

Summary
Klebsiella pneumoniae has become the number one bacterial pathogen that causes high mortality in clinical settings worldwide. Clinical K. pneumoniae strains with carbapenem resistance and/or hypervirulent phenotypes cause higher mortality comparing with classical K. pneumoniae strains. Rapid differentiation of clinical K. pneumoniae with high resistance/hypervirulence from classical K. pneumoniae would allow us to develop rational and timely treatment plans. In this study, we developed a convolution neural network (CNN) as a prediction method using Raman spectra raw data for rapid identification of ARGs, hypervirulence‐encoding factors and resistance phenotypes from K. pneumoniae strains. A total of 71 K. pneumoniae strains were included in this study. The minimum inhibitory concentrations (MICs) of 15 commonly used antimicrobial agents on K. pneumoniae strains were determined. Seven thousand four hundred fifty‐five spectra were obtained using the InVia Reflex confocal Raman microscope and used for deep learning‐based and machine learning (ML) algorithms analyses. The quality of predictors was estimated in an independent data set. The results of antibiotic resistance and virulence‐encoding factors identification showed that the CNN model not only simplified the classification system for Raman spectroscopy but also provided significantly higher accuracy to identify K. pneumoniae with high resistance and virulence when compared with the support vector machine (SVM) and logistic regression (LR) models. By back‐testing the Raman‐CNN platform on 71 K. pneumoniae strains, we found that Raman spectroscopy allows for highly accurate and rationally designed treatment plans against bacterial infections within hours. More importantly, this method could reduce healthcare costs and antibiotics misuse, limiting the development of antimicrobial resistance and improving patient outcomes.
By back‐testing the Raman‐CNN platform on 71 Klebsiella pneumoniae strains, we found that Raman spectroscopy allows for highly accurate and rationally designed treatment plans against bacterial infections within hours. This method could reduce healthcare costs, antibiotics misuse, limiting the development of antimicrobial resistance, and improving patient outcomes.

Introduction
Klebsiella pneumoniae, belonging to the family Enterobacteriaceae, forms part of the normal flora colonizing mucosal surfaces in healthy humans and animals (Navon‐Venezia et al., 2017). Its infections account for a significant proportion of serious community‐acquired (CA) infections worldwide (Magill et al., 2014). This is because that, under certain conditions, Klebsiella could disseminate to other tissues and cause life‐threatening infections including pneumonia, wound, soft tissue or urinary tract infections, which are particularly problematic among neonates, the elderly and the immunocompromised.
Virulence factors including various iron acquisition molecules, specific capsular polysaccharide and the hypermucoidy‐related rmpA and rmpA2 genes are known to be associated with invasive CA K. pneumoniae infections characterized by high morbidity and high mortality (Russo and Marr, 2019). Reported mortality rates of K. pneumoniae bacteraemia in China ranged from 8.7% to 24.6% (Zhang et al., 2018; Liu et al., 2019; Li et al., 2020). K. pneumoniae is also the most common multidrug‐resistant (MDR) pathogens and one of the six nosocomial pathogens “ESKAPE” (Boucher et al., 2009). Gu et al. (2018) reported a fatal outbreak of MDR, in which carbapenem‐resistant K. pneumoniae (CRKP) were found to be highly transmissible and posed a substantial threat to public health. Polymyxins can be used to treat CRKP infections. Nevertheless, transferable colistin resistance mcr gene has been identified in such strains in our recent study, which can cause the invalidation of polymyxins (Lu et al., 2020a, 2020b,2020a, 2020b). What is more, K. pneumoniae strains undergo active horizontal transfer of antimicrobial resistance genes (ARGs) and act as the reservoir of such genes. Current studies showed that a large proportion of ARGs were identified in K. pneumoniae for their first time (Nordmann and Poirel, 2014; Zheng et al., 2020; Hu et al., 2021).
A rapid and high specificity identification method is urgently needed to differentiate MDR K. pneumoniae from the classic non‐resistant strains. This is because such a method could allow clinicians to select appropriate antibiotics at the beginning of the treatment course and to reduce the prevalence of drug resistance. Conventional antimicrobial‐susceptibility testing (AST) methods were based on the observation of bacterial colonies in the presence of antibiotics, which typically take a few hours to produce results (van Belkum et al., 2019). Polymerase chain reaction (PCR)‐based ARGs detection methods require prior knowledge of the strains and is time‐consuming, which normally require several hours to run (Tadesse et al., 2020). Although matrix‐assisted laser desorption/Ionization time‐of‐flight mass spectrometry (MALDI‐TOF MS) is a fast and effective method, it is hard to be optimized due to the heterogeneity of analyte/matrix mixture (Dortet et al., 2018; Furniss et al., 2019). Hence, the initial antimicrobial therapy for acute infections is often empirical (Rhodes et al., 2017). The Centers for Disease Control and Prevention reported that over 30% of patients are treated unnecessarily (Fleming‐Dutra et al., 2016). Such unnecessary treatment may select and promote the dissemination of MDR K. pneumoniae strains.
Raman spectroscopy is a fast, noninvasive and cost‐effective technology, which requires a minimum sample preparation process to generate a molecular fingerprint of the chemical constituents of the sample (Pahlow et al., 2015; Butler et al., 2016; Teng et al., 2016). When combined with machine learning (ML), Raman spectroscopy has the potential to identify the bacterial species and depict their antibiotic resistance status (Uysal Ciloglu et al., 2020). However, previous studies focused on the pathogen identification and resistance phenotype prediction using deep learning or ML algorithms (Pahlow et al., 2015; Ho et al., 2019; Uysal Ciloglu et al., 2020). The ability of Raman spectroscopy combined with a deep neural network for ARGs or virulence genes prediction remains unknown. In this study, we used the convolution neural network (CNN) as a deep learning strategy to classify bacterial spectra according to the antibiotic resistance and virulence encoding factors.
