

SUMMARY
Neocortical circuits consist of stereotypical motifs that must self-assemble during development. Recent evidence suggests that the subtype identity of both excitatory projection neurons (PNs) and inhibitory interneurons (INs) is important for this process. We knocked out the transcription factor Satb2 in PNs to induce those of the intratelencephalic (IT) type to adopt a pyramidal tract (PT)-type identity. Loss of IT-type PNs selectively disrupted the lamination and circuit integration of INs derived from the caudal ganglionic eminence (CGE). Strikingly, reprogrammed PNs demonstrated reduced synaptic targeting of CGE-derived INs relative to controls. In control mice, IT-type PNs targeted neighboring CGE INs, while PT-type PNs did not in deep layers, confirming this lineage-dependent motif. Finally, single-cell RNA sequencing revealed that major CGE IN subtypes were conserved after loss of IT PNs, but with differential transcription of synaptic proteins and signaling molecules. Thus, IT-type PNs influence CGE-derived INs in a non-cell-autonomous manner during cortical development.
In Brief
Neocortical circuits are comprised of intermingled excitatory projection neuron and inhibitory interneuron subtypes. Wester et al. show that fate-switching corticocortical projection neurons to a subcerebral type selectively influences the lamination, circuit integration, and gene transcription of CGE-derived interneurons.
INTRODUCTION
Neocortical circuits, for all their complexity, demonstrate a stereotypical organization across regions, with many repeated motifs (Harris and Shepherd, 2015). This suggests there may be general principles that guide the self-assembly of these circuits during development. A particularly interesting problem for the developing cortex is how to integrate inhibitory interneurons (INs), which are produced by progenitors outside the telencephalon, into circuits among locally generated excitatory projection neurons(PNs) (Bartolini et al., 2013; Kepecs and Fishell, 2014; Lodato and Arlotta, 2015; Pelkey et al., 2017). Upon reaching the nascent cortex, INs must coordinate with their new host environment to establish proper positioning, wiring, and total numbers. There is accumulating evidence this process involves subtype-specific interactions between INs and neighboring PNs (Lodato et al., 2011a; Miyoshi and Fishell, 2011; Pla et al., 2006; Priya et al., 2018; Vogt et al., 2014; Ye et al., 2015). This is daunting given the large diversity among INs; however, it is possible these interactions are organized within primary classes of PNs and INs.
In the cortex, both PNs and INs can be parsed into generally non-overlapping subgroups with matched laminar distributions. PNs are classified according to their efferent targets: intratelencephalic (IT) type project to other cortical areas and the striatum, pyramidal tract (PT) type project to the thalamus, midbrain, brainstem, and spinal cord, and corticothalamic (CT) type project to ipsilateral thalamic nuclei (Harris and Shepherd, 2015). Importantly, while IT-type PNs are found throughout the cortical depth, PT-type PNs are restricted to layer 5, and CT-type PNs to layer 6. INs can also be segregated into major groups, based on their embryonic lineage from progenitors of either the medial or caudal ganglionic eminence (MGE or CGE) (Pelkey et al., 2017; Rudy et al., 2011; Tremblay et al., 2016). MGE-derived INs include fast-spiking parvalbumin (PV)-expressing basket and axo-axonic cells, and somatostatin (SST)-expressing cells, which target dendrites. Cortical CGE-derived INs are principally represented by reelin-expressing neurogliaform cells and vasoactive intestinal peptide (VIP)-expressing cells that target other INs. Similar to PNs, CGE- and MGE-derived INs are differentially biased in their laminar distributions, toward superficial layers 1–3 and deep layer 5, respectively (Miyoshi et al., 2007, 2010). However, in contrast to PNs, they are initially unsorted throughout the cortical depth and only acquire their final laminar positions over the course of the first postnatal week (Miyoshi and Fishell, 2011).
These correlations in laminar profile, coupled with the delayed sorting of INs relative to PNs, suggest preferential interactions between major classes of these cell types. Indeed, PT-type PNs appear to selectively influence the circuit integration and lamination of MGE-derived INs. In layer 5, PV+ INs preferentially target PT-type PNs, and SST+ Martinoti cells preferentially mediate disynaptic inhibition among PT-type PNs relative to IT type (Le Bé et al., 2007; Lee et al., 2014; Morishima et al., 2017; Silberberg and Markram, 2007; Wu et al., 2016). Furthermore, manipulating master regulator transcription factors to fate-switch PN subtypes has shown that converting PNs from IT-to-PT attracts PV+ axon terminals (Ye et al., 2015), while converting them from PT-to-IT disrupts the lamination of both PV+ and SST+ INs away from layer 5 (Lodato et al., 2011a). Cortical circuit motifs involving CGE-derived INs and PNs are far less well understood, and synaptic connections from local PNs to VIP INs have only recently been described (Karnani et al., 2016). However, such circuits are essential to normal function, as compromising glutamatergic synaptic input onto VIP+ INs disrupts cortical state transitions and the receptive field properties of PNs in visual cortex (Batista-Brito et al., 2017). Because CGE-derived INs are biased to superficial layers, where all PNs are of the IT type, we hypothesized that these INs may be selectively influenced by IT-type PNs during development and form preferential microcircuits with these PNs relative to PT-type. Here, we show that IT- but not PT-type PNs target CGE-derived INs of all subtypes throughout the cortical depth. Furthermore, fate-switching PNs from IT to PT identity by conditional knockout of the transcription factor Satb2 disrupts this circuit motif and results in the mislamination of CGE- but not MGE-derived IN subtypes. Finally, loss of IT-type PNs alters the transcription of several synaptic proteins and adhesion molecules in cardinal CGE-derived IN classes.
RESULTS
To study the influence of IT-type PNs on IN migration and circuit development, we used a strategy initially described by Srinivasan et al. (2012) to conditionally knockout (cKO) the transcription factor Satb2 from PNs during embryogenesis, which disrupts their subtype specification. Mice carrying a Satb2 allele with loxP sites flanking exons 4 and 5 (Leone et al., 2015; Srinivasan et al., 2012) were crossed to the Emx1IRES-Cre line (Gorski et al., 2002), which results in expression of non-functional Satb2 protein in PNs as they become postmitotic (Britanova et al., 2008). On this genetic background, we further crossed the 5HT3A-GFP mouse line, which labels INs derived primarily from the CGE, as well as a subset of neurogliaform cells from the preoptic area (Chittajallu et al., 2013; Lee et al., 2010; Niquille et al., 2018; Vucurovic et al., 2010) (Figure 1A). Because graded expression of Satb2 may influence PN identity (Harb et al., 2016; McKenna et al., 2015), control mice in this study completely lacked Cre-recombinase, loxP sites, or both. Furthermore, for consistency, all experiments were performed in primary visual cortex (V1).
Figure 1. Reprogramming IT-Type Projection Neurons by Conditional Loss of Satb2.
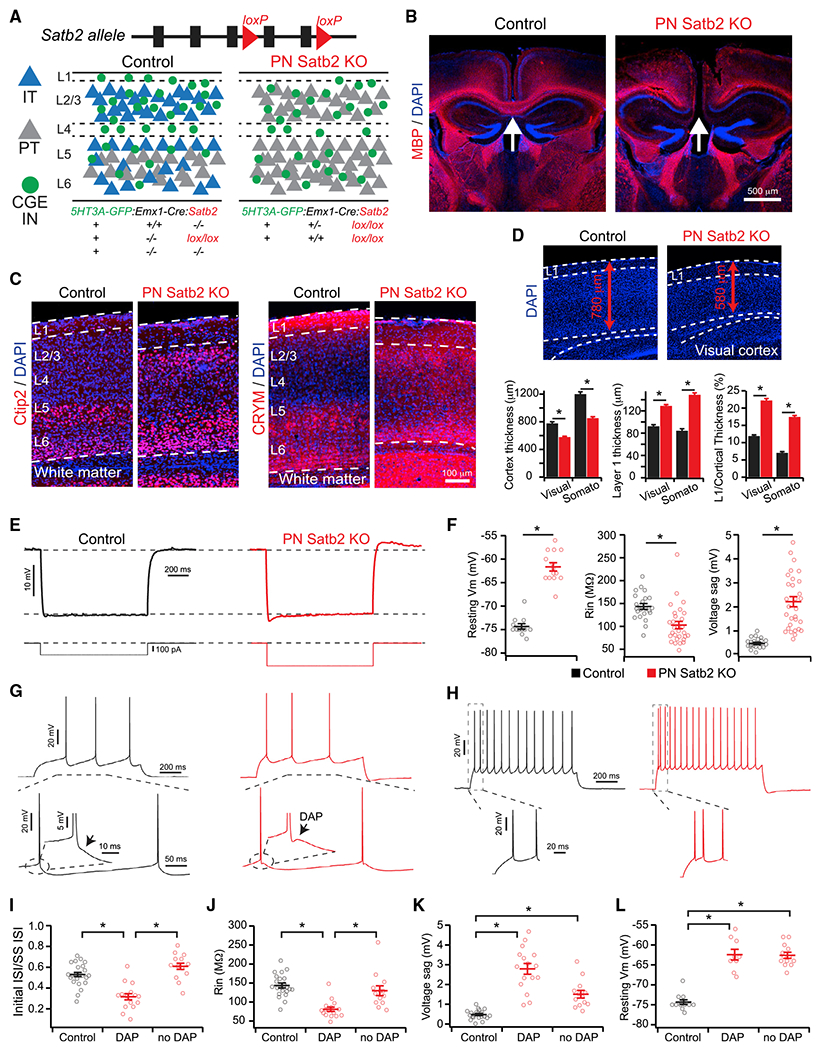
(A) Experimental strategy.
(B) Loss of callosal axons (arrows) in Satb2 cKO mice. Myelin basic protein (MBP).
(C) Ectopic superficial expression of Ctip2 and CRYM in Satb2 cKO mice.
(D) Changes in cortical and layer 1 thickness in Satb2 cKO. *p < 0.001, Wilcoxon rank tests.
(E) Responses to hyperpolarizing current pulses for control (black) and Satb2 KO (red) PNs.
(F) Intrinsic membrane properties. Resting Vm: control (n = 12 cells) versus cKO (n = 14 cells), t test. Rin: control (n = 22 cells) versus cKO (n = 29 cells), Wilcoxon rank test. Voltage sag: control (n = 22 cells) versus cKO (n = 29 cells), Wilcoxon rank test. *p < 0.001.
(G) DAPs in Satb2 cKO (red) but not control (black) PNs (arrows).
(H) Initial doublet spikes in Satb2 cKO (red) but not control (black) PNs.
(I) Doublet spikes as a function of the presence of DAP. Control (n = 22 cells) versus cKO (DAP) (n = 15 cells) versus cKO (No DAP) (n = 13 cells), *p < 0.001, Kruskal-Wallis test, Dunn-Holland-Wolfe multiple comparisons test.
(J) Input resistance as a function of the presence of DAP. Control (n = 22 cells) versus cKO (DAP) (n = 16 cells) versus cKO (No DAP) (n = 13 cells). *p < 0.001, Kruskal-Wallis test, Dunn-Holland-Wolfe multiple comparisons test.
(K) Voltage sag as a function of the presence of DAP. Control (n = 22 cells) versus cKO (DAP) (n = 16 cells) versus cKO (No DAP) (n = 13 cells). *p < 0.001, Kruskal-Wallis test, Dunn-Holland-Wolfe multiple comparisons test.
(L) Resting Vm as a function of the presence of DAP. Control (n = 12 cells) versus cKO (DAP) (n = 9 cells) versus cKO (No DAP) (n = 13 cells). *p < 0.001, Kruskal-Wallis test, Dunn-Holland-Wolfe multiple comparisons test.
Control (black), cKO (red). Error bars represent ± SEM.
As expected, loss of Satb2 disrupted IT-type PN specification, leading to loss of axon-targeting across the corpus callosum and ectopic expression of deep-layer PT-type markers in superficial layers, such as the transcription factor Ctip2 and molecular marker CRYM (Arlotta et al., 2005; Chen et al., 2008; Rouaux and Arlotta, 2013) (Figures 1B and 1C). Earlier work using a global Satb2 KO model showed reduced cortical plate thickness during late embryogenesis (Alcamo et al., 2008). In the conditional KO, reduced cortical thickness persisted into postnatal development and coincided with an increase in the thickness of layer 1 (Figure 1D). Thus, we do not attempt a one-to-one mapping of cortical layers between control and mutant mice but consider the upper half of the cortex to be superficial.
Loss of Satb2 Reprograms IT-Type Projection Neurons in a Heterogeneous Manner
In the superficial cortex, where all PNs are of the IT type under control conditions, PNs lacking Satb2 demonstrated reprogrammed electrophysiological properties characteristic of deep-layer PT-type, including lower input resistance, greater voltage sag, and depolarized resting membrane potential relative to controls (Figures 1E and 1F) (Brown and Hestrin, 2009; Dembrow et al., 2010; Hattox and Nelson, 2007; Morishima and Kawaguchi, 2006; Yamamuro et al., 2017). Interestingly, half of these mutant PNs (16/29 cells) also exhibited a depolarizing afterpotential (DAP) following action potentials and initial doublet spiking, additional PT-type characteristics (Harb et al., 2016; Hattox and Nelson, 2007; Yamamuro et al., 2017) (Figures 1G–1I). While all mutant PNs exhibited comparable changes in voltage sag and resting membrane potential, those lacking DAPs had higher input resistances, which were not different from controls (Figures 1J–1L). These data are consistent with Ye et al. (2015), who reprogrammed superficial layer PNs by overexpressing Fezf2. They observed comparable electrophysiological changes in mutant PNs but also found that some retained properties similar to controls and were considered “non-reprogrammed” (Ye et al., 2015). Together, our data suggest heterogeneity among superficial PNs regarding their regulation by Satb2, regardless of being generally of the IT type.
