

Abstract
Background
Inhaled corticosteroids (ICS) are anti‐inflammatory drugs that have proven benefits for people with worsening symptoms of chronic obstructive pulmonary disease (COPD) and repeated exacerbations. They are commonly used as combination inhalers with long‐acting beta2‐agonists (LABA) to reduce exacerbation rates and all‐cause mortality, and to improve lung function and quality of life. The most common combinations of ICS and LABA used in combination inhalers are fluticasone and salmeterol, budesonide and formoterol and a new formulation of fluticasone in combination with vilanterol, which is now available. ICS have been associated with increased risk of pneumonia, but the magnitude of risk and how this compares with different ICS remain unclear. Recent reviews conducted to address their safety have not compared the relative safety of these two drugs when used alone or in combination with LABA.
Objectives
To assess the risk of pneumonia associated with the use of fluticasone and budesonide for COPD.
Search methods
We identified trials from the Cochrane Airways Group Specialised Register of trials (CAGR), clinicaltrials.gov, reference lists of existing systematic reviews and manufacturer websites. The most recent searches were conducted in September 2013.
Selection criteria
We included parallel‐group randomised controlled trials (RCTs) of at least 12 weeks' duration. Studies were included if they compared the ICS budesonide or fluticasone versus placebo, or either ICS in combination with a LABA versus the same LABA as monotherapy for people with COPD.
Data collection and analysis
Two review authors independently extracted study characteristics, numerical data and risk of bias information for each included study.
We looked at direct comparisons of ICS versus placebo separately from comparisons of ICS/LABA versus LABA for all outcomes, and we combined these with subgroups when no important heterogeneity was noted. After assessing for transitivity, we conducted an indirect comparison to compare budesonide versus fluticasone monotherapy, but we could not do the same for the combination therapies because of systematic differences between the budesonide and fluticasone combination data sets.
When appropriate, we explored the effects of ICS dose, duration of ICS therapy and baseline severity on the primary outcome. Findings of all outcomes are presented in 'Summary of findings' tables using GRADEPro.
Main results
We found 43 studies that met the inclusion criteria, and more evidence was provided for fluticasone (26 studies; n = 21,247) than for budesonide (17 studies; n = 10,150). Evidence from the budesonide studies was more inconsistent and less precise, and the studies were shorter. The populations within studies were more often male with a mean age of around 63, mean pack‐years smoked over 40 and mean predicted forced expiratory volume of one second (FEV1) less than 50%.
High or uneven dropout was considered a high risk of bias in almost 40% of the trials, but conclusions for the primary outcome did not change when the trials at high risk of bias were removed in a sensitivity analysis.
Fluticasone increased non‐fatal serious adverse pneumonia events (requiring hospital admission) (odds ratio (OR) 1.78, 95% confidence interval (CI) 1.50 to 2.12; 18 more per 1000 treated over 18 months; high quality), and no evidence suggested that this outcome was reduced by delivering it in combination with salmeterol or vilanterol (subgroup differences: I2 = 0%, P value 0.51), or that different doses, trial duration or baseline severity significantly affected the estimate. Budesonide also increased non‐fatal serious adverse pneumonia events compared with placebo, but the effect was less precise and was based on shorter trials (OR 1.62, 95% CI 1.00 to 2.62; six more per 1000 treated over nine months; moderate quality). Some of the variation in the budesonide data could be explained by a significant difference between the two commonly used doses: 640 mcg was associated with a larger effect than 320 mcg relative to placebo (subgroup differences: I2 = 74%, P value 0.05).
An indirect comparison of budesonide versus fluticasone monotherapy revealed no significant differences with respect to serious adverse events (pneumonia‐related or all‐cause) or mortality. The risk of any pneumonia event (i.e. less serious cases treated in the community) was higher with fluticasone than with budesonide (OR 1.86, 95% CI 1.04 to 3.34); this was the only significant difference reported between the two drugs. However, this finding should be interpreted with caution because of possible differences in the assignment of pneumonia diagnosis, and because no trials directly compared the two drugs.
No significant difference in overall mortality rates was observed between either of the inhaled steroids and the control interventions (both high‐quality evidence), and pneumonia‐related deaths were too rare to permit conclusions to be drawn.
Authors' conclusions
Budesonide and fluticasone, delivered alone or in combination with a LABA, are associated with increased risk of serious adverse pneumonia events, but neither significantly affected mortality compared with controls. The safety concerns highlighted in this review should be balanced with recent cohort data and established randomised evidence of efficacy regarding exacerbations and quality of life. Comparison of the two drugs revealed no statistically significant difference in serious pneumonias, mortality or serious adverse events. Fluticasone was associated with higher risk of any pneumonia when compared with budesonide (i.e. less serious cases dealt with in the community), but variation in the definitions used by the respective manufacturers is a potential confounding factor in their comparison.
Primary research should accurately measure pneumonia outcomes and should clarify both the definition and the method of diagnosis used, especially for new formulations and combinations for which little evidence of the associated pneumonia risk is currently available. Similarly, systematic reviews and cohorts should address the reliability of assigning 'pneumonia' as an adverse event or cause of death and should determine how this affects the applicability of findings.
Plain language summary
Do inhaled steroids increase the risk of pneumonia in people with chronic obstructive pulmonary disease (COPD)?
Why is this question important? Inhaled corticosteroids (ICS) are drugs that can reduce the occurrence of COPD flare‐ups and improve quality of life. In COPD, ICS are commonly used alongside long‐acting beta2‐agonists (LABA). The most common combinations of ICS and LABA inhalers are fluticasone and salmeterol, and budesonide and formoterol, but fluticasone furoate is also used once daily with a new LABA called vilanterol. Lots of studies have shown benefits of ICS, but they can also increase the risk of pneumonia. Added to this concern, pneumonia can be difficult to diagnose, and the severity of pneumonia can be poorly reported in trials. Therefore even though we have reviews on inhaled steroids for COPD, we wanted to do a review exclusively on pneumonia, so we could take a closer look at the evidence.
The overall aim of this review is to assess the risk of pneumonia for people with COPD taking fluticasone or budesonide.
How did we answer the question? We looked for all studies comparing budesonide or fluticasone versus a dummy inhaler (placebo), and all studies comparing their use in combination with a LABA (i.e. budesonide/formoterol, fluticasone propionate/salmeterol, and fluticasone furoate/vilanterol) versus the same dose of LABA alone. This allowed us to assess the risk of ICS used alone or in combination with LABA.
What did we find? We found 43 studies including more than 30,000 people with COPD. More studies used fluticasone (26 studies; 21,247 people) than budesonide (17 studies; 10,150 people). A higher proportion of people in the studies were male (around 70%), and their COPD was generally classed as severe. The last search for studies to include in the review was done in September 2013.
We compared each drug against controls and assessed separately the results of studies that compared ICS versus placebo, and an ICS/LABA combination versus LABA alone. We also conducted an indirect comparison of budesonide and fluticasone based on their effects against placebo, to explore whether one drug was safer than the other.
Fluticasone increased 'serious' pneumonias (requiring hospital admission). Over 18 months, 18 more people of every 1000 treated with fluticasone were admitted to hospital for pneumonia.
Budesonide also increased pneumonias that were classed as 'serious'. Over nine months, six more hospital admissions were reported for every 1000 individuals treated with budesonide. A lower dose of budesonide (320 mcg) was associated with fewer serious pneumonias than a higher dose (640 mcg).
No more deaths overall were reported in the ICS groups compared with controls, and deaths related to pneumonia were too rare to tell either way.
When we compared fluticasone and budesonide versus each other, the difference between them was not clear enough to tell whether one was safer (for pneumonia, requiring a hospital stay, general adverse events and death). The risk of any pneumonia event (i.e. less serious cases that could be treated without going to hospital) was higher with fluticasone than with budesonide.
Evidence was rated to be of high or moderate quality for most outcomes. When an outcome is rated of high quality, further research is very unlikely to change our confidence in the estimate of effect, but moderate ratings reflect some uncertainty in the findings. Results from the budesonide studies were generally less clear because they were based on fewer people, and the studies were shorter.
Conclusion Budesonide and fluticasone, delivered alone or in combination with LABA, can increase serious pneumonias that result in hospitalisation of people. Neither has been shown to affect the chance of dying compared with not taking ICS. Comparison of the two drugs revealed no difference in serious pneumonias or risk of death. Fluticasone was associated with a higher risk of any pneumonia (i.e. cases that could be treated in the community) than budesonide, but potential differences in the definition used by the respective drug manufacturers reduced our confidence in this finding. These concerns need to be balanced with the known benefits of ICS (e.g. fewer exacerbations, improved lung function and quality of life).
Researchers should remain aware of the risks associated with ICS and should make sure that pneumonia is properly diagnosed in studies.
Summary of findings
Summary of findings for the main comparison. Fluticasone for chronic obstructive pulmonary disease.
| Fluticasone for chronic obstructive pulmonary disease | |||||
|
Patient or population: patients with chronic obstructive pulmonary disease
Intervention: fluticasone (alone or with LABA co‐intervention) Comparison: placebo or LABA monotherapy (dependent upon whether fluticasone was given with LABA in the intervention group) Setting: community | |||||
|
Outcomes Follow‐ups presented as weighted means |
Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| Control | Fluticasone | ||||
| Non‐fatal, serious adverse pneumonia events (requiring hospital admission) Follow‐up: 18 months | 25 per 1000 | 43 per 1000 (37 to 51) | OR 1.78 (1.50 to 2.12) | 19,504 (17 studies) | ⊕⊕⊕⊕ high |
| Mortality, all‐cause Follow‐up: 19 months | 58 per 1000 | 58 per 1000 (51 to 65) | OR 0.99 (0.87 to 1.13) | 20,861 (22 studies) | ⊕⊕⊕⊕ high |
| Mortality, due to pneumonia Follow‐up: 18 months | 2 per 1000 | 3 per 1000 (2 to 5) | OR 1.23 (0.70 to 2.15) | 19,532 (18 studies) | ⊕⊕⊕⊝ moderate1 |
| Non‐fatal, serious adverse events (all) Follow‐up: 19 months | 227 per 1000 | 237 per 1000 (225 to 251) | OR 1.06 (0.99 to 1.14) | 20,381 (19 studies) | ⊕⊕⊕⊕ high |
| All pneumonia events Follow‐up: 22 months | 72 per 1000 | 116 per 1000 (104 to 129) | OR 1.68 (1.49 to 1.90) | 15,377 (11 studies) | ⊕⊕⊕⊝ moderate2 |
| Withdrawals Follow‐up: 18 months | 343 per 1000 | 297 per 1000 (286 to 310) | OR 0.81 (0.77 to 0.86) | 21,243 (26 studies) | ⊕⊕⊕⊕ high |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI).
CI: Confidence interval; OR: Odds ratio. Unless otherwise stated, subgroup differences between monotherapy studies (fluticasone versus placebo) and combination therapy studies (fluticasone/LABA versus LABA) were not significant. | |||||
| GRADE Working Group grades of evidence. High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1Wide confidence intervals include significant benefit and harm, based on very few events (‐1 for imprecision). 2More than half the studies did not report the outcome (‐1 for publication bias).
Summary of findings 2. Budesonide for chronic obstructive pulmonary disease.
| Budesonide for chronic obstructive pulmonary disease | |||||
|
Patient or population: patients with chronic obstructive pulmonary disease
Intervention: budesonide (alone or with LABA co‐intervention) Comparison: placebo or LABA monotherapy (dependent upon whether fluticasone was given with LABA in the intervention group) Setting: community | |||||
|
Outcomes Follow‐ups presented as weighted means |
Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| Control | Budesonide | ||||
| Non‐fatal, serious adverse pneumonia events (requiring hospital admission) Follow‐up: 9 months | 9 per 1000 | 15 per 1000 (9 to 24) | OR 1.62 (1.00 to 2.62) | 6472 (7 studies) | ⊕⊕⊕⊝ moderate1,2 |
| Mortality, all‐cause Follow‐up: 14 months | 17 per 1000 | 16 per 1000 (11 to 21) | OR 0.90 (0.65 to 1.24) | 10,009 (12 studies) | ⊕⊕⊕⊝ moderate3 |
| Mortality, due to pneumonia Follow‐up: 12 months | 0 per 1000 | 0 per 1000 (0 to 0) | OR 4.46 (0.07 to 286.99) | 1511 (3 studies) | ⊕⊝⊝⊝ very low2,4 |
| Non‐fatal, serious adverse events (all) Follow‐up: 14 months | 145 per 1000 | 146 per 1000 (124 to 172) | OR 1.01 (0.83 to 1.22) | 10,009 (12 studies) | ⊕⊕⊕⊝ moderate5 |
| All pneumonia events Follow‐up: 10 months | 28 per 1000 | 31 per 1000 (23 to 41) | OR 1.12 (0.83 to 1.51) | 7011 (6 studies) | ⊕⊕⊕⊝ moderate1,2 |
| Withdrawals Follow‐up: 14 months | 280 per 1000 | 232 per 1000 (216 to 248) | OR 0.78 (0.71 to 0.85) | 10150 (15 studies) | ⊕⊕⊕⊕ high |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI).
CI: Confidence interval; OR: Odds ratio. Unless otherwise stated, subgroup differences between monotherapy studies (budesonide versus placebo) and combination therapy studies (budesonide/LABA versus LABA) were not significant. | |||||
| GRADE Working Group grades of evidence. High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1Confidence intervals are quite wide but are not considered serious enough to downgrade. 2More than half the studies did not report the outcome (‐1 for publication bias). 3Confidence interval includes significant benefit and potential harm. 4Very wide confidence intervals. Only one death observed over the three studies (‐2 for imprecision). 5I2 = 59%, P value 0.002 (‐1 for inconsistency).
Background
Description of the condition
Chronic obstructive pulmonary disease (COPD) is a respiratory disease characterised by chronic and progressive breathlessness, cough, sputum production and airflow obstruction, which leads to restricted activity and poor quality of life (GOLD 2013). The World Health Organization (WHO 2012) has estimated that COPD is the fourth or fifth most common single cause of death worldwide, and that the treatment and management costs present a significant burden to public health. In the UK the annual cost of COPD to the National Health Service (NHS) is estimated to be GBP 1.3 million per 100,000 people (NICE 2011). Furthermore, because of its slow onset and under‐recognition of the disease by patients and healthcare professionals, COPD is heavily under diagnosed (GOLD 2013). COPD comprises a combination of bronchitis and emphysema and involves chronic inflammation and structural changes in the lung. Cigarette smoking is the most important risk factor, but air pollution and occupational dust and chemicals are also recognised risk factors. COPD is a progressive disease that leads to decreased lung function over time, even with the best available care. Currently no cure is known for COPD, although the condition is both preventable and treatable. As yet, apart from smoking cessation and non‐pharmacological treatments such as long‐term oxygen therapy in hypoxic patients and pulmonary rehabilitation, no intervention has been shown to reduce mortality (GOLD 2013;Puhan 2011). Management of the disease is multi‐faceted and includes interventions for smoking cessation (Van der Meer 2001), pharmacological treatments (GOLD 2013), education (Effing 2007) and pulmonary rehabilitation (Lacasse 2006; Puhan 2011). Pharmacological therapy is aimed at relieving symptoms, improving exercise tolerance and quality of life, improving lung function and preventing and treating exacerbations.
Description of the intervention
Pharmacological management for COPD is generally a stepwise process, commencing with therapy for symptoms, which is followed by introduction of additional therapeutic agents as needed to achieve control and to reduce the frequency and severity of exacerbations (GOLD 2013). Often the first step is to use a short‐acting bronchodilator for control of breathlessness when needed: a short‐acting beta2‐agonist (SABA) (e.g. salbutamol) or the short‐acting muscarinic antagonist (SAMA) ipratropium. For persistent or worsening breathlessness associated with lung function decline, long‐acting bronchodilators may be introduced (GOLD 2013). These comprise twice‐daily long‐acting beta2‐agonists (LABA), such as salmeterol or formoterol; once‐daily beta2‐agonists, such as indacaterol; and the long‐acting anticholinergic agent tiotropium. For patients with severe or very severe COPD (forced expiratory volume in one second (FEV1) < 50% predicted) and with repeated exacerbations, the Global initiative for chronic Obstructive Lung Disease (GOLD 2013) recommends the addition of inhaled corticosteroids (ICS) to bronchodilator treatment. ICS are anti‐inflammatory drugs that are licensed as combination inhalers for use with LABA. The most common ICS and LABA components in combination inhalers are fluticasone propionate and salmeterol, budesonide and formoterol and a new formulation of fluticasone furoate in combination with vilanterol, which is now available for once‐daily use. Patients with severe COPD may also be treated with the phosphodiesterase 4 (PDE4) inhibitor roflumilast, which may reduce the risk of exacerbations (GOLD).
