

SUMMARY
The duality of liquid-liquid phase separation (LLPS) of cellular components into membraneless organelles defines the nucleation of both normal and disease processes including stress granule (SG) assembly. From mounting evidence of LLPS utility by viruses, we discover that HIV-1 nucleocapsid (NC) protein condenses into zinc-finger (ZnF)-dependent LLPSs that are dynamically influenced by cytosolic factors. ZnF-dependent and Zinc (Zn2+)-chelation-sensitive NC-LLPS are formed in live cells. NC-Zn2+ ejection reverses the HIV-1 blockade on SG assembly, inhibits NC-SG assembly, disrupts NC/Gag-genomic RNA (vRNA) ribonucleoprotein complexes, and causes nuclear sequestration of NC and the vRNA, inhibiting Gag expression and virus release. NC ZnF mutagenesis eliminates the HIV-1 blockade of SG assembly and repositions vRNA to SGs. We find that NC-mediated, Zn2+-coordinated phase separation is conserved among diverse retrovirus subfamilies, illustrating that this exquisitely evolved Zn2+-dependent feature of virus replication represents a critical target for pan-antiretroviral therapies.
Graphical Abstract
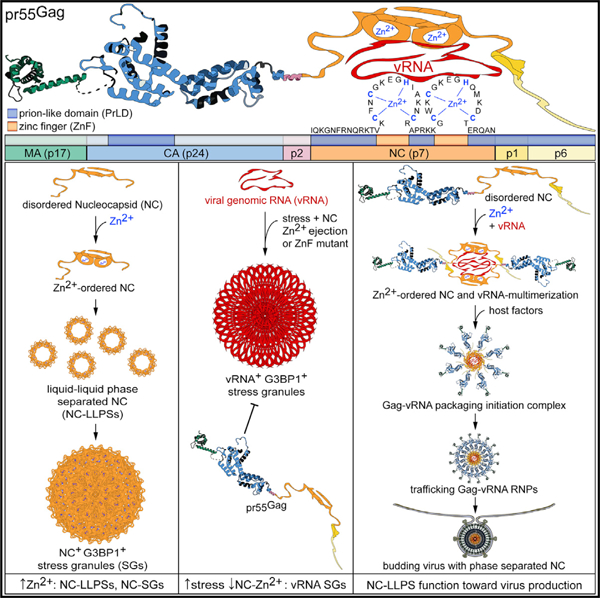
In Brief
Monette et al. discover a high degree of conservation of zinc-finger embedded, intrinsically disordered prion-like domains across retrovirus Gag proteins. These domains within the Gag Nucleocapsid regulate the formation of zinc-dependent liquid-liquid phase condensates and stress granules in HIV-1-expressing cells to induce repositioning of the viral genomic RNA.
INTRODUCTION
Eukaryotic cells have liquid-like but spatially organized, membraneless compartments such as stress granules (SGs), P bodies (PBs), and nucleoli, enriched in multivalent proteins having disordered and modular domains, and which are concentrated away from the aqueous environment by a process called liquid-liquid phase separation (LLPS) (Ambadipudi et al., 2017; Bergeron-Sandoval et al., 2016). Phase separation initiates LLPS and is emerging as one of the underlying principles of cell organization governing cell function and survival (Uversky, 2017), such that protective SGs assemble in response to environmental and genotoxic stressors (Franzmann and Alberti, 2019; Kroschwald and Alberti, 2017). Following stress withdrawal, LLPS and SGs rapidly disassemble, and are thus centers of exquisite control in normal cell physiology.
Viruses depend on an exhaustive array of host cell components and like liquid droplets (Camus et al., 2013), LLPSs represent ideal platforms for many virus replication stages (Alenquer et al., 2019; Heinrich et al., 2018; Liu et al., 2014; Nikolic et al., 2017). In addition to co-opting cellular proteins, LLPS may promote proximity-dependent interactions and the assembly of viral factories for virus expression (Nikolic et al., 2017; Novoa et al., 2005). Numerous viruses, however, inhibit SG assembly to evade the inherently antiviral nature of these ribonucleoprotein assemblies required for protein synthesis (Poblete-Durán et al., 2016). SG blockade by viruses is achieved by modification, cleavage, sequestering, or redistribution of SG-proteins and translation initiation factors (Zhang et al., 2019). HIV-1 has evolved multiple mechanisms to block the assembly of canonical and non-canonical SGs. For example, HIV-1 blocks SG assembly induced by heavy metals as well as SG assembly induced by the expression of key SG-inducing proteins, G3BP1 and TIAR (Valiente-Echeverría et al., 2014). HIV-1 achieves this blockade via interactions between the viral structural precursor polyprotein (pr55Gag), and several host factors such as eukaryotic elongation factor 2, G3BP1, Cyclophilin A, and eIF4E (Cinti et al., 2016; Valiente-Echeverría et al., 2014). A single point mutation, in fact, in the C-terminal capsid (CA) domain of pr55Gag, prevents host factor binding and disables the blockade to SG assembly, likely by changing the conformation of this pr55Gag domain (Valiente-Echeverría et al., 2014). Complete regulation of this mechanism is increasingly puzzling from demonstrations that the nucleocapsid (NC) domain of pr55Gag promotes Gag- and CA-resistant SGs (Rao et al., 2018), and suggests that an equilibrium exists between SG assembly and disassembly during HIV-1 replication.
NC is among the viral protease (PR) cleavage products generated from pr55Gag, also yielding matrix (MA) and capsid (CA) proteins. This 55-amino acid (7-kDa) protein is a major component of infectious virus cores, with ~2,000 HIV-1 NC molecules coating the dimeric, viral genomic RNA (vRNA) (Darlix et al., 1995). NC has two zinc-coordinating finger (ZnF) domains in CCHC conformation controlling several steps of retroviral replication from transcription to vRNA selection for packaging (Muriaux and Darlix, 2010). The highly conserved amino acids essential for the many functions of retroviral NC ZnFs, along with their inability to produce viable escape mutants from drug targeting, poise them as ideal therapeutic targets (Goebel et al., 2001; Rice et al., 1995, 1997b).
NC is characterized as a nucleic acid chaperone from its ZnF-dependent roles in nucleic acid binding (single-stranded DNA and RNA), condensing, annealing, and strand transfer (Muriaux and Darlix, 2010). It localizes to the cytoplasm, and to the nucleus on account of its nuclear localization signals (Anton et al., 2015; Lochmann et al., 2013). Like NC, other cellular RNA-binding proteins and chaperones (e.g., G3BP1, TIAR, TIA-1, DDX6, TDP-43, FUS/TLS, and hnRNPs) are SG and PB components (Kedersha et al., 2016; Poblete-Durán et al., 2016), may be Zn2+-regulated (Garnier et al., 2017; Rayman et al., 2018), and form LLPSs due to their low-complexity, intrinsically disordered prion-like domains (PrLDs) (Kedersha et al., 2016; Maharana et al., 2018). The entire NC is a PrLD at the C terminus of pr55Gag, where NC exerts its functions via its ZnFs (Darlix et al., 2011; Dick and Vogt, 2014). Retroviral HIV-1, SIV, and MuLV NCs are also important for virion structure, whereby NC-mutated virions are immature and have aberrant particle morphology (Cimarelli et al., 2000; Muriaux et al., 2004; Tanchou et al., 1998; Yovandich et al., 2001). Rabies virus (RABV), vesicular stomatitis virus (VSV), Ebola, and measles virus proteins have PrLDs and use LLPS mechanisms for host-defense shielding replication dynamics in membraneless inclusions within viroplasms (Heinrich et al., 2018; Nikolic et al., 2017).
From demonstrations that other viral proteins form LLPS, and from shared similarities that NC has with other proteins that bind RNA, possess PrLDs, and induce SG assembly and LLPS, we investigated the possibility that NC function may be triggered by its ability to phase separate. We performed biochemical and cellular experiments to show that NC condenses into LLPSs in vitro and in living cells. NC-LLPSs displayed fundamental properties of a liquid phase, including a spherical shape from surface tension, an ability to fuse and separate, and rapid internal and cellular diffusion (Heinrich et al., 2018). We observed that NC-LLPS and the HIV-1 blockade to SG assembly are ZnF-dependent, blocked by NC ZnF mutagenesis or Zn2+ chelation, to promote aberrant cellular localization of NC, Gag, and the vRNA, retention of Gag, and reduced virus production. In silico methods mapped conserved Gag protein PrLD and ZnF positioning across retroviruses. We demonstrated that both HIV-1 Gag and NC from several retroviral subfamily members undergo Zn2+-dependent LLPS. Finally, we have observed that NC is generated in cells by PR. Our results point to a pan-retrovirus NC-specific, membraneless, LLPS mechanism nucleating virus assembly, and controlled by reversible, Zn2+-dependent secondary structures supported by PrLDs. In this report, we provide insight on how Zn2+-dependent NC-LLPSs promote vRNA trafficking, vRNA packaging, and virus release.
RESULTS
NC Condenses into ZnF-Dependent LLPSs Dynamically Regulated by the Cellular Environment
To establish that HIV-1 NC condensed into LLPSs, increasing concentrations of purified Zn2+-loaded and green-fluorescence-labeled NC protein (Wu et al., 1996) were mixed with buffers containing Ficoll or dextran as crowding agents (Alberti et al., 2019; Boehning et al., 2018; Maharana et al., 2018). Laser microscopy and differential interference contrast (DIC) validated that green fluorescent NC-LLPSs had expected spherical morphology. NC-LLPSs could be seen at 5 µM of NC protein with Ficoll, but were more numerous and spherical at the optimal concentration of 10 µM, with homogeneous NC-LLPS sizes ranging from 0.5 to 2 µm (Alberti et al., 2019) (5 versus 10 µM of NC, p < 0.0001) (Figure 1A).
Figure 1. Zn2+-Dependent HIV-1 NC Phase Separation Is Dynamically Influenced by the Cellular Microenvironment.
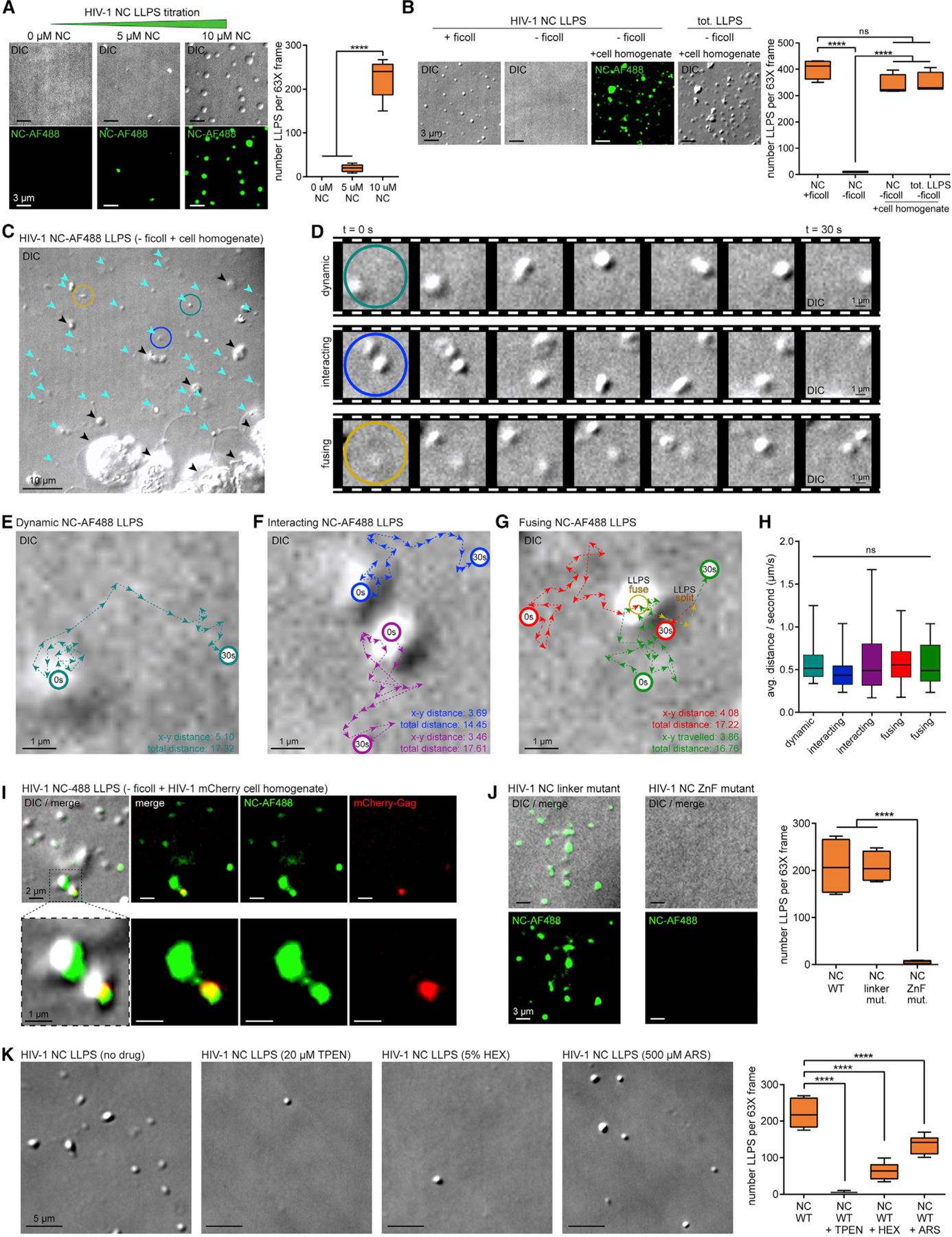
(A) LLPS titration of NC protein (green) mixed with phase separation buffer and Ficoll, visualized by laser microscopy and DIC, with corresponding boxplots (n = 3).
(B) Ficoll-substituted cell homogenates inducing NC-LLPSs (green), with corresponding boxplots (n = 3).
(C) NC-LLPSs (cyan arrows) versus cellular RNP aggregates (black arrows) from cell homogenate experiments (n = 3).
(D) Dynamic (strong cyan), interacting (blue), and fusing (yellow) behaviors of cell homogenate-induced NC-LLPS (n = 3). (C and D) Colored circles define examples of LLPS behaviors.
(E–G) Traced distances traveled by (E) dynamic (strong cyan), (F) interacting (blue, purple), and (G) (red, green) fusing (yellow) NC-LLPS (n = 3); x-y distance (µm), linear distance traveled; total distance (µm), sum total distances traveled.
(H) Boxplots of NC-LLPS travel rates colored as in (E) to (G) (n = 3).
(I) Co-condensation of NC-LLPS and mCherry-Gag from cell homogenate (n = 3).
(J) Effect of linker or ZnF mutant NC proteins on NC-LLPSs, with corresponding boxplots (right) (n = 3).
(K) Effect of drugs on NC-LLPSs, with supporting boxplots (right) (n = 3).
Statistical analysis details are described in STAR Methods. Boxplot horizontal lines indicate median, and whiskers are minimum to maximum.
NC, nucleocapsid; NC-AF488, AlexaFluor 488-labeled NC; DIC, differential interference contrast; tot, total; t, time; s, second; avg., average; ns, non-significant.
LLPSs can be produced in vitro using synthetic macromolecular crowding agent Ficoll, hypothesized to mimic the crowded cellular environment (Alberti et al., 2019). To predict whether NC could initiate formation of LLPS in cells, we performed in vitro experiments replacing Ficoll with cellular homogenate with estimated equimolar concentration of total protein to NC. Cell homogenate readily induced formation of NC-LLPSs (Figure 1B), with these being more diverse in size (ranging from 0.5 to 3 µm) and less spherical, supporting that these NC assemblies may represent NC-LLPS and NC-RNPs containing cytosolic proteins or nucleic acids. With NC proteins greatly outnumbering any single cellular protein from homogenates in these formulations, comparison of visible DIC aggregates with green NC aggregates confirmed that 97.63% of these were NC-positive and distinguishable from cell debris (Figures 1B and 1C).
Unlike static NC-LLPSs generated by in vitro Ficoll-induced LLPS experiments, those formed by cell homogenates were dynamic, exhibiting liquid-phase properties including round shape induced by surface tension, fluidity, rapid internal diffusion, and interaction and fusion forming novel droplets (Baase et al., 2010; Brangwynne et al., 2009; Heinrich et al., 2018) (Figures 1C–1G; Video S1). Live cell footage was used to track NC-LLPS movement, where the homogenate induced these to travel total distances of 16.67 ± 1.28 µm and linear distances of 4.04 ± 0.65 µm in 30 s (Figures 1E–1G). NC-LLPS displacements had average speeds of 0.56 ± 0.04 µm/s, ranging from 0.17 to 1.67 µm/s, thus moving at rates proportional to their sizes (Figure 1H).
We have recently demonstrated that HIV-1 NC induces SG assembly (Rao et al., 2018), despite NC being cleaved from pr55Gag during virus maturation, and therefore not typically found as an abundant entity in cells. To test if the NC moiety within full-length Gag inhibits or participates in LLPS in cells, mCherry-Gag expressing cellular homogenates were used to observe that albeit at lower abundance, mCherry-Gag co-condensed with NC-LLPSs, suggesting that NC within Gag may stimulate LLPS activity prior to its cleavage by PR (Figure 1I).
Low complexity PrLDs and ZnFs are hallmarks of phase-separating proteins (Maharana et al., 2018). Like NC, PrLD-containing proteins are intrinsic to SGs and can multimerize into Zn2+-dependent LLPS processes of aggregate-prone neurodegenerative disease markers TIA-1, TDP-43, FUS/TLS, Tau, amyloid-β, synucleins, and SOD1 (Caragounis et al., 2010; Garnier et al., 2017; Pfaender and Grabrucker, 2014; Rayman et al., 2018; Shelkovnikova et al., 2012). Gag multimerization requires NC with intact ZnFs and an RNA scaffold (Burniston et al., 1999; Campbell and Vogt, 1995; El Meshri et al., 2015). To test whether NC ZnFs were responsible for NC-LLPS, we performed in vitro experiments comparing wild-type (WT) NC to those with mutated ZnF motifs (SSHS-SSHS) (Guo et al., 2000) or the mutated linker region (R7A/R10A/K11A) (Wu et al., 2014a). Although linker mutant LLPSs were not as spherically shaped as WT NC-LLPSs, their overall numbers did not change (Figure 1J). In contrast, the ZnF NC mutant could not form LLPS (p < 0.0001), suggesting that the NC-LLPS may be Zn2+ dependent. Therefore, we used the Zn2+ chelator TPEN (N,N,N′,N′-tetrakis(2-pyridinylmethyl)-1,2-ethanediamine), which inhibits LLPS and SG by TIA-1 (Rayman et al., 2018), and 1,6-Hexanediol (HEX), which inhibits LLPS by hnRNP A1 and TDP-43 (Babinchak et al., 2019; Molliex et al., 2015) and should interfere with maintenance of the Zn2+-induced globular, hydrophobic plateau of NC ZnFs. We also tested the oxidizer arsenite (ARS), which causes release of Zn2+ from ZnFs, thus disrupting SG assembly in cells (Rayman et al., 2018), and which selectively binds and unfolds C3H ZnFs (Zhao et al., 2012). All three chemicals disrupted NC-LLPSs (p < 0.0001) (Figure 1K), confirming that NC-LLPSs are Zn2+-dependent.
Zn2+ Chelation Inhibits NC-SG Assembly and Disrupts NC-vRNA-Gag RNP Formation
We have previously reported that NC induces translational arrest and Gag- and CA-resistant SGs containing TIAR-1, G3BP1, eIF3, PABP, and poly(A) mRNAs (Rao et al., 2018). We have herein recapitulated that Gag did not disrupt NC-LLPS granules, but it rather colocalized with NC-LLPS granules, perhaps through Gag/NC-NC interactions (Figure 1I). From observations that NC-LLPS were Zn2+ dependent (Figures 1J and 1K), and demonstrations that NC ZnF mutants have reduced SG assembly (Yu et al., 2016), we tested NC-SG sensitivity to Zn2+ chelation, as previously shown for TIA-1 (Rayman et al., 2018). Dose-escalating TPEN treatment (0–40 µM) of HeLa cells transfected with the NC-Renilla Luciferase (RLuc) expression constructs showed that TPEN reversed both NC-RLuc-G3BP1 colocalization and NC-induced SGs with no effect on expression of NC-RLuc (Figure 2A). TPEN also modified the formerly diffused cytoplasmic NC-RLuc localization to one mostly localizing closer to the plasma membrane (Figure 2A).
Figure 2. HIV-1 NC-SGs and NC-vRNA-Gag RNPs Are Disrupted by Zn2+ Chelation.