Results
Antimicrobial susceptibility profiles
Antimicrobial susceptibility testing results indicated that all of the 71 K. pneumonia strains exhibited MDR phenotypes (Fig. 1), with all carbapenemase‐producing strains being resistant to meropenem (MICs ≥ 4 mg l−1), ertapenem (MICs ≥ 2 mg l−1), ceftazidime (MICs ≥ 16 mg l−1), cefotaxime (MICs ≥ 4 mg l−1), cefoperazone/sulbactam (MICs ≥ 64 mg l−1) and cefepime (MICs ≥ 16 mg l−1). All but two carbapenemase‐producing strains were resistant to aztreonam (MICs ≥ 16 mg l−1). Moreover, it was also found that all but one noncarbapenemase‐producing strains were sensitive to imipenem, meropenem and ertapenem, whereas all were sensitive to ceftazidime/avibactam. The number of these 71 K. pneumoniae strains resistant to imipenem, meropenem, ertapenem, cefmetazole, ceftazidime, cefotaxime, piperacillin/tazobactam, cefoperazone/sulbactam, ceftazidime/avibactam, cefepime, colistin, tigecycline, ciprofloxacin, amikacin and aztreonam was 31 (43.7%), 42 (59.2%), 42 (59.2%), 31 (43.7%), 54 (76.1%), 59 (83.1%), 42 (59.2%), 46 (64.8%), 21 (29.6%), 53 (74.7%), 23 (32.4%), 6 (8.2%), 43 (60.6%), 20 (28.2%) and 53 (74.7%) respectively.
Fig. 1.
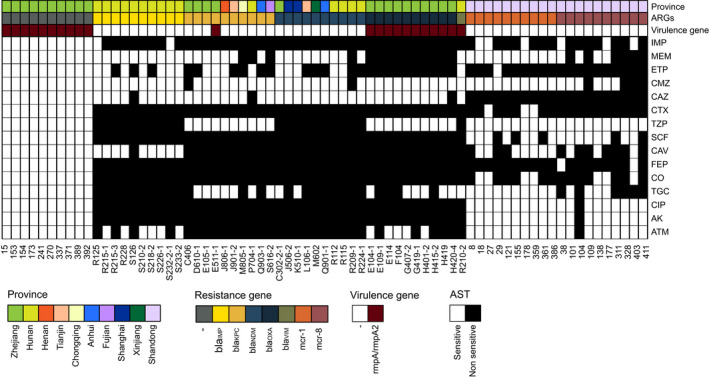
The heat map of the origin, resistance genes and the results of antibiotic susceptibility testing of all isolates for convolution neural network training. The origin, resistance genes, virulence genes and the result of antibiotic susceptibility testing are given as coloured annotations at the bottom of the heat map. AMK, amikacin; ATM, aztreonam; CAV, ceftazidime/avibactam; CAZ, ceftazidime; CIP, ciprofloxacin; CMZ, cefmetazole; CO, colistin; CTX, cefotaxime; ETP, ertapenem; FEP, cefepime; IPM, imipenem; MEM, meropenem; SCF, cefoperazone/sulbactam; TGC, tigecycline; TZP, piperacillin/tazobactam.
Data preprocessing
Before the modelling process, preprocessing of spectral data is indispensable. Raman spectra contain useful information related to a sample, but it is often accompanied by interfering information such as background fluorescent signal, cosmic rays and random noise. Therefore, eliminating noise and removing fluorescence through spectral preprocessing is important and necessary for the information receiving. We removed the background of the spectrum, used a smoothing filter for noise minimization and performed baseline correction and area normalization via the R program. The corrected spectra were displayed in Fig. 2. Differences in intensities were displayed among different groups of K. pneumoniae strains. Table 1 showed the main Raman bands that were observed in K. pneumoniae strains and their corresponding assignments.
Fig. 2.

Typical Ramanome of K. pneumoniae with diverse antimicrobial resistance genes. The spectra were shown after background subtraction and normalization for a measurement time of 5 s. The spectra were averaged spectra for each antimicrobial resistance gene group and vertically shifted to increase the visibility of details. The mean Raman spectra were shown by the solid line, and the 95% confident intervals were presented by the shadow.
Table 1.
Molecular assignment of the Raman peaks found in this study.
