

SUMMARY
Tbet+CD11c+ B cells arise during type 1 pathogen challenge, aging, and autoimmunity in mice and humans. Here, we examined the developmental requirements of this B cell subset. In acute infection, T follicular helper (Tfh) cells, but not Th1 cells, drove Tbet+CD11c+ B cell generation through proximal delivery of help. Tbet+ CD11c+ B cells developed prior to germinal center (GC) formation, exhibiting phenotypic and transcriptional profiles distinct from GC B cells. Fate tracking revealed that most Tbet+CD11c+ B cells developed independently of GC entry and cell-intrinsic Bcl6 expression. Tbet+CD11c+ and GC B cells exhibited minimal repertoire overlap, indicating distinct developmental pathways. As the infection resolved, Tbet+CD11c+ B cells localized to the marginal zone where splenic retention depended on integrins LFA-1 and VLA-4, forming a competitive memory subset that contributed to antibody production and secondary GC seeding upon rechallenge. Therefore, Tbet+CD11c+ B cells comprise a GC-independent memory subset capable of rapid and robust recall responses.
In brief
Tbet+CD11c+ B cells arise during viral infection and accumulate in aging and in settings of autoimmunity. Song et al. find that Tbet+CD11c+ B cells require interactions with T follicular helper cells but develop outside of germinal centers, forming instead a distinct memory subset at the splenic marginal zone that can substantially contribute to recall responses.
Graphical Abstract
INTRODUCTION
Humoral immunity is mediated by diverse B cell subsets contributing to pathogen clearance and autoimmune-mediated tissue injury through antigen presentation, cytokine secretion, and antibody production. Age-associated B cells (ABCs), marked by expression of T-box transcription factor (Tbet) and the surface marker CD11c, have been described in aging mice (Hao et al., 2011; Rubtsov et al., 2011). B cells exhibiting the Tbet+CD11c+ phenotype in nonaging conditions also arise in chronic infections and autoimmune diseases, as well as following vaccination and acute viral infections (Barnett et al., 2016; Hao et al., 2011; Jenks et al., 2018; Lau et al., 2017; Nellore et al., 2019; Racine et al., 2008; Rubtsov et al., 2011; Rubtsova et al., 2013). Characteristics of these cells vary depending on the inflammatory environment, but dual expression of Tbet and CD11c is a congruent feature. Tbet expression in B cells is necessary for immunoglobulin (Ig) class switching to IgG2a/c and antibody-secreting cell differentiation (Peng et al., 2002; Stone et al., 2019; Wang et al., 2012), whereas the role of CD11c on these cells is unclear. ABCs have high MHC II expression, and upon transfer, migrate to the T-B cell border (Rubtsov et al., 2015), suggesting that they interact with T cells, possibly leading to the Ig somatic hypermutations (Russell Knode et al., 2017). Tbet+CD11c+ B cells are enriched for autoreactive clones and secrete antibodies in response to cytokine and Toll-like receptor (TLR) agonists ex vivo, in contrast to conventional memory B cells that respond effectively to B cell receptor (BCR) stimulation (Jenks et al., 2018). However, their functional contributions in vivo are incompletely understood. On the one hand, Tbet+CD11c+ B cells may contribute to pathogen clearance through the production of protective antibodies (Nellore et al., 2019; Racine et al., 2008; Yates et al., 2013); on the other hand, they are implicated in the progression of autoimmunity (Jenks et al., 2018; Liu et al., 2017; Rubtsova et al., 2017; Wang et al., 2018; Woodruff et al., 2020a, 2020b). Therefore, elucidating Tbet+CD11c+ B cell development along with the regulation of their balance between protective and autoreactive function is necessary for their therapeutic manipulation upon pathogen challenge and in autoimmunity.
The signals driving Tbet+CD11c+ B cell development are being elucidated; nonetheless, the in vivo source(s) of their stimuli remains unclear. IFN-γ induces Tbet expression in in vitro-cultured B cells (Naradikian et al., 2016), whereas IL-21 induces CD11c in both in vitro B cell systems and lupus-prone mice (Manni et al., 2018; Wang et al., 2018). These cytokines inducing Tbet+CD11c+ B cells could be secreted in vivo by T follicular helper (Tfh) and T helper 1 (Th1) cells (Miyauchi et al., 2016; Weinstein et al., 2018). Tfh cells orchestrate germinal center (GC) responses through delivery of cytokines and contact-dependent help such as CD40 ligand (CD40L) (Linterman et al., 2010; Weinstein et al., 2018; Xu et al., 1994; Zotos et al., 2010). Generation of Tbet+CD11c+ B cells is impaired in CD40L- or in SLAM-associated protein (SAP, encoded by Sh2d1a)-deficient mice (Manni et al., 2018; Russell Knode et al., 2017), both of which have defects in Tfh cell differentiation and GC formation (Qi et al.,2008; Xu et al., 1994), suggesting that Tbet+CD11c+ B cell development is Tfh cell and GC dependent (Karnell et al., 2017; Knox et al., 2019). However, T cell expression of Bcl6, the canonical Tfh cell transcription factor (Johnston et al., 2009; Nurieva et al., 2009; Yu et al., 2009), may not be required for Tbet+CD11c+ B cell formation (Levack et al., 2020). Furthermore, Th1 cells secrete IFN-γ and IL-21, express CD40L, and aid B cell production of protective antibodies in the absence of Tfh cells and GCs (Miyauchi et al., 2016; Weinstein et al., 2018). Therefore, the CD4+ T cell subset responsible for the development of Tbet+CD11c+ B cells is incompletely understood, as is the developmental relationship of the latter to GC B cells. Addressing these questions is crucial for developing therapeutic strategies targeting Tbet+CD11c+ B cells while preserving protective GC functions including the generation of affinity matured long-lived memory and plasma cells (PCs).
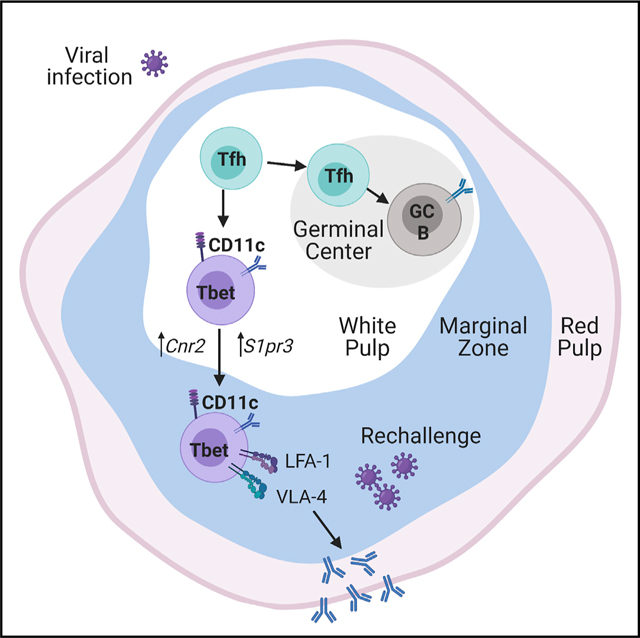
We adopted an acute lymphocytic choriomeningitis virus (LCMV) infection mouse model previously described to induce Tbet+CD11c+ B cells (Rubtsova et al., 2013). We examined their in vivo requirements for generation, relationship with other B cell subsets, localization, and function. We found that Tfh, not Th1, cells colocalize with and drive the generation of Tbet+CD11c+ B cells. Tbet+CD11c+ B cells developed before GC formation, exhibiting phenotypic and transcriptional profiles distinct from GC B cells. Genetic fate tracking and perturbations to GCs showed Tbet+CD11c+ B cell formation to be predominantly GC-independent, with Ig repertoire analysis revealing a minimal clonal overlap between Tbet+CD11c+ and GC B cells. Furthermore, at the peak of GC response, Tbet+CD11c+ B cells were retained via the integrins LFA-1 and VLA-4 at the marginal zone, where they awaited reactivation to become a substantial contributor to the recall response. Thus, Tbet+CD11c+ B cell development in response to viral infection is independent of the GC response, yet still requires help delivered by Tfh cells.
RESULTS
Tbet+CD11c+ B cells expand and contract following an acute viral infection
Tbet+CD11c+ B cells produce antibodies in response to infections and in autoimmune diseases (Isnardi et al., 2010; Liu et al., 2017; Racine et al., 2008; Rubtsov et al., 2013; Rubtsova et al., 2013), yet their developmental requirements in vivo remain unclear. To this end, we infected wild-type (WT) C57BL/6 mice with LCMV-Armstrong to investigate the induction of Tbet+CD11c+ B cells (Rubtsova et al., 2013), a subset found in the spleen but rare in the lymph nodes and blood at days 3, 5, 8, 10, and 14 postinfection (p.i.) (Figure S1A and data not shown). A distinct population of Tbet+CD11c+ B cells emerged by day 5, with its frequency and number continuing to increase until day 8 and 10 p.i., respectively (Figure 1A). Most Tbet+CD11c+ B cells were Ki67hi between days 5 and 8, indicative of cell cycle activation, with a Ki67lo population emerging at day 10 p.i. (Figure 1B). The latter became the predominant Tbet+CD11c+ B cell subset by day 14 p.i., consistent with its decline in cell number (Figures 1A and 1B). These data indicated that Tbet+CD11c+ B cell development and expansion occur in the first 10 days following acute viral infection.
Figure 1. Kinetics of Tbet+CD11c+ B cells in LCMV-Armstrong infection.

(A) Frequencies and numbers of Tbet+CD11c+ B cells among B220+ CD19+ CD44hi splenocytes from WT mice following LCMV infection.
(B) B220+ CD19+ CD44hi CD11c+ splenocytes’ expression of Ki67 and Tbet is shown at top. Frequencies of Ki67lo and Ki67hi Tbet+CD11c+ B cells from individual mice are shown at bottom.
*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 (one-way ANOVA). Data are representative of three independent experiments with 3–5 mice per group. Bars represent means ± SEM.
Tfh cells drive in vivo generation of Tbet+CD11c+ B cells
The kinetics of Tbet+CD11c+ B cell development correlated with those of Th1 and Tfh cells following LCMV infection (Weinstein et al., 2018). To confirm Tbet+CD11c+ B cells’ developmental dependence on CD4+ T cells, we infected Tcrb−/− animals with or without having received Smarta (Stg) CD4+ T cells, which express a transgenic TCR specific for the LCMV peptide Gp66–77 (Oxenius et al., 1998). At day 10 p.i., mice receiving Stg CD4+ T cells had increased induction in total splenic activated B cells as well as Tbet+CD11c+ B cells compared with Tcrb−/− mice receiving no cells and to uninfected controls (Figures 2A and 2B). These data suggest that in vivo, CD4+ T cells drive Tbet+CD11c+ B cell generation.
Figure 2. Tfh cells drive in vivo Tbet+CD11c+ B cell generation.

(A–D) CD44 and Ki67 expression in B220+ CD19+ splenocytes and CD11c and Tbet expression in B220+ CD19+ CD44hi Ki67+ splenocytes from day 10 p.i. Tcrb−/− recipients of transferred Thy1.1 Stg cells and control Tcrb−/− mice (A and B) or day 10 p.i. WT and Icos−/− mice (C and D). Frequencies and numbers of Tbet+CD11c+ B cells (B220+ CD19+ CD44hi Ki67+ CD11+ Tbet+) are shown.
(E) Experimental design of sorted Tfh or Th1 cell transfer into Tcrb−/− mice and quantification of Tbet+CD11c+ B cell frequencies and numbers.
(F and G) Confocal microscopy of spleens from Tbet-AmCyan reporter mice day 8 p.i. (left); anti-CD4 (cyan), anti-B220 (magenta), and anti-IgD (light gray). Shown is a histocytometric recreation of microscopy image plotting location of single cells gated as CD4+, B220+, or IgD+ (middle) as well as a plot showing the location of Tbet+CD11c+ B, Tfh, and Th1 cells, gated as CD4− B220+ CD11c+ Tbet-AmCyan+, CD4+ B220− PSGL1lo PD-1hi, and CD4+ B220− PSGL1hi PD-1lo in (G), respectively (right).
(H) Summary Ripley’s multitype K function (black) calculated using Tbet+CD11c+ B with Tfh cells (left) and with Th1 cells (right) with pointwise 95% confidence interval (gray). The theoretical K function of a homogeneous Poisson process is shown in red.
*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 (one-way ANOVA). Data are representative of three independent experiments with 3–5 mice per group (A–E) or pooled from two independent infections with five mice (F–H). Bars represent means ± SEM.