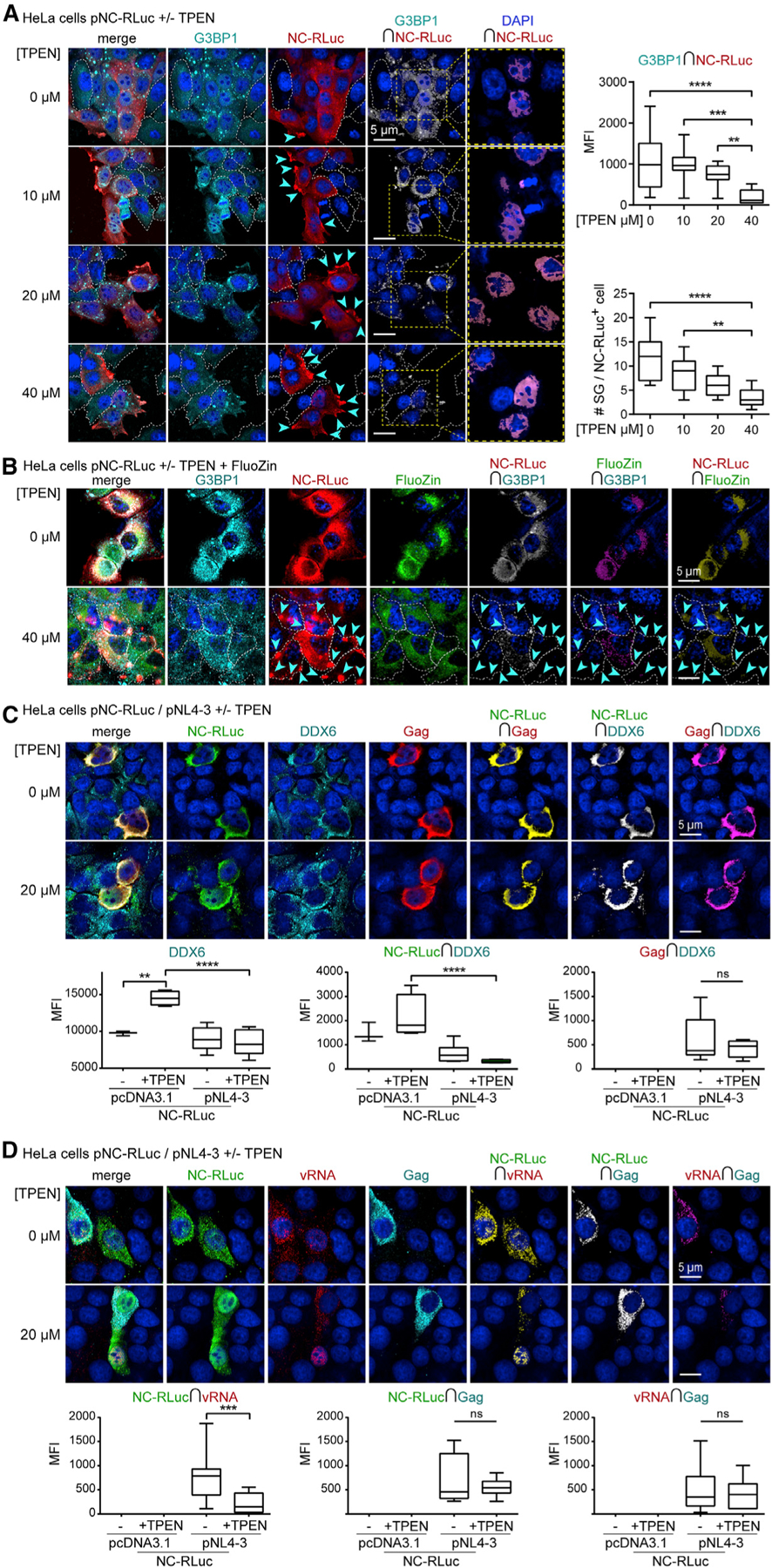
(A) HeLa cells transfected with pNC-RLuc (p2-p1/Rluc) (red) and treated with TPEN (0–40 µM), reversing NC-mediated G3BP1+ SGs (cyan), with corresponding boxplots (right) of decreased NC-RLuc-G3BP1 colocalization by TPEN (top right) (n = 3), and decreased NC-induced SGs by TPEN (bottom right) (n = 3). White dashed cell outlines indicate cells that do not express NC-RLuc. Yellow dashed boxes and corresponding close-up images (right) demonstrate nuclear NC-RLuc via NC-RLuc-DAPI colocalization.
(B) HeLa cells transfected with pNC-RLuc (red) and treated with TPEN (40 µM), with effects on Zn2+ distribution (green, FluoZin-3) and reversal of NC-mediated G3BP1+ SGs (cyan) (n = 3).
(C) HeLa cells co-transfected with pNC-RLuc and proviral construct pNL4–3, and treated with TPEN (20 µM), for effects on NC (green), Gag (red), and DDX6 (cyan) localization, with corresponding boxplots (bottom) (n = 4).
(D) HeLa cells co-transfected with pNC-RLuc and pNL4–3, and treated with TPEN (20 µM), for effects on NC (green), Gag (cyan), and vRNA (red) localization, with corresponding boxplots (bottom) showing decreased NC-vRNA colocalization by TPEN (n = 4). Statistical analysis details described in STAR Methods. Boxplot horizontal lines indicate median, and whiskers are minimum to maximum. µm, micron; RLuc, Renilla Luciferase; ∩, colocalization (intersection); MFI, mean fluorescence intensity; ns, non-significant.
To visualize effects of TPEN on NC and cellular Zn2+, we loaded HeLa cells with a Zn2+-fluorescent probe FluoZin prior to TPEN treatment. This provided evidence that Zn2+ strongly localized with NC-RLuc and G3BP1 in absence of TPEN, and that while TPEN did not diminish overall cellular Zn2+ content, it resulted in dispersion and reduced colocalization with NC-RLuc (−drug versus 40 µM of TPEN (Figure 2B; STAR Methods for statistics). These data provide evidence that TPEN successfully chelates Zn2+ from NC and NC-G3BP1 SGs but does not deplete it from cells. From observations that G3BP1-FluoZin colocalization was also diminished by TPEN treatment−despite G3BP1 not containing ZnFs, and no reports supporting the possibility of it binding Zn2+, where G3BP1 rather assembles into SGs via RNA binding and acetylation dynamics (Gal et al., 2019; Irvine et al., 2004)−we performed dose-escalating TPEN treatment (0–40 µM) of HeLa cells transfected with pG3BP1-GFP (Valiente-Echeverría et al., 2014). These experiments demonstrated that TPEN treatment had no significant effects on G3BP1-GFP-induced SGs also populated by SG marker TIAR (Figure S1), confirming earlier demonstrations that ARS-induced SGs are insensitive to TPEN (Rayman et al., 2018).
We sought to investigate the effects of TPEN on NC, vRNA, and Gag distribution in WT HIV-1 expressing cells. HeLa cells were cotransfected with NC-RLuc and the WT proviral construct pNL4–3, then treated with 20 µM of TPEN. We first examined the effect of TPEN on association of NC with DDX6, since DDX6 is a component of the HIV-1 Gag-vRNA packaging initiation complex (Barajas et al., 2018). Despite TPEN causing differences in DDX6 signal intensity and its colocalization with NC-RLuc in HIV-1 negative conditions, TPEN had no effect on NC-RLuc-DDX6 or Gag-DDX6 colocalization in HIV-1 expressing cells (Figure 2C). These results are in agreement with reports proposing that association of NC or Gag with DDX6 in early virion assembly intermediates are indirect, where the DDX6 helicase may rather unwind the vRNA for Gag accessibility (Reed et al., 2012). We did observe, however, that coupled HIV-1 expression and Zn2+ chelation by TPEN led to a fraction of NC-RLuc localizing to nuclei (Figure 2C).
Interactions between PrLDs and RNA are believed to drive LLPS (Maharana et al., 2018), and NC ZnFs are required for selectivity and packaging of the vRNA (Gorelick et al., 1990). Thus, to investigate whether TPEN treatment could alter the interactions between the vRNA and NC, we cotransfected HeLa cells with pNC-RLuc and WT proviral pNL4–3. Combined immunofluorescence/fluorescence in situ hybridization of the full-length vRNA (Vyboh et al., 2012) and NC-RLuc showed reduced colocalization (p = 0.0002) (Figure 2D). This effect was not seen for NC-RLuc-Gag or vRNA-Gag colocalization, perhaps because these associations are assisted by members of larger RNPs or because Gag is itself expressed from the vRNA. TPEN treatment also caused nuclear retention of the vRNA and NC-RLuc (Figure 2D), suggesting that the Zn2+-loaded NC domain may promote nuclear egress of NC-vRNA RNPs for virus production. Protein sequence analysis of components of HIV-1 vRNA export complexes revealed that they do not possess ZnFs and are thus not expected to be affected by Zn2+ chelation.
Zn2+ Chelation Inhibits the HIV-1 Blockade to SG Assembly and Induces vRNA Repositioning to SGs
Numerous divergent viruses including HIV-1, Ebola, and Zika block SG assembly (Amorim et al., 2017; Cinti et al., 2016; Le Sage et al., 2017; Valiente-Echeverría et al., 2014). For HIV-1, the CA domain may be responsible for the SG blockade, whereas WT NC, but not NC ZnF mutants, promotes SGs (Rao et al., 2018; Yu et al., 2016). The Zn2+-loaded Gag NC domain may thus also promote LLPS toward growth of Gag-vRNA RNPs for virus assembly, whereas the CA domain and other cellular proteins interacting with NC-ZnF may keep SG assembly in check to guarantee that viral RNPs do not grow uncontrolled, producing dysfunctional viral particles. Thus, we sought to determine whether Zn2+ chelation could reverse the HIV-1 blockade of SG assembly. Infected Jurkat T cells treated with 20 µM of TPEN showed that Zn2+ chelation caused increased G3BP1 signal intensity in ARS-positive and -negative conditions (p < 0.0001), increased vRNA-G3BP1 colocalization (p < 0.0001), and increased Gag expression (ARS+ versus ARS+TPEN+; p = 0.0017, 95% confidence interval (CI) −1,139.0 to −220.1, 1-way ANOVA, Tukey post-test) (Figure 3A). vRNA was also visualized in HeLa cells transfected with pNL4–3 and treated with ARS and TPEN (20 or 40 µM). TPEN consistently increased the G3BP1 signal intensity in ARS+ and negative conditions (p < 0.0001), increased vRNA-G3BP1 colocalization (p < 0.0001), and increased Gag expression (ARS+ versus ARS+TPEN+; p = 0.0010, 95% CI −2,483.0 to −469.3, 1-way ANOVA, Tukey post-test) (Figure 3B). As shown in earlier work (Abrahamyan et al., 2010; Valiente-Echeverría et al., 2014), HIV-1 reduced the size and number of ARS-induced G3BP1+ SGs (pcDNA3.1+ARS versus pNL4–3+ARS, p < 0.0001, two-tailed t test), whereas TPEN treatment abrogated the SG blockade by HIV-1 (pNL4–3+ARS versus pNL4–3+ARS+TPEN (20 µM), p = 0.0037, two-tailed t test) (Figure 3B). Importantly, TPEN also caused vRNA localization to SGs in ARS-treated cells (Figure 3B). Conversely, TPEN treatment had a lesser effect on G3BP1-Gag colocalization (p = 0.0261), and little influence on Gag-vRNA colocalization (Figure 3B) in both HeLa and Jurkat T cells. Together, these results show that Zn2+ chelation of NC abrogates the SG blockade by HIV-1, leading to vRNA repositioning to SGs.
Figure 3. Zn2+ Chelation Reverses the HIV-1 SG-Blockade and Induces vRNA Localization to SGs.
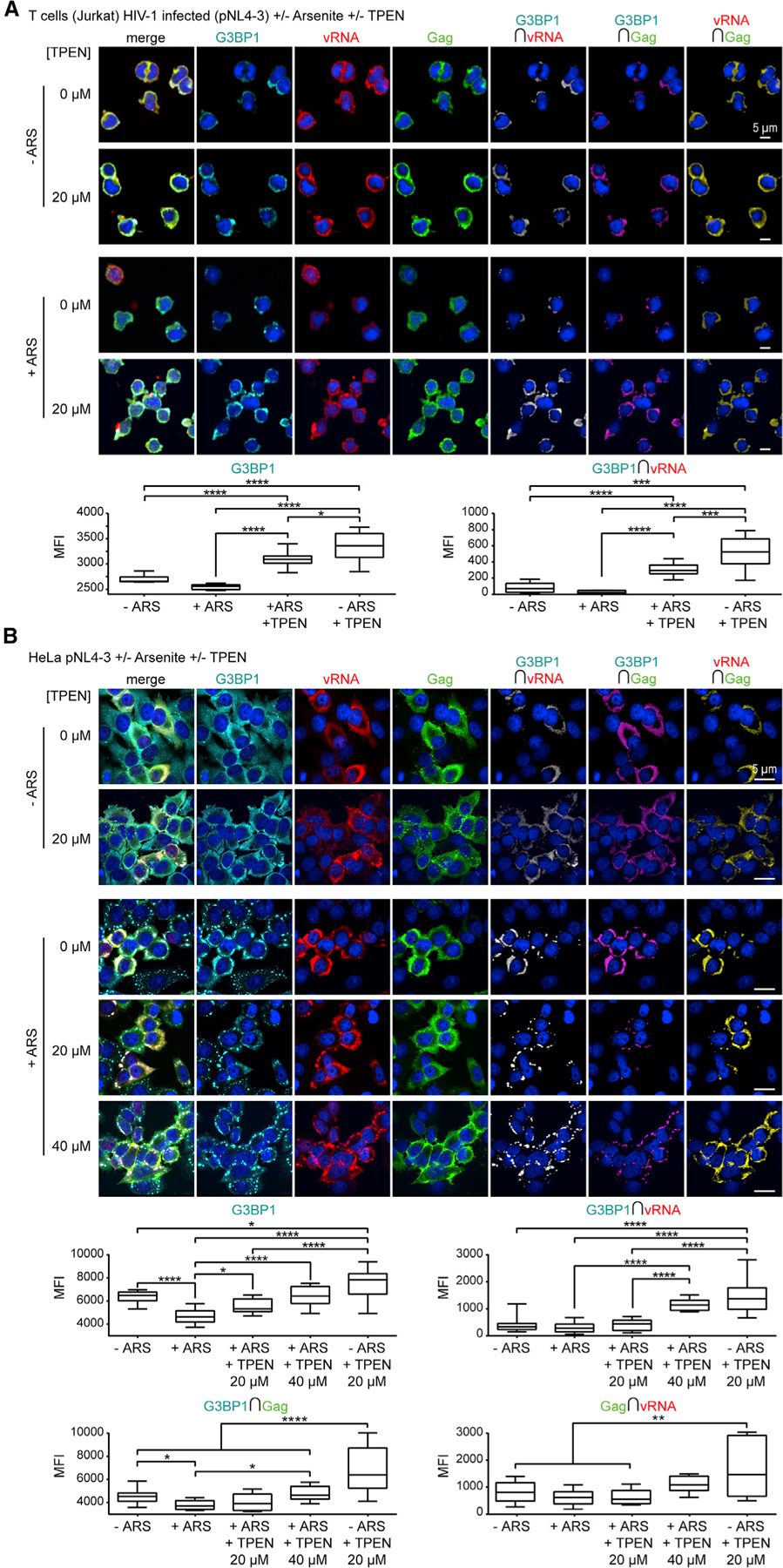
(A) Infected Jurkat T cells treated with ARS (500 µM) and/or TPEN (20 µM) for effects on SG assembly and localization of G3BP1 (cyan), vRNA (red), and Gag (green), with corresponding boxplots (bottom), showing TPEN increases G3BP1 signal intensity (ARS + versus ARS+TPEN+), and vRNA-G3BP1 colocalization (ARS + versus ARS+TPEN+) (n = 3).
(B) HeLa cells transfected with pNL4–3 and treated with ARS (500 µM) and/or TPEN (20 or 40 µM) for effects on SG assembly and localization of G3BP1 (cyan), vRNA (red), and Gag (green), with corresponding boxplots (bottom), showing TPEN increases G3BP1 signal intensity (ARS+ versus ARS+TPEN, 40 µM), vRNA-G3BP1 colocalization (ARS + versus ARS+TPEN, 40 µM), but that TPEN in ARS-treated cells has a lesser effect on G3BP1-Gag colocalization (ARS + versus ARS+TPEN, 40 µM) or Gag-vRNA colocalization (ns, non-significant) (n = 4). Statistical analysis details described in STAR Methods. Boxplot horizontal lines indicate median, and whiskers are minimum to maximum. RLuc, Renilla Luciferase; ∩, colocalization (intersection); MFI, mean fluorescence intensity.
Zn2+ Chelation Causes Cellular Retention of Gag and Decreased Virus Production
Our observations that Zn2+ chelation abrogated the SG blockade by HIV-1, vRNA accumulation in SGs, but concomitant increased Gag expression, appeared confounding since SGs usually contain translationally silent mRNAs (Anderson and Kedersha, 2009). To address this, we transfected HeLa and HEK293T cells with pNL4–3, or cotransfected with pNL4–3 and NC-RLuc and treated with TPEN, ARS, or HEX, and collected cell lysates for western blotting and viral supernatants for virus quantification. NC-RLuc caused decreased Gag expression (pNL4–3 versus pNL4–3/NC-RLuc; p = 0.0008, 95% CI 27.12 to 122.2, 1-way ANOVA, Tukey post-test) (Figure S2; Figure 4A), supporting that NC induces SG assembly and represses vRNA translation favoring its encapsidation (Rao et al., 2018). As previously observed (Figures 3A and 3B), and consistent between HeLa and HEK293T cells, TPEN caused a striking increase in Gag expression (p < 0.0001) (Figure S2; Figure 4A). Consistent with previous work (Rao et al., 2018), NC-RLuc expression in both cell types resulted in decreased virus production (p < 0.0001) (Figure 4B). Decreased virus production from TPEN treatment was also observed in HIV-1 infected Jurkat T cells (pNL4–3 versus pNL4–3+TPEN; p = 0.0003, 95% CI 9.012 to 18.84, 1-way ANOVA, Tukey post-test, data not shown). Decreased virus production by TPEN suggests that Zn2+ is required for NC-LLPS, NC-SGs, and limiting Gag expression for virus production. These findings suggest that Zn2+-dependent NC-LLPSs may act by binding and arresting vRNA translation favoring egress of packaging initiation complexes slated for encapsidation and release as infectious viral particles.
Figure 4. Zn2+ Chelation Leads to Cellular Retention of HIV-1 Gag and Decreased Virus Production.
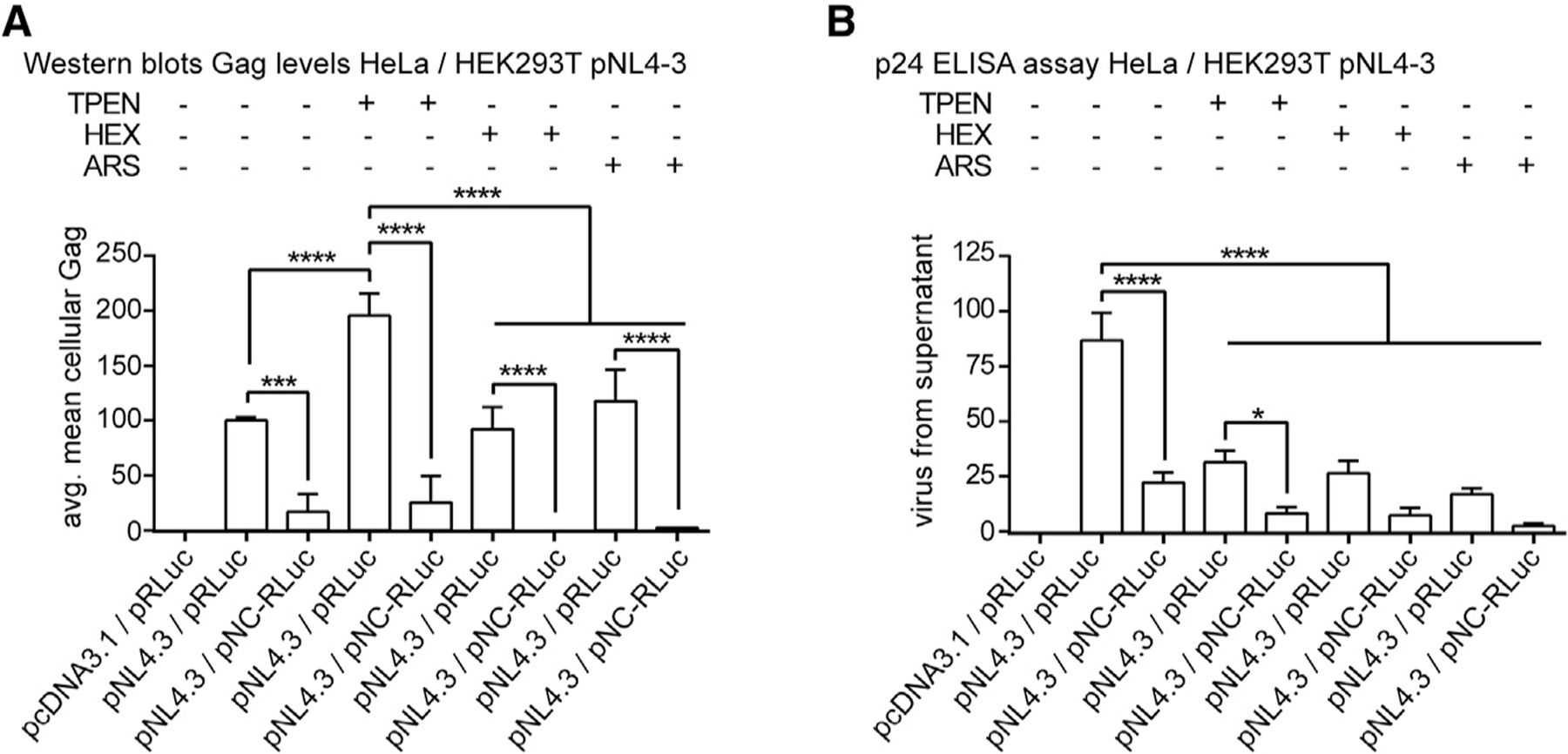
(A) Bar graph from western blots (Figure S2) of effects of drugs on Gag expression, showing increased Gag by TPEN treatment (pNL4–3 versus pNL4–3+TPEN) (n = 3).
(B) Bar graph of effects of drugs on virus production or on pNC-RLuc expression (pNL4–3 versus pNL4–3+TPEN; pNL4–3 versus pNL4–3+HEX; pNL4–3 versus pNL4–3+ARS; pNL4–3/pRLuc versus pNL4–3/pNC-RLuc) (n = 4); avg, average. Statistical analysis details described in STAR Methods.
NC ZnF Mutants Reverse HIV-1 Blockade on SG Assembly and Induce vRNA Repositioning to SGs
Our observations that the HIV-1 blockade to ARS-induced SG assembly was consistently abrogated by Zn2+ chelation (Figures 3A and 3B), and led to vRNA repositioning (Figure 3B), prompted us to determine if Zn2+-chelation phenotypes were specific to NC ZnF function. HeLa cells transfected with the NC ZnF-mutated pNL4–3 proviral constructs (i.e., pNL4–3 NC-C15S-C49S) (Guo et al., 2000) were treated with ARS and TPEN. Strikingly, mutated NC ZnF led to a complete reversal of HIV-1 blockade of SG assembly that was unaltered by TPEN treatment (p < 0.0001) (Figures 5A and 5B, where Figure 5B compares conditions tested to those in Figure 3). As formerly observed from TPEN treatment (Figure 3B), NC ZnF mutants also caused the vRNA to be restricted to G3BP1+ SGs (Figure 5A). As observed with SG-inducing NC-RLuc (Figure 2D), vRNA was localized to the nucleus in untreated cells, and this was further pronounced in ARS-treated cells (Figure 5A), providing evidence that NC ZnFs are required for nuclear export and/or cytoplasmic retention of the vRNA for virus assembly. Other prominent NC ZnF mutant phenotypes were loss of G3BP1-vRNA, G3BP1-Gag, and vRNA-Gag colocalization in ARS-negative conditions (Figure 5A), supporting that NC ZnF nucleates HIV-1 Gag-vRNA RNPs.