Loss of IT-Type Projection Neurons Selectively Disrupts the Lamination of CGE-Derived Interneurons
To investigate how loss of IT-type PNs impacts neighboring INs, we first asked whether this mutation affects their lamination, parsing INs broadly based on their embryonic origin from the MGE or CGE (Pelkey et al., 2017; Rudy et al., 2011). As shown previously, CGE INs in control mice were strongly biased toward superficial layers (Lee et al., 2010; Miyoshi et al., 2010; Vucurovic et al., 2010); however, this lamination pattern was strikingly disrupted in PN Satb2 KOs (Figures 2A and S1B). To quantify these data, we calculated a normalized position value for each cell body relative to the dorsal and white matter surfaces (Figure S1A) and plotted histograms of their laminar distributions (Figure 2B). As a population, CGE INs were redistributed in mutants from superficial to deep halves of cortex relative to controls (control versus mutant: superficial, 80% versus 63%; deep, 20% versus 37%; p < 0.01, χ2 test.) (Figures 2B and 2C). Importantly, this shift was due to loss of IT-type PNs, per se, rather than because of changes in cortical structure observed in Satb2 conditional mutants: as a separate control, we observed that CGE INs laminate normally in a mouse model of cortical malformation and disorganization, type I lissencephaly (Fleck et al., 2000) (Figure S1F).
Figure 2. Reprogramming IT-Type PNs Disrupts the Lamination of CGE-but Not MGE-Derived INs.
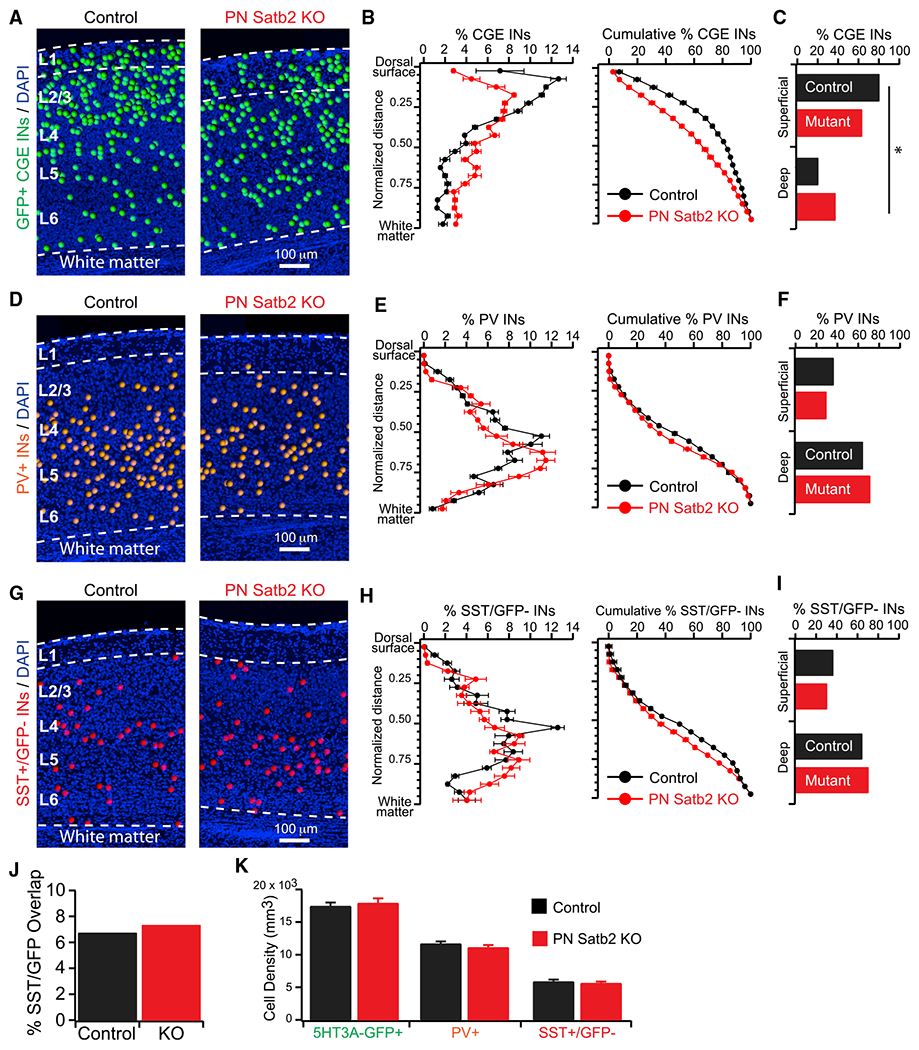
(A) CGE IN cell body positions.
(B) (Left) Percentage of CGE INs as a function of normalized cortical depth. (Right) Cumulative percentage.
(C) Total percentage of CGE INs in the top 50% (first 10 bins: Superficial) versus bottom 50% (last 10 bins: Deep) of cortex. *p < 0.01, χ2 test.
(D) PV+ IN cell body positions.
(E) (Left) Percentage of PV+ INs as a function of normalized cortical depth. (Right) Cumulative percentage.
(F) Total percentage of PV+ INs in the top 50% (Superficial) versus bottom 50% (Deep) of cortex. p = 0.29, χ2.
(G) SST+(GFP−) IN cell body positions.
(H) (Left) Percentage of SST+(GFP−) INs as a function of normalized cortical depth. (Right) Cumulative percentage.
(I) Total percentage of SST+(GFP−) INs in the top 50% (Superficial) versus bottom 50% (Deep) of cortex. p = 0.37, χ2.
(J) Overlap of SST (IHC) and 5HT3A-GFP signal. p = 0.47, χ2 test.
(K) Cell densities. 5HT3A-GFP+ INs: control (n = 23 sections) versus cKO (n = 24 sections), p = 0.62. PV+ INs: control (n = 36 sections) versus cKO (n = 33 sections), p = 0.29. SST+(GFP−) INs: control (n = 35 sections) versus cKO (n = 36 sections), p = 0.57. t tests.
Error bars represent ± SEM.
We next asked how loss of IT-type PNs affects the lamination of MGE-derived INs by performing immunohistochemistry (IHC) for PV and SST (Miyoshi et al., 2010; Nery et al., 2002; Rudy et al., 2011; Xu et al., 2004) (Figures S1C–S1E). Because a subset of 5HT3A-GFP+ INs also express SST (Chittajallu et al., 2013; Lee et al., 2010), we counted SST+/GFP− INs as representing MGE-derived SST+ cells; there was no difference in the overlap of SST and GFP between control and mutant mice (Figure 2J). Importantly, PV+ and SST+/GFP− INs maintained laminar distributions with a deep-layer bias that resembled controls after loss of IT-type PNs (Figures 2D–2I). In control mice, both PV+ and SST+/GFP− INs reached a stereotypical peak at just below 50% of the cortical depth that was absent in mutants, likely accounting for their slight shift toward white matter in the deep half of cortex (Figures 2E and 2H). However, their percentages in the superficial and deep halves of the cortex were not significantly different between control and mutant mice (Figures 2F and 2I) (control versus mutant: PV+ superficial, 36% versus 29%; deep, 64% versus 71%; p = 0.29, χ2 test. SST+/GFP− superficial, 36% versus 30%; deep, 64% versus 70%; p = 0.37, χ2 test). Our data provide an important complement to those of Lodato et al. (2011a), who reprogrammed PT-type PNs to an IT type by KO of the transcription factor Fezf2. This converse mutation resulted in the accumulation of MGE-derived PV+ and SST+ INs in superficial layers at the expense of deep. Together, our data strongly suggest that PNs influence the lamination of INs dependent on both their own projection type and the embryonic lineage of the INs.
Finally, the densities of all three IN groups were not different between mutant and control mice (Figure 2K). Thus, despite the radical change in PN subtype composition, postnatal mechanisms that regulate the final number of INs proceeded largely normally (López-Bendito et al., 2004; Priya et al., 2018; Southwell et al., 2012; Tricoire et al., 2011; Verney et al., 2000; Wong et al., 2018).
Disrupted Lamination of CGE-Derived INs Is Attributable to VIP+ but Not reelin+ Subtypes
In neocortex CGE INs can be generally segregated into two non-overlapping groups based on differential expression of VIP and reelin (lacking SST) (Wamsley and Fishell, 2017). Thus, we performed co-immunostaining for these markers to investigate effects of PN Satb2 KO on these two CGE-derived subsets. Because overlap between the 5HT3A-GFP signal and SST staining was comparably low between controls and mutants (Figure 2J), we considered reelin+/GFP+ cells to be CGE-derived (i.e., SST−).
The lamination of VIP+ INs in both controls and mutants was reflective of the general 5HT3A-GFP+ population, with a bias toward superficial layers that was disrupted in mutants: there was a significantly lower percentage of VIP+ INs in the superficial half of the cortex, and significantly higher in the deep half, relative to controls (control versus mutant: superficial, 82% versus 67%; deep, 18% versus 33%; p < 0.02, χ2 test) (Figures 3A and 3B). Furthermore, there was no difference in the density of VIP+ INs in control versus mutant mice, also consistent with our observation for 5HT3A-GFP+ INs as a group (Figure 3C). In contrast, reelin+/GFP+ INs were not redistributed to deep layers and remained concentrated in or near layer 1 (Figures 3D and 3E). The small difference between the distributions of control and mutant mice (Figure 3E) is largely attributable to the increase in layer 1 thickness in mutants (Figure 1D). There was a small but significant increase in the overall density of reelin+/GFP+ INs in mutants (Figure 3F), potentially due to the expansion of layer 1, or related to data presented in the next section. Finally, VIP and reelin expression among 5HT3A-GFP+ INs remained non-overlapping (Figures 3J and S2A–S2D); thus, the global loss of IT-type PNs did not alter the differential expression of these molecular markers.
Figure 3. Loss of IT-Type PNs Disrupts VIP+ but Not reelin+ IN Lamination and Results in Aberrant CCK+ INs.
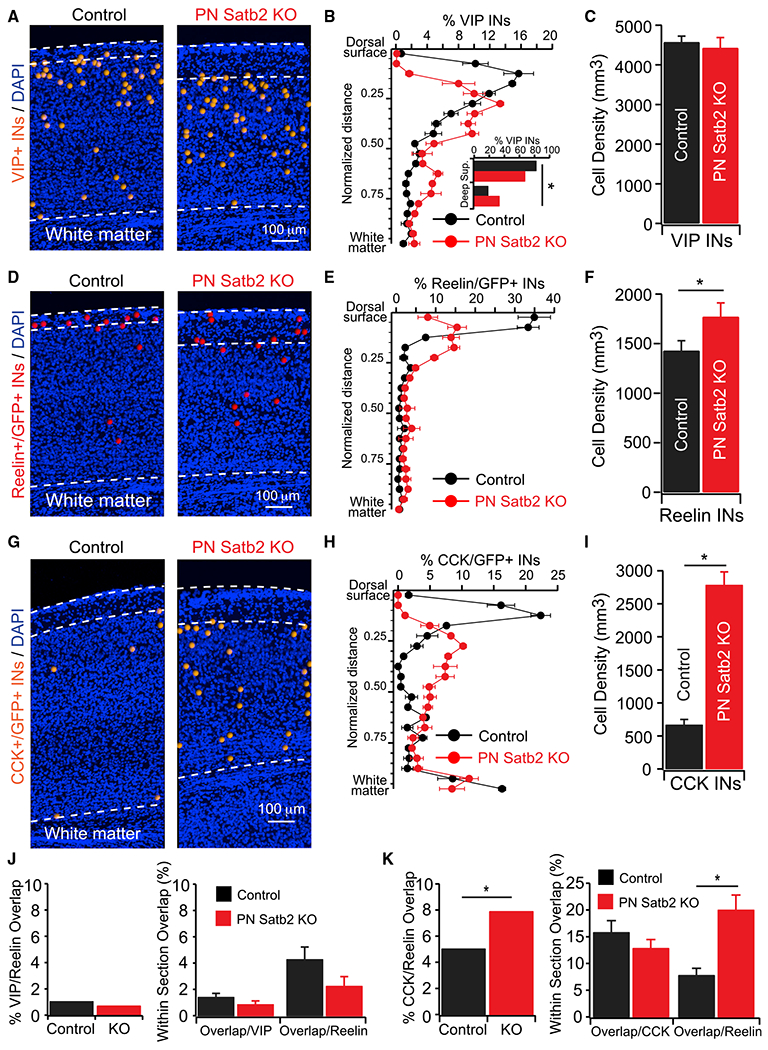
(A) VIP IN cell body positions.
(B) Percentage of VIP INs as a function of normalized cortical depth. Inset, total percentage of VIP INs in the top 50% (first 10 bins: Superficial) versus bottom 50% (last 10 bins: Deep) of cortex. *p < 0.02, χ2 test.
(C) VIP IN densities. Control (n = 35 sections) versus cKO (n = 33 sections). p = 0.7, Wilcoxon rank test.
(D) Reelin+(GFP+) IN cell body positions.
(E) Percentage of reelin+(GFP+) INs as a function of normalized cortical depth.
(F) Reelin+(GFP+) IN densities. Densities: control (n = 35 sections) versus cKO (n = 33 sections). p < 0.05, t test.
(G) CCK+(GFP+) IN cell body positions.
(H) Percentage of CCK+(GFP+) INs as a function of normalized cortical depth.
(I) CCK+(GFP+) IN densities. Densities: control (n = 35 sections) versus cKO (n = 34 sections). p < 0.001, Wilcoxon Rank test.
(J) (Left) Total overlap of VIP and reelin(GFP+). Control versus cKO: p = 0.29, χ2 test. (Right) Within-section overlap. Overlap/VIP total: control (n = 35 sections) versus cKO (n = 33 sections), p = 0.15; overlap/reelin total: control (n = 35 sections) versus cKO (n = 33 sections), p = 0.1. Wilcoxon rank tests.
(K) (Left) Total overlap of CCK(GFP+) and reelin(GFP+). Control versus cKO: *p < 0.05, χ2 test. (Right) Within-section overlap. Overlap/CCK total: control (n = 35 sections) versus cKO (n = 34 sections), p = 0.28, t test; overlap/reelin total: control (n = 35 sections) versus cKO (n = 34 sections), *p < 0.001, Wilcoxon rank test.
Error bars represent ± SEM.
