipid species altered in the APOE3/4 vs. APOE3/3 EC were also significantly affected in the APOE4/4 vs. APOE3/3 EC. In the PVC, we detected 2 and 9 significantly altered species in APOE3/4 or APOE4/4 mice, respectively (Fig. 1c).
Of the significantly altered species in the EC, DAG 28:0, 30:0, 32:0, and 32:1 were decreased in APOE3/4 and also ranked within the top-10 most significantly altered in APOE4/4 mice (Fig. 1b and 2a). Since no significant differences were detected in these species from the APOE4/4 vs. APOE3/4 EC paired comparison, these findings suggest a dominant effect of APOE4 on DAG levels (Fig. 2a). Box-plot representations of the average fold-change of lipid levels across each of the three genotypes confirm the dominant effect of APOE4 in DAG levels (Fig. 2a). Additionally, we found a two-step additive effect in specific BMP species (Fig. 2b), as well as Cer 16:0, 18:1, and more complex glycosphingolipids, such as HexCer 16:0, 16:1, 18:0 (Fig. 2c, d), and GM3 22:0, in which significant changes were only detected in the APOE4/4 vs. APOE3/3 EC. The only decreased species in APOE4/4 vs. APOE3/4 EC were dhSM 16:1 and LPI 16:0. Altogether, our findings indicate a dominant effect of APOE4 on DAG levels, with a semi-dominant and additive effect on Cer, glycosylated sphingolipids, and BMP.
Fig. 2. Dominant and semi-dominant effects of APOE4 expression on lipid composition in the EC in aged APOE mice.

a–c Normalized average mol% (fold-change) of (a) DAG, (b) BMP, (c) Hex and (d) Cer lipid species from the EC of aged APOE targeted replacement mice. Lipids were annotated per total acyl carbons and degree of unsaturation. Data represent median ± max/min. One-way ANOVA followed by Tukey’s post-hoc test for multiple comparisons was performed; n = 8, 6, and 8 for EC and n = 7, 8, and 7 for PVC in APOE3/3, APOE3/4, and APOE4/4 mice, respectively. Kruskal–Wallis H test confirmed statistically significant difference in levels of BPM 32:0 (p = 0.03), HexCer 16:0 (p = 0.003) and Hex Cer 16:1 (p = 0.003). *p < 0.05, **p < 0.01, and ***p < 0.001.
Acyl chain profile is not affected by APOE4 genotype in the EC and PVC
The chemical structures of lipids are a determinant of their physical/chemical properties, which in turn determines the biological roles that they play in cells and tissues. While lipid categorization is largely determined by its polar head group, its acyl chain structure further contributes to membrane compositional diversity, which differs greatly between tissues and subcellular compartments [47]. Importantly, lipid-metabolizing enzymes and transporters present preferred affinity to certain acyl chains; as such, acyl chain profiles may indicate selective modulation of specific metabolic pathways. For these reasons, we analyzed the effect of APOE4 alleles on the acyl chain profile of DAG/glycerophospholipids (GPL) and sphingolipids (SL) in the EC and PVC.
Despite the dominant effect of APOE4 on decreasing the levels of short-length DAG species in the EC, no global impact was observed in the acyl chain length for DAG/GPL or for SL in APOE3/4 or APOE4/4 mice, as compared to APOE3/3 mice (Supplementary Fig. 1a, b). Similarly, no overall impact was observed on the degree of saturation of either lipid category. While we have previously reported an effect at the level of free fatty acids in the EC of APOE4 mice, including an increase in docosahexaenoic acid (22:6) in the EC [34], our results do not support a preferential acyl chain enrichment in the molecular signature of DAG/GPL and SL.
Effects of APOE astrocyte conditioned media on neuronal lipid composition
A present limitation of performing whole tissue lipidomic analysis is the inability to discern the corresponding contribution of each cell-type present within that tissue. To overcome this constraint, we used the paradigm of in vitro conditioned media transfer to analyze the effect of humanized APOE4/4 vs. APOE3/3 expression in astrocytes (the primary source of apoE protein in the brain) on the lipid composition of wild-type, B6 cortical neurons. Specifically, ACM was collected from immortalized astrocytes generated from post-natal day 1-2 (P1-2) pups of APOE3/3 or APOE4/4 targeted replacement mice [41]. Primary neurons derived from embryonic day 17 (E17) B6 mice were plated in either no ACM, APOE3/3 ACM, or APOE4/4 ACM (n = 6 wells per group) and incubated for 7 days, before harvesting for lipidomic analysis.
In comparison to untreated cells, the ACM dramatically altered neuronal lipid composition, with overlapping effects regardless of APOE isoform (Supplementary Fig. 2a and Extended Data File). We observed a 2-fold increase in DAG levels and a more modest elevation of SL levels, including SM, dhSM, HexCer, and LacCer (Supplementary Fig. 2b). For GPL, PA, and phosphatidylglycerol (PG) levels were decreased, whereas the products of PG transformation, acyl-PG and BMP, were increased by 1.3-fold and a dramatic 8-fold average, respectively (Supplementary Fig. 1c). Also, ether phosphatidylcholines (PCe) and LPCe were increased by APOE ACM, although to a different extent by each isoform. Thus, ACM alone induces significant remodeling of neuronal membranes, a fundamental characteristic to account for in future co-culture experiments.
Next, we compared the differential effects of APOE4/4 vs. APOE3/3 ACM on neuronal lipid signatures (Fig. 3a, b). We opted for the use of homozygous astrocytes for each isoform to increase the sensitivity of any additive effects of APOE4. Importantly, we found a 30% increase in free cholesterol levels in APOE4/4 vs. APOE3/3 ACM-treated neurons. Several CE species (20:1, 20:2, 20:3, and 22:3) and TAG species (52:1/18:0, 54:2/18:0, 56:3/18:1, 56:4/20:4, 56:5/20:4, 56:6/20:4) were increased (Fig. 3c, d). On the other hand, short-length DAG species (30:1/14:0, 32:1/16:0, 32:2/16:1, and 34:2/16:1) were decreased in APOE4/4 vs. APOE3/3 ACM-treated neurons (Fig. 3e), confirming an inverse effect of APOE4 expression on DAG levels comparatively to neutral lipids. Concerning SL, we found increased levels of Cer 18:1/16:0 and SM 18:1/16:1 in the presence of APOE4, while longer SM 18:1/22:1 levels were decreased, as were multiple species of more complex and glycosylated sphingolipids, including HexCer, Sulf, LacCer, and GM3.
