

Abstract
Glioblastoma (GBM), as a very aggressive cancer of central nervous system, is very challenging to completely cure by the conventional combination of surgical resection with radiotherapy and chemotherapy. The success of emerging immunotherapy in hot tumors has attracted considerable interest for the treatment of GBM, but the unique tumor immunosuppressive microenvironment (TIME) of GBM leads to the failure of immunotherapy. Here, we show the significant improvement of the immunotherapy efficacy of GBM by modulating the TIME through novel all-in-one biomimetic nanoparticles (i.e. CS-I/J@CM NPs). The nanoparticles consist of utrasmall Cu2-xSe nanoparticles (NPs) with outstanding intrinsic properties (e.g., photo-responsive Fenton-like catalytic property for inducing immunogenic cell death (ICD) and alleviating the hypoxia of tumor), indoximod (IND, an inhibitor of indoleamine-2,3-dioxygenease in tumor), JQ1 (an inhibitor for reducing the expression of PD-L1 by tumor cells), and tumor cell membrane for improving the targeting capability and accumulation of nanoparticles in tumor. We reveal that these smart CS-I/J@CM NPs could drastically activate the immune responses through remodeling TIME of GBM by multiple functions. They could (1) increase M1-phenotype macrophages at tumor site by promoting the polarization of tumor-associated macrophages through the reactive oxygen species (ROS) and oxygen generated from the Fenton-like reaction between nanoparticles and H2O2 within tumor under NIR II irradiation; (2) decrease the infiltration of Tregs cells at tumor site through the release of IND; (3) decrease the expression of PD-L1 on tumor cells through JQ1. The notable increments of anti-tumor CD8+T cells in the tumor and memory T cells (TEM) in the spleen show excellent therapy efficacy and effectively prevent the recurrence of GBM after modulation of the TIME. This work demonstrates the modulation of TIME could be a significant strategy to improve the immunotherapy of GBM and other cold tumors.
Keywords: Glioblastoma, Immunotherapy, Ultrasmall copper selenide nanoparticles, Tumor immune microenvironment, Immune checkpoint blockade therapy
Graphical abstract
Highlights
-
•
Biomimetic nanoparticles could both remodel tumor immunosuppressive microenvironment and serve as an excellent checkpoint inhibitor to decrease the expression of PD-L1 for glioblastoma immunotherapy.
-
•
The smart nanoparticle could efficiently cross blood-brain barrier and target at tumor site under the assistance of noninvasively focused ultrasound.
-
•
The nanoparticles significantly reversed glioblastoma from “cold” tumor into “hot” tumor under the NIR II light irradiation.
-
•
Immunological memory was efficiently induced to prevent the recurrence of glioblastoma.
1. Introduction
Glioblastoma (GBM) as the most common and aggressive tumor in the brain is one of the hardest challenges in oncotherapy [1,2]. Despite of advances in the surgical resection and the combined radiotherapy and chemotherapy, the five-year survival rate of GMB patients is still very low (<5%) [3]. Therefore, development of innovative treatments is urgently important. The emerging immunotherapy has attracted considerable interest in the therapy of GBM. However, the immunotherapy does not work well for GBM, which is a typical “cold” tumor due to the very strong and complicated tumor immunosuppressive microenvironment (TIME). The severe TIME could not only lead to large amount of tumor-associated macrophages (TAMs) and regulatory (Treg) cells for tumor immune escape, but also inhibit the activity of M1 macrophages, dendritic cells (DCs) and cytotoxic T lymphocytes (CTLs) in the recurred tumors and tumor draining lymph nodes (TDLNs) [[4], [5], [6], [7]]. How to effectively and precisely modulate the TIME is a crucial issue for improving immunotherapy of GBM due to its significant roles in immunosuppression.
In the GBM TIME, the amount of TAMs is up to ∼50% of tumor mass. They could seriously impair antitumor immunity by depriving the nutrients of lymphocytes, promoting the recruitment or induction of regulatory T cells (Tregs), etc. [[8], [9], [10]] Therefore, regulation of TAMs is of great importance and the first choice for decreasing the immunosuppression in tumor. However, there is rare report on the regulation of TAMs to improve the immunotherapy of GBM, although repolarization of TAMs from M2 phenotype into M1 phenotype has been demonstrated to show great potential in colon adenocarcinoma and B16F10 melanoma [11]. As the hypoxic microenvironment benefits to the polarization of macrophages into M2-like TAMs to suppress immune responses, relieving the hypoxia through advanced nanoparticles has become an important strategy to reprogram M2-like TAMs into M1 phenotype for improving immunotherapy [12,13], which has been successfully used to treat other tumors. For example, Liu et. used hollow MnO2 nanoparticles to generate oxygen and relieve tumor hypoxia to reduce the M2-like TAMs amount in 4T1 solid tumor, and combined with anti-PD-L1 to improve the efficacy of immunotherapy [14]. In addition to oxygen, the reactive oxygen species (ROS) could also promote polarization of M2-like TAMs into M1 phenotype [15]. Chen et al. co-encapsulated photosensitizers indocyanine green (ICG) and titanium dioxide (TiO2) with or without NH4HCO3 in mannose-modified PLGA NPs to increase the generation of ROS to reprogram M2-like TAMs into M1 phenotype for therapy of breast cancer [16].
The above examples demonstrate the great potential of regulation of TAMs in tumor immunotherapy, which could be applied to GBM immunotherapy. Besides the TAMs, another important regulator in the GBM TIME is different types of enzymes. It has been demonstrated that the upregulated indoleamine 2,3-dioxygenase (IDO) in the TIME of GBM is an important suppressor of CTLs [[17], [18], [19]]. IDO could accelerate the degradation of tryptophan (Try) and increase the accumulation of kynurenine (Kyn) in tumor [20], which led to the suppression of T cells infiltration and the recruitment of Tregs cells at tumor site [[21], [22], [23]]. Therefore, inhibiting the expression of IDO by targeting agents is another important strategy to relieve the TIME. As previously reported, the immunosuppressive effects could be reduced by 1-methyl-d-tryptophan (also known as indoximod, IND) [24], which has been used in the clinical trials with a high dose due to the lack of targeting capability and the low accumulation in tumor [25]. To increase the therapeutic efficacy and reduce the potential side effects of IND, nanoparticles were used as carriers to deliver IND for increasing its accumulation in tumor through the passive targeting effect of nanoparticles [26].
The immunosuppressive-programmed cell death ligand-1 (PD-L1) on the tumor cells in the TIME is another crucial impediment to the therapeutic efficacy of GBM. The immune checkpoint inhibitors (ICIs) such as PD-L1 monoclonal antibodies have shown excellent efficacy for immunotherapy of non-small cell lung cancer and melanoma [27]. However, the blood-brain barrier (BBB) prevent large size monoclonal antibodies entering into the brain tumor, leading to their very poor effects on the GBM [10,[28], [29], [30], [31], [32]]. To achieve successfully therapeutic effects on GBM, the immune checkpoint blocking antibodies should be able to cross the BBB and target the tumor efficiently. Ljubimova et al. used poly(β-l-malic acid)-conjugated Angiopep-2 (AP-2) peptide with the capability of crossing BBB to deliver a-PD-1 and a-CTLA-4 for GBM immunity after intravenous injection [33]. Alternatively, small-molecule immunodulators could provide a new way for GBM immunity if they can cross the BBB and target tumor [34].
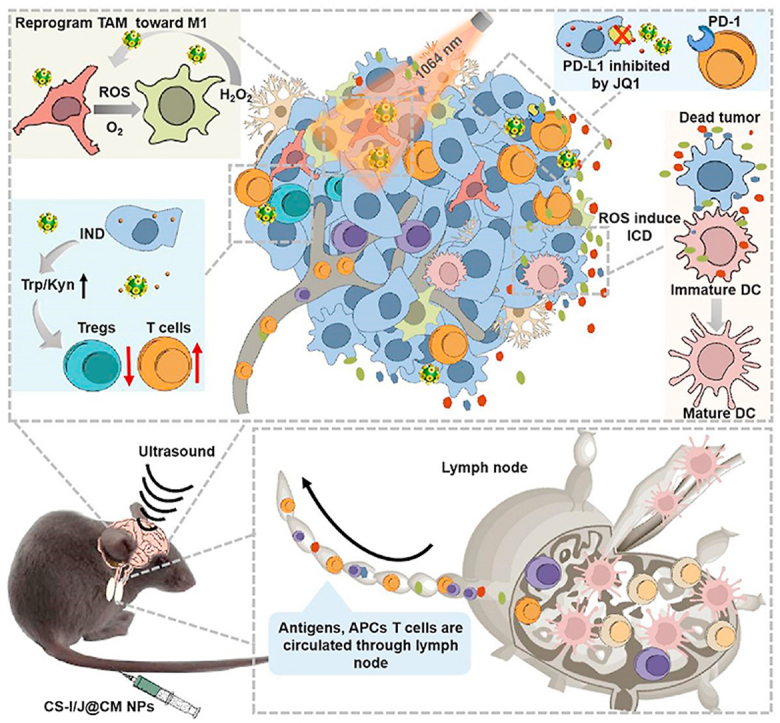
Herein, we report the improvement of immunotherapy of GBM (Scheme 1) by modulating the TIME through robust biomimetic nanoparticles (i.e. CS-I/J@CM NPs), which were efficiently delivered to the tumor site with assistance of focused ultrasound to activate strong immune responses. The smart CS-I/J@CM NPs could remodel TIME of GBM by increasing M1-phenotype macrophages and decreasing the Tregs cell infiltration at tumor site. They could also serve as a checkpoint inhibitor to decrease the expression of PD-L1 on tumor cells and activate anti-tumor immune response for the treatment. Their multiple functions led to the notable increments in the CD8+T cells in the tumor to show the excellent immunotherapy efficacy of GBM. More importantly, the immunological memory to prevent the recurrence of tumor in the treated mice was induced after treatment with our smart nanoparticles. Our work shows great potential of modulation of TIME in immunotherapy of GBM and other cold tumors.
Scheme 1.

Schematic illustration of remodeling the tumor immunosuppressive microenvironment by the all-in-one CS-I/J@CM NPs to improve the immunotherapy of glioblastoma.
2. Experimental methods
2.1. Materials
CuCl2·2H2O (≥99%), Se powder (≥99.5%), sodium borohydride (NaBH4, 99%), mercaptosuccinic acid (MSA, 99%) were purchased from Sigma-Aldrich. Mono-(6-mercapto-6-deoxy)-β-cyclodextrin (CD) was purchased from Shangdong Binzhou Zhiyuan Biotechnology Co. Ltd. JQ1 was purchased from Shanghai Selleck Chemicals Co. Ltd. Indoximod (IND) was purchased from Beijing annoron Co. Ltd. 2,7-dichlorofluorescein diacetate (DCFH-DA) was purchased from AAT Bioquest Inc., Thermo Fisher Scientific. Milli-Q water (>18 MΩ cm) was used in the experiments. All chemicals and reagents were used as received without any further purification.