An Aberrant Population of CCK+ INs Is Observed after Loss of IT-Type PNs
Although VIP+ and reelin+/SST− subsets account for a majority of neocortical CGE-derived INs (Miyoshi et al., 2010), additional markers can further specify subtypes, such as the neuropeptide cholecystokinin (CCK). In cortex, these INs include small basket and bi-tufted cells that co-express VIP, and large basket cells that do not (Gonchar et al., 2008; Karube et al., 2004; Kawaguchi and Kubota, 1997; Kubota, 2014; Kubota et al., 2011; Wang et al., 2002). In control mice, we observed a small number of CCK+/GFP+ INs primarily restricted to two bands: one in the superficial cortex and the other deep near the white matter (Figures 3G and S2E). In stark contrast, CCK+/GFP+ INs in mutants were scattered throughout the cortical depth, with a striking increase in their density relative to controls (Figures 3G–3I and S2G). Costaining for reelin revealed a significant increase in the percentage of CGE INs expressing both markers in mutants (Figures 3K, S2F, and S2H) (antibody limitations precluded VIP and CCK co-staining). The percentage of overlapping cells was significantly greater among reelin+/GFP+ INs, but not among CCK+/GFP+ INs (Figure 3K), suggesting that a subset of reelin+/GFP+ INs misexpress CCK or aberrantly survive postnatal apoptosis after loss of IT-type PNs.
Reprogrammed Projection Neurons Demonstrate Reduced Synaptic Targeting of CGE-Derived Interneurons in Superficial Layers
Despite ectopic CCK expression in some cells, basic CGE IN subtypes in the superficial cortex appeared unaltered after loss of IT-type PNs. In mutants, INs exhibited electrophysiological and morphological characteristics matching those previously described, including late spiking (LS) neurogliaform cells (putative reelin+) and non-LS subtypes that likely express VIP (Kawaguchi and Kubota, 1996; Lee et al., 2010; Miyoshi et al., 2010) (Figure 4A). Furthermore, we observed similar proportions of these cells in both controls and mutants (Figure 4B). Thus, we next asked how reprogramming PNs impacts their synaptic connectivity with these different CGE IN subtypes, using dual whole-cell recordings in the superficial cortex where all PNs are of the IT type under control conditions (Figures 4C1 and 4D1). Notably, in control mice excitatory synaptic connections from PNs to CGE INs were very common and targeted all observed subtypes (Figures 4C2, 4E, and 4F). In stark contrast, mutant PNs demonstrated significantly reduced connection probabilities with all IN subtypes relative to controls (but with similar synaptic dynamics), with a concomitant reduction in reciprocally connected pairs (Figures 4D2–4F, S3A, and S3B). Importantly, this was not due to axon retraction among PNs after loss of Satb2, which maintained local collaterals throughout cortex and a primary axon that entered the underlying white matter (Figures 4G and S4). Thus, our data strongly suggest that local excitatory synaptic input from PNs to CGE INs is a circuit motif that depends on PNs being of IT-subtype identity. This motif is expected to shape computations in the superficial cortex via interactions with well-described long-range input to CGE INs, such as cholinergic fibers from the basal forebrain (Fu et al., 2014). Indeed, we found that cholinergic modulation not only depolarized VIP INs (Kawaguchi, 1997) but enhanced the efficacy of PN-to-VIP IN synaptic connections in control mice (Figure S5).
Figure 4. Loss of IT-Type Identity Disrupts Synaptic Connectivity between PNs and CGE-Derived INs in the Superficial Cortex.
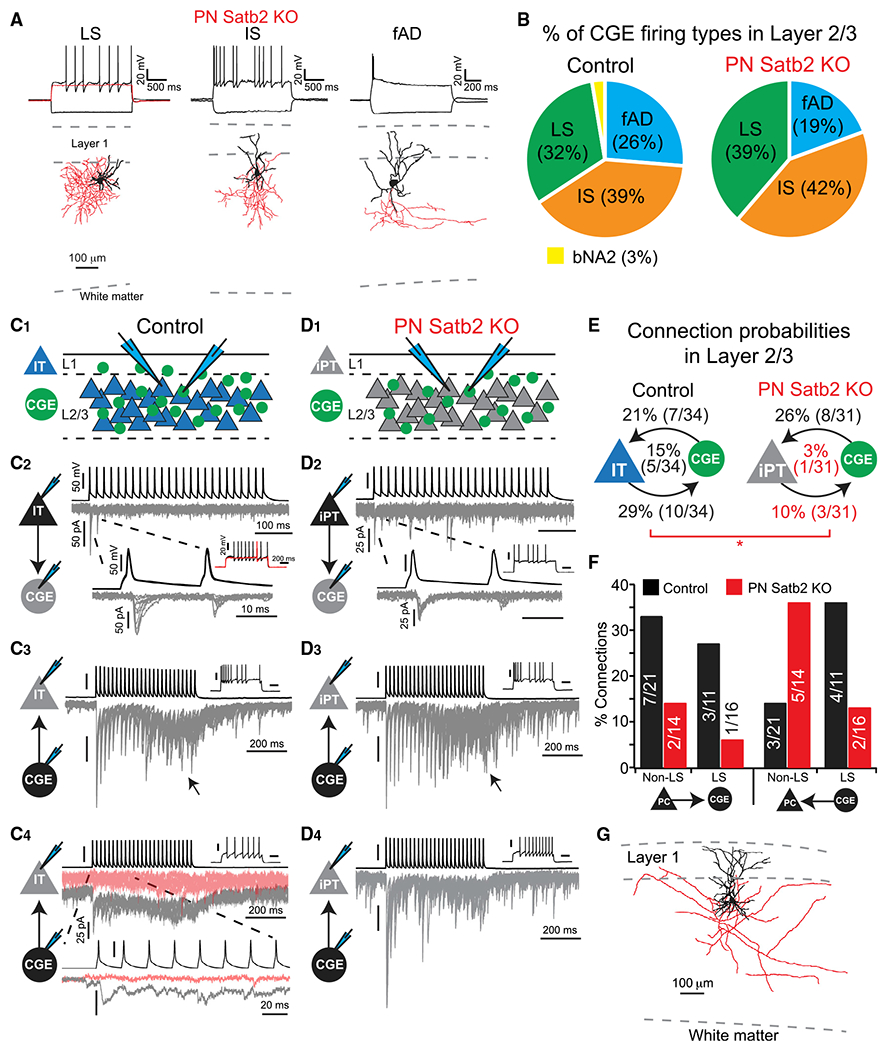
(A) CGE INs in the superficial cortex of cKO mice: late spiking (LS) neurogliaform, irregular spiking (IS) and fast adapting (fAD) bi-tufted. LS cell red trace: depolarizing Vm ramp. Dendrites and soma in black, axon in red.
(B) Percentage of IN firing types. Control (n = 38 cells) versus cKO mice (n = 31 cells). bNA2, burst nonadapting 2.
(C) Paired whole-cell recordings of synaptic connections in control mice. (1) Recording configuration. (2) PN-to-IN; postsynaptic LS IN. (3) IS-type IN-to-PN. Arrow: asynchronous transmitter release. (4) LS-type IN-to-PN synaptic connection. Gray traces: first 5 trials; pink traces: subsequent failures. Scale bars correspond to (2) unless otherwise noted. 10 overlaid trials in each.
(D) Paired whole-cell recordings of synaptic connections in Satb2 cKO mice. (1) Recording configuration. iPT = induced-PT. (2) PN-to-IN; postsynaptic IS IN. (3) IS-type IN-to-PN. Arrow: asynchronous transmitter release. (4) LS-type IN-to-PN synaptic connections lack failures after many trials (cf. Figure 4C4). Scale bars correspond to (2) unless otherwise noted. 10 overlaid trials in each.
(E) Connection probabilities (reciprocal in middle). Satb2 cKO PNs denoted iPT. *p < 0.05, χ2 test.
(F) Connection probabilities by IN firing type.
(G) Reconstructed Satb2 KO PN. Dendrites and soma in black, axon in red.
Among inhibitory connections, we observed synaptic input from both LS and non-LS INs to PNs in both control and mutant mice (Figures 4C and 4D). However, while the overall connectivity rate from INs to PNs was not different in mutants (Figure 4E), we observed fewer LS inputs with a concomitant increase in non-LS connections (Figure 4F). Furthermore, while synaptic connections from LS INs faded over multiple trials in controls, this was not observed in mutants (Figures 4C4 and 4D4). Thus, loss of IT-type identity generally disrupts the circuit integration of PNs among neighboring CGE-derived INs in the superficial cortex.
Selective Targeting of CGE-Derived INs by IT-Type Projection Neurons Is a General Circuit Motif in Visual Cortex
To directly test the hypothesis that IT- but not PT-type PNs selectively target CGE-derived INs, we performed experiments in deep cortical layers of control mice, where these PN subtypes are intermingled. In 5HT3A-GFP mice, we injected red retrograde tracer into the contralateral visual cortex or ipsilateral superior colliculus to identify IT- and PT-type PNs, respectively, and then performed dual whole-cell recordings between GFP+ INs and identified PN subtypes to test for synaptic connections (Figure 5A). As in the superficial cortex, we observed LS neurogliaform cells and heterogeneous non-LS subtypes in deep layers (Figures S6A and S6B). Strikingly, deep IT-type PNs targeted both with equal probability, while no connections were observed from PT-type PNs (Figures 5B and 5C). These synapses mostly depressed regardless of postsynaptic IN type, but with greater variably than in superficial layers (Figures S3C–S3F). Interestingly, non-LS (putative VIP+) INs could be further parsed based on their subthreshold membrane properties: those with linear current-voltage relationships were targeted by IT-type PNs at a significantly higher rate than those with inward rectification (Figures 5D and S6D–S6H). Thus, IT-type PNs further refine their synaptic targeting to a specific subset of VIP+ INs in deep layers. Finally, among inhibitory connections, input from LS INs to IT-type PNs was the most common (like in superficial layers), exhibiting asynchronous transmitter release that faded after multiple trials (Figures 5C and S6C).
Figure 5. IT-Type but Not PT-Type PNs Target CGE-Derived INs in Deep Cortical Layers.
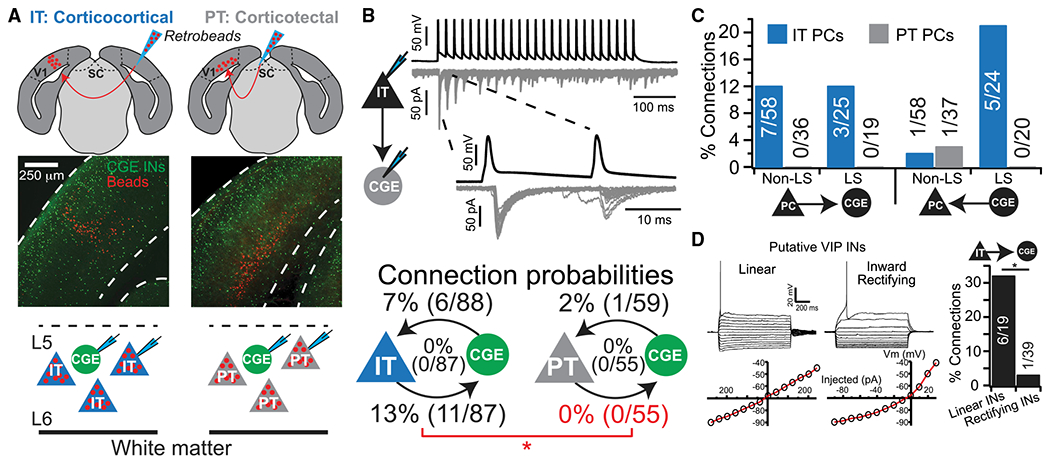
(A) Experimental design. SC, superior colliculus.
(B) IT PN to CGE IN synaptic connection and connection probabilities (reciprocal in middle). *p < 0.01, χ2 test.
(C) Connection probabilities by IN firing type.
(D) Putative VIP+ INs with a linear a current-voltage relationship receive synaptic connections at a significantly higher rate than those with inward rectification. *p < 0.01, χ2 test.
These data support our hypothesis that IT-type PNs, relative to PT-type, selectively target CGE-derived INs. However, deep-layer PNs project horizontally over long distances, raising the possibility these INs do receive input from PT-type PNs, but not locally in our paired recordings (Larsen et al., 2008; Oberlaender et al., 2011; Wester and Contreras, 2012). Thus, we used optogenetics to stimulate populations of IT- or PT-type PNs across our slice preparation. PT-type PNs were targeted by retrograde infection with G-deleted rabies encoding ChR2 and mCherry (Osakada et al., 2011), with which we could evoke spikes using short duration, low-power light stimuli (1 ms, 3.4 mW) (Figure 6A and 6B). However, excitatory postsynaptic currents (EPSCs) in neighboring GFP+ INs were rare and weak, and only observed with higher power and longer duration stimuli (11–26 mW power, 5– 10 ms duration; 4/14 INs with 8.5 ± 2.5 pA EPSCs; Figures 6B and 6C). To selectively target populations of deep layer IT-type PNs, we injected an adeno-assoicated virus (AAV) encoding Cre-dependent channelrhodopsin (Chrimson) and tdTomato into the cortex of 5HT3A-GFP:Tlx3(PL56)-Cre mice, a layer 5 IT-type PN driver line (Gerfen et al., 2013; Kim et al., 2015; Klapoetke et al., 2014) (Figure 6D). Low-power stimuli (0.6 mW) reliably evoked EPSCs in GFP+ INs (8/8 INs), which grew in amplitude with increasing light intensity (Figure 6E). Thus, PT-type PNs rarely target CGE-derived INs in deep layers, while IT-type PNs integrate these cells into the local recurrent excitatory network.
Figure 6. Population Input from PNs to CGE INs.