How the intervention might work
ICS are anti‐inflammatory drugs. They reduce the rate of exacerbation and improve quality of life, but they have not been found to have an effect on overall mortality or on the long‐term decline in FEV1 (Agarwal 2010; GOLD 2013; Yang 2009). ICS and LABA combination inhalers reduce exacerbation rates and all‐cause mortality and improve lung function and quality of life (Nannini 2013). These effects are thought to be greater for combination inhalers than for the component preparations (GOLD 2013; Nannini 2013). ICS, alone or in combination with LABA, however, have been associated with increased risk of pneumonia (GOLD 2013; Singh 2009). Several mechanisms have been proposed by which ICS could increase the risk of pneumonia; these mechanisms are principally related to the immunosuppressive effects of ICS and include ICS reaching the lung in high concentrations. Particularly, inhibition of nuclear factor kappa B (NF‐κB) by ICS in COPD, one of the proposed mechanisms for their therapeutic effect, could lead to the suppression of normal host responses to bacterial infection (Singanayagam 2010).
Why it is important to do this review
Use of ICS for treatment of COPD may be beneficial, at least for some COPD patients. But the role of ICS therapy in patients with stable COPD is controversial, especially as an elevated risk of pneumonia has been found in studies of ICS use. Pneumonia in COPD is associated with high morbidity and mortality (Ernst 2007) and worsening quality of life and pulmonary function, so it is important to understand the strength and nature of the association between ICS use and this adverse event.
Several systematic reviews published in the last few years have looked at the risk of pneumonia with ICS use (Drummond 2008; Halpin 2011; Sin 2009; Singh 2009). Of these, only one compared different ICS versus each other and as combination inhaler therapy together with a LABA (Halpin 2011). Although ICS are usually administered in a combination inhaler in clinical practice, we are interested in the most comprehensive evidence on the risk of pneumonia with ICS. Differences in the molecular structures of ICS formulations are known to alter their relative potency ratios and durations of action (Johnson 1998; Rossios 2011), but potential differences between formulations in the magnitude of pneumonia risk remains unclear. It is also uncertain whether the association with pneumonia is altered by LABA in combination inhalers. We therefore included studies that examined ICS treatment both alone and in combination with a LABA. We focused on the risk of pneumonia with the two most frequently prescribed ICS—fluticasone and budesonide—compared with control, and on the difference in risk of pneumonia between these ICS. When there was a paucity of head‐to‐head trials directly comparing fluticasone and budesonide, we planned to complement the direct comparisons with an adjusted indirect comparison of budesonide and fluticasone using placebo as a common comparator (Figure 1). Indirect comparisons are considered valid if 'clinical and methodological homogeneity' is present between the budesonide and fluticasone studies (Cipriani 2013). The indirect comparison of budesonide and fluticasone when taken in a combination inhaler with different LABA assumes that the LABA salmeterol, formoterol and vilanterol do not have an important effect on the risk of pneumonia.
1.
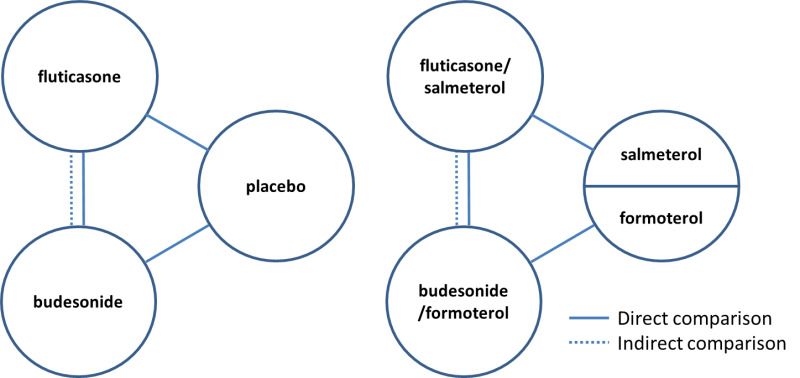
Direct and indirect comparisons of fluticasone and budesonide covered in the review.
Objectives
To assess the risk of pneumonia associated with the use of fluticasone and budesonide for COPD.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs) with a parallel‐group design of at least 12 weeks' duration. We did not exclude studies on the basis of blinding. Cross‐over trials were not included, as ICS can have long‐acting effects, and because the primary outcome is an adverse event.
Types of participants
We included RCTs that recruited participants with a diagnosis of COPD (e.g. based on criteria recommended by the American Thoracic Society and the European Respiratory Society) (ATS/ERS 2004).
Forced expiratory volume after one second (FEV1)/forced vital capacity (FVC) ratio < 0.7, which confirms the presence of persistent airflow limitation.
-
One or more of the following key indicators.
Progressive and/or persistent dyspnoea.
Chronic cough.
Chronic sputum production.
History of exposure to risk factors (tobacco smoke, smoke from home cooking and heating fuels, occupational dusts and chemicals).
Types of interventions
We included studies that performed any of the following comparisons.
Fluticasone versus placebo.
Budesonide versus placebo.
Fluticasone/salmeterol versus salmeterol.
Fluticasone/vilanterol versus vilanterol.
Budesonide/formoterol versus formoterol.
Fluticasone versus budesonide.
Fluticasone/salmeterol versus budesonide/formoterol.
Fluticasone/vilanterol versus budesonide/formoterol.
We allowed ICS/LABA combination treatment in a single inhaler and in separate inhalers. Participants were allowed to take other concomitant COPD medications as prescribed by their healthcare practitioner provided they were not part of the trial treatment under study. For example, we excluded studies that compared triple therapy of budesonide/formoterol combination inhaler plus tiotropium versus formoterol plus tiotropium.
Types of outcome measures
We were interested in events of pneumonia. Pneumonia is usually defined as an acute lower respiratory tract infection that generally includes symptoms and signs from the respiratory tract and noted in the general health of the patient, but the specific definition/diagnosis varies. We recorded the basis of diagnosis, specifically, radiological confirmation, and planned to conduct a subgroup analysis. One example of the definition of diagnostic criteria for pneumonia is found in BTS 2009.
Symptoms of an acute lower respiratory tract illness (cough and at least one other lower respiratory tract symptom).
New focal chest signs on examination.
At least one systemic feature (either a symptom complex of sweating, fevers, shivers, aches and pains and/or temperature of 38°C or higher).
No other explanation for the illness.
We primarily looked at pneumonia events leading to hospital admissions (i.e. serious adverse pneumonia events), which usually are better documented and diagnosed by imaging studies and laboratory investigations than pneumonia events of any severity, and are associated with substantial morbidity and mortality. One example of the definition of diagnostic criteria for pneumonia in hospital is found in BTS 2009.
Symptoms and signs consistent with an acute lower respiratory tract infection associated with new radiographic shadowing for which no other explanation is known (e.g. not pulmonary oedema or infarction).
The illness is the primary reason for hospital admission and is managed as pneumonia.
We used end of study as the time of analysis for all studies, which ranged from three to 36 months in duration.
Primary outcomes
Non‐fatal, serious adverse pneumonia events (requiring hospital admission).
We chose serious adverse pneumonia events as the primary outcome because of the increased burden these events have on the individual and on healthcare systems.
Secondary outcomes
Mortality: all‐cause and due to pneumonia.
Non‐fatal serious adverse events: all‐cause.
All pneumonia events.
Withdrawals.
Search methods for identification of studies
Electronic searches
We identified trials from the Cochrane Airways Group Specialised Register of trials (CAGR), which is maintained by the Trials Search Co‐ordinator for the Group. The Register contains trial reports identified by systematic searches of bibliographic databases including the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, EMBASE, CINAHL, AMED and PsycINFO, and conference abstracts found through handsearching (see Appendix 1 for further details). We searched all records in the CAGR coded 'COPD' using the following terms.
((steroid* or corticosteroid*) and inhal*) or ICS or budesonide or fluticasone or pulmicort or flovent or flixotide or symbicort or viani or seretide or advair.
We also searched ClinicalTrials.gov using search terms provided in Appendix 2. We searched all databases with no restriction on date or language of publication up to September 2013.
Searching other resources
We checked reference lists of all primary studies and review articles for additional references. We searched the manufacturers' websites (AstraZeneca and GlaxoSmithKline) for additional information on studies identified through the electronic searches.
Data collection and analysis
Selection of studies
Two review authors (AS and CK) independently screened the titles and abstracts of citations retrieved through literature searches and obtained those deemed to be potentially relevant. We assigned all references to a study identifier and assessed them against the inclusion criteria of this protocol. We resolved disagreements by consensus. Subsequent search updates were screened by AS and KMK.
Data extraction and management
Two review authors (KMK and AS or CK) independently extracted information from each included study (recording the data source) for the following characteristics.
Design (study design, total duration of study, number of study centres and locations).
Participants (number randomly assigned to each treatment, mean age, gender, baseline lung function, smoking history, inclusion criteria, exclusion criteria).
Interventions (run‐in, intervention and control treatment including concentration and formulation).
Outcomes (definitions of pneumonia events and data on the numbers of participants with one or more events with onset during the treatment period).
We resolved discrepancies in the data by discussion, or by consultation with a third party when necessary.
Assessment of risk of bias in included studies
We assessed the risk of bias according to recommendations outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) for the following items.
Random sequence generation.
Allocation concealment.
Blinding of participants and personnel.
Blinding of outcome assessment.
Incomplete outcome data.
Selective outcome reporting.
We graded each potential source of bias as high, low or unclear according to recommendations outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
Measures of treatment effect
Direct comparisons (fluticasone vs placebo; budesonide vs placebo; fluticasone/LABA vs LABA; budesonide/LABA vs LABA)
We analysed direct pair‐wise comparisons using Mantel‐Haenszel odds ratios (ORs) with 95% confidence intervals (CIs). When events were rare, we employed the Peto odds ratio. When count data were available as rate ratios, we transformed them into log rate ratios and analysed them using generic inverse variance (GIV). For the primary outcome, the number needed to treat for an additional harmful outcome was calculated from the pooled odds ratio and its confidence interval and was applied to appropriate levels of baseline risk.
Indirect comparisons (monotherapy: fluticasone vs budesonide; combination therapy: fluticasone/LABA vs budesonide/LABA)
We also conducted indirect comparisons of fluticasone and budesonide treatments using odds ratios with a 95% CI (Bucher 1997). When available, we planned to combine the indirect evidence with randomly assigned head‐to‐head comparisons of fluticasone and budesonide.
Assessing transitivity and similarity
To permit valid indirect comparisons of fluticasone and budesonide, the sets of trials for each drug must be similar in their distribution of effect modifiers (Cipriani 2013). Before conducting indirect comparisons, we constructed summary tables for monotherapy and combination therapy separately to compare the following characteristics between budesonide and fluticasone trials.
Inclusion and exclusion criteria (including allowed co‐medications).
Baseline characteristics (smoking history, % predicted FEV1, age, percentage male).
Intervention characteristics (dose distribution, inhaler device).
Methodology (risk of bias, study duration, sample size, funding).
Control group event rates.
Unit of analysis issues
The unit of analysis was the individual participant for dichotomous outcomes, but events were used to compare rates of exacerbation.
Dealing with missing data
When pneumonia data or key study characteristics were not reported in the primary publication, we searched clinical trial reports and contacted study authors and sponsors for additional information. We used intention‐to‐treat (ITT) analysis on outcomes of all randomly assigned participants when possible. We considered the impact of the unknown status of participants who withdrew from the trials as part of the sensitivity analysis.
Assessment of heterogeneity
We assessed clinical and methodological heterogeneity by recording differences in study design, participant characteristics, study sponsorship and pneumonia definition between individual studies. We assessed the extent of statistical variation among study results by using the I2 measurement. We tested for inconsistency between direct and indirect data by calculating the log ratio of direct and indirect odds ratios (ROR) (Song 2008).
Assessment of reporting biases
We tried to minimise reporting bias from non‐publication of studies or selective outcome reporting by using a broad search strategy, checking references of included studies and relevant systematic reviews and contacting study authors for additional outcome data. We visually inspected funnel plots when 10 or more studies were included.
Data synthesis
We looked at direct comparisons of ICS versus placebo separately from comparisons of ICS/LABA versus LABA for all outcomes (Figure 1). When no important discrepancy was noted between the analyses with and without LABA, we combined the results. When a study comparing ICS/LABA versus LABA included arms for both a single inhaler (ICS/LABA) and separate inhalers (ICS + LABA), we split the control group (LABA) in half to avoid double‐counting.
The decision whether to perform indirect comparisons of studies was based on our assessment of their clinical and methodological differences.
If both direct and indirect comparison data were available, we planned to combine the estimates using a fixed‐effect model, but when statistical heterogeneity was evident (I2 > 30%), we used a random‐effects model to analyse the data and explore the heterogeneity (see below). When no important discrepancy was noted between direct and indirect estimates, we combined the resulting odds ratio and 95% CI from the indirect comparison with any direct pair‐wise data for the same comparison using inverse variance weighting (Glenny 2005).
We presented the findings of all outcomes in 'Summary of findings' tables using GRADEPro software and recommendations provided in the Cochrane Handbook for Systematic Reviews of Interventions.
Subgroup analysis and investigation of heterogeneity
When appropriate, we explored heterogeneity between studies by analysing data for the primary outcome by looking at the following subgroups.
ICS dose (separate subgroups for each of the following drugs and doses: fluticasone 500 and 1000 mcg; budesonide 320, 640 and 1280 mcg).
Duration of ICS therapy (≤ one year; > one year).
Diagnostic criteria of pneumonia.
Disease severity at baseline (FEV1 < 50% predicted; FEV1 ≥ 50% predicted).
Sensitivity analysis
We assessed the robustness of our analyses by performing sensitivity analyses, while systematically excluding studies from the overall analysis:
of high risk of bias; or
with high and or uneven withdrawal rates.
Results
Description of studies
Results of the search
Two thousand forty‐seven citations were identified by searching electronic databases. Twenty‐seven additional citations were found by searching reference lists, clinicaltrials.gov and drug company websites. Forty‐two duplicates were removed, and the remaining 2032 titles and abstracts were sifted. Two review authors excluded 1806 references that did not meet the inclusion criteria. Full texts were obtained and scrutinised for the final 226 references, and 195 (representing 43 studies) met all of the inclusion criteria. Figure 2 shows this information as a flow diagram and gives reasons for exclusion of the 31 references that were excluded after the full text was reviewed.
2.
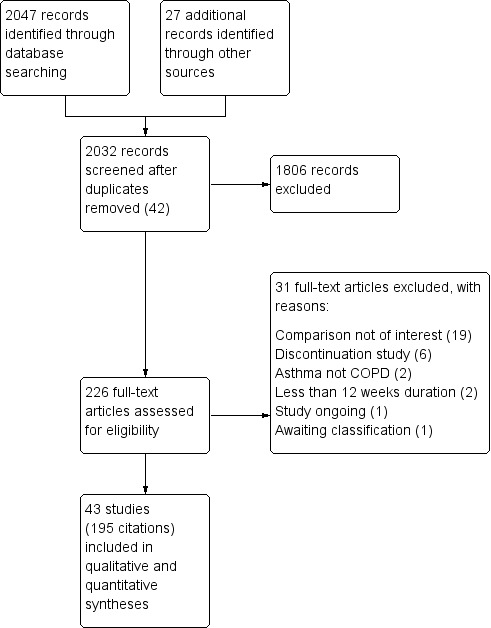
Study flow diagram.
Included studies
Forty‐three studies met all of the inclusion criteria and were included in the review: 26 using fluticasone and 17 using budesonide as the inhaled steroid. The fluticasone studies included more than twice as many people, with 21,247 people randomly assigned to the treatments of interest compared with 10,067 in the budesonide studies. Two of the included budesonide studies reported no data that could be used in the analyses and are not included in these numbers (Laptseva 2002; Senderovitz 1999).