Figure 5. HIV-1 NC ZnF Mutants Reverse the HIV-1 SG-Blockade and Induce vRNA Localization to SGs.
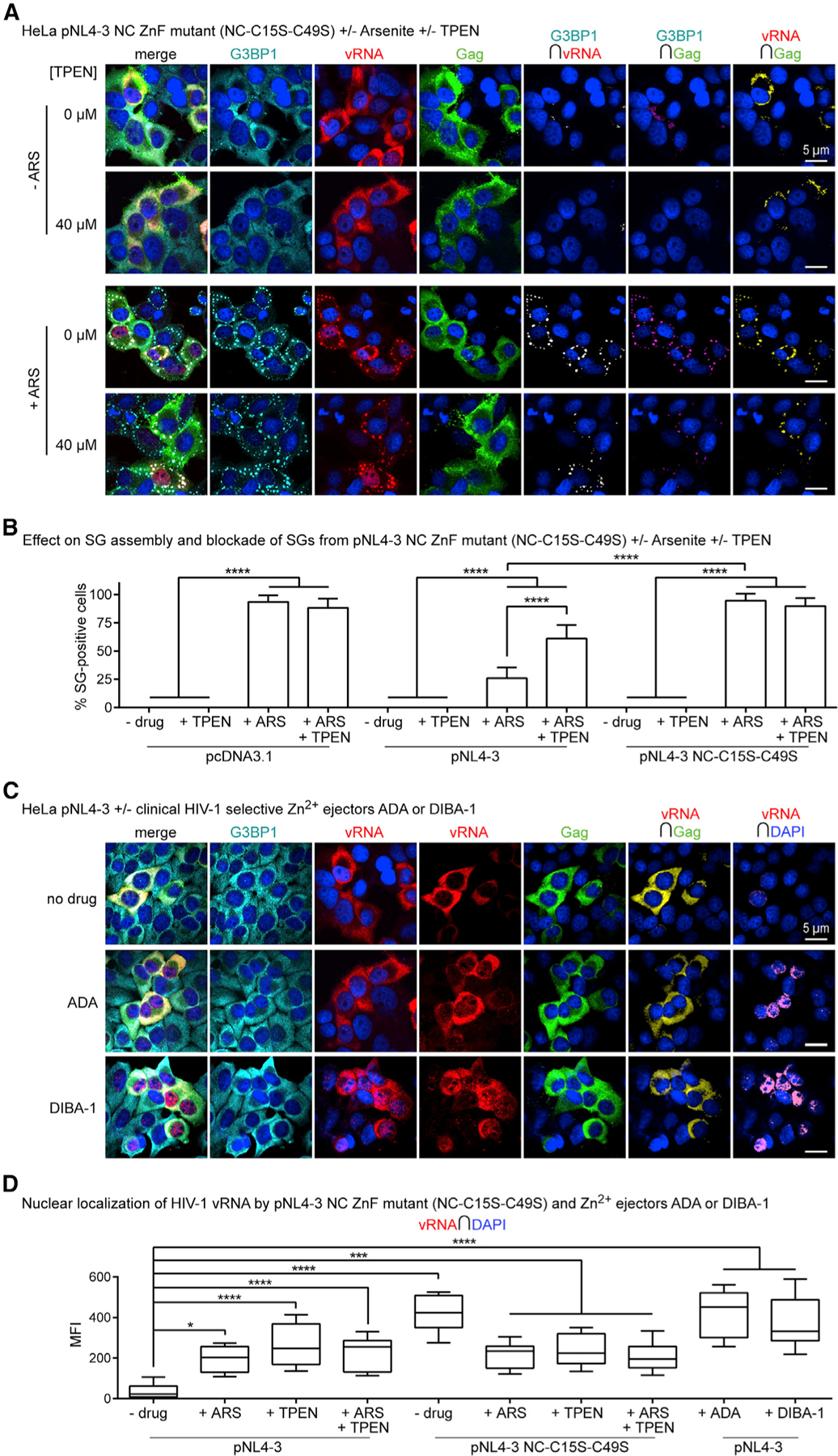
(A) HeLa cells transfected with pNL4–3 NC ZnF mutant (NC-C15S-C39S) and treated with ARS (500 µM) and/or TPEN (40 µM) for effects on SG assembly and localization of G3BP1 (cyan), vRNA (red), and Gag (green), where NC ZnF mutant cannot block SG assembly and restricts the vRNA to SGs and nuclei (n = 4).
(B) Bar graph comparing SG numbers in HeLa cells across conditions tested, including data presented in Figure 3B; demonstrating that the pNL4–3 NC ZnF mutant cannot block SG assembly relative to WT pNL4–3 transfected cells, which is unaltered by TPEN (40 µM) (n = 4).
(C) HeLa cells transfected with WT pNL4–3 and treated with ADA (100 µM) or DIBA-1 (50 µM) for their effects on localization of G3BP1 (cyan), vRNA (red), and Gag (green), where both drugs cause the nuclear retention of the vRNA (n = 3).
(D) Boxplots of vRNA-DAPI colocalization comparing effects of ADA or DIBA-1 with other conditions tested; demonstrating that all Zn2+ drugs cause nuclear retention of the vRNA (−drug versus +ADA; −drug versus DIBA; −drug versus TPEN; pNL4–3 versus pNL4–3 NC-C15S-C49S) (n = 3). Statistical analysis details described in STAR Methods. Boxplot horizontal lines indicate median, and whiskers are minimum to maximum; ∩, colocalization (intersection); MFI, mean fluorescence intensity.
Clinical HIV-1 Zn2+ Ejectors Induce the Nuclear Retention of the vRNA
From our observations that TPEN treatment caused nuclear localization of NC and the vRNA (Figures 2C, 2D, and 3B), as also validated by TPEN treatment of the NC-ZnF mutant (Figure 5A), we examined 19 essential and associated Rev-RRE-CRM1-vRNA nuclear export proteins (e.g., Rev, CRM1, DDX1, DDX3, DHX9 (RHA), eIF5a, RanGTP, RIP, Sam68) (Hofmann et al., 2001; Maares and Haase, 2016; Yedavalli et al., 2004), to find that none possessed ZnFs and should not be affected by TPEN treatment. We then tested whether the nuclear vRNA phenotype could be repeated using clinical HIV-1 NC ZnF-selective targeting Zn2+ ejectors. From numerous clinical HIV-1 Zn2+ ejectors, we chose Azodicarbonamide (ADA) and 2,2′-dithiobisbenzamide-1 (DIBA-1) (Rice et al., 1995, 1997b) from their entry into Phase I/II testing, specific NC-selectivity with no affinity for cellular protein ZnFs (Goebel et al., 2001; Huang et al., 1998), and from interests in their repurposing as microbicides (Mori et al., 2015). HeLa cells transfected with pNL4–3 were treated with 100 µM of ADA or 50 µM of DIBA-1. Indeed, both drugs led to nuclear retention of the HIV-1 vRNA (both −drug versus +drug, p < 0.0001) (Figure 5C), in similar proportions to ZnF mutant proviral constructs (pNL4–3 versus pNL4–3 NC-C15S-C49S, p < 0.0001) (Figures 5A and 5D). Increased nuclear localization of the vRNA by ADA and DIBA-1 suggests that vRNA localization and trafficking is in part NC-Zn2+ dependent.
Pan-retrovirus NC Proteins Have Overlapping ZnFs and PrLDs and Undergo Zn2+-Dependent LLPS
HIV-1 NC overexpression thus triggers Zn2+-dependent SGs (Figures 2A and 2B), and purified HIV-1 NC protein undergoes Zn2+-dependent LLPS (Figures 1A and 1J). Envisaging this phenomenon exists across retrovirus subfamily NCs, we used predictive software to identify ZnF-containing PrLDs, because we expected that their proximities would regulate NC-LLPSs. This notion is supported by studies of PrLDs across eukaryotic viruses with a high proportion identified in retroviruses (Tetz and Tetz, 2018). To map proximity of retrovirus Gag PrLDs and ZnFs, PONDR and PLAAC algorithms were used to generate PrLD alignment maps of pan-retrovirus NC and Gag amino acid sequences (Lancaster et al., 2014; Peng et al., 2006, 2005; Romero et al., 2001). Predicted PrLDs for HIV-1 Gag and NC were validated by previous studies (Xue et al., 2012), while others were validated by the MobiDB database (Piovesan et al., 2018). The accuracy of predictive software programs was tested using proteins with established presence or absence of PrLDs (Figure S3A) (Baase et al., 2010; Wang et al., 2018; Ward et al., 2004). All retrovirus Gag proteins displayed two conserved PrLDs of similar length and location: one encompassing the NC domain and extending to C-terminal end of Gag, and another within the Gag N-terminal CA domain and late (L) domains, as supported by previous reports (Figure 6A) (Deshmukh et al., 2015; Freed, 2002; Liang et al., 2003).
Figure 6. Pan-Retrovirus NC Proteins Condense into Zinc-Dependent LLPSs.
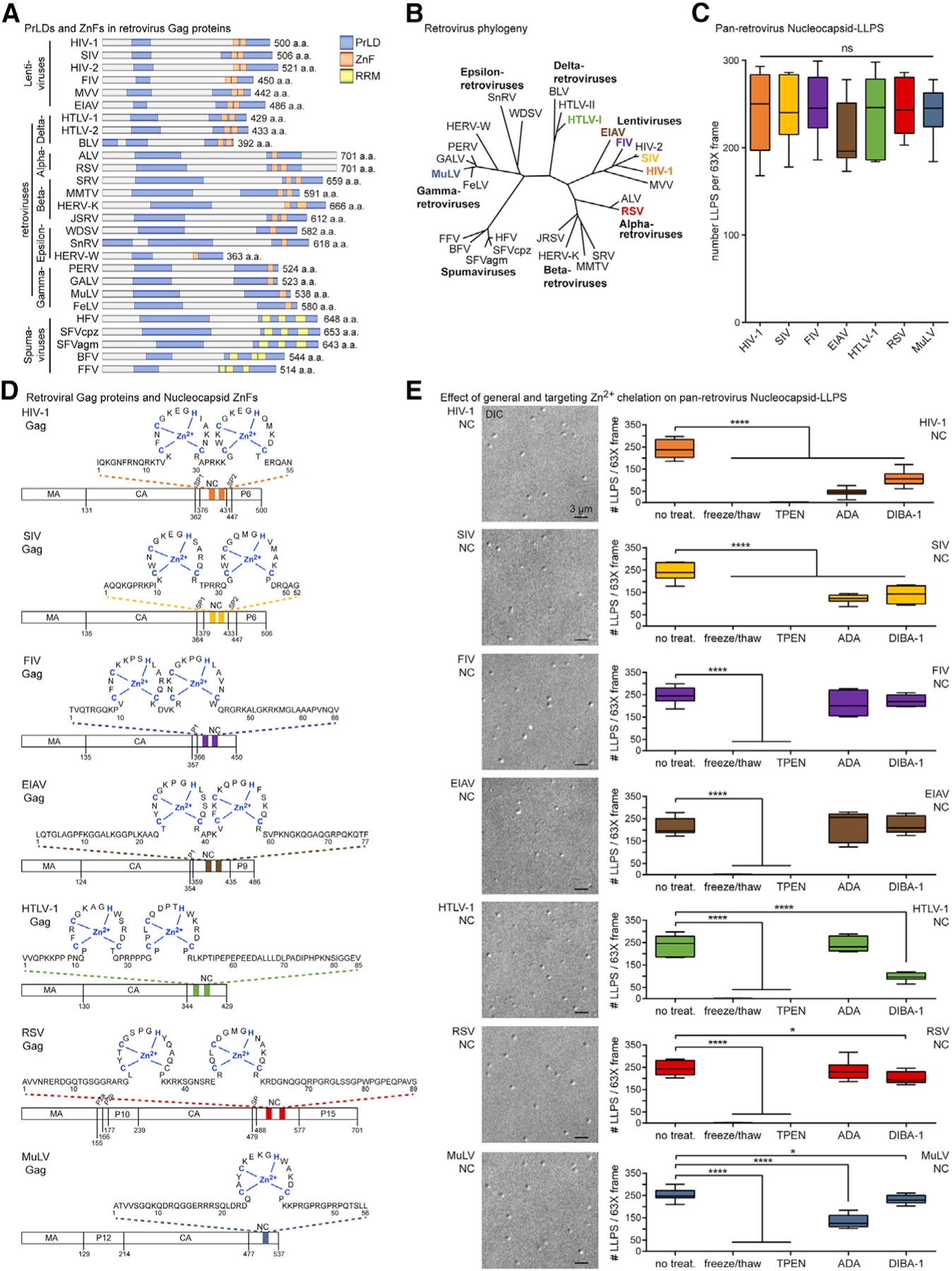
(A) Predicted Gag PrLDs (blue), mapped ZnFs (orange), and RRMs (yellow).
(B) Phylogeny of retrovirus subfamily members, NC-LLPS tested, colored; modified from Linial (1999) and Weiss (2006).
(C) Boxplots of pan-retrovirus NC-LLPSs (see also Figure S3B) (n = 4).
(D) Sequences, lengths, and ZnF-Zn2+ positions (blue) of NCs tested for LLPS.
(E) Pan-retrovirus NC-LLPSs (left), with boxplots (right) of their disruption by Zn2+ depletion treatments, with all highly sensitive to freeze/thaw or TPEN treatment (all p < 0.0001), but some differently affected by ADA and DIBA-1 (HIV-1, −drug versus ADA; HIV-1, −drug versus DIBA; SIV, −drug versus ADA; SIV, −drug versus DIBA; MuLV, −drug versus ADA; MuLV −drug versus DIBA; HTLV-1, −drug versus DIBA; RSV, −drug versus DIBA) (n = 3). Statistical analysis details described in STAR Methods. Boxplot horizontal lines indicate median, and whiskers are minimum to maximum. Retroviruses are defined in the Key Resources Table; PrLDs, prion-like domains; ZnFs, zinc fingers; RRMs, RNA-recognition motifs; aa, amino acid; DIC, differential interference contrast; ns, non-significant.
The molecular composition of membraneless organelles includes ZnFs or Zn2+-binding domains also found in disordered proteins forming amyloidogenic segments (Alberti et al., 2019; Garnier et al., 2017; Gomes and Shorter, 2019). We mapped retroviral ZnFs onto predicted PrLDs to find them positioned within predicted C-terminal PrLDs, with exception of Spumaviruses lacking traditional ZnFs, instead having functionally equivalent RNA recognition motifs (RRMs) (Linial, 1999; Müllers, 2013), also concentrated within predicted PrLDs (Figure 6A). Thus, proximity of ZnFs and PrLDs are highly conserved throughout Retroviridae Gag proteins, suggesting that all could form Zn2+-dependent NC-LLPSs.
We tested purified Zn2+-loaded NC proteins from several closely and distantly related retroviral genera for their ability to LLPS (Weiss, 2006), including HIV-1, simian immunodeficiency virus (SIV), feline immunodeficiency virus (FIV), equine infectious anemia virus (EIAV), human T-lymphotropic virus type 1 (HTLV-1), Rous sarcoma virus (RSV), and murine leukemia virus (MuLV) (Guo et al., 2000; Post et al., 2016; Stewart-Maynard et al., 2008; Wu et al., 2014b, 1996; Yovandich et al., 2001) (Figure 6B). As expected, all pan-retroviral NC proteins could initiate LLPSs at similar sizes of ~1 µm (Figure 6C), despite differences in their lengths, sequences, linker regions, or number of ZnFs (Figure 6D).
From our observations that NC proteins produced the most spherical LLPS at first thaw, we next used mechanical and chemical methods to test if pan-retrovirus NC-LLPS were also Zn2+ dependent (Figure 1K), including Zn2+-oxidizing freeze/thaw cycles, and Zn2+ drugs TPEN, ADA, and DIBA-1. Both freeze/thaw cycles and 20 µM of TPEN disabled NC-LLPSs (p < 0.0001 for all cases) (Figure 6E; Figure S3B). Both ADA (100 µM) and DIBA-1 (50 µM) potently disabled HIV-1 and SIV NC-LLPSs (all, p < 0.0001) (Figure 6E; Figure S3B), as supported by reports of these inhibiting multiple replication steps of HIV-1, HIV-2, and SIV (Rice et al., 1997a, 1995, 1997b). Both drugs also disabled single-ZnF-containing MuLV NC-LLPSs (−drug versus ADA, p < 0.0001; −drug versus DIBA, p = 0.0300) (Figures 6D and 6E; Figure S3B), as supported by reports that the DIBA-2 congener inactivates HIV-1 and MuLV (Figure 6A) (Rein et al., 1996; Rice et al., 1995). DIBA-1 also potently disabled HTLV-1 NC-LLPS (p < 0.0001), and modestly disabled RSV NC-LLPS (p = 0.0294) (Figure 6E; Figure S3B), although to our knowledge, DIBA drug variants have yet to be tested on either retrovirus. Finally, to address whether the NC domain within full-length Gag also contributes to LLPS, we tested purified Gag protein for its ability to form condensates, where titration experiments demonstrated Gag-LLPS to form starting at 10 µM, but optimally at 20 µM (p < 0.0001), and where Gag-LLPSs were also observed to be sensitive to mechanical (freeze/thaw; p < 0.0001), general (TPEN; p < 0.0001), and specific (ADA and DIBA; p < 0.0001) Zn2+ ejection treatments (Figure S3C). These data demonstrate that divergent pan-retroviral NC and full-length HIV-1 Gag proteins undergo Zn2+-dependent LLPS.
ZnF-Dependent and Zn2+-Chelation-Sensitive NC-LLPS Are Dynamically Formed in Live Cells
NC-LLPSs are thus not specific to HIV-1 (Figures 6C and 6D), and rather represent a possible conserved pan-retrovirus replication mechanism. Based on our findings that cell homogenates induce dynamic NC-LLPSs (Figures 1C–1H), we then performed experiments to observe Zn2+-dependent NC-LLPS in real-time. HeLa cells were transfected with WT or mutated ZnF NC-YFP for live cell imaging 4 h later, where 3–4 µm \WT NC-YFP cytoplasmic structures were already apparent in some cells at this time point (Figure 7A; Video S2). In other cells, we observed de novo formation of WT NC-YFP structures, growing from 0.5 µm to 1.5–2 µm during 9 min of video capture (Figure 7A; Video S2). NC-YFP-LLPSs demonstrated dynamics similar to in vitro cell homogenate-stimulated NC-LLPSs (Figures 1C–1H), observed to travel, interact, and fuse with each other (Figure 7A; Video S2).
Figure 7. ZnF-Dependent Dynamically Formed Cellular NC-LLPSs Are Zn2+-Chelation Sensitive.
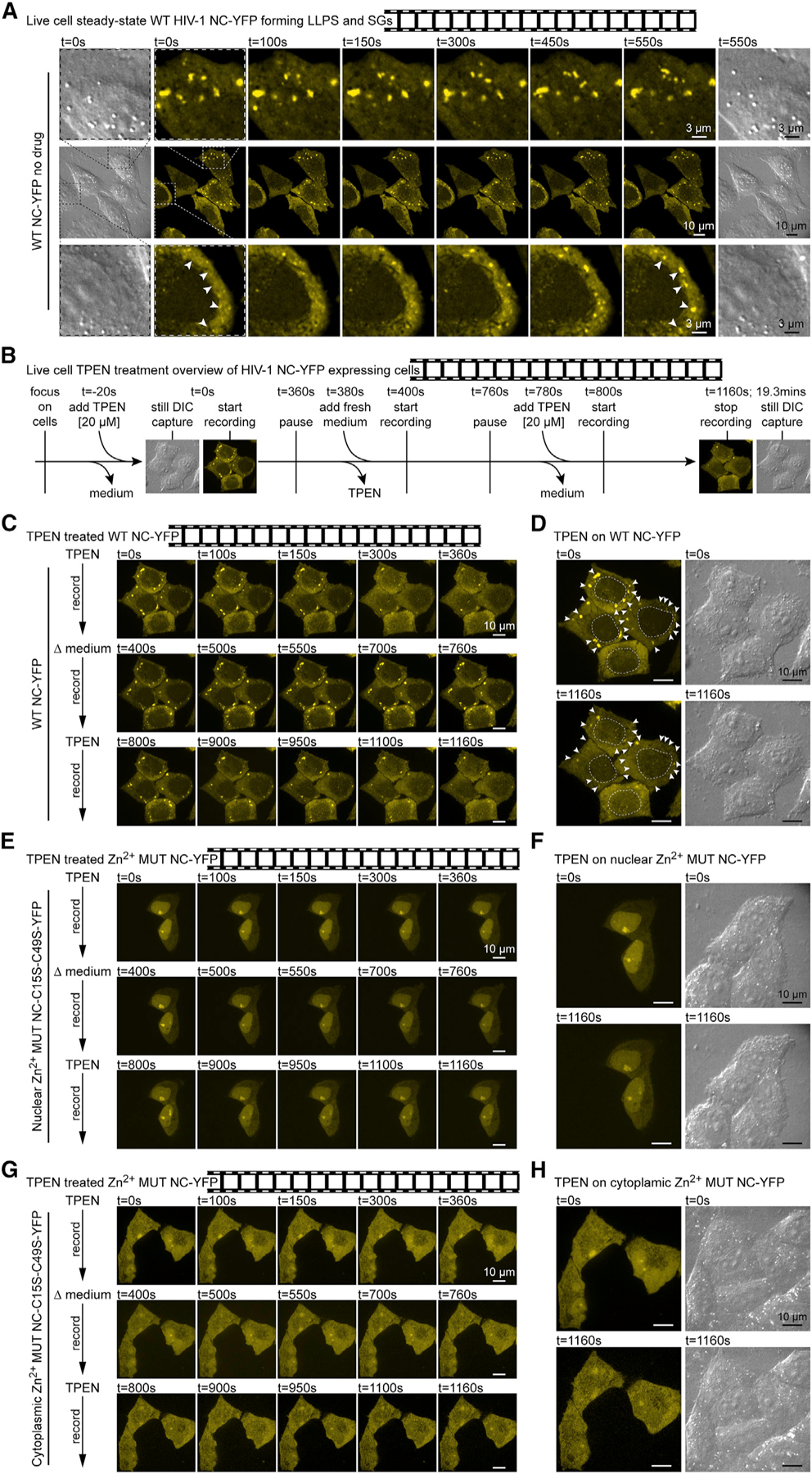
(A) Still images of NC-YFP experiment videos (see Videos S1, S2, S3, S4, S5, S6, S7, S8, S9, S10, S11, and S12) of HeLa cells transfected with WT NC-YFP, with established NC-SGs (top), and de novo NC-LLPS assemblies (white arrows, bottom) (n = 3).