LCMV infection induces robust CD4+ T cell differentiation into both Th1 and Tfh cell subsets coproducing IFN-γ and IL-21 (Weinstein et al., 2018), two cytokines that promote the Tbet+ CD11c+ B cell phenotype in vitro (Naradikian et al., 2016; Wang et al., 2018). To separate the capability of each Th cell subset in driving the Tbet+CD11c+ B cell development in vivo, we examined LCMV-infected Icos−/− mice, which have defective Tfh cell differentiation and consequently more Th1 cells than their WT counterparts (Figure S1B). At day 10 p.i., B cell activation was reduced in Icos−/− mice compared with WT or uninfected mice, as was the generation of Tbet+CD11c+ B cells (Figures 2C and 2D), suggesting their dependence on Tfh cells for differentiation. We additionally infected Sh2d1a−/− and CD4Cre Bcl6fl/fl mice, two Tfh-cell-targeting mouse models that had little to no reduction in Th1 cell numbers (Figures S1C and S1D). We observed decreased percentages and numbers of Tbet+CD11c+ B cells compared with WT and Bcl6fl/fl controls, respectively (Figures S1C and S1D). To confirm these findings, we adoptively transferred Thy1.1+ Stg+ CD4+ T cells into congenic WT mice and sorted Th1 (Thy1.1+ PSGL-1hi Ly6chi) or Tfh (Thy1.1+ PSGL-1lo Ly6clo PD-1+ CXCR5+) cells (Marshall et al., 2011) at day 5 p.i. with their retransfer into infection-matched Tcrb−/− animals (Figure 2E). Recipients of Th1 cells and uninfected controls failed to generate a substantial Tbet+ CD11c+ B cell response, whereas mice receiving Tfh cells had significant induction of Tbet+CD11c+ B cells (Figure 2E). Together, these data suggest that Tfh cells, not Th1 cells, are the effector CD4+ T cell subset responsible for driving Tbet+ CD11c+ B cell formation in vivo.
Developing Tbet+CD11c+ B cells are found proximal to Tfh cells
Given that the development of Tbet+CD11c+ B cells is promoted by Tfh cells, we hypothesized that they would be enriched in proximity to the latter cells. To test this hypothesis, we analyzed spleen sections obtained from Tbet-AmCyan reporter mice at day 8 p.i. with LCMV, the proliferative phase of Tbet+CD11c+ B cell response (Figure 1B), by multiplexed confocal imaging with histocytometric analysis (Gerner et al., 2012; Yu et al., 2015) (Figure S2A). This technique allowed quantitative analysis of immunofluorescent images on a single-cell level and identification of Tbet+CD11c+ B cells, Tfh cells, and, as a control, Th1 cells in each spleen section (Figures 2F and 2G). We confirmed that plasmacytoid dendritic cells (pDCs) were Tbet− at this time point and, therefore, not misidentified as Tbet+CD11c+ B cells (Figure S2B). Because of the early disruption of splenic architecture by LCMV infection (Mueller et al., 2007; Scandella et al., 2008), most microanatomical structures could not be delineated to precisely quantify the distribution of Tbet+CD11c+ B, Tfh, and Th1 cells. However, by plotting the location of the three populations, we found Tfh cells mostly around B cell follicles and Th1 cells dispersed within red pulp (Figure 2F). Moreover, we observed clusters of Tbet+CD11c+ B cells at the edge of follicles adjacent to Tfh cells (Figure 2F). To quantify the clustering of Tbet+CD11c+ B cells around Tfh or Th1 cells, we used a multitype Ripley’s K function to analyze the distribution pattern of Tbet+CD11c+ B cells in relation to either effector CD4+ T cell subset. We found no significant difference between the computed K function for Th1 cells and the theoretical K function assuming Poisson distribution (Figure 2H), suggesting random distribution. In contrast, the calculated K function for Tfh cells exceeded that of the theoretical K function (Figure 2H), consistent with clustering between Tbet+CD11c+ B cells and Tfh cells. Combined with their role in driving Tbet+CD11c+ B cell formation (Figures 2C–2E), these findings suggest that Tfh cells deliver help in proximity to promote Tbet+CD11c+ B cell differentiation, which could include cytokines such as IL-21 and IFN-γ and the contact-dependent help CD40L (Keller et al., 2021; Manni et al., 2018; Naradikian et al., 2016; Russell Knode et al., 2017; Wang et al., 2018).
Most Tbet+CD11c+ B cells develop independently of GCs
Tbet+CD11c+ B cells have been speculated to be GC-derived because they undergo Ig somatic mutation and class switch recombination (Karnell et al., 2017; Knox et al., 2019); however, both processes can occur outside GCs (Di Niro et al., 2015; Roco et al., 2019; William et al., 2002). To assess whether Tbet+CD11c+ B cell differentiation is dependent on GCs, we first examined the expression of canonical GC B cell markers throughout their development (Laidlaw et al., 2017; Lu et al., 2017). At 6, 10, and 15 days p.i. with LCMV, we found minimal coexpression of GL-7 and EphrinB1 on Tbet+CD11c+ B cells, indicating phenotypic distinction from GC B cells (Figure 3A). We next performed RNA-sequencing on sorted Tbet+CD11c+, GC, and naive follicular B cells at day 12 p.i. with LCMV (Figures S3A and S3B). Principal-component analysis (PCA) of total transcriptome demonstrated that Tbet+CD11c+ B cells were a subset distinct from GC and naive B cells (Figure 3B). Moreover, differential gene expression analysis of Tbet+CD11c+ and GC B cells, performed postnormalization to naive B cells, revealed that GC B cell signature genes, such as Bcl6, S1pr2, and Aicda, were downregulated in Tbet+CD11c+ B cells. Conversely, expression of Tbet+CD11c+ B cell signature genes such as Itgax (encoding CD11c), Tbx21 (encoding Tbet), and Fcrl5 were lower in GC B cells (Figure 3C).
Figure 3. Tbet+CD11c+ B cells develop independently of GC B cells.

(A) GL-7 and EphrinB1 expression on B220+ IgDlo (black) and B220+ CD19+ CD44hi CD11c+ Tbet+ (blue) splenocytes from WT mice at days 6, 10, and 15 p.i. with gating for GC (B220+ IgDlo GL-7+ EphrinB1+) B cells.
(B) PCA of naive follicular (B220+ CD19+ IgDhi CD23+, green), GC (B220+ IgDlo CD95+ GL-7+, gray), and Tbet+CD11c+ (B220+ CD19+ CD44hi CD11c+ Tbet-AmCyanhi, blue) B cells sorted from Tbet-AmCyan reporter mice day 12 p.i.
(C) Heatmap of selected significantly (q < 0.05) differentially expressed genes.
(D) Confocal microscopy of spleens from Tbet-AmCyan reporter mice day 12 p.i. (top left); anti-CD4 (cyan), anti-B220 (orange), anti-IgD (yellow), and PNA (magenta). Shown is a histocytometric recreation of microscopy image plotting location of single cells gated as CD4+, B220+, IgD+, or PNA+ (top right). Location and quantification of Tbet+CD11c+ B cells (CD4− B220+ CD11c+ Tbet-AmCyan+) in splenic white pulp compartments: follicular mantle (FM), germinal center (GC), and T cell zone (TZ) on bottom.
(E) TdTomato expression of splenic GL-7+ (GL-7+ B220+ IgDlo) and Tbet+CD11c+ B cells at day 10 p.i.
(F) Experimental design of chimeric mice generated from CD45.1 CD19Cre Bcl6+/+ mixed with CD45.2 CD19Cre Bcl6fl/fl donor bone marrow cells. Frequencies and ratios of CD45.1 to CD45.2 cells in naive, GC, and Tbet+CD11c+ B cells in the spleens of mixed bone marrow chimeric mice at day 12 p.i. are shown.
**p < 0.01; ***p < 0.001; ****p < 0.0001 (D and F, one-way ANOVA; E, Student’s t test). Data are pooled from two independent infections with seven mice (D), are pooled from one experiment with nine mice (E), or are representative of two independent experiments with five mice per group (F). Bars represent means ± SEM.
To determine whether Tbet+CD11c+ B cells developed within GCs, we performed histocytometry in Tbet-AmCyan reporter mouse spleens at day 12 p.i. with LCMV when architecture has been mostly restored and early GCs have formed (Figures 3D and S4A) (Scandella et al., 2008). Within splenic white pulp outlined by areas with IgD and CD4 staining, we found that Tbet+CD11c+ B cells were largely absent from PNA- and B220-labeled GCs (Figure 3D). Most white pulp Tbet+CD11c+ B cells were found at the outer edge of the follicle, the follicular mantle (Figure 3D). This was consistent with our observation that at day 14 p.i., Tbet+CD11c+ B cells have uniformly downregulated Ki67 (Figure 1B), suggesting that they do not participate in the GC response.
The development of Tbet+CD11c+ B cells preceded that of GC B cells in response to LCMV infection (Figure S3C); however, the former could be early emigrants from GCs that have downregulated their GC signatures. To test this hypothesis, we utilized S1PR2CreERT2 Rosa26Lox-Stop-Lox-tdTomato mice which allow GC-specific permanent cell labeling (Green et al., 2011; Shinnakasu et al., 2016). Mice were infected and administered tamoxifen starting from day 4 p.i. to fate track GC output. At day 10 p.i., more than 80% of Tbet+CD11c+ B cells lacked TdTomato expression, whereas the majority of GL-7+ B cells enriched for early GC B cells expressed TdTomato, suggesting that Tbet+CD11c+ B cells rarely originate from GCs (Figure 3E). To determine whether Tbet+CD11c+ B cell differentiation diverges from that of GC B cells, we next generated mixed bone marrow chimeras of 50% CD45.2-marked CD19CreBcl6fl/fl and 50% CD45.1-marked CD19CreBcl6+/+ control cells such that the latter would give rise to B cells capable of supporting Tfh cell development and GC formation (Dent et al., 1997; Ye et al., 1997). Mice were infected with LCMV, and at day 12 p.i., the ratio of splenic CD45.1 to CD45.2 cells within the naive, GC, and Tbet+CD11c+ B cell subsets was assessed. Compared with GC B cells whose development requires cell-intrinsic Bcl6 expression, CD45.2 CD19CreBcl6fl/fl cells populated about half of the Tbet+CD11c+ B cell subset in a competitive setting (Figure 3F). Thus, Tbet+CD11c+ B cells are phenotypically and transcriptionally distinct from GC B cells and predominantly develop independently of GCs.
GC-independent generation of Tbet+CD11c+ B cells following influenza virus infection
LCMV infection leads to architectural disruption in lymphoid tissues, a common feature among infections that induce accumulation of Tbet+CD11c+ B cells (Austin et al., 2019; Racine et al., 2008; Rivera-Correa et al., 2017). To investigate whether GC-independent generation of Tbet+CD11c+ B cells is conserved in infections that preserve splenic architecture, we infected Tbet-AmCyan reporter mice with the mouse-adapted strain of influenza virus, A/Puerto Rico/8/1934 (PR8), which induces antigen-specific Tbet+CD11c+ B cells in the spleen but rarely the draining mediastinal lymph node (mLN) (Johnson et al., 2020) (Figure S3D). On days 7, 10, and 15 p.i., HA-specific GC B cells in the spleen and mLN frequently expressed low amounts of reporter AmCyan but did not phenotypically overlap with the Tbet+CD11c+ B cells that were high in AmCyan expression (Johnson et al., 2020) (Figures 4A–4C). Intracellular staining of Tbet resulted in a loss of AmCyan reporter expression in the GL-7+ population consistent with its low expression of Tbet protein (Figure 4B). On the other hand, the canonical GC markers GL-7 and EphrinB1 were rarely expressed or coexpressed by the splenic Tbet+CD11c+ B cells throughout the course of infection, regardless of whether they were identified through reporter or intracellular staining (Figure 4C and data not shown). Therefore, influenza infection generates Tbet+CD11c+ B cells with minimal phenotypic overlap with GC B cells.
Figure 4. GC-independent development of influenza HA-specific Tbet+CD11c+ B cells.

(A) Flow cytometry verification of HA-specific B cell detection in mediastinal lymph node (mLN) following PR8 infection.
(B) Expression of AmCyan (left, right) and Tbet protein by intracellular staining (center) against GL-7 in the spleen of a representative Tbet-AmCyan reporter mouse (gray and magenta) and a nonreporter WT mouse (orange) at day 10 p.i.