No studies directly comparing fluticasone with budesonide met the inclusion criteria (either as monotherapy or in their combination preparations), so only indirect evidence was available for these comparisons.
Design and duration
All studies were randomised, double‐blind, parallel‐group trials of at least 12 weeks' duration. Most were funded by pharmaceutical companies, predominantly GlaxoSmithKline for the fluticasone studies and AstraZeneca for the budesonide studies. Duration ranged from three to 36 months for both drugs, but mean duration weighted by sample size was longer for the fluticasone studies (fluticasone, 18 months; budesonide, 14 months). A summary of each study and baseline characteristics can be found in Table 3 and Table 4.
1. Fluticasone—summary of studies and baseline characteristics.
| Study ID | Duration (m) | N Rand | Funder | ICS dose (mcg) | % Male | Mean age | Pack‐years | % pred FEV1 |
| Fluticasone versus placebo (n = 18) | ||||||||
| Bourbeau 2007 | 3 | 41 | GSK | 1000 | 78 | 65 | 53 | 57 |
| Burge 2000 | 36 | 740 | GSK | 1000 | 75 | 64 | 44 | 50 |
| Calverley 2003 TRISTANa | 12 | 763 | GSK | 1000 | 73 | 63 | 43 | 45 |
| Calverley 2007 TORCHa | 36 | 3097 | GSK | 1000 | 76 | 65 | 49 | 44 |
| Choudhury 2005 | 12 | 260 | Indep. | 1000 | 52 | 67 | 39 | 54 |
| GSK FLTA3025 2005 | 6 | 640 | GSK | 500, 1000 | 69 | 64 | ‐ | ‐ |
| GSK SCO104925 2008a | 3 | 84 | GSK | 1000 | 77 | 64 | ‐ | ‐ |
| GSK SCO30002 2005 | 12 | 256 | GSK | 1000 | 82 | 65 | ‐ | ‐ |
| Hanania 2003a | 6 | 368 | GSK | 500 | 63 | 64 | 57 | 42 |
| Hattotuwa 2002 | 3 | 36 | GSK | 1000 | 87 | 65 | 63 | 46 |
| Kerwin 2013a,b | 6 | 413 | GSK | 100 | 66 | 62 | 46 | 42 |
| Lapperre 2009 | 30 | 55 | GSK | 1000 | 86 | 60 | 43 | 55 |
| Mahler 2002a | 6 | 349 | GSK | 1000 | 66 | 65 | 55 | 41 |
| Martinez 2013a,b | 6 | 612 | GSK | 100, 200 | 72 | 62 | 43 | 48 |
| Paggiaro 1998 | 6 | 281 | ‐ | 1000 | 74 | 63 | ‐ | 57 |
| Schermer 2009 | 36 | 190 | Indep. | 1000 | 71 | 59 | 28 | 64 |
| van Grunsven 2003 | 24 | 48 | GSK | 500 | 52 | 47 | 9 | 97 |
| Verhoeven 2002 | 6 | 23 | GSK | 1000 | 82 | 55 | 26 | 63 |
| WM 22 m | 459 | ‐ | ‐ | 72 | 62 | 43 | 54 | |
| Fluticasone/LABA combination versus LABA monotherapy (n = 15) | ||||||||
| Anzueto 2009 | 12 | 797 | GSK | 500 | 54 | 65 | 57 | 34 |
| Calverley 2003 TRISTANa | 12 | 731 | GSK | 1000 | 73 | 63 | 43 | 45 |
| Calverley 2007 TORCHa | 36 | 3088 | GSK | 1000 | 76 | 65 | 49 | 44 |
| Dal Negro 2003 | 12 | 12 | ‐ | 500 | 92 | ‐ | 42 | 50 |
| Dransfield 2013b | 12 | 3255 | GSK | 50, 100, 200 | 57 | 64 | ‐ | 45 |
| Ferguson 2008 | 12 | 782 | GSK | 500 | 55 | 65 | 56 | 33 |
| GSK FCO30002 2005 | 3 | 140 | GSK | 1000 | 66 | 62 | ‐ | ‐ |
| GSK SCO100470 2006 | 6 | 1050 | GSK | 500 | 78 | 64 | ‐ | ‐ |
| GSK SCO104925 2008a | 3 | 77 | GSK | 1000 | 77 | 64 | ‐ | ‐ |
| GSK SCO40041 2008 | 36 | 186 | GSK | 500 | 61 | 66 | ‐ | ‐ |
| Hanania 2003a | 6 | 355 | GSK | 500 | 63 | 64 | 57 | 42 |
| Kardos 2007 | 10 | 994 | GSK | 1000 | 76 | 64 | 37 | 40 |
| Kerwin 2013a,b | 6 | 617 | GSK | 100 | 67 | 63 | 46 | 43 |
| Mahler 2002a | 6 | 325 | GSK | 1000 | 66 | 65 | 55 | 41 |
| Martinez 2013a,b | 6 | 610 | GSK | 100, 200 | 72 | 62 | 43 | 48 |
| WM 16 m | 867 | ‐ | ‐ | 69 | 64 | 53 | 42 | |
aMulti‐arm studies making both comparisons of interest (ICS vs placebo and ICS/LABA vs LABA).
bStudies using vilanterol as the LABA combination and monotherapy comparator, with fluticasone furoate.
Dose is given as the total received per day (i.e. 500 signifies 250 morning and evening).
WM = weighted mean.
2. Budesonide—summary of studies and baseline characteristics.
| Study ID | Duration (m) | N Rand | Funder | ICS dose (mcg) | % Male | Mean age | Pack‐years | % pred FEV1 |
| Budesonide versus placebo (n = 13) | ||||||||
| Bourbeau 1998 | 6 | 79 | AZ | 640 | 79 | 66 | 51 | 37 |
| Calverley 2003ba | 12 | 513 | GSK | 640 | 76 | 64 | 35 | 36 |
| Laptseva 2002 | 6 | 49 | NR | 640 | NR | NR | NR | NR |
| Mirici 2001 | 3 | 50 | NR | 640 | 75 | 53 | 27 | 62 |
| Ozol 2005 | 6 | 26 | NR | 640 | 69 | 65 | 45 | 59 |
| Pauwels 1999 | 36 | 1277 | AZ | 640 | 73 | 52 | 39 | 77 |
| Renkema 1996 | 24 | 39 | AZ | 1280 | 100 | 55 | NR | 64 |
| Senderovitz 1999 | 6 | 26 | NR | 640 | 54 | 61 | NR | NR |
| Shaker 2009 | 36 | 254 | AZ | 640 | 58 | 64 | 56 | 52 |
| Szafranski 2003a | 12 | 403 | AZ | 640 | 79 | 64 | 45 | 36 |
| Tashkin 2008 SHINEa | 6 | 575 | AZ | 640 | 68 | 63 | 41 | 40 |
| Vestbo 1999 | 36 | 290 | AZ | 640 | 88 | 59 | NR | 87 |
| Yildiz 2004 | 3 | 38 | ? | 1280 | 100 | 67 | 51 | 46 |
| WM 23 m | 278 | ‐ | ‐ | 77 | 61 | 43 | 54 | |
| Budesonide/LABA combination versus LABA monotherapy (n = 7) | ||||||||
| Calverley 2003ba | 12 | 509 | GSK | 640 | 76 | 64 | 35 | 36 |
| Calverley 2010 | 11 | 481 | Chiesi | 640 | 81 | 64 | 39 | 42 |
| Fukuchi 2013 | 3 | 1293 | AZ | 640 | 89 | 65 | 44 | 41 |
| Rennard 2009 | 12 | 1483 | AZ | 320, 640 | 63 | 63 | NR | 39 |
| Sharafkhaneh 2012 | 12 | 1219 | AZ | 320, 640 | 62 | 63 | 44 | 38 |
| Szafranski 2003a | 12 | 409 | AZ | 640 | 79 | 64 | 45 | 36 |
| Tashkin 2008 SHINEa | 6 | 1129 | AZ | 320, 640 | 68 | 63 | 41 | 40 |
| WM 9 m | 932 | ‐ | ‐ | 75 | 64 | 41 | 39 | |
aMulti‐arm studies making both comparisons of interest (ICS vs placebo and ICS/LABA vs LABA).
Dose is given as the total received per day (i.e. 640 signifies 320 morning and evening).
WM = weighted mean.
Participant inclusion and exclusion criteria
Full details of the inclusion and exclusion criteria for each trial can be found in Characteristics of included studies. Inclusion and exclusion criteria were largely similar across trials, with the exception of which medications participants were allowed to continue taking during the study period. In most studies, participants were required to be over the age of 40 and to have a smoking history of at least 10 pack‐years. In terms of lung function, most studies required values consistent with a GOLD diagnosis of COPD. Studies excluded participants if they had asthma or any other respiratory disorder. Other common exclusion criteria included recent lower respiratory tract infection, the need for long‐term or nocturnal oxygen therapy and recent use of antibiotics or oral corticosteroids (usually within four to six weeks of screening).
Baseline characteristics of participants
Baseline data are given for individual trial arms in Characteristics of included studies tables and are summarised across fluticasone and budesonide studies in Table 3 and Table 4, respectively. All of the trials recruited more men than women, with a mean of around 70% and 75% in the fluticasone and budesonide studies, respectively (range 52% to 100%). Mean age within the trials ranged from 47 to 67 years, and the overall mean was similar in the fluticasone and budesonide studies (˜63 years). Smoking history as measured by overall pack‐years (one pack‐year = one pack of 20 cigarettes per day for one year) was reported in three‐quarters of the studies, and the mean was higher in the fluticasone combination therapy studies (53 pack‐years) than in the other sets of trials; all were between 41 and 43 pack‐years overall. The range across all trials was 27 to 63 pack‐years. Percentage predicted FEV1, an indicator of disease severity, was reported in most trials. Overall means for fluticasone and budesonide were very similar (47% and 48%, respectively). One outlier in the fluticasone monotherapy studies recruited a much less severe population (those showing early signs and symptoms of COPD; van Grunsven 2003), with nine mean pack‐years and percentage predicted FEV1 of 97. Two budesonide monotherapy studies also recruited less severe populations, with percentage predicted FEV1 of 77 and 87 (Pauwels 1999; Vestbo 1999). None of these three studies reported the primary outcome.
Characteristics of the interventions
Of the 26 fluticasone studies, 18 compared fluticasone monotherapy versus placebo, and 15 compared fluticasone/LABA combination versus LABA monotherapy (12 using salmeterol and three using the new LABA, vilanterol). Seven trials used multi‐arm double‐dummy designs that performed both comparisons of interest (Calverley 2003 TRISTAN; Calverley 2007 TORCH; GSK SCO104925 2008; Hanania 2003; Mahler 2002), including two newly published trials using vilanterol and fluticasone furoate (Kerwin 2013; Martinez 2013). Fifteen fluticasone studies used fluticasone propionate at a total daily dose of 1000 mcg, and seven used 500 mcg. One further study, GSK FLTA3025 2005, included both doses. The three vilanterol studies used fluticasone furoate at total daily doses of 50, 100 and 200 mcg, and 25 mcg of vilanterol.
Of the 17 budesonide studies, 13 compared budesonide monotherapy versus placebo, and seven compared budesonide/formoterol combination versus formoterol monotherapy. Three studies had four or more arms and performed both comparisons of interest (Calverley 2003b; Szafranski 2003; Tashkin 2008 SHINE). Twelve studies used a total daily budesonide dose of 640 mcg, and two studies used a daily dose of 1280 mcg (Renkema 1996; Yildiz 2004). Three studies used more than one dose (Rennard 2009; Sharafkhaneh 2012), including one that had a total of six arms (Tashkin 2008 SHINE): three combination arms, budesonide 640 mcg, formoterol 18 mcg and placebo. Formoterol as monotherapy control or in combination with budesonide was given at a total daily dose of 18 mcg in all studies.
Table 5 presents the beclomethasone dipropionate (BDP) equivalent doses for the included treatments. As shown, higher‐dose budesonide (1280 mcg/d = 1280 BDP) is more similar to the lower dose of fluticasone (500 mcg/d = 1000 BDP). Fluticasone furoate doses of 100 mcg and 200 mcg daily are equivalent to fluticasone propionate 250 mcg twice daily (1000 BDP) and 500 mcg twice daily (2000 BDP) respectively, and the lowest dose of fluticasone furoate is equivalent to 500 BDP. Only the 100 mcg dose of fluticasone furoate is currently licensed for use in COPD
3. BDP equivalent doses.
| Drug | Daily dose (mcg) | BDP equivalent (mcg) |
| Budesonide | 320 | 320 |
| 640 | 640 | |
| 1280 | 1280 | |
| Fluticasone | 500 (propionate) | 1000 |
| 1000 (propionate) | 2000 | |
| 50 (furoate) | 500 | |
| 100 (furoate) | 1000 | |
| 200 (furoate) | 2000 |
In most studies, participants were allowed short‐acting bronchodilators and treatment for acute exacerbations. Most studies also allowed people to continue on some long‐acting treatments that were not the treatments under study (usually theophylline, mucolytics, anticholinergics). Run‐in periods varied somewhat in length and nature. Most ranged from two to eight weeks; some required all bronchodilator treatment, ICS alone or ICS and LABA treatment to be tapered off; others used placebo and oral corticosteroids; and a subset did not describe the procedures used.
Transitivity and similarity
Inclusion and exclusion criteria: Inclusion and exclusion criteria, as described above, were considered comparable between the two sets of trials; although variation was noted in the allowed co‐medications between individual trials, this was not systematically different between the fluticasone and budesonide studies.
Baseline characteristics: Although variation between trials was seen, we did not consider that baseline characteristics systematically differed between budesonide monotherapy and fluticasone monotherapy trials, or between budesonide combination therapy and fluticasone combination therapy trials (see above and Table 3; Table 4).
Intervention characteristics: Budesonide and fluticasone studies most often used the respective commonly used twice‐daily dose, although the once‐daily fluticasone furoate studies introduced a potential source of heterogeneity. More important, the fluticasone studies generally used the Diskus or Accuhaler, and the budesonide studies used the Turbuhaler device; this may have confounded the common placebo comparator.
Methodology: We were concerned that funding for the fluticasone and budesonide trials was systematically different, but in light of similar inclusion criteria, baseline characteristics and study designs, we believed that funding alone was not a reason to believe that the transitivity assumption did not hold. However, although the monotherapy trials were of a comparable duration (weighted means of 22 and 23 months), the fluticasone combination therapy trials were a lot longer than the budesonide combination therapy trials (16 and nine months, respectively). Risk of bias was similar across all studies and did not differ systematically between those funded by the two main drug companies. Similarly, although fluticasone monotherapy trials had somewhat larger sample sizes than budesonide monotherapy trials, this finding was not deemed significant. The two sets of combination therapy trials had very similar mean sample sizes.
Control group event rates: Event rates for placebo monotherapy comparisons and for LABA combination therapy comparisons are presented in Table 6. Differences were noted between fluticasone and placebo for both monotherapy and combination therapy comparisons, with fluticasone studies consistently showing higher control group event rates. Inspection of control events showed that Calverley 2007 TORCH, which observed a large population over a longer time scale than most other studies (three years), was skewing the event rates. Although four other long‐term fluticasone monotherapy studies (two to three years) were identified, they had smaller populations, did not contribute to all of the outcomes and did not observe the same magnitude of event rates. With this study removed, control group events were much more similar between the two drugs (presented in brackets). Overall, considered in light of similar baselines and inclusion criteria, it is unlikely that the figures represent true differences in baseline event rates of the two populations.
4. Control group event rates.
| Monotherapy comparison—Placebo control events | Combination comparison—LABA control events | |||
| Fluticasone | Budesonide | Fluticasone | Budesonide | |
| Pneumonia‐related serious adverse events | 2.5%, 77/310 | 0.9%, 4/445 | 2.5%, 134/5420 | 0.9%, 19/2079 |
| 0.5% without TORCH | 0.7% without TORCH | |||
| All‐cause mortality | 7.6%, 282/3713 | 2.1%, 37/1763 | 5.1%, 254/5489 | 1.5%, 37/2534 |
| 2.4% without TORCH | 1.2% without TORCH | |||
| All‐cause serious adverse events | 25%, 882/3471 | 15%, 268/1763 | 21%, 1152/5489 | 14%, 356/2534 |
| 14% without TORCH | 14% without TORCH | |||
For the fluticasone control groups with and without the large 3‐year TORCH study.