2.2. Characterization
The morphology of the nanoparticles was characterized by transmission electron microscopy (TEM, FEI Tecnai F20) at an acceleration voltage of 200 kV. The ultraviolet-visible-near-infrared (UV-vis-NIR) absorbance was recorded with a PerkinElmer Lambda 750 UV-vis-NIR spectrophotometer.
2.3. Synthesis of CS-I/J@CM NPs
0.5 mmol (39.48 mg) Se powder was reduced rapidly by 1.5 mmol (56.75 mg) NaBH4 at room temperature in 50 mL H2O under magnetic stirring and the protection of N2. Then, 5 mL mixed solution of 1 mmol (170 mg) CuCl2·2H2O and 6.66 mmol (1 g) MSA was added into the selenium precursor solution to form a black solution immediately. After stirring at room temperature for 1.5 h under N2 protection, the solution was purified by ultrafiltration to result in MSA-capped Cu2-xSe NPs.
1 g CD was added into the Cu2-xSe NPs solution and stirred at 4 °C for 12 h. Then 0.2 g HS-PEG-SH was introduced into the above solution and stirred at 0 °C for 12 h to improve their biocompatibility. The mixed solution was also purified by ultrafiltration, and the resultant PEGylated nanoparticles (referred as CS NPs) were stored for use.
10 mL CS NPs solution (400 μg/mL) and 440 μL IND (20 mg/mL in DMSO) were mixed and stirred at 0 °C for 4 h, and then free IND was removed by ultrafiltration with Milli-Q water. The resultant nanoparticles were referred to as CS-IND NPs (abbreviated into CS-I NPs). The JQ1-modified CS NPs were prepared and purified by the same method, and the obtained nanoparticles were referred to as CS-JQ1 NPs (abbreviated into CS-J NPs).
To improve the homologous adhesion of nanoparticles, GL261 cell membranes were used to coat the mixture of CS-I and CS-J NPs (the ratio of CS-I and CS-J was 10:1) to form CS-I/J@CM NPs. GL261 cells were firstly harvested and washed with PBS three times and resuspended in a cold Tris buffer (pH = 7.4) (the buffer solution consisted of 10 mM MgCl2, 10 mM Tris and 1 × ethylenediamine tetra-acetic acid (EDTA)-free protease inhibitor) at 4 °C for 1 h, before being sonicated for 10 min in an ultrasonicator at 0 °C. The solution was then centrifuged at 600 rpm for 10 min at 4 °C to remove cell nucleus, and the resultant supernatants were centrifuged at 11480 rpm for 10 min to separate other organelle. The obtained supernants were further centrifuged at 110,000 rpm for 30 min to collect cell membranes, which were resuspended and extruded through 400 nm polycarbonate membranes for 5 cycles. Afterwards, the mixture of CS-I and CS-J NPs solution and cell membranes was extruded through 200 nm polycarbonate membranes for at least 5 cycles. The resultant sample was denoted as CS-I/J@CM NPs.
Sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was used to characterize the membrane proteins and demonstrate the successful coating of nanoparticles with cell membrane. The GL261 cell lysate, GL261 cell membrane vesicles and CS-I/J@CM NPs were mixed with loading buffer (lithium dodecyl sulfate, LDS) respectively, and heated for 10 min at 90 °C and then loaded into each well of NuPAGE Novex 4–12% Bis-Tris minigel (20 μL sample for each well). 3-(N-morpholino) propane sulfonic acid (MOPS) and sodium dodecyl sulfate (SDS) were used as a running buffer solution in an Electrophoresis System, stained by Coomassie Blue, and then destained overnight before imaging.
2.4. Fenton-like reaction of CS-I/J@CM NPs under irradiation
The degradation of H2O2 by CS-I/J@CM NPs was characterized by an indicator containing 24% Ti(SO4)2 (1.33 mL) and 8.33 mL H2SO4 in 50 mL H2O. H2O2 (400 μM) was mixed with CS-I/J@CM NPs (12.5 μg/mL) in 1 mL H2O, and then irradiated with a pulsed 1064 nm laser for 5 min. The residual H2O2 was detected by measuring the absorbance of reaction solution at 405 nm. The generation of total ROS was measured by the fluorescence of dichlorofluorescein diacetate (DCFH-DA) after reaction with ROS. The generated O2 was characterized with a dissolve oxygen meter (JPBJ-608).
2.5. In vitro cytotoxicity
GL261 cells were seeded in the 96-well plates with a density of 8 × 103 - 1 × 104 cells/well for 24 h, and CS-I/J@CM NPs in Dulbecco's modified Eagle medium (DMEM) culture medium with different concentrations (0.4, 0.8, 1.6, 3.1, 6.2, 12.5 μg/mL) were added. After cultured for 4 h, the medium was removed and washed twice by PBS, and then the cytoxicity of CS-I/J@CM NPs was characterized by the standard MTT assay.
2.6. Hypoxia condition at the cell level
Three dimensional multicellular tumor spheroids (3D MCTSs) of GL261 cells were fabricated and cultured by the liquid overlay method. The GL261 cells at the phase of exponential growth were dissociated as individual cells by EDTA and washed by PBS for three times. 4 × 103 GL261 cells were seeded on 15 mg/mL agarose-coated 96-well plates with 100 μL of DMEM and matrigel. The single GL261 3D MCTS could grow to around 700 μm in diameter about 7 d under the culture conditions (37 °C, 5% CO2). MCTSs were removed to glass bottomed dishes after formed, and media containing CS-I/J@CM NPs (12.5 μg/mL) and hypoxia probe (50 nM) were added, respectively. They were cultured at 37 °C under 5% CO2 for 8 h. The non-endocytosed CS-I/J@CM NPs were removed by washing three times with PBS. After that, MCTSs were irradiated with the pulsed 1064 nm laser (0.75 W/cm2, 5 min) and then stained with Hoechst 33342 for 20 min, characterized by confocal laser scanning microscopy (CLSM, λex = 596 nm, λem = 670 nm).
2.7. Monitoring of intracellular total ROS
2,7-dichlorofluorescin diacetate (DCFH-DA) was used to detect total ROS. GL261 cells were seeded on glass-bottom dishes with a density of 8 × 103 − 1 × 104 cells per well for 24 h. After incubation with or without CS-I/J@CM NPs (12.5 μg/mL) for 4 h, GL261 cells were washed twice with PBS, and then DCFH-DA was introduced at 37 °C under 5% CO2 for 30 min. They were then irradiated or not with the pulsed 1064 nm laser (0.75 W/cm2) for 5 min, and then incubated for 2 h under the culture conditions (37 °C, 5% CO2) again. Afterwards, the culture medium was removed, and the GL261 cells were stained with Hoechst 33342 for 15 min for characterization by CLSM or flow cytometry analysis.
2.8. Flow cytometry analysis of M1-and M2-Macrophages
GL261 cells (8 × 104 − 1 × 105) were seeded in the lower chambers of Transwell cell culture plate, and same amounts of RAW 264.7 cells were placed in the upper chambers at 37 °C under 5% CO2 for 24 h in DMEM containing 10% FBS. After incubation with or without CS-I/J@CM NPs (12.5 μg/mL) for 4 h, the upper RAW 264.7 cells were removed. The GL261 cells in the lower chambers were irradiated with or without the pulsed 1064 nm laser (0.75 W/cm2) for 5 min, and the upper RAW 264.7 cells were put back and co-cultured with GL261 cells in the Transwell setup at 37 °C under 5% CO2. After co-culturing for 24 h, the RAW 264.7 cells were collected, washed twice with PBS, and then re-dispersed in 100 μL of anti-CD86-FITC (eBioscience) and anti-CD-206-APC (eBioscience) solution and incubated for 20 min on ice. The cells were washed twice with PBS, and then analyzed with flow cytometry analysis.
3. IDO inhibitory effect of CS-I/J@CM NPs in vitro
To exploit the effect of IDO inhibitor of CS-I/J@CM NPs, GL261 cells were seeded in the 96-well plates of 4 × 103 - 5 × 103 cells/well for 24 h under the culture conditions (37 °C, 5% CO2). Then, the GL261 cells were treated with or without solutions of CS-I/J@CM NPs at different concentrations containing IFN-γ (100 ng/mL), the cell culture medium of each well was collected and added with dimethylamine benzaldehyde and glacial acetic acid at room temperature for 10 min, and the absorbance of medium at 490 nm was measured to calculate the concentration of Kyn.
3.1. PD-L1 expression of GL261 cells co-cultured with CS-I/J@CM NPs
The GL261 cells were seeded in the 12-well plates (8 × 104 − 1 × 105) and incubated for 24 h. The cells were then cultured with or without CS-I/J@CM NPs (12.5 μg/mL) at 37 °C under 5% CO2 for 12 h, washed twice with PBS and re-dispersed in 100 μL of anti-PD-L1-APC (Biolegend) for 20 min on ice. Afterwards, the cells were washed twice with PBS, and then analyzed with flow cytometry analysis.
3.2. Immunogenic and DC mature induced by CS-I/J@CM NPs
Immunofluorescence was used to assess the calreticulin (CRT) exposure onto the plasma membrane surface. GL261 cells (8 × 104 − 1 × 105) were seeded on glass-bottom dishes for 24 h to allow them attaching to the surface of wells. After being treated with or without CS-I/J@CM NPs (12.5 μg/mL) for 4 h incubation, irradiated with or without pulsed 1064 nm laser (0.75 W/cm2) for 5 min, the exposure of CRT was detected by using calreticulin antibody (PE) (Fitzgerald). The membranes of GL261 cells were stained with 3, 3′-dioctadecyloxacarbocyanineperchlorate (DiO) for colocalization.
The extracellularly released adenine nucleoside triphosphate (ATP) and high-mobility group box 1 (HMGB1) were detected by ATP detection Kit (A22066) (Molecular Probes) and HMGB1 detection Kit (Chondrex), respectively. GL261 cells were cultured similarly by the above method and irradiated with or without pulsed 1064 nm laser (0.75 W/cm2) for 5 min. The released ATP and HMGB1 in the supernatant of GL261 cells were detected by the provided protocols with the above Kits, respectively.
The bone marrow-derived dendritic cells obtained from Balb/c mice were incubated with GL261 cells, which were pre-treated with or without CS-I/J@CM NPs for 24 h and then irradiated with or without pulsed 1064 nm laser. Cells were washed twice with PBS and then re-dispersed in 100 μL of the mixture solution containing anti-CD11c-PE (Biolegend), anti-CD80-APC (Biolegend) and anti-CD86-FITC (Biolegend) for 20 min on ice. The cells were washed twice with PBS, and then analyzed with flow cytometry.
3.3. Orthotopic glioblastoma model
All animal experiments were carried out according to the guidelines approved by the ethics committee of Soochow University (Soochow, China). Male C57BL/6 mice, aged 6–8 weeks, were supplied by laboratory animal centre of Soochow University. They were divided randomly into four groups, and each group consists of ten mice.