(C) Expression of AmCyan in splenic HA-specific GC B cells (left) and GL-7 in HA-specific Tbet+CD11c+ B cells (right) along with gating controls.
(D) TdTomato expression of HA-specific GC (B220+ CD19+ IgDlo HA+ GL-7+ EphrinB1+) and Tbet+ CD11c+ (B220+ CD19+ HA+ CD44hi Ki67+ Tbet+ CD11c+) B cells in the spleens and mLNs at day 10 p.i. with PR8.
(E) Frequencies and ratios of naive, HA-specific GC, and Tbet+CD11c+ B cells in spleens of 50/50 CD45.1 CD19Cre Bcl6+/+ to CD45.2 CD19Cre Bcl6fl/fl mixed bone marrow chimeric mice at day 10 p.i. with PR8.
**p < 0.01; ***p < 0.001; ****p < 0.0001 (one-way ANOVA). Data are from one experiment with seven mice (D) or are representative of two independent experiments with 4–5 mice per group (A–C and E). Bars represent means ± SEM.
We next investigated the potential GC origin of Tbet+CD11c+ B cells in influenza virus challenge using S1PR2CreERT2 Rosa26Lox-Stop-Lox-tdTomato mice. Similar to our observation with LCMV, influenza infection generated Tbet+CD11c+ B cells in the spleen that predominantly lacked TdTomato expression at day 10 p.i., in contrast to the majority of the GC B cells expressing TdTomato, suggesting that Tbet+CD11c+ B cells rarely originated from GCs (Figure 4D). We next infected mixed bone marrow chimeras of 50% CD45.2-marked CD19CreBcl6fl/fl and 50% CD45.1-marked CD19CreBcl6+/+ control cells. HA-specific Tbet+CD11c+ B cells contained roughly equal fractions of Bcl6-deficient and Bcl6-sufficient cells at day 10 p.i., compared with their GC counterpart that was dominated by Bcl6-sufficient cells (Figure 4E). Altogether, these data suggest that the GC-independent generation pathway of Tbet+CD11c+ B cells is conserved among acute viral infections.
Infrequent clonal sharing between Tbet+CD11c+ and GC B cells
We next evaluated class switch recombination, somatic mutation, and clonal relationships by performing next-generation Ig sequencing of sorted Tbet+CD11c+, GC, and naive follicular B cells from individual Tbet-AmCyan reporter mice at day 12 p.i. with LCMV. Libraries were prepared by amplifying full-length Igs with unique molecular bar coding of transcripts for high sequence accuracy. Each biological sample yielded on average 248,116 unique high-quality assembled reads, from which the heavy-chain sequences were filtered for analysis. As expected, naive follicular B cells were predominantly of IgM and IgD isotypes, whereas about 80% of GC B cell sequences had class switched to IgG (Figure 5A). Isotype usage of Tbet+CD11c+ B cells was not different from that of GC B cells (Figure 5A), consistent with previous reports of class switch recombination occurring outside GCs (Chan et al., 2009; Di Niro et al., 2015; Roco et al., 2019; William et al., 2002). Quantification of overall mutation frequency within full-length Igs revealed that on average 61.5% of GC B cell sequences were mutated compared with 41.6% of Tbet+CD11c+ B cell sequences. In addition, Tbet+ CD11c+ B cells had a lower total mutation load of 0.64% on average compared with GC B cells with 0.99% of total mutation frequency (Figure 5B). Analysis of mutations within the complementarity-determining region revealed that GC B cells had a higher load of replacement mutations (1.21%) compared with Tbet+CD11c+ B cells (0.66%) despite having similar frequencies of silent mutations (Figure 5C), suggesting reduced affinity maturation in the latter subset. These data indicate that although their GC entry is not required for somatic mutation, affinity maturation may be inefficient in Tbet+CD11c+ B cells outside the GC.
Figure 5. Ig repertoire analysis of Tbet+CD11c+ and GC B cells.

(A) Isotype distribution in Ig sequences of naive follicular (B220+ CD19+ IgDhi CD23+), GC (B220+ IgDlo CD95+ GL-7+), and Tbet+CD11c+ (B220+ CD19+ CD44hi CD11c+ Tbet-AmCyanhi) sorted from individual Tbet-AmCyan reporter mice day 12 p.i. with LCMV.
(B) Total mutation frequency within the Ighv gene.
(C) Frequencies of silent and replacement mutations within the complementary determining regions (CDRs).
(D) Clonal overlap of naive follicular (green), GC (gray), and Tbet+CD11c+ B cells (blue) sequences. Chords show clones shared by naive and GC (gray), naive and Tbet+CD11c+ (light green), GC and Tbet+CD11c+ (light blue), and all three subsets (purple) with chord width corresponding to clonal population size.
(E) Venn diagram showing numbers of shared clones between naive follicular (green), GC (gray), and Tbet+CD11c+ B cells (blue) sequences.
(F) Reconstructed lineage trees representative of most expanded clones (left three) and of shared clones between GC and Tbet+CD11c+ B cells (right three). Number of unique sequences at node with more than one unique sequence is shown to which the node size is proportional. Acquisition of somatic mutations is denoted by numbers in blue indicating number of mutations and branching, with the exception of the node directly connected to germline in each tree, which has the germline sequence.
**p < 0.01; ****p < 0.0001 (two-sided Dunn’s multiple comparisons test). Data are from one mouse representative of three biological replicates (A–C and F).
To determine clonal relationships between Tbet+CD11c+ and GC B cells, we examined clonal overlaps between B cell subsets (Figure 5D). Clones shared between Tbet+CD11c+ and GC B cells accounted for less than 10% of total sequences in the respective populations (Figures 5D and 5E). Next, we reconstructed lineage trees for clonally related sequences and found clonal lineages predominantly derived from either Tbet+CD11c+ or GC B cells (Figure 5F). Analysis of shared clones revealed early bifurcation of Tbet+CD11c+ and GC B cells and infrequent interconversions, consistent with the GC-independent development of most Tbet+CD11c+ B cells (Figures 3, 4, and 5F). These data collectively reveal that Tbet+CD11c+ and GC B cell development occurs through largely separate processes, resulting in distinct clones.
Tbet+CD11c+ B cells accumulate at the splenic marginal zone
Tbet+CD11c+ B cells developed independently of GCs and localized both within and outside the white pulp (Figure 3F). To locate Tbet+CD11c+ B cells outside of white pulp, we performed histocytometry of day 12 p.i. LCMV-infected Tbet-AmCyan reporter mouse spleens with the red pulp marker F4/80 (Figures 6A and S4B). We observed clusters of Tbet+CD11c+ B cells located close to the white and red pulp border (Figure 6A). By day 15 p.i., many of these B cells were localized to the marginal zone as identified by MadCam-1 expressed on sinus-lining cells (Figure 6B). To confirm this distribution pattern, we performed intravenous (i.v.) injection with anti-CD45 antibodies, preferentially labeling compartments exposed to open blood circulation including the marginal zone (Cinamon et al., 2008). A significant fraction of Tbet+CD11c+ B cells was labeled at day 12 p.i. and continued to increase at day 15 p.i. compared with naive and GC B cells (Figure 6C). These data suggest that Tbet+CD11c+ B cells accumulate at the marginal zone as the infection resolves.
Figure 6. Integrin-dependent Tbet+CD11c+ B cell retention at the marginal zone.

(A) Confocal microscopy of spleens from Tbet-AmCyan reporter mice day 12 p.i. (top left), showing staining of anti-CD4 (magenta), anti-B220 (yellow), and anti-F4/80 (yellow). Shown is a histocytometric recreation of confocal microscopy image plotting location of single cells gated as CD4+, B220+, or F4/80+ (top right). Locations of Tbet+CD11c+ B cells (CD4− B220+ CD11c+ Tbet-AmCyan+) are shown in blue (clusters highlighted in magenta), along with splenic white pulp (WP) and red pulp (RP).
(B) Confocal microscopy of spleens from Tbet-AmCyan reporter mice day 15 p.i., showing staining of anti-MadCam1 (yellow), anti-F4/80 (green), anti-CD11c (cyan), and anti-B220 (orange), with Tbet-AmCyan (magenta). Tbet+CD11c+ B cells are identified through histocytometry (white), and the splenic compartments WP, RP, and marginal zone (MZ) were marked.
(C) I.v.-administered anti-CD45 antibody labeling of naive (B220+ CD19+ IgDhi CD23+), GC (B220+ IgDlo CD95+ GL-7+), and Tbet+CD11c+ (B220+ CD19+ CD44hi CD11c+ Tbet-AmCyanhi) at days 12 and 15 p.i. in Tbet-AmCyan mice.
(E) Volcano plot comparing gene expression of Tbet+CD11c+ B cells sorted and sequenced on days 8 and 15 p.i.
(F) Temporal expression of S1pr3 and Cnr2 from RNA-seq data in (E).
(G) S1P transwell assay measuring ratio of migrating to total input Tbet+CD11c+ B cells from Tbet-AmCyan reporter mice pretreated with inhibitors.
(H) VLA-4 and integrin αL expression on naive and Tbet+CD11c+ B cells at 15 p.i. in Tbet-AmCyan mice.
(I) Percentage and number of Tbet+CD11c+ B cells among total B cells after injection of LFA-1/VLA-4 blocking or isotype control antibodies.
*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 (C and G, one-way ANOVA; I, Student’s t test). Data are pooled from two independent infections (A, B, and I) or are representative of three independent experiments with three or four mice per group (C, H, and G). Bars represent means ± SEM.
Integrin-dependent splenic retention of Tbet+CD11c+ B cells
To assess transcriptional changes accompanying the development of Tbet+CD11c+ B cells, we next examined their gene expression at days 8, 10, and 15 p.i., which corresponded to their proliferation, peak, and contraction phases, respectively. Analysis of Tbet+CD11c+ B cell transcriptomes revealed 1,433 differentially expressed genes between days 8 and 15, 762 of which continued to be differentially expressed by day 10, suggesting that most transcriptional changes occurred during the resolution phase of the infection (Figures S5A and 6E). Expressions of S1pr3, a S1P receptor family member, and cannabinoid receptor 2 (Cnr2), both previously reported to be involved in migration to the marginal zone, were increased as the infection resolved (Figures 6F and S5B) (Basu et al., 2011; Cinamon et al., 2004, 2008; Muppidi et al., 2011). CXCR3, whose ligands are produced at the marginal zone (Hirako et al., 2016; Kurachi et al., 2011), was expressed higher on Tbet+CD11c+ B cells than their naive follicular and GC counterparts (Figure S5C), reflecting its transcriptional regulation by Tbet (Beima et al., 2006; Li et al., 2016; Moir et al., 2008; Rakhmanov et al., 2009; Weiss et al., 2009). To investigate whether S1PR3 is involved in the positioning of splenic Tbet+CD11c+ B cells, we assayed their migration toward S1P ex vivo at day 15 p.i. in the presence of inhibitors. Addition of FTY720, a S1P analog with high and moderate affinity to S1PR1 and S1PR3, respectively, limited Tbet+CD11c+ B cell chemotaxis (Figure 6G). TY52156, a selective inhibitor for S1PR3, completely abolished Tbet+CD11c+ B cell migration (Figure 6G). However, mixed bone marrow chimeras of CD45.1-marked WT and CD45.2-expressing S1pr3−/− or S1pr3+/− cells demonstrated no advantage of S1pr3+/− over S1pr3−/− cells at day 15 p.i. in populating the i.v.-labeled Tbet+CD11c+ B cell compartment. This suggested that despite the role of S1PR3 in driving Tbet+CD11c+ B cell migration, its singular loss was insufficient to disrupt their localization to the marginal zone, possibly because of compensation by other factors such as Cnr2 and CXCR3 (Figure S5D).
The integrins LFA-1 (αLβ2, encoded by Itgal and Itgb2, respectively) and VLA-4 (α4β1, encoded by Itga4 and Itgb1, respectively) facilitate splenic retention of marginal zone B cells through interactions with ICAM-1 and VCAM-1 therein (Lu and Cyster, 2002; Tedford et al., 2017). Tbet+CD11c+ B cells showed elevated transcriptional and surface protein expression of both LFA-1 and VLA-4, compared with naive follicular B cells (Figures 3C and 6H). To examine whether these integrins are required for Tbet+CD11c+ B cell retention, we treated Tbet-AmCyan reporter mice at day 15 p.i. with antibodies blocking the alpha subunits of LFA-1 and VLA-4 or isotype control for 3 h. LFA-1 and VLA-4 blockade resulted in a significant loss of Tbet+CD11c+ B cells in the spleens with concomitant increase in the blood, suggesting their release into circulation (Figure 6I). We conclude that Tbet+CD11c+ B cell retention in the marginal zone is dependent on LFA-1 and VLA-4 activity.