Fig. 3. Comparative effect of APOE3/3 versus APOE4/4 astrocyte conditioned media (ACM) on the lipid profile of wild-type (WT) neurons.

a Heatmap of lipids significantly altered in WT neurons after treatment with ACM derived from immortalized APOE3/3 and APOE4/4 astrocytes. Results expressed as Z-score [(average mol% of individual lipid species per genotype−average mol% of lipid in all genotypes)/standard deviation of average mol% lipid] represented in gradient color; blue and red indicate negative and positive Z-score, respectively. One-way ANOVA was performed and results threshold by p < 0.05; n = 6. b Volcano plots of differentially modulated lipid species under same conditions as in (a). Colored plots denote significant alterations (Tukey’s post-hoc p < 0.05), black plots denote unaltered species (p > 0.05). c–h Normalized average mol% (fold-change) of (c) CE, (d) TAG, (e) DAG, (f) PA (g) PC, (h) BMP lipid species from WT neurons incubated with ACM. Lipids were annotated per total acyl carbons and degree of unsaturation. Data represent median ± max/min. One-way ANOVA followed by Tukey’s post-hoc test for multiple comparisons was performed. *p < 0.05, **p < 0.01, and ***p < 0.001.
GPL were also differentially modulated by the presence of humanized APOE4/4 vs. APOE3/3 ACM, including increases in multiple PA species (30:0, 32:0, 34:0, 36:1, 38:3, and 40:6) (Fig. 3f), whereas PC species (32:0, 32:1, 34:1, 34:2, and 36:3) were decreased (Fig. 3g), suggesting direct conversion of these metabolically linked lipids. Notably, longer PC species (38:4, 40:6, and 40:7) were increased in APOE4/4 vs. APOE3/3 ACM (Fig. 3g). We also observed a prominent decrease in several PCe species, spanning several degrees of acyl-chain length (30–38 C) and unsaturation (1-6 double bonds), while only PE 34:2, 36:2, and PEp 36:3 were decreased among ethanolamine-containing glycerophospholipids. Another synthetic pathway upregulated upon exposure to APOE4/4 vs. APOE3/3 ACM was PG (30:0, 32:1, 36:1, and 36:2), Acyl-PG (34:1, 34:2, 36:1, 36:2, 36:3, 36:4, 38:4, and 38:5), and BMP (36:1, 38:2, and 40:7), similar to what was observed in APOE4/4 brains, while BMP 34:1 levels were decreased (Fig. 3h). Finally, LPC levels (18:0, 20:0 and 20:1) were increased, whereas LPCe (16:0 and 18:1) and LPI (16:0, 18:1, and 20:3) were decreased. Altogether, while we confirm that the previously observed effect of APOE4 on DAG and BMP levels also occurs in ACM-treated neurons, we note an inverse effect of APOE4 between PC and PCe levels and metabolically linked PA specifically in ACM-treated neurons (Fig. 4).
Fig. 4. APOE isoform expression differentially modulates lipid profiles.
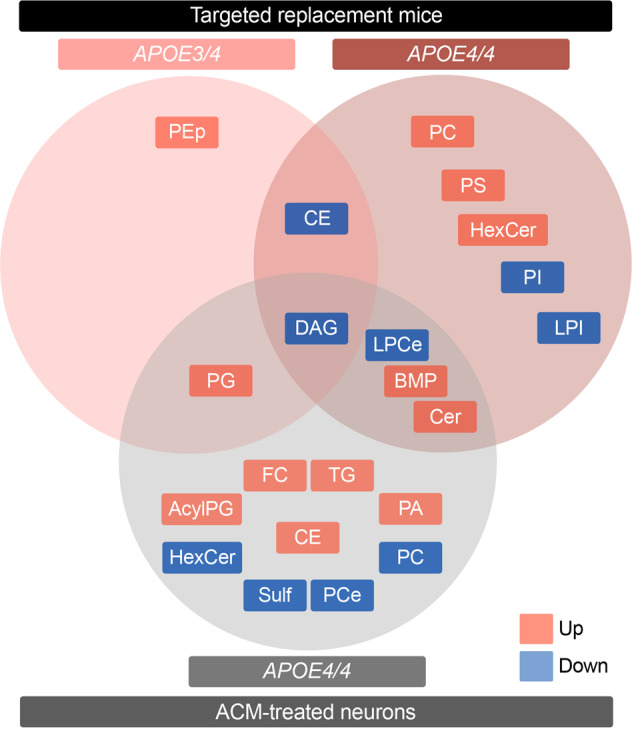
Venn diagram generated from lipid species significantly altered in the EC in APOE4/4 and APOE3/4 targeted replacement mice versus APOE3/3 mice (upper row) and in WT cortical neurons treated with APOE4/4 versus APOE3/3 ACM (lower row). Only lipid classes are represented; refer to Figs. 1–3 for specific lipid acyl-chain composition per class, respectively. Red denotes increased levels; blue denotes decreased levels.
