

Sir,
The spectrum of clinical manifestations in children with inborn errors of metabolism (IEMs) is highly variable. Despite the rapid advances in biochemical and genetic testing, early and specific diagnosis of IEMs often pose a challenge for clinicians. Also, early diagnosis offers an opportunity for early initiation of treatment. In this case report, we describe a 18-month-old child genetically proven to have D-2-hydroxyglutaric aciduria (D-2-HGA) type II, a rare neurometabolic disorder manifesting with developmental delay, autistic behavior, epilepsy, hypotonia, and neonatal hypoglycemia. A high index of suspicion and screening for IEMs must be considered in children with unexplained developmental delay, epilepsy, or movement disorders.
A one-and-half-year-old boy, first born to a nonconsanguineous couple, presented for the evaluation of delayed milestones. He was born full-term with a birth weight of 2000 g and had recurrent hypoglycemia in the neonatal period for which he was managed elsewhere. There was a history of recurrent respiratory tract infections requiring repeated hospitalizations in infancy. At ten months, he developed generalized tonic-clonic seizures, mostly occurring in sleep, once to twice per month.
At 18 months of age, he sat with support but he could not reach for toys. He had not attained a social smile and had no reciprocative babbling. On examination, he had a head circumference of 43.8 cm (microcephaly) with a depressed nasal bridge, down-slanting palpebral fissures, short philtrum, and a high-arched palate. Poor eye contact, motor motor stereotypies, and auditory agnosia were observed. He did not fix or follow the light. Bilateral nystagmus, right eye divergent squint, hypotonia, brisk deep tendon reflexes, and flexor plantar responses were also observed. Brain magnetic resonance imaging (MRI) done at eight months of age revealed delayed myelination with open opercula bilaterally [Figure 1a-d]. Despite neonatal risk factors such as low birth weight and recurrent hypoglycemia, there were no areas of gliosis on imaging. Considering the possibility of an inborn error of metabolism, urine organic acid analysis by gas chromatography-mass spectrometry (GC-MS) was performed, which revealed a markedly increased excretion of 2-hydroxy-glutarate and 2-hydroxygluatric lactone. Chiral differentiation using a an O-acetyl-di-(D)-2-butyl ester derivative revealed a specific elevation of the D isomer of 2-hydroxyglutarate, consistent with the diagnosis of D-2-hydroxyglutaric aciduria. Electroencephalography and nerve conduction studies were normal. Visual evoked potential and brainstem auditory evoked responses were normal. Clinical exome identified a likely pathogenic heterozygous missense variation in exon 4 of the IDH2 gene (Chr15:90631934C > T; Depth: 95×), which results in the substitution of glutamine for arginine at codon 140 (p.Arg140Gln; ENST00000330062) confirming a diagnosis of D-2-hydroxyglutaric aciduria type II. Mother was heterozygous and father was wild type for the same variant. Genetic counseling was provided to the family. Further evaluation of our patient revealed normal echocardiography. Seizures were managed with levetiracetam and clobazam. While occupational therapy, physiotherapy, and speech therapy were initiated, the child shows poor gains in socio-cognitive skills and has persistent autistic traits.
Figure 1.