Tbet+CD11c+ B cells produce virus-specific antibodies upon acute infection
The relationship between the Tbet+CD11c+ B cells with non-GC B cell subsets remains unclear. Therefore, we examined the differentially expressed genes between Tbet+CD11c+, GC, and naive B cells to identify signatures indicative of other functional B cell subsets. Among the highly expressed genes in the Tbet+CD11c+ compartment were Prdm1 (encoding Blimp-1), Xbp1, and Sdc1 (encoding CD138) suggestive of the capability for antibody secretion, along with Mndal, Zeb2, and Tle3 associated with memory B cells (Figure 7A) (Bhattacharya et al., 2007; Laidlaw et al., 2020). The surface phenotype of Tbet+CD11c+ B cells resembled that of memory B cells and not PCs having increased expression of CD38 but not CD138 at days 10, 12, and 15 p.i. (Figure 7B and data not shown). However, gene set enrichment analysis revealed that day 12 p.i. Tbet+CD11c+ B cells did not exhibit transcriptional signatures of conventional memory B cells in existing molecular signature databases (data not shown), possibly because of the low frequency of this subset in the memory B cell pool. To test whether Tbet+CD11c+ B cells showed functional similarities with other B cell subsets, we assayed Tbet-AmCyanhi CD11c+ B cells, PCs, and IgDlo CD38+ GL-7− Tbet-AmCyanlo/− containing prememory B cells and naive B cells sorted from reporter mice at day 10 p.i. for anti-LCMV IgG secretion by ELISPOT. B cells were cultured in the presence or absence of R848, a TLR7 agonist that drives robust ex vivo differentiation of memory B cells into antibody-secreting cells (Richard et al., 2008; Weinstein et al., 2013). Antibody production by IgDlo CD38+ GL-7− B cells was reliant on TLR stimulation, whereas that of PCs was independent (Figure 7C). Tbet+CD11c+ B cells contained antibody-secreting cells producing anti-LCMV antibodies prior to TLR stimulation, with their number increasing after stimulation (Figure 7C). This suggests that Tbet+CD11c+ B cells have the potential to contribute to the primary humoral response against viral infection.
Figure 7. Tbet+CD11c+ B cells produce antiviral antibodies in both primary and recall responses.

(A) Heatmap of selected significantly (q < 0.05) differentially expressed genes in naive follicular, GC, and Tbet+CD11c+ B cells from RNA-seq data in Figure 3B.
(B) CD138 and CD38 expression of PCs (CD19int CD138+, pink), GC (gray), and CD11c+ Tbet+ B cells (solid line) in the spleens of Tbet-AmCyan reporter mice at day 10 p.i.
(C) Representative ELISPOT with number of cells plated per well and its quantification of sorted naive (B220+ CD19+ IgDhi Tbet-AmCyan−), IgDlo CD38+ GL-7− (B220+ CD19+ IgDlo CD38+ GL-7− Tbet-AmCyanlo/−), PCs (CD19int CD138+), and Tbet+CD11c+ B cells from spleens of Tbet-AmCyan reporter.
(D) BrdU and CD38 staining among B220+ CD19+ splenocytes from Tbet-AmCyan reporter mice at day 30 p.i. I.v.-administered anti-CD45 antibody labeling of BrdU+ memory (gray) and BrdU+ Tbet+CD11c+ B cells (black line).
(E) Representative anti-LCMV IgG ELISPOT (top) with number of cells plated per well and its quantification (bottom) of sorted naive, memory (B220+ CD19+ IgDlo CD38hi GL-7lo Tbet-AmCyanlo/−), PCs, and Tbet+CD11c+ B cells from spleens of Tbet-AmCyan reporter mice.
(F) Experimental design of polyclonal CD11c-DTR B cell transfer into MD4 mice. ELISA quantification of serum anti-LCMV IgG antibody of MD4 mice at days 0, 3, and 5 postrechallenge.
(G) Experimental design showing the transfer of all sorted naive, memory, and Tbet+CD11c+-B cells into MD4 mice. ELISA quantification of serum anti-LCMV IgG antibody of MD4 mice at days 5 and 10 p.i.
*p < 0.05; ***p < 0.001 (C–F, Student’s t test; G, one-way ANOVA). Data are representative of three (C and E) independent experiments or pooled from two independent experiments (F and G). Bars represent means ± SEM.
Tbet+CD11c+ B cells have been implicated in contributing to circulating autoantibodies in response to acute viral infection (Woodruff et al., 2020b, 2020c). We next examined whether they could produce autoantibodies in addition to virus-specific antibodies. We cultured Tbet+CD11c+, IgDlo CD38+ GL-7− B cells, and PCs sorted from day 10 p.i. Tbet-AmCyan reporter mice in the presence of R848 and assessed their secreted self-reactive Igs by autoantibody microarray. Tbet+CD11c+ and IgDlo CD38+ GL-7− B cells secreted similar repertoires of autoantigen-binding IgG compared with that from PCs, suggesting that the two former populations are enriched for autoreactive clones (Figure S6A). To test whether Tbet+CD11c+ B cells produce autoantibodies without TLR-driven differentiation ex vivo, we performed anti-double stranded DNA and anti-chromatin IgG ELISPOTs. Despite containing a higher frequency of autoantibody producers compared with IgDlo CD38+ GL-7− B cells, Tbet+CD11c+ B cells rarely secreted autoantibodies in the absence of R848, suggesting that further differentiation toward antibody-secreting cells is necessary for their contribution to serum autoreactivity (Figure S6B).
The extent of in vivo contribution from Tbet+CD11c+ B cells to humoral responses against acute viral infection has not been properly evaluated. Transient Tbet expression is required for class switching to IgG2c (Peng et al., 2002) and is critical for antibody-secreting cell differentiation (Stone et al., 2019), which obfuscate experimental results obtained using animals genetically deficient in Tbet. To circumvent this caveat, we designed a specific depletion model in which purified polyclonal B cells from CD11c-diphtheria toxin receptor (DTR)/GFP mice were adoptively transferred into congenically marked MD4 recipients expressing a transgenic BCR against lysozyme (Jung et al., 2002; Mason et al., 1992), which mount minimal endogenous B cell responses to LCMV (Figure S7A and data not shown). Activated B cells unexpectedly increased the expression of DTR independently of Itgax expression in vitro and CD11c protein expression in vivo, leading to global ablation of proliferating B cells on diphtheria toxin (DT) administration (Figures S7B–S7D). These data indicate that the CD11c-DTR system is unsuitable for depleting CD11c-expressing B cells immediately following viral challenge.
Tbet+CD11c+ B cells form a rare yet functionally competitive memory B cell subset
Tbet+CD11c+ B cells populate the memory compartment, and we observed that these cells maintained the IgDlo CD38+ GL-7− memory phenotype to until at least day 35 p.i. (data not shown) (Johnson et al., 2020; Nellore et al., 2019; Sutton et al., 2021). Calculation of Tbet+CD11c+ B cell frequency among IgDlo CD38+ GL-7− B cells throughout the course of the infection is unlikely to reflect contribution of the former population to the memory pool, as the latter also contained memory B cells generated prior to LCMV infection. To confirm that acute infection generated Tbet+ CD11c+ memory B cells and assess their prevalence, we administered BrdU in the drinking water of LCMV-infected Tbet-AmCyan reporter mice starting from day 4 p.i. for 10 days (Figure 7D). At 30 days p.i., Tbet+CD11c+ B cells represented around 5% of BrdU+ IgDlo CD38+ GL-7− memory B cells, suggesting that they are a small fraction within the latter compartment. I.v. injection of anti-CD45 antibody labeled the majority of Tbet+ CD11c+ memory B cells, but only 30% of total memory B cells, suggesting that the former population maintained their localization to the marginal zone (Figure 7D). To assess the function of Tbet+CD11c+ memory B cells, LCMV-specific ELISPOT assays were performed on sorted memory B cell subsets as well as PCs and naive B cells at day 30 p.i. Similar to memory B cells, Tbet+CD11c+ B cells were largely unable to secrete anti-LCMV IgG antibodies in the absence of ex vivo TLR7 stimulation, suggesting that they represent a memory subset in both phenotype and function. Despite being less common within the IgDlo CD38+ GL-7− memory B cells, a large number of Tbet+CD11c+ B cells were capable of differentiation into anti-LCMV IgG producers (Figure 7E). Therefore, Tbet+CD11c+ B cells are a memory subset with considerable functional potential, likely against blood borne pathogens, given their localization.
To determine the in vivo contribution of Tbet+CD11c+ B cells as a memory B cell subset, we modified our previous CD11c-DTR transfer system to circumvent its caveats (Figure 7F). Two doses of DT were administered at the memory phase, days 28 and 30 p.i., with transfer recipients rechallenged 5 days after the last dose to ensure that reactivation was not affected by DT. By day 35 p.i., there was a universal increase in LCMV-specific serum antibodies in MD4 mice receiving transferred polyclonal B cells compared with those receiving no cells and uninfected controls (data not shown). Serum anti-LCMV IgG titers following LCMV recall demonstrated a trend toward being elevated in PBS-treated animals compared with their DT-treated counterparts, suggesting that CD11c-expressing memory B cells rapidly differentiated into antibody-secreting cells upon rechallenge (Figure 7F). At day 5 of recall, we examined the phenotype of transferred B cells and found them to be more prominent participants of the secondary GC response in PBS-treated animals compared with mice receiving DT (Figure S7E). In contrast, endogenous B cells from MD4 recipients showed comparable percentages of secondary GC B cells, suggesting minimal effect from global DT toxicity (Figure S7E). To confirm these observations, we sorted and transferred all naive or memory B cells, the latter with or without the Tbet+CD11c+ phenotype, from spleens of day 30 p.i. infected Tbet-AmCyan reporter mice into naive MD4 recipients. Animals receiving the small number of Tbet+CD11c+ B cells generated rapid and robust antibody response against LCMV at days 5 and 10 p.i., with serum IgG titers higher than mice receiving naive B cells and comparable to those receiving memory B cells lacking the Tbet+CD11c+ phenotype (Figure 7G). In contrast, these latter two recipients produced more anti-LCMV IgM antibodies (Figure S7F). We additionally found at day 10 p.i. that all three transferred subsets gave rise to GC B cells, confirming the ability of Tbet+CD11c+ memory B cells to enter GCs upon antigenic reencounter (Figure S7G). Altogether, these observations suggest that Tbet+CD11c+ memory B cells are competitive within the memory B cell compartment for reactivation, entry into the secondary GC, and antibody production.
DISCUSSION
The developmental dependence of Tbet+CD11c+ B cells on T cells and their relationship to GC B cells have been unclear, as has their function in acute viral infections. We showed that Tbet+CD11c+ B cells are developmentally dependent on help delivered by Tfh cells in proximity. Yet, most of these B cells developed prior to and independently of GCs, indicating a separate developmental pathway. Tbet+CD11c+ B cells were retained at the marginal zone via integrins LFA-1 and VLA-4, with corresponding increased expression of genes suggestive of their migration during the resolution phase of infection. Functional comparisons between memory B cells expressing and lacking Tbet and CD11c uncovered the substantial contribution of the former to recall responses.
B cell responses mediated by Tfh cells outside of GCs (extra-GC Tfh cells) have not been well characterized. Previous reports have described Tfh-cell-dependent, extrafollicular plasma cell responses that arise when GC formation is inhibited (Di Niro et al., 2015; Lee et al., 2011; Levack et al., 2020; Odegard et al., 2008; William et al., 2002); however, it is unclear to what extent this occurs when GCs are intact. We found that extra-GC Tfh cells are necessary for the Tbet+CD11c+ B cell response. Tbet+CD11c+ B cells expressed IgG2 isotype, with more than one-third of mutated sequences in their repertoire, which is likely a consequence of CD40L-dependent Tfh cell help inducing AID expression in B cells (Dedeoglu et al., 2004; Zhou et al., 2003). However, Tbet+CD11c+ B cells lacked signs of affinity maturation and rapidly downregulated Aicda expression, suggesting that they do not engage in serial interactions with Tfh cells, in contrast to GC B cells.