| Peak position (Raman shift cm−1) | Band assignment | References |
|---|---|---|
| ~ 495–550 | Disulphide bond (S–S) | Devitt et al. (2018) |
| ~ 786 | Nucleic acid | Notingher and Hench (2006) |
| ~ 726 | C–S/cysteine | Devitt et al. (2018) |
| ~ 843 | Glucose | Devitt et al. (2018) |
| ~ 923–958 | C–C | Devitt et al. (2018) |
| ~ 1035 | Phenylalanine (the in‐plane C–H bending mode) | Fan et al. (2011) |
| ~ 1060–1095 | Nucleic acid, lipid and carbohydrates | Notingher and Hench (2006) |
| ~ 1101 | Nucleic acid | Teng et al. (2016) |
| ~ 1121–1145 | C–N | Devitt et al. (2018) |
| ~ 1158 | C–C/C–N | Notingher and Hench (2006) |
| ~ 1220–1240 | Nucleic acid, protein and lipid | Notingher and Hench (2006) |
| ~ 1333–1350 | Nucleic acid and protein | Notingher and Hench (2006) and Navon‐Venezia et al. (2017) |
| ~ 1420–1480 | Nucleic acid, protein, lipid and carbohydrates | Notingher and Hench (2006) |
Deep learning for three identification tasks from Raman spectra
To gather a training data set, we measured 71 K. pneumoniae strains using confocal microscopic Raman spectrometer in short measurement time. Only one strain carried bla VIM gene and 10 strains each carrying the bla NDM, bla KPC, bla IMP, bla OXA, mcr‐1, mcr‐8 and no above genes (Fig. 1). We constructed our data sets of 7455 spectra from 71 K. pneumoniae isolates, and three strains were measured repeatedly (3×). The results prove a relative consistency of spectra over time in Raman spectroscopy (Fig. S1). We then trained the neural network on three identification tasks, where the CNN outputs a probability distribution across the seven ARGs, two virulence genes (rampA and rampA2) and the drug‐resistant phenotypes (sensitive or non‐sensitive) among 15 commonly used antimicrobial agents; the maximum was taken as the predicted models (Fig. 3). The models were only trained for 20 epochs as accuracy did not increase significantly for greater epochs (Fig. S2). We then evaluated and reported the accuracy of these three types of models on the test data set, which was gathered from independently cultured and prepared samples. The performance breakdown of CNN models for ARGs and the identification tasks of rampA/rampA2 genes were displayed in the confusion matrixes in Figs 4A and 5A. For the identification of isolates, which harboured neither carbapenemase‐encoding genes (bla NDM, bla KPC, bla IMP, bla OXA and bla VIM) nor mcr gene, the accuracy was 99.2%. The prediction accuracies for isolates, which harboured bla IMP, mcr‐1, mcr‐8, bla NDM and bla OXA, were higher than 92% by our deep learning model. As for other ARGs such as bla KPC and bla VIM, the identification accuracies were 88.4% and 84.0% respectively. The performance of our CNN model for virulence genes prediction delivers similarly good results, reaching 98.4% prediction accuracy with non‐rmpA/rmpA2‐carrying strains and 83.3% prediction accuracy with rmpA/rmpA2‐carrying strains. The ROC curve was shown in Fig. 5B, and the area under the curve (AUC) is 0.979. For predicting the resistance phenotypes, the identification accuracies of meropenem, ertapenem, ceftazidime, cefotaxime, cefoperazone/sulbactam, ceftazidime/avibactam, cefepime, tigecycline and ciprofloxacin were more than 85% (Fig. 6). However, the identification accuracy of cefmetazole was only 81.7 ± 1.1%, which was the lowest accuracy in the resistance prediction by our CNN models. The average accuracies for prediction of ARGs, virulence‐related genes and resistance are 94.2 ± 1.1%, 95.3 ± 0.5% and 81.7–96.2% respectively.
Fig. 3.
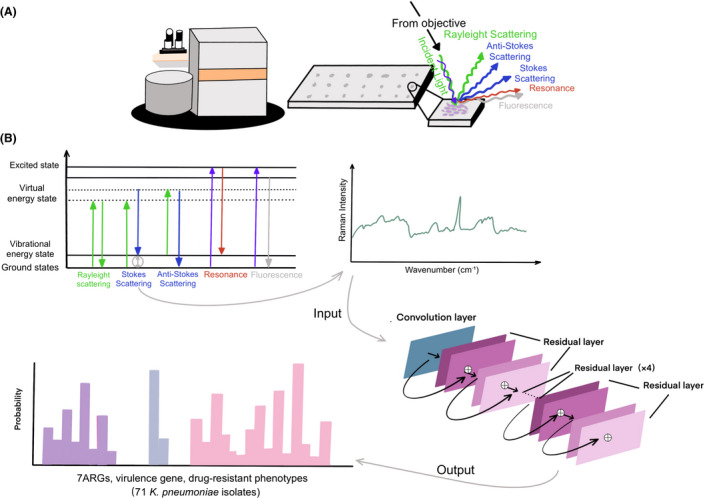
Summary schematic of confocal Raman microscope techniques including sample preparation to spectral analysis and the construction of ResNet taxonomic model.
A. The K. pneumoniae lawn was smeared onto the stainless‐steel plate for Raman spectral collection. The schematic identifying light scattering after laser exposure on a sample surface. When the electrons are excited to virtual energy levels, it can return to the original energy level by emitting a photon of light, known as Rayleigh scattering, or it can undergo an energy shift, known as Stokes scattering or anti‐Stokes scattering. Resonance Raman scattering and fluorescence can occur when electrons are excited to electronic energy levels.
B. The antimicrobial resistance genes (ARGs), virulence genes and antibiotic susceptibility are identified and analysed using convolutional neural network. Using a one‐dimensional residual network with 25 total convolutional layers (see Section 2 for details), Raman spectra are analysed to predict the existence of ARGs and virulence genes or the drug‐resistant phenotypes.
Fig. 4.
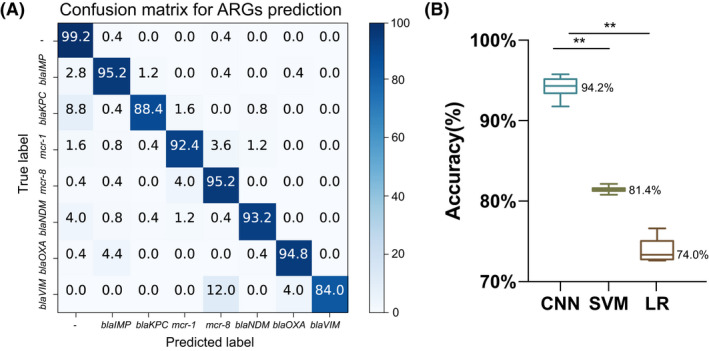
Three model performance breakdown by antimicrobial resistance genes (ARGs).