(B) Methodology of NC-YFP TPEN treatments.
(C) Video of HeLa cells transfected with WT NC-YFP, where TPEN treatment disrupts NC-SGs and is reversible (n = 3).
(D) Locations of TPEN-sensitive NC-SGs (white arrows), and DIC images demonstrating morphology is unaffected by treatments.
(E) As in (C), for nuclear phenotype of ZnF-mutated NC-YFP unaffected by TPEN treatment (n = 3).
(F) As in (D), where TPEN has no effect on ZnF mutant NC-YFP or morphology.
(G) As in (C) and (E), for cytoplasmic phenotype of ZnF-mutated NC-YFP unaffected by TPEN treatment (n = 3).
(H) As in (D) and (F), where TPEN has no effect on ZnF mutant NC-YFP or morphology.
WT, wild-type; MUT, mutant; YFP, yellow fluorescent protein; t, time; s, second.
To test the Zn2+ dependency of NC-YFP-LLPSs, and to test whether Zn2+ chelation was reversible, we performed live cell experiments alternating regular medium and TPEN treatment (Figure 7B). We observed that 20 µM of TPEN dissolved pre-established NC-LLPSs and NC-SGs in 6 min, and that replacement of TPEN medium with unconditioned medium restored NC-LLPSs and NC-SGs in 20 s, indicating that effects of Zn2+ chelation were rapidly reversible (Figure 7C), as supported by TPEN displacing but not depleting cellular Zn2+ (Figure 2B). Importantly, in unconditioned medium, NC-YFP localized primarily to cytoplasmic LLPSs and SGs, whereas TPEN treatment not only reduced the size and abundance of NC-LLPSs/SGs, but also led to a diffuse phenotype and nuclear localization of NC-YFP (Figure 7D), as supported by earlier experiments (Figures 2C and 2D). Conversely, the ZnF mutant NC-YFP construct (i.e., NC-C15S-C49S-YFP) mostly localized to nuclei and nucleoli, and could not form LLPSs or SGs at steady state or under any treatment tested (Figures 7E and 7F). Adjusting microscope objectives permitted us to observe that in cells expressing higher levels of mutant ZnF NC-YFP, also localizing to the cytoplasm, LLPSs or SGs could not form at steady state or under any treatment tested (Figures 7G and 7H). Altogether, these results demonstrate that de novo-expressed NC undergoes dynamic LLPS and SG assembly in cells, and that these structures and NC localization are ZnF- and Zn2+ dependent.
The Gag SP1/NC cleavage site is the most sensitive to PR, and mutations in NC/SP2 and SP1/NC cleavage sites are responsible for PR inhibitor (PI) resistance (Côté et al., 2001; Könnyũ et al., 2013). Several reports provide evidence of active PR and GagPol polyprotein proteolytic products in the cytoplasm of infected cells in the absence of virus production and in high viral load patient-derived T cells (Freed et al., 1994; Hu et al., 2005; Kaplan and Swanstrom, 1991; Nie et al., 2007; Park and Morrow, 1991; Wen et al., 2016). Our observations of rapidly forming NC-LLPSs in live cell experiments suggest that basal NC cleavage by PR in cells may represent another event in HIV-1 replication. The unprocessed PR is 10,000-fold less sensitive to PIs than the targeted mature PR dimer (Pettit et al., 2004). Therefore, we overexpressed PR-GFP in cells cotransfected with pNL4–3 or pNC-RLuc, where we observed that NC and PR were extensively colocalized, and this was unaffected by the PI saquinavir (SAQ), suggesting that PR association with NC persists independently of proteolytic activity. SAQ treatment also produced PR- and NC-positive 0.5–1 µm of puncta co-staining for G3BP1, but not PB marker Dcp1 (Figures S4A and S4B). We examined HIV-1 PR protein sequence to find it devoid of predicted PrLDs, indicating that PR should not itself undergo LLPS, promoting the more likely scenario that its affinity with NC-SGs or NC-LLPSs is likely derived from its role in NC cleavage or perhaps in a mechanism protecting cells from PR-derived toxicities. Finally, to gain evidence warranting future studies of cellular NC cleavage by active PR, we treated pNL4–3 transfected cells with SAQ, and used two different Gag-specific antibodies to observe that SAQ treatment caused decreased puncta resembling viruses (Figure S4C). Supernatants were collected and production of infectivity was tested using an X-Gal staining assay in TZM-bl cells, demonstrating a complete loss of infectivity by SAQ treatment (p = 0.0001). Finally, to assess the effect of SAQ treatment on active PR in cells generating Gag-free NC, western blots of cytosolic Gag proteins were analyzed from cellular lysates extensively washed and treated with trypsin and EDTA removing bound, noninternalized virus particles (Yao et al., 1998), demonstrating that SAQ treatment efficiently blocks NC cleavage in cells, while imaging analysis demonstrates that SAQ treatment increases NC-Gag colocalization (Gag(rabbit), p = 0.0106; Gag(mouse), p = 0.0064) (Figure S4C), providing further evidence that NC is cleaved from Gag in untreated cells. These findings warrant future studies on possible roles for NC-LLPSs in the late stages of HIV-1 replication.
DISCUSSION
We are first to discover that both purified HIV-1 NC and Gag proteins induce LLPS and that both this phenomenon and NC-induced SGs are Zn2+ dependent. NC-LLPS was induced in vitro with crowding agents or cytosol but also in cellulo. In live cell experiments, de novo-synthesized NC efficiently bound Zn2+ to induce LLPSs and SGs. The rapid reestablishment of NC-SGs following TPEN washout highlights NC’s high affinity for Zn2+. Indeed, zinc deficiency is the most prevalent micronutrient abnormality in HIV-1 infected and treated patients (Cárcamo et al., 2006; Koch et al., 1996a, 1996b; Visser et al., 2003; Bunupuradah et al., 2012; Jones et al., 2006; Wellinghausen et al., 2000) and it correlates with decreased CD4+ T cells, high viral load, and mortality (Baum et al., 2003; Bunupuradah et al., 2012; Cunningham-Rundles et al., 2005; Fufa et al., 2009; Graham et al., 1991; Irlam et al., 2010; Lai et al., 2001; Visser et al., 2003; Wellinghausen et al., 2000). While zinc supplementation delays disease progression (Baum et al., 2010; Cárcamo et al., 2006; Mocchegiani et al., 1999, 1995; Read et al., 2019; Zeng and Zhang, 2011), an excess also causes disease progression and mortality (Tang et al., 1993, 1996). This duality of Zn2+ homeostasis is also linked to the onset of neurodegeneration (Szewczyk, 2013). Our work highlights Zn2+ homeostasis duality, where virus-promoting NC-LLPSs require Zn2+, but an excess of NC leads to the assembly of translationally silent NC-SGs (Rao et al., 2018). Likewise, heavy metal neurotoxins cause transition of ‘‘protective’’ LLPS to pathological SGs and permanent aggregates in neurological diseases (Ash et al., 2019; Rayman et al., 2018; Trojsi et al., 2013).
Viruses have evolved a finely tuned requirement for Zn2+. We observed that pan-retroviral NC-LLPSs were sensitive to Zn2+ chelation. Indeed, Zn2+ is the most common metal binding to viral proteins and is a key cofactor for DNA and RNA viruses (Chaturvedi and Shrivastava, 2005; Lazarczyk and Favre, 2008). Aside from NC, HIV-1 Integrase (IN), Tat, and Vif also require Zn2+ for folding, varied functions, and host resistance (Frankel et al., 1988; Garber et al., 1998; Huang and Wang, 1996; Lazarczyk and Favre, 2008; Lee and Han, 1996; Lee et al., 1997; Luo et al., 2005; McEuen et al., 1992; Misumi et al., 2004; Paul et al., 2006; Xiao et al., 2007). Viruses outcompete cellular proteins for cellular Zn2+ via their highly conserved (CCHC) anisotropic ZnFs distinct from canonical eukaryotic (CCHH) ZnFs (Bess et al., 1992; Laity et al., 2001), providing the opportunity to target viral ZnFs and not host protein ZnFs (Beerheide et al., 1999; Rice et al., 1993).
We observed dramatic effects on vRNA positioning, Gag expression, and virus production by Zn2+ chelators and ejectors, highlighting the importance of Zn2+ bioavailability for retroviral NC function. As the second most abundant metal essential for >300 enzymes, metalloproteins (MTs), and Zn2+ transporters (ZNTs), Zn2+ is tightly and unevenly controlled across organs (Chaturvedi and Shrivastava, 2005; Colvin et al., 2008; Friedman et al., 1984; Lazarczyk and Favre, 2008). Indeed, MT and ZNT expression and functions are modified by numerous viruses (Ilbäck et al., 2004; Mindaye et al., 2017; Raymond et al., 2010; Read et al., 2018; Zilliox et al., 2006) and HIV-1 shifts cell and organ Zn2+ bioavailability for inflammation and apoptotic resistance (Joshi and Guidot, 2011; Raymond et al., 2010).
This work also demonstrates that Zn2+ chelation/ejection mediates the relocalization NC and the vRNA to the nucleus. NC-G3BP1 SGs were found to be enriched in Zn2+, and TPEN treatment shows that NC-SGs are Zn2+ dependent. Despite nuclear repositioning of both NC and vRNA by TPEN in the proviral context, this chemical also stunts their interaction, suggesting it may be Zn2+-dependent and nuclear in origin. Although components of the Rev-RRE-CRM1 export complex do not possess ZnFs, we could not exclude the possibility that vRNA retention by TPEN was specific for NC and therefore tested the NC-Zn2+-selective clinical compounds, ADA and DIBA-1. The presence of nuclear NC is not without precedence, as it possesses a nuclear localization signal (Yu et al., 2016), and is responsible for early Zn2+-dependent events stimulating integration (Levin et al., 2010; Poljak et al., 2003), and mediates nucleolar retroviral Gag localization (Lochmann et al., 2013). In its apo form, NC remains disordered like FG-repeat-containing nucleoporins (Yu et al., 2016), providing yet another passport for its nucleocytoplasmic shuttling (Yamada et al., 2010).
Viral infection imposes cellular stress. To maintain a non-hostile environment hosting replication, various viruses have evolved different strategies to block SG assembly (Poblete-Durán et al., 2016), and usurping SG proteins for their own functions (Abrahamyan et al., 2010; Thomas et al., 2009). We show that NC does not cause SG assembly when HIV-1 is co-expressed, suggesting that accessory viral proteins may chelate Zn2+ from NC, or that vRNA binding to NC outcompetes its propensity to multimerize. We also find that HIV-1 NC ZnF mutants cannot block SG assembly, where vRNA accumulates in ARS-induced SGs. This establishes that the Zn2+-dependent NC-vRNA association is required for SG blockade by HIV-1, otherwise possibly initiated by expression of this foreign vRNA.
Indeed, numerous studies on a broad range of viruses have long demonstrated that most induce phase separation for replication. Across time and virus type- or family-centric literature, widely varying terms and functions have been ascribed to classify these phase-separating viral replication compartments, including virus factories, viroplasm, or mini-organelles generating subcellular microenvironments concentrating viral components into membrane-bound or membraneless inclusions for replication and protection against virus defenses (Netherton and Wileman, 2011; Novoa et al., 2005). Cytoplasmic inclusion bodies produced later by aggregation of structural proteins or nucleocapsids are also importantly similar to pathologic aggresomes causing neuronal dysfunction (Novoa et al., 2005; Wileman, 2006). Negri body viral factories described for RABV and VSV have been re-examined for their LLPS properties (Heinrich et al., 2018; Nikolic et al., 2017). Viral proteins from divergent influenza A, hendra, measles, and herpes simplex viruses also appear to use LLPS for virus replication (Alenquer et al., 2019; McSwiggen et al., 2019; Zhou et al., 2019). In a greater perspective, the association between viral nucleoproteins and vRNA appears responsible for LLPS (Alberti et al., 2019; Kondo et al., 2013; Lifland et al., 2012; Nikolic et al., 2019; Zhou et al., 2019), and the association between ZnF and RING finger proteins lead to the assembly of virus factories, virus-like particles, and inclusion bodies (Brick et al., 1998; Fehling et al., 2012; Hanslip et al., 2006; Hoenen et al., 2012; Mathur et al., 2014; Nerenberg et al., 2005; Senkevich et al., 1995). Our work demonstrates that Gag and pan-retrovirus NC proteins induce Zn2+-dependent LLPS and SGs, supporting the notion that translational silencing of vRNA ushers it into trafficking cytoplasmic RNPs slated for virus assembly.
A fundamental principle underlying biological molecules undergoing LLPS is multivalency and the capacity to interact with multiple nucleic acids and proteins simultaneously (Li et al., 2012). This is in line with the pleiotropic, flexible, disordered retroviral NC nucleic acid chaperones performing a myriad of functions during early and late stages of viral replication (Darlix et al., 2014). Similar to NC composition and function, it is widely accepted that proteins that undergo LLPS typically contain PrLDs and can bind multivalent DNA and RNA scaffolds through ZnFs or RNA-recognition motifs (RRMs). From our investigation of previously mapped domains of proteins undergoing LLPS, we observed that all have juxtaposed or overlapping PrLDs and RRMs and that the longest proteins, like Gag, which we also find to form LLPS, have both RNA-binding RRMs and ZnFs within PrLDs (Wang et al., 2018).
Large in silico and meta-analyses show disproportionately higher PrLDs in viruses relative to eukaryotes and in retroviruses and Gag proteins relative to other viruses (Pushker et al., 2013; Tetz and Tetz, 2018). It is speculated that viral protein PrLDs facilitate multiple inter-protein interactions for LLPS viral protein condensates, providing a competitive advantage over host RNPs requiring the same machineries for replication (Tarakhovsky and Prinjha, 2018). Other speculations based on degree of outer virus shell disorder, where HIV-1 MA, CA, and NC are more disordered than their orthologs, suggest that disorder assists “shapeshifting” immune evasion strategies (Goh et al., 2019). From our discovery that HIV-1 NC undergoes Zn2+-dependent LLPS, we conducted computational studies to map conserved and overlapping PrLDs and ZnFs. These analyses revealed that NC proteins from divergent retrovirus subfamilies also generate Zn2+-dependent LLPS.
HIV-1 resistance to RT, PR, and integrase (IN) targeting drugs creates an urgent need for new drug strategies (Das and Arnold, 2013). From its high conservation among all viral clades and its many essential functions during replication, NC ZnF is maintained as a prime target for therapeutic intervention (Mori et al., 2015). Despite many Zn2+ ejectors showing strong antiviral activity against a spectrum of strains without eliciting resistance, the difficulty in identifying those whose selectivity outweighs toxicity drives their repurposing as potent microbicides and ongoing efforts to identify non-covalent NC inhibitors (Bernacchi et al., 2007; Breuer et al., 2012; de Rocquigny et al., 2008; Goebel et al., 2001; Goudreau et al., 2013; Mori et al., 2012; Musah, 2004; Pustowka et al., 2003; Raja et al., 2006; Rice et al., 1993; Shvadchak et al., 2009; Srivastava et al., 2004; Stephen et al., 2002; Turpin et al., 2008; Vercruysse et al., 2012; Wallace et al., 2009; Warui and Baranger, 2009). Our discovery of NC- and Gag-induced LLPS sheds new light on the molecular basis for selection and packaging of vRNA and expands our understanding of mechanisms governing NC activity to assist in perfecting promising NC-ZnF-targeting compounds.
STAR☆METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
This study did not generate new unique reagents. Further request for information on methods and reagents should be directed to Lead Contact, Andrew J. Mouland (andrew.mouland@mcgill.ca).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
Adherent HeLa cells and HeLa-derived TZM-bl cells originated from a human female cervix, while adherent HEK293T cells originated from a human female embryonic kidney. HeLa and HEK293T cells [American Type Culture Collection (ATCC)], and TZM-bl cells (NIH AIDS Reagent Program) were grown and maintained in Dulbecco’s Modified Eagle Medium (DMEM) (GIBCO Thermo Fisher Scientific #) supplemented with 10% fetal bovine serum (FBS) (Hyclone), 1% penicillin/streptomycin (Invitrogen) at 37°C and 5% CO2. The CD4+/CXCR4+ Jurkat CE6.1 T cell line (ATCC) originates from peripheral blood and was grown and maintained as suspension culture in RPMI 1640 (Life Technologies) supplemented with 10% FBS (Hyclone) and 1% penicillin/streptomycin (Life Technologies) at 37°C and 5% CO2.
METHOD DETAILS
Recombinant DNA
pNL4–3 was obtained from NIH AIDS Reference and Reagent Program (ARRP). pNL4–3 NC C15S/C49S has been previously described (Guo et al., 2000). Gag-mCherry was provided by Paul Bieniasz (Rockefeller University, USA) (Jouvenet et al., 2008). pRluc-N1 (Packard BioScience/Perkin-Elmer Life Sciences) and the construction of p2-p1/Rluc and NC-p1R7-YFP and NC-p1C15–49S-YFP have been previously described (Chatel-Chaix et al., 2008, 2004). pG3BP1-GFP was provided by Imed Gallouzi (McGill University, Canada) (Tourriére et al., 2001). pcDNA3.1 was purchased from Invitrogen. pRF-EGFP was provided by Rongtuan Lin (Solis et al., 2011), and pEGFP-C1 was purchased from Clonetech.
Antibodies and fluorescent probes
Primary antibodies used were as follows: mouse anti-p24 (IF, 1:250; WB, 1:10,000; NIH AIDS ARRP); rabbit anti-DDX6 (IF, 1:200; Bethyl Laboratories #A300–461A), rabbit anti-G3BP1 (IF, 1:1,000; WB, 1:1,000; (provided by Imed Eddine Gallouzi, McGill University, Canada (Gallouzi et al., 1998)), goat anti-TIAR (1:200; Santa Cruz Biotechnology #sc-1749), rabbit anti-Renilla Luciferase (IF, 1:200; WB, 1:1000; MBL International #PM047), sheep anti-Digoxigenin-AP, Fab fragments (IF, 1:250; Roche #11093274910), rabbit anti-phospho-eIF2α (Ser51) (WB, 1:1,000; Cell Signaling Technology #9721), rabbit anti-eIF2 α (WB, 1:1,000; Cell Signaling Technology #9722), rabbit anti-cleaved caspase-3 (Asp175) (WB, 1:1,000; Cell Signaling Technology #9661), rabbit anti-beta Actin (1:5,000; Abcam #ab8227), and rabbit anti-GFP (WB for YFP, 1:5000 Novus Biologicals # NB600–308). For IF, secondary antibodies used were as follows: Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed, Alexa Fluor® 488 (1:500; Invitrogen-Thermo Fisher Scientific #A-21202); Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor® 488 (1:500; Invitrogen-Thermo Fisher Scientific #A-21206); Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed, Alexa Fluor® 594 (1:500; Invitrogen-Thermo Fisher Scientific #A-21207); Donkey anti-Sheep IgG (H+L) Cross-Adsorbed, Alexa Fluor® 594 (1:500; Invitrogen-Thermo Fisher Scientific #A-31573); Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed, Alexa Fluor® 647 (1:500; Invitrogen-Thermo Fisher Scientific #A-31573); Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor® 647 (1:500; Invitrogen-Thermo Fisher Scientific #A-21447). For western blotting secondary antibodies used were as follows, goat anti-mouse, goat anti-rabbit, or donkey anti-goat IgG polyclonal antibodies conjugated to horseradish peroxidase (HRP) (Rockland Immunochemicals). FluoZin-3, AM, cell permeant (Thermo Fisher Scientific # F24195) was used to visualize Zn2+ in cells.
Proteins
Methods and analyses of NC protein purification and preparation have been previously described for WT and mutant NC HIV-1 (Guo et al., 2000; Wu et al., 2014a, 1996), and WT RSV, MuLV, HTLV-1 (Stewart-Maynard et al., 2008), SIV (Post et al., 2016), FIV (Wu et al., 2014b), and EIAV (Stewart-Maynard et al., 2008). NC proteins lyophilized from acetronitrile, water and trifluoroacetic acid, and containing 1 equivalent of Zn2+ per finger were dissolved in commercial D-PBS (Wisent), aliquoted and stored at −80°C until further use. Recombinant HIV-1 Gag protein purchased from Abcam (ab109969) was diluted in D-PBS (Wisent), as recommended by the manufacturer. Purified Gag was produced using Escherichia coli expression systems and purified using nickel-affinity columns, which maintain purified protein Zn2+ content and secondary structures permitting functional and structural studies of Zn2+ binding proteins (Colombo et al., 2013; Zhang et al., 2017).