For orthotopic glioblastoma implantation, a solution of GL261 cells (3 × 105) in PBS (5 μL) was injected into the mice's striatum (bregma was 1.0 mm, right lateral was 2.0 mm), and the depth was 2.5 mm. The glioblastoma-bearing mice were treated with different therapy methods after tumor cells were inoculated for 7 d.
3.4. Opening of the blood-brain barrier (BBB)
US transducer (0.5 MHz and 30 mm diameter) was used to open the BBB of mice bearing orthotopic glioblastoma, driven by a function generator connected to a power amplifier. A removable cone filled with water was employed as transducer and guide the US beam into the brain. The acoustic parameters were 0.6 MPa acoustic pressure, 1 ms pulse interval, 0.5 MHz frequency, and 90 s sonication duration. Microbubbles (50 μL) were injected intravenously into the mice before sonication.
3.5. In vivo immunotherapy of orthotopic glioblastoma
The GL261 mice bearing orthotopic glioblastoma were classified randomly into four groups, in which each group had ten mice. The four groups were (1) Control group, (2) Control + NIR group, (3) CS-I/J@CM group, and (4) CS-I/J@CM + NIR group, respectively. The injection dose of CS-I/J@CM NPs was 5 mg/kg, and the power density of the1064 nm laser was 0.75 W/cm2. After 8 h post-injection of PBS or CS-I/J@CM NPs solution, the tumor site of mice was irradiated with the pulsed 1064 nm laser (0.75 W/cm2, 5 min).
The different therapeutic efficacy of mice was monitored by magnetic resonance imaging (MRI). Their brains were also collected for H&E staining to examine the antitumor efficacy.
TUNEL, MBP and GFAP staining. The GL261 tumor-bearing mice were injected with a solution of PBS or CS-I/J@CM NPs (dose: 5 mg/kg) via their tail veins. After 8 h post-injection, the mice were irradiated with a 1064 nm laser (0.75 W/cm2, 5 min). After 1 d treatment, the tumors from various groups of mice were harvested and stained with TUNEL, MBP or GFAP, respectively, and examined with CLSM.
3.6. Flow cytometry
All the antibodies were purchased from Biolegend for flow cytomertry experiments. The treated tumor-bearing mice were sacrificed and their cervical lymph nodes and spleen were harvested after 3 d different treatments. The cells were isolated by Collagenase Type I (1 mg/mL, purchased from Gibico) for 1 h at 37 °C, after washed by PBS twice, the cell pellets were suspended in 3 mL of Ack lysis buffer for lysing red blood cells at 4 °C for 5 min. After centrifugation for 5 min (1000 rpm), the single cell suspensions were washed again by PBS for twice. Then the CD16/32 were used to block nonspecitic binding at 4 °C for 10 min. The cells were detected by flow cytometry analysis: (1) CD8+T cells (CD3+CD8+), (2) Tregs (CD3+CD4+ CD25+Foxp3+), (3) TEM (CD3+CD44+CD62Llow), (4) DC (CD11c+CD80+CD86+).
After intracranial implantation of GL261-Luciferase cells (3 × 105) for 30 days, long-term survivors from the CS-I/J@CM + NIR group and native healthy mice were injected subcutaneously in the hind limbs with GL261-Luciferase cells (2.5 × 106) in 50 μL of PBS and Matrigel (BD, Biosciences). Tumor growth was monitored by measuring bioluminescence through the IVIS Lumina XRMS Series Imaging System.
3.7. Statistical analysis
All results of experiments were expressed as mean ± SD. One-way ANOVA statistical was used to calculate the experimental data. The date classified by the values of p and denoted by (*) for p < 0.05, (**) for p < 0.01, (***) for p < 0.001, (****) for p < 0.0001.
4. Results and discussion
4.1. CS-I/J@CM NPs synthesis
The ultrasmall Cu2-xSe nanoparticles were synthesized under ambient conditions in an aqueous solution [35], and then modified with β-cyclodextrin (CD) (named as Cu2-xSe-CD nanoparticles, CS NPs) for loading hydrophobic drug IND or JQ1. To maximally load IND and JQ1 on our ultrasmall nanoparticles, we loaded them respectively onto CS NPs, and then mixed together for cell-membrane encapuslation. As shown in Figs. S1a and b, IND was successfully loaded onto CS NPs (named as CS-I) and the ratio of CS (Cu weight) and IND was determined to be 4.2:1 by measuring their ultraviolet-visible-near infrared (UV-vis-NIR) absorbance. The zeta potential of CS NPs (Fig. S1c) was slightly fluctuated between −7.0 mV and −8.5 mV after modification with IND. The similar modification of CS NPs with JQ1 (tumorous PD-L1 inhibitor) was carried out, and the optimal ratio of CS (Cu weight) and JQ1 was 2.6:1 (Figs. S2a and b). The zeta potential of resulting CS-J NPs was about −4.5 mV (Fig. S2c). Apart from measuring the UV absorbance, we also used the thermogravimetric analysis (TGA) to determine the loading amounts of small molecular inhibitors. As shown in Fig. S1d, there was about 6% more weight loss of CS-IND NPs than that of CS NPs. In addition, about 10% more weight loss of CS-JQ1 NPs than that of CS NPs was observed (Fig. S2d). The above results illustrate that about 6 wt% IND and 10 wt% JQ-1 were loaded onto the surface of CS NPs, respectively. The ratios of IND and JQ1 loaded onto CS NPs determined by TGA are consistent with those determined by UV-vis-NIR measurement.
We mixed CS-I NPs and CS-J NPs with a ratio of 10:1 because of much higher toxicity of JQ1 than IND. The mixed CS-IND/JQ1 nanoparticles (referred to CS-I/J NPs) were then coated with GL261 cell membranes (CM) to improve their homologous adhesion. The resultant nanoparticles were denoted as CS-I/J@CM NPs. The CS-I/J NPs were characterized with transmission electron microscope (TEM, Figs. S3a and b) to be 3.8 ± 0.6 nm. Their typical high-resolution TEM image was inserted in Fig. S3a, which clearly diplays lattice fringes with an interplanar spacing of 0.20 nm, matching well with that of the (220) planes of cubic berzelianite (Cu2-xSe). Furthermore, as shown in Fig. S3d, the crystal stracture of CS NPs were determined by poweder X-ray diffraction (XRD), which shows the characteristic diffraction peaks of cubic berzeliante (Cu2-xSe, JCPDS no. 06–0680). The broad diffraction peaks are attributed to their ultrasmall size. After being coated with cancer cell membranes, the size of nanoparticles (i.e. CS-I/J@CM NPs) were increased to 38.2 ± 6.8 nm (Fig. 1a and Fig. S3c), each CS-I/J@CM NP consisted of tens of small CS-I/J NPs. The variation of particle size determined by TEM is rather consistent with that obtained from dynamic light scattering (DLS) measurement (Fig. S3e).
Fig. 1.

Preparation and characterization of CS-I/J@CM NPs. (a) TEM image of CS-I/J@CM NPs (scale bar: 50 nm). (b) Sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis and Western-blot analysis of specific protein markers on the membrane, including EpCAM (Epc) and N-cadherin(N–C) of proteins from (I) GL261 cell lysate, (II) GL261 cell membrane vesicles, and (III) CS-I/J@CM nanoparticles. Samples were stained with Coomassie Blue for 12 h. Charaterization of Fenton-like reaction between CS-I/J@CM NPs (12.5 μg/mL) and H2O2 (400 μM). (c) Fluorescence spectra of 2,7-dichlorofluorescin diacetate (DCFH-DA) mixed with a solution of CS-I/J@CM NPs and H2O2, with or without 1064 nm laser irradiation. The fluorescence spectra were obtained under 488 nm excitation. (d) Generation of O2 with a solution of CS-I/J@CM NPs and H2O2 under the irradiation with or without a pulsed 1064 nm laser. Characterization of Fenton-like reaction performance of CS-I/J@CM NPs at cellular level. (e) Viability of GL261 cells cultured with different concentrations of CS-I/J@CM NPs for 4 h. (f) Hypoxia of GLSM images of GL261 MCTSs cultured with CS-I/J or CS-I/J@CM NPs (12.5 μg/mL), and irradiated with or without a pulsed 1064 nm laser (0.75 W/cm2, 5 min) (scale bar: 200 μm). (g) Flow cytometry analysis of intracellular hypoxia relief. (h) Flow cytometry analysis of intracellular total ROS radicals by using DCFH-DA as a probe. (i) Cell apoptosis ratios determined by flow cytometry analysis. (j) Corresponding statistical percentage of apoptotic GL261 cells. (n = 3).
It should be noted that CS NPs are not very stable in the PBS due to the slow release of copper ions as reported in our previous work [36]. They could be degraded in the PBS to cause the decrease of their hydrodynamic size. As shown in Fig. S3f, their hydrodynamic size did not change within 2 days. However, their hydrodynamic size was slowly decreased after stored in PBS for 2 days because of the degradation of nanoparticles, which illustrates that our nanoparticles could be degraded in mouse after tail vein injection. To show the successful coating of cancer cell membrane onto the nanoparticles, we used the sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) to characterize the proteins on the nanoparticles in comparison with those of cancer cell membrane and cell membrane vesicles (Fig. 1b). The similar protein profiles illustrate the good retention of proteins and their lower damage during coating process [37,38]. To further confirm the specific homologous-binding adhesion molecules on CS-I/J@CM NPs, we detected the cellular adhesion molecules (EpCAM (EpC) and N-cadherin (N–C)) on cancer cell membrane. As shown in Fig. 1b, the GL261 cell lysate, GL261 cell membrane vesicles and CS-I/J@CM NPs possessed the similar EpC and N–C profiles on the cell membrane for adhesion. It should be noted that the coated cell membrane did not prevent the release of IND and JQ1 from nanoparticles. Figs. 1g and h, show that about 50% IND or JQ1 was released from CS-I@CM NPs or CS-J@CM NPs.
To evaluate the Fenton-like property of CS-I/J@CM NPs, their catalytic degradation of H2O2 (400 μM) under the pulsed irradiation of the second near-infrared (NIR II) light (1064 nm, 0.75 W/cm2, 5 min) was carried out (Fig. S3i). The pulsed irradiation was used to minimize the influence of temperature variation on the Fenton-like reaction. The results show that more than 90% of H2O2 was degraded within 150 min by CS-I/J@CM NPs under the 1064 nm laser irradiation, however, less than 62% H2O2 was degraded without laser irradiation. Therefore, laser irradiation could speed up the degradation of H2O2 catalyzed by CS-I/J@CM NPs through the Fenton-like reaction, [39] which was further proved by the generation of ROS and O2. As shown in Figs. 1c and d, very strong fluorescence at 529 nm was observed in the case with the NIR II laser irradiation in comparison without pulsed laser irradiation, which supports that laser irradiation could boost the catalytic performance of CS-I/J@CM NPs to degrade H2O2 to generate ROS (Fig. 1c). Furthermore, there was about 10.8 ppm O2 generated from the degradation of H2O2 catalyzed by CS-I/J@CM NPs under the pulsed irradiation of 1064 nm laser (Fig. 1d), which was more than that (5.1 ppm O2) produces without laser irradiation.