In light of all of the information collected, we decided to calculate the indirect comparison of fluticasone and budesonide monotherapy via placebo because the only potential confound was the inhaler device used, and all other moderating factors were considered comparable. For the reasons outlined regarding control group event rates, we conducted a sensitivity analysis excluding Calverley 2007 TORCH.
For the combination therapy comparison, we considered the common LABA comparison to systematically differ between the two sets of combination studies; fluticasone studies used either salmeterol or vilanterol, which differed in their delivery and dosing schedules, and budesonide was always compared with formoterol. In addition, the fluticasone studies were much longer, and the same funding and device issues existed as for the monotherapy studies. As such, we did not perform an indirect comparison to compare fluticasone/LABA versus budesonide/LABA.
Outcomes and analysis structure
Unless otherwise stated, all of the analyses were conducted as proposed with fixed‐effect models using Mantel‐Haenszel methods. Several outcomes included some zero cells, but estimates were barely affected in sensitivity analyses using the Peto method. Therefore, Peto odds ratios were used only for 'mortality due to pneumonia', because events for this outcome were very rare. The quality of evidence for each outcome was rated using GRADEPro software, and this information is presented in Table 1 (fluticasone) and Table 2 (budesonide).
Indirect comparisons performed to compare fluticasone with budesonide monotherapy are presented for the primary outcome and for three secondary outcomes (all‐cause mortality, all non‐fatal serious adverse events and all pneumonia events). Although we did not foresee the inclusion of fluticasone furoate when we conceived of the protocol, we decided to combine these data with the fluticasone propionate data on the basis of consistency observed in the monotherapy subgroup of each of the relevant direct analyses (Analysis 1.1; Analysis 1.2; Analysis 1.4).
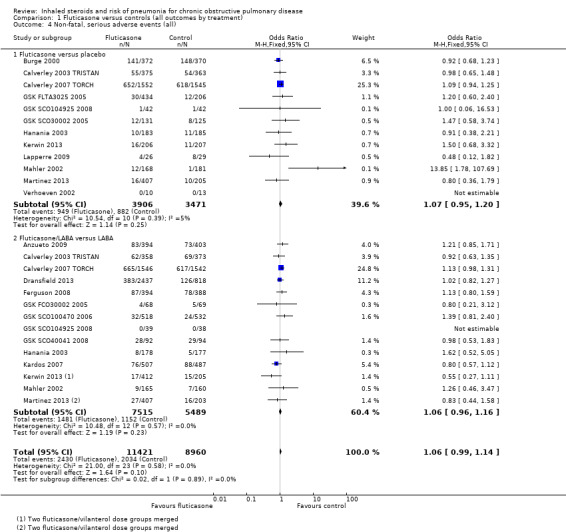
1.1. Analysis.
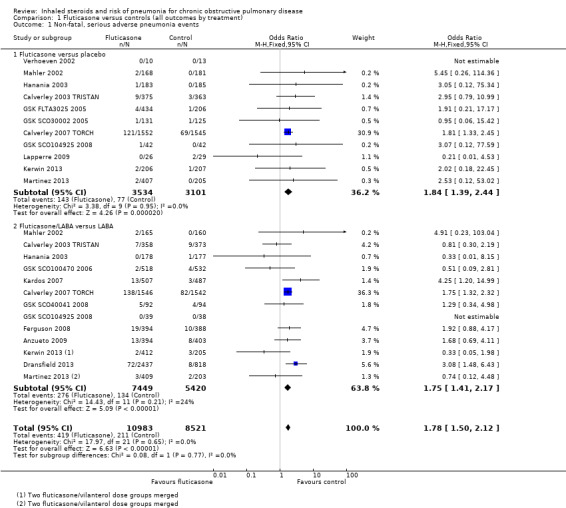
Comparison 1 Fluticasone versus controls (all outcomes by treatment), Outcome 1 Non‐fatal, serious adverse pneumonia events.
1.2. Analysis.
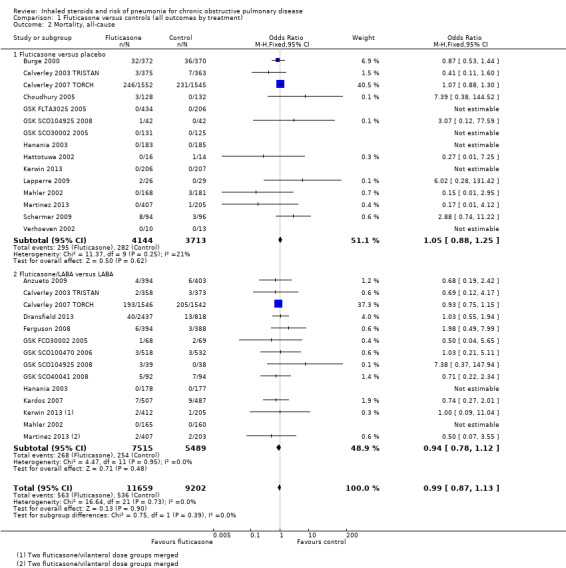
Comparison 1 Fluticasone versus controls (all outcomes by treatment), Outcome 2 Mortality, all‐cause.
1.4. Analysis.
Comparison 1 Fluticasone versus controls (all outcomes by treatment), Outcome 4 Non‐fatal, serious adverse events (all).
For the primary outcome, 'non‐fatal serious adverse pneumonia events', data were organised into three sets of subgroups to explore the effects of three prespecified potential moderators. Subgroup analyses for daily dose, duration of ICS therapy (≤ one year and > one year) and baseline severity (< 50% FEV1 predicted and ≥ 50% FEV1 predicted) are presented separately for fluticasone and budesonide in comparisons three and four, respectively.
We also conducted a sensitivity analysis while removing studies judged to be at high risk of bias because of high or uneven levels of dropout. Of the 17 studies that fell into this category, eight reported the primary outcome and were removed from the analysis (fluticasone: Calverley 2003 TRISTAN; Ferguson 2008; GSK SCO40041 2008; Lapperre 2009; Mahler 2002; budesonide: Rennard 2009; Shaker 2009; Sharafkhaneh 2012).
Excluded studies
Thirty‐one references were excluded after full texts were consulted. Reasons for exclusion were 'wrong comparison' (n = 19), discontinuation of study (n = 6), 'asthma diagnosis' (n = 2) and 'treatment period less than 12 weeks (n = 2). One study is awaiting classification (only abstract is available), and recruitment for one study was ongoing in March 2014. Full details are listed in Characteristics of excluded studies.
Risk of bias in included studies
Studies generally were well conducted and were rated as low or unclear risk of bias for all four of the allocation and blinding parameters. However, in almost half of the studies, potential for bias was due to attrition and selective outcome reporting. Full details of our judgements for each study, as well as supporting information for each judgement, can be found in Characteristics of included studies. A summary of risk of bias across all studies is shown in Figure 3.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Studies were rated for potential biases introduced by the method of sequence generation (e.g. computerised random number generator) and by the methods used to conceal the allocation sequence from those recruiting people into the studies.
Most studies were judged to be at low risk of bias for sequence generation (n = 39). Although not all of these studies adequately described sequence generation methods, all were funded by pharmaceutical companies that had previously confirmed their methods. The remaining four studies were rated 'unclear' because they did not describe their methods in detail and did not appear to be funded by a pharmaceutical company (Dal Negro 2003; Senderovitz 1999; Yildiz 2004).
Allocation concealment was not well reported, and only 17 studies were given a rating of low risk of bias because they adequately described the methods used. However, no studies were considered to be at high risk of bias, and the remaining 26 studies were given an 'unclear' rating.
Blinding
The risk of bias introduced by methods of blinding was rated separately for blinding of participants and personnel and for blinding of the people assessing outcomes.
Most studies stated that double‐blind procedures were used, and trial reports or registrations usually confirmed that this approach included both participants and investigators. For this reason, most studies were rated as low risk of bias (n = 39), and the remaining four were rated as 'unclear'. No studies were open‐label or used inadequate blinding procedures, so none were judged to be at high risk of bias.
Quite often, it was difficult to ascertain from the study reports who the outcome assessors were and for which outcomes the blinding applied. Only 10 studies gave enough information to allow a judgement of low risk of bias to be made, and the remaining 33 were rated as 'unclear'.
Incomplete outcome data
Around half of the studies were rated as low risk of bias because of incomplete outcome data (n = 21), either because the number of dropouts per group was low and even, or because the quantity and distribution of missing data were deemed acceptable given the method of imputation (e.g. intention‐to‐treat analysis using last observation carried forward). Sixteen studies were rated as high risk of bias, usually because dropout was very high in both groups, or because dropout was much higher in one group than in another. In the remaining six studies, authors considered that the information regarding attrition was not sufficient to permit judgement of whether dropout and methods of data imputation were likely to have affected the results.
Selective reporting
More than half of the studies were rated as low risk of bias for selective outcome reporting (n = 24), either because reported outcomes could be checked against the outcomes stated in a prospectively registered protocol, or because study authors provided additional data through personal communication. Five studies were rated as unclear, usually because no clear evidence on missing outcomes was available, but no trial registration could be found to confirm that all prespecified outcomes were properly reported. In all cases, attempts were made to contact trial authors for clarification; this is detailed in each study's risk of bias table in Characteristics of included studies. The remaining 14 studies were judged to be at high risk of bias, either because outcomes stated in the trial registration were missing or poorly reported in the published report, or because several key outcomes analysed in this review were not reported.
Other potential sources of bias
No additional sources of bias were identified.
Effects of interventions
In comparisons one and three, studies are pooled for fluticasone and budesonide, respectively, and are subgrouped in each case in terms of whether each randomly assigned group also received a long‐acting beta‐agonist. Comparisons two and four show additional subgroup analyses for the primary outcome of non‐fatal serious adverse events for fluticasone and budesonide, respectively.
Comparison five presents results for the indirect comparison of fluticasone and budesonide via the common placebo comparator (Figure 1). Comparison six refers to the equivalent comparison of fluticasone with budesonide given as combination therapy with a long‐acting beta‐agonist. As described above, no indirect comparison was undertaken for combination therapy because of violations of transitivity and similarity.
For illustration and visual comparison of the two drugs, effects of all fluticasone studies (comparison one) and all budesonide studies (comparison three) are presented together for each outcome in Figure 4. No statistical heterogeneity was noted between the pooled effect for budesonide and that of fluticasone for all outcomes except 'all pneumonia events'; for this reason the effects are not pooled for this outcome.
4.
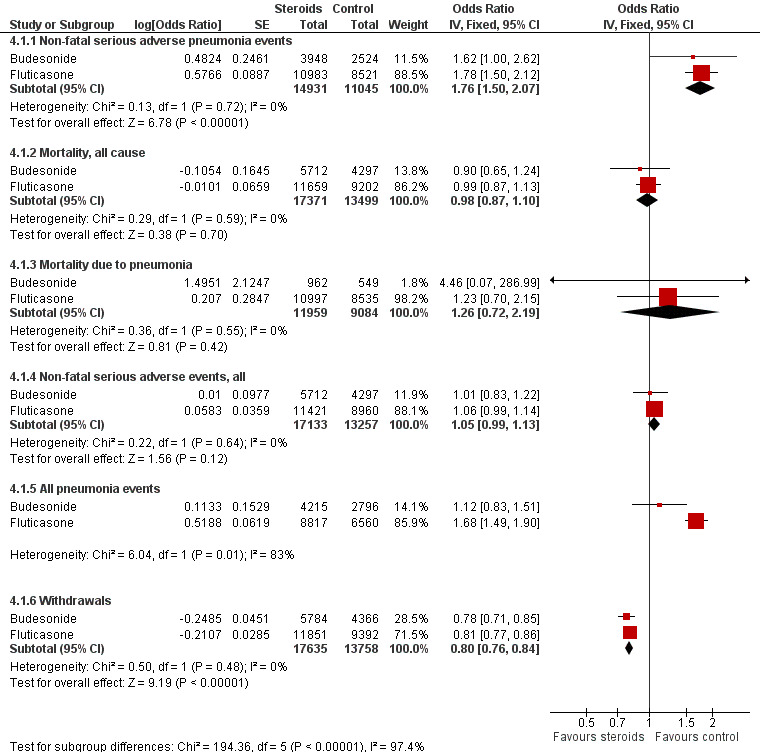
Summary of pooled effects of trials comparing ICS versus placebo and combination versus LABA. Non‐fatal serious adverse pneumonia events were those requiring hospital admission. Data for all pneumonia events were not pooled because of heterogeneity.
Comparison one: fluticasone versus controls (all outcomes subgrouped to compare ICS vs placebo with ICS/LABA vs LABA)
1.1 Primary outcome: non‐fatal, serious adverse pneumonia events (requiring hospital admission)
Twenty‐four comparisons in 17 studies were analysed (n = 19,504), with seven studies contributing data to both the 'fluticasone versus placebo' and 'fluticasone/LABA versus LABA' subgroups. Fluticasone increased the incidence of non‐fatal serious adverse pneumonia events (OR 1.78, 95% CI 1.50 to 2.12; I2 = 0%, P value 0.65), with no significant heterogeneity noted between studies. No significant evidence indicated that the odds of having a serious adverse pneumonia event were differentially affected by fluticasone alone (against placebo) compared with fluticasone/LABA combination (against LABA alone) (I2 = 0%, P value 0.77). The outcome was rated of high quality.
1.2 Mortality, all‐cause
Twenty‐nine comparisons in 22 fluticasone studies were included in the analysis (n = 20,861), although seven comparisons did not contribute to the pooled estimate because no events were reported in either group. No evidence suggested a difference between fluticasone and controls (OR 0.99, 95% CI 0.87 to 1.13; I2 = 0%, P value 0.73), and a test for subgroup differences between 'fluticasone versus placebo' and 'fluticasone/LABA versus LABA' comparisons was not significant (I2 = 0%, P value 0.39). Evidence was rated of high quality.
1.3 Mortality, due to pneumonia
Data for pneumonia‐related deaths were available for 25 comparisons in 18 studies (n = 19,532). However, all but five comparisons observed no events, and Calverley 2007 TORCH accounted for 80% of the analysis weight across its two comparisons. No difference was detected between fluticasone and control overall (Peto OR 1.23, 95% CI 0.70 to 2.15; I2 = 0%, P value 0.95), and no observable subgroup differences were reported between the monotherapy and combination therapy subgroups (I2 = 0%, P value 0.44). Evidence was rated of moderate quality, being downgraded once for imprecision because so few events were reported.
1.4. Non‐fatal serious adverse events, all‐cause
Nineteen studies across 26 comparisons reported all‐cause serious adverse events (n = 20,381). The odds of a serious adverse event were higher with fluticasone than with control (OR 1.06, 95% CI 0.99 to 1.16; I2 = 0%, P value 0.66). The lower confidence just crossed the line of no effect, but no significant heterogeneity was noted between studies. No evidence suggested a difference between monotherapy and combination therapy subgroups (I2 = 0%, P value 0.58). Evidence was rated of high quality.
1.5. All pneumonia events
Fifteen studies making 11 comparisons reported the outcome (n = 15,377), and Calverley 2007 TORCH carried almost 80% of the weight across the analysis. The odds of any pneumonia event were significantly greater with fluticasone than with control (OR 1.68, 95% CI 1.49 to 1.90), with no important heterogeneity observed between studies (I2 = 0%, P value 0.76) or subgroups (I2 = 0%, P value 0.64). The outcome was underreported across the studies, so evidence was downgraded for publication bias, and was rated of moderate quality.
1.6. Withdrawals
Data from 26 studies (33 comparisons, n = 21,243) show that withdrawals were much less common on fluticasone than on control, with no significant heterogeneity noted (OR 0.81, 95% CI 0.77 to 0.86; I2 = 3%, P value 0.43). The effect was larger for fluticasone monotherapy, but the difference between subgroups was not statistically significant (I2 = 66%, P value 0.09). No reasons suggested the need to downgrade the evidence from high quality.