A. Confusion matrix for convolution neural network (CNN) models. Each row of this matrix represents the percentage of spectra in an actual class, whereas each column represents the percentage of spectra in a predicted class. The diagonal elements of this matrix show the percentage of correctly classified spectra, whereas the off‐diagonal elements indicate the percentage of misclassified ones.
B. The accuracy box plot to three types of models; *P < 0.05; **P < 0.001. LR, logistic regression; SVM, support vector machine.
Fig. 5.

Three model performance breakdown by virulence genes.
A. Confusion matrix for convolution neural network (CNN) models; values are listed as percentages.
B. The receiver operating characteristic curves and area under curves of different models for rampA/rampA2 identification.
C. Bar diagrams of accuracy to three types of models; *P < 0.05; **P < 0.001. LR, logistic regression; SVM, support vector machine.
Fig. 6.
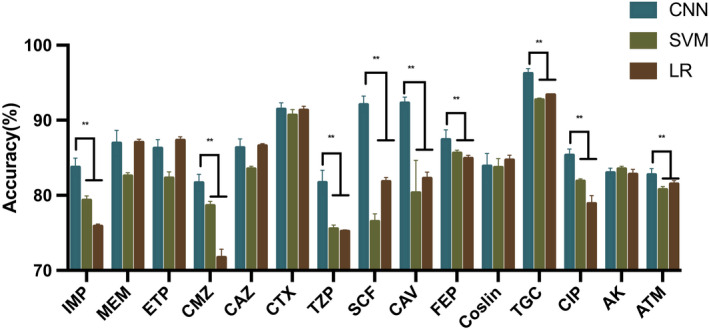
Bar diagrams of accuracy ± standard deviation values for drug‐resistant phenotypes prediction. *P < 0.05; **P < 0.001. AMK, amikacin; ATM, aztreonam; CAV, ceftazidime/avibactam; CAZ, ceftazidime; CIP, ciprofloxacin; CMZ, cefmetazole; CNN, convolution neural network; CO, colistin; CTX, cefotaxime; ETP, ertapenem; FEP, cefepime; IPM, imipenem; LR, logistic regression; MEM, meropenem; SCF, cefoperazone/sulbactam; SVM, support vector machine; TGC, tigecycline; TZP, piperacillin/tazobactam.
ML for three tasks identification from Raman spectra
We also predicted the ARGs, virulence genes and drug‐resistant phenotypes with classical analysis techniques including LR and SVM based on the test data set. The mean accuracies with a standard deviation of both LR and SVM models were calculated. According to these analyses, the SVM classifier exhibited a higher‐level accuracy (81.4 ± 0.3%), whereas LR model showed a slightly lower classification accuracy of 74.0 ± 1.4% in ARGs identification (Fig. 4B). When identifying the existence of virulence genes, SVM and LR models achieve 88.1 ± 0.3% and 89.6 ± 0.5% accuracies respectively (Fig. 5C). For predicting drug‐resistant phenotypes, lower accuracies were observed in the SVM model, with the accuracy ranging from 75.5% to 92.7%. The accuracy of LR model for phenotypes identification ranged between 71.7% and 91.3% (Fig. 6).
Statistical comparisons
The sensitivity, specificity and accuracy, including standard deviation values for each classifier model, were presented in Table S1. The CNN classifier achieved better accuracy of the ARGs and virulence genes prediction, comparing with the ML algorithms (P ≤ 0.01). In resistance phenotypes identifying, the prediction accuracies of CNN models for all 15 antimicrobial agents were higher than the ones in both two ML models. Nine of the comparisons were statistically significant (Fig. 6).
Discussion
Klebsiella pneumoniae has the potential to carry a wide range of ARGs that render current options for treatment of K. pneumoniae infections ineffective (Boucher et al., 2009). During the last decades, the incidence of CRKP has markedly increased worldwide and posed an urgent threat to public health. The polymyxins (colistin and polymyxin B) are antibiotics used for treating CRKP infections. With an increasing prevalence of CRKP infection, the use of polymyxins has been rising. However, since the discovery of the plasmid‐mediated polymyxin resistance gene (mcr) at the end of 2015, the treatment of K. pneumoniae infections has become ineffective thus eliciting worldwide attentions (Liu et al., 2016). Therefore, effective methods for screening ARGs and virulence genes are demanded urgently in clinical practice.
In this work, we collected the Raman spectra from 71 K. pneumoniae isolates. The band assignments (Table 1) were performed based on the literature. These spectroscopic vibrations were primarily related to the skeletal structure of nucleic acid and the proteins they express as these strains harboured a diverse range of ARGs, virulence genes or resistance elements. Thus, we considered these peaks as the discrimination of different resistant genes and proteins, which may be related to the degradation of antibacterial drugs, alteration in antimicrobial targets and changes in membrane permeability to antibiotics. However, it was extremely difficult to distinguish them by the naked eyes (Fig. 2). Thus, we employed the ResNet architecture to produce predictors of ARGs, virulence genes and resistance phenotypes based on the Raman spectra. We attempted to compare the CNN model with other classical ML algorithms like SVM and LR, which are the most preferred models for bacteria detection studies with Raman spectroscopy (Beier et al., 2010; Uysal Ciloglu et al., 2020). The results of ARGs identification showed that the CNN model could identify both carbapenemase‐encoding genes (bla NDM, bla KPC, bla IMP, bla OXA and bla VIM) and mobile colistin resistance genes with 94.24 ± 1.14% accuracy, which is better than SVM (81.44 ± 0.33%) and LR (73.97 ± 1.41%) (Fig. 4B). These differences were statistically significant (P < 0.01). Moreover, 7455 spectra were then used to train a CNN model for rapid virulence‐related genes detection. The results in the test data set were nearly the same as ARGs prediction task, indicating that CNN model provided better detection accuracy than ML algorithms (Fig. 5). The accuracy of phenotype‐based identification is important to guide the efficient therapies for clinical infections. In this study, we found significant differences in the accuracy of prediction of phenotypic resistance to nine commonly used antimicrobial agents by the CNN model and ML algorithms. Our CNN predictive model demonstrated reasonably good discrimination. The LR and SVM classifier exhibits lower accuracies than CNN when used in resistance prediction.