The above all results highlight the potential of CS-I/N@CM NPs in the degradation of tumor endogenous H2O2 to generate ROS and O2 for remodeling the TIME of GBM through the Fenton-like reaction under the NIR II irradiation. Before in vivo experiments, their performance was investigated on the cellular level. Methyl thiazolyl tetrazolium (MTT) assay was used to investigate their cytotoxicity toward G261 cells in comparison with IND and JQ1 compounds [40]. As shown in Fig. S4a, the IND cytotoxicity was relatively lower when the concentration was below 50 μg/mL. In contrast, JQ1 had higher toxicity when its concentration was 25 μg/mL (Fig. S4b). Compared with IND and JQ1 compounds, there was no obvious toxicity to GL261 cells when the CS-I/J@CM NPs concentration was lower than 12.5 μg/mL (Fig. 1e).
To detect the toxicity of CS-I/J@CM NPs toward the normal brain cells, we co-cultured SH-SY5Y cells or BV2 cells with different concentrations of CS-I/J@CM NPs for 4 h. Figs. S4c and d shows that the viability of SH-SY5Y cells and BV2 cells were high than that of GL261 cells under the same concentration of CS-I/J@CM NPs. The results further prove that CS-I/J@CM NPs could effectively target the tumor cells. To further characterized the in vitro targeting capability of CS-I/J@CM NPs, the confocal laser scanning microscopy (CLSM) was used. Three types of cells (BV2, SH-SY5Y and GL261 cells) were cultured with 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO)-labeled CS-I/J@CM NPs (12.5 μg/mL) for 4 h. As shown in Fig. S4e, the stronger green fluorescence (FL) observed in GL261 cells than those of BV2 and SH-SY5Y cells suggests that CS-I/J@CM NPs were efficiently taken up by GL261 cells. These results demonstrate that CS-I/J@CM NPs can be efficiently and specifically recognized by the same cell lines in vitro, and suggest excellent targeting ability to the homologous tumor cells in vivo. This homologous adhesion effect ensures that CS-I/J@CM NPs exhibited good capability of targeting glioblastoma to reduce their harm to normal brain cells, which is consistent with the results in Figs. S4a to d.
In addition, when the NIR II irradation was applied to the GL261 cells, their viability was decreased almost 2-flod compared with those cells without NIR II irradiation at the concentration of 12.5 μg/mL. These results prove that the 1064 nm irradiation could enhance the generation of ROS by CS-I/J@CM NPs to induce cancer cell apoptosis (Fig. S5a). To explore the capability of CS-I/J@CM NPs generating O2 for alleviating the tumor hypoxia under the irradiation of 1064 nm laser (0.75 W/cm2, 5 min), we chosed 12.5 μg/mL CS-I/J@CM NPs for experiments based on the cytotoxicity results. We used GL261 multicellular tumor spheroids (MCTSs) heterogeneous cellular aggregates as a 3D model to characterize the relief of intracellular hypoxia by CS-I/J@CM NPs. The hypoxia/oxidative stress detection kit was used, and the fluorescence intensity in the confocal laser scanning microscopy (CLSM) images indicated the degree of hypoxia [41]. As shown in Fig. 1f, MCTSs cultured without any NPs presented strong red fluorescence whether they were irradiated with or without the 1064 nm laser. After they were treated with CS-I/J NPs, the red fluorescence in their CLSM images became weaker than that in the control group, especially after they were irradiated with the 1064 nm laser, because of the O2 generated from the Fenton-like reaction catalyzed by Cu2-xSe NPs. For the MCTSs treated with CS-I/J@CM NPs, their red fluorescence was much weaker than that of MCTSs treated with CS-I/J NPs under the same irradiation. The quantification of fluorescence intensity indicated that the fluorescence intensity of MCTSs treated with CS-I/J NPs was 3.4-fold stronger than that of the CS-I/J@CM group (Fig. S5b), which illustrate that the cell membrane coated nanoparticles could infiltrate into the MCTSs to relieve the hypoxia due to the excellent self-targeting ability of the homologous tumor. Their above excellent ability to generate O2 under irradiation and homologous adhesion effect of CS-I/J@CM NPs make them promising for relieving the hypoxic tumor microenvironment (TME), especially in the deep site of tumor. Furthermore, flow cytometry analysis shows the similar results with CLSM images in Fig. 1g and Fig. S6a. Moreover, the total ROS detected by flow cytometry analysis (Fig. 1h) shows the similar results in Fig. S6b, which proves that CS-I/J@CM NPs could effectively generate much more ROS under the 1064 nm pulsed laser irradiation than CS-I/J NPs without irradiation. In contrast, IND and JQ1 alone couldn't generate obvious ROS without NIR II irradiation, which further pove that the ROS was mainly generated by the Fenton-like property of CS-I/J@CM NPs under the NIR II irradiation. These results are consistent with those shown in Figs. 1c and d, and again demonstrate the enhanced Fenton-like reaction of CS-I/J@CM NPs under laser irradiation.
To determine the ability of our nanoparticles to kill GL261 cells under the 1064 nm laser irradiation, the apoptosis of cancer cells was analyzed. As shown in Figs. 1i and j, without laser irradiation, the apoptosis rates of GL261 cells cultured without any NPs and with CS-I/J@CM NPs were only 0.04% and 14.10%, respectively. In contrast, after irradiation with pulsed 1064 nm laser (0.75 W/cm2, 5 min), the corresponding apoptosis rates were notably increased up to 0.1% and 46.45%, respectively. As shown in Fig. 1j, more than 60% apoptotic GL261 cells were observed in the CS-I/J@CM + NIR group, which was almost 2-fold that of CS-I/J@CM group. Therefore, combination of CS-I/J@CM NPs with NIR II irradiation could induce apoposis of GL261 cells to release damage-associated molecular patterns (DAMPs). These results are quite consistent with that in Fig. S5a, and demonstrate the excellent enhanced performance of our CS-I/J@CM NPs under the NIR II laser irradiation. Furthermore, we also stained the living and dead GL261 cells to show the excellent performance of CS-I/J@CM NPs under the 1064 nm irradiation (Fig. S7). All these results demonstrate that CS-I/J@CM NPs could efficiently degrade H2O2 to generate abundant ROS to kill cancer cells and O2 to alleviate hypoxic environment, and their performance could be drastically enhanced under the irradiation of NIR II light.
4.2. The reprogramming of TIME by CS-I/J@CM NPs in vitro
All the above results highlight the potential of CS-I/J@CM NPs in generating ROS and O2 under the 1064 nm laser irradiation. In addition to killing cancer cells and alleviating hypoxia, whether the generated ROS and O2 could reprogram TAMs toward M1 phenotype effectively [42,43], as illustrated in Fig. 2a, is an opening question. After co-cultured RAW 264.7 cells with GL261 cells in a transwell setup (Fig. S8a), we detected the biomarkers CD86 (a marker of M1 phenotype of macrophages) and CD206 (a marker of M2 phenotype of macrophages) through flow cytometry analysis (Fig. 2b and Fig. S8b). Clearly, after RAW 264.7 cells were co-incubated with GL261 cells in the presence of CS-I/J@CM NPs (12.5 μg/mL) in a transwell setup, M1-macrophages were increased from 2.16% up to 5.53%, in comparison with the RAW 264.7 cells co-cultured with GL261 cells only. Furthermore, after being irradiated with the 1064 nm laser, the M1-macrophages in the CS-I/J@CM + NIR group were increased to 11.4%, which suggests that the generated ROS and O2 could reprogram TAMs into M1 phenotype for remodeling the TIME of GBM. Furthermore, the biomarkers iNOS (a marker of M1 phenotype of macrophages) and Arg1 (a marker of M2 phenotype of macrophages) were also detected by Western-blot (Figs. S8c and d) [44]. The variation of iNOS and Arg1 is consist with that of CD86 and CD206, which suppots the polarization of TAMs by our nanoparticles.
Fig. 2.

In vitro remodeling of tumor immune microenvironment (TIME) by CS-I/J@CM NPs. (a) Schematic illustration of ROS and O2 generated from the Fenton-like reaction to polarize the tumor-associated macrophages (TAMs). (b) Flow cytometry analysis of macrophages after GL261 cells were incubated with or without CS-I/J@CM NPs (12.5 μg/mL) and then irradiated with or without pulsed 1064 nm laser (0.75 W/cm2, 5 min) (M1 (CD86) and M2 (CD206)). (c) Schematic illustration of CS-I/J@CM NPs to decrease the expression of IDO for suppressing the infiltration of Tregs cells in tumor, and decrease the expression of PD-L1 on GL261 cells. (d) The inhibitory effect of IND and CS-I/J@CM NPs on IDO enzyme activity was evaluated by examining the amount of kynurenine (Kyn) in GL261 cell medium. GL261 cells were treated with IFN-γ to induce IDO expression, and subsequently incubated with IND or CS-I/J@CM NPs (12.5 μg/mL) at different concentrations. The amount of Kyn in the culture medium was determined by the absorbance at 490 nm. (e) Detection of IDO expression in the GL261 cells from different groups. (f) Flow cytometry analysis of PD-L1 expression on the GL261 cells after co-cultured with JQ1 or CS-I/J@CM NPs (12.5 μg/mL) for 12 h. (g) Detection of PD-L1 expression in the GL261 cells of different groups. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n = 3).
Apart from TAMs, the immunosuppression of GBM was also induced by tumor-intrinsic factors such as the expressed IDO enzyme to catalyze the conversion of tryptophan (Try) into kynurenine (Kyn), which was involved in the infiltration of T cells and activation of Tregs cells [45,46]. Therefore, combination of immune checkpoint blockade with IDO enzyme inhibitor could induce efficient immunotherapy. In addition, the programmed death ligand 1 (PD-L1, CD274) was also involved in the immunotherapy of GBM. Fig. 2c shows the application of CS-I/J@CM NPs to reprogram the immunosuppressive TIME caused by the overexpression of IDO and PD-L1. The CS-I/J@CM NPs showed the similar inhibiting effect on IDO activity in comparison with free IND at the same concentration (Fig. 2d). The inhibiting effect of free IND or CS-I/J@CM NPs on the IDO expression was charicterized by Western-blot (Fig. 2e). The IDO expression was greatly decreased after treatment with CS-I/J@CM NPs or free IND at the same concentration, which is consistent with the result shown in Fig. 2d. The expression of PD-L1 by GL261 cells was characterized by both flow cytometry (Fig. 2f) and Western-blot (Fig. 2g and Fig. S9a). The IND did not effectively reduce the PD-L1 expression in comparison with JQ1, which illustrates that the PD-L1 was blocked by JQ1 rather than IND. As the JQ1 was loaded onto Cu2-xSe NPs, the CS-I/J@CM NPs could successfully decrease the PD-L1 expression of tumor cells. More importantly, CS-I/J@CM NPs showed better performance in reducing the PD-L1 expression than JQ1 alone under the same JQ1 concentration, because of the improved enrichment and stability of JQ1 by Cu2-xSe nanoparticles. As shown in Fig. S9b, we changed the ratio of CS-IND NPs and CS-JQ1 NPs from 40:1 to 5:1. When the ratio of CS-IND NPs and CS-JQ1 NPs was decreased to 10:1, the PD-L1 expression was decreased. However, when their ratio was further decreased to 5:1, the decrease of PD-L1 was not significant in comparison with that obtained from the ratio of 10:1, which could be due to the toxicity of JQ1 to GL261 cells. On the basis of our optimization, we chose the ratio of CS-IND and CS-JQ1 10:1 for our subsequent experiments. When GL261 cells were irradiated with 1064 nm laser (0.75 W/cm2, 5 min), the PD-L1 expression was also decreased, which illustrates that the NIR II irradiation have no influnce on the PD-L1 block property (Fig. S9c). These results prove that the CS-I/J@CM NPs could act as checkpoint blockade agents for boosting the immunotherapy efficacy of GBM.