Comparison two: subgroup analyses—fluticasone versus controls
Dose: Combining all studies and organising by fluticasone dose did not reveal significant subgroup differences between doses (I2 = 0%, P value 0.90; Analysis 2.1). Pooled effects for the three furoate dose subgroups (50 mcg, 100 mcg and 200 mcg once a day) contained fewer data and therefore had much wider confidence intervals than the more widely used propionate preparation doses. Higher‐dose fluticasone propionate was the most widely studied and hence has the most precise estimate, but the pooled effect was not statistically different from the other dose subgroups.
2.1. Analysis.
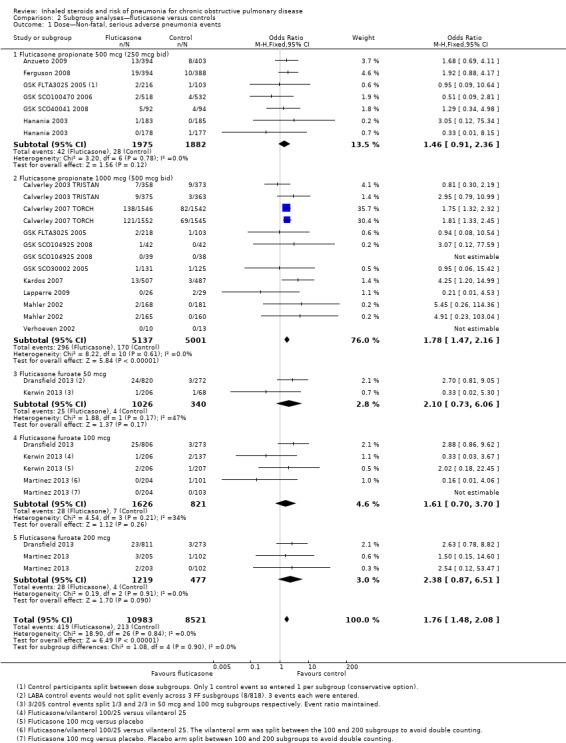
Comparison 2 Subgroup analyses—fluticasone versus controls, Outcome 1 Dose—Non‐fatal, serious adverse pneumonia events.
Trial duration: No evidence shows significant differences between the trials with duration of one year or less and the three trials (four comparisons) that followed participants for three years (I2 = 0%, P value 0.61; Analysis 2.2). No significant heterogeneity between individual studies was noted within either subgroup (I2 = 0% in both cases).
2.2. Analysis.
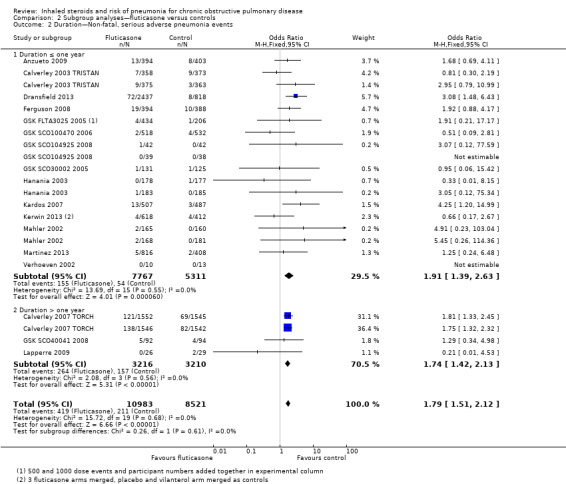
Comparison 2 Subgroup analyses—fluticasone versus controls, Outcome 2 Duration—Non‐fatal, serious adverse pneumonia events.
Baseline percentage predicted FEV1 : Studies with a mean baseline percentage predicted FEV1 of less than 50% accounted for 99% of the analysis weight (Analysis 2.3), so no conclusions could be drawn regarding the moderating effect of baseline severity.
2.3. Analysis.
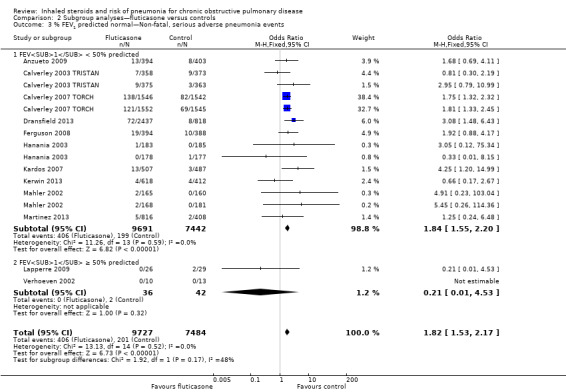
Comparison 2 Subgroup analyses—fluticasone versus controls, Outcome 3 % FEV1 predicted normal—Non‐fatal, serious adverse pneumonia events.
Comparison three: budesonide versus controls (all outcomes subgrouped to compare ICS vs placebo with ICS/LABA vs LABA)
3.1 Primary outcome: non‐fatal, serious adverse pneumonia events (requiring hospital admission)
Data for eight comparisons in seven studies were analysed (n = 6472), with Tashkin 2008 SHINE contributing data to both 'budesonide versus placebo' and 'budesonide/LABA versus LABA' subgroups. Budesonide increased non‐fatal serious adverse pneumonia events (OR 1.62, 95% CI 1.00 to 2.62), and, although a degree of variation was noted between study results, it was not significant (I2 = 28%, P value 0.21). Heterogeneity was evident between the monotherapy and combination subgroups, but the test for differences was not statistically significant (I2 = 55%, P value 0.14). The confidence intervals around the pooled estimate were quite wide but were not considered serious enough to warrant downgrading. However, because two‐thirds of the budesonide studies did not appear in the analysis, the outcome was downgraded once for publication bias and was rated of moderate quality.
3.2. Mortality, all‐cause
Budesonide did not significantly affect all‐cause mortality relative to control interventions (OR 0.90, 95% CI 0.65 to 1.24), based on 15 comparisons in 12 studies (n = 10,009). Heterogeneity was not significant across studies (I2 = 0%, P value 0.76), and no statistically significant difference was noted between the monotherapy and combination therapy subgroups (I2 = 0%, P value 0.75). Evidence was rated of moderate quality after being downgraded once for imprecision because the confidence intervals included significant benefit and harm.
3.3. Mortality, due to pneumonia
Only three budesonide studies reported the outcome (n = 1511), of which two studies observed no events in either group. No conclusions could be made from Sharafkhaneh 2012, which observed one event in the budesonide/LABA group. Evidence was rated of very low quality, being downgraded twice for imprecision and once for publication bias.
3.4. Non‐fatal serious adverse events, all‐cause
Fifteen comparisons in 12 studies were analysed (n = 10,009). Budesonide was not found to increase the odds of a serious adverse event (OR 1.01, 95% CI 0.83 to 1.22), although significant heterogeneity was noted between studies (I2 = 59%, P value 0.002), so a random‐effects analysis was used and the outcome was downgraded for inconsistency to moderate quality. No heterogeneity was observed between the subgroups (I2 = 0%, P value 0.68).
An outlier in the budesonide versus placebo subgroup was removed in a post hoc sensitivity analysis (Vestbo 1999), which changed the effect for the subgroup to favour the control (OR 1.27, 95% CI 1.04 to 1.55) with no within‐subgroup heterogeneity (previously I2 = 72%). The study recruited a less severe population, which might explain the difference in effect.
3.5. All pneumonia events
Not enough evidence was obtained to rule out a significant increase or a potential reduction in pneumonia events on budesonide compared with controls (OR 1.12, 95% CI 0.83 to 1.51; eight comparisons in six studies; n = 7011). A degree of unexplained heterogeneity was observed between studies (I2 = 13%, P value 0.33) and between treatment subgroups (I2 = 14%, P value 0.28), neither of which was significant. Although confidence intervals were quite wide, findings were not deemed serious enough to warrant downgrading of the evidence. However, because most of the budesonide studies did not appear in the analysis, the outcome was downgraded for publication bias and was rated of moderate quality.
3.6. Withdrawals
When 18 comparisons in 15 studies were combined (n = 10,150), withdrawals were seen to be less common in the budesonide groups than in the control groups (OR 0.78, 95% CI 0.71 to 0.85). No important heterogeneity was noted between individual studies (I2 = 12%, P value 0.31) or between monotherapy and combination therapy subgroups (I2 = 0%, P value 0.57). Evidence was rated of high quality.
Comparison four: subgroup analyses—budesonide versus controls
Dose: When all budesonide studies were subgrouped according to daily dose, the difference between 320 mcg and 640 mcg was significant (I2 = 74%, P value 0.05; Analysis 4.1); the higher dose increased non‐fatal serious adverse pneumonia events (OR 2.02, 95% CI 1.15 to 3.57), and no significant difference was observed for the lower dose (OR 0.68, 95% CI 0.27 to 1.71). The only study using the highest dose of 1280 mcg, Yildiz 2004, did not contribute data to the analysis because no events occurred in either group.
4.1. Analysis.
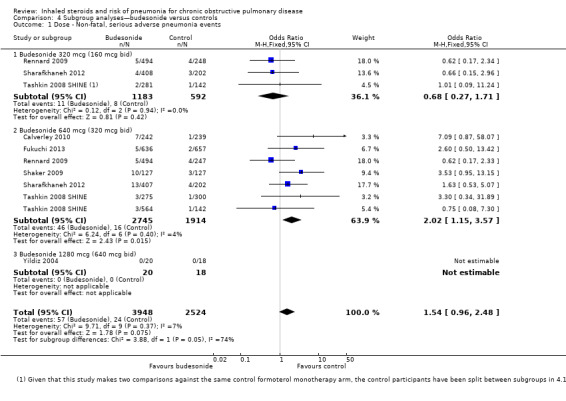
Comparison 4 Subgroup analyses—budesonide versus controls, Outcome 1 Dose ‐ Non‐fatal, serious adverse pneumonia events.
Trial duration: The difference between five studies lasting a year or less and Shaker 2009 (which followed participants for a minimum of two years) was not significant (I2 = 39%, P value 0.20; Analysis 4.2).
4.2. Analysis.
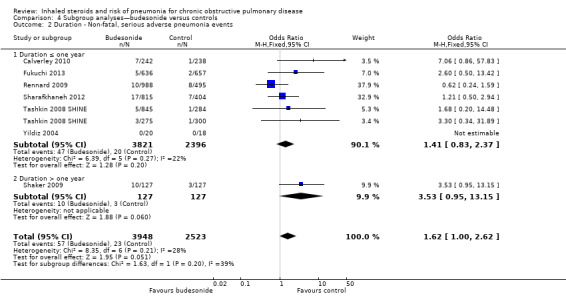
Comparison 4 Subgroup analyses—budesonide versus controls, Outcome 2 Duration ‐ Non‐fatal, serious adverse pneumonia events.
Baseline percentage predicted FEV1: When studies were subgrouped according to baseline severity, differences were not significant (I2 = 40%, P value 0.20; Analysis 4.3), and only one study reported a baseline mean FEV1 above 50% predicted (Shaker 2009).
4.3. Analysis.
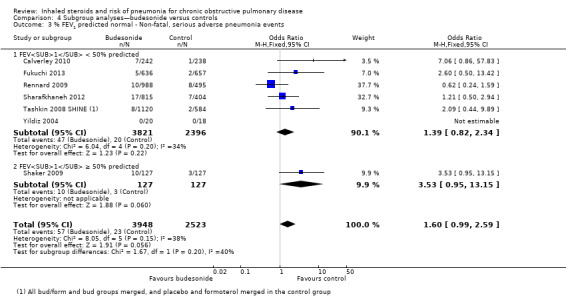
Comparison 4 Subgroup analyses—budesonide versus controls, Outcome 3 % FEV1 predicted normal ‐ Non‐fatal, serious adverse pneumonia events.
Comparison five: sensitivity analysis—risk of bias
5.1 Non‐fatal serious adverse pneumonia events
5.1.1 Fluticasone versus controls
Four fluticasone studies representing six comparisons rated at high risk for attrition were removed from the primary outcome in a sensitivity analysis. The estimate gave a slightly larger effect of fluticasone on pneumonia than the main analysis (15 RCTs; n = 16,338; OR 1.82, 95% CI 1.52 to 2.19).
5.1.2 Budesonide versus controls
Three studies judged to be at high risk of bias due to high or uneven levels of attrition were removed from the primary outcome in a sensitivity analysis. The effect from the remaining four studies was larger but more imprecise because far fewer events were included in the analysis (5 RCTs; n = 3515; OR 3.28, 95% CI 1.22 to 8.81).
Comparison six: indirect comparison of fluticasone and budesonide monotherapy
We calculated the relative effects of fluticasone and budesonide for four outcomes by comparing their effects against placebo (see Figure 1). No studies directly comparing the two drugs met this review's inclusion criteria, but cohort data are summarised in Agreements and disagreements with other studies or reviews.
All four outcomes were downgraded for indirectness because no direct evidence was found and the estimate was obtained purely from indirect comparisons. None of the outcomes were downgraded for risk of bias or inconsistency. The indirect comparisons are presented in Figure 5 and are summarised below, and the sensitivity analysis removing Calverley 2007 TORCH is shown in Figure 6.
5.
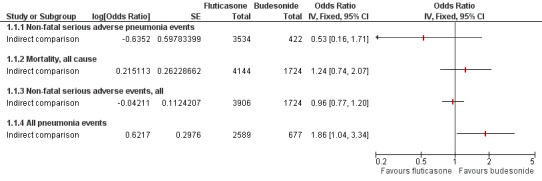
Indirect comparisons of fluticasone and budesonide monotherapy.
Non‐fatal serious adverse pneumonia events were defined as those requiring hospital admission.
6.
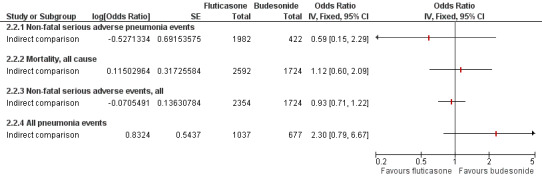
Indirect comparisons of fluticasone and budesonide monotherapy—Sensitivity analysis removing (Calverley 2007 TORCH).
6.1 Non‐fatal serious adverse pneumonia events
The point estimate favoured fluticasone, but the difference was not significant and the confidence intervals were very wide (OR 0.53, 95% CI 0.16 to 1.71). In addition to being downgraded for indirectness, the outcome was downgraded twice for imprecision and was rated as very low quality.
6.2 Mortality, all‐cause
The point estimate favoured budesonide, but the difference was not significant and the confidence intervals were wide (OR 1.24, 95% CI 0.74 to 2.07). Evidence was also downgraded once for imprecision and was rated of low quality.
6.3 All non‐fatal serious adverse events
The difference between fluticasone and budesonide was not significant, and the confidence intervals were much tighter than for the other two indirect comparisons (OR 0.96, 95% CI 0.77 to 1.20). The evidence was not downgraded for any other reason and was rated of moderate quality.
6.4 All pneumonia events
A significant difference was noted between fluticasone and budesonide, although the confidence interval was quite wide (OR 1.86, 95% CI 1.04 to 3.34). The evidence was not downgraded for any other reason and was rated of moderate quality. When Calverley 2007 TORCH was removed from the sensitivity analysis, the difference was larger in magnitude but was much less precise and was not statistically significant (OR 2.30, 95% CI 0.79 to 6.67).
Comparison seven: indirect comparison of fluticasone/LABA and budesonide/LABA combination therapy
For the reasons described in 'Transitivity and similarity', we chose not to calculate an indirect comparison for combination therapy. Recent studies directly comparing fluticasone/salmeterol with budesonide/formoterol were not comparable with the design or time scales used in the rest of this review (Blais 2010; Janson 2013 [PATHOS]; Partridge 2009; Roberts 2011), but their findings are discussed in Agreements and disagreements with other studies or reviews.
Discussion
Summary of main results
We found a total of 43 studies that met the inclusion criteria for this review, with more evidence for fluticasone (26 studies, n = 21,247) than budesonide (17 studies, n = 10,150). Evidence from the budesonide studies was more inconsistent and less precise, and the mean duration of trials was shorter.