Discussion
AD is a multi-faceted disease caused by both genetic and environmental factors. While hallmark AD pathology includes Aβ and tau accumulation, dysregulated lipid homeostasis has also been implicated in AD [42]. Furthermore, APOE4, the primary genetic risk factor for late-onset AD, is itself a regulator of lipid transport and metabolism. Our findings suggest that the EC, one of the first brain regions to develop AD pathology, presents increased susceptibility to APOE4-associated lipid alterations, as many more lipids were altered by APOE4 expression in the EC than the PVC (Fig. 1). Interestingly, this corroborates findings from our previous transcriptomic analysis of APOE3/4 vs. APOE3/3 mice, where far more genes were differentially expressed by APOE4 expression in the EC than in the PVC [33]. This differential effect may in part be related to the increased expression of APOE observed in the medial temporal lobe compared to other brain regions [49, 50]. Nonetheless, regional microstructure differences, such as cell-type distribution, fiber tract density or local enzyme expression, likely contribute as well.
Our data indicates that APOE4 expression produces a dose-dependent accumulation of several lipids associated with endosomal-lysosomal trafficking, such as HexCer and BMP, in both the brain and in ACM-treated neurons (Figs. 1, 3, 4). Given that the number of APOE4 alleles associates with increased risk for AD, we hypothesize that the additive effect on accumulation of lipids related to the endosomal-lysosomal system is a critical driver of prodromal neuronal pathology. In fact, these effects of APOE4 are reminiscent of that observed in lysosomal storage disorders [54, 55]. Not only is accumulation of BMP a hallmark of Nieman-Pick type C (NPC), it is also observed in AD specimens and upon disruption of endolysosomal trafficking, reflecting expansion of late endosome volume and cholesterol storage [48, 56, 57]. Also, homozygous mutations in GBA and GALC impair clearance of HexCer, namely gluco- and galactosylceramides, and cause Gaucher’s and Krabbe’s disease, respectively. Notably, heterozygous GBA carriers present the largest genetic risk factor for developing idiopathic Parkinson’s disease (PD) [58]. Since APOE4 also increases the risk for dementia in pure synucleinopathies and specifically exacerbates α-synuclein pathology [59, 60], we propose the disruption of lysosomal metabolism of glycosphingolipids as a common contributing factor in both forms of dementia. This association bears great therapeutical potential, as allosteric activators of glucocerebrosidase restore lysosomal function [61] and have recently been proven safe in humans [62], underscoring the possibility of extending their application to management of AD in APOE4 carriers.
In keeping with a prominent effect of APOE4 in endosomal-lysosomal trafficking, we previously showed increased expression levels of Rab GTPase genes, such as Rab5b, Rab7 and Rab9, in the EC of aged APOE4 mice [33], similar to that observed in cholinergic basal forebrain neurons in MCI and AD [63]. Importantly, Rab5 activation recapitulates prodromal features of AD, including endosomal enlargement and synaptic dysfunction [64], Rab7b granules are found in the EC of Lewy Body dementia patients [65], and altered Rab7 colocalizes with Aβ in apoE treated microglia [66]; that said, altered Rab GTPase levels are thus far correlative in APOE4 background, and their individual contributions to pathology or therapeutic potential remain elusive [67]. Also, we previously reported increased expression of several V-type ATPase subunits [33], possibly in response to altered lysosomal pH and membrane composition [68, 69]. Since neurons upregulate the secretion of exosomes to mitigate lysosomal lipid and cargo burden [56], the inhibitory effect of APOE4 expression in exosomes release [70] may also contribute to neuronal vulnerability. An association between endolysosomal defects and AD is additionally supported by recent genome wide association studies (GWAS), which identified novel loci related to endosomal-lysosomal trafficking, including WDR81, SNX1, CSTB, and GRN [71]. Thus, our lipidomic findings strengthen the association of APOE4 to endosomal-lysosomal dysfunction in contributing to increased susceptibility to AD and other forms of dementia.
Another key finding we observed was a dominant effect of APOE4 on DAG levels, which was recapitulated when exposing neurons to APOE4 vs. APOE3 ACM (Fig. 4). Interestingly, our previous transcriptomic analysis revealed decreased expression of Lpin3, Plc1, and Plcg2 in the EC of APOE3/4 vs. APOE3/3 mice [33], which could potentially contribute to decreased DAG levels via decreased dephosphorylation of PA and decreased cleavage of PI. Concurrently, we observed an increase in multiple CE and TAG species upon APOE4 ACM treatment, which may also implicate DAG consumption, as inhibition of DAG transferases rescues TAG burden in APOE4 expressing astrocytes [72]. Interestingly, lipid droplet number and volume have previously been shown to increase in neurons treated with APOE4 vs APOE3 ACM as a result of both increased lipid droplet synthesis and decreased mobilization [73, 74]. Higher cholesterol levels in neurons also likely reflect decreased secretion, which instead accumulates in distinct subcellular compartments, such as lysosomes [75, 76]. As sequestration of excessive fatty acids into lipid droplets is critical to neuroprotection [77], decreased DAG availability may reduce the capacity to buffer metabolic challenges. As such, a high reliance of neurons on incorporation of fatty acids into lipid droplets could justify the dominant effect of APOE4 on DAG levels versus other lipids. Furthermore, the previously observed decrease in the ability to mobilize lipid droplets in the presence of APOE4 ACM [74], which the authors were not able to rescue by supplementation with recombinant apoE4 protein, likely reflects a loss-of-function effect against other APOE isoforms. This loss-of-function possibility is supported by the description of CE and BMP accumulation in the forebrain of Apoe−/− mice [78], as well as increased HexCer levels in the corpus callosum (CC) upon Apoe ablation [79]. However, DAG levels are increased in the CC of Apoe−/− mice, which may reflect regional specificity (e.g., enrichment for commissural tracts) or co-existing gain-of-function effects (e.g., downstream of neuronal receptor signaling) [80]. The milder effect of APOE4 ACM on the accumulation of BMP, comparatively to APOE4 mice, may result from the interaction with endogenous neuronal murine apoE within the endolysosomal compartments.