Limited overlap was found between the Tbet+CD11c+ and GC B cells in phenotype, transcriptome, Ig repertoire, and localization despite developmental reliance of both on Tfh cells, suggesting that they are opposing cell fates regulated by disparate signals. This seemingly contradicts a previous report suggesting extensive overlap between the two populations (Johnson et al., 2020), but most likely resulted from our exclusion of Tbet reporterlo cells that at the time of detection failed to maintain Tbet expression. We instead focused on Tbet reporterhi cells, which contained the subset that coexpressed CD11c and were uniquely retained in the spleen (Johnson et al., 2020). Separation in the development of the Tbet+CD11c+ and GC subsets may occur early after activation and was maintained by rare interconversion, as their shared clones originated from common precursors that lacked extensive somatic mutation. Although previous work provided evidence for an extrafollicular origin for Tbet+CD11c+ B cells, our study directly elucidated their extent of developmental divergence from the GC pathway (Jenks et al., 2018).
Tbet+CD11c+ B cells undergo transcriptional changes as infection resolves, with accumulation at the splenic marginal zone, a site exposed to blood circulation. Tbet+ B cells were found at the marginal zone in an Ehrlichia muris infection model (Trivedi et al., 2019) and were resident in the spleen after influenza infection (Johnson et al., 2020). Our observation of increased expression of S1pr3, Cnr2, and Cxcr3 suggest that multiple factors could be involved in Tbet+CD11c+ B cell migration to the marginal zone, whereas their integrin-dependent retention may be the mechanism underlying their splenic residency. Unique homing of Tbet+CD11c+ B cells potentially enables a protective mechanism through which an antigen-experienced population is placed at the marginal zone to sample antigen and mount rapid response to systemic reinfection.
The relative functional contributions of Tbet+CD11c+ B cells within the memory pool have been incompletely understood. Our findings suggested that the small subset of Tbet+CD11c+ B cells could account for a considerable fraction of the antibody production and secondary GC seeding by memory B cells on rechallenge. Although their differential localization may contribute to their competitiveness within the memory pool, other factors such as their BCR affinity, threshold for activation, and proliferative capabilities may be at play. The observation of Tbet+CD11c+ B cells entering secondary GCs conceptually follows previous studies demonstrating competitive advantage of presumably GC-independent memory cells over GC-derived ones in this aspect (Dogan et al., 2009; Pape et al., 2011; Zuccarino-Catania et al., 2014). However, memory cells could be outnumbered and outcompeted by naive B cells in populating secondary GCs (Mesin et al., 2020; Viant et al., 2020). Therefore, to what extent Tbet+CD11c+ B cells contribute to seeding secondary GCs remains a question.
Although our data elucidated the development and function of Tbet+CD11c+ B cells in response to acute viral infection, it is unclear whether and how this may differ in chronic infection or autoimmunity. Chronic exposure to cytokines could result in transcriptional remodeling of Tbet+CD11c+ B cells, which may explain the different ABC-like cellular phenotypes in various inflammatory conditions (Isnardi et al., 2010; Karnell et al., 2017; Rubtsov et al., 2011; Trivedi et al., 2019). Continuous activation of B and T cells would create a less synchronized cellular response, which may promote aberrant interactions rarely observed in acute infections. Reactivated Tbet+CD11c+ B cells could give rise to secondary GC B cells and plasmablasts, despite having been largely absent from those responses in the primary phase. To this point, our observation of nonoverlapping phenotypes of Tbet+CD11c+ B cells with GC B cells and PCs in acute infection stood in stark contrast to the intermixed phenotype of these subsets seen in autoimmune animals (Ricker et al., 2021). These transitional populations may point to a shift toward a GC-dependent pathway of Tbet+CD11c+ B cells in autoimmunity but could instead be a sign of their role as GC B cell and PC precursors. Continuous reactivation of Tbet+CD11c+ B cells may lead to increased autoreactive clones populating the two latter B cell compartments. Thus, developmental pathways and functional axes of Tbet+CD11c+ B cells in acute versus chronic inflammatory conditions need to be further explored.
Limitations of the study
We established the necessity of Tfh cell help in generation of Tbet+CD11c+ B cells; however, Tfh cell effector molecules promoting generation of these cells were not assessed. We demonstrated that most Tbet+CD11c+ B cells developed independently of GC passage, although a small fraction of them were labeled by S1pr2-based fate tracking, suggesting that their GC development is possible under inflammatory conditions other than acute viral infections investigated here. Most Tbet+CD11c+ B cells were proximal to the marginal zone after infection as assessed by i.v.-labeling; however, we are uncertain whether the unlabeled fraction localized within the white pulp. We suggested that the lack of in vivo function of S1PR3 on Tbet+CD11c+ B cells was due to compensation by other factors such as Cnr2 and CXCR3, which we found to be highly expressed, but this was not further investigated.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Jason Weinstein (jason.weinstein@rutgers.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The RNA-seq and Ig-seq data have been deposited at GEO: GSE150124, GSE150139, and GSE192765 and are publicly available as of the date of publication. This paper does not report original code. Other data reported in this paper will be shared by the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mice were housed in pathogen-free conditions at the Yale School of Medicine, New Haven, CT, Rutgers New Jersey Medical School, Newark, NJ, or Washington University School of Medicine, St. Louis, MO. C57BL/6N (B6) animals were purchased from Charles River. Icos−/− (004859), Sh2d1a−/− (025754), Tcrb−/− (002118), and CD11c-DTR/GFP (004509) were purchased from Jackson Laboratories. SMARTA (Stg) (Oxenius et al., 1998), MD4, and S1pr3−/− (Cinamon et al., 2004) mice were bred in-house. CD4Cre Bcl6fl/fl mice were obtained from Stephanie Eisenbarth (Yale University). S1PR2CreERT2 Rosa26Lox-Stop-Lox-tdTomato mice were obtained from Takaharu Okada (RIKEN). Tbet-AmCyan reporter mice were obtained from Jinfang Zhu (National Institutes of Health). Both sexes of mice were used in experiments with sex-matched controls. All animals were used at 6–8 weeks of age, with approval for all procedures given by the Institutional Animal Care and Use Committee of Yale University, Rutgers New Jersey Medical School or Washington University in Saint Louis.
Cell transfers and viral infections
2.5 × 105 sorted Tfh or Th1 cells or 5 × 104 Stg CD4 T cells were resuspended in sterile phosphate-buffered saline (PBS) and transferred to recipient mice via retroorbital injection (Weinstein et al., 2018). 5 × 106 B cells were purified using a magnetic negative selection kit (EasyStep, StemCell Technologies) from spleens and lymph nodes of CD11c-DTR/GFP mice and resuspended in sterile PBS prior to retroorbital injection. Mice were infected intraperitoneally (i.p.) with 2 × 105 PFU LCMV-Armstrong 24 hours after transfer. For rechallenge, mice were infected i.p. with 8 × 105 PFU LCMV-Armstrong 35 days after the initial infection (Schweier et al., 2019). Animals were sacrificed at different time points post infection, and spleens were harvested and processed for downstream applications. For transfers of cells from Tbet-AmCyan reporter mice, all splenic naïve and memory B cells were sorted (12.4 million naïve, 0.5 million Tbet CD11c, and 4.9 million memory B cells on average) and transferred into recipients regardless of total cell number. F1 progeny resulted from crossing of MD4 and Tbet-AmCyan reporter mice expressing the MD4 transgene were used as recipients to prevent rejection. Influenza virus A/Puerto Rico/8/1934 H1N1 (PR8) was a kind gift from A. Iwasaki (Yale University, New Haven, CT) and was given to mice at 10 PFUs in 30 μl of PBS intranasally.
Flow cytometry and cell sorting
Tissues were homogenized by crushing with the head of a 1ml syringe in a Petri dish followed by straining through a 40μm nylon filter. Red blood cells were lysed with ACK buffer and remaining splenocytes were stained for flow cytometry or sorting using antibodies listed in key resources table. Surface staining was performed at room temperature (25°C) with 35 minutes of incubation. Intracellular staining for Tbet and Ki67 was performed using Foxp3/Transcription Factor Staining kit (eBioscience) according to the manufacturer’s protocol. Staining for HA-specific B cells were performed on ice for 1 hour using recombinant HA bound to streptavidin APC or PE (Prozyme) generated as previously described (Angeletti et al., 2017). Both or one of the two HA-probes were used for detecting HA-specific B cells due to the low frequency of HA-APC and HA-PE single-binding cells. For flow cytometric analysis, stained and rinsed cells were analyzed using a multilaser cytometer (LSRII; or Fortessa X-20; BD Biosciences). For sorting of Stg cells, CD4+ T cells were enriched using a magnetic negative selection kit (EasyStep, StemCell Technologies) before cell surface staining, with specific populations sorted using a FACSAria (BD Biosciences). Cells for adoptive transfer were sorted into complete RPMI media and washed with sterile PBS. Cells for RNA and Ig sequencing were sorted into RNAprotect Cell Reagent (Qiagen).
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| CD4 (RM4–5) | BD Biosciences | Cat# 563726, 563151, 563106; RRID:AB_2738389, AB_2687549, AB_2687550 |
| CD4 (RM4–5) | Biolegend | Cat# 100532, 100536; RRID:AB_493373, AB_493701 |
| CD44 (IM7) | Biolegend | Cat# 103057; RRID:AB_2564214 |
| CD44 (IM7) | eBioscience | Cat# 47–0441-82; RRID:AB_1272244 |
| CD11c (HL3) | BD Biosciences | Cat# 553802; RRID:AB_395061 |
| CD11c (N418) | Biolegend | Cat# 117314, 117329; RRID:AB_492850, AB_10897814 |
| GL-7 (GL-7) | BD Biosciences | Cat# 553666; RRID:AB_394981 |
| GL-7 (GL-7) | eBioscience | Cat# 50–5902-82; RRID:AB_2574252 |
| CD95 (Jo2) | BD Biosciences | Cat# 557653; RRID:AB_396768 |
| CD23 (B3B4) | Biolegend | Cat# 101619; RRID:AB_2563438 |
| CD38 (T10) | Biolegend | Cat# 102717; RRID:AB_2072892 |
| CD138 (281–2) | Biolegend | Cat# 142504; RRID:AB_10916119 |
| B220 (RA3–6B2) | BD Biosciences | Cat# 561878, 553093, 561102, 561880, 558108; RRID:AB_10893353, AB_394622, AB_10561687, AB_10897020, AB_397031 |
| B220 (RA3–6B2) | Biolegend | Cat# 103254; RRID:AB_2563229 |
| CD19 (6D5) | Biolegend | Cat# 115524, 115519; RRID:AB_493339, AB_313654 |
| IgD (11–26c.2a) | Biolegend | Cat# 405727, 405729; RRID:AB_2562887, AB_2563340 |
| CXCR5 (2G8) | BD Biosciences | Cat# 551959; RRID:AB_394300 |
| CXCR5 (L138D7) | Biolegend | Cat# 145513; RRID:AB_2562208 |
| PD-1 (RMP1–30) | Biolegend | Cat# 109117, 109109; RRID:AB_2566549, AB_572016 |
| PD-1 (J43) | eBioscience | Cat# 25–9985-82; RRID:AB_10853805 |
| Thy1.1 (OX-7) | Biolegend | Cat# 202526; RRID:AB_1595470 |
| PSGL-1 (2PH1) | BD Biosciences | Cat# 562807, 563448; RRID:AB_2737808, AB_2738211 |
| Tbet (eBio4B10) | eBioscience | Cat# 50–5825-82; RRID:AB_10596655 |
| Tbet (4B10) | Biolegend | Cat# 644815; RRID:AB_10896427 |
| Ki67 (16A8) | Biolegend | Cat# 652417, 652419; RRID:AB_2564236, AB_2564284 |
| CD45.1 (A20) | Biolegend | Cat# 110741; RRID:AB_2563378 |
| CD45.2 (104) | Biolegend | Cat# 109831, 109823; RRID:AB_10900256, AB_830788 |
| Ly6C (HK1.4) | Biolegend | Cat# 128021; RRID:AB_10640820 |
| CD21/35 (7G6) | BD Biosciences | Cat# 553818; RRID:AB_395070 |
| Pdca-1 (927) | Biolegend | Cat# 127014; RRID:AB_1953289 |
| CXCR3 (CXCR3–173) | Biolegend | Cat# 126511; RRID:AB_1088994 |