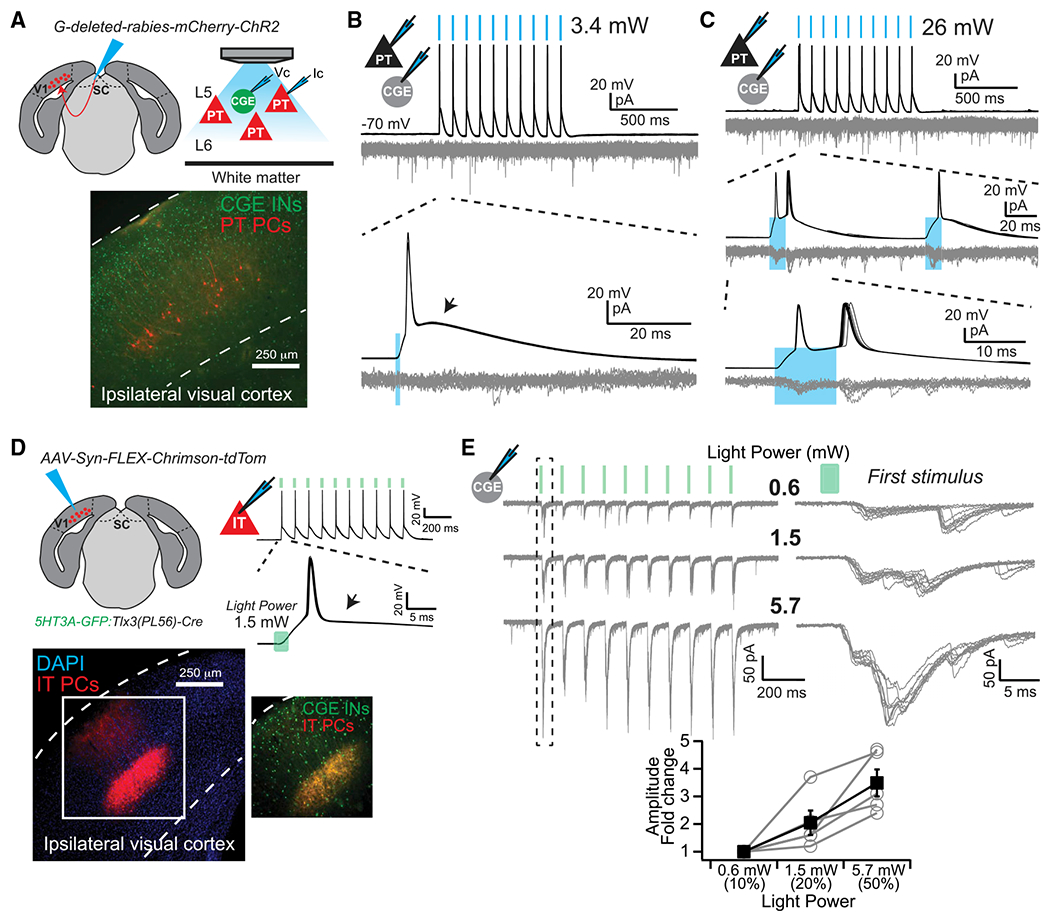
(A) Experimental design. Vc, voltage clamp; Ic, current clamp.
(B) Light stimuli (blue bars) evoked action potentials in infected PT PNs, without postsynaptic responses in INs. Depolarizing afterpotential (arrow). 10 overlaid trials.
(C) Long-duration (10 ms), high-power (26 mW) light stimuli (blue bars) were required to evoke rare, low amplitude postsynaptic responses in 5HT3A-GFP+ INs. Note doublet spiking in PT PN. 10 overlaid trials.
(D) Experimental design and example of 1.5 mW, 2 ms light stimuli evoked spikes. Lack of a depolarizing afterpotential (arrow). 10 overlaid trials.
(E) Amplitude of postsynaptic responses in CGE INs increase as a function of light power. 10 overlaid trials. Plotted are amplitudes of the first postsynaptic response evoked during a stimulus train, normalized to the lowest intensity (0.6 mW) (n = 5 cells). Error bars represent ± SEM.
Major CGE IN Subtypes Are Conserved after Loss of IT-Type PNs but Demonstrate Non-Cell-Autonomous Changes in Gene Expression Related to Synapses
To better understand how loss of IT-type PNs impacts the molecular identity and postnatal development of CGE INs, we investigated their transcriptomic profiles in control versus mutant mice. We microdissected V1 from juvenile mice (~3 weeks), dissociated the cells, and performed single-cell RNA sequencing on fluorescence-sorted GFP+ INs (Figures 7A, S7A, and S7B). We collected comparable numbers of sequenced CGE INs from both controls and mutants (~5,500 from each; Table S1) and used the R toolkit Seurat to integrate and align the datasets for differential gene expression analysis (Butler et al., 2018) (Figures 7B and S7C–S7H). Values for differential expression explicitly mentioned in the text are provided in Table S2; the entire analysis is available in Table S3.
Figure 7. Single-Cell RNA Sequencing of CGE INs after Loss of IT-Type PNs.
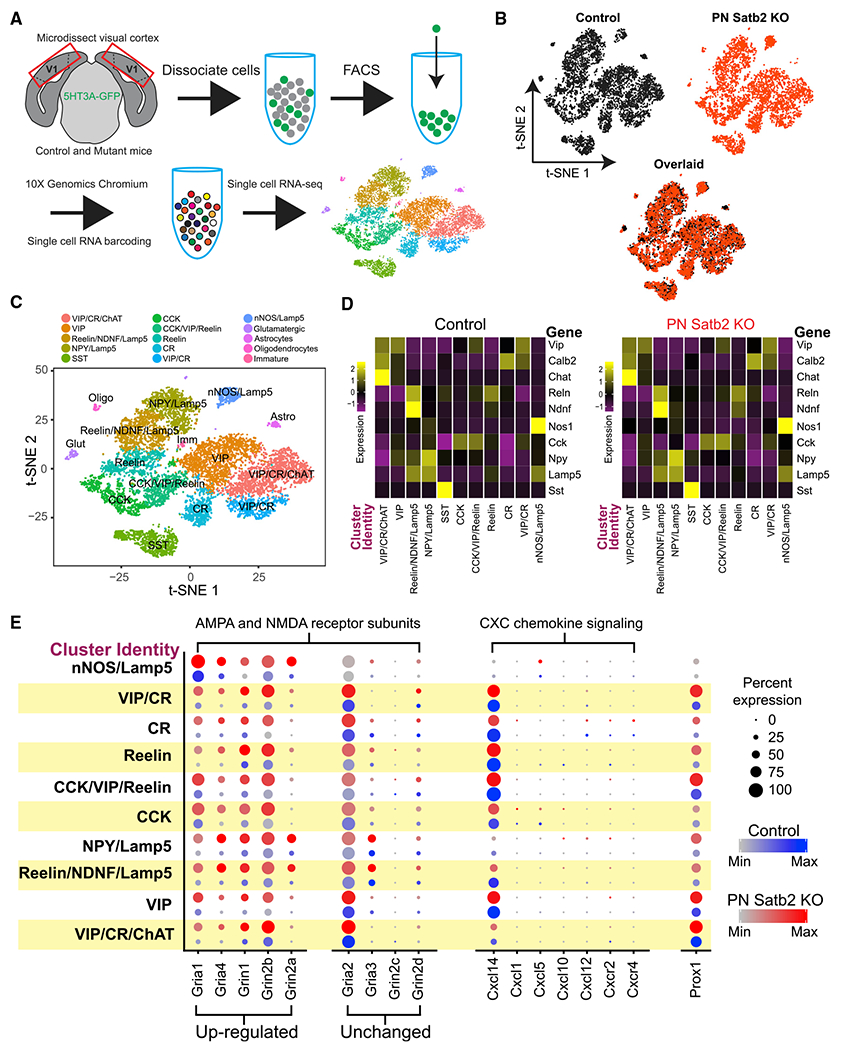
(A) Experimental design.
(B) t-distributed stochastic neighbor embedding (t-SNE) plots of CGE INs from control and cKO mice after integration and alignment of transcriptomic datasets.
(C) t-SNE plot (overlaid control and cKO datasets) with color-coded clusters identified by unbiased analysis. IN cluster names were manually curated based on prevalence of major molecular markers. Small contaminant clusters also identified (Glutamatergic, Astrocytes, Oligodendrocytes, Immature).
(D) Heatmaps of transcription for major molecular markers across IN clusters. Colors represent log normalized average expression values relative to each gene across clusters.
(E) Differential expression of selected genes across IN clusters from control and cKO mice. Colors are average expression values within a cluster; dot size is percentage of cells within a cluster expressing the gene.
Within the aligned datasets, we performed unbiased clustering, which segregated populations of INs (and a small number of clusters containing other cell types) that we labeled according to expression of previously described unique molecular markers and transcripts (Cahoy et al., 2008; Tasic et al., 2018; Tremblay et al., 2016; Wamsley and Fishell, 2017) (Figures 7C and S8). Importantly, major CGE IN subtypes identified by these combinatorial markers were preserved in the mutant mice (Figures 7C, 7D, and S8). Thus, while a small number of cells ectopically expressed (or retained) CCK and reelin in mutants (Figures 3G–3I), the molecular identity (by transcription) of cardinal CGE interneuron subtypes was undisturbed. This is congruent with our IHC data that VIP and reelin remained non-overlapping (Figure 3J) and our observation of comparable IN types identified by electrophysiology and morphology in controls and mutants (Figures 4A and 4B). We note that a single cluster (calretinin [CR]) revealed significant upregulation of Cck and Reln transcription (by percentage of expressing cells) and may include the aberrant CCK+ population.
Within the clusters of conserved CGE IN subtypes, we observed important changes in transcription after loss of IT-type PNs. Strikingly, despite observing lower PN-to-IN connection probabilities from paired recordings in mutants (Figure 4), transcripts for multiple glutamatergic AMPA and NMDA receptor subunits were upregulated in IN clusters, and none were reduced (Figure 7E). We also found upregulation of the transcription factor Prox1 and the tyrosine kinase receptor ErbB4, both of which were previously described as important for promoting excitatory synaptic input onto VIP INs (Batista-Brito et al., 2017; Miyoshi et al., 2015) (Figures 7E and 8C). Indeed, collectively, many transcripts for synaptic proteins and adhesion molecules were upregulated (Figure 8), suggesting that CGE INs may be attempting to compensate for a lack of expected excitatory presynaptic input from mutant PNs.
Figure 8. CGE INs Upregulate Many Genes Relevant to Migration and Synaptic Connectivity in Mice Lacking IT-Type PNs.
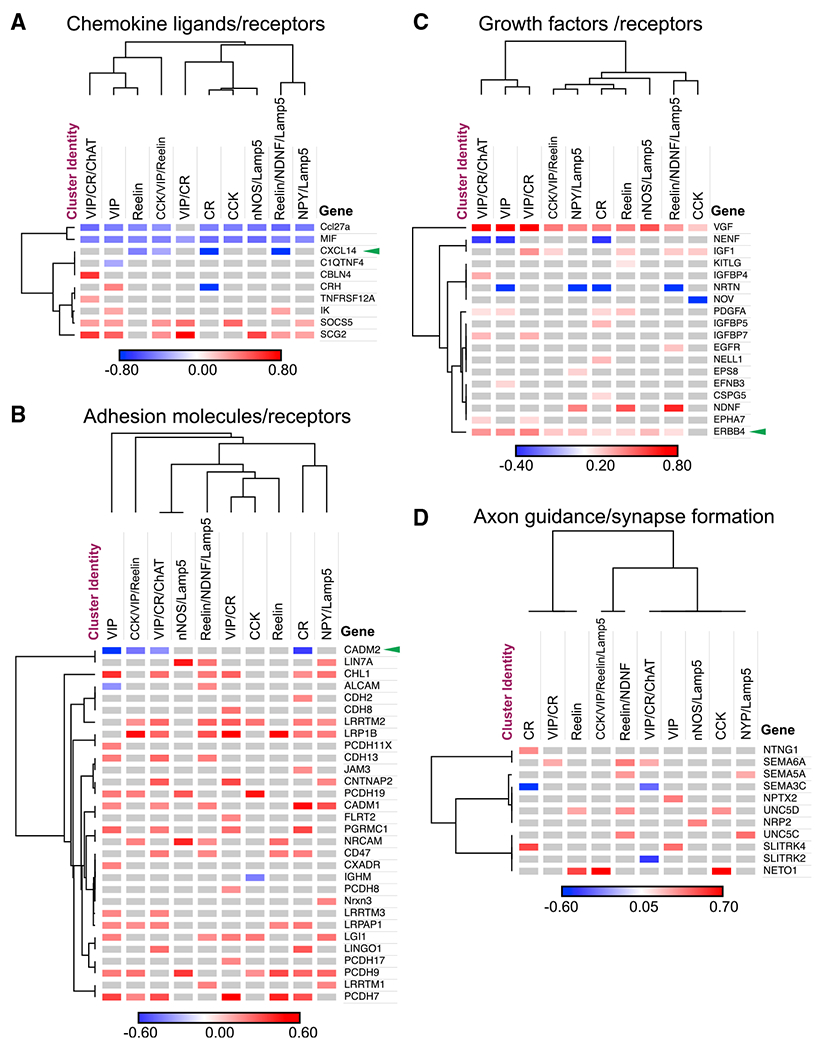
Selected genes with significant differential expression after loss of IT-type PNs in manually curated categories: (A) chemokine ligands and receptors, (B) adhesion molecules and receptors, (C) growth factors, and (D) molecules involved in axon guidance and synapse formation. Green arrowheads indicate genes mentioned in the text. Genes and clusters sorted by Euclidean distance. Colors denote log fold change of average expression within each cluster.
To identify a candidate mechanism underlying reduced synaptic targeting of CGE INs by PNs in mutants, we focused on C-X-C motif chemokines, as previous work identified this family of signaling molecules as a mediating cell-type-specific circuit motifs between PT-type PNs and PV+ INs (Wu et al., 2016). Furthermore, Cxcl14 was of special interest due to its selective transcription in CGE-but not MGE-derived interneuron subtypes (Tasic et al., 2016). Interestingly, while we found high Cxcl14 transcription across most of our IN clusters (including both VIP and reelin subtypes), transcription of other CXC ligands and receptors was uniformly low or undetected (Figure 7E). Importantly, Cxcl14 was significantly downregulated in mutants within IN clusters identified primarily by reelin transcription, but unchanged within IN clusters identified by Vip (Figure 8A). However, within Vip IN clusters we found selective downregulation of Cadm2 (Figure 8B), which encodes the adhesion protein SynCAM 2, important for the attraction of functional excitatory presynaptic terminals (Fogel et al., 2007). Thus, after loss of IT-type PNs, cardinal CGE IN classes remained undisturbed but exhibited differential changes in transcription for chemokine signaling and transsynaptic adhesion molecules.
DISCUSSION
Across cortical regions, common organizing principles among microcircuits are beginning to emerge, providing a framework for investigating their function (Harris and Shepherd, 2015; Kepecs and Fishell, 2014; Wester and McBain, 2014). Across cortical areas, excitatory PNs form preferential synaptic connections according to their projection class: IT- and PT-type form recurrent connections within their class, however, IT PNs provide output to PT PNs that is largely unreciprocated (Brown and Hestrin, 2009; Kiritani et al., 2012; Morishima and Kawaguchi, 2006; Morishima et al., 2011). Recent work has shown PN connectivity among different IN subtypes is also class dependent. In deep cortical layers, PT-type PNs, relative to IT type, preferentially target SST+ INs and are preferentially targeted by PV+ INs, both of which are derived from the MGE during embryonic development (Lee et al., 2014; Morishima et al., 2017; Silberberg and Markram, 2007; Wu et al., 2016; Ye et al., 2015). Here, we show that IT-type PNs, relative to PT type, preferentially target IN subtypes derived from the CGE. Furthermore, loss of IT-type identity among PNs disrupts this circuit motif and the lamination of CGE-derived INs. This complements previous work demonstrating that PT-type PNs influence the lamination and synaptic targeting of MGE-derived PV+ and SST+ INs (Lodato et al., 2011a; Wu et al., 2016; Ye et al., 2015). Collectively, these data suggest that many circuit motifs, and the rules for their self-assembly, may be organized within primary classes of PNs and INs, dependent on their projection type and embryonic lineage, respectively.