Fluticasone increased non‐fatal serious adverse pneumonia events (i.e. those requiring a hospital admission) (OR 1.78, 95% CI 1.50 to 2.12; high‐quality evidence), and no significant evidence suggested that this was different when fluticasone was delivered in combination with salmeterol or vilanterol (subgroup differences, I2 = 0%, P value 0.51). We did not find that different doses, trial duration or baseline severity significantly affected this estimate. Budesonide also increased non‐fatal serious adverse pneumonia events, but the effect was less precise (OR 1.62, 95% CI 1.00 to 2.62; moderate‐quality evidence). Some of the variation in the budesonide data could be explained by a significant difference between the two commonly used doses: 640 mcg showed a larger increase than 320 mcg (subgroup differences, I2 = 74%, P value 0.05). An indirect comparison of budesonide and fluticasone using placebo as a common comparator showed no significant differences with respect to serious adverse events (pneumonia‐related or all‐cause) or mortality.
No difference in overall mortality rates was noted between either of the inhaled steroids and the control interventions (both high‐quality evidence), but no conclusions could be drawn regarding deaths that were pneumonia‐related, as so few events were reported. Evidence of total pneumonia events on fluticasone was dominated by the three‐year Calverley 2007 TORCH study, and far fewer budesonide studies reported the outcome.
Indirect comparisons of budesonide and fluticasone monotherapy showed no significant differences with respect to serious adverse events (pneumonia‐related or all‐cause) or mortality. The risk of any pneumonia event (i.e. less serious cases treated in the community) was higher with fluticasone than with budesonide (OR 1.86, 95% CI 1.04 to 3.34); this was the only significant difference found between the two drugs. However, this finding should be interpreted with caution because of possible differences in assignment of a diagnosis of pneumonia, and because no trials directly compared the two drugs.
Fewer people in the inhaled steroid groups withdrew from study medication; this was true for both monotherapy versus placebo and combination therapy versus LABA trials. It is possible that this was a result of lack of efficacy in the control groups, but it was not formally evaluated. High or uneven dropout was considered to show a high risk of bias in almost 40% of the trials, but conclusions for the primary outcome did not change when the trials at high risk of bias were removed in a sensitivity analysis.
Overall completeness and applicability of evidence
Efforts were made to contact all trial authors to obtain additional data when outcomes did not appear in the available reports. Although this resulted in some additional data being provided—both numerical and related to study conduct—most studies do not appear in every analysis; only around a third of the studies contribute data to the primary outcome.
We chose to look at fluticasone and budesonide as the two most widely used inhaled steroids that are also commonly prescribed in combination with the long‐acting beta2‐agonists salmeterol and formoterol, respectively, and we did not include trials studying other available inhaled steroids (e.g. mometasone, triamcinolone, ciclesonide, beclomethasone, flunisolide). As such, the study results can be applied only to the inhaled steroids in question, not to the class in general.
The plan to indirectly compare budesonide/formoterol and fluticasone/salmeterol combination therapies was more complicated than it was for monotherapy, and by choosing not to conduct the indirect comparison, we were unable to draw conclusions about the relative safety of budesonide and fluticasone when used in combination with a LABA. Even without the addition of the fluticasone furoate/vilanterol studies, we considered the LABA comparator to differ systematically between fluticasone and budesonide combination therapy studies, hence violating the transitivity assumption. Variation in the LABA used within the fluticasone trials further reduced our confidence in the indirect comparison.
Inhaled corticosteroids are not equal in strength, and their doses are normally expressed as equivalents to budesonide dipropionate (BDP) doses (Table 5). By pooling data for all doses of fluticasone for comparison with all doses of budesonide, the indirect comparison could not account for the dose‐related effects that we found with budesonide, or for differences in dose equivalence between the two sets of data.
Quality of the evidence
We wrote to all study authors when numerical outcome data or details related to risk of bias could not be obtained from available reports. Of all the studies, nine reported all information and did not need to be contacted; we failed to find contact details for a further six. Of the remaining 28 studies, 13 study authors did not reply, six provided additional data or confirmed that all measured outcomes were reported in the original reports, four forwarded the request to the drug company that held the data, four were requests sent directly to GlaxoSmithKline and one could not provide data. No drug company data were provided by the time the review was published, and the application for data is ongoing and may be incorporated in future updates. In addition, nearly half of the studies were conducted 10 or more years ago; this made it difficult for review authors to locate contact details and for study authors to locate the data requested.
The definition of pneumonia and the method of diagnosis were routinely missing from trial reports, and this information could not be obtained to conduct a subgroup analysis on this basis. We were unsure whether pneumonia was radiographically confirmed in most cases, and the trials were not designed for the purpose of measuring the incidence of pneumonia. In addition, the extent to which pneumonia events are misclassified as acute exacerbations of COPD (and vice versa) is somewhat unclear (Marzoratti 2013). We could not obtain information to judge whether ascertainment of pneumonia was systematically different between funding drug companies or between healthcare systems in which studies were conducted; this is a potential source of bias.
Despite a relatively low rate of response from trial authors, all outcomes based on direct evidence were rated as high or moderate quality with GRADE. All trials were generally of good methodological quality, having been conducted by large pharmaceutical companies. In addition, more than 40 studies were found that contributed data to at least one analysis. Some imprecision was noted for rare events such as mortality due to pneumonia; we were unable to draw conclusions in these cases, especially for budesonide, for which less evidence was available. For this reason, we were generally less confident in the budesonide outcomes.
We identified two potential confounds, which we did not consider to be sufficient to downgrade the quality of the evidence. First, most of the budesonide studies were funded by AstraZeneca, and most of the fluticasone studies by GlaxoSmithKline. However, systematic differences in study conduct were not identified in the assessments of bias. Second, budesonide monotherapy and combination therapy are delivered via a Turbuhaler, and fluticasone is given via a Diskus or Accuhaler device. We assumed that participants in the trials were given adequate training for the device used, and that this did not systematically affect the amount of drug delivered or the likelihood of compliance between the two drugs. For this reason, and because in each double‐blind trial the placebo device matched that of the active drug, we did not consider different drug devices a reason not to conduct the indirect comparison of budesonide versus fluticasone.
When the protocol for this review was written, we did not anticipate the inclusion of a new preparation of fluticasone furoate in combination with the long‐acting beta2‐agonist vilanterol. Including these studies may have introduced heterogeneity into the direct fluticasone versus placebo comparison, and into the fluticasone/LABA versus LABA comparison, although a recent randomised trial comparing fluticasone/vilanterol with fluticasone/salmeterol found no significant differences in their efficacy or safety profiles (Agusti 2013). Inclusion of these studies may also have reduced the reliability of the indirect comparisons by increasing variation within the fluticasone monotherapy node and the fluticasone/salmeterol node (by including fluticasone/vilanterol studies in a combined fluticasone/LABA node), but the absence of statistical heterogeneity in the analyses did not suggest that this was the case. The expected publication of further RCTs of fluticasone furoate and other new preparations will increase the likelihood of detecting any differences which may exist between their safety profiles and more established corticosteroids. This may warrant dealing with different preparations separately in future updates of this review, which was not possible with the current evidence base.
Our decision to conduct the monotherapy indirect comparison was based on statistical consistency within the two sets of monotherapy trials (budesonide vs placebo and fluticasone vs placebo) and on a comprehensive assessment of transitivity across these two sets of trials. As such, we considered the indirect comparison to be valid, but the quality of the evidence remains limited by the observational nature of indirect comparisons, and by the lack of head‐to‐head trials comparing fluticasone and budesonide.
Potential biases in the review process
We made every effort to adhere to Cochrane methods during the review process. As stated in the protocol, all numerical data were extracted by two review authors, and discrepancies were resolved through discussion. The same was true for the risk of bias ratings. Neither of the review authors have conflicting interests.
It is unlikely that the review is biased as a result of published studies missed during study selection. The electronic searches were relatively broad, and review authors searched additional resources to locate any studies that might have been missed (e.g. trial registration websites, drug company databases, reference lists of included studies). In addition, review authors attempted to contact all study authors when data or details of study methodology were not reported in the published reports.
Agreements and disagreements with other studies or reviews
Two Cochrane reviews that are related to this safety review have recently been updated: Yang 2012, looking at the safety and efficacy of any inhaled corticosteroid versus placebo, and Nannini 2012, focusing on ICS/LABA combination therapy versus LABA alone. Both reviews are consistent with the findings and conclusions of this review; no effect was observed for mortality, and increased rates of pneumonia seen with inhaled steroids were not significantly different between types of inhaled steroids. Nannini 2012 called for more evidence regarding differential safety of different doses of inhaled steroids, which this review has helped to clarify. Both author teams correctly point out in their reviews that evidence for harms associated with these medications needs to be assessed in conjunction with good evidence of the clinical benefit of inhaled steroids (notably, fewer exacerbations and improved quality of life). In contrast to other previous meta‐analyses (Ruiz 2011; Sin 2009), this review found evidence for an association between budesonide and serious adverse pneumonia events, and indirect comparison with fluticasone yielded little evidence for differential safety between the two drugs. It is problematic to incorporate evidence for dose‐related safety with efficacy data to assess whether a particular dose of a drug is preferable to another; evidence from Calverley 2007 TORCH suggests that the trade‐off between reducing exacerbations and increasing pneumonia changes over time for fluticasone, but it is not yet clear whether this is true for different doses and products (Cates 2013).
Evidence from cohorts corroborates the data from randomised trials, showing increased rates of pneumonia in those treated with inhaled steroids and more clearly demonstrating a dose‐related effect (Ernst 2007; Janson 2013 [PATHOS]; Suissa 2013; Yawn 2013). Evidence from one of the cohorts, which observed more than 20,000 serious pneumonias, revealed increased risk with fluticasone (RR 2.01, 95% CI 1.93 to 2.10) compared with budesonide (RR 1.17, 95% CI 1.09 to 1.26) (Suissa 2013); this conclusion could not be drawn from the randomised trial evidence. Although we believe that it was not viable to make an indirect comparison of budesonide combination therapy versus fluticasone combination therapy, cohort data suggest that the fluticasone combination is associated with higher rates of pneumonia, deaths related to pneumonia and admissions to hospital than budesonide/formoterol, but the duration of admissions for pneumonia and overall mortality were similar (Janson 2013 [PATHOS]). We found no indirect comparisons of budesonide/formoterol and fluticasone/salmeterol using a LABA control group, but indirect evidence based on randomised trials using placebo as the common comparator supported the cohort findings (Halpin 2011).
Authors' conclusions
Implications for practice.
Safety concerns related to inhaled steroids highlighted in this review should be balanced with recent cohort data and established randomised evidence of efficacy regarding exacerbations and quality of life. Budesonide and fluticasone, delivered as monotherapy or in combination with a LABA, are associated with increased risk of serious adverse pneumonia events, but neither significantly affected mortality compared with controls. Comparison of the two drugs showed no statistical difference in serious pneumonias, mortality or serious adverse events, but the risk of any pneumonia (i.e. less serious cases dealt with in the community) was higher with fluticasone than with budesonide. However, definitions used by the respective manufacturers is a potential confound in their comparison.
Implications for research.
Primary research into the use of inhaled steroids for COPD in particular should accurately measure pneumonia outcomes and clarify both the definition and the method of diagnosis used. This is particularly important for studies conducted on emerging corticosteroid preparations and combinations , for which little evidence is currently available on associated pneumonia risk. Similarly, systematic reviews, meta‐analyses and cohorts should address the reliability of assigning 'pneumonia' as an adverse event or a cause of death and should explain how this affects the applicability of findings.
Feedback
Response regarding fluticasone furoate, 1 April 2014
Summary
I was very interested to read the recent Cochrane review of inhaled steroids and risk of pneumonia in COPD. This is an important issue for patient safety and therefore for GSK. We have been looking at all of the data from our own trials, from meta‐analyses, systematic reviews and database studies in great detail. The view we have come to is that many of the previous systematic reviews were flawed because of inappropriate comparisons. The recently published Cochrane review is a huge improvement on the previous reviews and deals with many of the issues that we had identified as problems. There are, however a number of issues, which I would be grateful that you consider: 1. Your recent review analyses fluticasone propionate and fluticasone furoate together. I would like to emphasise that these are distinct inhaled steroid molecules with completely different potency ratios and I strongly feel that in the review these need to be separated. If you require any papers on their pharmacology to support this please let me know. 2. The review includes both licensed and unlicensed doses of fluticasone furoate. This means that currently the review is including doses which are lower than the licensed dose (50 mcg) and higher than the licensed dose (200 mcg). Would you consider analysing the results for the licensed and unlicensed doses separately? I would be very interested in your views. As you know GSK has supported the initiative to provide data for meta‐analysis and systematic reviews. If there is any further data that you require, we will do our very best to provide this in order to advance the scientific and clinical understanding on this important question.
Reply
We thank you for your comments and the papers which we have now referenced. Our responses to the issues you have raised are as follows.
1) Combining fluticasone furoate with fluticasone propionate
When the review protocol was written (September 2012), we did not foresee the inclusion of fluticasone furoate (FF). As such, we were faced with the decision to either exclude these studies, or include them in the review alongside fluticasone propionate (FP) evidence. We chose to include the studies, taking justification for their combination with FP from tests to assess the statistical similarity of their effects (test for subgroup differences). Analysis 2.1, which has now been adjusted to show each fluticasone furoate dose separately, showed no significant difference between the effects for the five doses (two FP and three FF; Analysis 2.1; I2=0%, P=0.90), and there was substantial imprecision in the FF estimates compared with FP. There was no statistical difference when we compared only the studies which used the licensed dose of FF (100 mcg) to studies using the BDP equivalent dose of FP (250 mcg twice daily) (I2=0%, P=0.85), and the same was true for FF 200 mcg once daily compared with FP 500 mcg twice daily (I2=0%, P=0.58).
That said, we tried to remain aware of the possible difficulty of combining the two preparations, and attempted to make the reader aware of this narratively throughout the review. We accept that, as further RCTs of FF are published, this is likely to increase the precision of the evidence and hence our confidence in any difference that might exist between FP and FF. This may warrant separating FF from FP in future updates of this review.
2) Licensing and BDP equivalent doses
Cochrane reviews are intended to be used internationally and hence do not generally exclude studies on the basis of local licenses. As a result, it would be post‐hoc to exclude or isolate particular doses of FF, especially as we did not set out to do so for FP or budesonide.
Both authors searched for information regarding the BDP equivalence of FF to present in Table 3 (BDP equivalent doses). Since the publication of the review, information from NICE and the eMC has been published which states that 100 mcg FF daily is approximately equivalent to twice daily FP 250 mcg (i.e. 1000 mcg BDP) and 200 mcg is equivalent to FP 500 mcg twice daily (i.e. 2000 mcg BDP). We have added this information to the review to help readers make sense of the evidence and comparability of the two preparations to each other, and to budesonide. The lowest dose of 50 mcg FF is not mentioned, but since this is roughly equivalent to the lower included budesonide doses, it does not seem necessary or consistent to re‐analyse separately or exclude it entirely.
Contributors
Neil Barnes Medical Head, Global Respiratory Franchise, GSK COI statement: I am employed by GSK and submitted these comments on behalf of GSK
What's new
| Date | Event | Description |
|---|---|---|
| 4 December 2014 | Amended | Typo corrected in analysis 2.1 |
History
Protocol first published: Issue 9, 2012 Review first published: Issue 3, 2014
| Date | Event | Description |
|---|---|---|
| 25 April 2014 | Feedback has been incorporated | Feedback added to the review along with a response from the review authors. The following changes were made
|
Acknowledgements
We are grateful to Elizabeth Stovold for her help in designing the search strategy and to Charlotta Karner for her role in study selection and data extraction.
CRG Funding Acknowledgement: The National Institute for Health Research (NIHR) is the largest single funder of the Cochrane Airways Group.
Disclaimer: The views and opinions expressed therein are those of the review authors and do not necessarily reflect those of NIHR, NHS or the Department of Health.
Julia Walters was the Editor for this review and commented critically on the review.