4.3. Tumor immunotherapy caused by CS-I/J@CM NPs
Based on the reprogramming of TIME in GBM, the release of cancer antigens through immunogenic cell death (ICD) is quitely necessary for the effective innate and adaptive antitumor immunity [47]. This is because ICD could induce the dying tumor cells to release damage-associated molecular patterns (DAMPs), which could activate antigen-presenting cells (APCs), especially DC cells to stimulate tumor-specific effector T cells (Fig. 3a). The CRT exposure, ATP secretion and HMGB1 release of GL261 cells were detected after they were incubated with CS-I/J@CM NPs and irradiated with or without the 1064 nm laser (0.75 W/cm2, 5 min). As presented in Fig. 3b, GL261 cells incubated without CS-I/J@CM NPs had no CRT exposure on cell membrane because of the very weak red fluorescence on the cell membrane no matter with or without laser irradiation. However, after they were incubated with CS-I/J@CM NPs and irradiated with the 1064 nm NIR II laser (0.75 W/cm2, 5 min), the GL261 cells presented much strong red fluorescence on cell membrane. These results illustrate that very high cell-surface CRT exposure was induced by the abundant ROS generated by CS-I/J@CM NPs under the irradiation of the NIR II laser. The co-localization of fluorescence of CRT (bright red fluorescence) and cell membrane (bright green fluorescence) in Figs. 3b and c clearly showed the distinct characteristics of CRT exposure on cell membrane induced by ICD. Furthermore, we also used flow cytometry to detect the CRT exposure of GL261 cells after different treatments (Fig. 3d). The obtained results were consistent with those in Fig. 3b. We then used the luciferin-based ATP assay to detect the release of ATP from GL261 cells after they were treated with CS-I/J@CM NPs and irradiated with or without the NIR II laser. As shown in Fig. 3e, ATP secretion from the GL261 cells was enhanced significantly after co-incubation with CS-I/J@CM NPs and irradiation with the 1064 nm laser, which was about 2.9-fold that of GL261 cells without irradiation, and 4.7 times that of cells without any treatment. Furthermore, we used enzyme-linked immunosorbent assay (ELISA) to analyze the released HMGB1 (Fig. 3f). The GL261 cells treated with CS-I/J@CM NPs and the NIR II irradiation released the highest HMGB1 compared with other groups, which was almost 3-fold higher than that without the NIR II irradiation, and similar to the CRT exposure and ATP secretion. All these results illustrate that CS-I/J@CM NPs could trigger the ICD of GL261 cells through the massive ROS generated under the laser irradiation.
Fig. 3.

In vitro immune cell death (ICD) induced by ROS generated from the Fenton-like reaction under the NIR II irradiation. (a) Schematic illustration of the DAMPs (CRT, ATP and HMGB1) released from the dying tumor cells to promote the DC mature, triggered by large amount of ROS generated from the degradation of H2O2 by CS-I/J@CM NPs under irradiation. (b) CLSM of CRT (red fluorescence) exposed on the cytomembrane (green fluorescence) of GL261 cells, after different treatment with or without CS-I/J@CM NPs (12.5 μg/mL) (scale bar: 20 μm). (c) Colocalization of CRT and cytomembrane of GL261 cells. (d) Flow cytometry analysis of CRT expose on GL261 cells after co-cultured with or without CS-I/J@CM NPs (12.5 μg/mL). (e) Detection of extracellular ATP secreted. (f) Detection of extracellular HMGB1 released. (g) Flow cytometry analysis of the maturation of DC (CD11c+CD80+CD86+). (h) Normalization of the maturation of DC in Fig. 3g. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n = 5).
The maturation of DC cells induced by the ICD of GL261 cells was also evaluated. Figs. 3g and h show that the bone marrow-derived DCs (bone marrow dendritic cells, BMDCs) co-incubated with GL261 cells, which were pretreated with CS-I/J@CM NPs and irradiation, had the strongest immunogenicity and induced the highest matured DCs (CD11c+CD80+CD86+, 19.8%), compared to 3.0% matured DCs obtained from BMDCs cultured with CS-I/J@CM NPs only without the NIR II laser irradiation, and 1.16% from the Control group. Obviously, the results illustrate that the maturation of DCs could be significantly enhanced by the ICD induced by CS-I/J@CM NPs under the laser irradiation, which is significant for tumor immunotherapy.
4.4. Targeted immunotherapy of GBM with CS-I/J@CM NPs
The above in vitro results demonstrate that CS-I/J@CM NPs could remodel the TIME and induce the ICD to promote the maturation of DCs by the quick generation of ROS and O2 under the 1064 nm laser irradiation. Since Cu2-xSe NPs exhibited very strong NIR absorbance and could convert the NIR light into heat efficiently for photoacoustic (PA) imaging, we used PA imaging to characterize the accumulation of NPs at the tumor site in the brain [48,49]. The PA images of brain were collected at different times after orthotopic GL261 tumor-bearing mice were intravenously injected with CS-I/J@CM NPs, followed by immediate ultrasound sonication (Fig. 4a). The PA signals at the tumor site of mice injected with CS-I/J @CM NPs and treated with sonication reached their maximum at 8 h post injection, which are much higher than the mice from the CS-I/J NPs group at the same time, and demonstrate the homologous adhesion effect of tumor cell membranes for improving nanoparticles accumulation and retention at the tumor site [50]. The accumulated nanoparticles in tumor were also quantified by inductively coupled plasma–mass spectrometry (ICP-MS) and presented in Fig. S10a. The copper concentration in the GBM of mice administrated with CS-I/J@CM NPs and then treated with ultrasound was almost 1.6-fold that of mice injected with CS-I/J NPs. The results further demonstrate that coating CS-I/J NPs with GL261 cell membranes could significantly improve the accumulation and retention of NPs at tumor site because of the homologous adhesion effect. Furthermore, the copper concentration in normal mice after administrated with CS-I/J@CM NPs and treated with ultrasound was also detected. As shown in Fig. S10b, the accumulation of CS-I/J@CM NPs in tumor was 1.4-fold that of normal brain of healthy mice. The results prove that CS-I/J@CM NPs held a better homologous adhesion effect to decrease the damage to normal brain cells.
Fig. 4.

In vivo therapy of GBM by different treatments: (a) Photoacoustic (PA) imaging of tumors from orthotopic GL261 tumor-bearing mice collected before and after tail vein injection of CS-I/J NPs or CS-I/J@CM NPs (dose: 5 mg/kg) at different time points (tumor is highlighted by red circles). (b) H&E staining images of brain slices from different treatment groups of 13 d (scale bar: 1 mm). (c) The relative size of the tumor in brain from Fig. S10c. (d) Survival rates of mice. Immunofluorescence images of tumor slices from GL261 tumor-bearing mice that received different treatments, **p < 0.01 (n = 10). (e) The terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) (green fluorescence) was used to detect the apoptotic cells, in which the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI, blue fluorescence) (scale bar: 100 μm). (f) Myelin basic protein (MBP, red fluorescence) and glial fibrillary acidic protein (GFAP, green fluorescence) in 5 μm immunofluorescence (normal brain (N) and tumor (T) tissue) (scale bar: 25 μm).
Based on the above results, we evaluated the in vivo anticancer effect by using four groups of mice bearing the GL261 tumors. Two groups of mice were only injected with PBS solution (200 μL, Control group) and CS-I/J@CM nanoparticle solution (dose: 5 mg/kg, CS-I/J@CM group) through their tail veins, respectively, followed by immediate ultrasound sonication. They were denoted as Control group and CS-I/J@CM group. Another two groups of mice were also similarly injected with PBS solution and CS-I/J@CM NP solution, followed by ultrasound sonication, and then irradiated with the NIR II laser (0.75 W/cm2, 5 min) at 8 h post-injection. These two groups were denoted as Control + NIR group and CS-I/J@CM + NIR group. As shown in Fig. 4b, the H&E staining slices of brains from four different groups of mice after 13 days treatment illustrate that the mouse tumor from the CS-I/J@CM + NIR group was the smallest among the four groups of mice. Furthermore, the treatment efficacy was also monitored by the magnetic resonance imaging (MRI, Fig. S10c) and their tumor size was decreased by almost 75% in comparison with untreated mice from the Control group (Fig. 4c), which is consistent with the H&E results in Fig. 4b. Obviously, the results demonstrate that CS-I/J@CM NPs could effectively prevent the growth of GBM after the NIR II irradiation. In addition, the survival rates of mice were recorded in Fig. 4d, which shows that the survival rate of the mice from the CS-I/J@CM + NIR group was 80% within 40 days after orthotopic glioblastoma implantation, much higher than that of other groups, in which mice were died eventually. Their body weights were also recorded every day (Fig. S10d), and the weights of all mice were gradually decreased with the progression of tumor, even for the mice from the CS-I/J@CM + NIR group.
To detect the biodistribution of CS-I/J@CM NPs after intravenous administration into glioblastoma-bearing mice, the mice were sacrificed at 1, 3, 5, 7 and 15 days to harvest their major organs for measuring the copper concentrations by ICP-MS. As shown in Fig. S10e, the CS-I/J@CM NPs were mainly distributed in reticuloendothelial organs such as liver, spleen and lung. In addition, the high copper concentration the in kidney suggests that some CS-I/J@CM NPs could be excreted through renal clearance.
To further demonstrate the ability of CS-I/J@CM NPs to kill the cancer cells at tumor site, the terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) analysis (Fig. 4e) was performed. The stronger green fluorescence in TUNEL analysis indicates that the apoptotic tumor cells were increased significantly after the mice were administrated with CS-I/J@CM NPs and then irradiated with the NIR II laser, which further supports that combination of CS-I/J@CM NPs with NIR II irradiation could effectively inhibit tumor growth. In addition, almost no green fluorescence in the tumor from the mice injected with PBS and with or without the NIR II irradiation, which further demonstrate the excellent therapy performance of CS-I/J@CM NPs under the NIR II irradiation.