High accuracy of the Raman‐CNN system indicates that we can utilize Raman spectroscopy to produce a fine‐grained and reliable genotype and phenotype signature characteristic for each K. pneumoniae strains. Raman spectroscopy has the unique potential to identify bacterial phenotypes without requiring additional markers and generate spectral data within a few seconds. The advantages of this method for bacterial species identification and phenotyping include more rapid and easier to interpret when comparing with other culture‐free methods such as single‐cell sequencing and fluorescence or magnetic tagging (Wang and Navin, 2015; Martynenko et al., 2019; Tadesse et al., 2020). It also allows the prediction at both genotype and phenotype levels. When compared with MALDI‐TOF MS, Raman spectroscopy is non‐destructive because no living cells need to be broken for DNA or protein extraction. Besides, MALDI‐TOF MS requires the use of additional materials or chemicals and time for sample preparation such as sample deposition onto a plate, drying, adding matrix, drying again and even semiextraction or extraction step. These processes may result in variable analyte signal intensities (AlMasoud et al., 2021). Several Raman data analysis approaches based on ML algorithms, including decision tree learning, LR analysis and SVM, have been developed for discriminative analysis (Ho et al., 2019; Uysal Ciloglu et al., 2020). However, when creating models for discriminating analyses, some valuables are likely to get lost during data transformation and variable selection, such as dimensionality reduction from the raw spectral data or selecting a variable from a large number of variables. In this work, we use CNN as the prediction method which utilize Raman spectra as raw data for analysis. Unlike other conventional Raman analysis methods, CNN combines feature extraction and classification in a single network architecture. In other words, it requires a lower level of manual supervision. Our results showed that the CNN model not only simplifies the classification system for Raman spectroscopy but also provides significantly higher accuracy than SVM and LR models.
Some limitations in this study should be realized. First, due to the difficulties in obtaining sufficient carbapenemase and mcr‐producing K. pneumoniae, only one VIM‐positive K. pneumoniae strain was used for modelling. Besides, the same strains were used to obtain the test data set. Although other researchers have employed the same approach to obtain a test data set, it should be noted, more clinical strains are required for validation in further studies. Second, as shown in Fig. 2, our model encountered serious generalization problems due to the small differences between the spectra. In addition, the accuracy of the models in our study needs to be improved. Further studies using optimized models are suggested to improve the practicability of deep learning in Raman spectrum application.
In conclusion, Raman spectroscopy is a promising technique for a fast, noninvasive, culture‐independent, single‐cell level identification of microbial genotype and phenotype. Combined with artificial intelligence and large data sets, the method provides results with high accuracy. The Raman–CNN platform allows for highly accurate and targeted treatment of bacterial infections within hours. This will reduce turnaround time, healthcare costs and antibiotics misuse. In the meantime, it will limit antimicrobial resistance and improve patient outcomes.
Experimental procedures
Bacterial isolates
A total of 51 K. pneumonia strains collected from January 2018 to December 2018 in a Chinese clinical microbiology laboratory and 20 animal‐borne K. pneumonia strains collected in November 2019 were included in this study (Fig. 1). The clinical strains were recovered from 16 hospitals in 10 Chinese provinces (Anhui, Fujian, Henan, Hunan, Shandong, Shanghai, Tianjin, Xinjiang, Zhejiang and Chongqing provinces). The 20 K. pneumoniae strains of animal origin were isolated from chicken cloaca in Shandong province. A total of 61 K. pneumonia strains were found to carry diverse carbapenemase‐encoding genes (bla NDM, bla KPC, bla IMP, bla OXA and bla VIM), mobile colistin resistance genes (mcr‐1 and mcr‐8) and virulence genes (rampA and rampA2). The remaining 10 K. pneumonia strains did not carry carbapenemase genes or mcr gene, but the rampA was detected in these strains. In total, 22 K. pneumoniae strains were found to harbour the rampA or rampA2 genes. All the ARGs and virulence genes were verified by PCR analysis.
All the K. pneumonia strains were recovered on the Mueller–Hinton agar (MH, Oxoid, Basingstoke, UK) for 24 ± 2 h at 35°C as described in a previous study (Uysal Ciloglu et al., 2020). Species identity of 71 isolates was confirmed by matrix‐assisted laser desorption/ionization time‐of‐flight mass spectrometry (MALDI Biotyper; Bruker Daltonik GmbH, Bremen, Germany), followed by antimicrobial susceptibility tests, whole‐genome sequencing and Raman spectroscopy analysis.
Antimicrobial susceptibility testing
The minimum inhibitory concentrations (MICs) of 15 commonly used antimicrobial agents (imipenem, meropenem, ertapenem, ceftazidime/avibactam, cefepime, cefmetazole, ceftazidime, colistin, tigecycline, cefotaxime, ciprofloxacin, piperacillin/tazobactam, amikacin, cefoperazone/sulbactam and aztreonam) on K. pneumoniae strains were determined by the broth microdilution method, using K. pneumoniae strain ATCC 13883 as a quality control. Susceptibility results for tigecycline were interpreted using the EUCAST breakpoints (www.eucast.org). The results of the other agents were interpreted according to Clinical and Laboratory Standards Institute guidelines (www.clsi.org).