Creation and imaging of liquid-liquid phase condensates
Formation of NC-LLPS protein samples was monitored by DIC and fluorescence microscopy. For examination of fluorescently labeled NC-LLPS, purified NC proteins were labeled using the Alexa Fluor 488 Microscale Protein Labeling Kit (Thermo Fisher Scientific, #A30006) according to manufacturer’s instructions. Freshly labeled Retroviral NC and HIV-1 Gag proteins were mixed to final concentrations of 0, 1, 5, 10, and 20 µM, using buffers containing 20 mM HEPES, 220mM NaCl, pH 7.4, and with either 150 mg/ml ficoll (Lymphocyte Separation Medium, Corning, #25–072-CV), or 10% dextran (Sigma-Aldrich #9004–54-0) added as molecular crowding agents. A technical consideration is that both labeled and non labeled NC proteins can only produce perfectly spherical and homogeneously sized LLPS after one round of freezing at 80°C, and thereafter gradually loose this ability as a result of their loss of Zn2+ produced by freeze thaw cycles. For the induction of in vitro LLPS using cellular homogenate instead of ficoll as crowning agent, HeLa cells were grown in 12-well plates, and 9 × 106 cells were washed with PBS, then collected from plates using cell scrapers and spun down at 1,500 rpm for 10 mins at 4’C. Cell pellets in Eppendorf tubes were resuspended in 50 µL of PBS, and were homogenized using Eppendorf® micropestles (Sigma-Aldrich #Z317314). For LLPS experiments, 1 µL of cellular homogenate was added to formulations, for an estimated cellular contents of 15,000 cells, and thus approximately 1 × 1014 proteins per reaction (based on predictions of 101′ proteins per mammalian cell). The number of NC proteins per reaction was estimated to be 3.5 × 1013. For imaging of LLPSs from purified proteins in LLPS formulations, 6 µL of sample mixtures were loaded onto 25 × 75 mm × 1mm thick glass slides (Thermo Scientific, # 640–004T, and covered with 18 mm ø No. 1 cover glasses (VWR VistaVision™, VWR International), and were sealed with clear enamel (Revlon). NC-LLPSs were observed by microscopy 10 mins later, for both green fluorescence coinciding with clearly discernible spheres using differential interference contrast. This was performed using a Leica DM16000B laser confocal microscope equipped with a WaveFX spinning disk confocal head (Quorum Technologies), HCX PL APO / 63×, Oil / 0.60–1.40 NA BL objective, 525/50 nm ex/em filter to detect Alexa Fluor 488, and a Hamamatsu EM-CCD digital camera for image capture. Scanning was performed and digitized at a resolution 1,024 × 1,024 pixel. Image processing and analyses were performed by Imaris software v. 8.1.2 (Bitplane/Andor). For imaging of dynamic LLPS created using cellular homogenate, 525/50 nm ex/em filter was used to detect Alexa Fluor 488 positive NC-LLPS spheres, and then DIC and time-lapse microscopy videos were recorded to capture their motion and surrounding cell lysate. Imaris imaging software (Bitplane/Andor) was used for measurements of LLPS particle sizes, shapes, and displacement distances.
Cell transfection
For adherent HeLa and HEK293T cells, 4 × 105 cells were seeded directly into 12-well plates (VWR) for western blotting, and 1.5 × 105 cells were seeded onto sterile coverslips (18 mm ø No. 1 German cover glasses, VWR VistaVision™, VWR International) deposited into 12-well plates for imaging. 0.8 × 105 cells were seeded per chamber of 4-chamber wells (Lab-Tek®II Chambered #1.5 German Coverglass System; ThermoFisher) for live cell imaging microscopy. Cells were transfected 24 hr later with 2 µg plasmid DNA per well using JetPrime (PolyPlus) according to the manufacturer’s instructions. pcDNA3.1 was used as control for transfections with pNL4–3, and pRluc-N1 was used as control for experiments using p2-p1/Rluc (i.e., NC-RLuc). Jurkat T cells were transfected with 3 µg of plasmid DNA per 1 × 106 cells using JetPrime (PolyPlus) for 12 days prior to treatments or collection.
Reagents used for treatment and Zn2+ chelation and detection
Cells were treated with 500 µM sodium arsenite (ARS) (NaAsO2; Sigma-Aldrich # S7400) for 1 hr prior to collection or fixing onto cover glasses. Cells were treated with indicated concentrations of TPEN (N,N,N′,N′-Tetrakis(2-pyridylmethyl)ethylenediamine) (10, 20, or 40 µM) (Sigma-Aldrich # P4413), azodicarbonamide (ADA; 50µM, Sigma-Aldrich #A96606) or 2,2′-dithiobisbenzamide-1 (DIBA-1; 100µM, Sigma-Aldrich #PZ0634) for 1hr prior to cell harvest. For visualization of Zn2+, cells were loaded with 1 µM FluoZin-3 AM (Thermo Fisher Scientific) and 0.02% Pluronic F-127 (Thermo Fisher Scientific # P3000MP) for 40 min, washed, and given fresh media. NC-LLPSs were treated with 500 µM ARS, 20 µM TPEN, or 3.5% 1,6-hexanediol (HEX) (Sigma-Aldrich #240117). The HIV-1 PR inhibitor saquinavir (SAQ) was obtained from the Division of AIDS, NIH through the NIH AIDS Research Reference and Reagent Program, and was used from the time of transfection to the time of collection.
Fluorescence in situ hybridization, immunofluorescence
FISH/IF co-analyses on adherent and suspension cells were performed exactly as described previously (Monette et al., 2009; Vyboh et al., 2012). Briefly, for suspension cells, sterile 18 mm ø No. 1 cover glasses (VWR) were treated with 0.1% poly-L-lysine solution (Sigma) overnight at 4°C. Cover glasses were dropped into wells, and cells were allowed to settle onto these for 1 h at 37°C prior to fixing cells onto cover glasses. For fixing or cells onto cover glasses, cells were washed once in D-PBS (Wisent) and fixed with 4% paraformaldehyde for 20 min. Fixed cells were then washed with D-PBS, quenched in 0.1 M glycine for 10 min, washed with D-PBS, permeabilized in 0.2% Triton X-100 for 5 min and washed twice with D-PBS. A digoxigenin-labeled RNA probe was synthesized in vitro in presence of digoxigenin-labeled UTP (Roche). To stain the vRNA, cells were DNase (Invitrogen) treated for 15 min (25 U per coverslip), then incubated in hybridization solution for 16–18 h at 42°C (50% formamide, 1 mg/ml tRNA, 2 × SSPE, 5 × Denharts, 5 U RNaseOut (Invitrogen), 50 ng probe). Cells were then incubated in 50% formamide for 15 min at 42°C and incubated twice in 2 × SSPE for 5 min each at 42°C. Cells were briefly washed in PBS before being blocked in 1 × blocking solution (Roche). Primary antibodies were applied for 1 h at 37°C, and then washed for 10 min in PBS followed by secondary antibodies for 1 h. Cells were washed for 20 min in PBS before being mounted on glass slides using ProLong Gold Antifade Reagent with DAPI (Life Technologies). Negative isotype-matched antibodies were used to control staining specificity.
Microscopy and imaging analyses
Laser confocal microscopy was performed using a Leica DM16000B microscope equipped with a WaveFX spinning disk confocal head (Quorum Technologies) and HCX PL APO / 40 ×, Oil / 0.75–1.25 NA CS and HCX PL APO / 63 ×, Oil / 0.60–1.40 NA BL objectives, and images were acquired with a Hamamatsu EM-charge coupled device digital camera. Scanning was performed and digitized at a resolution 1,024 × 1,024 pixel. For multi-color image capture, AlexaFluor-647, −594, 488, conjugated secondary antibody emissions were sequentially captured with 665–715, 570–620, and 500–550, bandpass filters, followed by 435–485 nm [for 4′,6-diamidino-2-phenylindole (DAPI) staining], and then DIC image capture. For live cell experiments, imaging capture experiments of NC-YFP transfected cells began 4 hr post transfection. Multiwell chambered cell culture dishes (LabTek) were transferred to a closed stage-top incubator, preheated to 37°C and infused with 5% CO2. Transfected cells were located, and still images of YFP (YFP ET540/30 m filter) and DIC were captured. To avoid bleaching YFP signal during video recording, signal intensity was first observed to set live cell imaging capture to low laser power (i.e., 47%) and exposure time (i.e., 561 ms), and boosted sensitivity (i.e., 255). Videos were captured using a heated 63 × NA 1.40 objective. Images were recorded from laser-scanned cell layers with a thickness of 1 µm and were digitized at a resolution of 1024 × 1024 pixels. Raw .liff files were exported by the Volocity software (Perkin Elmer) for import into Imaris and ImarisColoc software v. 8.1.2 (Bitplane/Andor) used for generation of new colocalization channels, and .csv exports of quantitative measurements of mean signal intensity values used for downstream data harmonizing and statistical analyses using Excel (Microsoft) and GraphPad v6.1 (Prism), respectively. The steady state localization of NC-YFP was first recorded for 9 minutes. For TPEN experiments, we first, focused on live cells having established NC-YFP structures, and took still capture DIC images of starting cell populations, then rapidly changed unconditioned medium for medium containing 20 µM TPEN, and begun recording live cell videos to observe NC-YFP behavior for 360 s. TPEN containing medium was then replaced with unconditioned medium, to again record NC-YFP behavior for another 360 s. Unconditioned medium was again replaced with medium containing 20 µM TPEN, and NC-YFP behavior was recorded again for 360 s. Finally, DIC image were recorded. Videos were exported as .avi files from Volocity software (Perkin Elmer).
Western blot analysis
Cells were collected at indicated times post transfection. Cells were first washed with D-PBS, then lysed in ice-cold lysis buffer (100 mM NaCl, 10 mM Tris, pH 7.5, 1 mM EDTA, 0.5% Nonidet P-40, protease and phosphatase inhibitor cocktail [Roche]). For cell free virus analyses, cells were washed twice with ice-cold D-PBS, treated with 0.1% trypsin–0.02% EDTA 2 × for 5 min at 37°C, and washed twice with D-PBS prior lysis buffer (Yao et al., 1998). Cell lysates were quantified using the Bradford assay (Bio-Rad), used according to manufacturer instructions, and 20 µg of total cell lysates were denatured in Laemmli sample buffer and incubated for 5 min at 95°C prior to loading into SDS-PAGE protein separating gels. Gels were transferred onto polyvinylidene difluoride membranes (company). Membranes were blocked with 5% non-fat milk in Tris-buffered saline and 0.5% Tween 20 (TBST) prior to incubation with primary antibodies listed. Membranes were washed 3 times with TBST, and then probed with secondary HRP-conjugated secondary antibodies listed. Proteins of interest were detected using the Western Lightning Plus-ECL reagent kit (Perkin-Elmer). Signal intensity was quantified by ImageJ (NIH).
Virus quantification from supernatants
Culture supernatants from HeLa, HEK293T and Jurkat T cells were harvested and passed through a 0.2 µm filter (VWR) to remove cellular debris and centrifuged at 3,000 rpm for 20 mins. HIV-1 virus-p24 antigen levels in culture supernatants were assessed using the 96-well plate format HIV-1 p24 Antigen Capture Assay ELISA kit (Advanced Biosciences Laboratories, #5421).
Infectivity assay
Viral titer in cell supernatants was quantified using the X-gal staining assay in TZM-bl cells as described previously (Rao et al., 2019; Xing et al., 2016), where TZM-bl cells seeded onto 96-well plates were subjected to dilutions of supernatants from each condition for 48 hours, and were then fixed using 1% paraformaldehyde, washed, and treated with X-Gal for the detection of β-galactosidase.
Informatics
A Predictor of prion-like domains (PLAAC; http://plaac.wi.mit.edu/), a predictor of Natural Disordered Regions (PONDR; http://www.pondr.com/), and a protein disorder database (MobiDB; http://mobidb.bio.unipd.it/) were used to identify and validate the positions of PrLDs cellular proteins, HIV-1 PR protein, and Gag proteins from various retroviruses, and to examine their distributions relative to NC ZnF positioning. For PLAAC, the Gag FASTA sequences were obtained from the NCBI protein sequence database and applied to the software using default parameters. From software outputs, predicted PrLDs having > 0.8 score were mapped out. For PONDR, the Gag FASTA sequences were obtained the same way as PLAAC, however the name of the retrovirus was manually typed out in the ‘Protein name’ section. In the ‘Predictor’ section, VLXT and VSL2 were selected, but the ‘Output options’ remained untouched. For a more stringent cut-off, only the regions that peaked above 0.8 on the PONDR Score were considered during the mapping process. Several HIV-1 sequences from different strains were analyzed using both PLAAC and PONDR. These predictions were subsequently validated using MobiDB. The full name of the retrovirus in addition to the key word ‘Gag’ was typed into the search engine. The consensus PrLDs in the overview tab were used to validate the PrLD maps that were scaled and finalized using Illustrator. Finally, the ZnF placements were determined using the NCBI protein sequence database and scaled for each Retrovirus. For Spumaviruses lacking traditional ZnF motif, predefined RGG/RG motifs were mapped instead using a protein sequence analysis tool perry.
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiments were performed in triplicate with similar results, unless otherwise indicated in figure legends as n = #. Statistical analyses for each figure panel are presented below. Three independent observers validated phenotypes resulting from all experimental conditions tested. Cellular imaging statistics reported for MFI and calculated SG-positive cell numbers are from observation of average of n = 120 cells per condition tested. SGs were identified as G3BP1+ foci, and cells were scored as SG-positive when they had at least three G3BP1+ foci ranging in size from 200 to 300 nm in diameter (Jain et al., 2016; Thomas et al., 2009; Wheeler et al., 2016), as measured using Imaris software. LLPS imaging statistics result from counting an average of n = 2,000 LLPS per condition tested, or 10 63× frames where LLPSs were diminished or absent as a result of applied treatments. LLPS dynamics statistics were calculated from the analysis of an average of n = 20 LLPS per condition tested. Statistical analyses were performed using Prism V6.01, GraphPad, where one-way ANOVA (with Tukey’s post-test) and 95% CI was used for multiple comparisons, and an unpaired 2-tailed Student’s t test with 95% CI was used to compare two groups. Data are presented as mean ± SEM or SD, as indicate in figure legends, and P values of less than 0.05 were considered to indicate a statistically significant difference.
Figure 1A, boxplot: p < 0.0001, 95% CI −250.7 to −161.7
Figure 1B, boxplot: NC -ficoll versus NC -ficoll+cell lysate: p < 0.0001, 95% CI −399.0 to −260.5 Figure 1J, boxplot: NC WT versus NC ZnF mutant, p < 0.0001, 95% CI 141.3 to 264.7
Figure 1K, boxplot: all p < 0.0001, no drug versus TPEN, 95% CI 178.6 to 253.2; no drug versus HEX, 95% CI 122.3 to 196.8; no drug versus ARS, 95% CI 53.6 to 117.1. ****, p < 0.0001, 1-way ANOVA, Tukey post-test.
Figure 2A: boxplots (right) (p < 0.0001; no drug versus 40 µM TPEN; 95% CI 445.3 to 1271.0); boxplots (bottom right) (p < 0.0001; no drug versus 40 µM TPEN; 95% CI 4.586 to 12.14).
Figure 2B: drug versus 40 µM TPEN, p = 0.0045, 95% CI −612.2 to −73.33, 1-way ANOVA, Tukey post-test)
Figure 2D: boxplot (p = 0.0002, 95% CI 218.8 to 785.6).
**, p < 0.01; ***, p < 0.001; ****, p < 0.0001; 1-way ANOVA, Tukey post-test.
Figure 3A: G3BP1 signal intensity (ARS + versus ARS+TPEN +; p < 0.0001, 95% CI −737.8 to −380.7, 1-way ANOVA, Tukey post-test), and vRNA-G3BP1 colocalization (ARS + versus ARS + TPEN +; p < 0.0001, 95% CI −385.8 to −170.9).
Figure 3B: TPEN increases G3BP1 signal intensity (ARS+ versus ARS+TPEN[40 µM]+; p < 0.0001, 95% CI −2624.0 to −827.3); vRNA-G3BP1 colocalization (ARS+ versus ARS+TPEN[40 µM]+; p < 0.0001, 95% CI −1219.0 to −494.1); (ARS+ versus ARS+TPEN[40 µM]+; p = 0.0261, 95% CI −911.4305 to −38.66125). *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; 1-way ANOVA, Tukey post-test.
Figure 4A: Gag expression induced by TPEN treatment (pNL4–3 versus pNL4–3+TPEN; p < 0.0001, 95% CI −143.1 to −47.97). Data are mean ± SEM.
Figure 4B: Bar graph pNL4–3 versus pNL4–3+TPEN; p < 0.0001, 95% CI 33.61 to 76.79; pNL4–3 versus pNL4–3+HEX, p < 0.0001, 95% CI 33.63 to 81.98; pNL4–3 versus pNL4–3+ARS, p < 0.0001, 95% CI 48.20 to 91.55; pNL4–3/pRLuc versus pNL4–3/pNC-RLuc; p < 0.0001, 95% CI 42.90 to 86.24). Data are mean ± SD. *, p < 0.05; ***, p < 0.001; ****, p < 0.0001; 1-way ANOVA, Tukey post-test.
Figure 5B: Bar graph NC ZnF mutant cannot block SG assembly relative to WT pNL4–3 (p < 0.0001, 95% CI −81.97 to −54.97); bar graph (+40 µM TPEN; p < 0.0001, 95% CI −49.91 to −20.33) Data are mean ± SD.
Figure 5D: boxplots nuclear retention of the vRNA (-drug versus +ADA, p < 0.0001, 95% CI −510.0 to −262.3; -drug versus DIBA, p < 0.0001, 95% CI −430.4 to −260.8; -drug versus TPEN, p < 0.0001, 95% CI −354.3 to −116.8; pNL4–3 versus pNL4–3 NC-C15S-C49S, p < 0.0001, 95% CI −523.1 to −247.1). *, p < 0.05; ***, p < 0.001; ****, p < 0.0001; 1-way ANOVA, Tukey post-test.
Figure 6E: boxplots (right) (HIV-1, -drug versus ADA, p < 0.0001, 95% CI 169.8 to 217.8; HIV-1, -drug versus DIBA, p < 0.0001, 95% CI 105.6 to 159.7; SIV, -drug versus ADA, p < 0.0001, 95% CI 89.49 to 157.0; SIV, -drug versus DIBA, p < 0.0001, 95% CI 71.54 to 141.5; MuLV, -drug versus ADA, p < 0.0001, 95% CI 93.35 to 147.1; MuLV -drug versus DIBA, p = 0.0300, 95% CI 2.012 to 55.77; HTLV-1, -drug versus DIBA, p < 0.0001, 95% CI 97.48 to 179.1; RSV, -drug versus DIBA, p = 0.0294, 95% CI 3.053 to 82.06). *, p < 0.05; ****, p < 0.0001; 1-way ANOVA, Tukey post-test.