| Ephrin B1 (polyclonal) | R&D Systems | Cat# BAF473, AF473; RRID:AB_2293418, AB_2293419 |
| APC-Cy7 Streptavidin | Biolegend | 405208 |
| Alexa Fluor 647 Streptavidin | Thermo Fisher | S21374 |
| Alexa Fluor 594 anti-Goat IgG (H+L) | Thermo Fisher | Cat# A-11058; RRID:AB_2534105 |
| APC Streptavidin | Prozyme | PJ27S |
| PE Streptavidin | Prozyme | PJRS25 |
| Peanut Agglutinin (PNA) Cy3 | Vector Labs | CL-1073–1 |
| F4/80 (BM8) | Thermo Fisher | Cat# 53–4801-82; RRID:AB_469915 |
| MadCam1 (MECA-367) | Biolegend | Cat# 120705; RRID:AB_493396 |
| InVivoMAb anti-mouse LFA-1α | Bio X Cell | Cat# BE0006; RRID:AB_1107578 |
| InVivoMAb anti-mouse/human VLA-4 | Bio X Cell | Cat# BE0071; RRID:AB_1107657 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| LCMV-Armstrong | N/A | N/A |
| Influenza A/Puerto Rico/8/1934 H1N1 (PR8) | Laboratory of A. Iwasaki, Yale University | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| TO-PRO-1 Iodide | Thermo Fisher | T3602 |
| Recombinant PR8 hemagglutinin (HA) | Laboratory of Davide Angeletti, University of Gothenburg |
Angeletti et al., 2017 |
| TY 52156 | Sigma | 5330620001 |
| FTY720 | MilliporeSigma | 344597 |
| Sphingosine-1-Phosphate | Avanti Polar Lipids | 860492 |
| Tamoxifen | Sigma–Aldrich | T5648 |
| TAM diet (irradiated) | Envigo | TD.130859 |
| Corn Oil | Sigma–Aldrich | C8267 |
| Diphtheria toxin | Sigma–Aldrich | D0564 |
| BrdU | Sigma–Aldrich | B5002 |
| R848 | Invivogen | tlrl-r848 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Foxp3/Transcription Factor Staining kit | eBioscience | 00–5523-00 |
| EasySep™ Mouse CD4+ T Cell Isolation Kit | StemCell | 19852 |
| RNeasy Micro Kit | Qiagen | 74004 |
| NEBNext AbSeq immune sequencing kit | New England Biolabs | |
|
| ||
| Deposited data | ||
|
| ||
| RNA-seq of B cell subsets | This paper | GSE150124 |
| RNA-seq of Tbet+CD11c+ B cell kinetics | This paper | GSE150139 |
| Ig-seq of B cell subsets | This paper | GSE192765 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6 | Charles River | N/A |
| Mouse: Icos−/− | Jackson Laboratories | 004859 |
| Mouse: Sh2d1a−/− | Jackson Laboratories | 025754 |
| Mouse: Tcrb−/− | Jackson Laboratories | 002118 |
| Mouse: CD11c-DTR/GFP | Jackson Laboratories | 004509 |
| Mouse: Tbet-AmCyan | Jinfang Zhu (National Institutes of Health) | Yu et al., 2015 |
| Mouse: S1PR2CreERT2 Rosa26Lox-Stop-Lox-tdTomato |
Takaharu Okada (RIKEN) | Shinnakasu et al., 2016 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Hprt qPCR primers | Keck Oligo Synthesis Resource | (F) CTGGTGAAAAGGACCTCTCG (R) TGAAGTACTCATTATAGTCAAGGGCA |
| Gfp qPCR primers | Keck Oligo Synthesis Resource | (F) AAGCTGACCCTGAAGTTCATCTGC (R) CTTGTAGTTGCCGTCGTCCTTGAA |
| Itgax qPCR primers | Keck Oligo Synthesis Resource | (F) TGCCAGGATGACCTTAGTGTCG (R) CAGAGTGACTGTGGTTCCGTAG |
|
| ||
| Software and algorithms | ||
|
| ||
| Huygen’s Essential | Scientific Volume Imaging | N/A |
| Imaris | Bitplane Scientific Software | N/A |
| Flowjo | TreeStar FlowJo LLC. | N/A |
| GraphPad Prism | GraphPad Software | N/A |
| Spatstat | Baddeley and Turner, 2005 | N/A |
| pRESTO | Vander Heiden et al., 2014 | N/A |
| Immcantation | Gupta et al., 2015 | N/A |
Microscopy and histo-cytometry
Spleens were fixed and frozen as previously described (Weinstein et al., 2016). In brief, spleens fixed in PLP buffer were incubated in 30% sucrose solution prior to being snap frozen in OCT tissue-freezing solution and stored at −80°C. 8μm sections were cut and blocked with 5% rat serum, 3% BSA and, 0.1% Tween. Histo-cytometry was performed as described(Gerner et al., 2012) with the following modifications. Sections were stained with directly conjugated antibodies listed in key resources table for 16 to 18 hours in a dark, humidifier chamber at 4°C. Cellular nuclei were stained with TO-PRO-1 Iodide (Thermo Fisher) at 0.1μM for 20 minutes at room temperature (25°C). Images were obtained from a Leica SP5 confocal microscope with a motorized stage for tiled imaging at 63x magnification, 1024 by 1024 pixels resolution. Images were deconvolved using Huygen’s Essential software (Scientific Volume Imaging). Segmentation was performed on Imaris software (Bitplane Scientific Software) using the surface creation module with the TO-PRO-1 nuclear stain channel brightened with the linear stretch function and gamma set to 1.5. All other channels were not altered for histo-cytometric analysis. The channel statistics, including mean fluorescence intensity, X and Y location, for all surfaces were exported into a.csv file in Excel (Microsoft) and plotted in Flowjo software. All presented images were enhanced with the linear stretch function in Imaris to increase channel intensity for visual clarity only. Multitype K function between cell types of interest was calculated in the R package Spatstat using the Kcross function and the pool function (Baddeley and Turner, 2005).
Transwell migration assay
Chemotaxis assays were performed as described previously (Beck et al., 2014). 1.5 × 106 splenocytes were incubated for 1 hour with 1× DMEM containing 0.5% fatty acid–free BSA (Sigma-Aldrich), 5% antibiotics, L-glutamine (Cellgro), and Hepes with 1μM FTY720 (Sigma), 10μM TY 52156 (Tocris) or media alone. Cells were then allowed to migrate through 5-μm-pore–sized transwells (Corning) toward S1P (Aventis Polar Lipids)-containing media or media alone for 3 hours at 37°C. Cells were collected, stained, and resuspended in 25μl of staining buffer and analyzed by flow cytometry.
Ig-sequencing and analysis
RNA from sorted B cell populations was extracted using the RNeasy Plus Micro kit (Qiagen) with quantification and quality control performed by Yale Keck Biotechnology Resource Laboratory on a TapeStation 2200 (Agilent Technologies). Library preparation for sequencing was performed using the NEBNext Immune-seq library preparation kit for Mouse IG (New England Biolabs) according to the manufacturer’s protocol. Quality verification of libraries and sequencing were performed at the Yale Center for Genomic Analysis on a Miseq (Illumina) using a 600-cycle V3 reagent kit with dual-index. Raw reads were quality controlled, assembled and filtered using pRESTO (Vander Heiden et al., 2014) before germline gene annotations were assigned using IgBLAST (Ye et al., 2013). Sequences were processed and analyzed using the Immcantation pipeline (Gupta et al., 2015). In brief, productively rearranged heavy-chain sequences were clustered and assigned to clonally related lineages, and full-length clonal-consensus germline sequences were reconstructed for each clone. Mutation frequency and selection pressure were calculated using SHazaM. Using Alakazam, lineage trees were reconstructed after collapsing duplicate sequences within each clone.
RNA-Seq and analysis
B cell populations were sorted by flow cytometry, with three separate sorts performed on different days, pooling spleens of four to six mice at each timepoint. Quality verification, library preparation, and sequencing were performed at the Rutgers Genomic Core or Genewiz. Samples were sequenced on an Illumina HiSeq 2500 using 150-bp paired end reads (Figures 6E and 6F) or a NovaSeq 6000 and S1 flow cell using 150-bp paired end reads (Figures 3B and 3C). RNA-seq analysis was performed in accordance with the NF-Core RNA-seq guidelines (version 1.4.2). Briefly the reads were aligned to the GRCm38 genome using STAR, followed by gene count generation using featureCounts. Read count normalizations and differential gene expression calculations between groups were performed using DESeq2 with significance thresholds set at FDR adjusted p <0.05. For heatmaps, expression values or fold changes were normalized per gene.
Antibody injection and treatment
For labeling of B cells subsets proximal to vasculature, anesthetized animals were administered 6μg of PE-conjugated anti-CD45 antibody in 200ul of sterile PBS via retroorbital injection 5 minutes before euthanasia. To block LFA-1 and VLA-4 activity, 100μg of anti-LFA-1α (clone M17/4, Bio X Cell) and anti-VLA-4 (clone PS/2, Bio X Cell) or isotype control (Sigma–Aldrich) blocking antibodies in 200ul of sterile PBS were administered via retroorbital injection 3 hours before euthanasia. Tamoxifen (Sigma–Aldrich) was dissolved in Corn Oil (Sigma–Aldrich) and administered via intraperitoneal injection at 100 mg/kg day day 4 and 6 p.i.. TAM diet (Envigo) containing chow replaced normal chow on the day after the final tamoxifen dose. To overcome initial taste aversion, additional crushed TAM diet was placed in the cage at the time of the first TAM diet feeding.
Mixed bone marrow chimeras
Recipient WT CD45.1+ mice were irradiated with a single dose of 1000 rads and reconstituted 3 hours later with either 6 × 106 bone marrow cells from a 50:50 mix of CD45.2 Cd19Cre Bcl6fl/fl mice and CD45.1 Cd19Cre Bcl6+/+ mice or 6 × 106 bone marrow cells from a 50:50 mix of WT CD45.1mice and from S1pr3−/− mice via retroorbital injection. Bone marrow chimeric mice were infected 6 to 8 weeks after reconstitution.
Diphtheria toxin (DT) treatment
For depletion of CD11c-expressing B cells, mice were i.p. injected with 200ng of DT (Sigma-Aldrich) diluted in PBS on day 6 and 8 p.i. or day 28 and 30 p.i. with LCMV-Armstrong infection.
BrdU treatment
Mice received BrdU (Sigma-Aldrich) dissolved in sterile drinking water to 0.8 mg/ml continuously for 10 days starting from day 4 p.i. with LCMV-Armstrong infection. BrdU water was shielded from light and refreshed every other day.
ELISPOT
MultiScreen HTS plates (Millipore) were coated with sonicated cell lysate from LCMV-infected BHK-21 cells. Sorted B cell subsets were plated in media only overnight or with R848 (10ng/ml; Invivogen) for 72 hours followed by addition of alkaline phosphatase-conjugated anti-mouse IgG antibodies (Southern Biotech). Spots were developed with Vector Blue (Vector Laboratories) and read using an ImmunoSpot Analyzer (Cellular Technology Limited) or counted blinded.
ELISA
Anti-LCMV antibodies in mouse sera were measured on Nunc PolySorp 96 well plates (Thermo Fisher) coated with sonicated cell lysate from LCMV-infected BHK-21 cells diluted in carbonate buffer (Sigma). Alkaline phosphatase-conjugated anti-mouse IgG antibodies (Southern Biotech) and phosphatase substrate (Sigma) were used for detection. ODs were read at 405nm on a SpectraMax Microplate Reader (Molecular Devices). Two-fold serial dilutions of the serum from a WT day 14-infefcted mouse was used to calculate relative ELISA units.
Autoantigen microarray
Sorted B cell subsets were cultured for 72 hours in complete media containing R848 before supernatant collection. Reactivity against a panel of 95 autoantigens were performed as previous described (UT-Southwestern (UTSW) Microarray Core Facility, Dallas, TX) (Li et al., 2005).
Statistics
Data were analyzed using the Student’s t, One-way multiple-comparison ANOVA, Mann-Whitney, or Dunn’s multiple comparisons test with Prism 8 (GraphPad Software).
Supplementary Material
Highlights.
T follicular helper cells are necessary for the generation of Tbet+CD11c+ B cells
Tbet+CD11c+ B cells form outside of germinal centers upon LCMV and flu infection
Integrins LFA-1 and VLA-4 retain Tbet+CD11c+ B cells at the splenic marginal zone
Tbet+CD11c+ B cells are functionally competitive within the memory compartment
ACKNOWLEDGMENTS
The authors acknowledge members of our departments for critical review of the manuscript. W.S. is the recipient of the Edward L. Tatum Fellowship from the Graduate School of Arts & Sciences and the Gershon-Trudeau Fellowship from the Department of Immunobiology of Yale University. J.S.W. and the laboratory were supported in part by New Jersey Health Foundation and National Institutes of Health (NIH) grants K01 AR067892 and R01 AR073912. J.C. and laboratory support came in part from the Lupus Research Alliance and NIH grants R37 AR40072 and R01 AR074545. Correspondence and requests for materials should be addressed to J.C. (joseph.craft@yale.edu) and J.S.W. (jason.weinstein@rutgers.edu).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.immuni.2022.01.002.