Raman microscopy
All experiments were performed using a InVia Reflex confocal Raman microscope (Renishaw, Wotton‐under‐Edge, UK). Bacterial samples were excited with a near infrared 785‐nm diode laser in a range of 390.79–1552.14 cm−1 at ~ 150‐mW laser power. The wavelength of the instrument was calibrated automatically using a silicon wafer by setting the silicon peak to 520 cm–1. One thousand two hundred litres per millimetres grating was used to maximize signal strength while minimizing background signal from autofluorescence. Spectral resolution was < 1 cm−1. The 50 × 0.5‐NA objective lens (Leica, Wetzlar, Germany) generates a diffraction‐limited spot size, ~ 1.9 μm in diameter, the spacing between which was set at 14.5 μm to avoid overlap between spectra. The spectra were generated with five areas of scanning in each sample (technical replicates). At each area, 21 scans were obtained; a total of 105 scans were obtained for each sample (5‐s integration time per scan). These 7455 spectra were used for ResNet and ML learning models training. Meanwhile, another 1775 spectra gathered from separately cultured 71 strains were used as an independent test data set to evaluate the accuracy of the models referring to the method of Lu et al. (2020a, 2020b,2020a, 2020b). Strain 270, 104 and R210 were measured repeatedly (3×) to ensure the consistency of spectrum for each at different measurements, according to previous studies (Rebrošová et al., 2017).
Raman spectral preprocessing
Nearest neighbour algorithm was used to remove cosmic rays. The spectra are preprocessed in three steps: (1) background subtraction, (2) smoothing and (3) normalizing according to a previous existing method (Lu et al., 2020a, 2020b,2020a, 2020b). The polynomial baseline fitting was applied for background fluorescence subtraction, and the Savitzky–Golay filter was used for smoothing. For the vector normalization method, the zero‐mean normalization (Z‐score) was applied. These preprocessing steps were all conducted using the R language (v3.6.2) with the packages of “prospectr” and “baseline.”
Identification by deep learning
The CNN architecture is adapted from the ResNet architecture, which has been shown to be successful in a range of computer vision tasks (He et al., 2020). The model consists of an initial convolution layer, followed by six residual layers and a final fully connected classification layer (Fig. 3). Each residual layer contains four convolutional layers, and the total depth of the network is 26 layers. Shortcut connections are added throughout the residual layers between the input and output of each residual block for better gradient propagation and stable training. The initial convolution layer contains 64 convolutional filters, whereas each of the hidden layers has 100 filters. The architecture of our network was similar to the previous study by Ho et al. (2019).
We first trained the network on the seven diverse ARGs identification tasks, where output of the CNN is a vector of probabilities across the seven ARGs and the maximum probability is taken as the predicted class. The binary differentiation of drug‐resistant phenotypes (sensitive or resistant) among 15 commonly used antimicrobial agents and the virulence genes have the same architecture as the ARGs identification task, except for the number of classes in the final classification layer. We trained the network with the Stochastic Gradient Descent (SDG) optimizer across all experiments at a learning rate 0.001, betas (0.5 and 0.999) and batch size 100 to minimize the categorical cross entropy loss. We utilized a 10‐fold cross‐validation to evaluate the classifying ability of these ResNet models. The 10‐fold cross‐validation consists of 10 separate runs with different sets of training data (used for fitting the model) and test data (used for testing model performance). This process was repeated 10 times to make sure that each of the 10 sets acted as test data once.
We next used our trained ResNet models to identify seven diverse ARGs, drug‐resistant phenotypes and virulence genes in a test data set. The test data set is independent of the training data set used for constructing the deep learning models. The diagnostic performance of these identification models was assessed using confusion matrixes and plotting a receiver operating characteristic (ROC) curve. The accuracy, sensitivity and specificity of the deep learning models were calculated. All procedures were implemented with the PyTorch deep learning framework in Python programming language in the NVIDIA GeForce RTX 3070 Ti platform.
Identification by ML algorithms
The identification of seven diverse ARGs, drug‐resistant phenotypes and the virulence genes was also accomplished by using the ML algorithms, including logistic regression (LR) and support vector machine (SVM). The data set was trained using 10‐fold cross‐validation where 90% was used for training and 10% was used for cross‐validation. The predictive performance of these ML models was calculated in terms of accuracy, sensitivity, specificity and the area under the ROC curve in the test data set. All calculations were implemented in Python (v3.7.3, package sklearn) and r software (package pROC).
Statistical comparisons
The mean accuracy for the CNN, SVM and LR models was tested for equal variances using Levene's test. The Student's t test or Welch's t test were used to test whether the differences in mean accuracy between the CNN and ML algorithms were statistically significant. A P‐value < 0.05 was statistically significant. Multicomparison correction was performed by Bonferroni correction.
Ethics
Bacterial isolates used in this study were approved by the Ethics Committee of Second Affiliated Hospital of Zhejiang University, School of Medicine (Number: 2018‐039). All specimens were processed and anonymized in accordance with ethical and legal standards, and patients were not physically involved in this study. Informed consent was not needed for this study.
Conflict of interest
All authors report no potential conflicts of interest with respect to the research, authorship and publication of this article.
Ethical approval
Bacterial isolates used in this study were approved by the Ethics Committee of Second Affiliated Hospital of Zhejiang University, School of Medicine (Number: 2019‐074).
Supporting information
Fig. S1. 2‐D‐scores plots of principle component relation (PC1and PC2) for three K. pneumoniae strains (270, 104, R210). The dots in different color and shapes represent the spectra gathered in different measurement times.
Fig S2. The model was created at each epoch to select the most optimal epoch. The accuracy of the models did not increase significantly after 20 epochs training.