DATA AND CODE AVAILABILITY
This study did not generate any unique datasets or code. The retroviral Gag and other protein sequences examined for PrLDs and ZnFs were obtained through the NCBI Reference Sequence Database (RefSeq) and GenBank: HIV-1: AAD39400.1; SIV: AEK79593.1; FIV: CAA48157.1; EIAV: ACT31322.1; HTLV-1: AAB20767.1; RSV: CAA48534.1; MuLV, RefSeq: NP_040332; HTLV-2: CAA73488.1; BLV: AAA42784.1; MVV: AAB25459.1; HIV-2: AAA76840.1; ALV: AJG42160.1; SRV: BAD89356.1; MMTV: AAF31472.1; FFV: AGC11912.1; SFVagm = RefSeq: YP_001956721.2; SFVcpz: AAA19977.1; HFV: GenBank: CAA68998.1; FeLV: AAA43054.1; GALV: ALV83305.1; PERV: CAA76581.1; BFV: AWK77106.1; SnRV = RefSeq: NP_043925.1; WDSV, RefSeq: NP_045938.1; HERV-W: AAF74213.1; HERV-K: CAA69289.1; JSRV: CAA01899.1; TIA1, GenBank: EAW99824.1; hnRNPA2B1, RefSeq: XP_005249786.1; hnRNPA0, RefSeq: NP_006796.1; hnRNPA1L2, RefSeq: NP_001011725.1; DAZAP1, RefSeq: NP_061832.2; RNApolII, GenBank: AAT12524.1; HEWL, PDB: 1LSZ_A; BPT1, RefSeq: NP_001001554.2; CALM1, GenBank: AAD45181.1; UB, GenBank: AA36789.1; REV, GenBank: AYF56141.1; CRM1, GenBank: CAA69905.2; DDX1, RefSeq: NP_004930.1; DDX11, RefSeq: NP_085911.2; DDX17, GenBank: CAG30318.1; DDX18, GenBank: CAG33341.1; DDX21, RefSeq: NP_004719.2; DDX24, RefSeq: NP_065147.1; DDX3, GenBank: AAC34298.1; DDX4, RefSeq: NP_077726.1; DDX5, RefSeq: NP_001307524.1; DHX36, GenBank: AAH36035.1; DHX9, RefSeq: NP_001348.2; Eif5A, GenBank: AAH80196.1; PIMT, GenBank: AAH07501.1; RanGTP, GenBank: CAG29343.1; RHA, GenBank: AML93444.1; RIP, GenBank: AAH46349.1; Sam68, RefSeq: NP_006550.1.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-HIV-1 p24 Monoclonal | NIH AIDS Reagent Program | Cat#3537 |
| Anti-HIV-1 SF2 p24 Polyclonal | NIH AIDS Reagent Program | Cat#4250 |
| Goat antiserum against NCp7 | Robert Gorelick, Institute National Cancer | N/A |
| Rabbit anti-DDX6 | Bethyl Laboratories | Cat#A300–461A, RRID:AB_2277216 |
| Rabbit anti-G3BP1 | Imed Gallouzi, McGill University | Gallouzi et al., 1998 |
| Goat anti-G3BP1 | Santa Cruz Biotechnology | Cat#sc-70283, RRID:AB_2107230 |
| Goat anti-TIAR | Santa Cruz Biotechnology | Cat#sc-1749,RRID:AB_632508 |
| Rabbit anti-Renilla Luciferase | MBL International | Cat#PM047, RRID:AB_1520866 |
| Sheep anti-Digoxigenin-AP, Fab fragments | Sigma | Cat#11093274910, RRID:AB_514497 |
| rabbit anti-phospho-eIF2α | Cell Signaling Technology | Cat#9721, RRID:AB_330951 |
| rabbit anti-eIF2α | Cell Signaling Technology | Cat#9722, RRID:AB_2230924 |
| rabbit anti-cleaved caspase-3 | Cell Signaling Technology | Cat#9661, RRID:AB_2341188 |
| rabbit anti-beta Actin | Abcam | Cat#ab8227, RRID:AB_2305186 |
| rabbit anti-GFP | Novus Biologicals | Cat#NB600–308 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed, Alexa Fluor® 488 | Invitrogen | Cat#A-21202, RRID:AB_10003058 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor® 488 | Invitrogen | Cat#A-21206, RRID:AB_2535792 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed, Alexa Fluor® 594 | Invitrogen | Cat#A-21207, RRID:AB_141637 |
| Donkey anti-Sheep IgG (H+L) Cross-Adsorbed, Alexa Fluor® 594 | Invitrogen | Cat#A-31573, RRID:AB_2716768 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed, Alexa Fluor® 647 | Invitrogen | Cat#A-31573, RRID:AB_2536183 |
| Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor® 647 | Invitrogen | Cat#A-21447, RRID:AB_141844 |
| goat anti-mouse IgG (H+L) Polyclonal Antibody (HRP (Horseradish Peroxidase)) | Rockland Immunochemicals | Cat#ROCK610–1319, RRID:AB_219659 |
| goat anti-rabbit IgG (H+L) Polyclonal Antibody (HRP (Horseradish Peroxidase))l | Rockland Immunochemicals | Cat#ROCK611–1322, RRID:AB_219723 |
| donkey anti-goat IgG (H+L) Polyclonal Antibody (HRP (Horseradish Peroxidase)) | Rockland Immunochemicals | Cat#ROCK711–703, RRID:AB_840935 |
|
Chemicals, Peptides, and Recombinant Proteins | ||
| HIV-1/NL4–3 (NCp7-WT) | Robert Gorelick, National Cancer Institute | Wu et al., 1996 |
| HIV-1/NL4–3 (NCp7-SSHS/SSHS mutant) | Robert Gorelick, National Cancer Institute | Guo et al., 2000 |
| HIV-1/NL4–3 (NCp7-linker mutant) | Robert Gorelick, National Cancer Institute | Wu et al., 2014a |
| EIAV (NCp11-WT) | Robert Gorelick, National Cancer Institute | Stewart-Maynard et al., 2008 |
| FIV (NCp10-WT) | Robert Gorelick, National Cancer Institute | Wu et al., 2014b |
| HTLV-1 (NCp15-WT) | Robert Gorelick, National Cancer Institute | Stewart-Maynard et al., 2008 |
| MuLV (NCp10-WT) | Robert Gorelick, National Cancer Institute | Stewart-Maynard et al., 2008 |
| RSV (NCp12-WT) | Robert Gorelick, National Cancer Institute | Stewart-Maynard et al., 2008 |
| SIV/Mne (NCp8-WT) | Robert Gorelick, National Cancer Institute | Post et al., 2016 |
| HIV-1 Gag protein | Abcam | Cat#ab109969 |
| FluoZin-3, AM, cell permeant | Thermo Fisher Scientific | Cat#F24195 |
| Alexa Fluor 488 Microscale Protein Labeling Kit | Thermo Fisher Scientific | Cat#A30006 |
| DMEM | GIBCO / Thermo Fisher Scientific | Cat#11965–118 |
| RPMI 1640 | Invitrogen | Cat#11875–119 |
| FBS | Wisent | Cat#080–150 |
| Penicillin-streptomycin | Wisent | Cat#450–201-EL |
| HEPES | VWR | Cat#CA-EM5320 |
| Ficoll (Lymphocyte Separation Medium) | Corning | Cat#25–072-CV |
| Dextran | Sigma | Cat#1179741 |
| JetPrime (PolyPlus) | VWR | Cat#CA89129–924 |
| Sodium Arsenite (NaAsO2) | Sigma | Cat#S7400 |
| TPEN (N,N,N′,N′-Tetrakis(2-pyridylmethyl) ethylenediamine) | Sigma | Cat#P4413 |
| 1,6-hexanediol | Sigma | Cat#240117 |
| Saquinavir | NIH AIDS Reagent Program | Cat#4658 |
| Trypsin/EDTA | Thermo Fisher Scientific | Cat#25200056 |
| Pluronic F-127 | Thermo Fisher Scientific | Cat#P3000MP |
| Poly-L-lysine solution 0.1% (w/v) in H2O | Sigma | Cat#P8920 |
| D-PBS, 1X | Wisent | Cat#311–425-CL |
| Formamide | VWR | Cat#CAFX0420–8 |
| Sodium Chloride | EMD Chemicals | Cat#Sx0420–5 |
| Tris | Bioshop Canada | Cat#TRS001.5 |
| EDTA-free Protease inhibitor tablets | Roche | Cat#11873580001 |
| Paraformaldehyde | Polysciences, Inc. | Cat#18814 |
| tRNA | Invitrogen | Cat#15401–021 |
| Triton-X | OmniPur, EMD Millipore | Cat#9400 |
| Denhardt’s Solution (50X) | Thermo Fisher Scientific | Cat#750018 |
| DNasel | Invitrogen | Cat#18047–019 |
| RNase OUT | Invitrogen | Cat#10777019 |
| Fluorescent Antibody Enhancer Set for DIG Detection #4 (blocking solution) | Roche | Cat#1768506 |
| ProLong Gold Antifade Mountant with DAPI | Invitrogen / Thermo Fisher Scientific | Cat#P36931 |
| DIG RNA Labeling Kit | Sigma | Cat#11277073910 |
| Transcription T7 RNA Polymerase | Invitrogen | Cat#18033–019 |
|
Critical Commercial Assays | ||
| Pierce ECL Plus western blotting Substrate | Thermo Fisher Scientific | Cat#32132 |
| Bradford protein assay | BioRad | Cat#500–0006 |
| HIV-1 p24 Antigen Capture Assay ELISA kit | Advanced Biosciences Laboratories | Cat#5421 |
|
Deposited Data | ||
| HIV-1 | NCBI protein sequence database | GenBank: AD39400.1 |
| SIV | NCBI protein sequence database | GenBank: AEK79593.1 |
| FIV | NCBI protein sequence database | GenBank: AA48157.1 |
| EIAV | NCBI protein sequence database | GenBank: ACT31322.1 |
| HTLV-1 | NCBI protein sequence database | GenBank: AAB20767.1 |
| RSV | NCBI protein sequence database | GenBank: AA48534.1 |
| MuLV | NCBI protein sequence database | RefSeq: NP_040332. |
| HTLV-2 | NCBI protein sequence database | GenBank: AA73488.1 |
| BLV | NCBI protein sequence database | GenBank: AAA42784.1 |
| MVV | NCBI protein sequence database | GenBank: AAB25459.1 |
| HIV-2 | NCBI protein sequence database | GenBank: AAA76840.1 |
| ALV | NCBI protein sequence database | GenBank: AJG42160.1 |
| SRV | NCBI protein sequence database | GenBank: AD89356.1 |
| MMTV | NCBI protein sequence database | GenBank: AAF31472.1 |
| FFV | NCBI protein sequence database | GenBank: GC11912.1 |
| SFVagm | NCBI protein sequence database | RefSeq: YP_001956721.2 |
| SFVcpz | NCBI protein sequence database | GenBank: AAA19977.1 |
| HFV | NCBI protein sequence database | GenBank: CAA68998.1 |
| FeLV | NCBI protein sequence database | GenBank: AAA43054.1 |
| GALV | NCBI protein sequence database | GenBank: ALV83305.1 |
| PERV | NCBI protein sequence database | GenBank: CAA76581.1 |
| BFV | NCBI protein sequence database | GenBank: AWK77106.1 |
| SnRV | NCBI protein sequence database | RefSeq: NP_043925.1 |
| WDSV | NCBI protein sequence database | RefSeq: NP_045938.1 |
| HERV-W | NCBI protein sequence database | GenBank: AAF74213.1 |
| HERV-K | NCBI protein sequence database | GenBank: CAA69289.1 |
| JSRV | NCBI protein sequence database | GenBank: CAA01899.1 |
| TIA1 | NCBI protein sequence database | GenBank: EAW99824.1 |
| hnRNPA2B1 | NCBI protein sequence database | RefSeq: XP_005249786.1 |
| hnRNPA0 | NCBI protein sequence database | RefSeq: NP_006796.1 |
| hnRNPA1L2 | NCBI protein sequence database | RefSeq: NP_001011725.1 |
| DAZAP1 | NCBI protein sequence database | RefSeq: NP_061832.2 |
| RNApolII | NCBI protein sequence database | GenBank: AAT12524.1 |
| HEWL | NCBI protein sequence database | PDB: 1LSZ_A |
| BPT1 | NCBI protein sequence database | RefSeq: NP_001001554.2 |
| CALM1 | NCBI protein sequence database | GenBank: AAD45181.1 |
| UB | NCBI protein sequence database | GenBank: AAA36789.1 |
| REV | NCBI protein sequence database | GenBank: AYF56141.1 |
| CRM1 | NCBI protein sequence database | GenBank: CAA69905.2 |
| DDX1 | NCBI protein sequence database | RefSeq: NP_004930.1 |
| DDX11 | NCBI protein sequence database | RefSeq: NP_085911.2 |
| DDX17 | NCBI protein sequence database | GenBank: CAG30318.1 |
| DDX18 | NCBI protein sequence database | GenBank: CAG33341.1 |
| DDX21 | NCBI protein sequence database | RefSeq: NP_004719.2 |
| DDX24 | NCBI protein sequence database | RefSeq: NP_065147.1 |
| DDX3 | NCBI protein sequence database | GenBank: AAC34298.1 |
| DDX4 | NCBI protein sequence database | RefSeq: NP_077726.1 |
| DDX5 | NCBI protein sequence database | RefSeq: NP_001307524.1 |
| DHX36 | NCBI protein sequence database | GenBank: AAH36035.1 |
| DHX9 | NCBI protein sequence database | RefSeq: NP_001348.2 |
| Eif5A | NCBI protein sequence database | GenBank: AAH80196.1 |
| PIMT | NCBI protein sequence database | GenBank: AAH07501.1 |
| RanGTP | NCBI protein sequence database | GenBank: CAG29343.1 |
| RHA | NCBI protein sequence database | GenBank: AML93444.1 |
| RIP | NCBI protein sequence database | GenBank: AAH46349.1 |
| Sam68 | NCBI protein sequence database | RefSeq: NP_006550.1 |
|
Experimental Models: Cell Lines | ||
| HeLa (ATCC® CCL-2) | American Type Culture Collection | N/A |
| HEK293T/17 (ATCC® CRL-11268) | American Type Culture Collection | N/A |
| Jurkat, CE6–1 (ATCC® TIB-152) | American Type Culture Collection | N/A |
| TZM-bl | NIH AIDS Reagent Program | Cat#8129 (Platt et al., 2009; Takeuchi et al., 2008; Wei et al., 2002; Derdeyn et al., 2000; Platt et al., 1998) |
|
Recombinant DNA | ||
| pcDNA3.1 | Thermo Fisher Scientific | Cat#V79020 |
| HIV-1 NL4–3 Infectious Molecular Clone (pNL4–3) | NIH AIDS Reagent Program | Cat#114 |
| Gag-mCherry | Paul Bieniasz, Rockefeller University, USA | Jouvenet et al., 2008 |
| pG3BP1-GFP | Imed Gallouzi, McGill University, Canada | Tourriére et al., 2001 |
| pNL4–3 NC-C15S-C49S | Robert Gorelick, National Cancer Institute | Guo et al., 2000 |
| pRluc-N1 | BioSignal Packar | Cat#631000 |
| p2-p1/Rluc | Mouland Lab | Chatel-Chaix et al., 2004 |
| NC-p1R7-YFP | Mouland Lab | Chatel-Chaix et al., 2008 |
| NC-p1C15–49S-YFP | Mouland Lab | Chatel-Chaix et al., 2008 |
| pPR-EGFP | Rongtuan Lin, McGill University | Solis et al., 2011 |
| pEGFP-C1 | Clontech | Cat#6084–1 |
|
Software and Algorithms | ||
| Imaris software v. 8.1.2 | Oxford Instruments / Imaris | https://imaris.oxinst.com/ |
| Volocity v. 6.3 | Perkin Elmer | https://www.perkinelmer.com/ |
| ImageJ | National Institutes of Health (NIH) | https://imagej.nih.gov/ij/ |
| GraphPad v. 6.1 | Prism | https://www.graphpad.com/scientific-software/prism/ |
| Predictor of prion-like domains (PLAAC) | Lancaster et al., 2014 | http://plaac.wi.mit.edu/ |
| Predictor of Natural Disordered Regions (PONDR) | Molecular Kinetics, Inc.(Peng et al., 2006, 2005; Romero et al., 2001) | http://www.pondr.com/ |
| MobiDB: a database of protein disorder and mobility annotations | Piovesan et al., 2018 | http://mobidb.bio.unipd.it/ |
| Protein sequence analysis tool | Philipp O. Tsvetkov | Garnier, et al. (2017) |
|
Other | ||
| Leica DM16000B laser confocal microscope | Leica | N/A |
| WaveFX spinning disk confocal head | Quorum Technologies | N/A |
| Hamamatsu EM-CCD digital camera | Hamamatsu | N/A |
| Hybridization Oven | Boekel Scientific | Model 24100 |
| Nitrocellulose membranes 0.2 µm | Bio-Rad | Cat#1620112 |
| Quick Spin Columns | Roche | Cat#11814427001 |
| 4-chamber Lab-Tek®II Chambered #1.5 German Coverglass System | Thermo Fisher Scientific | Cat#155382 |
| 12-well plates | VWR | Cat#CA62406–165 |
| Boil proof 1.5 mL tubes | DIAMED | Cat#DIATE610–3167 |
| Eppendorf® micropestles | Sigma | Cat#Z317314 |
| 25 × 75 mm x 1mm thick glass slides | Thermo Fisher Scientific | Cat#640–004T |
| 18 mm ø No. 1 cover glasses | VWR | Cat#16004–300 |
| 0.2 µm Sterile syringe filters | VWR | Cat#28143–310 |
Highlights.
Retroviral Gag proteins have conserved intrinsically disordered prion-like domains
Pan-retrovirus family nucleocapsid proteins induce liquid-liquid phase separation
Nucleocapsid protein phase separation and stress granule assembly is Zn2+ dependent
Zn2+ chelators and ejectors induce nuclear repositioning of the genomic RNA
ACKNOWLEDGMENTS
We thank members of the late Mark Wainberg’s lab for p24 ELISA support; Donald Johnson and Cathy Hixson for producing recombinant NC; Imed Gallouzi, Colin Crist, Solene Jamet, Henry Yu, Stéphane Richard, and Rongtuan Lin for reagents and advice; and Chia-Hao Chang and Christian Young for microscopy assistance. The following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: TZM-bl cells (Cat#8129) from Dr. John C. Kappes and Dr. Xiaoyun Wu. M.N. was supported by generous funding from the Lady Davis Institute at the Jewish General Hospital (Montreal, Canada). This work was supported by operating grants FRN-56974 and FRN-162447 from the Canadian Institutes of Health Research (to A.J.M.) and by U.S. Federal funds from the NCI, NIH, directly and under contract no. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S.A. Government. The funders had no role in study design, data collection and interpretation, or the decision to submit for publication.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.03.084.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abrahamyan LG, Chatel-Chaix L, Ajamian L, Milev MP, Monette A, Clément JF, Song R, Lehmann M, DesGroseillers L, Laughrea M, et al. (2010). Novel Staufen1 ribonucleoproteins prevent formation of stress granules but favour encapsidation of HIV-1 genomic RNA. J. Cell Sci 123, 369–383. [DOI] [PubMed] [Google Scholar]
- Alberti S, Gladfelter A, and Mittag T (2019). Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell 176, 419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alenquer M, Vale-Costa S, Etibor TA, Ferreira F, Sousa AL, and Amorim MJ (2019). Influenza A virus ribonucleoproteins form liquid organelles at endoplasmic reticulum exit sites. Nat. Commun 10, 1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambadipudi S, Biernat J, Riedel D, Mandelkow E, and Zweckstetter M (2017). Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat. Commun 8, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amorim R, Temzi A, Griffin BD, and Mouland AJ (2017). Zika virus inhibits eIF2a-dependent stress granule assembly. PLoS Negl. Trop. Dis 11, e0005775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P, and Kedersha N (2009). RNA granules: post-transcriptional and epigenetic modulators of gene expression. Nat. Rev. Mol. Cell Biol 10, 430–436. [DOI] [PubMed] [Google Scholar]
- Anton H, Taha N, Boutant E, Richert L, Khatter H, Klaholz B, Rondé P, Réal E, de Rocquigny H, and Mély Y (2015). Investigating the cellular distribution and interactions of HIV-1 nucleocapsid protein by quantitative fluorescence microscopy. PLoS ONE 10, e0116921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash PEA, Dhawan U, Boudeau S, Lei S, Carlomagno Y, Knobel M, Al Mohanna LFA, Boomhower SR, Newland MC, Sherr DH, and Wolozin B (2019). Heavy metal neurotoxicants induce ALS-linked TDP-43 pathology. Toxicol. Sci 167, 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baase WA, Liu L, Tronrud DE, and Matthews BW (2010). Lessons from the lysozyme of phage T4. Protein Sci 19, 631–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babinchak WM, Haider R, Dumm BK, Sarkar P, Surewicz K, Choi JK, and Surewicz WK (2019). The role of liquid-liquid phase separation in aggregation of the TDP-43 low-complexity domain. J. Biol. Chem 294, 6306–6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barajas BC, Tanaka M, Robinson BA, Phuong DJ, Chutiraka K, Reed JC, and Lingappa JR (2018). Identifying the assembly intermediate in which Gag first associates with unspliced HIV-1 RNA suggests a novel model for HIV-1 RNA packaging. PLoS Pathog 14, e1006977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum MK, Campa A, Lai S, Lai H, and Page JB (2003). Zinc status in human immunodeficiency virus type 1 infection and illicit drug use. Clin. Infect. Dis 37 (Suppl 2), S117–S123. [DOI] [PubMed] [Google Scholar]
- Baum MK, Lai S, Sales S, Page JB, and Campa A (2010). Randomized, controlled clinical trial of zinc supplementation to prevent immunological failure in HIV-infected adults. Clin. Infect. Dis 50, 1653–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerheide W, Bernard HU, Tan YJ, Ganesan A, Rice WG, and Ting AE (1999). Potential drugs against cervical cancer: zinc-ejecting inhibitors of the human papillomavirus type 16 E6 oncoprotein. J. Natl. Cancer Inst 91, 1211–1220. [DOI] [PubMed] [Google Scholar]
- Bergeron-Sandoval LP, Safaee N, and Michnick SW (2016). Mechanisms and consequences of macromolecular phase separation. Cell 165, 1067–1079. [DOI] [PubMed] [Google Scholar]
- Bernacchi S, Freisz S, Maechling C, Spiess B, Marquet R, Dumas P, and Ennifar E (2007). Aminoglycoside binding to the HIV-1 RNA dimerization initiation site: thermodynamics and effect on the kissing-loop to duplex conversion. Nucleic Acids Res 35, 7128–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bess JW Jr., Powell PJ, Issaq HJ, Schumack LJ, Grimes MK, Henderson LE, and Arthur LO (1992). Tightly bound zinc in human immunodeficiency virus type 1, human T-cell leukemia virus type I, and other retroviruses. J. Virol 66, 840–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehning M, Dugast-Darzacq C, Rankovic M, Hansen AS, Yu T, Marie-Nelly H, McSwiggen DT, Kokic G, Dailey GM, Cramer P, et al. (2018). RNA polymerase II clustering through carboxy-terminal domain phase separation. Nat. Struct. Mol. Biol 25, 833–840. [DOI] [PubMed] [Google Scholar]
- Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, Jülicher F, and Hyman AA (2009). Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 324, 1729–1732. [DOI] [PubMed] [Google Scholar]
- Breuer S, Chang MW, Yuan J, and Torbett BE (2012). Identification of HIV-1 inhibitors targeting the nucleocapsid protein. J. Med. Chem 55, 4968–4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brick DJ, Burke RD, Schiff L, and Upton C (1998). Shope fibroma virus RING finger protein N1R binds DNA and inhibits apoptosis. Virology 249, 42–51. [DOI] [PubMed] [Google Scholar]
- Bunupuradah T, Ubolyam S, Hansudewechakul R, Kosalaraksa P, Ngam-piyaskul C, Kanjanavanit S, Wongsawat J, Luesomboon W, Pinyakorn S, Kerr S, et al. ; PREDICT Study Group (2012). Correlation of selenium and zinc levels to antiretroviral treatment outcomes in Thai HIV-infected children without severe HIV symptoms. Eur. J. Clin. Nutr 66, 900–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burniston MT, Cimarelli A, Colgan J, Curtis SP, and Luban J (1999). Human immunodeficiency virus type 1 Gag polyprotein multimerization requires the nucleocapsid domain and RNA and is promoted by the capsid-dimer interface and the basic region of matrix protein. J. Virol 73, 8527–8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell S, and Vogt VM (1995). Self-assembly in vitro of purified CA-NC proteins from Rous sarcoma virus and human immunodeficiency virus type 1. J. Virol 69, 6487–6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camus G, Vogt DA, Kondratowicz AS, and Ott M (2013). Lipid droplets and viral infections. Methods Cell Biol 116, 167–190. [DOI] [PubMed] [Google Scholar]
- Caragounis A, Price KA, Soon CP, Filiz G, Masters CL, Li QX, Crouch PJ, and White AR (2010). Zinc induces depletion and aggregation of endogenous TDP-43. Free Radic. Biol. Med 48, 1152–1161. [DOI] [PubMed] [Google Scholar]
- Cárcamo C, Hooton T, Weiss NS, Gilman R, Wener MH, Chavez V, Meneses R, Echevarria J, Vidal M, and Holmes KK (2006). Randomized controlled trial of zinc supplementation for persistent diarrhea in adults with HIV-1 infection. J. Acquir. Immune Defic. Syndr 43, 197–201. [DOI] [PubMed] [Google Scholar]