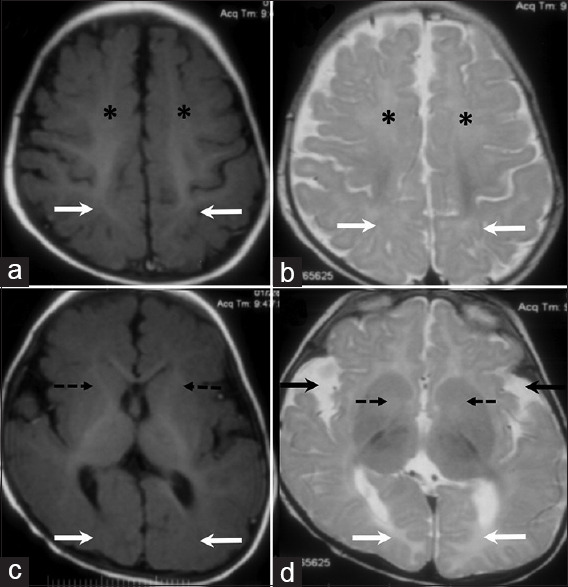
MRI brain images of an 8-month-old child. Axial T1 (A) and T2 weighted images (B) at the level of the perirolandic region shows delayed myelination in the frontal (asterisk) and parietal (arrows) white matter bilaterally. In comparison to the normally myelinated perirolandic white matter, there is mild T2 hyperintensity and relative T1 iso to hypointensity in these regions. Axial T1 (C) and T2 weighted images (D) at the level of the basal ganglia shows similar changes at the level of the anterior limbs of internal capsule (dashed arrows) and calcarine sulcus white matter (white solid arrows). Bilateral open opercula (black solid arrows, D) also noted
D-2-hydroxyglutaric aciduria is a rare neurometabolic disorder that manifests with developmental delay, hypotonia, and epilepsy.[1] D-2-HGA type I is caused by autosomal recessive mutations in the D2HGDH gene that encodes D-2-hydoxyglutarate dehydrogenase while D-2-HGA type II is caused by autosomal dominant heterozygous mutations in the IDH2 gene that encodes mitochondrial isocitrate dehydrogenase-2.[2] Developmental delay was found to be more severe in patients with D-2-HGA type II.[2]
Our patient with D-2-HGA type II has the cardinal clinical features of developmental delay, epilepsy, and hypotonia. Phenotypic heterogeneity has been documented in patients with D-2-HGA with additional findings including epileptic encephalopathy, speech delay, microcephaly, macrocephaly, facial dysmorphism, cerebral visual insufficiency, stridor, peripheral neuropathy, episodic vomiting, apnoea, and cardiomyopathy.[1,2,3,4,5,6] Facial dysmorphism, microcephaly, autistic traits, and cortical visual impairment were observed in our patient, but there was no cardiomyopathy. Neonatal hypoglycemia has not been documented previously in D-2-HGA cases. It might not be reliable to conclude that recurrent neonatal hypoglycemia detected in our case was strictly due to inadequate glycogen stores resulting from low birth weight. Clinicians must always exclude an underlying inborn error of metabolism in neonates with recurrent or refractory hypoglycemia. Animal models have shown that the cytotoxic and neurotoxic effects in D-2-HGA are mediated by the intracellular accumulation of D-2-HG that is speculated to lead to energy depletion, oxidative stress, N-methyl-D-aspartate (NMDA) receptor activation, and increased glutamate uptake.[2] Neuroimaging findings identified in patients with D-2-HGA include delayed myelination, enlarged ventricles and subarachnoid spaces, sub-ependymal cysts and signal changes involving white matter, basal ganglia, thalami, hypothalamus, substantia nigra, and periaqueductal grey matter.[1,5,7,8] Delayed myelination and open opercula were observed in our case as described previously.[8] Our patient has a pathogenic mutation p.Arg140Gln, which has been reported previously.[9] A majority of them were de novo cases while somatic mosaicism was identified in the blood of one patient's mother.[9] Another case of D-2-HGA due to heterozygous mutation in IDH2 gene with an unaffected mother being a mosaic carrier has also been reported.[10] In our case, mother was found to be a heterozygous for the IDH2 gene mutation in blood and could possibly be a mosaic carrier. In IDH2 mosaicism, threshold and tissue-specificity of mosaicism may determine the presence or absence of symptoms.[10] It would be worthwhile to test different tissues such as skin or buccal cells in the mother to look for the grade of mosaicism. However, further evaluation of mother in our case was not possible due to financial constraints. Another possible but less likely explanation for the absence of symptoms in the mother of our patient could be reduced penetrance though not reported previously. No effective intervention exists for the management of D-2-HGA. The course of illness and life expectancy in these patients are often variable.[2]