REFERENCES
- Angeletti D, Gibbs JS, Angel M, Kosik I, Hickman HD, Frank GM, Das SR, Wheatley AK, Prabhakaran M, Leggat DJ, et al. (2017). Defining B cell immunodominance to viruses. Nat. Immunol. 18, 456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin JW, Buckner CM, Kardava L, Wang W, Zhang X, Melson VA, Swanson RG, Martins AJ, Zhou JQ, Hoehn KB, et al. (2019). Overexpression of T-bet in HIV infection is associated with accumulation of B cells outside germinal centers and poor affinity maturation. Sci. Transl. Med. 11, eaax0904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baddeley A, and Turner R (2005). spatstat: an R package for analyzing spatial point patterns. J. Stat. Softw. 12, 1–42. [Google Scholar]
- Barnett BE, Staupe RP, Odorizzi PM, Palko O, Tomov VT, Mahan AE, Gunn B, Chen D, Paley MA, Alter G, et al. (2016). Cutting edge: B cell-intrinsic T-bet expression is required to control chronic viral infection. J. Immunol. 197, 1017–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S, Ray A, and Dittel BN (2011). Cannabinoid receptor 2 is critical for the homing and retention of marginal zone B lineage cells and for efficient T-independent immune responses. J. Immunol. 187, 5720–5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck TC, Gomes AC, Cyster JG, and Pereira JP (2014). CXCR4 and a cell-extrinsic mechanism control immature B lymphocyte egress from bone marrow. J. Exp. Med. 211, 2567–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beima KM, Miazgowicz MM, Lewis MD, Yan PS, Huang TH-M, and Weinmann AS (2006). T-bet binding to newly identified target gene promoters is cell type-independent but results in variable context-dependent functional effects. J. Biol. Chem. 281, 11992–12000. [DOI] [PubMed] [Google Scholar]
- Bhattacharya D, Cheah MT, Franco CB, Hosen N, Pin CL, Sha WC, and Weissman IL (2007). Transcriptional profiling of antigen-dependent murine B cell differentiation and memory formation. J. Immunol. 179, 6808–6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan TD, Gatto D, Wood K, Camidge T, Basten A, and Brink R (2009). Antigen affinity controls rapid T-dependent antibody production by driving the expansion rather than the differentiation or extrafollicular migration of early plasmablasts. J. Immunol. 183, 3139–3149. [DOI] [PubMed] [Google Scholar]
- Cinamon G, Matloubian M, Lesneski MJ, Xu Y, Low C, Lu T, Proia RL, and Cyster JG (2004). Sphingosine 1-phosphate receptor 1 promotes B cell localization in the splenic marginal zone. Nat. Immunol. 5, 713–720. [DOI] [PubMed] [Google Scholar]
- Cinamon G, Zachariah MA, Lam OM, Foss FW Jr., and Cyster JG (2008). Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat. Immunol. 9, 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedeoglu F, Horwitz B, Chaudhuri J, Alt FW, and Geha RS (2004). Induction of activation-induced cytidine deaminase gene expression by IL-4 and CD40 ligation is dependent on STAT6 and NFkappaB. Int. Immunol. 16, 395–404. [DOI] [PubMed] [Google Scholar]
- Dent AL, Shaffer AL, Yu X, Allman D, and Staudt LM (1997). Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science 276, 589–592. [DOI] [PubMed] [Google Scholar]
- Di Niro R, Lee S-J, Vander Heiden JA, Elsner RA, Trivedi N, Bannock JM, Gupta NT, Kleinstein SH, Vigneault F, Gilbert TJ, et al. (2015). Salmonella infection drives promiscuous B cell activation followed by extrafollicular affinity maturation. Immunity 43, 120–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan I, Bertocci B, Vilmont V, Delbos F, Mégret J, Storck S, Reynaud C-A, and Weill J-C (2009). Multiple layers of B cell memory with different effector functions. Nat. Immunol. 10, 1292–1299. [DOI] [PubMed] [Google Scholar]
- Gerner MY, Kastenmuller W, Ifrim I, Kabat J, and Germain RN (2012). Histo-cytometry: a method for highly multiplex quantitative tissue imaging analysis applied to dendritic cell subset microanatomy in lymph nodes. Immunity 37, 364–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green JA, Suzuki K, Cho B, Willison LD, Palmer D, Allen CD, Schmidt TH, Xu Y, Proia RL, Coughlin SR, and Cyster JG (2011). The sphingosine 1-phosphate receptor S1P2 maintains the homeostasis of germinal center B cells and promotes niche confinement. Nat. Immunol. 12, 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta NT, Vander Heiden JA, Uduman M, Gadala-Maria D, Yaari G, and Kleinstein SH (2015). Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 31, 3356–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, O’Neill P, Naradikian MS, Scholz JL, and Cancro MP (2011). A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood 118, 1294–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirako IC, Ataide MA, Faustino L, Assis PA, Sorensen EW, Ueta H, Araújo NM, Menezes GB, Luster AD, and Gazzinelli RT (2016). Splenic differentiation and emergence of CCR5+CXCL9+CXCL10+ monocyte-derived dendritic cells in the brain during cerebral malaria. Nat. Commun. 7, 13277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isnardi I, Ng Y-S, Menard L, Meyers G, Saadoun D, Srdanovic I, Samuels J, Berman J, Buckner JH, Cunningham-Rundles C, and Meffre E (2010). Complement receptor 2/CD21- human naive B cells contain mostly autoreactive unresponsive clones. Blood 115, 5026–5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenks SA, Cashman KS, Zumaquero E, Marigorta UM, Patel AV, Wang X, Tomar D, Woodruff MC, Simon Z, Bugrovsky R, et al. (2018). Distinct effector B cells induced by unregulated toll-like receptor 7 contribute to pathogenic responses in systemic lupus erythematosus. Immunity 49, 725–739.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JL, Rosenthal RL, Knox JJ, Myles A, Naradikian MS, Madej J, Kostiv M, Rosenfeld AM, Meng W, Christensen SR, et al. (2020). The transcription factor T-bet resolves memory B cell subsets with distinct tissue distributions and antibody specificities in mice and humans. Immunity 52, 842–855.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, Dent AL, Craft J, and Crotty S (2009). Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 325, 1006–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, et al. (2002). In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity 17, 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnell JL, Kumar V, Wang J, Wang S, Voynova E, and Ettinger R (2017). Role of CD11c(+) T-bet(+) B cells in human health and disease. Cell. Immunol. 321, 40–45. [DOI] [PubMed] [Google Scholar]
- Keller B, Strohmeier V, Harder I, Unger S, Payne KJ, Andrieux G, Boerries M, Felixberger PT, Landry JJM, Nieters A, et al. (2021). The expansion of human T-bet high CD21 low B cells is T cell dependent. Sci. Immunol. 6, eabh0891. [DOI] [PubMed] [Google Scholar]
- Knox JJ, Myles A, and Cancro MP (2019). T-bet(+) memory B cells: generation, function, and fate. Immunol. Rev. 288, 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurachi M, Kurachi J, Suenaga F, Tsukui T, Abe J, Ueha S, Tomura M, Sugihara K, Takamura S, Kakimi K, et al. (2011). Chemokine receptor CXCR3 facilitates CD8(+) T cell differentiation into short-lived effector cells leading to memory degeneration. J. Exp. Med. 208, 1605–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laidlaw BJ, Duan L, Xu Y, Vazquez SE, and Cyster JG (2020). The transcription factor Hhex cooperates with the corepressor Tle3 to promote memory B cell development. Nat. Immunol. 21, 1082–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laidlaw BJ, Schmidt TH, Green JA, Allen CD, Okada T, and Cyster JG (2017). The Eph-related tyrosine kinase ligand ephrin-B1 marks germinal center and memory precursor B cells. J. Exp. Med. 214, 639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau D, Lan LY-L, Andrews SF, Henry C, Rojas KT, Neu KE, Huang M, Huang Y, DeKosky B, Palm A-KE, et al. (2017). Low CD21 expression defines a population of recent germinal center graduates primed for plasma cell differentiation. Sci. Immunol. 2, eaai8153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SK, Rigby RJ, Zotos D, Tsai LM, Kawamoto S, Marshall JL, Ramiscal RR, Chan TD, Gatto D, Brink R, et al. (2011). B cell priming for extrafollicular antibody responses requires Bcl-6 expression by T cells. J. Exp. Med. 208, 1377–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levack RC, Newell KL, Popescu M, Cabrera-Martinez B, and Winslow GM (2020). CD11c(+) T-bet(+) B cells require IL-21 and IFN-γ from type 1 T follicular helper cells and intrinsic Bcl-6 expression but develop normally in the absence of T-bet. J. Immunol. 205, 1050–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Borrego F, Nagata S, and Tolnay M (2016). Fc receptor–like 5 expression distinguishes two distinct subsets of human circulating tissue–like memory B cells. J. Immunol. 196, 4064–4074. [DOI] [PubMed] [Google Scholar]
- Li Q-Z, Xie C, Wu T, Mackay M, Aranow C, Putterman C, and Mohan C (2005). Identification of autoantibody clusters that best predict lupus disease activity using glomerular proteome arrays. J. Clin. Invest. 115, 3428–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, Verma NK, Smyth MJ, Rigby RJ, and Vinuesa CG (2010). IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J. Exp. Med. 207, 353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhou S, Qian J, Wang Y, Yu X, Dai D, Dai M, Wu L, Liao Z, Xue Z, et al. (2017). T-bet(+)CD11c(+) B cells are critical for antichromatin immunoglobulin G production in the development of lupus. Arthritis Res. Ther. 19, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Shih C, and Qi H (2017). Ephrin B1-mediated repulsion and signaling control germinal center T cell territoriality and function. Science 356. [DOI] [PubMed] [Google Scholar]
- Lu TT, and Cyster JG (2002). Integrin-mediated long-term B cell retention in the splenic marginal zone. Science 297, 409–412. [DOI] [PubMed] [Google Scholar]
- Manni M, Gupta S, Ricker E, Chinenov Y, Park SH, Shi M, Pannellini T, Jessberger R, Ivashkiv LB, and Pernis AB (2018). Regulation of age-associated B cells by IRF5 in systemic autoimmunity. Nat. Immunol. 19, 407–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall HD, Chandele A, Jung YW, Meng H, Poholek AC, Parish IA, Rutishauser R, Cui W, Kleinstein SH, Craft J, and Kaech SM (2011). Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4(+) cell properties during viral infection. Immunity 35, 633–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason DY, Jones M, and Goodnow CC (1992). Development and follicular localization of tolerant B lymphocytes in lysozyme/anti-lysozyme IgM/IgD transgenic mice. Int. Immunol. 4, 163–175. [DOI] [PubMed] [Google Scholar]
- Mesin L, Schiepers A, Ersching J, Barbulescu A, Cavazzoni CB, Angelini A, Okada T, Kurosaki T, and Victora GD (2020). Restricted clonality and limited germinal center reentry characterize memory B cell reactivation by boosting. Cell 180, 92–106.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyauchi K, Sugimoto-Ishige A, Harada Y, Adachi Y, Usami Y, Kaji T, Inoue K, Hasegawa H, Watanabe T, Hijikata A, et al. (2016). Protective neutralizing influenza antibody response in the absence of T follicular helper cells. Nat. Immunol. 17, 1447–1458. [DOI] [PubMed] [Google Scholar]
- Moir S, Ho J, Malaspina A, Wang W, DiPoto AC, O’Shea MA, Roby G, Kottilil S, Arthos J, Proschan MA, et al. (2008). Evidence for HIV-associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV-infected viremic individuals. J. Exp. Med. 205, 1797–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SN, Hosiawa-Meagher KA, Konieczny BT, Sullivan BM, Bachmann MF, Locksley RM, Ahmed R, and Matloubian M (2007). Regulation of homeostatic chemokine expression and cell trafficking during immune responses. Science 317, 670–674. [DOI] [PubMed] [Google Scholar]