- Chatel-Chaix L, Clément JF, Martel C, Bériault V, Gatignol A, DesGroseillers L, and Mouland AJ (2004). Identification of Staufen in the human immunodeficiency virus type 1 Gag ribonucleoprotein complex and a role in generating infectious viral particles. Mol. Cell. Biol 24, 2637–2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatel-Chaix L, Boulay K, Mouland AJ, and Desgroseillers L (2008). The host protein Staufen1 interacts with the Pr55Gag zinc fingers and regulates HIV-1 assembly via its N-terminus. Retrovirology 5, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi UC, and Shrivastava R (2005). Interaction of viral proteins with metal ions: role in maintaining the structure and functions of viruses. FEMS Immunol. Med. Microbiol 43, 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimarelli A, Sandin S, Höglund S, and Luban J (2000). Basic residues in human immunodeficiency virus type 1 nucleocapsid promote virion assembly via interaction with RNA. J. Virol 74, 3046–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinti A, Le Sage V, Ghanem M, and Mouland AJ (2016). HIV-1 Gag blocks selenite-induced stress granule assembly by altering the mRNA capbinding complex. MBio 7, e00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo J, Provazzi PJS, Calmon MF, Pires LC, Rodrigues NC, Petl P Jr., Fossey MA, de Souza FP, Canduri F, and Rahal P (2013). Expression, purification and molecular analysis of the human ZNF706 protein. Biol. Proced Online 15, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin RA, Bush AI, Volitakis I, Fontaine CP, Thomas D, Kikuchi K, and Holmes WR (2008). Insights into Zn2+ homeostasis in neurons from experimental and modeling studies. Am. J. Physiol. Cell Physiol 294, C726–C742. [DOI] [PubMed] [Google Scholar]
- Côté HC, Brumme ZL, and Harrigan PR (2001). Human immunodeficiency virus type 1 protease cleavage site mutations associated with protease inhibitor cross-resistance selected by indinavir, ritonavir, and/or saquinavir. J. Virol 75, 589–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham-Rundles S, McNeeley DF, and Moon A (2005). Mechanisms of nutrient modulation of the immune response. J. Allergy Clin. Immunol 115, 1119–1128, quiz 1129. [DOI] [PubMed] [Google Scholar]
- Darlix JL, Godet J, Ivanyi-Nagy R, Fossé P, Mauffret O, and Mély Y (2011). Flexible nature and specific functions of the HIV-1 nucleocapsid protein. J. Mol. Biol 410, 565–581. [DOI] [PubMed] [Google Scholar]
- Darlix JL, de Rocquigny H, Mauffret O, and Mély Y (2014). Retrospective on the all-in-one retroviral nucleocapsid protein. Virus Res 193, 2–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darlix JL, Lapadat-Tapolsky M, de Rocquigny H, and Roques BP (1995). First glimpses at structure-function relationships of the nucleocapsid protein of retroviruses. J. Mol. Biol 254, 523–537. [DOI] [PubMed] [Google Scholar]
- Das K, and Arnold E (2013). HIV-1 reverse transcriptase and antiviral drug resistance. Part 1. Curr. Opin. Virol 3, 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rocquigny H, Shvadchak V, Avilov S, Dong CZ, Dietrich U, Darlix JL, and Mély Y (2008). Targeting the viral nucleocapsid protein in anti-HIV-1 therapy. Mini Rev. Med. Chem 8, 24–35. [DOI] [PubMed] [Google Scholar]
- Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O’Brien WA, Ratner L, Kappes JC, Shaw GM, and Hunter E (2000). Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J. Virol 74, 8358–8367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh L, Ghirlando R, and Clore GM (2015). Conformation and dynamics of the Gag polyprotein of the human immunodeficiency virus 1 studied by NMR spectroscopy. Proc. Natl. Acad. Sci. USA 112, 3374–3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick RA, and Vogt VM (2014). Membrane interaction of retroviral Gag proteins. Front. Microbiol 5, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Meshri SE, Dujardin D, Godet J, Richert L, Boudier C, Darlix JL, Didier P, Mély Y, and de Rocquigny H (2015). Role of the nucleocapsid domain in HIV-1 Gag oligomerization and trafficking to the plasma membrane: a fluorescence lifetime imaging microscopy investigation. J. Mol. Biol 427 (6 Pt B), 1480–1494. [DOI] [PubMed] [Google Scholar]
- Fehling SK, Lennartz F, and Strecker T (2012). Multifunctional nature of the arenavirus RING finger protein Z. Viruses 4, 2973–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel AD, Chen L, Cotter RJ, and Pabo CO (1988). Dimerization of the tat protein from human immunodeficiency virus: a cysteine-rich peptide mimics the normal metal-linked dimer interface. Proc. Natl. Acad. Sci. USA 85, 6297–6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzmann TM, and Alberti S (2019). Protein phase separation as a stress survival strategy. Cold Spring Harb. Perspect. Biol 11, a034058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EO (2002). Viral late domains. J. Virol 76, 4679–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EO, Orenstein JM, Buckler-White AJ, and Martin MA (1994). Single amino acid changes in the human immunodeficiency virus type 1 matrix protein block virus particle production. J. Virol 68, 5311–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RL, Manly SP, McMahon M, Kerr IM, and Stark GR (1984). Transcriptional and posttranscriptional regulation of interferon-induced gene expression in human cells. Cell 38, 745–755. [DOI] [PubMed] [Google Scholar]
- Fufa H, Umeta M, Taffesse S, Mokhtar N, and Aguenaou H (2009). Nutritional and immunological status and their associations among HIV-infected adults in Addis Ababa, Ethiopia. Food Nutr. Bull 30, 227–232. [DOI] [PubMed] [Google Scholar]
- Gal J, Chen J, Na DY, Tichacek L, Barnett KR, and Zhu H (2019). The acetylation of lysine-376 of G3BP1 regulates RNA binding and stress granule dynamics. Mol. Cell. Biol 39, 00052–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallouzi IE, Parker F, Chebli K, Maurier F, Labourier E, Barlat I, Capony JP, Tocque B, and Tazi J (1998). A novel phosphorylation-dependent RNase activity of GAP-SH3 binding protein: a potential link between signal transduction and RNA stability. Mol. Cell. Biol 18, 3956–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber ME, Wei P, KewalRamani VN, Mayall TP, Herrmann CH, Rice AP, Littman DR, and Jones KA (1998). The interaction between HIV-1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes Dev 12, 3512–3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnier C, Devred F, Byrne D, Puppo R, Roman AY, Malesinski S, Golovin AV, Lebrun R, Ninkina NN, and Tsvetkov PO (2017). Zinc binding to RNA recognition motif of TDP-43 induces the formation of amyloid-like aggregates. Sci. Rep 7, 6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel FD, Hemmer R, Schmit JC, Bogner JR, de Clercq E, Witvrouw M, Pannecouque C, Valeyev R, Vandevelde M, Margery H, and Tassignon JP (2001). Phase I/II dose escalation and randomized withdrawal study with add-on azodicarbonamide in patients failing on current antiretroviral therapy. AIDS 15, 33–45. [DOI] [PubMed] [Google Scholar]
- Goh GK, Dunker AK, Foster JA, and Uversky VN (2019). HIV vaccine mystery and viral shell disorder. Biomolecules 9, E178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes E, and Shorter J (2019). The molecular language of membraneless organelles. J. Biol. Chem 294, 7115–7127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelick RJ, Nigida SM Jr., Bess JW Jr., Arthur LO, Henderson LE, and Rein A (1990). Noninfectious human immunodeficiency virus type 1 mutants deficient in genomic RNA. J. Virol 64, 3207–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudreau N, Hucke O, Faucher AM, Grand-Maître C, Lepage O, Bonneau PR, Mason SW, and Titolo S (2013). Discovery and structural characterization of a new inhibitor series of HIV-1 nucleocapsid function: NMR solution structure determination of a ternary complex involving a 2:1 inhibitor/NC stoichiometry. J. Mol. Biol 425, 1982–1998. [DOI] [PubMed] [Google Scholar]
- Graham NM, Sorensen D, Odaka N, Brookmeyer R, Chan D, Willett WC, Morris JS, and Saah AJ (1991). Relationship of serum copper and zinc levels to HIV-1 seropositivity and progression to AIDS. J. Acquir. Immune Defic. Syndr 4, 976–980. [PubMed] [Google Scholar]
- Guo J, Wu T, Anderson J, Kane BF, Johnson DG, Gorelick RJ, Henderson LE, and Levin JG (2000). Zinc finger structures in the human immunodeficiency virus type 1 nucleocapsid protein facilitate efficient minus- and plus-strand transfer. J. Virol 74, 8980–8988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanslip SJ, Zaccai NR, Middelberg AP, and Falconer RJ (2006). Assembly of human papillomavirus type-16 virus-like particles: multifactorial study of assembly and competing aggregation. Biotechnol. Prog 22, 554–560. [DOI] [PubMed] [Google Scholar]
- Heinrich BS, Maliga Z, Stein DA, Hyman AA, and Whelan SPJ (2018). Phase transitions drive the formation of vesicular stomatitis virus replication compartments. MBio 9, e02290–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenen T, Shabman RS, Groseth A, Herwig A, Weber M, Schudt G, Dolnik O, Basler CF, Becker S, and Feldmann H (2012). Inclusion bodies are a site of ebolavirus replication. J. Virol 86, 11779–11788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann W, Reichart B, Ewald A, Müller E, Schmitt I, Stauber RH, Lottspeich F, Jockusch BM, Scheer U, Hauber J, and Dabauvalle MC (2001). Cofactor requirements for nuclear export of Rev response element (RRE)- and constitutive transport element (CTE)-containing retroviral RNAs. An unexpected role for actin. J. Cell Biol 152, 895–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu K, Clément JF, Abrahamyan L, Strebel K, Bouvier M, Kleiman L, and Mouland AJ (2005). A human immunodeficiency virus type 1 protease biosensor assay using bioluminescence resonance energy transfer. J. Virol. Methods 128, 93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HW, and Wang KT (1996). Structural characterization of the metal binding site in the cysteine-rich region of HIV-1 Tat protein. Biochem. Biophys. Res. Commun 227, 615–621. [DOI] [PubMed] [Google Scholar]
- Huang M, Maynard A, Turpin JA, Graham L, Janini GM, Covell DG, and Rice WG (1998). Anti-HIV agents that selectively target retroviral nucleocapsid protein zinc fingers without affecting cellular zinc finger proteins. J. Med. Chem 41, 1371–1381. [DOI] [PubMed] [Google Scholar]
- Ilbäck NG, Glynn AW, Wikberg L, Netzel E, and Lindh U (2004). Metallothionein is induced and trace element balance changed in target organs of a common viral infection. Toxicology 199, 241–250. [DOI] [PubMed] [Google Scholar]
- Irlam JH, Visser MM, Rollins NN, and Siegfried N (2010). Micronutrient supplementation in children and adults with HIV infection. Cochrane Database Syst. Rev (12), CD003650. [DOI] [PubMed] [Google Scholar]
- Irvine K, Stirling R, Hume D, and Kennedy D (2004). Rasputin, more promiscuous than ever: a review of G3BP. Int. J. Dev. Biol 48, 1065–1077. [DOI] [PubMed] [Google Scholar]
- Jain S, Wheeler JR, Walters RW, Agrawal A, Barsic A, and Parker R (2016). ATPase-modulated stress granules contain a diverse proteome and substructure. Cell 164, 487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CY, Tang AM, Forrester JE, Huang J, Hendricks KM, Knox TA, Spiegelman D, Semba RD, and Woods MN (2006). Micronutrient levels and HIV disease status in HIV-infected patients on highly active antiretroviral therapy in the Nutrition for Healthy Living cohort. J. Acquir. Immune Defic. Syndr 43, 475–482. [DOI] [PubMed] [Google Scholar]
- Joshi PC, and Guidot DM (2011). HIV-1 transgene expression in rats induces differential expression of tumor necrosis factor alpha and zinc transporters in the liver and the lung. AIDS Res. Ther 8, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvenet N, Bieniasz PD, and Simon SM (2008). Imaging the biogenesis of individual HIV-1 virions in live cells. Nature 454, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan AH, and Swanstrom R (1991). Human immunodeficiency virus type 1 Gag proteins are processed in two cellular compartments. Proc. Natl. Acad. Sci. USA 88, 4528–4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Panas MD, Achorn CA, Lyons S, Tisdale S, Hickman T, Thomas M, Lieberman J, McInerney GM, Ivanov P, and Anderson P (2016). G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. J. Cell Biol 212, 845–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch J, Neal EA, Schlott MJ, Garcia-Shelton YL, Chan MF, Weaver KE, and Cello JP (1996a). Serum zinc and protein levels: lack of a correlation in hospitalized patients with AIDS. Nutrition 12, 511–514. [DOI] [PubMed] [Google Scholar]
- Koch J, Neal EA, Schlott MJ, Garcia-Shelton YL, Chan MF, Weaver KE, and Cello JP (1996b). Zinc levels and infections in hospitalized patients with AIDS. Nutrition 12, 515–518. [DOI] [PubMed] [Google Scholar]
- Kondo H, Chiba S, Andika IB, Maruyama K, Tamada T, and Suzuki N (2013). Orchid fleck virus structural proteins N and P form intranuclear viroplasm-like structures in the absence of viral infection. J. Virol 87, 7423–7434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Könnyű B, Sadiq SK, Turányi T, Hírmondó R, Müller B, Kräusslich HG, Coveney PV, and Müller V (2013). Gag-Pol processing during HIV-1 virion maturation: a systems biology approach. PLoS Comput. Biol 9, e1003103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroschwald S, and Alberti S (2017). Gel or die: phase separation as a survival strategy. Cell 168, 947–948. [DOI] [PubMed] [Google Scholar]
- Lai H, Lai S, Shor-Posner G, Ma F, Trapido E, and Baum MK (2001). Plasma zinc, copper, copper:zinc ratio, and survival in a cohort of HIV-1-in-fected homosexual men. J. Acquir. Immune Defic. Syndr 27, 56–62. [DOI] [PubMed] [Google Scholar]
- Laity JH, Lee BM, and Wright PE (2001). Zinc finger proteins: new insights into structural and functional diversity. Curr. Opin. Struct. Biol 11, 39–46. [DOI] [PubMed] [Google Scholar]
- Lancaster AK, Nutter-Upham A, Lindquist S, and King OD (2014). PLAAC: a web and command-line application to identify proteins with prion-like amino acid composition. Bioinformatics 30, 2501–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarczyk M, and Favre M (2008). Role of Zn2+ ions in host-virus interactions. J. Virol 82, 11486–11494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Sage V, Cinti A, McCarthy S, Amorim R, Rao S, Daino GL, Tramontano E, Branch DR, and Mouland AJ (2017). Ebola virus VP35 blocks stress granule assembly. Virology 502, 73–83. [DOI] [PubMed] [Google Scholar]
- Lee SP, and Han MK (1996). Zinc stimulates Mg2+-dependent 3′-processing activity of human immunodeficiency virus type 1 integrase in vitro. Biochemistry 35, 3837–3844. [DOI] [PubMed] [Google Scholar]
- Lee SP, Xiao J, Knutson JR, Lewis MS, and Han MK (1997). Zn2+ promotes the self-association of human immunodeficiency virus type-1 integrase in vitro. Biochemistry 36, 173–180. [DOI] [PubMed] [Google Scholar]
- Levin JG, Mitra M, Mascarenhas A, and Musier-Forsyth K (2010). Role of HIV-1 nucleocapsid protein in HIV-1 reverse transcription. RNA Biol 7, 754–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Banjade S, Cheng HC, Kim S, Chen B, Guo L, Llaguno M, Hollingsworth JV, King DS, Banani SF, et al. (2012). Phase transitions in the assembly of multivalent signalling proteins. Nature 483, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C, Hu J, Whitney JB, Kleiman L, and Wainberg MA (2003). A structurally disordered region at the C terminus of capsid plays essential roles in multimerization and membrane binding of the gag protein of human immunodeficiency virus type 1. J. Virol 77, 1772–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifland AW, Jung J, Alonas E, Zurla C, Crowe JE Jr., and Santangelo PJ (2012). Human respiratory syncytial virus nucleoprotein and inclusion bodies antagonize the innate immune response mediated by MDA5 and MAVS. J. Virol 86, 8245–8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linial ML (1999). Foamy viruses are unconventional retroviruses. J. Virol 73, 1747–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Sae-Ueng U, Li D, Lander GC, Zuo X, Jönsson B, Rau D, Shefer I, and Evilevitch A (2014). Solid-to-fluid-like DNA transition in viruses facilitates infection. Proc. Natl. Acad. Sci. USA 111, 14675–14680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochmann TL, Bann DV, Ryan EP, Beyer AR, Mao A, Cochrane A, and Parent LJ (2013). NC-mediated nucleolar localization of retroviral gag proteins. Virus Res 171, 304–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo K, Xiao Z, Ehrlich E, Yu Y, Liu B, Zheng S, and Yu XF (2005). Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5-E3 ligase through a HCCH motif to suppress APOBEC3G. Proc. Natl. Acad. Sci. USA 102, 11444–11449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maares M, and Haase H (2016). Zinc and immunity: an essential interrelation. Arch. Biochem. Biophys 611, 58–65. [DOI] [PubMed] [Google Scholar]
- Maharana S, Wang J, Papadopoulos DK, Richter D, Pozniakovsky A, Poser I, Bickle M, Rizk S, Guillén-Boixet J, Franzmann TM, et al. (2018). RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science 360, 918–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur C, Mohan K, Usha Rani TR, Krishna Reddy M, and Savithri HS (2014). The N-terminal region containing the zinc finger domain of tobacco streak virus coat protein is essential for the formation of virus-like particles. Arch. Virol 159, 413–423. [DOI] [PubMed] [Google Scholar]
- McEuen AR, Edwards B, Koepke KA, Ball AE, Jennings BA, Wolstenholme AJ, Danson MJ, and Hough DW (1992). Zinc binding by retroviral integrase. Biochem. Biophys. Res. Commun 189, 813–818. [DOI] [PubMed] [Google Scholar]
- McSwiggen DT, Hansen AS, Teves SS, Marie-Nelly H, Hao Y, Heckert AB, Umemoto KK, Dugast-Darzacq C, Tjian R, and Darzacq X (2019). Evidence for DNA-mediated nuclear compartmentalization distinct from phase separation. eLife 8, e47098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindaye ST, Ilyushina NA, Fantoni G, Alterman MA, Donnelly RP, and Eichelberger MC (2017). Impact of Influenza A virus infection on the proteomes of human bronchoepithelial cells from different donors. J. Proteome Res 16, 3287–3297. [DOI] [PubMed] [Google Scholar]
- Misumi S, Takamune N, Ohtsubo Y, Waniguchi K, and Shoji S (2004). Zn2+ binding to cysteine-rich domain of extracellular human immunodeficiency virus type 1 Tat protein is associated with Tat protein-induced apoptosis. AIDS Res. Hum. Retroviruses 20, 297–304. [DOI] [PubMed] [Google Scholar]
- Mocchegiani E, Veccia S, Ancarani F, Scalise G, and Fabris N (1995). Benefit of oral zinc supplementation as an adjunct to zidovudine (AZT) therapy against opportunistic infections in AIDS. Int. J. Immunopharmacol 17, 719–727. [DOI] [PubMed] [Google Scholar]
- Mocchegiani E, Muzzioli M, Gaetti R, Veccia S, Viticchi C, and Scalise G (1999). Contribution of zinc to reduce CD4+ risk factor for ‘severe’ infection relapse in aging: parallelism with HIV. Int. J. Immunopharmacol 21, 271–281. [DOI] [PubMed] [Google Scholar]
- Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, and Taylor JP (2015). Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monette A, Ajamian L, López-Lastra M, and Mouland AJ (2009). Human immunodeficiency virus type 1 (HIV-1) induces the cytoplasmic retention of heterogeneous nuclear ribonucleoprotein A1 by disrupting nuclear import: implications for HIV-1 gene expression. J. Biol. Chem 284, 31350–31362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Schult-Dietrich P, Szafarowicz B, Humbert N, Debaene F, Sanglier-Cianferani S, Dietrich U, Mély Y, and Botta M (2012). Use of virtual screening for discovering antiretroviral compounds interacting with the HIV-1 nucleocapsid protein. Virus Res 169, 377–387. [DOI] [PubMed] [Google Scholar]
- Mori M, Kovalenko L, Lyonnais S, Antaki D, Torbett BE, Botta M, Mirambeau G, and Mély Y (2015). Nucleocapsid protein: a desirable target for future therapies against HIV-1. Curr. Top. Microbiol. Immunol 389, 53–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müllers E (2013). The foamy virus Gag proteins: what makes them different? Viruses 5, 1023–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muriaux D, and Darlix JL (2010). Properties and functions of the nucleocapsid protein in virus assembly. RNA Biol 7, 744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muriaux D, Costes S, Nagashima K, Mirro J, Cho E, Lockett S, and Rein A (2004). Role of murine leukemia virus nucleocapsid protein in virus assembly. J. Virol 78, 12378–12385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musah RA (2004). The HIV-1 nucleocapsid zinc finger protein as a target of antiretroviral therapy. Curr. Top. Med. Chem 4, 1605–1622. [DOI] [PubMed] [Google Scholar]
- Nerenberg BT, Taylor J, Bartee E, Gouveia K, Barry M, and Früh K (2005). The poxviral RING protein p28 is a ubiquitin ligase that targets ubiquitin to viral replication factories. J. Virol 79, 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netherton CL, and Wileman T (2011). Virus factories, double membrane vesicles and viroplasm generated in animal cells. Curr. Opin. Virol 1, 381–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z, Bren GD, Vlahakis SR, Schimnich AA, Brenchley JM, Trushin SA, Warren S, Schnepple DJ, Kovacs CM, Loutfy MR, et al. (2007). Human immunodeficiency virus type 1 protease cleaves procaspase 8 in vivo. J. Virol 81, 6947–6956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolic J, Le Bars R, Lama Z, Scrima N, Lagaudriére-Gesbert C, Gaudin Y, and Blondel D (2017). Negri bodies are viral factories with properties of liquid organelles. Nat. Commun 8, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolic J, Lagaudriére-Gesbert C, Scrima N, Blondel D, and Gaudin Y (2019). Structure and Function of Negri Bodies. Adv. Exp. Med. Biol 1215, 111–127. [DOI] [PubMed] [Google Scholar]