The effects of ultrasound combined with CS-I/J@CM NPs and the NIR II irradiation on the tumor surrounding brain tissues were assessed by analyzing myelin basic protein (MBP, an oligodendrocyte integrity index) and glial fibrillary acid protein (GFAP, an astrocyte integrity index) [51]. As shown in Fig. 4f, the red fluorescence from MBP analysis indicates that our experimental operation had a negligible effect on the integrity of oligodendrocyte. The green fluorescence of GFAP had no change in the four groups of mice, which means no apparent damage to the astrocyte integrity. These results demonstrate the excellent safety of sonication to the brain.
The above results demonstrate that combination of CS-I/J@CM NPs with the NIR II irradiation had rather good therapeutic effect on GBM in vitro and in vivo. It is important to reveal whether the excellent therapeutic efficacy was achieved through the remodeling of TIME and inducing of ICD to activate immune in vivo by the CS-I/J@CM NPs [52]. We built orthotopic GBM mice (Fig. 5a), which were injected with PBS solution or CS-I/J@CM NP solution, and divided into four groups (i.e., Control group, Control + NIR group, CS-I/J@CM group, and CS-I/J@CM + NIR group). After 1 d therapy, we collected tumor tissues for detection of TIME and ICD-associated factors. To demonstrate the alleviation of tumor hypoxic environment, the hypoxia-inducible factor-1 (HIF-1α) as an important indicator of hypoxic environment was detected [53]. As shown in Fig. S11a, CS-I/J@CM NPs could alleviate the tumors’ hypoxic environment by generation of O2 from degradation of H2O2 in tumor through the Fenton-like reaction under the 1064 nm laser irradiation. In addition, the biomarkers of TAMs were analyzed by immunofluorescence (Fig. 5b). Obviously, the stronger fluorescence of F4/80+INOS+ in the tumor tissues from the mice in the group of CS-I/J@CM + NIR indicates that their M1 phenotype cells were increased in comparison with the mice from the other groups. Figs. S11b and d present the quantification of M1 phenotype cells (F4/80+CD80+) and M2 phenotype cells (F4/80+CD206+) from different groups of mice, which showed that about 21.4% M1 phenotype cells were found in the CS-I/J@CM group, and about 10.9-fold higher than that of CS-I/J@CM group. Furthermore, the M2 phenotype cells in the Control group are 17.9%, which was decreased to 5.98% in the CS-I/J@CM + NIR group. These results illustrate the generation of O2 and ROS through Fenton-like reaction under NIR II irradiation could remodel immunosuppression of TIME caused by M2 macrophages in GBM. Secondly, the capability of IND from CS-I/J@CM NPs preventing the catalytic conversion of Trp into Kyn in tumor is presented in Fig. 5c, more than 20% Trp was inhibited to form Kyn, which was much higher than the groups without IND. The results illustrate that these CS-I/J@CM NPs could inhibit the IDO activity in the GBM and remodel the immunosuppression of TIME associated with IDO. Thirdly, the expression of PD-L1 in GBM was also detected by flow cytometry analysis (Fig. 5d), obviously, the mice treated with CS-I/J@CM nanoparticles and irradiated with or without NIR II laser could significantly decrease the expression of PD-L1 in the tumor tissues. The result further illustrates that CS-I/J@CM NPs could remodel TIME by decreasing the overexpression of PD-L1 on tumor cells.
Fig. 5.

In vivo remodeling tumor immune microenvironment (TIME) and anti-tumor immune response induced by CS-I/J@CM NPs (dose: 5 mg/kg). (a) Schematic illustration of the remodeling of TIME of orthotopic GL261 therapy with CS-I/J@CM NPs under the 1064 nm laser irradiation. (b) Immunofluorescence images of M1 macrophages (F4/80+INOS+) in tumor site (scale bar: 50 μm). (c) The Kyn inhibition. (d) Flow cytometry analysis of PD-L1 expression on GL261 cells in tumor. (e) Immunofluorescence images of CRT exposed in tumor. (scale bar: 100 μm). (f) Flow cytometry analysis of matured DC (CD11c+CD80+CD86+) in lymph nodes of neck. (g) Normalization of the matured DC in Fig. 5f. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n = 5).
The above all results prove that CS-I/J@CM NPs have excellent ability to remodel the immunosuppressive TIME in GBM to improve the immunotherapy. As demonstrated in vitro, CS-I/N@CM NPs could induce ICD of tumor cells by the generated ROS [54]. We also explored whether they could induce ICD in vivo to initiate the immune response (Figs. 5e to g). The CRT exposure was detected by the immunofluorescence (Fig. 5e), and the significantly increased red fluorescence of CRT in the tumor tissue from the mice in the CS-I/J@CM + NIR group illustrate that under the NIR II irradiation, CS-I/J@CM NPs could cause ICD to induce the exposure of CRT of apoptotic tumor cells to activate the antitumor immunity. The flow cytometry analysis in Fig. S12 presents the quantification of CRT exposure obtained from different groups, which shows that the CRT obtained from the CS-I/J@CM + NIR group was 2.9-fold more than that from the Control group, and 1.4-flod more than that from the CS-I/J@CM group. The result further illustrates that CS-I/J@CM NPs could induce serious ICD of tumor cells under the 1064 nm irradiation to expose their CRT, which is consistent with the results shown in Fig. 5e. Furthermore, the matured DC cells (CD11c+CD86+CD80+) in the lymph nodes of neck from four groups of mice are shown in Fig. 5f and Fig. S13, in which about 19.11% of matured DC cells were found in the CS-I/J@CM + NIR group, and they are 5.2- and 1.5-fold higher than those from the Control group and the CS-I/J@CM group, respectively (Fig. 5g).
The effect of immunity was also characterized by the infiltration profiles of both CD8+T cells and immunosuppressive Tregs cells in the tumor and spleen tissues after 3 d post-injection of PBS or CS-I/J@CM NPs. The immunocompetent GL261 tumor mice were also divided into four groups, i.e. Control, Control + NIR, CS-I/J@CM and CS-I/J@CM + NIR groups as schematically shown in Fig. 6a. As shown in Fig. 6b that more than 11.11% CD8+T cells had been activated and infiltrated into the tumor in the CS-I/J@CM + NIR group, which were almost 2.7-fold higher than that from the Control group. In contrast, the CD8+T cells from the CS-I/J@CM group were only 1.4-fold higher than those from the Control group, and were quite lower than the CS-I/J@CM + NIR group, further demonstrating that CS-I/J@CM NPs with the NIR II irradiation could effectively elicit the infiltration of CD8+T cells into tumor (Fig. 6b). The remarkable red fluorescence of CD8+T cells in the tumor tissues from the CS-I/J@CM + NIR group (Fig. 6c), in comparison with the absence of red fluorescence from other groups, further demonstrating the enhancement of the activated and infiltrated intratumoral CD8+T cells through the CS-I/J@CM NPs and the NIR II irradiation. Furthermore, the cytokines of tumor necrosis factor alpha (TNF-α) secreted in serum by the activated T cells were analyzed through enzyme-linked immunosorbent assay (ELISA, Fig. 6d). The TNF-α from the CS-I/J@CM + NIR group was about 4.3-fold and 1.6-fold higher than that obtained from the Control group and the CS-I/J@CM group, respectively, which proves that those CS-I/J@CM NPs could efficiently induce the infiltration and activation of T cells under the assistance of NIR II irradiation. The immunosuppressive Treg cells (CD3+CD4+CD25+Foxp3+cells) in tumors from the four groups of mice were also detected and shown in Fig. 6e and Fig. S14. There were 45.61% and 42.78% of Treg cells in the tumors of mice injected with PBS solution and irradiated with or without NIR II light. However, after the GBM-bearing mice were treated by intravenous injection of CS-I/J@CM NPs and then irradiation without and with NIR II laser, the Tregs cells in their tumors were decreased to 12.46% and 9.03%, respectively, which were only one third and one fifth of those obtained from the Control group and Control + NIR group. These results further illustrate that CS-I/J@CM NPs could not only induce the infiltration of both CD8+T cells into tumor, but also could decrease the immunosuppressive Treg cells, both which are beneficial to tumor immunotherapy.
Fig. 6.

In vivo infiltration profiles of immune cells in the brain and spleen. (a) Schematic illustration of the therapy of orthotopic GBM with CS-I/J@CM NPs under the irradiation of 1064 nm laser (dose: 5 mg/kg). (b) Flow cytometry analysis of CD8+T cells (CD3+CD8+) in tumor and the normalization. (c) Immunofluorescence images of CD8+T cells in tumor tissue (scale bar: 100 μm). (d) Tumor necrosis factor alpha (TNF-α) secreted in serum analyzed by enzyme-linked immunosorbent assay (ELISA). (e) Flow cytometry analysis of Tregs cells (CD3+CD25+CD4+Foxp3+) in tumor and the normalization. (f) Flow cytometry analysis of CD8+T cells (CD3+CD8+) in the spleen and the normalization. (g) Flow cytometry analysis of TEM cells (CD3+CD44+CD62Llow) in the spleen after 30 days orthotopic glioblastoma implantation. (h) Bioluminescent images collected on 10 d after flank tumor implantation, the naive mice (left) and long-term survivors (right) were inoculated subcutaneously with 2.5 × 106 GL261-Luciferase cells in the right flank. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n = 5).
The CD8+T cells in their spleens were also detected by flow cytometry analysis to further evidence the activation of antitumor immunity. As shown in Fig. 6f, the CD8+T cells were increased from 10.3% in the spleen of Control group to 15.4% in the CS-I/J@CM + NIR group. This results again prove that CS-I/J@CM NPs could activate the anti-tumor immune response.
Although the immune response of GBM-bearing mice was activated by CS-I/J@CM NPs with NIR II irradiation, whether the treated mice held a memory immunity to prevent the recurrence of tumor is extremely important. We detected the memory T cells (TEM, CD3+CD44+CD62Llow) in the spleen of mice by flow cytometry analysis and showed in Fig. 6g and Fig. S15. The TEM cells in the spleen of mice from the Control group were 7.20%, which was increased to 29.4% in the CS-I/J@CM + NIR group. The TEM cells from the CS-I/J@CM + NIR group were 4 times of that from the Control group. The results prove the effective immunotherapy in inducing much more memory T cells.
To further evaluate the immunotherapy efficacy conveniently, we built the glioblastoma by using GL261-Luciferase cells. The bioluminescence imaging (Fig. S16) was used to evaluate the efficacy of treatment at day 7, 10, 20 and 30. The bioluminescence intensity could reflect the growth of tumor and treatment efficacy. To further demonstrate the effect of memory immunity, the surviving GBM-bearing mice after orthotopic glioblastoma implantation 30 d were subcutaneously injected with GL261-Luciferase cells into the right flank. The healthy mice were also subcutaneously injected with the same cells for comparison. Their bioluminescent images recorded on day 10 after the flank tumor implantation were shown in Fig. 6h. The strong bioluminescence was observed in the right flank of native mice. The absence of bioluminescence in the treated GBM-bearing survivors demonstrates their excellent anti-tumor memory immunity.