Table S1 Sensitivity, specificity and accuracy of the CNN models and the ML models in three identification tasks.
Acknowledgements
We sincerely thank Zhaofen Li and Jiulong Yang of Renishaw (Shanghai) Trading Co. Ltd. and Kun Zhu of Bruker (Beijing) Scientific Technology Co. Ltd. for the technical support. This work was partially funded by the National Natural Science Foundation of China (grant nos. 81861138052 and 31761133004) and the Basic Research Fund of Shenzhen (20170410160041091). The funders had no role in study design, data collection and interpretation or the decision to submit the work for publication.
Microb. Biotechnol. (2022) 15(4), 1270–1280
Funding Information
This work was partially funded by the National Natural Science Foundation of China (grant nos. 81861138052 and 31761133004) and the Basic Research Fund of Shenzhen (20170410160041091).
References
- AlMasoud, N. , Muhamadali, H. , Chisanga, M. , AlRabiah, H. , Lima, C.A. , and Goodacre, R. (2021) Discrimination of bacteria using whole organism fingerprinting: the utility of modern physicochemical techniques for bacterial typing. The Analyst 146: 770–788. [DOI] [PubMed] [Google Scholar]
- Beier, B.D. , Quivey, R.G. , and Berger, A.J. (2010) Identification of different bacterial species in biofilms using confocal Raman microscopy. J Biomed Opt 15: 66001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Belkum, A. , Bachmann, T.T. , Lüdke, G. , Lisby, J.G. , Kahlmeter, G. , Mohess, A. , et al. (2019) Developmental roadmap for antimicrobial susceptibility testing systems. Nat Rev Microbiol 17: 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher, H.W. , Talbot, G.H. , Bradley, J.S. , Edwards, J.E. , Gilbert, D. , Rice, L.B. , et al. (2009) Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 48: 1–12. [DOI] [PubMed] [Google Scholar]
- Butler, H.J. , Ashton, L. , Bird, B. , Cinque, G. , Curtis, K. , Dorney, J. , et al. (2016) Using Raman spectroscopy to characterize biological materials. Nat Protoc 11: 664–687. [DOI] [PubMed] [Google Scholar]
- Devitt, G. , Howard, K. , Mudher, A. , and Mahajan, S. (2018) Raman spectroscopy: an emerging tool in neurodegenerative disease research and diagnosis. ACS Chem Neurosci 9: 404–420. [DOI] [PubMed] [Google Scholar]
- Dortet, L.‐A.‐O. , Potron, A. , Bonnin, R.‐A.‐O. , Plesiat, P. , Naas, T.‐A.‐O. , Filloux, A.‐A.‐O. , and Larrouy‐Maumus, G. (2018) Rapid detection of colistin resistance in Acinetobacter baumannii using MALDI‐TOF‐based lipidomics on intact bacteria. Sci Rep 8: 16910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, C. , Hu, Z. , Mustapha, A. , and Lin, M. (2011) Rapid detection of food‐ and waterborne bacteria using surface‐enhanced Raman spectroscopy coupled with silver nanosubstrates. Appl Microbiol Biotechnol 92: 1053–1061. [DOI] [PubMed] [Google Scholar]
- Fleming‐Dutra, K.E. , Hersh, A.L. , Shapiro, D.J. , Bartoces, M. , Enns, E.A. , File, T.M. , et al. (2016) Prevalence of inappropriate antibiotic prescriptions among US ambulatory care visits, 2010–2011. JAMA 315: 1864–1873. [DOI] [PubMed] [Google Scholar]
- Furniss, R.C.D. , Dortet, L. , Bolland, W. , Drews, O. , Sparbier, K. , Bonnin, R.A. , et al. (2019) Detection of Colistin resistance in Escherichia coli by use of the MALDI Biotyper Sirius mass spectrometry system. J Clin Microbiol 57: e01427‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, D. , Dong, N. , Zheng, Z. , Lin, D. , Huang, M. , Wang, L. , et al. (2018) A fatal outbreak of ST11 carbapenem‐resistant hypervirulent Klebsiella pneumoniae in a Chinese hospital: a molecular epidemiological study. Lancet Infect Dis 18: 37–46. [DOI] [PubMed] [Google Scholar]
- He, F. , Liu, T. , and Tao, D. (2020) Why ResNet works? Residuals generalize. IEEE Trans Neural Netw Learn Syst 31: 5349–5362. [DOI] [PubMed] [Google Scholar]
- Ho, C.‐S. , Jean, N. , Hogan, C.A. , Blackmon, L. , Jeffrey, S.S. , Holodniy, M. , et al. (2019) Rapid identification of pathogenic bacteria using Raman spectroscopy and deep learning. Nat Commun 10: 4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, Y. , Anes, J. , Devineau, S. , and Fanning, S. (2021) Klebsiella pneumoniae: prevalence, reservoirs, antimicrobial resistance, pathogenicity, and infection: a hitherto unrecognized zoonotic bacterium. Foodborne Pathog Dis 18: 63–84. [DOI] [PubMed] [Google Scholar]
- Li, Y. , Li, J. , Hu, T. , Hu, J. , Song, N. , Zhang, Y. , and Chen, Y. (2020) Five‐year change of prevalence and risk factors for infection and mortality of carbapenem‐resistant Klebsiella pneumoniae bloodstream infection in a tertiary hospital in North China. Antimicrob Resist Infect Control 9: 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, B. , Yi, H. , Fang, J. , Han, L. , Zhou, M. , and Guo, Y. (2019) Antimicrobial resistance and risk factors for mortality of pneumonia caused by Klebsiella pneumoniae among diabetics: a retrospective study conducted in Shanghai, China. Infect Drug Resist 12: 1089–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y.‐Y. , Wang, Y. , Walsh, T.R. , Yi, L.‐X. , Zhang, R. , Spencer, J. , et al. (2016) Emergence of plasmid‐mediated colistin resistance mechanism mcr‐1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis 16: 161–168. [DOI] [PubMed] [Google Scholar]