- Novoa RR, Calderita G, Arranz R, Fontana J, Granzow H, and Risco C (2005). Virus factories: associations of cell organelles for viral replication and morphogenesis. Biol. Cell 97, 147–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, and Morrow CD (1991). Overexpression of the gag-pol precursor from human immunodeficiency virus type 1 proviral genomes results in efficient proteolytic processing in the absence of virion production. J. Virol 65, 5111–5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul I, Cui J, and Maynard EL (2006). Zinc binding to the HCCH motif of HIV-1 virion infectivity factor induces a conformational change that mediates protein-protein interactions. Proc. Natl. Acad. Sci. USA 103, 18475–18480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK, and Obradovic Z (2005). Optimizing long intrinsic disorder predictors with protein evolutionary information. J. Bioinform. Comput. Biol 3, 35–60. [DOI] [PubMed] [Google Scholar]
- Peng K, Radivojac P, Vucetic S, Dunker AK, and Obradovic Z (2006). Length-dependent prediction of protein intrinsic disorder. BMC Bioinformatics 7, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit SC, Everitt LE, Choudhury S, Dunn BM, and Kaplan AH (2004). Initial cleavage of the human immunodeficiency virus type 1 GagPol precursor by its activated protease occurs by an intramolecular mechanism. J. Virol 78, 8477–8485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaender S, and Grabrucker AM (2014). Characterization of biometal profiles in neurological disorders. Metallomics 6, 960–977. [DOI] [PubMed] [Google Scholar]
- Piovesan D, Tabaro F, Paladin L, Necci M, Micetic I, Camilloni C, Davey N, Dosztányi Z, Mészáros B, Monzon AM, et al. (2018). MobiDB 3.0: more annotations for intrinsic disorder, conformational diversity and interactions in proteins. Nucleic Acids Res 46 (D1), D471–D476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, and Kabat D (1998). Effects of CCR5 and CD4 cell surface concentrations on infections by macro-phagetropic isolates of human immunodeficiency virus type 1. J. Virol 72, 2855–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt EJ, Bilska M, Kozak SL, Kabat D, and Montefiori DC (2009). Evidence that ecotropic murine leukemia virus contamination in TZM-bl cells does not affect the outcome of neutralizing antibody assays with human immunodeficiency virus type 1. J. Virol 83, 8289–8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poblete-Durán N, Prades-Pérez Y, Vera-Otarola J, Soto-Rifo R, and Valiente-Echeverría F (2016). Who regulates whom? An overview of RNA granules and viral infections. Viruses 8, E180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poljak L, Batson SM, Ficheux D, Roques BP, Darlix JL, and Käs E (2003). Analysis of NCp7-dependent activation of HIV-1 cDNA integration and its conservation among retroviral nucleocapsid proteins. J. Mol. Biol 329, 411–421. [DOI] [PubMed] [Google Scholar]
- Post K, Olson ED, Naufer MN, Gorelick RJ, Rouzina I, Williams MC, Musier-Forsyth K, and Levin JG (2016). Mechanistic differences between HIV-1 and SIV nucleocapsid proteins and cross-species HIV-1 genomic RNA recognition. Retrovirology 13, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushker R, Mooney C, Davey NE, Jacqué JM, and Shields DC (2013). Marked variability in the extent of protein disorder within and between viral families. PLoS ONE 8, e60724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pustowka A, Dietz J, Ferner J, Baumann M, Landersz M, Königs C, Schwalbe H, and Dietrich U (2003). Identification of peptide ligands for target RNA structures derived from the HIV-1 packaging signal psi by screening phage-displayed peptide libraries. ChemBioChem 4, 1093–1097. [DOI] [PubMed] [Google Scholar]
- Raja C, Ferner J, Dietrich U, Avilov S, Ficheux D, Darlix JL, de Rocquigny H, Schwalbe H, and Mély Y (2006). A tryptophan-rich hexapeptide inhibits nucleic acid destabilization chaperoned by the HIV-1 nucleocapsid protein. Biochemistry 45, 9254–9265. [DOI] [PubMed] [Google Scholar]
- Rao S, Cinti A, Temzi A, Amorim R, You JC, and Mouland AJ (2018). HIV-1 NC-induced stress granule assembly and translation arrest are inhibited by the dsRNA binding protein Staufen1. RNA 24, 219–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S, Amorim R, Niu M, Breton Y, Tremblay MJ, and Mouland AJ (2019). Host mRNA decay proteins influence HIV-1 replication and viral gene expression in primary monocyte-derived macrophages. Retrovirology 16, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayman JB, Karl KA, and Kandel ER (2018). TIA-1 self-multimerization, phase separation, and recruitment into stress granules are dynamically regulated by Zn2. Cell Rep 22, 59–71. [DOI] [PubMed] [Google Scholar]
- Raymond AD, Gekonge B, Giri MS, Hancock A, Papasavvas E, Chehimi J, Kossenkov AV, Nicols C, Yousef M, Mounzer K, et al. (2010). Increased metallothionein gene expression, zinc, and zinc-dependent resistance to apoptosis in circulating monocytes during HIV viremia. J. Leukoc. Biol 88, 589–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read SA, Parnell G, Booth D, Douglas MW, George J, and Ahlenstiel G (2018). The antiviral role of zinc and metallothioneins in hepatitis C infection. J. Viral Hepat 25, 491–501. [DOI] [PubMed] [Google Scholar]
- Read SA, Obeid S, Ahlenstiel C, and Ahlenstiel G (2019). The role of zinc in antiviral immunity. Adv. Nutr 10, 696–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JC, Molter B, Geary CD, McNevin J, McElrath J, Giri S, Klein KC, and Lingappa JR (2012). HIV-1 Gag co-opts a cellular complex containing DDX6, a helicase that facilitates capsid assembly. J. Cell Biol 198, 439–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rein A, Ott DE, Mirro J, Arthur LO, Rice W, and Henderson LE (1996). Inactivation of murine leukemia virus by compounds that react with the zinc finger in the viral nucleocapsid protein. J. Virol 70, 4966–4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice WG, Schaeffer CA, Graham L, Bu M, McDougal JS, Orloff SL, Villinger F, Young M, Oroszlan S, and Fesen MR (1993). The site of antiviral action of 3-nitrosobenzamide on the infectivity process of human immunodeficiency virus in human lymphocytes. Proc. Natl. Acad. Sci. USA 90, 9721–9724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice WG, Supko JG, Malspeis L, Buckheit RW Jr., Clanton D, Bu M, Graham L, Schaeffer CA, Turpin JA, Domagala J, et al. (1995). Inhibitors of HIV nucleocapsid protein zinc fingers as candidates for the treatment of AIDS. Science 270, 1194–1197. [DOI] [PubMed] [Google Scholar]
- Rice WG, Baker DC, Schaeffer CA, Graham L, Bu M, Terpening S, Clanton D, Schultz R, Bader JP, Buckheit RW Jr., et al. (1997a). Inhibition of multiple phases of human immunodeficiency virus type 1 replication by a dithiane compound that attacks the conserved zinc fingers of retroviral nucleocapsid proteins. Antimicrob. Agents Chemother 41, 419–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice WG, Turpin JA, Huang M, Clanton D, Buckheit RW Jr., Covell DG, Wallqvist A, McDonnell NB, DeGuzman RN, Summers MF, et al. (1997b). Azodicarbonamide inhibits HIV-1 replication by targeting the nucleocapsid protein. Nat. Med 3, 341–345. [DOI] [PubMed] [Google Scholar]
- Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, and Dunker AK (2001). Sequence complexity of disordered protein. Proteins 42, 38–48. [DOI] [PubMed] [Google Scholar]
- Senkevich TG, Wolffe EJ, and Buller RM (1995). Ectromelia virus RING finger protein is localized in virus factories and is required for virus replication in macrophages. J. Virol 69, 4103–4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelkovnikova TA, Kulikova AA, Tsvetkov FO, Peters O, Bachurin SO, Bukhman VL, and Ninkina NN (2012). [Proteinopathies−forms of neurodegenerative disorders with protein aggregation-based pathology]. Mol. Biol. (Mosk.) 46, 402–415. [PubMed] [Google Scholar]
- Shvadchak V, Sanglier S, Rocle S, Villa P, Haiech J, Hibert M, van Dorsselaer A, Mély Y, and de Rocquigny H (2009). Identification by high through put screening of small compounds inhibiting the nucleic acid destabilization activity of the HIV-1 nucleocapsid protein. Biochimie 91, 916–923. [DOI] [PubMed] [Google Scholar]
- Solis M, Nakhaei P, Jalalirad M, Lacoste J, Douville R, Arguello M, Zhao T, Laughrea M, Wainberg MA, and Hiscott J (2011). RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J. Virol 85, 1224–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava P, Schito M, Fattah RJ, Hara T, Hartman T, Buckheit RW Jr., Turpin JA, Inman JK, and Appella E (2004). Optimization of unique, uncharged thioesters as inhibitors of HIV replication. Bioorg. Med. Chem 12, 6437–6450. [DOI] [PubMed] [Google Scholar]
- Stephen AG, Worthy KM, Towler E, Mikovits JA, Sei S, Roberts P, Yang QE, Akee RK, Klausmeyer P, McCloud TG, et al. (2002). Identification of HIV-1 nucleocapsid protein: nucleic acid antagonists with cellular anti-HIV activity. Biochem. Biophys. Res. Commun 296, 1228–1237. [DOI] [PubMed] [Google Scholar]
- Stewart-Maynard KM, Cruceanu M, Wang F, Vo MN, Gorelick RJ, Williams MC, Rouzina I, and Musier-Forsyth K (2008). Retroviral nucleocapsid proteins display nonequivalent levels of nucleic acid chaperone activity. J. Virol 82, 10129–10142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szewczyk B (2013). Zinc homeostasis and neurodegenerative disorders. Front. Aging Neurosci 5, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanchou V, Decimo D, Péchoux C, Lener D, Rogemond V, Berthoux L, Ottmann M, and Darlix JL (1998). Role of the N-terminal zinc finger of human immunodeficiency virus type 1 nucleocapsid protein in virus structure and replication. J. Virol 72, 4442–4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang AM, Graham NM, Kirby AJ, McCall LD, Willett WC, and Saah AJ (1993). Dietary micronutrient intake and risk of progression to acquired immunodeficiency syndrome (AIDS) in human immunodeficiency virus type 1 (HIV-1)-infected homosexual men. Am. J. Epidemiol 138, 937–951. [DOI] [PubMed] [Google Scholar]
- Tang AM, Graham NM, and Saah AJ (1996). Effects of micronutrient intake on survival in human immunodeficiency virus type 1 infection. Am. J. Epidemiol 143, 1244–1256. [DOI] [PubMed] [Google Scholar]
- Takeuchi Y, McClure MO, and Pizzato M (2008). Identification of gammaretroviruses constitutively released from cell lines used for human immunodeficiency virus research. J. Virol 82, 12585–12588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarakhovsky A, and Prinjha RK (2018). Drawing on disorder: how viruses use histone mimicry to their advantage. J. Exp. Med 215, 1777–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetz G, and Tetz V (2018). Prion-like domains in eukaryotic viruses. Sci. Rep 8, 8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MG, Martinez Tosar LJ, Desbats MA, Leishman CC, and Boccaccio GL (2009). Mammalian Staufen 1 is recruited to stress granules and impairs their assembly. J. Cell Sci 122, 563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourriére H, Gallouzi IE, Chebli K, Capony JP, Mouaikel J, van der Geer P, and Tazi J (2001). RasGAP-associated endoribonuclease G3Bp: selective RNA degradation and phosphorylation-dependent localization. Mol. Cell. Biol 21, 7747–7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojsi F, Monsurró MR, and Tedeschi G (2013). Exposure to environmental toxicants and pathogenesis of amyotrophic lateral sclerosis: state of the art and research perspectives. Int. J. Mol. Sci 14, 15286–15311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turpin JA, Schito ML, Jenkins LM, Inman JK, and Appella E (2008). Topical microbicides: a promising approach for controlling the AIDS pandemic via retroviral zinc finger inhibitors. Adv. Pharmacol 56, 229–256. [DOI] [PubMed] [Google Scholar]
- Uversky VN (2017). The roles of intrinsic disorder-based liquid-liquid phase transitions in the ‘‘Dr. Jekyll-Mr. Hyde’’ behavior of proteins involved in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Autophagy 13, 2115–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiente-Echeverría F, Melnychuk L, Vyboh K, Ajamian L, Gallouzi IE, Bernard N, and Mouland AJ (2014). eEF2 and Ras-GAP SH3 domain-binding protein (G3BP1) modulate stress granule assembly during HIV-1 infection. Nat. Commun 5, 4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercruysse T, Basta B, Dehaen W, Humbert N, Balzarini J, Debaene F, Sanglier-Cianférani S, Pannecouque C, Mély Y, and Daelemans D (2012). A phenyl-thiadiazolylidene-amine derivative ejects zinc from retroviral nucleocapsid zinc fingers and inactivates HIV virions. Retrovirology 9, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser ME, Maartens G, Kossew G, and Hussey GD (2003). Plasma vitamin A and zinc levels in HIV-infected adults in Cape Town, South Africa. Br. J. Nutr 89, 475–482. [DOI] [PubMed] [Google Scholar]
- Vyboh K, Ajamian L, and Mouland AJ (2012). Detection of viral RNA by fluorescence in situ hybridization (FISH). J. Vis. Exp 5, e4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace GS, Cheng-Mayer C, Schito ML, Fletcher P, Miller Jenkins LM, Hayashi R, Neurath AR, Appella E, and Shattock RJ (2009). Human immunodeficiency virus type 1 nucleocapsid inhibitors impede trans infection in cellular and explant models and protect nonhuman primates from infection. J. Virol 83, 9175–9182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Choi JM, Holehouse AS, Lee HO, Zhang X, Jahnel M, Maharana S, Lemaitre R, Pozniakovsky A, Drechsel D, et al. (2018). A molecular grammar governing the driving forces for phase separation of prion-like RNA binding proteins. Cell 174, 688–699 e616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, and Jones DT (2004). Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol 337, 635–645. [DOI] [PubMed] [Google Scholar]
- Warui DM, and Baranger AM (2009). Identification of specific small molecule ligands for stem loop 3 ribonucleic acid of the packaging signal Psi of human immunodeficiency virus-1. J. Med. Chem 52, 5462–5473. [DOI] [PubMed] [Google Scholar]
- Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, and Kappes JC (2002). Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother 46, 1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss RA (2006). The discovery of endogenous retroviruses. Retrovirology 3, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellinghausen N, Kern WV, Jöchle W, and Kern P (2000). Zinc serum level in human immunodeficiency virus-infected patients in relation to immunological status. Biol. Trace Elem. Res 73, 139–149. [DOI] [PubMed] [Google Scholar]
- Wen J, Yan M, Liu Y, Li J, Xie Y, Lu Y, Kamata M, and Chen IS (2016). Specific elimination of latently HIV-1 infected cells using HIV-1 protease-sensitive toxin nanocapsules. PLoS ONE 11, e0151572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler JR, Matheny T, Jain S, Abrisch R, and Parker R (2016). Distinct stages in stress granule assembly and disassembly. eLife 5, e18413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wileman T (2006). Aggresomes and autophagy generate sites for virus replication. Science 312, 875–878. [DOI] [PubMed] [Google Scholar]
- Wu W, Henderson LE, Copeland TD, Gorelick RJ, Bosche WJ, Rein A, and Levin JG (1996). Human immunodeficiency virus type 1 nucleocapsid protein reduces reverse transcriptase pausing at a secondary structure near the murine leukemia virus polypurine tract. J. Virol 70, 7132–7142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Mitra M, Naufer MN, McCauley MJ, Gorelick RJ, Rouzina I, Musier-Forsyth K, and Williams MC (2014a). Differential contribution of basic residues to HIV-1 nucleocapsid protein’s nucleic acid chaperone function and retroviral replication. Nucleic Acids Res 42, 2525–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Wang W, Naiyer N, Fichtenbaum E, Qualley DF, McCauley MJ, Gorelick RJ, Rouzina I, Musier-Forsyth K, and Williams MC (2014b). Single aromatic residue location alters nucleic acid binding and chaperone function of FIV nucleocapsid protein. Virus Res 193, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Ehrlich E, Luo K, Xiong Y, and Yu XF (2007). Zinc chelation inhibits HIV Vif activity and liberates antiviral function of the cytidine deaminase APOBEC3G. FASEB J 21, 217–222. [DOI] [PubMed] [Google Scholar]
- Xing L, Wang S, Hu Q, Li J, and Zeng Y (2016). Comparison of three quantification methods for the TZM-bl pseudovirus assay for screening of anti-HIV-1 agents. J. Virol. Methods 233, 56–61. [DOI] [PubMed] [Google Scholar]
- Xue B, Mizianty MJ, Kurgan L, and Uversky VN (2012). Protein intrinsic disorder as a flexible armor and a weapon of HIV-1. Cell. Mol. Life Sci 69, 1211–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada J, Phillips JL, Patel S, Goldfien G, Calestagne-Morelli A, Huang H, Reza R, Acheson J, Krishnan VV, Newsam S, et al. (2010). A bimodal distribution of two distinct categories of intrinsically disordered structures with separate functions in FG nucleoporins. Mol. Cell. Proteomics 9, 2205–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao XJ, Mouland AJ, Subbramanian RA, Forget J, Rougeau N, Bergeron D, and Cohen EA (1998). Vpr stimulates viral expression and induces cell killing in human immunodeficiency virus type 1-infected dividing Jurkat T cells. J. Virol 72, 4686–4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yedavalli VS, Neuveut C, Chi YH, Kleiman L, and Jeang KT (2004). Requirement of DDX3 DEAD box RNA helicase for HIV-1 Rev-RRE export function. Cell 119, 381–392. [DOI] [PubMed] [Google Scholar]
- Yovandich JL, Chertova EN, Kane BP, Gagliardi TD, Bess JW Jr., Sowder RC 2nd, Henderson LE, and Gorelick RJ (2001). Alteration of zinc-binding residues of simian immunodeficiency virus p8(NC) results in subtle differences in gag processing and virion maturation associated with degradative loss of mutant NC. J. Virol 75, 115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu KL, Lee SH, Lee ES, and You JC (2016). HIV-1 nucleocapsid protein localizes efficiently to the nucleus and nucleolus. Virology 492, 204–212. [DOI] [PubMed] [Google Scholar]
- Zeng L, and Zhang L (2011). Efficacy and safety of zinc supplementation for adults, children and pregnant women with HIV infection: systematic review. Trop. Med. Int. Health 16, 1474–1482. [DOI] [PubMed] [Google Scholar]
- Zhang K, Zhang Y, Zi J, Xue X, and Wan Y (2017). Production of human Cu,Zn SOD with higher activity and lower toxicity in E. coli via mutation of free cysteine residues. BioMed Res. Int 2017, 4817376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Sharma NR, Zheng ZM, and Chen M (2019). Viral regulation of RNA granules in infected cells. Virol. Sin 34, 175–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Chen S, Jia L, Shu S, Zhu P, and Liu Y (2012). Selectivity of arsenite interaction with zinc finger proteins. Metallomics 4, 988–994. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Su JM, Samuel CE, and Ma D (2019). Measles virus forms inclusion bodies with properties of liquid organelles. J. Virol 93, e00948–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilliox MJ, Parmigiani G, and Griffin DE (2006). Gene expression patterns in dendritic cells infected with measles virus compared with other pathogens. Proc. Natl. Acad. Sci. USA 103, 3363–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code. The retroviral Gag and other protein sequences examined for PrLDs and ZnFs were obtained through the NCBI Reference Sequence Database (RefSeq) and GenBank: HIV-1: AAD39400.1; SIV: AEK79593.1; FIV: CAA48157.1; EIAV: ACT31322.1; HTLV-1: AAB20767.1; RSV: CAA48534.1; MuLV, RefSeq: NP_040332; HTLV-2: CAA73488.1; BLV: AAA42784.1; MVV: AAB25459.1; HIV-2: AAA76840.1; ALV: AJG42160.1; SRV: BAD89356.1; MMTV: AAF31472.1; FFV: AGC11912.1; SFVagm = RefSeq: YP_001956721.2; SFVcpz: AAA19977.1; HFV: GenBank: CAA68998.1; FeLV: AAA43054.1; GALV: ALV83305.1; PERV: CAA76581.1; BFV: AWK77106.1; SnRV = RefSeq: NP_043925.1; WDSV, RefSeq: NP_045938.1; HERV-W: AAF74213.1; HERV-K: CAA69289.1; JSRV: CAA01899.1; TIA1, GenBank: EAW99824.1; hnRNPA2B1, RefSeq: XP_005249786.1; hnRNPA0, RefSeq: NP_006796.1; hnRNPA1L2, RefSeq: NP_001011725.1; DAZAP1, RefSeq: NP_061832.2; RNApolII, GenBank: AAT12524.1; HEWL, PDB: 1LSZ_A; BPT1, RefSeq: NP_001001554.2; CALM1, GenBank: AAD45181.1; UB, GenBank: AA36789.1; REV, GenBank: AYF56141.1; CRM1, GenBank: CAA69905.2; DDX1, RefSeq: NP_004930.1; DDX11, RefSeq: NP_085911.2; DDX17, GenBank: CAG30318.1; DDX18, GenBank: CAG33341.1; DDX21, RefSeq: NP_004719.2; DDX24, RefSeq: NP_065147.1; DDX3, GenBank: AAC34298.1; DDX4, RefSeq: NP_077726.1; DDX5, RefSeq: NP_001307524.1; DHX36, GenBank: AAH36035.1; DHX9, RefSeq: NP_001348.2; Eif5A, GenBank: AAH80196.1; PIMT, GenBank: AAH07501.1; RanGTP, GenBank: CAG29343.1; RHA, GenBank: AML93444.1; RIP, GenBank: AAH46349.1; Sam68, RefSeq: NP_006550.1.