PN class is largely determined by interactions among the transcription factors Satb2, Fezf2, and Ctip2 during embryonic development (Leone et al., 2008). Fezf2 suppresses Satb2 while promoting Ctip2 expression, which in turn directs PN axons toward subcerebral targets rather than across the corpus callosum (Chen et al., 2005; Chen et al., 2008; Molyneaux et al., 2005). Correspondingly, loss of Fezf2 causes PNs to acquire electrophysiological, morphological, and axonal projections characteristic of IT-type PNs (Chen et al., 2008; Lodato et al., 2011a). Furthermore, overexpression of Fezf2 or Ctip2 in superficial layers causes IT-type PNs to redirect their axons toward the thalamus, midbrain, and brainstem, and to acquire PT-type intrinsic membrane properties (Chen et al., 2008; Ye et al., 2015). In turn, Satb2 suppresses Ctip2, promotes the expression of IT-type genes and is necessary for axon targeting across the corpus callosum (Alcamo et al., 2008; Britanova et al., 2008; McKenna et al., 2015). However, it is becoming clear that the interactions among these transcription factors can be combinatorial, dependent on the timing and levels of expression, and influenced by other proteins yet to be identified. Indeed, transient, embryonic expression of Satb2 promotes Fezf2 expression among future PT-type PNs and appears necessary for extension of their axons past the cerebral peduncle (Leone et al., 2015; McKenna et al., 2015). However, it is unclear why Satb2 fails to activate Fezf2 expression among future IT-type PNs. Furthermore, a subset of deep-layer PNs coexpress Satb2 and Ctip2 late into postnatal development; however, these PNs remain segregated as IT or PT projecting (Harb et al., 2016). Our data show that loss of Satb2 in superficial layers causes variable reprogramming of PN intrinsic membrane properties. Thus, the role of Satb2 in specifying IT-type characteristics is not uniform across PNs of this class. It is possible that Satb2 differentially instructs the development of IT PNs according to their projection targets within the cortex, or according their location within the superficial layers (Molyneaux et al., 2009).
Major IN classes are also defined by embryonic transcriptional programs, which are unique between MGE and CGE progenitors, and evolve temporally during development (Mayer et al., 2018; Mi et al., 2018). Thus, as INs invade the cortex, their subtype identities are largely predetermined as they seek to establish their proper positions and synaptic connections (Mayer et al., 2018; Mi et al., 2018). Indeed, here we show that cardinal CGE-derived IN classes defined by morphology, electrophysiology, and molecular expression are largely undisturbed by loss of a major subset of PNs. However, our data contribute to accumulating evidence that as these IN subtypes invade the developing cortex they differentially engage with local PNs to establish laminar positions and local circuit motifs (Bartolini et al., 2013; Krook-Magnuson et al., 2012; Lodato and Arlotta, 2015; Pelkey et al., 2017). Recent work has begun to identify potential molecular mechanisms mediating such interactions. For example, PNs express Neuregulin 3, which acts as a chemoattractant to influence the lamination of MGE-derived, PV+ INs (Bartolini et al., 2017). Furthermore, PT-type PNs preferentially express the cytokine CXCL12, which instructs the synaptic targeting of PV+ INs, and may also influence their lamination (Vogt et al., 2014; Wu et al., 2016). Among CGE-derived INs, we identified CXCL14 and SynCAM 2 as potential candidates involved in selective IT-type PN targeting of reelin+ and VIP+ INs, respectively, due to selective downregulation in mutant mice. However, we also observed upregulation of AMPA and NMDA receptor subunits, despite observing lower probabilities of synaptic connections from neighboring PNs in mutants. Furthermore, previous work found that KO of Prox1 resulted in VIP+ INs receiving fewer excitatory inputs and being mislaminated from superficial to deep layers (Miyoshi et al., 2015); however, we observed upregulation of Prox1 in mutants. This suggests that CGE-derived INs activated compensatory mechanisms after loss of expected IT-type PN input in Satb2 KO mice. Thus, our observation of selective upregulation of specific cadherins, protocadherins, and other synaptic adhesion proteins may implicate them as alternative candidates for future studies.
Among cortical CGE-derived INs, circuit motifs involving VIP+ cells in superficial layers are among the best studied. In particular, VIP INs receive long-range cholinergic input during locomotion and principally target neighboring SST+ INs, leading to a hypothesized role of PN disinhibition during active behavior (Kepecs and Fishell, 2014). However, SST-to-VIP IN connections are also common and neighboring PNs target both VIP and SST INs, but with distinct synaptic dynamics (depressing PN-to-VIP and facilitating PN-to-SST connections) (Karnani et al., 2016; Pfeffer et al., 2013), likely leading to more complicated circuit interactions (Dipoppa et al., 2018). Recently, it was shown that under cholinergic modulation excitatory PN-to-SST IN connections remain facilitating but are potentiated (Urban-Ciecko et al., 2018). We found that cholinergic modulation also facilitates PN-to-VIP excitatory synaptic transmission for the first presynaptic spike in a train. Thus, during active behaviors, the synaptic dynamics of PN-to-VIP and PN-to-SST IN connections should remain matched and potentiated, which in turn would be expected to influence the reciprocal inhibition of these IN subtypes, and circuit function (Figure S6).
We found that conditional loss of Satb2 among PNs across the cortex did not affect the overall densities of MGE- or CGE-derived INs. This was surprising, given that the composition of PN classes was dramatically altered and that the structure of the cortex was changed, with a reduction in total thickness and expansion of layer 1. Both MGE- and CGE-derived INs undergo programmed cell death during the first two postnatal weeks to determine their final numbers (Denaxa et al., 2018; Priya et al., 2018; Southwell et al., 2012). Recent work has shown that neuronal activity, evoked either cell-autonomously or by neighboring PNs, engages signaling mechanisms that promote IN survival (Denaxa et al., 2018; Priya et al., 2018; Wong et al., 2018). Thus, in PN Satb2 KO mice, homeostatic plasticity mechanisms might scale the excitability of the cortical network, with a concomitant balancing of IN numbers (Turrigiano, 2008). However, VIP+ INs are notably exempt from this activity-dependent survival mechanism (Priya et al., 2018). Thus, although we observed reduced synaptic targeting of all CGE-derived IN subtypes in our mutant mice, this cannot explain the scaling of VIP IN numbers. There is also evidence that loss of MGE-derived INs can lead to a compensatory increase in the number of CGE-derived INs, and vice versa, maintaining a constant overall number of INs in the cortex (Denaxa et al., 2018; Lodato et al., 2011b). However, while genetic ablation of VIP+ INs leads to an increase in the survival of PV+ INs (Lodato et al., 2011b), ablation of MGE-derived subtypes leads to an increase in CGE-derived reelin+ INs but not VIP+ cells (Denaxa et al., 2018). Thus, although VIP+ IN numbers are clearly regulated along with other subtypes after loss of IT-type PNs, the mechanisms are far from clear.
Finally, multiple psychiatric disorders are associated with disruptions of IN circuit function (Marín, 2012). However, as PNs importantly influence IN maturation and circuit integration, it is necessary to consider their potential interactions when investigating causes and therapies for these diseases. For example, autism spectrum disorder is associated with thinning of the corpus callosum (Frazier and Hardan, 2009), suggesting disruptions among IT-type PNs. Indeed, Satb2 is a candidate gene potentially disrupted by chromosomal abnormalities leading to autism (Marshall et al., 2008). Our data suggest that disturbance of Satb2 function may be associated with aberrant circuit integration of VIP+ and reelin+ INs. Such considerations are also important in context of recent advances in cell transplantation-based therapies, which aim to introduce class-specific PN and IN precursors into diseased host tissue to induce neuronal repair (Southwell et al., 2014; Wuttke et al., 2018). A clearer understanding of how specific PN and IN subtypes self-assemble will allow for proper assays of these techniques and guide more precise therapeutic targets.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Chris McBain (mcbainc@mail.nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All experiments were conducted in accordance with animal protocols approved by the National Institutes of Health. Both male and female mice were used without bias. Emx1IRES-Cre mice were obtained from The Jackson Laboratory (stock no. 005628). Satb2lox/lox mice (Srinivasan et al., 2012) were obtained from the lab of Susan McConnell (Stanford University) with the permission of Rudolf Grosschedl (Max Planck Institute) and backcrossed at least 4 generations to a C57BL/6 background. Tlx3(PL56)-Cre mice were generated by the GENSAT project (Gerfen et al., 2013) and obtained from the Janiela Research Campus (Ashburn, Virginia). Emx1IRES-Cre: Satb2lox/lox mice and Tlx3(PL56)-Cre mice were further crossed to the 5HT3A-GFP mouse line (Chittajallu et al., 2013). Lis1lox/lox mice were obtained from the lab of Anthony Wynshaw-Boris (Case Western Reserve University) (Gambello et al., 2003), and crossed to female Sox2-Cre mice (Jax: 008454) to generate Lis1 germline mutants. Lis1 mutants were further crossed to the 5HT3A-GFP line.
For IN lamination experiments, we primarily used littermates as controls, however, this was occasionally not possible due to our criteria that control mice completely lack Cre-recombinase or floxed Satb2. Paired whole-cell recordings in control mice in were performed in 5HT3A-GFP mice. All experiments were performed in primary visual cortex (V1).
METHOD DETAILS
Slice preparation
Juvenile mice (p15 – 23; p21 – 44 for optogenetics) were used. All mice were anesthetized with isoflurane and then decapitated. The brain was dissected out in ice-cold artificial cerebrospinal fluid (ACSF) containing (in mM): 100 sucrose, 80 NaCl, 3.5 KCl, 24 NaHCO3, 1.25 NaH2PO4, 4.5 MgCl, 0.5 CaCl2, and 10 glucose, saturated with 95% O2 and 5% CO2. Coronal slices of visual cortex (300 mm) were cut using a VT-1000S vibratome (Leica Microsystems) and incubated in the above solution at 35°C for 30 – 45 minutes. Slices were maintained at room temperature until use in recording ACSF consisting of (in mM): 130 NaCl, 3.5 KCl, 24 NaHCO3, 1.25 NaH2PO4, 1.5 MgCl, 2.5 CaCl2, and 10 glucose, saturated with 95% O2 and 5% CO2.
Electrophysiology
For recording, slices were transferred to an upright microscope (Zeiss Axioskop 2), perfused at 2 mL/min, and maintained at a temperature of 32 – 34°C. Electrodes were pulled from borosilicate glass (World Precision Instruments) to a resistance of 3 – 5 MΩ using a vertical pipette puller (Narishige, PP-830). Whole-cell patch-clamp recordings were made using a Multiclamp 700A amplifier (Molecular Devices), filtered at 3 kHz (Bessel filter) and digitized at 20 kHz (Digidata 1322A or 1550 and pClamp 9.2 or 10.6 software; Molecular Devices). Recordings were not corrected for a liquid junction potential. Uncompensated series resistance during recordings ranged from 10 – 25 MΩ and was monitored continuously throughout with −5 mV voltage steps. In current-clamp mode, cells were biased to a membrane potential (Vm) of −70 mV; in voltage-clamp mode, a holding potential of −70 mV was applied. For whole-cell recordings, internal solutions used were as follows. Glutamatergic currents and potentials were recorded with electrodes containing (in mM): 130 K-gluconate, 5 KCl, 3 MgCl, 2 Na2ATP, 0.3 NaGTP, 10 HEPES,0.6 EGTA, and 0.2% biocytin. The calculated ECl-for this solution was −67 mV. To record GABA(A)-mediated currents in voltage-clamp at a holding potential of −70 mV, electrodes contained (in mM): 100 K-gluconate, 45 KCl, 3 MgCl, 2 Na2ATP, 0.3 NaGTP, 10 HEPES, 0.6 EGTA, and 0.2% biocytin, for a calculated ECl- of −27 mV. During paired recordings, biocytin was left out of the internal solution when recording pyramidal cells. Osmolarity of internal solutions was adjusted to ~290 mOsm with sucrose; the pH was adjusted to 7.4 with KOH.
For tests of synaptic connectivity using paired recordings, presynaptic cells in current-clamp were made to fire trains of action potentials (25 at 50 Hz) using 1 – 2 nA current steps of 1 – 2 ms duration, every 10 s for at least 10 trials. Post-synaptic cells were recorded in voltage-clamp at −70 mV. For experiments with carbachol (20 μM), synaptic connections were assayed before and after 5 minutes of drug wash in. Only recordings with stable access resistance in the postsynaptic neuron during the entire recording session were included for analysis. For optogenetic experiments, we focused light on the region of interest through a 40X water immersion objective. Light wavelengths and intensity were generated and controlled by a CoolLED pE-4000 (Andover, United Kingdom). Channelrhodopsin-2 was stimulated with a wavelength of 460 nm and Chrimson was stimulated with a wavelength of 550 nm. Light power was measured using a PM100D Optical Power Meter (Thorlabs).
Antibodies
Primary antibodies included: rabbit anti-VIP (1:000; ImmunoStar, #20077); mouse anti-reelin (1:1000; MBL, #D223-3); mouse anti-PV (1:3000; Sigma, #P3088); rat anti-SST (1:1000; MilliporeSigma, #MAB354); chicken anti-MBP (1:100; MilliporeSigma, #AB9348); rat anti-Ctip2 (1:200; Abcam, #ab18465); mouse anti-CRYM (1:100; Abcam, #ab54669); rabbit anti-CCK (1:1000; Frontier Institute, Japan, #AB2571674). Secondary antibodies were conjugated with Alexa Fluor dyes 488, 555, or 633 (1:1000; Molecular Probes).