Appendices
Appendix 1. Sources and search methods for the Cochrane Airways Group Specialised Register (CAGR)
Electronic searches: core databases
| Database | Frequency of search |
| MEDLINE (Ovid) | Weekly |
| EMBASE (Ovid) | Weekly |
| CENTRAL | Quarterly (four issues per year) |
| PSYCINFO (Ovid) | Monthly |
| CINAHL (EBSCO) | Monthly |
| AMED (EBSCO) | Monthly |
Handsearches: core respiratory conference abstracts
| Conference | Years searched |
| American Academy of Allergy, Asthma and Immunology (AAAAI) | 2001 onwards |
| American Thoracic Society (ATS) | 2001 onwards |
| Asia Pacific Society of Respirology (APSR) | 2004 onwards |
| British Thoracic Society Winter Meeting (BTS) | 2000 onwards |
| Chest Meeting | 2003 onwards |
| European Respiratory Society (ERS) | 1992, 1994, 2000 onwards |
| International Primary Care Respiratory Group Congress (IPCRG) | 2002 onwards |
| Thoracic Society of Australia and New Zealand (TSANZ) | 1999 onwards |
MEDLINE search strategy used to identify trials for the CAGR
COPD search
1. Lung Diseases, Obstructive/
2. exp Pulmonary Disease, Chronic Obstructive/
3. emphysema$.mp.
4. (chronic$ adj3 bronchiti$).mp.
5. (obstruct$ adj3 (pulmonary or lung$ or airway$ or airflow$ or bronch$ or respirat$)).mp.
6. COPD.mp.
7. COAD.mp.
8. COBD.mp.
9. AECB.mp.
10. or/1‐9
Filter to identify RCTs
1. exp "clinical trial [publication type]"/
2. (randomised or randomised).ab,ti.
3. placebo.ab,ti.
4. dt.fs.
5. randomly.ab,ti.
6. trial.ab,ti.
7. groups.ab,ti.
8. or/1‐7
9. Animals/
10. Humans/
11. 9 not (9 and 10)
12. 8 not 11
The MEDLINE strategy and RCT filter are adapted to identify trials in other electronic databases.
Appendix 2. Search terms ClinicalTrials.gov
intervention: fluticasone OR budesonide OR corticosteroid OR steroid
condition: COPD
study type: interventional studies
Data and analyses
Comparison 1. Fluticasone versus controls (all outcomes by treatment).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Non‐fatal, serious adverse pneumonia events | 17 | 19504 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.78 [1.50, 2.12] |
| 1.1 Fluticasone versus placebo | 11 | 6635 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.84 [1.39, 2.44] |
| 1.2 Fluticasone/LABA versus LABA | 13 | 12869 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.75 [1.41, 2.17] |
| 2 Mortality, all‐cause | 22 | 20861 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.87, 1.13] |
| 2.1 Fluticasone versus placebo | 15 | 7857 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.88, 1.25] |
| 2.2 Fluticasone/LABA versus LABA | 14 | 13004 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.78, 1.12] |
| 3 Mortality, due to pneumonia | 18 | 19532 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.23 [0.70, 2.15] |
| 3.1 Fluticasone versus placebo | 12 | 6665 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.20 [0.52, 2.77] |
| 3.2 Fluticasone/LABA versus LABA | 13 | 12867 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.25 [0.59, 2.65] |
| 4 Non‐fatal, serious adverse events (all) | 19 | 20381 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.99, 1.14] |
| 4.1 Fluticasone versus placebo | 12 | 7377 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.95, 1.20] |
| 4.2 Fluticasone/LABA versus LABA | 14 | 13004 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.96, 1.16] |
| 5 All pneumonia events | 11 | 15377 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.68 [1.49, 1.90] |
| 5.1 Fluticasone versus placebo | 6 | 4971 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.62 [1.33, 1.97] |
| 5.2 Fluticasone/LABA versus LABA | 9 | 10406 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.72 [1.47, 2.01] |
| 6 Withdrawals | 26 | 21243 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.77, 0.86] |
| 6.1 Fluticasone versus placebo | 18 | 8227 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.70, 0.84] |
| 6.2 Fluticasone/LABA versus LABA | 15 | 13016 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.79, 0.92] |
1.3. Analysis.
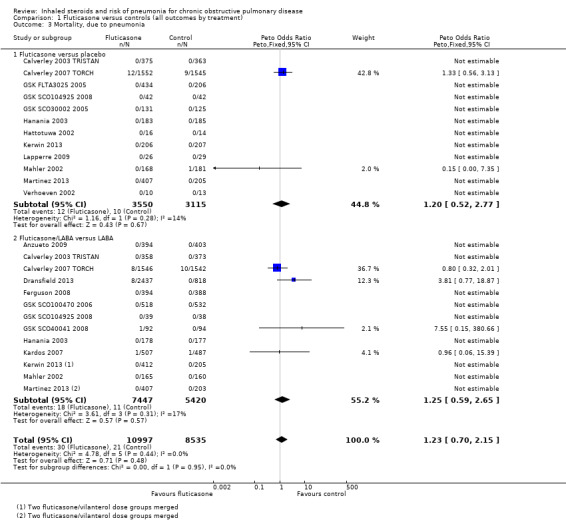
Comparison 1 Fluticasone versus controls (all outcomes by treatment), Outcome 3 Mortality, due to pneumonia.
1.5. Analysis.
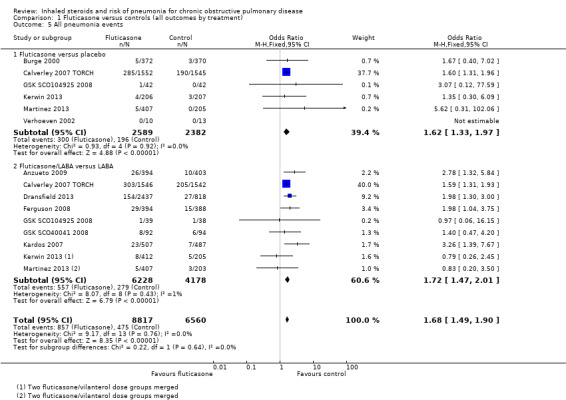
Comparison 1 Fluticasone versus controls (all outcomes by treatment), Outcome 5 All pneumonia events.
1.6. Analysis.
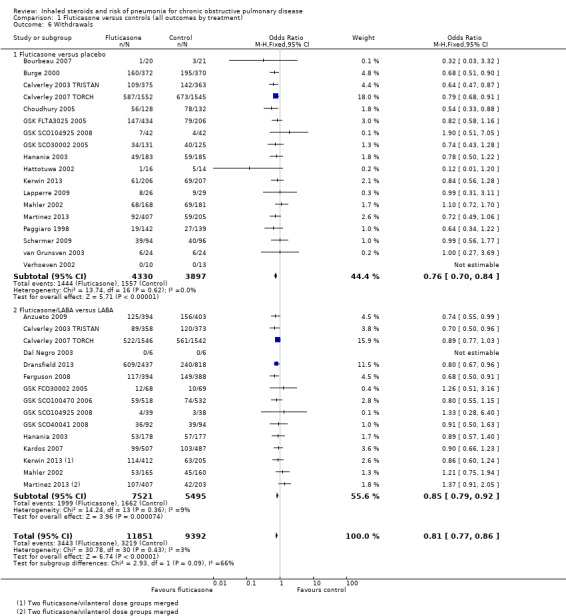
Comparison 1 Fluticasone versus controls (all outcomes by treatment), Outcome 6 Withdrawals.
Comparison 2. Subgroup analyses—fluticasone versus controls.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Dose—Non‐fatal, serious adverse pneumonia events | 17 | 19504 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.76 [1.48, 2.08] |
| 1.1 Fluticasone propionate 500 mcg (250 mcg bid) | 6 | 3857 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.46 [0.91, 2.36] |
| 1.2 Fluticasone propionate 1000 mcg (500 mcg bid) | 9 | 10138 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.78 [1.47, 2.16] |
| 1.3 Fluticasone furoate 50 mcg | 2 | 1366 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.10 [0.73, 6.06] |
| 1.4 Fluticasone furoate 100 mcg | 3 | 2447 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.61 [0.70, 3.70] |
| 1.5 Fluticasone furoate 200 mcg | 2 | 1696 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.38 [0.87, 6.51] |
| 2 Duration—Non‐fatal, serious adverse pneumonia events | 17 | 19504 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.79 [1.51, 2.12] |
| 2.1 Duration ≤ one year | 14 | 13078 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.91 [1.39, 2.63] |
| 2.2 Duration > one year | 3 | 6426 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.74 [1.42, 2.13] |
| 3 % FEV1 predicted normal—Non‐fatal, serious adverse pneumonia events | 12 | 17211 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.82 [1.53, 2.17] |
| 3.1 FEV1 < 50% predicted | 10 | 17133 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.84 [1.55, 2.20] |
| 3.2 FEV1 ≥ 50% predicted | 2 | 78 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.21 [0.01, 4.53] |
Comparison 3. Budesonide versus controls (all outcomes by treatment).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Non‐fatal, serious adverse pneumonia events | 7 | 6472 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.62 [1.00, 2.62] |
| 1.1 Budesonide versus placebo | 3 | 867 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.47 [1.11, 10.83] |
| 1.2 Budesonide/formoterol versus formoterol | 5 | 5605 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.78, 2.28] |
| 2 Mortality, all‐cause | 12 | 10009 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.65, 1.24] |
| 2.1 Budesonide versus placebo | 8 | 3487 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.52, 1.37] |
| 2.2 Budesonide/formoterol versus formoterol | 7 | 6522 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.61, 1.46] |
| 3 Mortality, due to pneumonia | 3 | 1511 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.46 [0.07, 286.99] |
| 3.1 Budesonide versus placebo | 2 | 292 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3.2 Budesonide/formoterol versus formoterol | 1 | 1219 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.46 [0.07, 286.99] |
| 4 Non‐fatal, serious adverse events (all) | 12 | 10009 | Odds Ratio (M‐H, Random, 95% CI) | 1.01 [0.83, 1.22] |
| 4.1 Budesonide versus placebo | 8 | 3487 | Odds Ratio (M‐H, Random, 95% CI) | 1.02 [0.69, 1.50] |
| 4.2 Budesonide/formoterol versus formoterol | 7 | 6522 | Odds Ratio (M‐H, Random, 95% CI) | 0.93 [0.78, 1.11] |
| 5 All pneumonia events | 6 | 7011 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.83, 1.51] |
| 5.1 Budesonide versus placebo | 3 | 1378 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.87 [0.50, 1.50] |
| 5.2 Budesonide/formoterol versus formoterol | 5 | 5633 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.24 [0.87, 1.77] |
| 6 Withdrawals | 15 | 10150 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.71, 0.85] |
| 6.1 Budesonide versus placebo | 11 | 3627 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.69, 0.93] |
| 6.2 Budesonide/formoterol versus formoterol | 7 | 6523 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.67, 0.86] |
3.1. Analysis.
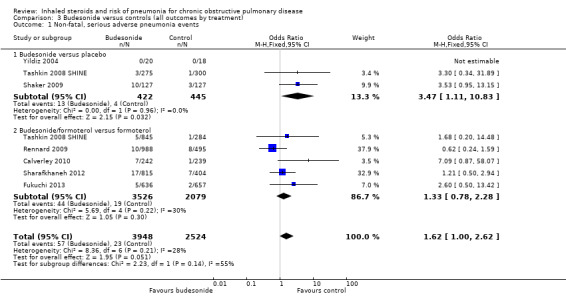
Comparison 3 Budesonide versus controls (all outcomes by treatment), Outcome 1 Non‐fatal, serious adverse pneumonia events.
3.2. Analysis.
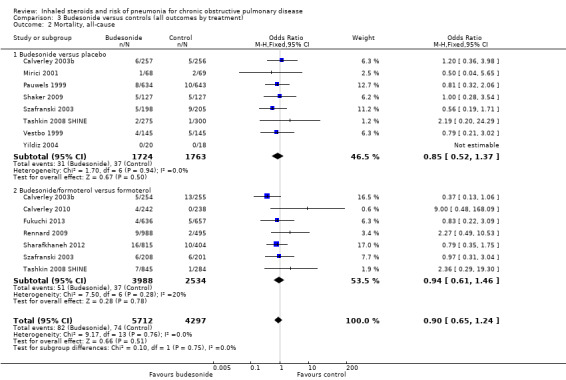
Comparison 3 Budesonide versus controls (all outcomes by treatment), Outcome 2 Mortality, all‐cause.
3.3. Analysis.
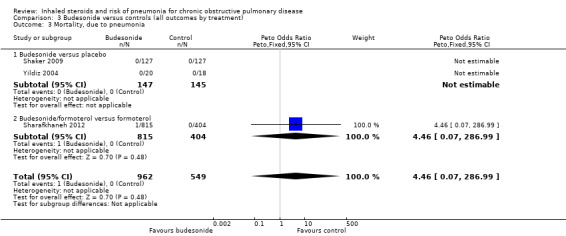
Comparison 3 Budesonide versus controls (all outcomes by treatment), Outcome 3 Mortality, due to pneumonia.
3.4. Analysis.
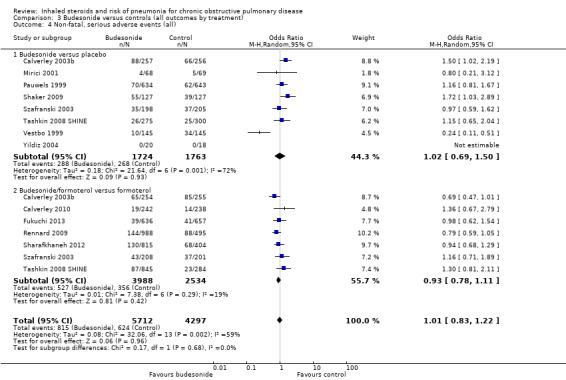
Comparison 3 Budesonide versus controls (all outcomes by treatment), Outcome 4 Non‐fatal, serious adverse events (all).
3.5. Analysis.
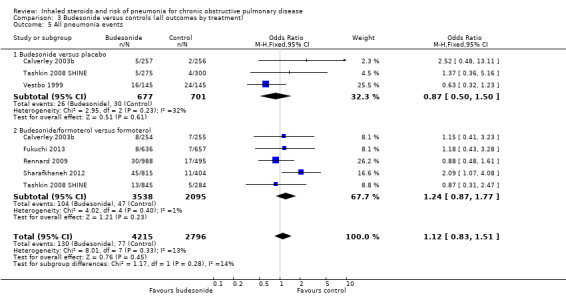
Comparison 3 Budesonide versus controls (all outcomes by treatment), Outcome 5 All pneumonia events.
3.6. Analysis.
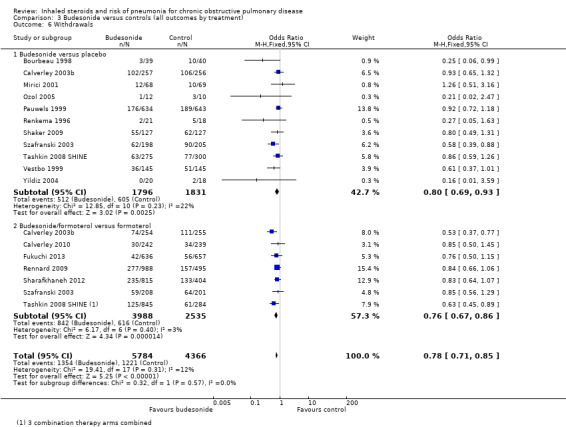
Comparison 3 Budesonide versus controls (all outcomes by treatment), Outcome 6 Withdrawals.