We also observed an effect of APOE4 ACM on increasing PA and decreasing PC and PCe levels in the ACM-treated neurons (Fig. 3). PA is tightly linked to DAG metabolism via synthesis and catalysis by DAG kinase and lipin/PA phosphatase [81] and has been implicated in synaptic vesicle recycling and in neuronal plasticity mechanisms [82], which may underlie neuronal hyperactivity in aged APOE4 mice [32]. Also, PC is converted to PA by phospholipase D1 (PLD1) or PLD2 isoenzymes [83], both of which have been implicated in AD [84] and shown to signal downstream of Aβ [85, 86] and APOE [87]. Since PC can similarly be converted to LPC through the action of phospholipase A2 (PLA2), the inverse relationship between PC and LPC levels upon exposure to APOE4/4 ACM suggests upregulation of this pathway, also observed in several transgenic AD models [88, 89]. Interestingly, supplementation with choline, a precursor of PC, restores APOE4-induced lipid defects and highlights the therapeutical potential of dietary interventions targeting lipid metabolism [72]. Additionally, we observed a neuron-specific decrease in multiple PCe lipid species, which is also observed in the prefrontal cortex [45] and ventral hippocampus [44] of rodents exposed to chronic stress, a known risk factor for AD [90]. As PCe species are natural scavengers of reactive oxygen species [91], this alteration may result in decreased neuroprotection. Recently, Fitz et al. showed that APOE particles in ACM from APOE4 targeted replacement mouse primary astrocytes presented overall decreased levels of most GPL classes compared to APOE3 particles [92]. This suggests that the more complex GPL changes we observe in neurons are not simply due to differences in the amounts of lipids received from the ACM, but to lipid metabolism occurring in APOE4 vs. APOE3 ACM-treated neurons.
While our results complement previous studies on APOE biology and AD, we acknowledge the limitations of our lipidomic snapshot, taken from a single age in male APOE mice and a single APOE treatment condition in cultured WT neurons. We anticipate that additional complexity in APOE4-associated lipid signatures will likely be uncovered when investigating the effects of differing ages, sex, and brain regions in vivo, or additional cell-types or neuronal environments in vitro. Indeed, aging has been shown to be associated with blood brain barrier (BBB) breakdown in the hippocampus [93], and BBB leakage is also found to be an early marker of dysfunction in both APOE4 carriers [94] and mice [95]. Whether the lipid regional changes induced by APOE4 are a cause or a consequence of BBB breakdown is still an intriguing unanswered question. Further caution is also warranted due to the observation that lipidome complexity increases from mice to humans [96], and APOE lipid effects may diverge between mice and humans [76].
In summary, our findings demonstrate increased susceptibility of the EC to APOE4-associated lipid alterations and implicate endosomal-lysosomal lipid flux and lipid droplet regulation as potential factors in the increased risk of AD and other forms of dementia among APOE4 carriers. These findings support a role for lipid dyshomeostasis in selective vulnerability to AD pathology and highlight the therapeutic potential of lipid modulation in neurodegeneration.
Supplementary information
Author contributions
TN, TGO, GDP and KED designed and managed the study. HF conducted animal care and breeding. TN and AA performed sample preparation and RC, BZ and YX conducted the lipidomic analysis in the laboratories of GDP and EAG. AMM, TGO, LBM and TN performed bioinformatic analysis. AMM, TGO and TN wrote the manuscript with additional editing and input from RC, GDP, LBM and KED. All authors read and approved the final manuscript.
Funding
This work was supported by fellowships and career development awards to TN from NIA (AG047797 and AG061264) and the Brightfocus Foundation, as well as grants from the Alzheimer’s Association (AARG-19-619164) and Cure Alzheimer’s Fund to TN, grants from NIA and NINDS to KED (AG048408 and NS071836), and grants from NIA to LBM (AG062271, AG047954, and NS084328) and from the Alzheimer’s Association to LBM (2016-NIRG-397279). KED is also funded by Cure Alzheimer’s Fund and by the UK Dementia Research Institute, which receives its funding from DRI Ltd, funded by the UK Medical Research Council, Alzheimer’s Society and Alzheimer’s Research UK.
Data availability
The full lipidomic datasets are included as an additional Extended Data File and can also be found on the Metabolomics Workbench data repository at 10.21228/M82690.
Competing interests
GDP is a full-time employee and shareholder of Denali Therapeutics Inc. GDP and TGO are inventors on the patent WO2010138869A1 entitled “Modulation of phospholipase D for the treatment of neurodegenerative disorders”. KED serves on the board and KED and TGO are scientific advisors and shareholders of Ceracuity Inc.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Tiago Gil Oliveira, Tal Nuriel.
Contributor Information
Tiago Gil Oliveira, Email: tiago@med.uminho.pt.
Tal Nuriel, Email: tn2283@cumc.columbia.edu.
Supplementary information
The online version contains supplementary material available at 10.1038/s41398-022-01881-6.