In addition to their excellent antitumor efficacy, the in vivo biocompatibility of CS-I/J@CM NPs was assessed by hematoxylin-eosin (H&E) staining of major organs (i.e. heart, liver, spleen, lung, kidney) of mice, which were sacrificed at 7 days postinjection of CS-I/J@CM NPs. There was no obvious damage to the major organs after injection of CS-I/J@CM NPs, in comparison with healthy mice (Fig. S17), which demonstrates the good biocompatibility of CS-I/J@CM NPs.
5. Conclusion
In summary, we demonstrate the significant improvement of GBM immunotherapy by remodeling the immunosuppression via smart all-in-one biomimetic CS-I/J@CM NPs, which can effectively cross the BBB and target at tumor under the assistance of noninvasively focused ultrasound. The biomimetic nanoparticles are based on ultrasmall Cu2-xSe nanoparticles (3.1 ± 0.4 nm), which were prepared under ambient conditions and functionalized with IND (an inhibitor of indoleamine 2,3-dioxygeneasem in tumor) and JQ1 (an inhibitor of PD-L1), and then wrapped with cancer cell membrane to endow their targeting capability and improve their accumulation in tumor. The theranostic CS-I/J@CM NPs could generate large amounts of ROS and oxygen through the Fenton-like reaction to repolarize M2-macrophages into M1 type to relieve the suppression of TIME in GBM. Furthermore, the inhibitor IND released from CS-I/J@CM NPs could suppress Treg cells and increase the ratio of T cells/Treg cells at tumor site to remodel TIME. Furthermore, the small molecular inhibitor JQ1 released from CS-I/J@CM NPs acted as anti-programmed cell death ligand-1 (a-PD-L1) to reduce the express of PD-L1. The CS-I/J@CM NPs also could induce immunogenic cell death (ICD) responses to boost the adaptive anti-GBM immunity. All these effects lead to drastically enhanced GBM immunotherapy. This work demonstrates that our all-in-one CS-I/J@CM NPs could remodel TIME and simultaneously play roles in checkpoint blockade immunotherapy for glioblastoma. It brings new insights into the rational design of robust biomimetic nanoparticles for tumor immunotherapy, and could significantly influence the immunotherapy of other cold tumors in the future.
CRediT authorship contribution statement
Tingting Wang: Conceptualization, designed the experiments and wrote the manuscript. Hao Zhang: Performed the photoaccoustic imaging experiments and, Formal analysis. Weibao Qiu: provided the focused ultrasound sonicator and revised the manusicript. Yaobao Han: re-evaluated the anti-tumour activity. Hanghang Liu: re-evaluated the anti-tumour activity. Zhen Li: Conceptualization, designed the experiments and wrote the manuscript.
Declaration of competing interest
The authors declare no conflict of interest.
Acknowledgment
Z. Li acknowledges support from the Jiangsu Provincial Key Research Development Program (BE2019660), the National Natural Science Foundation of China (81971671), the National Key Research and Development Program of China (2018YFA0208800), and Suzhou Municipal Science and Technology Bureau (N312861019). T. Wang and H. Zhang are very appreciated for the support of China Postdoctoral Science Foundation (2020M671586, 2020M681720). The authors also are grateful for support from the Jiangsu Provincial Key Laboratory of Radiation Medicine and Protection, the Priority Academic Development Program of Jiangsu Higher Education Institutions (PAPD).
Footnotes
Peer review under responsibility of KeAi Communications Co., Ltd.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bioactmat.2021.12.029.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Chung E.J., Cheng Y., Morshed R., Nord K., Han Y., Wegscheid M.L. Fibrin-binding, peptide amphiphile micelles for targeting glioblastoma. Biomaterials. 2014;35(4):1249–1256. doi: 10.1016/j.biomaterials.2013.10.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim M.M., Parolia A., Dunphy M.P., Venneti S. Non-invasive metabolic imaging of brain tumours in the era of precision medicine. Nat. Rev. Clin. Oncol. 2016;13(12):725–739. doi: 10.1038/nrclinonc.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stupp R., Hegi M.E., Mason W.P., Van den Bent M.J., Taphoorn M.J.B., Janzer R.C., Ludwin S.K., Allgeier A., Fisher B., Belanger K., Hau P., Brandes A.A., Gijtenbeek J., Marosi C., Vecht C.J., Mokhtari K., Wesseling P., Villa S., Eisenhauer E., Gorlia T., Weller M., Lacombe D., Cairncross J.G., Mirimanoff R.O., European Org, R.; G Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 4.Trac N.T., Chung E.J. Peptide-based targeting of immunosuppressive cells in cancer. Bioact. Mater. 2020;5(1):92–101. doi: 10.1016/j.bioactmat.2020.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jackson C.M., Choi J., Lim M. Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat. Immunol. 2019;20(9):1100–1109. doi: 10.1038/s41590-019-0433-y. [DOI] [PubMed] [Google Scholar]
- 6.Zhang C., Pu K. Molecular and nanoengineering approaches towards activatable cancer immunotherapy. Chem. Soc. Rev. 2020;49(13):4234–4253. doi: 10.1039/c9cs00773c. [DOI] [PubMed] [Google Scholar]
- 7.Zhang J., Chen C., Li A., Jing W., Sun P., Huang X., Liu Y., Zhang S., Du W., Zhang R., Liu Y., Gong A., Wu J., Jiang X. Immunostimulant hydrogel for the inhibition of malignant glioma relapse post-resection. Nat. Nanotechnol. 2021;5(16) doi: 10.1038/s41565-020-00843-7. [DOI] [PubMed] [Google Scholar]
- 8.Zhang P., Miska J., Lee C.C., Rashidi A., Panek W.K., An S., Zannikou M., Lopez-Rosas A., Han Y., Xiao T., Pituch K.C., Kanojia D., Balyasnikova I.V., Lesniak M.S. Therapeutic targeting of tumor-associated myeloid cells synergizes with radiation therapy for glioblastoma. P. Natl. Acad. Sci. USA. 2019;116(47):23714–23723. doi: 10.1073/pnas.1906346116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishihara A., Ishihara J., Watkins E.A., Tremain A.C., Nguyen M., Solanki A., Katsumata K., Mansurov A., Budina E., Alpar A.T., Hosseinchi P., Maillat L., Reda J.W., Kageyama T., Swartz M.A., Yuba E., Hubbell J.A. Prolonged residence of an albumin-IL-4 fusion protein in secondary lymphoid organs ameliorates experimental autoimmune encephalomyelitis. Nat. Biomed. Eng. 2020;4(11):1117. doi: 10.1038/s41551-020-00649-x. [DOI] [PubMed] [Google Scholar]
- 10.Lim M., Xia Y., Bettegowda C., Weller M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018;15(7):422–442. doi: 10.1038/s41571-018-0003-5. [DOI] [PubMed] [Google Scholar]
- 11.Rodell C.B., Arlauckas S.P., Cuccarese M.F., Garris C.S., Ahmed R.L.M.S., Kohler R.H., Pittet M.J., Weissleder R. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2018;2(8):578–588. doi: 10.1038/s41551-018-0236-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gilkes D.M., Semenza G.L., Wirtz D. Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat. Rev. Cancer. 2014;14(6):430–439. doi: 10.1038/nrc3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taylor C.T., Colgan S.P. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat. Rev. Immunol. 2017;17(12):774–785. doi: 10.1038/nri.2017.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang G., Xu L., Chao Y., Xu J., Sun X., Wu Y., Peng R., Liu Z. Hollow MnO2 as a tumor-microenvironment-responsive biodegradable nano-platform for combination therapy favoring antitumor immune responses. Nat. Commun. 2017;8:902. doi: 10.1038/s41467-017-01050-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu X., Gong X., Wang Y., Li J., Wang H., Wang J., Sha X., Li Y., Zhang Z. Reprogramming tumor associated macrophages toward M1 phenotypes with nanomedicine for anticancer immunotherapy. Adv. Ther. 2020;3(5):1900181. [Google Scholar]
- 16.Shi C., Liu T., Guo Z., Zhuang R., Zhang X., Chen X. Reprogramming tumor-associated macrophages by nanoparticle-based reactive oxygen species photogeneration. Nano Lett. 2018;18(11):7330–7342. doi: 10.1021/acs.nanolett.8b03568. [DOI] [PubMed] [Google Scholar]
- 17.Feng B., Hou B., Xu Z., Saeed M., Yu H., Li Y. Self-amplified drug delivery with light-inducible nanocargoes to enhance cancer immunotherapy. Adv. Mater. 2019;31(40):1902960. doi: 10.1002/adma.201902960. [DOI] [PubMed] [Google Scholar]
- 18.He S., Li J., Lyu Y., Huang J., Pu K. Near-infrared fluorescent macromolecular reporters for real-time imaging and urinalysis of cancer immunotherapy. J. Am. Chem. Soc. 2020;142(15):7075–7082. doi: 10.1021/jacs.0c00659. [DOI] [PubMed] [Google Scholar]
- 19.Yin J.Q., Zhu J., Ankrum J.A. Manufacturing of primed mesenchymal stromal cells for therapy. Nat. Biomed. Eng. 2019;3(2):90–104. doi: 10.1038/s41551-018-0325-8. [DOI] [PubMed] [Google Scholar]
- 20.Zeng Z., Zhang C., Li J., Cui D., Jiang Y., Pu K. Activatable polymer nanoenzymes for photodynamic immunometabolic cancer therapy. Adv. Mater. 2021;33(4):2007247. doi: 10.1002/adma.202007247. [DOI] [PubMed] [Google Scholar]
- 21.Muller A.J., DuHadaway J.B., Donover P.S., Sutanto Ward E., Prendergast G.C. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat. Med. 2005;11(3):312–319. doi: 10.1038/nm1196. [DOI] [PubMed] [Google Scholar]
- 22.Sheridan C. Ido inhibitors move center stage in immuno-oncology. Nat. Biotechnol. 2015;33(4):321. doi: 10.1038/nbt0415-321. [DOI] [PubMed] [Google Scholar]