- Lu, J. , Dong, N. , Liu, C. , Zeng, Y.U. , Sun, Q. , Zhou, H. , et al. (2020a) Prevalence and molecular epidemiology of mcr‐1‐positive Klebsiella pneumoniae in healthy adults from China. J Antimicrob Chemother 75: 2485–2494. [DOI] [PubMed] [Google Scholar]
- Lu, W. , Chen, X. , Wang, L. , Li, H. , and Fu, Y.V. (2020b) Combination of an artificial intelligence approach and laser Tweezers Raman Spectroscopy for microbial identification. Anal Chem 92: 6288–6296. [DOI] [PubMed] [Google Scholar]
- Magill, S.S. , Edwards, J.R. , Bamberg, W. , Beldavs, Z.G. , Dumyati, G. , Kainer, M.A. , et al. (2014) Multistate point‐prevalence survey of health care‐associated infections. N Engl J Med 370: 1198–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynenko, I.V. , Kusić, D. , Weigert, F. , Stafford, S. , Donnelly, F.C. , Evstigneev, R. , et al. (2019) Magneto‐fluorescent microbeads for bacteria detection constructed from superparamagnetic FeO nanoparticles and AIS/ZnS quantum dots. Anal Chem 91: 12661–12669. [DOI] [PubMed] [Google Scholar]
- Navon‐Venezia, S. , Kondratyeva, K. , and Carattoli, A. (2017) Klebsiella pneumoniae: a major worldwide source and shuttle for antibiotic resistance. FEMS Microbiol Rev 41: 252–275. [DOI] [PubMed] [Google Scholar]
- Nordmann, P. , and Poirel, L. (2014) The difficult‐to‐control spread of carbapenemase producers among Enterobacteriaceae worldwide. Clin Microbiol Infect 20: 821–830. [DOI] [PubMed] [Google Scholar]
- Notingher, I. , and Hench, L.L. (2006) Raman microspectroscopy: a noninvasive tool for studies of individual living cells in vitro. Expert Rev Med Devices 3: 215–234. [DOI] [PubMed] [Google Scholar]
- Pahlow, S. , Meisel, S. , Cialla‐May, D. , Weber, K. , Rösch, P. , and Popp, J. (2015) Isolation and identification of bacteria by means of Raman spectroscopy. Adv Drug Deliv Rev 89: 105–120. [DOI] [PubMed] [Google Scholar]
- Rebrošová, K. , Šiler, M. , Samek, O. , Růžička, F. , Bernatová, S. , Ježek, J. , et al. (2017) Differentiation between Staphylococcus aureus and Staphylococcus epidermidis strains using Raman spectroscopy. Future Microbiol 12: 881–890. [DOI] [PubMed] [Google Scholar]
- Rhodes, A. , Evans, L.E. , Alhazzani, W. , Levy, M.M. , Antonelli, M. , Ferrer, R. , et al. (2017) Surviving Sepsis Campaign: International guidelines for management of sepsis and septic shock: 2016. Intensive Care Med 43: 304–377. [DOI] [PubMed] [Google Scholar]
- Russo, T.A. , and Marr, C.M. (2019) Hypervirulent Klebsiella pneumoniae . Clin Microbiol Rev 32: e00001‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadesse, L.F. , Safir, F. , Ho, C.‐S. , Hasbach, X. , Khuri‐Yakub, B.P. , Jeffrey, S.S. , et al. (2020) Toward rapid infectious disease diagnosis with advances in surface‐enhanced Raman spectroscopy. J Chem Phys 152: 240902. [DOI] [PubMed] [Google Scholar]
- Teng, L. , Wang, X. , Wang, X. , Gou, H. , Ren, L. , Wang, T. , et al. (2016) Label‐free, rapid and quantitative phenotyping of stress response in E. coli via ramanome. Sci Rep 6: 34359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uysal Ciloglu, F. , Saridag, A.M. , Kilic, I.H. , Tokmakci, M. , Kahraman, M. , and Aydin, O. (2020) Identification of methicillin‐resistant Staphylococcus aureus bacteria using surface‐enhanced Raman spectroscopy and machine learning techniques. Analyst 145: 7559–7570. [DOI] [PubMed] [Google Scholar]
- Wang, Y. , and Navin, N.E. (2015) Advances and applications of single‐cell sequencing technologies. Mol Cell 58: 598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Guo, L.‐Y. , Song, W.‐Q. , Wang, Y. , Dong, F. , and Liu, G. (2018) Risk factors for carbapenem‐resistant K. pneumoniae bloodstream infection and predictors of mortality in Chinese paediatric patients. BMC Infect Dis 18: 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, B. , Xu, H. , Lv, T. , Guo, L. , Xiao, Y.U. , Huang, C. , et al. (2020) Stool samples of acute diarrhea inpatients as a reservoir of ST11 hypervirulent KPC‐2‐producing Klebsiella pneumoniae . mSystems 5: e00498‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. 2‐D‐scores plots of principle component relation (PC1and PC2) for three K. pneumoniae strains (270, 104, R210). The dots in different color and shapes represent the spectra gathered in different measurement times.
Fig S2. The model was created at each epoch to select the most optimal epoch. The accuracy of the models did not increase significantly after 20 epochs training.
Table S1 Sensitivity, specificity and accuracy of the CNN models and the ML models in three identification tasks.