References
- 1.Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, et al. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet. 1997;17:263–4. doi: 10.1038/ng1197-263. [DOI] [PubMed] [Google Scholar]
- 2.Castano EM, Prelli F, Wisniewski T, Golabek A, Kumar RA, Soto C, et al. Fibrillogenesis in Alzheimer’s disease of amyloid beta peptides and apolipoprotein E. Biochem J. 1995;306:599–604. doi: 10.1042/bj3060599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rebeck GW, Reiter JS, Strickland DK, Hyman BT. Apolipoprotein E in sporadic Alzheimer’s disease: allelic variation and receptor interactions. Neuron. 1993;11:575–80. doi: 10.1016/0896-6273(93)90070-8. [DOI] [PubMed] [Google Scholar]
- 4.Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:9649–53. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma J, Yee A, Brewer HB, Jr., Das S, Potter H. Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature. 1994;372:92–94. doi: 10.1038/372092a0. [DOI] [PubMed] [Google Scholar]
- 6.Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med. 2011;3:89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2000;97:2892–7. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hunsberger HC, Pinky PD, Smith W, Suppiramaniam V, Reed MN. The role of APOE4 in Alzheimer’s disease: strategies for future therapeutic interventions. Neuronal Signal. 2019;3:NS20180203. doi: 10.1042/NS20180203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Safieh M, Korczyn AD, Michaelson DM. ApoE4: an emerging therapeutic target for Alzheimer’s disease. BMC Med. 2019;17:64. doi: 10.1186/s12916-019-1299-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang Y. Abeta-independent roles of apolipoprotein E4 in the pathogenesis of Alzheimer’s disease. Trends Mol Med. 2010;16:287–94. doi: 10.1016/j.molmed.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 11.Wolf AB, Valla J, Bu G, Kim J, Ladu MJ, Reiman EM, et al. Apolipoprotein E as a beta-amyloid-independent factor in alzheimer’s disease. Alzheimers Res Ther. 2013;5:38. doi: 10.1186/alzrt204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahley RW, Rall SC., Jr Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;1:507–37. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- 13.Tudorache IF, Trusca VG, Gafencu AV. Apolipoprotein E - a multifunctional protein with implications in various pathologies as a result of its structural features. Comput Struct Biotechnol J. 2017;15:359–65. doi: 10.1016/j.csbj.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019;15:501–18. doi: 10.1038/s41582-019-0228-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamanaka H, Katoh-Fukui Y, Suzuki K, Kobayashi M, Suzuki R, Motegi Y, et al. Altered cholesterol metabolism in human apolipoprotein E4 knock-in mice. Hum Mol Genet. 2000;9:353–61. doi: 10.1093/hmg/9.3.353. [DOI] [PubMed] [Google Scholar]
- 16.Chouinard-Watkins R, Plourde M. Fatty acid metabolism in carriers of apolipoprotein E epsilon 4 allele: is it contributing to higher risk of cognitive decline and coronary heart disease? Nutrients. 2014;6:4452–71. doi: 10.3390/nu6104452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeong W, Lee H, Cho S, Seo J. ApoE4-induced cholesterol dysregulation and its brain cell type-specific implications in the pathogenesis of alzheimer’s disease. Mol Cells. 2019;42:739–46. doi: 10.14348/molcells.2019.0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han X. The role of apolipoprotein E in lipid metabolism in the central nervous system. Cell Mol Life Sci. 2004;61:1896–906. doi: 10.1007/s00018-004-4009-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dupuy AM, Mas E, Ritchie K, Descomps B, Badiou S, Cristol JP, et al. The relationship between apolipoprotein E4 and lipid metabolism is impaired in Alzheimer’s disease. Gerontology. 2001;47:213–8. doi: 10.1159/000052801. [DOI] [PubMed] [Google Scholar]
- 20.Di Paolo G, Kim TW. Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat Rev Neurosci. 2011;12:284–96. doi: 10.1038/nrn3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mapstone M, Cheema AK, Fiandaca MS, Zhong X, Mhyre TR, MacArthur LH, et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat Med. 2014;20:415–8. doi: 10.1038/nm.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van der Kant R, Langness VF, Herrera CM, Williams DA, Fong LK, Leestemaker Y, et al. Cholesterol Metabolism Is a Druggable Axis that Independently Regulates Tau and Amyloid-beta in iPSC-Derived Alzheimer’s Disease Neurons. Cell Stem Cell. 2019;24:363–75 e369. doi: 10.1016/j.stem.2018.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hutter-Paier B, Huttunen HJ, Puglielli L, Eckman CB, Kim DY, Hofmeister A, et al. The ACAT inhibitor CP-113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer’s disease. Neuron. 2004;44:227–38. doi: 10.1016/j.neuron.2004.08.043. [DOI] [PubMed] [Google Scholar]
- 24.Zhao N, Ren Y, Yamazaki Y, Qiao W, Li F, Felton LM, et al. Alzheimer’s risk factors age, APOE genotype, and sex drive distinct molecular pathways. Neuron. 2020;106:727–42 e726. doi: 10.1016/j.neuron.2020.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Foley KE, Hewes AA, Garceau DT, Kotredes KP, Carter GW, Sasner M, Howell GR. The APOEε3/ε4 genotype drives distinct gene signatures in the cortex of young mice. Front Aging Neurosci 2022;14. [DOI] [PMC free article] [PubMed]
- 26.Sharman MJ, Shui G, Fernandis AZ, Lim WL, Berger T, Hone E, et al. Profiling brain and plasma lipids in human APOE epsilon2, epsilon3, and epsilon4 knock-in mice using electrospray ionization mass spectrometry. J Alzheimers Dis. 2010;20:105–11. doi: 10.3233/JAD-2010-1348. [DOI] [PubMed] [Google Scholar]