Surprisingly, there have been no cases of either form of D-2-HGA reported from India. Although this disorder has a variable clinical phenotype, our patient presented with cardinal clinical features of developmental delay, hypotonia, and epilepsy. Evaluation of high-risk neonates or infants for IEMs must, therefore, be considered even in resource-limited settings. While there is no specific treatment for either form of D-2-HGA, early diagnosis will help to prognosticate long-term outcomes as well as provide a risk assessment for future pregnancies and for other family members.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.van der Knaap MS, Jakobs C, Hoffmann GF, Duran M, Muntau AC, Schweitzer S, et al. D-2-hydroxyglutaric aciduria: Further clinical delineation. J Inherit Metab Dis. 1999;22:404–13. doi: 10.1023/a:1005548005393. [DOI] [PubMed] [Google Scholar]
- 2.Kranendijk M, Struys EA, Salomons GS, Van der Knaap MS, Jakobs C. Progress in understanding 2-hydroxyglutaric acidurias. J Inherit Metab Dis. 2012;35:571–87. doi: 10.1007/s10545-012-9462-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mahfoud A, Domínguez CL, Rashed M, Durán M, Rodríguez T, Rodríguez D, et al. [D-2-hydroxyglutaric aciduria. Report of two cases] Invest Clin. 2009;50:369–75. [PubMed] [Google Scholar]
- 4.Phillips E, Sasarman F, Sinasac DS, Al-Hertani W. D-2-hydroxyglutaric aciduria in a patient with speech delay due to a novel homozygous deletion in the D2HGDH gene. Mol Genet Metab Rep. 2019;20:100482. doi: 10.1016/j.ymgmr.2019.100482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Knaap MS, Jakobs C, Hoffmann GF, Nyhan WL, Renier WO, Smeitink JA, et al. D-2-Hydroxyglutaric aciduria: Biochemical marker or clinical disease entity? Ann Neurol. 1999;45:111–9. doi: 10.1002/1531-8249(199901)45:1<111::aid-art17>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 6.Haliloglu G, Temucin CM, Oguz KK, Celiker A, Coskun T, Sass JO, et al. Peripheral neuropathy in a patient with D-2-hydroxyglutaric aciduria. J Inherit Metab Dis. 2009;32(Suppl 1):S21–5. doi: 10.1007/s10545-009-0933-2. [DOI] [PubMed] [Google Scholar]
- 7.Wajne M, Vargas CR, Funayama C, Fernandez A, Elias ML, Goodman SI, et al. D-2-Hydroxyglutaric aciduria in a patient with a severe clinical phenotype and unusual MRI findings. J Inherit Metab Dis. 2002;25:28–34. doi: 10.1023/a:1015165212965. [DOI] [PubMed] [Google Scholar]
- 8.van der Knaap M, Valk J. D-2-Hydroxyglutaric Aciduria. In: Ute Hteilmann., editor. Magnetic resonance of myelination and myelin disorders. 3rd ed. Berlin Heidelberg New York: Springer; 2005. pp. 338–41. [Google Scholar]
- 9.Kranendijk M, Struys EA, van Schaftingen E, Gibson KM, Kanhai WA, van der Knaap MS, et al. IDH2 mutations in patients with D-2-hydroxyglutaric aciduria. Science. 2010;330:336. doi: 10.1126/science.1192632. [DOI] [PubMed] [Google Scholar]
- 10.Nota B, Hamilton EM, Sie D, Ozturk S, van Dooren SJ, Ojeda MR, et al. Novel cases of D-2-hydroxyglutaric aciduria with IDH1 or IDH2 mosaic mutations identified by amplicon deep sequencing. J Med Genet. 2013;50:754–9. doi: 10.1136/jmedgenet-2013-101961. [DOI] [PubMed] [Google Scholar]