Immunohistochemistry
Mice were deeply anesthetized and tissue was fixed via transcardial perfusion with 4% paraformaldehyde. Brains were post-fixed for at least 1 hour when preparing for immunostaining for PV, SST, MBP, Ctip2, or CRYM, but not for more than 1 hour when preparing for VIP, CCK, and reelin. Brains were cryopreserved in 30% sucrose. Sections were cut on a freezing microtome at 50 μm when preparing for immunohistochemistry (IHC) or 100 μm when only preparing to image GFP+ cells from 5HT3A-GFP mice. Sections were rinsed in phosphate buffered saline (PBS), blocked for 1 – 2 hours in 10% normal goat serum with 0.5% Triton X-100, and then incubated in primary antibody for 2 hours at room temperature or overnight at 4°C. Sections were then rinsed with PBS and incubated in secondary antibodies (1:1000) and DAPI (1:2000) for 1 – 2 hours at room temperature or overnight at 4°C. All antibodies were diluted in carrier solution consisting of PBS with 1% BSA, 1% normal goat serum, and 0.5% Triton X-100. Sections were then rinsed, mounted on slides, and coverslipped.
Stereotaxic injections
For injections of retrograde tracer and viral vectors, mice were anesthetized with 5% isoflurane and mounted in a stereotax (David Kopf Instruments Model 1900,Tujunga, CA, USA). Topical lidocaine/prilocaine cream (2.5%/2.5%) and buprenorphine (0.1 mg/kg via subcutaneous injection) were provided for post-operative analgesia. Mice were provided with topical lidocaine/antibiotic ointment and ketoprofen daily for at least 3 days following surgery. 5HT3A-GFP mice were injected with red retrobeads (Lumafluor) into visual cortex or superior colliculus; G-deleted-rabies-mCherry-ChR2 (3.26 × 108 virus molecules/ml Salk Institute Gene, Transfer, Targeting and Therapeutics Core) was injected into superior colliculus. 5HT3A-GFP:Tlx3(PL56)-Cre mice were injected with AAV9-Syn-FLEX-Chrimson-tdTomato (5.6 × 1012 virus molecules/ml, UNC Vector Core, contributed by Edward Boyden) into visual cortex. Retrobeads and viral vectors were delivered via a glass micropipette attached to a syringe (Hamilton Company Inc., Reno, NV, USA) and back filled with light mineral oil. Visual cortex was targeted using the following coordinates: 3.7 (±0.2) mm caudal and 2.7 mm lateral to bregma, and 0.7 mm deep from the pia. Superior colliculus was targeted using the following coordinates: 4.0 (±0.2) mm caudal and 0.3 mm lateral to bregma, and 0.7 mm deep from the dura. At both injection sites, 100 nl of retrobeads or viral vector were injected. Retrobeads and G-deleted-rabies-mCherry-ChR2 were injected at 100 nl/min; rAAV9-Syn-FLEX-Chrimson-tdTomato was injected at 50 nl/min. In all cases, the pipette was left in place for 5 min following the injection before removal. For retrobead injections, mice were sacrificed after at least 2 days post-injection; for G-deleted-rabies-mCherry-ChR2, at least 7 days post-injection; for AAV9-Syn-FLEX-Chrimson-tdTomato at least 14 days post-injection.
Image acquisition and analysis of IHC and GFP somatic fluorescence
For 5HT3A-GFP+ interneuron analysis we imaged 7 – 8 100 μm thick hemisphere sections per mouse per condition. For all immunostained sections, we imaged 10 – 12 50 μm thick hemisphere sections per mouse (due to use of thinner sections for IHC). For all comparisons of laminar position, we used n = 3 control mice and n = 3 mutant mice. Sections were imaged using a Zeiss LSM 780 confocal microscope. Confocal stacks were stitched and then cropped to a 500 μm width centered on primary visual cortex using Zen Black software (Zeiss). These confocal stacks were imported into Imaris (Bitplane) software to identify interneuron cell body locations, which were marked as “spots.” 5HT3A-GFP+ and PV+ IN cell bodies were amenable to automatic detection by Imaris software. For 5HT3A-GFP+ INs, we set a signal detection radius of 8 μm to detect cell bodies, and for PV+ INs a detection radius of 12 μm. We then used the Imaris “Quality” filter (intensity center of detected regions after Gaussian filtering by 3/4 of spot radius with background subtraction (Gaussian filter of 8/9 the detection radius)) above an empirically determined threshold. The detection threshold was chosen to maximize the number of detected cells while minimizing observed false-positives. In some cases, artifactual signal on the pial surface, or contamination from neuronal fibers in layer 1, required manual deletion of incorrectly identified cell bodies in these locations. For detection of SST+, SST+/GFP-, VIP+, CCK+, and reelin+/GFP+ interneurons, cell bodies were marked with spots manually. After marking interneuron cell bodies with spots, we traced planes that followed the contours of the dorsal and white matter surfaces, which were then converted into 3-dimensional objects by Imaris (Figure S1A). Imaris then calculated “distance gradients” for each point in space relative to the inner surface planes of these objects (i.e., the dorsal and white matter surfaces). In this manner, the normalized position value of every spot relative to these planes could be calculated. These data were imported into Igor Pro (WaveMetrics) to generate histograms for analysis. We plotted histograms of laminar distribution with high granularity (20 equal sized bins; 5% of cortical depth per bin). For each histogram, we pooled all the interneuron position data from one mouse for one condition (e.g., 8 hemisphere sections for one control mouse) and normalized the data to the total number of cells counted. For each condition, we averaged 3 histograms (1 per mouse, 3 mice per condition). For calculations of cell density, we drew a 3-dimensional region of interest from the dorsal surface to the white matter (variable height and depth depending on the section) with a constant width of 230 μm (chosen to minimize artifacts from dorsal or white matter curvature). We then calculated cell density as the number of spots normalized to this region of interest volume. To analyze within-section overlap of interneuron markers (e.g., Figure 3J), we counted the number of cells that co-express two markers and then divided that value by the of total number of cells expressing each of the individual markers. This was calculated for every section, from which we calculated averages.
A small number of images (Figures 1B, 5A, 6A, 6D) were acquired with a QImaging Retiga 4000R CCD camera mounted to an Olympus AX70 microscope.
Image brightness and contrast were adjusted using Imaris, Fiji, or Zen Black software to highlight cell body locations in example images.
Electrophysiology data analysis
All data were analyzed in Igor Pro (WaveMetrics) using custom routines. To import pClamp files into Igor we used NeuroMatic (Rothman and Silver, 2018).
To analyze postsynaptic currents in paired recordings, we first zeroed the data by subtracting the baseline and then performed 10 repetitions of binomial (Gaussian) smoothing. All postsynaptic currents (PSCs) were then analyzed relative to the timing of the peak of each presynaptic spike during the train. PSCs were detected by threshold crossing (−7 to −10 pA); if this threshold was already crossed at the instant of spike peak, the PSC data associated with that spike of that trial were discarded as being contaminated by spontaneous events. The proportion of failures was calculated as the ratio of evoked PSCs to the total number of non-contaminated trials for each presynaptic spike. The PSC potency was calculated as the average peak amplitude of all successfully evoked PSCs for each presynaptic spike (i.e., failures were not included in the average).
Projection neuron input resistance (Rin) was measured using a linear regression of voltage deflections (±15 mV from −70 mV resting potential) in response to 1 s current steps. To calculate voltage sag, we injected 1 s long negative current steps of sufficient amplitude to achieve a steady state Vm (last 200 ms of response) of −90 mV. We then compared the peak hyperpolarized voltage deflection to the steady state. To quantify doublet spiking we injected depolarizing current steps (1 s duration) at an amplitude twice that necessary to evoke an action potential. We then took the ratio of the initial inter-spike interval (ISI) (first two spikes) to the steady state ISI (average remaining spiking responses). To segregate INs according to possessing a linear versus inward rectifying current-voltage relationship, we injected positive and negative current pulses of sufficient amplitude to result in +15 mV and – 15 mV membrane potential deflections from resting potential (−70 mV), respectively. We then calculated the Rin during the last 200 ms of these responses (during steady state), and took the ratio of the Rin during depolarizing to hyperpolarizing responses. Ratios of < 1.5 were used to classify INs as linear.
Anatomical reconstruction
After biocytin filling during whole-cell recordings, slices were fixed with 4% paraformaldehyde and stored at 4°C. After at least 24 hours, slices were transferred to PBS. To recover the cells, slices were permeabilized with 0.3% Triton X-100 and incubated with secondary antibodies conjugated with Alexa Fluor dye 555 (1:1000; Molecular Probes). Slices were re-sectioned (100 μm) and mounted on gelatin-coated slides using Mowiol mounting medium. Cells were visualized using epifluorescence microscopy (Olympus AX70) and imaged using a confocal microscope (Leica TCS SP2RS or Zeiss LSM 780). Reconstructions were made using Neurolucida (MicroBrightField) or Simple Neurite Tracer (Longair et al., 2011) for Fiji (Schindelin et al., 2012).
Single cell dissociation and FACS
P20 juvenile 5HT3A-GFP (Control) and Emx1IRES-Cre: Satb2Fl/Fl: 5HT3A-GFP (Mutant) mice were used for single-cell sequencing experiments. All mice were anesthetized with isoflurane and then decapitated. Brain dissection, slicing and FACS sorting were carried out as described (Harris et al., 2018), with slight modifications. NMDG-HEPES–based solution was used in all steps to enable better recovery of the cells (Tanaka et al., 2008) during FACS sorting and single-cell bar coding. The NMDG-HEPES-based high-Mg2+ cutting solution contained 93 mM NMDG, 2.5 mM KCl, 1.2 mM NaH2PO4, 30 mM NaHCO3, 20 mM HEPES, 25 mM glucose, 5 mM sodium ascorbate, 3 mM sodium pyruvate, 10 mM MgSO4*7H2O, and 0.5 mM CaCl2*2H2O; it was adjusted to pH 7.4 with 10N HCl, an osmolarity of 300-310 mOsm, and carbogenated (mix of 95% O2 and 5% CO2) before use.
Tissue from 4 5HT3A-GFP (Control) or 4 Satb2-KO:5HT3A-GFP (Mutant) mice were processed on consecutive days for single-cell sequencing experiments. 5HT3A-GFP negative animals (C57BL/6) were processed in parallel for setting FACS gate for the GFP channel. Coronal slices containing visual cortex (350 μM) were cut using VT-1000S vibratome (Leica Microsystems) in cold NMDG-HEPES-based high-Mg2+ cutting solution. Slices were recovered in the same solution at 20°C for 30 minutes, bollowed by micro-dissection of primary visual cortex. Cell dissociations were performed using the Worthington Papain Dissociation System (LK003150) according to manufacturer instructions with minor modifications. Briefly, single-cell suspensions of the micro-dissected visual cortices were prepared using sufficiently carbogenated dissociation solution (containing Papain, DNase in EBSS). After a 60 min enzymatic digestion at 37°C, followed by gentle manual trituration with fire-polished Pasteur pipettes, the cell dissociates were centrifuged at 300 g for 5 minutes at 20°C, and the supernatants were discarded. The albumin density gradient was only performed on the pellets, using a sufficiently carbogenated debris removal solution (containing albumin-ovomucoid inhibitor, DNase in EBSS). The resulting cell pellets were re-suspended in 1ml FACS buffer containing 10% FBS, 100 μM AP5 and 1 μM TTX in a 50:50 mix of carbogenated EBSS: NMDG-HEPES-based cutting saline (with 1mM MgSO4*7H2O).
For single cell sorting of GFP expressing cells by FACS, re-suspended cell dissociates were filtered through 35 mm cell strainer (Falcon 352235) to remove cell clumps. The single cell suspensions were then incubated with 1mg/ml DAPI (1:500, Thermo Scientific 62248) at 4°C for 5 minutes to label dead cells before loading each sample in a MoFlo Astrios EQ high speed cell sorter (Beckman Coulter). 5HT3A-GFP negative cells (from a C57BL/6 mouse) was used for setting FACS gate for the GFP channel. Flow data analysis and setting of sorting gates on live (DAPI-negative) and GFP-expressing cells were carried out using Summit software V6.3.016900 (Beckman Coulter). 20000 individual cells were sorted into a FBS-precoated, Eppendorf LoBind Microcentrifuge tube containing carbogenated 10 μL FACS buffer.
10x Genomics Chromium
The cells were inspected for viability, counted, and loaded on the 10X Genomics Chromium system, aiming to recover ~5000 cells per condition. 12 PCR cycles were conducted for cDNA amplification, and the subsequent library preparation and sequencing were carried out in accordance with the manufacturer recommendation (Chromium Single Cell 3′ Library & Gel Bead Kit v2, 16 rxns, Cat# 120237; Chromium Single Cell A Chip Kit, Cat# 120236). Sequencing of the libraries were performed on Illumina HiSeq2500, with the following recoveries for WT and Mutant genotypes respectively: cell estimates – 5565 and 5405; mean reads per cell (raw) - 35,295, and 37,008; median genes per cell – 2170 and 3181 (also see Table S1). Demultiplexed samples were aligned to the mouse reference genome (mm10), with the inclusion of EGFP sequence obtained from (Gong et al., 2003). The end definitions of genes were extended 4k bp downstream (or halfway to the next feature if closer), using a custom script and converted to mRNA counts using the Cell Ranger Version 2.1.1, provided by the manufacturer.
Single-cell RNA sequencing data processing, analyses and visualization
The R toolkit Seurat (Butler et al., 2018) (https://satijalab.org/seurat/) was employed to align, load and merge the WT and Mutant scRNA-seq dataset files. Dataset preprocessing, comparison of WT and Mutant GFP cells, canonical correlation analyses, and differential expression of genes (padj < 0.01) within the same cluster between WT and Mutant GFP cells were performed according to default Seurat parameters, unless otherwise noted (also see Figure S7 for data quality). Briefly, data was preprocessed by removing cells with less than 500 detectable genes and with reads mapping to mitochondrial genes comprising more than 0.05% of total reads. Gene expression counts were then log normalized and the resulting expression matrix was scaled and centered. Following preprocessing, the top 1000 variable genes were detected for WT and Mutant GFP cells and the intersection of these gene lists were used to run a canonical correlation analysis calculating 30 canonical vectors, followed by running RunTSNE function (from Seurat) with reduction.use = ”cca.aligned” and dim.use = 1:20 as well as the FindClusters function using reduction.use = ”cca.aligned,” resolution = 0.6 and the top 20 cca dimensions. Ingenuity Pathway Analyses were conducted on the differentially expressed genes and heatmaps were made using Morpheus online tool provided by the Broad Institute (https://software.broadinstitute.org/morpheus/).