- 27.Jenner AM, Lim WL, Ng MP, Wenk MR, Shui G, Sharman MJ, et al. The effect of APOE genotype on brain levels of oxysterols in young and old human APOE epsilon2, epsilon3 and epsilon4 knock-in mice. Neuroscience. 2010;169:109–15. doi: 10.1016/j.neuroscience.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lim WL, Lam SM, Shui G, Mondal A, Ong D, Duan X, et al. Effects of a high-fat, high-cholesterol diet on brain lipid profiles in apolipoprotein E epsilon3 and epsilon4 knock-in mice. Neurobiol aging. 2013;34:2217–24. doi: 10.1016/j.neurobiolaging.2013.03.012. [DOI] [PubMed] [Google Scholar]
- 29.Lefterov I, Wolfe CM, Fitz NF, Nam KN, Letronne F, Biedrzycki RJ, et al. APOE2 orchestrated differences in transcriptomic and lipidomic profiles of postmortem AD brain. Alzheimers Res Ther. 2019;11:113. doi: 10.1186/s13195-019-0558-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kariv-Inbal Z, Yacobson S, Berkecz R, Peter M, Janaky T, Lutjohann D, et al. The isoform-specific pathological effects of apoE4 in vivo are prevented by a fish oil (DHA) diet and are modified by cholesterol. J Alzheimers Dis. 2012;28:667–83. doi: 10.3233/JAD-2011-111265. [DOI] [PubMed] [Google Scholar]
- 31.Han X, Cheng H, Fryer JD, Fagan AM, Holtzman DM. Novel role for apolipoprotein E in the central nervous system. Modulation of sulfatide content. J Biol Chem. 2003;278:8043–51. doi: 10.1074/jbc.M212340200. [DOI] [PubMed] [Google Scholar]
- 32.Nuriel T, Angulo SL, Khan U, Ashok A, Chen Q, Figueroa HY, et al. Neuronal hyperactivity due to loss of inhibitory tone in APOE4 mice lacking Alzheimer’s disease-like pathology. Nat Commun. 2017;8:1464. doi: 10.1038/s41467-017-01444-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nuriel T, Peng KY, Ashok A, Dillman AA, Figueroa HY, Apuzzo J, et al. The endosomal-lysosomal pathway is dysregulated by APOE4 expression in vivo. Front Neurosci. 2017;11:702. doi: 10.3389/fnins.2017.00702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Area-Gomez E, Larrea D, Pera M, Agrawal RR, Guilfoyle DN, Pirhaji L, et al. APOE4 is associated with differential regional vulnerability to bioenergetic deficits in aged APOE mice. Sci Rep. 2020;10:4277. doi: 10.1038/s41598-020-61142-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sullivan PM, Mace BE, Maeda N, Schmechel DE. Marked regional differences of brain human apolipoprotein E expression in targeted replacement mice. Neuroscience. 2004;124:725–33. doi: 10.1016/j.neuroscience.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 36.Sullivan PM, Mezdour H, Aratani Y, Knouff C, Najib J, Reddick RL, et al. Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J Biol Chem. 1997;272:17972–80. doi: 10.1074/jbc.272.29.17972. [DOI] [PubMed] [Google Scholar]
- 37.Zhang X, Liu W, Zan J, Wu C, Tan W. Untargeted lipidomics reveals progression of early Alzheimer’s disease in APP/PS1 transgenic mice. Sci Rep. 2020;10:14509. doi: 10.1038/s41598-020-71510-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tajima Y, Ishikawa M, Maekawa K, Murayama M, Senoo Y, Nishimaki-Mogami T, et al. Lipidomic analysis of brain tissues and plasma in a mouse model expressing mutated human amyloid precursor protein/tau for Alzheimer’s disease. Lipids Health Dis. 2013;12:68. doi: 10.1186/1476-511X-12-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matyash V, Liebisch G, Kurzchalia TV, Shevchenko A, Schwudke D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J Lipid Res. 2008;49:1137–46. doi: 10.1194/jlr.D700041-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giavalisco P, Li Y, Matthes A, Eckhardt A, Hubberten HM, Hesse H, et al. Elemental formula annotation of polar and lipophilic metabolites using (13) C, (15) N and (34) S isotope labelling, in combination with high-resolution mass spectrometry. Plant J. 2011;68:364–76. doi: 10.1111/j.1365-313X.2011.04682.x. [DOI] [PubMed] [Google Scholar]
- 41.Morikawa M, Fryer JD, Sullivan PM, Christopher EA, Wahrle SE, DeMattos RB, et al. Production and characterization of astrocyte-derived human apolipoprotein E isoforms from immortalized astrocytes and their interactions with amyloid-beta. Neurobiol Dis. 2005;19:66–76. doi: 10.1016/j.nbd.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 42.Chan RB, Oliveira TG, Cortes EP, Honig LS, Duff KE, Small SA, et al. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J Biol Chem. 2012;287:2678–88. doi: 10.1074/jbc.M111.274142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liebisch G, Vizcaino JA, Kofeler H, Trotzmuller M, Griffiths WJ, Schmitz G, et al. Shorthand notation for lipid structures derived from mass spectrometry. J Lipid Res. 2013;54:1523–30. doi: 10.1194/jlr.M033506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miranda AM, Bravo FV, Chan RB, Sousa N, Di Paolo G, Oliveira TG. Differential lipid composition and regulation along the hippocampal longitudinal axis. Transl Psychiatry. 2019;9:144. doi: 10.1038/s41398-019-0478-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oliveira TG, Chan RB, Bravo FV, Miranda A, Silva RR, Zhou B, et al. The impact of chronic stress on the rat brain lipidome. Mol Psychiatry. 2016;21:80–8. doi: 10.1038/mp.2015.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 47.Harayama T, Riezman H. Understanding the diversity of membrane lipid composition. Nat Rev Mol Cell Biol. 2018;19:281–96. doi: 10.1038/nrm.2017.138. [DOI] [PubMed] [Google Scholar]
- 48.Gruenberg J. Life in the lumen: The multivesicular endosome. Traffic. 2020;21:76–93. doi: 10.1111/tra.12715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gryglewski G, Seiger R, James GM, Godbersen GM, Komorowski A, Unterholzner J, et al. Spatial analysis and high resolution mapping of the human whole-brain transcriptome for integrative analysis in neuroimaging. NeuroImage. 2018;176:259–67. doi: 10.1016/j.neuroimage.2018.04.068. [DOI] [PubMed] [Google Scholar]