- Muppidi JR, Arnon TI, Bronevetsky Y, Veerapen N, Tanaka M, Besra GS, and Cyster JG (2011). Cannabinoid receptor 2 positions and retains marginal zone B cells within the splenic marginal zone. J. Exp. Med. 208, 1941–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naradikian MS, Myles A, Beiting DP, Roberts KJ, Dawson L, Herati RS, Bengsch B, Linderman SL, Stelekati E, Spolski R, et al. (2016). Cutting edge: il-4, IL-21, and IFN-γ interact to govern T-bet and CD11c expression in TLR-activated B cells. J. Immunol. 197, 1023–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nellore A, Scharer CD, King RG, Tipton CM, Zumaquero E, Fucile C, Mousseau B, Bradley JE, Macon K, Mi T, et al. (2019). Fcrl5 and T-bet define influenza-specific memory B cells that predict long-lived antibody responses. bioRxiv. 10.1101/643973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, Wang Y-H, and Dong C (2009). Bcl6 mediates the development of T follicular helper cells. Science 325, 1001–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegard JM, Marks BR, DiPlacido LD, Poholek AC, Kono DH, Dong C, Flavell RA, and Craft J (2008). ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J. Exp. Med. 205, 2873–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxenius A, Bachmann MF, Zinkernagel RM, and Hengartner H (1998). Virus-specific MHC-class II-restricted TCR-transgenic mice: effects on humoral and cellular immune responses after viral infection. Eur. J. Immunol. 28, 390–400. [DOI] [PubMed] [Google Scholar]
- Pape KA, Taylor JJ, Maul RW, Gearhart PJ, and Jenkins MK (2011). Different B cell populations mediate early and late memory during an endogenous immune response. Science 331, 1203–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng SL, Szabo SJ, and Glimcher LH (2002). T-bet regulates IgG class switching and pathogenic autoantibody production. Proc. Natl. Acad. Sci. USA 99, 5545–5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H, Cannons JL, Klauschen F, Schwartzberg PL, and Germain RN (2008). SAP-controlled T-B cell interactions underlie germinal centre formation. Nature 455, 764–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine R, Chatterjee M, and Winslow GM (2008). CD11c expression identifies a population of extrafollicular antigen-specific splenic plasmablasts responsible for CD4 T-independent antibody responses during intracellular bacterial infection. J. Immunol. 181, 1375–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhmanov M, Keller B, Gutenberger S, Foerster C, Hoenig M, Driessen G, van der Burg M, van Dongen JJ, Wiech E, Visentini M, et al. (2009). Circulating CD21 low B cells in common variable immunodeficiency resemble tissue homing, innate-like B cells. Proc. Natl. Acad. Sci. USA 106, 13451–13456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard K, Pierce SK, and Song W (2008). The agonists of TLR4 and 9 are sufficient to activate memory B cells to differentiate into plasma cells in vitro but not in vivo. J. Immunol. 181, 1746–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricker E, Manni M, Flores-Castro D, Jenkins D, Gupta S, Rivera-Correa J, Meng W, Rosenfield AM, Pannellini T, Chinenov Y, et al. (2021). Sex-specific differences in the function and differentiation of ABCs mark TLR7-driven immunopathogenesis. bioRxiv. 10.1101/2021.01.20.427400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Correa J, Guthmiller JJ, Vijay R, Fernandez-Arias C, Pardo-Ruge MA, Gonzalez S, Butler NS, and Rodriguez A (2017). Plasmodium DNA-mediated TLR9 activation of T-bet(+) B cells contributes to autoimmune anaemia during malaria. Nat. Commun. 8, 1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roco JA, Mesin L, Binder SC, Nefzger C, Gonzalez-Figueroa P, Canete PF, Ellyard J, Shen Q, Robert PA, Cappello J, et al. (2019). Class-switch recombination occurs infrequently in germinal centers. Immunity 51, 337–350.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, and Marrack P (2011). Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c(+) B-cell population is important for the development of autoimmunity. Blood 118, 1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov AV, Rubtsova K, Kappler JW, Jacobelli J, Friedman RS, and Marrack P (2015). CD11c-expressing B cells are located at the T cell/B cell border in spleen and are potent APCs. J. Immunol. 195, 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov AV, Rubtsova K, Kappler JW, and Marrack P (2013). TLR7 drives accumulation of ABCs and autoantibody production in autoimmune-prone mice. Immunol. Res. 55, 210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsova K, Rubtsov AV, Thurman JM, Mennona JM, Kappler JW, and Marrack P (2017). B cells expressing the transcription factor T-bet drive lupus-like autoimmunity. J. Clin. Invest. 127, 1392–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsova K, Rubtsov AV, van Dyk LF, Kappler JW, and Marrack P (2013). T-box transcription factor T-bet, a key player in a unique type of B-cell activation essential for effective viral clearance. Proc. Natl. Acad. Sci. USA 110, E3216–E3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell Knode LM, Naradikian MS, Myles A, Scholz JL, Hao Y, Liu D, Ford ML, Tobias JW, Cancro MP, and Gearhart PJ (2017). Age-associated B cells express a diverse repertoire of VH and Vkappa genes with somatic hypermutation. J. Immunol. 198, 1921–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scandella E, Bolinger B, Lattmann E, Miller S, Favre S, Littman DR, Finke D, Luther SA, Junt T, and Ludewig B (2008). Restoration of lymphoid organ integrity through the interaction of lymphoid tissue-inducer cells with stroma of the T cell zone. Nat. Immunol. 9, 667–675. [DOI] [PubMed] [Google Scholar]
- Schweier O, Aichele U, Marx AF, Straub T, Verbeek JS, Pinschewer DD, and Pircher H (2019). Residual LCMV antigen in transiently CD4(+) T cell-depleted mice induces high levels of virus-specific antibodies but only limited B-cell memory. Eur. J. Immunol. 49, 626–637. [DOI] [PubMed] [Google Scholar]
- Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, Takahashi Y, Fukuyama H, Okada T, and Kurosaki T (2016). Regulated selection of germinal-center cells into the memory B cell compartment. Nat. Immunol. 17, 861–869. [DOI] [PubMed] [Google Scholar]
- Stone SL, Peel JN, Scharer CD, Risley CA, Chisolm DA, Schultz MD, Yu B, Ballesteros-Tato A, Wojciechowski W, Mousseau B, et al. (2019). T-bet transcription factor promotes antibody-secreting cell differentiation by limiting the inflammatory effects of IFN-γ on B cells. Immunity 50, 1172–1187.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton HJ, Aye R, Idris AH, Vistein R, Nduati E, Kai O, Mwacharo J, Li X, Gao X, Andrews TD, et al. (2021). Atypical B cells are part of an alternative lineage of B cells that participates in responses to vaccination and infection in humans. Cell Rep. 34, 108684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedford K, Steiner M, Koshutin S, Richter K, Tech L, Eggers Y, Jansing I, Schilling K, Hauser AE, Korthals M, and Fischer KD (2017). The opposing forces of shear flow and sphingosine-1-phosphate control marginal zone B cell shuttling. Nat. Commun. 8, 2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi N, Weisel F, Smita S, Joachim S, Kader M, Radhakrishnan A, Clouser C, Rosenfeld AM, Chikina M, Vigneault F, et al. (2019). Liver is a generative site for the B cell response to Ehrlichia muris. Immunity 51, 1088–1101.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden JA, Yaari G, Uduman M, Stern JN, O’Connor KC, Hafler DA, Vigneault F, and Kleinstein SH (2014). pRESTO: a toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics 30, 1930–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viant C, Weymar GHJ, Escolano A, Chen S, Hartweger H, Cipolla M, Gazumyan A, and Nussenzweig MC (2020). Antibody affinity shapes the choice between memory and germinal center B cell fates. Cell 183, 1298–1311.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang NS, McHeyzer-Williams LJ, Okitsu SL, Burris TP, Reiner SL, and McHeyzer-Williams MG (2012). Divergent transcriptional programming of class-specific B cell memory by T-bet and RORα. Nat. Immunol. 13, 604–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Wang J, Kumar V, Karnell JL, Naiman B, Gross PS, Rahman S, Zerrouki K, Hanna R, Morehouse C, et al. (2018). IL-21 drives expansion and plasma cell differentiation of autoreactive CD11c(hi)T-bet(+) B cells in SLE. Nat. Commun. 9, 1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein JS, Delano MJ, Xu Y, Kelly-Scumpia KM, Nacionales DC, Li Y, Lee PY, Scumpia PO, Yang L, Sobel E, et al. (2013). Maintenance of anti-Sm/RNP autoantibody production by plasma cells residing in ectopic lymphoid tissue and bone marrow memory B cells. J. Immunol. 190, 3916–3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein JS, Herman EI, Lainez B, Licona-Limón P, Esplugues E, Flavell R, and Craft J (2016). TFH cells progressively differentiate to regulate the germinal center response. Nat. Immunol. 17, 1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein JS, Laidlaw BJ, Lu Y, Wang JK, Schulz VP, Li N, Herman EI, Kaech SM, Gallagher PG, and Craft J (2018). STAT4 and T-bet control follicular helper T cell development in viral infections. J. Exp. Med. 215, 337–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss GE, Crompton PD, Li S, Walsh LA, Moir S, Traore B, Kayentao K, Ongoiba A, Doumbo OK, and Pierce SK (2009). Atypical memory B cells are greatly expanded in individuals living in a malaria-endemic area. J. Immunol. 183, 2176–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- William J, Euler C, Christensen S, and Shlomchik MJ (2002). Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science 297, 2066–2070. [DOI] [PubMed] [Google Scholar]
- Woodruff M, Ramonell R, Cashman K, Nguyen D, Saini A, Haddad N, Ley A, Kyu S, Howell JC, Ozturk T, et al. (2020a). Dominant extrafollicular B cell responses in severe COVID-19 disease correlate with robust viral-specific antibody production but poor clinical outcomes. medRxiv. 10.1101/2020.04.29.20083717. [DOI] [Google Scholar]
- Woodruff MC, Ramonell RP, Saini AS, Haddad NS, Anam FA, Rudolph ME, Bugrovsky R, Hom J, Cashman KS, and Nguyen DC (2020b). Relaxed peripheral tolerance drives broad de novo autoreactivity in severe COVID-19. medRxiv. 10.1101/2020.10.21.20216192. [DOI] [Google Scholar]
- Woodruff MC, Ramonell RP, Nguyen DC, Cashman KS, Saini AS, Haddad NS, Ley AM, Kyu S, Howell JC, Ozturk T, et al. (2020c). Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nat. Immunol. 21, 1506–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Foy TM, Laman JD, Elliott EA, Dunn JJ, Waldschmidt TJ, Elsemore J, Noelle RJ, and Flavell RA (1994). Mice deficient for the CD40 ligand. Immunity 1, 423–431. [DOI] [PubMed] [Google Scholar]
- Yates JL, Racine R, McBride KM, and Winslow GM (2013). T cell-dependent IgM memory B cells generated during bacterial infection are required for IgG responses to antigen challenge. J. Immunol. 191, 1240–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye BH, Cattoretti G, Shen Q, Zhang J, Hawe N, de Waard R, Leung C, Nouri-Shirazi M, Orazi A, Chaganti RS, et al. (1997). The BCL-6 protooncogene controls germinal-centre formation and Th2-type inflammation. Nat. Genet. 16, 161–170. [DOI] [PubMed] [Google Scholar]
- Ye J, Ma N, Madden TL, and Ostell JM (2013). IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 41, W34–W40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, Srivastava M, Linterman M, Zheng L, Simpson N, et al. (2009). The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity 31, 457–468. [DOI] [PubMed] [Google Scholar]
- Yu F, Sharma S, Edwards J, Feigenbaum L, and Zhu J (2015). Dynamic expression of transcription factors T-bet and GATA-3 by regulatory T cells maintains immunotolerance. Nat. Immunol. 16, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Saxon A, and Zhang K (2003). Human activation-induced cytidine deaminase is induced by IL-4 and negatively regulated by CD45: implication of CD45 as a Janus kinase phosphatase in antibody diversification. J. Immunol. 170, 1887–1893. [DOI] [PubMed] [Google Scholar]
- Zotos D, Coquet JM, Zhang Y, Light A, D’Costa K, Kallies A, Corcoran LM, Godfrey DI, Toellner K-M, Smyth MJ, et al. (2010). IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J. Exp. Med. 207, 365–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, Good-Jacobson KL, and Shlomchik MJ (2014). CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat. Immunol. 15, 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq and Ig-seq data have been deposited at GEO: GSE150124, GSE150139, and GSE192765 and are publicly available as of the date of publication. This paper does not report original code. Other data reported in this paper will be shared by the lead contact upon request.