- 23.Holmgaard R.B., Zamarin D., Li Y., Gasmi B., Munn D.H., Allison J.P., Merghoub T., Wolchok J.D. Tumor-expressed Ido recruits and activates MDSCs in a treg-dependent manner. Cell Rep. 2015;13(2):412–424. doi: 10.1016/j.celrep.2015.08.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lang T., Zheng Z., Huang X., Liu Y., Zhai Y., Zhang P., Li Y., Yin Q. Ternary regulation of tumor microenvironment by heparanase‐sensitive micelle‐loaded monocytes improves chemo‐immunotherapy of metastatic breast cancer. Adv. Funct. Mater. 2021;31(10):2007402. [Google Scholar]
- 25.Acharya A.P., Sinha M., Ratay M.L., Ding X., Balmert S.C., Workman C.J., Wang Y., Vignali D.A.A., Little S.R. Localized multi-component delivery platform generates local and systemic anti-tumor immunity. Adv. Funct. Mater. 2017;27(5):1604366. [Google Scholar]
- 26.Trac N., Chung E.J. Overcoming physiological barriers by nanoparticles for intravenous drug delivery to the lymph nodes. Exp. Biol. Med. 2021;246(22):2358–2371. doi: 10.1177/15353702211010762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madore J., Strbenac D., Vilain R., Menzies A.M., Yang J.Y.H., Thompson J.F., Long G.V., Mann G.J., Scolyer R.A., Wilmott J.S. PD-L1 negative status is associated with lower mutation burden, differential expression of immune-related genes, and worse survival in stage III melanoma. Clin. Cancer Res. 2016;22(15):3915–3923. doi: 10.1158/1078-0432.CCR-15-1714. [DOI] [PubMed] [Google Scholar]
- 28.Furtado D., Bjornmalm M., Ayton S., Bush A.I., Kempe K., Caruso F. Overcoming the blood-brain barrier: the role of nanomaterials in treating neurological diseases. Adv. Mater. 2018;30(46):1801362. doi: 10.1002/adma.201801362. [DOI] [PubMed] [Google Scholar]
- 29.Wainwright D.A., Chang A.L., Dey M., Balyasnikova I.V., Kim C.K., Tobias A. Durable therapeutic efficacy utilizing combinatorial blockade against Ido, CTLA-4, and PD-L1 in mice with brain tumors. Clin. Cancer Res. 2015;21(3):662. doi: 10.1158/1078-0432.CCR-14-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X., Tsibouklis J., Weng T., Zhang B., Yin G., Feng G., Cui Y., Savina I.N., Mikhalovska L.I., Sandeman S.R., Howel C.A., Mikhalovsky S.V. Nano carriers for drug transport across the blood-brain barrier. J. Drug Target. 2017;25(1):17–28. doi: 10.1080/1061186X.2016.1184272. [DOI] [PubMed] [Google Scholar]
- 31.Villasenor R., Ozmen L., Messaddeq N., Gruninger F., Loetscher H., Keller A., Betsholtz C., Freskgard P.O., Collin L. Trafficking of endogenous immunoglobulins by endothelial cells at the blood-brain barrier. Sci. Rep. 2016;6:25658. doi: 10.1038/srep25658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miyasato Y., Takashima Y., Takeya H., Yano H., Hayano A., Nakagawa T., Makino K., Takeya M., Yamanaka R., Komohara Y. The expression of PD-1 ligands and Ido1 by macrophage/microglia in primary central nervous system lymphoma. J. Clin. Exp. Neuropsychol. 2018;58(2):95–101. doi: 10.3960/jslrt.18001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galstyan A., Markman J.L., Shatalova E.S., Chiechi A., Korman A.J., Patil R., Klymyshyn D., Tourtellotte W.G., Israel L.L., Braubach O., Ljubimov V.A., Mashouf L.A., Ramesh A., Grodzinski Z.B., Penichet M.L., Black K.L., Holler E., Sun T., Ding H., Ljubimov A.V., Ljubimova J.Y. Blood-brain barrier permeable nano immunoconjugates induce local immune responses for glioma therapy. Nat. Commun. 2019;10:3850. doi: 10.1038/s41467-019-11719-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang J., Hu L. Immunomodulators targeting the PD-1/PD-L1 protein-protein interaction: from antibodies to small molecules. Med. Res. Rev. 2019;39(1):265–301. doi: 10.1002/med.21530. [DOI] [PubMed] [Google Scholar]
- 35.Zhang S., Sun C., Zeng J., Sun Q., Wang G., Wang Y., Wu Y., Dou S., Gao M., Li Z. Ambient aqueous synthesis of ultrasmall PEGylated Cu2-xSe nanoparticles as a multifunctional theranostic agent for multimodal imaging guided photothermal therapy of cancer. Adv. Mater. 2016;28(40):8927–8936. doi: 10.1002/adma.201602193. [DOI] [PubMed] [Google Scholar]
- 36.Han Y., Wang T., Liu H., Zhang S., Zhang H., Li Z. The release and detection of copper ions from ultrasmall theranostic Cu2-xSe nanoparticles. Nanoscale. 2019;11(24):11819–11829. doi: 10.1039/c9nr02884f. [DOI] [PubMed] [Google Scholar]
- 37.Wang T., Zhang H., Liu H., Yuan Q., Ren F., Han Y., Sun Q., Li Z., Gao M. Boosting H2O2‐guided chemodynamic therapy of cancer by enhancing reaction kinetics through versatile biomimetic fenton nanocatalysts and the second near‐infrared light irradiation. Adv. Funct. Mater. 2019;30(3):1906128. [Google Scholar]
- 38.Liu H., Han Y., Wang T., Zhang H., Xu Q., Yuan J., Li Z. Targeting microglia for therapy of Parkinson's disease by using biomimetic ultrasmall nanoparticles. J. Am. Chem. Soc. 2020;142(52):21730–21742. doi: 10.1021/jacs.0c09390. [DOI] [PubMed] [Google Scholar]
- 39.Wang T., Zhang H., Han Y., Liu H., Ren F., Zeng J., Sun Q., Li Z., Gao M. Light-enhanced O2-evolving nanoparticles boost photodynamic therapy to elicit antitumor immunity. ACS Appl. Mater. Interfaces. 2019;11(18):16367–16379. doi: 10.1021/acsami.9b03541. [DOI] [PubMed] [Google Scholar]
- 40.Zhang H., Wang T., Liu H., Ren F., Qiu W., Sun Q., Yan F., Zheng H., Li Z., Gao M. Second near-infrared photodynamic therapy and chemotherapy of orthotopic malignant glioblastoma with ultra-small Cu2-xSe nanoparticles. Nanoscale. 2019;11(16):7600–7608. doi: 10.1039/c9nr01789e. [DOI] [PubMed] [Google Scholar]
- 41.Zhou X., Liu H., Zheng Y., Han Y., Wang T., Zhang H., Sun Q., Li Z. Overcoming radioresistance in tumor therapy by alleviating hypoxia and using the HIF-1 inhibitor. ACS Appl. Mater. Interfaces. 2020;12(4):4231–4240. doi: 10.1021/acsami.9b18633. [DOI] [PubMed] [Google Scholar]
- 42.Wen M., Ouyang J., Wei C., Li H., Chen W., Liu Y.N. Artificial enzyme catalyzed cascade reactions: antitumor immunotherapy reinforced by NIR-II light. Angew. Chem. Int. Ed. 2019;58(48):17425–17432. doi: 10.1002/anie.201909729. [DOI] [PubMed] [Google Scholar]
- 43.Liu M., Connor R.S., Trefely S., Graham K., Snyder N.W., Beatty G.L. Metabolic rewiring of macrophages by CpG potentiates clearance of cancer cells and overcomes tumor-expressed CD47-mediated 'don't-eat-me' signal. Nat. Immunol. 2019;20(3):265–275. doi: 10.1038/s41590-018-0292-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng Y., Han Y., Wang T., Liu H., Sun Q., Hu S., Chen S.J., Chen J.Q., Li Z. Reprogramming tumor-associated macrophages via ROS-mediated novel mechanism of ultra-small Cu2-xSe nanoparticles to enhance anti-tumor immunity. Adv. Funct. Mater. 2021 doi: 10.1002/adfm.202108971. [DOI] [Google Scholar]
- 45.Li J., Cui D., Huang J., He S., Yang Z., Zhang Y., Luo Y., Pu K. Organic semiconducting pro-nanostimulants for near-infrared photoactivatable cancer immunotherapy. Angew. Chem. Int. Ed. 2019;58(36):12680–12687. doi: 10.1002/anie.201906288. [DOI] [PubMed] [Google Scholar]
- 46.Zhang C., Zeng Z., Cui D., He S., Jiang Y., Li J., Huang J.G., Pu K. Semiconducting polymer nano-PROTACs for activatable photo-immunometabolic cancer therapy. Nat. Commun. 2021;12:2934. doi: 10.1038/s41467-021-23194-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang F., Li F., Lu G.H., Nie W., Zhang L., Lv Y., Bao W., Gao X., Wei W., Pu K., Xie H.Y. Engineering magnetosomes for ferroptosis/immunomodulation synergism in cancer. ACS Nano. 2019;13(5):5662–5673. doi: 10.1021/acsnano.9b00892. [DOI] [PubMed] [Google Scholar]
- 48.Zhang H., Wang T., Qiu W., Han Y., Sun Q., Zeng J., Yan F., Zheng H., Li Z., Gao M. Monitoring the opening and recovery of the blood-brain barrier with noninvasive molecular imaging by biodegradable ultrasmall Cu2-xSe nanoparticles. Nano Lett. 2018;18(8):4985–4992. doi: 10.1021/acs.nanolett.8b01818. [DOI] [PubMed] [Google Scholar]
- 49.Zhang H., Zeng X., Li Z. Copper-chalcogenide-based multimodal nanotheranostics. ACS Applied. Bio. Materials. 2020;3(10):6529–6537. doi: 10.1021/acsabm.0c00937. [DOI] [PubMed] [Google Scholar]
- 50.Jiang Y., Krishnan N., Zhou J., Chekuri S., Wei X., Kroll A.V., Yu C.L., Duan Y., Gao W., Fang R.H., Zhang L. Engineered cell-membrane-coated nanoparticles directly present tumor antigens to promote anticancer immunity. Adv. Mater. 2020;32(30):2001808. doi: 10.1002/adma.202001808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kadiyala P., Li D., Nunez F.M., Altshuler D., Doherty R., Kuai R., Yu M., Kamran N., Edwards M., Moon J.J., Lowenstein P.R., Castro M.G., Schwendeman A. High-density lipoprotein-mimicking nanodiscs for chemo-immunotherapy against glioblastoma multiforme. ACS Nano. 2019;13(2):1365–1384. doi: 10.1021/acsnano.8b06842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pu K., Zhang Y. Transformable nanosensitizer with tumour microenvironment-activated sonodynamic process and calcium release for enhanced cancer immunotherapy. Angew. Chem. Int. Ed. 2021;60(25):14051–14059. doi: 10.1002/anie.202102703. [DOI] [PubMed] [Google Scholar]
- 53.Xu Q., Zhang H., Liu H., Han Y., Qiu W., Li Z. Inhibiting autophagy flux and DNA repair of tumor cells to boost radiotherapy of orthotopic glioblastoma. Biomaterials. 2021;280:121287. doi: 10.1016/j.biomaterials.2021.121287. [DOI] [PubMed] [Google Scholar]
- 54.Jiang Y., Huang J., Xu C., Pu K. Activatable polymer nanoagonist for second near-infrared photothermal immunotherapy of cancer. Nat. Commun. 2021;12(1):742. doi: 10.1038/s41467-021-21047-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.