- 50.Emrani S, Arain HA, DeMarshall C, Nuriel T. APOE4 is associated with cognitive and pathological heterogeneity in patients with Alzheimer’s disease: a systematic review. Alzheimers Res Ther. 2020;12:141. doi: 10.1186/s13195-020-00712-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eichmann TO, Lass A. DAG tales: the multiple faces of diacylglycerol-stereochemistry, metabolism, and signaling. Cell Mol Life Sci. 2015;72:3931–52. doi: 10.1007/s00018-015-1982-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hannun YA, Obeid LM. Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol. 2018;19:175–91. doi: 10.1038/nrm.2017.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Van Acker ZP, Bretou M, Annaert W. Endo-lysosomal dysregulations and late-onset Alzheimer’s disease: impact of genetic risk factors. Mol Neurodegener. 2019;14:20. doi: 10.1186/s13024-019-0323-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meikle PJ, Duplock S, Blacklock D, Whitfield PD, Macintosh G, Hopwood JJ, et al. Effect of lysosomal storage on bis(monoacylglycero)phosphate. Biochem J. 2008;411:71–78. doi: 10.1042/BJ20071043. [DOI] [PubMed] [Google Scholar]
- 55.Liu N, Tengstrand EA, Chourb L, Hsieh FY. Di-22:6-bis(monoacylglycerol)phosphate: a clinical biomarker of drug-induced phospholipidosis for drug development and safety assessment. Toxicol Appl Pharm. 2014;279:467–76. doi: 10.1016/j.taap.2014.06.014. [DOI] [PubMed] [Google Scholar]
- 56.Miranda AM, Lasiecka ZM, Xu Y, Neufeld J, Shahriar S, Simoes S, et al. Neuronal lysosomal dysfunction releases exosomes harboring APP C-terminal fragments and unique lipid signatures. Nat Commun. 2018;9:291. doi: 10.1038/s41467-017-02533-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Morel E, Chamoun Z, Lasiecka ZM, Chan RB, Williamson RL, Vetanovetz C, et al. Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat Commun. 2013;4:2250. doi: 10.1038/ncomms3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abeliovich A, Gitler AD. Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature. 2016;539:207–16. doi: 10.1038/nature20414. [DOI] [PubMed] [Google Scholar]
- 59.Tsuang D, Leverenz JB, Lopez OL, Hamilton RL, Bennett DA, Schneider JA, et al. APOE epsilon4 increases risk for dementia in pure synucleinopathies. JAMA Neurol. 2013;70:223–8. doi: 10.1001/jamaneurol.2013.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao N, Attrebi ON, Ren Y, Qiao W, Sonustun B, Martens YA, et al. APOE4 exacerbates alpha-synuclein pathology and related toxicity independent of amyloid. Sci Transl Med. 2020;12:eaay1809. [DOI] [PMC free article] [PubMed]
- 61.Mazzulli JR, Zunke F, Tsunemi T, Toker NJ, Jeon S, Burbulla LF, et al. Activation of beta-glucocerebrosidase reduces pathological alpha-synuclein and restores lysosomal function in Parkinson’s patient midbrain neurons. J Neurosci: Off J Soc Neurosci. 2016;36:7693–706. doi: 10.1523/JNEUROSCI.0628-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.den Heijer JM, Kruithof AC, van Amerongen G, de Kam ML, Thijssen E, Grievink HW, et al. A randomized single and multiple ascending dose study in healthy volunteers of LTI-291, a centrally penetrant glucocerebrosidase activator. Br J Clin Pharmacol. 2021;87:3561-3573. [DOI] [PMC free article] [PubMed]
- 63.Ginsberg SD, Mufson EJ, Alldred MJ, Counts SE, Wuu J, Nixon RA, et al. Upregulation of select rab GTPases in cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer’s disease. J Chem Neuroanat. 2011;42:102–10. doi: 10.1016/j.jchemneu.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pensalfini A, Kim S, Subbanna S, Bleiwas C, Goulbourne CN, Stavrides PH, et al. Endosomal dysfunction induced by directly overactivating Rab5 recapitulates prodromal and neurodegenerative features of alzheimer’s disease. Cell Rep. 2020;33:108420. doi: 10.1016/j.celrep.2020.108420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Higashi S, Moore DJ, Minegishi M, Kasanuki K, Fujishiro H, Kabuta T, et al. Localization of MAP1-LC3 in vulnerable neurons and Lewy bodies in brains of patients with dementia with Lewy bodies. J Neuropathol Exp Neurol. 2011;70:264–80. doi: 10.1097/NEN.0b013e318211c86a. [DOI] [PubMed] [Google Scholar]
- 66.Lee CY, Tse W, Smith JD, Landreth GE. Apolipoprotein E promotes beta-amyloid trafficking and degradation by modulating microglial cholesterol levels. J Biol Chem. 2012;287:2032–44. doi: 10.1074/jbc.M111.295451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kiral FR, Kohrs FE, Jin EJ, Hiesinger PR. Rab GTPases and membrane trafficking in neurodegeneration. Curr Biol. 2018;28:R471–86. doi: 10.1016/j.cub.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Larramona-Arcas R, Gonzalez-Arias C, Perea G, Gutierrez A, Vitorica J, Garcia-Barrera T, et al. Sex-dependent calcium hyperactivity due to lysosomal-related dysfunction in astrocytes from APOE4 versus APOE3 gene targeted replacement mice. Mol Neurodegener. 2020;15:35. doi: 10.1186/s13024-020-00382-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Prasad H, Rao R. Amyloid clearance defect in ApoE4 astrocytes is reversed by epigenetic correction of endosomal pH. Proc Natl Acad Sci USA. 2018;115:E6640–9. doi: 10.1073/pnas.1801612115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Peng KY, Perez-Gonzalez R, Alldred MJ, Goulbourne CN, Morales-Corraliza J, Saito M, et al. Apolipoprotein E4 genotype compromises brain exosome production. Brain. 2019;142:163–75. doi: 10.1093/brain/awy289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bellenguez C, Küçükali F, Jansen I, Andrade V, Moreno-Grau S, Amin N, et al. New insights on the genetic etiology of Alzheimer’s and related dementia. medRxiv 2020.