QUANTIFICATION AND STATISTICAL ANALYSIS
For two sample tests, a parametric t test was performed if samples were normally distributed (Jarque-Bera test) with equal variance (F test); otherwise we performed a nonparametric Wilcoxon rank test. For multiple comparisons, we performed a nonparametric Kruskal-Wallis test followed by a post hoc Dunn-Holland-Wolfe multiple comparisons test. Chi-square tests for laminar position were performed on whole-number percentages of INs to normalize for differences in total cell counts in control and mutant mice. Chi-square tests for IN marker overlap were run on the total number of cells counted in each condition. Chi-square tests for synaptic connectivity were run on the total number of connections tested in each category. For single-cell RNA-seq data, following cluster detection, differential expression analysis was performed using Wilcoxon Rank tests followed by Bonferroni correction for multiple tests. Statistical significance was set at p < 0.05, except for single- cell RNA-seq data which was set at padj < 0.01. Data in the text and graphs are reported as mean ± SEM.
DATA AND SOFTWARE AVAILABILITY
The accession number for single cell RNA-sequencing data in GEO is GSE127724.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-VIP | ImmunoStar | Cat# 20077; RRID: AB_572270 |
| Mouse anti-reelin | MBL International | Cat# D223-3; RRID: AB_843523 |
| Mouse anti-PV | Sigma-Aldrich | Cat# P3088; RRID: AB_477329 |
| Rat anti-SST | MilliporeSigma | Cat# MAB354; RRID: AB_2255365 |
| Chicken anti-MBP | MilliporeSigma | Cat# AB9348; RRID: AB_2140366 |
| Rat anti-Ctip2 | Abcam | Cat# ab18465; RRID: AB_1001525 |
| Mouse anti-CRYM | Abcam | Cat# ab54669; RRID: AB_943673 |
| Rabbit anti-CCK | Frontier Institute, Japan | Cat# AB2571674; RRID: AB_2571674 |
| Goat anti-rabbit Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A-21428; RRID: AB_141784 |
| Goat anti-rat Alexa Fluor 633 | Thermo Fisher Scientific | Cat# A-21094; RRID: AB_141553 |
| Goat anti-rat Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A-21434; RRID: AB_141733 |
| Goat anti-mouse Alexa Fluor 633 | Thermo Fisher Scientific | Cat# A-21052; RRID: AB_2535719 |
| Goat anti-mouse Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A-21422; RRID: AB_141822 |
| Goat anti-Chicken Alexa Flour 633 | Thermo Fisher Scientific | Cat# A-21103; RRID: AB_2535756 |
| Papain Dissociation System | Worthington Biochemical Corporation | Cat# LK003150 |
| Chromium Single Cell 3′ Library & Gel Bead Kit v2 | 10X Genomics | Cat# 120237 |
| Chromium Single Cell A Chip Kit | 10X Genomics | Cat# 120236 |
| Bacterial and Virus Strains | ||
| AAV9-Syn-FLEX-Chrimson-tdTomato | UNC Vector Core | N/A |
| G-deleted-rabies-mCherry-ChR2 | Salk Institute Gene, Transfer, Targeting and Therapeutics Core | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Carbachol | Sigma-Aldrich | Cat# 1092009 |
| Tetrodotoxin citrate | R&D Systems | Cat# 1078 |
| AP5 | Abcam | Cat# ab120271 |
| Deposited Data | ||
| Single cell RNA-sequencing | GEO: GSE127724 | |
| Experimental Models: Organisms/Strains | ||
| Mouse: 5HT3A-GFP | Charles Gerfen (Chittajallu et al., 2013) | N/A |
| Mouse: Tlx3(PL56)-Cre | Charles Gerfen (Gerfen et al., 2013) | N/A |
| Mouse: Emx1-IRES-Cre | The Jackson Laboratory | JAX: 005628 |
| Mouse: loxP flanked Satb2 | Rudolf Grosschedl (Srinivasan et al., 2012) | N/A |
| Mouse: LoxP flanked Lis1 | Anthony Wynshaw-Boris (Gambello et al., 2003) | N/A |
| Mouse: Sox2-Cre | The Jackson Laboratory | JAX: 008454 |
| Oligonucleotides | ||
| Primer: UNIV CRE 1: CAGAGACGGAAATCCATCGC | Invitrogen | N/A |
| Primer: UNIV CRE 2: GGTGCAAGTTGAATAACCGG | Invitrogen | N/A |
| Primer: Satb2 (flx fwd) AAGACTGCTGTGTGGGCTACAC | Invitrogen | N/A |
| Primer: Satb2 (flx rev) CACGTCCGTCCAAAGTTGCT | Invitrogen | N/A |
| Primer: Emx1-Cre: olMR1084 (MTfwd) GCG GTC TGG CAG TAA AAA CTA TC | Invitrogen | N/A |
| Primer: Emx1-Cre: olMR1085 (MTrvs) GTG AAA CAG CAT TGC TGT CAC TT | Invitrogen | N/A |
| Primer: Emx1-Cre: olMR4170 (WTfwd) AAG GTG TGG TTC CAG AAT CG | Invitrogen | N/A |
| Primer: Emx1-Cre: olMR4171 (WTrvs) CTC TCC ACC AGA AGG CTG AG | Invitrogen | N/A |
| Software and Algorithms | ||
| Seurat | Butler et al., 2018 | https://satijalab.org/seurat/ |
| Igor Pro | WaveMetrics | N/A |
| pClamp 10.6 | Molecular Devices | N/A |
| NeuroMatic | Rothman and Silver, 2018 | N/A |
| Morpheus | https://software.broadinstitute.org/morpheus/ | |
| Neurolucida | MicroBrightField | N/A |
| Simple Neurite Tracer for Fiji | Longair et al., 2011; Schindelin et al., 2012 | https://imagej.net/Simple_Neurite_Tracer |
| Imaris | Bitplane | N/A |
| Zen Black | Zeiss | N/A |
| Other | ||
| Retrobeads | Lumafluor | N/A |
| pE-4000 | CoolLED | N/A |
Highlights.
IT-type projection neurons influence the lamination of CGE-derived interneurons
Converting IT-type cells to PT type disrupts CGE interneuron synaptic connections
PT-type projection neurons do not target CGE interneuron subtypes in deep layers
Loss of IT-type cells alters gene transcription in CGE cells but not major subtypes
ACKNOWLEDGMENTS
This work was supported by a Eunice Kennedy Shriver National Institute of Child Health and Human Development Intramural Award to C.J.M. and a National Institute of Neurological Disorders and Stroke Competitive Fellowship Award to J.C.W. We would thank Susan McConnell (Stanford University) and Rudolf Grosschedl (Max Planck Institute) for providing us with floxed Satb2 mice, and the Janiela Research Campus for Tlx3(PL56)-Cre mice. We also thank Vincent Schram and Lynne Holtzclaw of the NICHD Microscopy and Imaging Core for imaging and Daniel Abebe for animal support. We thank Steven L. Coon and James R. Iben at the NICHD Molecular Genomics Core for RNA sequencing and support.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.neuron.2019.03.036.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alcamo EA, Chirivella L, Dautzenberg M, Dobreva G, Fariñas I, Grosschedl R, and McConnell SK (2008). Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron 57, 364–377. [DOI] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, and Macklis JD (2005). Neuronal subtype-specific genesthat control corticospinal motor neuron development in vivo. Neuron 45, 207–221. [DOI] [PubMed] [Google Scholar]
- Bartolini G, Ciceri G, and Marín O (2013). Integration of GABAergic interneurons into cortical cell assemblies: lessons from embryos and adults. Neuron 79, 849–864. [DOI] [PubMed] [Google Scholar]
- Bartolini G, Sánchez-Alcañiz JA, Osório C, Valiente M, García-Frigola C, and Marín O (2017). Neuregulin 3 Mediates Cortical Plate Invasion and Laminar Allocation of GABAergic Interneurons. Cell Rep. 18, 1157–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista-Brito R, Vinck M, Ferguson KA, Chang JT, Laubender D, Lur G, Mossner JM, Hernandez VG, Ramakrishnan C, Deisseroth K, et al. (2017). Developmental Dysfunction of VIP Interneurons Impairs Cortical Circuits. Neuron 95, 884–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britanova O, de Juan Romero C, Cheung A, Kwan KY, Schwark M, Gyorgy A, Vogel T, Akopov S, Mitkovski M, Agoston D, et al. (2008). Satb2 is a postmitotic determinant for upper-layer neuron specification in the neocortex. Neuron 57, 378–392. [DOI] [PubMed] [Google Scholar]
- Brown SP, and Hestrin S (2009). Intracortical circuits of pyramidal neurons reflect their long-range axonal targets. Nature 457, 1133–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, et al. (2008).Atranscriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci 28, 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Schaevitz LR, and McConnell SK (2005). Fezl regulates the differentiation and axon targeting of layer 5 subcortical projection neurons in cerebral cortex. Proc. Natl. Acad. Sci. USA 102, 17184–17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Wang SS, Hattox AM, Rayburn H, Nelson SB, and McConnell SK (2008). The Fezf2-Ctip2 genetic pathway regulates the fate choice of subcortical projection neurons in the developing cerebral cortex. Proc. Natl. Acad. Sci. USA 105, 11382–11387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittajallu R, Craig MT, McFarland A, Yuan X, Gerfen S, Tricoire L, Erkkila B, Barron SC, Lopez CM, Liang BJ, et al. (2013). Dual origins of functionally distinct O-LM interneurons revealed by differential 5-HT(3A)R expression. Nat. Neurosci 16, 1598–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dembrow NC, Chitwood RA, and Johnston D (2010). Projection-specific neuromodulation of medial prefrontal cortex neurons. J. Neurosci 30, 16922–16937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denaxa M, Neves G, Rabinowitz A, Kemlo S, Liodis P, Burrone J, and Pachnis V (2018). Modulation of Apoptosis Controls Inhibitory Interneuron Number in the Cortex. Cell Rep. 22, 1710–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dipoppa M, Ranson A, Krumin M, Pachitariu M, Carandini M, and Harris KD (2018). Vision and Locomotion Shape the Interactions between Neuron Types in Mouse Visual Cortex. Neuron 98, 602–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleck MW, Hirotsune S, Gambello MJ, Phillips-Tansey E, Suares G, Mervis RF, Wynshaw-Boris A, and McBain CJ (2000). Hippocampal abnormalities and enhanced excitability in a murine model of human lissencephaly. J. Neurosci 20, 2439–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel AI, Akins MR, Krupp AJ, Stagi M, Stein V, and Biederer T (2007). SynCAMs organize synapses through heterophilic adhesion. J. Neurosci 27, 12516–12530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier TW, and Hardan AY (2009). A meta-analysis ofthe corpus callosum in autism. Biol. Psychiatry 66, 935–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Tucciarone JM, Espinosa JS, Sheng N, Darcy DP, Nicoll RA, Huang ZJ, and Stryker MP (2014). A cortical circuit for gain control by behavioral state. Cell 156, 1139–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambello MJ, Darling DL, Yingling J, Tanaka T, Gleeson JG, and Wynshaw-Boris A (2003). Multiple dose-dependent effects of Lis1 on cerebral cortical development. J. Neurosci 23, 1719–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Paletzki R, and Heintz N (2013). GENSAT BAC cre-recombinase driver lines to study the functional organization of cerebral cortical and basal ganglia circuits. Neuron 80, 1368–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonchar Y, Wang Q, and Burkhalter A (2008). Multiple distinct subtypes of GABAergic neurons in mouse visual cortex identified bytriple immunostaining. Front. Neuroanat 1, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, and Heintz N (2003). A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature 425, 917–925. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, and Jones KR (2002). Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emxl-expressing lineage. J. Neurosci 22, 6309–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harb K, Magrinelli E, Nicolas CS, Lukianets N, Frangeul L, Pietri M, Sun T, Sandoz G, Grammont F, Jabaudon D, et al. (2016). Area-specific development of distinct projection neuron subclasses is regulated by postnatal epigenetic modifications. eLife 5, e09531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KD, and Shepherd GM (2015). The neocortical circuit: themes and variations. Nat. Neurosci 18, 170–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KD, Hochgerner H, Skene NG, Magno L, Katona L, Bengtsson Gonzales C, Somogyi P, Kessaris N, Linnarsson S, and Hjerling-Leffler J (2018). Classes and continua of hippocampal CA1 inhibitory neurons revealed by single-cell transcriptomics. PLoS Biol. 16, e2006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattox AM, and Nelson SB (2007). Layer V neurons in mouse cortex projecting to different targets have distinct physiological properties. J. Neurophysiol 98, 3330–3340. [DOI] [PubMed] [Google Scholar]
- Karnani MM, Jackson J, Ayzenshtat I, Tucciarone J, Manoocheri K, Snider WG, and Yuste R (2016). Cooperative Subnetworks of Molecularly Similar Interneurons in Mouse Neocortex. Neuron 90, 86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karube F, Kubota Y, and Kawaguchi Y (2004). Axon branching and synaptic bouton phenotypes in GABAergic nonpyramidal cell subtypes. J. Neurosci 24, 2853–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y (1997). Selective cholinergic modulation ofcortical GABAergic cell subtypes. J. Neurophysiol 78, 1743–1747. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, and Kubota Y (1996). Physiological and morphological identification of somatostatin- or vasoactive intestinal polypeptide-containing cells among GABAergic cell subtypes in rat frontal cortex. J. Neurosci 16, 2701–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, and Kubota Y (1997). GABAergic cell subtypes and theirsynaptic connections in rat frontal cortex. Cereb. Cortex 7, 476–486. [DOI] [PubMed] [Google Scholar]
- Kepecs A, and Fishell G (2014). Interneuron cell types are fit to function. Nature 505, 318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Juavinett AL, Kyubwa EM, Jacobs MW, and Callaway EM (2015). Three Types of Cortical Layer 5 Neurons That Differ in Brain-wide Connectivity and Function. Neuron 88, 1253–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiritani T, Wickersham IR, Seung HS, and Shepherd GM (2012). Hierarchical connectivity and connection-specific dynamics in the corticospinal-corticostriatal microcircuit in mouse motor cortex. J. Neurosci 32, 4992–5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapoetke NC, Murata Y, Kim SS, Pulver SR, Birdsey-Benson A, Cho YK, Morimoto TK, Chuong AS, Carpenter EJ, Tian Z, et al. (2014). Independent optical excitation of distinct neural populations. Nat. Methods 11 , 338–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook-Magnuson E, Varga C, Lee SH, and Soltesz I (2012). New dimensions of interneuronal specialization unmasked by principal cell heterogeneity. Trends Neurosci. 35, 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y (2014). Untangling GABAergic wiring in the cortical microcircuit. Curr. Opin. Neurobiol 26, 7–14. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Shigematsu N, Karube F, Sekigawa A, Kato S,Yamaguchi N, Hirai Y, Morishima M, and Kawaguchi Y (2011). Selective coexpression of multiple chemical markers defines discrete populations of neocortical GABAergic neurons. Cereb. Cortex 21, 1803–1817. [DOI] [PubMed] [Google Scholar]
- Larsen DD, Wickersham IR, and Callaway EM (2008). Retrograde tracing with recombinant rabies virus reveals correlations between projection targets and dendritic architecture in layer 5 of mouse barrel cortex. Front. Neural Circuits 1, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bé JV, Silberberg G, Wang Y, and Markram H (2007). Morphological, electrophysiological, and synaptic properties ofcorticocallosal pyramidal cells in the neonatal rat neocortex. Cereb. Cortex 17, 2204–2213. [DOI] [PubMed] [Google Scholar]
- Lee S, Hjerling-Leffler J, Zagha E, Fishell G, and Rudy B (2010). The largest group of superficial neocortical GABAergic interneurons expresses ionotropic serotonin receptors. J. Neurosci 30, 16796–16808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AT, Gee SM, Vogt D, Patel T, Rubenstein JL, and Sohal VS (2014). Pyramidal neurons in prefrontal cortex receive subtype-specific forms of excitation and inhibition. Neuron 81, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone DP, Srinivasan K, Chen B, Alcamo E, and McConnell SK (2008). The determination of projection neuron identity in the developing cerebral cortex. Curr. Opin. Neurobiol 18, 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone DP, Heavner WE, Ferenczi EA, Dobreva G, Huguenard JR, Grosschedl R, and McConnell SK (2015). Satb2 Regulates the Differentiation of Both Callosal and Subcerebral Projection Neurons in the Developing Cerebral Cortex. Cereb. Cortex 25, 3406–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodato S, and Arlotta P (2015). Generating neuronal diversity in the mammalian cerebral cortex. Annu. Rev. Cell Dev. Biol 31, 699–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodato S, Rouaux C, Quast KB, Jantrachotechatchawan C, Studer M, Hensch TK, and Arlotta P (2011a). Excitatory projection neuron subtypes control the distribution of local inhibitory interneurons in the cerebral cortex. Neuron 69,763–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodato S, Tomassy GS, De Leonibus E, Uzcategui YG, Andolfi G, Armentano M, Touzot A, Gaztelu JM, Arlotta P, Menendez de la Prida L, and Studer M (2011b). Loss of COUP-TFI alters the balance between caudal ganglionic eminence- and medial ganglionic eminence-derived cortical interneurons and results in resistancetoepilepsy. J. Neurosci 31,4650–4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longair MH, Baker DA, and Armstrong JD (2011). Simple NeuriteTracer: open source software for reconstruction, visualization and analysis of neuronal processes. Bioinformatics 27, 2453–2454. [DOI] [PubMed] [Google Scholar]
- López-Bendito G, Sturgess K, Erdélyi F, Szabó G, Molnár Z, and Paulsen O (2004). Preferential origin and layer destination of GAD65-GFP cortical interneurons. Cereb. Cortex 14, 1122–1133. [DOI] [PubMed] [Google Scholar]
- Marín O (2012). Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci 13, 107–120. [DOI] [PubMed] [Google Scholar]
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, et al. (2008). Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet 82, 477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer C, Hafemeister C, Bandler RC, Machold R, Batista Brito R, Jaglin X, Allaway K, Butler A, Fishell G, and Satija R (2018). Developmental diversification of cortical inhibitory interneurons. Nature 555, 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna WL, Ortiz-Londono CF, Mathew TK, Hoang K, Katzman S, and Chen B (2015). Mutual regulation between Satb2 and Fezf2 promotes subcerebral projection neuron identity in the developing cerebral cortex. Proc. Natl. Acad. Sci. USA 112, 11702–11707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi D, Li Z, Lim L, Li M, Moissidis M, Yang Y, Gao T, Hu TX, Pratt T, Price DJ, et al. (2018). Early emergence of cortical interneuron diversity in the mouse embryo. Science 360, 81–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi G, and Fishell G (2011). GABAergic interneuron lineages selectively sort into specific cortical layers during early postnatal development. Cereb. Cortex 21, 845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi G, Butt SJ, Takebayashi H, and Fishell G (2007). Physiologically distinct temporal cohorts of cortical interneurons arise from telencephalic Olig2-expressing precursors. J. Neurosci 27, 7786–7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi G, Hjerling-Leffler J, Karayannis T, Sousa VH, Butt SJ, Battiste J, Johnson JE, Machold RP, and Fishell G (2010). Genetic fate mapping reveals that the caudal ganglionic eminence produces a large and diverse population of superficial cortical interneurons. J. Neurosci 30, 1582–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi G, Young A, Petros T, Karayannis T, McKenzie Chang M, Lavado A, Iwano T, Nakajima M, Taniguchi H, Huang ZJ, et al. (2015). Prox1 Regulates the Subtype-Specific Development of Caudal Ganglionic Eminence-Derived GABAergic Cortical Interneurons. J. Neurosci 35,12869–12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Hirata T, Hibi M, and Macklis JD (2005). Fezl is required for the birth and specification of corticospinal motor neurons. Neuron 47,817–831. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Fame RM, MacDonald JL, MacQuarrie KL, and Macklis JD (2009). Novel subtype-specific genes identify distinct subpopulations of callosal projection neurons. J. Neurosci 29, 12343–12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima M, and Kawaguchi Y (2006). Recurrent connection patterns of corticostriatal pyramidal cells in frontal cortex. J. Neurosci 26, 4394–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima M, Morita K, Kubota Y, and Kawaguchi Y (2011). Highly differentiated projection-specific cortical subnetworks. J. Neurosci 31,10380–10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima M, Kobayashi K, Kato S, Kobayashi K, and Kawaguchi Y (2017). Segregated Excitatory-Inhibitory Recurrent Subnetworks in Layer 5 of the Rat Frontal Cortex. Cereb. Cortex 27, 5846–5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nery S, Fishell G, and Corbin JG (2002). The caudal ganglionic eminence is a source of distinct cortical and subcortical cell populations. Nat. Neurosci 5, 1279–1287. [DOI] [PubMed] [Google Scholar]
- Niquille M, Limoni G, Markopoulos F, Cadilhac C, Prados J, Holtmaat A, and Dayer A (2018). Neurogliaform cortical interneurons derive from cells in the preoptic area. eLife 7, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberlaender M, Boudewijns ZS, Kleele T, Mansvelder HD, Sakmann B, and de Kock CP (2011). Three-dimensional axon morphologies of individual layer 5 neurons indicate cell type-specific intracortical pathways for whisker motion and touch. Proc. Natl. Acad. Sci. USA 108, 4188–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osakada F, Mori T, Cetin AH, Marshel JH,Virgen B, and Callaway EM (2011). New rabies virus variants for monitoring and manipulating activity and gene expression in defined neural circuits. Neuron 71, 617–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkey KA, Chittajallu R, Craig MT,Tricoire L, Wester JC, and McBain CJ (2017). Hippocampal GABAergic Inhibitory Interneurons. Physiol. Rev 97, 1619–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer CK, Xue M, He M, Huang ZJ, and Scanziani M (2013). Inhibition of inhibition in visual cortex: the logic of connections between molecularly distinct interneurons. Nat. Neurosci 16, 1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pla R, Borrell V, Flames N, and Marín O (2006). Layer acquisition by cortical GABAergic interneurons is independent of Reelin signaling. J. Neurosci 26, 6924–6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priya R, Paredes MF, Karayannis T, Yusuf N, Liu X, Jaglin X, Graef I, Alvarez-Buylla A, and Fishell G (2018). Activity Regulates Cell Death within Cortical Interneurons through a Calcineurin-Dependent Mechanism. Cell Rep. 22, 1695–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman JS, and Silver RA (2018). NeuroMatic: An Integrated Open-Source Software Toolkit for Acquisition, Analysis and Simulation of Electrophysiological Data. Front. Neuroinform 12, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouaux C, and Arlotta P (2013). Direct lineage reprogramming of post-mitotic callosal neurons into corticofugal neurons in vivo. Nat. Cell Biol 15, 214–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy B, Fishell G, Lee S, and Hjerling-Leffler J (2011). Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev. Neurobiol 71, 45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberberg G, and Markram H (2007). Disynaptic inhibition between neocortical pyramidal cells mediated by Martinotti cells. Neuron 53, 735–746. [DOI] [PubMed] [Google Scholar]
- Southwell DG, Paredes MF, Galvao RP, Jones DL, Froemke RC, Sebe JY, Alfaro-Cervello C, Tang Y, Garcia-Verdugo JM, Rubenstein JL, et al. (2012). Intrinsically determined cell death of developing cortical interneurons. Nature 491, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwell DG, Nicholas CR, Basbaum AI, Stryker MP, Kriegstein AR, Rubenstein JL, and Alvarez-Buylla A (2014). Interneurons from embryonic development to cell-based therapy. Science 344, 1240622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan K, Leone DP, Bateson RK, Dobreva G, Kohwi Y, Kohwi-Shigematsu T, Grosschedl R, and McConnell SK (2012). A network of genetic repression and derepression specifies projection fates in the developing neocortex. Proc. Natl. Acad. Sci. USA 109, 19071–19078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Tanaka Y, Furuta T, Yanagawa Y, and Kaneko T (2008). The effects of cutting solutions on the viability of GABAergic interneurons in cerebral cortical slices of adult mice. J. Neurosci. Methods 171, 118–125. [DOI] [PubMed] [Google Scholar]
- Tasic B, Menon V, Nguyen TN, Kim TK, Jarsky T, Yao Z, Levi B, Gray LT, Sorensen SA, Dolbeare T, et al. (2016). Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci 19, 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Yao Z, Graybuck LT, Smith KA, Nguyen TN, Bertagnolli D, Goldy J, Garren E, Economo MN, Viswanathan S, et al. (2018). Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay R, Lee S, and Rudy B (2016). GABAergic Interneurons in the Neocortex: From Cellular Properties to Circuits. Neuron 91, 260–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tricoire L, Pelkey KA, Erkkila BE, Jeffries BW, Yuan X, and McBain CJ (2011). A blueprint for the spatiotemporal origins of mouse hippocampal interneuron diversity. J. Neurosci 31, 10948–10970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG (2008). The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135, 422–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban-Ciecko J, Jouhanneau JS, Myal SE, Poulet JFA, and Barth AL (2018). Precisely Timed Nicotinic Activation Drives SST Inhibition in Neocortical Circuits. Neuron 97, 611–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verney C, Takahashi T, Bhide PG, Nowakowski RS, and Caviness VS Jr. (2000). Independent controls for neocortical neuron production and histogenetic cell death. Dev. Neurosci 22, 125–138. [DOI] [PubMed] [Google Scholar]
- Vogt D, Hunt RF, Mandal S, Sandberg M, Silberberg SN, Nagasawa T, Yang Z, Baraban SC, and Rubenstein JL (2014). Lhx6 directly regulates Arx and CXCR7 to determine cortical interneuron fate and laminar position. Neuron 82, 350–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucurovic K, Gallopin T, Ferezou I, Rancillac A, Chameau P, van Hooft JA, Geoffroy H, Monyer H, Rossier J, and Vitalis T (2010). Serotonin 3A receptor subtype as an early and protracted marker of cortical interneuron subpopulations. Cereb. Cortex 20, 2333–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamsley B, and Fishell G (2017). Genetic and activity-dependent mechanisms underlying interneuron diversity. Nat. Rev. Neurosci 18, 299–309. [DOI] [PubMed] [Google Scholar]
- Wang Y, Gupta A, Toledo-Rodriguez M, Wu CZ, and Markram H (2002). Anatomical, physiological, molecular and circuit properties of nest basket cells in the developing somatosensory cortex. Cereb. Cortex 12, 395–410. [DOI] [PubMed] [Google Scholar]
- Wester JC, and Contreras D (2012). Columnar interactions determine horizontal propagation of recurrent network activity in neocortex. J. Neurosci 32, 5454–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wester JC, and McBain CJ (2014). Behavioral state-dependent modulation of distinct interneuron subtypes and consequences for circuit function. Curr. Opin. Neurobiol 29, 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong FK, Bercsenyi K, Sreenivasan V, Portalés A, Fernández-Otero M, and Marín O (2018). Pyramidal cell regulation of interneuron survival sculpts cortical networks. Nature 557, 668–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu PR, Cho KK, Vogt D, Sohal VS, and Rubenstein JL (2016). The Cytokine CXCL12 Promotes Basket Interneuron Inhibitory Synapses in the Medial Prefrontal Cortex. Cereb. Cortex 27, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuttke TV, Markopoulos F, Padmanabhan H,Wheeler AP, Murthy VN, and Macklis JD (2018). Developmentally primed cortical neurons maintain fidelity of differentiation and establish appropriate functional connectivity after transplantation. Nat. Neurosci 21, 517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Cobos I, De La Cruz E, Rubenstein JL, and Anderson SA (2004). Origins of cortical interneuron subtypes. J. Neurosci 24, 2612–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamuro K, Yoshino H, Ogawa Y, Makinodan M, Toritsuka M, Yamashita M, Corfas G, and Kishimoto T (2017). Social Isolation During the Critical Period Reduces Synaptic and Intrinsic Excitability of a Subtype of Pyramidal Cell in Mouse Prefrontal Cortex. Cereb. Cortex •••, 1–13. [DOI] [PubMed] [Google Scholar]
- Ye Z, Mostajo-Radji MA, Brown JR, Rouaux C, Tomassy GS, Hensch TK, and Arlotta P (2015). Instructing Perisomatic Inhibition by Direct Lineage Reprogramming of Neocortical Projection Neurons. Neuron 88, 475–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for single cell RNA-sequencing data in GEO is GSE127724.