- 72.Sienski G, Narayan P, Bonner JM, Kory N, Boland S, Arczewska AA, et al. APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci Transl Med. 2021;13:eaaz4564. [DOI] [PMC free article] [PubMed]
- 73.Tambini MD, Pera M, Kanter E, Yang H, Guardia-Laguarta C, Holtzman D, et al. ApoE4 upregulates the activity of mitochondria-associated ER membranes. EMBO Rep. 2016;17:27–36. doi: 10.15252/embr.201540614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Qi G, Mi Y, Shi X, Gu H, Brinton RD, Yin F. ApoE4 impairs neuron-astrocyte coupling of fatty acid metabolism. Cell Rep. 2021;34:108572. doi: 10.1016/j.celrep.2020.108572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J, et al. APOE4 causes widespread molecular and cellular alterations associated with alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron. 2018;98:1141–54 e1147. doi: 10.1016/j.neuron.2018.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.TCW J, Liang SA, Qian L, Pipalia NH, Chao MJ, Shi Y, et al. Cholesterol and matrisome pathways dysregulated in human APOE ε4 glia. bioRxiv 2019:713362.
- 77.Walther TC, Chung J, Farese RV., Jr Lipid Droplet Biogenesis. Annu Rev Cell Dev Biol. 2017;33:491–510. doi: 10.1146/annurev-cellbio-100616-060608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, et al. TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron. 2020;105:837–54 e839. doi: 10.1016/j.neuron.2019.12.007. [DOI] [PubMed] [Google Scholar]
- 79.Fitzner D, Bader JM, Penkert H, Bergner CG, Su M, Weil MT, et al. Cell-type- and brain-region-resolved mouse brain lipidome. Cell Rep. 2020;32:108132. doi: 10.1016/j.celrep.2020.108132. [DOI] [PubMed] [Google Scholar]
- 80.Huang YA, Zhou B, Nabet AM, Wernig M, Sudhof TC. Differential signaling mediated by ApoE2, ApoE3, and ApoE4 in human neurons parallels alzheimer’s disease risk. J Neurosci: Off J Soc Neurosci. 2019;39:7408–27. doi: 10.1523/JNEUROSCI.2994-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Smulan LJ, Ding W, Freinkman E, Gujja S, Edwards YJK, Walker AK. Cholesterol-Independent SREBP-1 maturation is linked to ARF1 inactivation. Cell Rep. 2016;16:9–18. doi: 10.1016/j.celrep.2016.05.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tu-Sekine B, Goldschmidt H, Raben DM. Diacylglycerol, phosphatidic acid, and their metabolic enzymes in synaptic vesicle recycling. Adv Biol Regul. 2015;57:147–52. doi: 10.1016/j.jbior.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Santa-Marinha L, Castanho I, Silva RR, Bravo FV, Miranda AM, Meira T, et al. Phospholipase D1 Ablation Disrupts Mouse Longitudinal Hippocampal Axis Organization and Functioning. Cell Rep. 2020;30:4197–208 e4196. doi: 10.1016/j.celrep.2020.02.102. [DOI] [PubMed] [Google Scholar]
- 84.Oliveira TG, Di Paolo G. Phospholipase D in brain function and Alzheimer’s disease. Biochimica Biophysica Acta. 2010;1801:799–805. doi: 10.1016/j.bbalip.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bravo FV, Da Silva J, Chan RB, Di Paolo G, Teixeira-Castro A, Oliveira TG. Phospholipase D functional ablation has a protective effect in an Alzheimer’s disease Caenorhabditis elegans model. Sci Rep. 2018;8:3540. doi: 10.1038/s41598-018-21918-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Oliveira TG, Chan RB, Tian H, Laredo M, Shui G, Staniszewski A, et al. Phospholipase d2 ablation ameliorates Alzheimer’s disease-linked synaptic dysfunction and cognitive deficits. J Neurosci: Off J Soc Neurosci. 2010;30:16419–28. doi: 10.1523/JNEUROSCI.3317-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang H, Kulas JA, Ferris HA, Hansen SB. Regulation of beta-amyloid production in neurons by astrocyte-derived cholesterol. bioRxiv 2020: 2020.2006.2018.159632. [DOI] [PMC free article] [PubMed]
- 88.Sanchez-Mejia RO, Newman JW, Toh S, Yu GQ, Zhou Y, Halabisky B, et al. Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer’s disease. Nat Neurosci. 2008;11:1311–8. doi: 10.1038/nn.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Granger MW, Liu H, Fowler CF, Blanchard AP, Taylor MW, Sherman SPM, et al. Distinct disruptions in Land’s cycle remodeling of glycerophosphocholines in murine cortex mark symptomatic onset and progression in two Alzheimer’s disease mouse models. J Neurochemistry. 2019;149:499–517. doi: 10.1111/jnc.14560. [DOI] [PubMed] [Google Scholar]
- 90.Sotiropoulos I, Catania C, Pinto LG, Silva R, Pollerberg GE, Takashima A, et al. Stress acts cumulatively to precipitate Alzheimer’s disease-like tau pathology and cognitive deficits. J Neurosci: Off J Soc Neurosci. 2011;31:7840–7. doi: 10.1523/JNEUROSCI.0730-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dean JM, Lodhi IJ. Structural and functional roles of ether lipids. Protein Cell. 2018;9:196–206. doi: 10.1007/s13238-017-0423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fitz NF, Nam KN, Wolfe CM, Letronne F, Playso BE, Iordanova BE, et al. Phospholipids of APOE lipoproteins activate microglia in an isoform-specific manner in preclinical models of Alzheimer’s disease. Nat Commun. 2021;12:3416. doi: 10.1038/s41467-021-23762-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85:296–302. doi: 10.1016/j.neuron.2014.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Montagne A, Nation DA, Sagare AP, Barisano G, Sweeney MD, Chakhoyan A, et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature. 2020;581:71–6. doi: 10.1038/s41586-020-2247-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–6. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bozek K, Wei Y, Yan Z, Liu X, Xiong J, Sugimoto M, et al. Organization and evolution of brain lipidome revealed by large-scale analysis of human, chimpanzee, macaque, and mouse tissues. Neuron. 2015;85:695–702. doi: 10.1016/j.neuron.2015.01.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The full lipidomic datasets are included as an additional Extended Data File and can also be found on the Metabolomics Workbench data repository at 10.21228/M82690.