Comparison 4. Subgroup analyses—budesonide versus controls.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Dose ‐ Non‐fatal, serious adverse pneumonia events | 7 | 6472 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.54 [0.96, 2.48] |
| 1.1 Budesonide 320 mcg (160 mcg bid) | 3 | 1775 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.68 [0.27, 1.71] |
| 1.2 Budesonide 640 mcg (320 mcg bid) | 6 | 4659 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.02 [1.15, 3.57] |
| 1.3 Budesonide 1280 mcg (640 mcg bid) | 1 | 38 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2 Duration ‐ Non‐fatal, serious adverse pneumonia events | 7 | 6471 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.62 [1.00, 2.62] |
| 2.1 Duration ≤ one year | 6 | 6217 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.41 [0.83, 2.37] |
| 2.2 Duration > one year | 1 | 254 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.53 [0.95, 13.15] |
| 3 % FEV1 predicted normal ‐ Non‐fatal, serious adverse pneumonia events | 7 | 6471 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.60 [0.99, 2.59] |
| 3.1 FEV1 < 50% predicted | 6 | 6217 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.39 [0.82, 2.34] |
| 3.2 FEV1 ≥ 50% predicted | 1 | 254 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.53 [0.95, 13.15] |
Comparison 5. Sensitivity analysis—risk of bias.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Non‐fatal serious adverse pneumonia events | 16 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Fluticasone versus control | 12 | 16338 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.82 [1.52, 2.19] |
| 1.2 Budesonide versus control | 4 | 3515 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.28 [1.22, 8.81] |
5.1. Analysis.
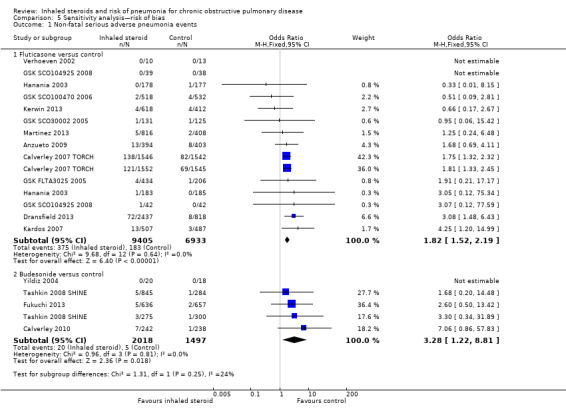
Comparison 5 Sensitivity analysis—risk of bias, Outcome 1 Non‐fatal serious adverse pneumonia events.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Anzueto 2009.
| Methods |
Design: randomised, double‐blind, parallel‐group study 12‐Month treatment period Conducted at 98 research sites in the United States and Canada |
|
| Participants |
Participants: 797 people were randomly assigned to fluticasone/salmeterol combination (394) and salmeterol alone (403) Baseline characteristics: Male %: flut/sal 51, sal 57 Mean age (SD), years: flut/sal 65.4 (9.1), sal 65.3 (8.8) Smoking history (mean (SD) pack‐years): flut/sal 57.8 (32.7), sal 56.5 (27.8) Mean % predicted pre‐FEV1 (SD): 34.1 (11.1), 33.9 (10.6) Inclusion criteria: ≥ 40 years of age with a diagnosis of COPD (chronic bronchitis and/or emphysema), a cigarette smoking history ≥ 10 pack‐years, a prealbuterol FEV1/FVC ≤ 0.70, an FEV1 ≤ 50% of predicted normal and a documented history of at least one COPD exacerbation the year before the study that required treatment with antibiotics or oral corticosteroids and/or hospitalisation Exclusion criteria: Individuals were excluded if they had a current diagnosis of asthma, a respiratory disorder other than COPD, historical or current evidence of a clinically significant uncontrolled disease or a COPD exacerbation that was not resolved at screening |
|
| Interventions |
Run‐in: Four‐week run‐in period during which participants received open‐label FSC 250/50 via DISKUS twice daily Treatments: 1. Fluticasone/salmeterol 250/50 mcg twice daily 2. Salmeterol 50 mcg twice daily Inhaler device: Diskus Co‐treatment: Concurrent use of inhaled long‐acting bronchodilators was not allowed during the study period. Oral corticosteroids and antibiotics were allowed for short‐term treatment of a COPD exacerbation |
|
| Outcomes |
Primary: annual rate of moderate/severe exacerbations Secondary: time to first moderate/severe exacerbation, annual rate of exacerbations requiring oral corticosteroids, predose FEV1, time to onset of each moderate/severe exacerbation, diary records of dyspnoea scores, night‐time awakenings due to COPD and use of supplemental albuterol |
|
| Notes |
Funding: GSK Clinicaltrials.gov: NCT00115492 Definition of pneumonia: confirmed by chest x‐ray |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Treatments were assigned in blocks using a centre‐based randomisation schedule. As bronchodilator response to FSC 250/50 is generally larger in individuals with COPD who demonstrate FEV1 reversibility to albuterol, assignment to blinded study medication was stratified on the basis of participants' FEV1 response to albuterol at screening to provide a similar distribution of albuterol‐responsive and non‐responsive participants in each treatment group |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind (assumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Withdrawal rates were very high compared with the numbers of events for different outcomes |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were reported and could be included |
Bourbeau 1998.
| Methods |
Design: double‐blind, randomised, parallel‐group trial of high‐dose inhaled budesonide versus placebo Six‐month treatment period (originally intended as 12 months) Conducted at a single centre in Canada |
|
| Participants |
Participants: 79 people were randomly assigned to budesonide (39) and placebo (40)
Baseline characteristics: Male %: bud 84.6, placebo 72.5 Mean age (SD), years: bud 66 (8), placebo 66 (8) Smoking history (mean (SD) pack‐years): bud 52 (27), placebo 50 (28) Mean % predicted pre‐FEV1 (SD): bud 36 (12), placebo 37 (10) Inclusion criteria: 40 years of age or older; smokers or ex‐smokers; absence of an exacerbation in respiratory symptoms during the two months before the study; pre‐bronchodilator FEV1 less than 65% of predicted and FEV1/forced vital capacity (FVC) less than 0.65; postbronchodilator FEV1 less than 80%; regular treatment with at least one bronchodilator Exclusion criteria: history of allergic asthma during childhood or as an adult; inhaled corticosteroids in the previous month or oral corticosteroids in the previous two months; any other active lung disease; diabetes, active peptic ulcer disease, uncontrolled high blood pressure or congestive heart failure; disease other than COPD that might interfere with quality of life |
|
| Interventions |
Run‐in: All participants were assessed in a single‐blind manner with a two‐week course of oral placebo followed by two weeks of prednisone 40 mg daily. The prednisone was subsequently tapered and was discontinued completely during the third week. Those who did not respond were randomly assigned Treatments: 1. Budesonide 400 mcg twice daily 2. Placebo twice daily Inhaler device: Turbohaler Co‐treatment: All medications needed for the well‐being of participants were permitted, except inhaled corticosteroids other than budesonide. In cases of treatment failure, rescue medication with beta2‐agonists or systemic steroids was available |
|
| Outcomes |
Primary: change in FEV1 Secondary: pre‐bronchodilator and postbronchodilator FEV1 and FVC, pre‐bronchodilator six‐minute walking test, dyspnoea with exercise, quality of life questionnaires, morning and evening PEFR, symptom scores and adverse events |
|
| Notes |
Funding: Astra Pharma Inc Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central computer‐generated list of random numbers |
| Allocation concealment (selection bias) | Low risk | Identification of individual treatment assignments was possible only in cases of emergency by breaking the sealed envelope kept by the investigator. The envelopes had to be kept with the case record forms and had to be returned unbroken at the end of the study |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | To ensure that outcomes were measured similarly in the treatment groups, both participants and investigators were blinded to the study treatment |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | To ensure that outcomes were measured similarly in the treatment groups, both participants and investigators were blinded to the study treatment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Uneven dropout. Much higher in placebo group (25% vs 7.7% in the ICS group) |
| Selective reporting (reporting bias) | High risk | Key outcomes missing (mortality, adverse events). No reply from study author |
Bourbeau 2007.
| Methods |
Design: randomised, double‐blind, parallel‐group, placebo‐controlled trial Three‐month treatment period Conducted at two respiratory centres in Canada |
|
| Participants |
Participants: 41 people were randomly assigned to fluticasone (20) and placebo (21)
Baseline characteristics: Male %: flut 84.6, placebo 72.5 Mean age (SD), years: flut 66 (8), placebo 66 (8) Smoking history (mean (SD) pack‐years): flut 52 (27), placebo 50 (28) Mean % predicted FEV1 (SD): flut 36 (12), placebo 37 (10) Inclusion criteria: Age > 40 and < 75 years; smoking history (> 10 pack‐years); postbronchodilator FEV1 > 25% of predicted value and FEV1/forced vital capacity (FVC) < 0.70 Exclusion criteria: history of asthma, atopy (as assessed by an allergy skin prick test during screening) or any other active lung disease. Individuals receiving home oxygen or with raised carbon dioxide tension (.44 mm Hg), α1‐antitrypsin deficiency, recent exacerbation (in the last 4 weeks), uncontrolled medical condition or hypersensitivity to inhaled corticosteroids and bronchodilators were not eligible for the study |
|
| Interventions |
Run‐in: unclear duration Treatments: 1. Fluticasone 500 mcg twice daily 2. Placebo twice daily Participants were also randomly assigned to fluticasone/salmeterol combination, but this was not included in the present review because no salmeterol arm was available for comparison Inhaler device: Diskus Co‐treatment: short‐acting bronchodilators, short‐ and long‐acting anticholinergics or theophylline was allowed throughout the study. Oral corticosteroids and/or antibiotics could be given only in short courses for exacerbation treatment |
|
| Outcomes |
Primary: treatment difference in the numbers of CD8+ T lymphocytes and CD68+ macrophages on bronchial biopsies Secondary: numbers of neutrophils and eosinophils in bronchial biopsies |
|
| Notes |
Funding: GSK Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central computer‐generated list of random numbers |
| Allocation concealment (selection bias) | Low risk | Set up by a data management/randomisation company (GEREQ, Montreal, Quebec). A procedure was established by GEREQ, which was in possession of the treatment code, to ensure that the treatment code would be broken only in accordance with the protocol and the criteria set up for unbinding of the study |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Observers and participants were blinded to drug treatment |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Observers were blinded to whether the biopsies were performed post‐treatment or pretreatment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropout uneven but low in both groups (5% ICS and 14% placebo) |
| Selective reporting (reporting bias) | High risk | Key outcomes not reported. No reply from study author |
Burge 2000.
| Methods |
Design: double‐blind, placebo‐controlled study Three‐year treatment period Conducted at 18 hospitals in the UK |
|
| Participants |
Participants: 740 people were randomly assigned to fluticasone (372) and placebo (370)Baseline characteristics: Male %: flut 75.0, placebo 74.1 Mean age (SD), years: flut 63.7 (7.1), placebo 63.8 (7.1) Smoking history (mean (SD) pack‐years): flut 44 (30), placebo 44 (34) Mean % predicted FEV1 (SD): flut 50.3 (14.9), placebo 50.0 (14.9) Inclusion criteria: current or former smokers 40 to 75 years of age with nonasthmatic chronic obstructive pulmonary disease. Baseline FEV1 after bronchodilator was at least 0.8 L but less than 85% of predicted normal, and the ratio of FEV1 to forced vital capacity was less than 70%. Previous use of inhaled and oral corticosteroids was permitted Exclusion criteria: Individuals were excluded if their FEV1 response to 400 mcg salbutamol exceeded 10% of predicted normal, if they had a life expectancy of less than five years from concurrent disease or if they used beta blockers |
|
| Interventions |
Run‐in: eight‐week runin period after withdrawal from any oral or inhaled corticosteroids Treatments: 1. Fluticasone propionate 500 mcg daily 2. Placebo twice daily Inhaler device: metered‐dose inhaler with a spacer device Co‐treatment: Nasal and ophthalmic corticosteroids, theophyllines and all other bronchodilators were allowed during the study |
|
| Outcomes |
Primary: decline (mL/y) in FEV1 after bronchodilator Secondary: frequency of exacerbations, changes in health status, withdrawals due to respiratory disease, morning serum cortisol concentrations and adverse events |
|
| Notes |
Funding: GlaxoWellcome Research and Development Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated allocation (block size of six) |
| Allocation concealment (selection bias) | Low risk | Participants were randomly assigned sequentially from a list comprising treatment numbers only |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Three‐year double‐blind phase using an identical placebo inhaler |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Very high dropout in both groups (43% and 53%) |
| Selective reporting (reporting bias) | Unclear risk | No outcomes appear to be missing, but protocol could not be located to ensure that all were reported. Author attempted contact with GSK statistician, but no data were provided in time for publication |
Calverley 2003 TRISTAN.
| Methods |
Design: multi‐centre, randomised, double‐blind, parallel‐group, placebo‐controlled study 12‐Month treatment period Conducted at 196 hospitals in 25 countries |
|
| Participants |
Participants: 1465 people were randomly assigned to fluticasone/salmeterol combination (358), fluticasone (374), salmeterol (372) and placebo (361)
Baseline characteristics: Male %: flut/salm 75, flut 70, salm 70, placebo 75 Mean age (SD), years: flut/salm 62.7 (8.7), flut 63.5 (8.5), salm 63.2 (8.6), placebo 63.4 (8.6) Smoking history (mean (SD) pack‐years): flut/salm 42.0 (22.4), flut 41.5 (20.7), salm 43.7 (21.9), placebo 43.4 (22.4) Mean % predicted FEV1 (SD): flut/salm 44.8 (14.7), flut 45.0 (13.6), salm 44.3 (13.8), placebo 44.2 (13.7) Inclusion criteria: All participants had a baseline FEV1 before bronchodilation that was 25% to 70% of that predicted, an increase of less than 10% of predicted FEV1 30 minutes after inhalation of 400 mcg salbutamol and a pre‐bronchodilator FEV1/forced vital capacity (FVC) ratio of 70% or less. Participants also had a history of at least 10 pack‐years of smoking, chronic bronchitis, at least one episode of acute COPD symptom exacerbation per year in the previous 3 years and at least one exacerbation in the year immediately before trial entry that required treatment with oral corticosteroids, antibiotics or both Exclusion criteria: We excluded individuals who had respiratory disorders other than COPD, required regular oxygen treatment or had received systemic corticosteroids, high doses of inhaled corticosteroids (> 1000 mcg daily beclomethasone dipropionate, budesonide or flunisolide, or > 500 mcg daily fluticasone) or antibiotics in the 4 weeks before the 2 week run‐in period before the trial began |
|
| Interventions |
Run‐in: During the 2‐week run‐in, participants stopped taking regular inhaled corticosteroids or long‐acting beta2‐agonists Treatments: 1. Fluticasone/salmeterol 500/50 mcg twice daily 2. Fluticasone 500 mcg twice daily 3. Salmeterol 50 mcg twice daily 4. Placebo twice daily Inhaler device: multi‐dose dry powder inhaler (Diskus or Accuhaler) Co‐treatment: Inhaled salbutamol was used as relief medication throughout the study, and regular treatment with anticholinergics, mucolytics and theophylline was allowed. All non‐COPD medications could be continued if the dose remained constant when possible, and if their use would not be expected to affect lung function |
|
| Outcomes |
Primary: FEV1 at least 6 and 12 hours after study medication Secondary: pretreatment FVC and post‐treatment FEV1 and FVC, daily record card symptoms, morning and evening PEF, use of relief medication, night‐time awakenings, acute exacerbations, health‐related quality of life (SGRQ), adverse events and electrocardiology |
|
| Notes |
Funding: GSK Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A randomisation schedule generated by the participant allocation for clinical trials (PACT) programme to assign participants to study treatment groups |
| Allocation concealment (selection bias) | Unclear risk | Every participating centre was supplied with a list of participant numbers (assigned to participants at their first visit) and a list of treatment numbers. Participants who satisfied the eligibility criteria were assigned the next sequential treatment number from the list |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Salmeterol and fluticasone combination, salmeterol, fluticasone and placebo were packaged in identical inhaler devices |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Adverse event information was obtained at each clinic visit by recording spontaneously reported complaints from participants and asking general questions about medical troubles and concomitant medications |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawal rates varied from 25% in the combination inhaler group to 39% in the placebo group. These numbers are substantially higher than the numbers of events reported for any of the outcomes. The unknown outcome of participant withdrawal from the study could have a major impact on the result |
| Selective reporting (reporting bias) | Low risk | All outcomes reported. Checked with study authors |
Calverley 2003b.
| Methods |
Design: multi‐centre, randomised, double‐blind, parallel‐group, placebo‐controlled study 12‐Month treatment period Conducted at 109 centres in 15 countries |
|
| Participants |
Participants: 1022 people were randomly assigned to budesonide/formoterol combination (254), budesonide (257), formoterol (255) and placebo (256)
Baseline characteristics: Male %: bud/form 78, bud 74, form 75, placebo 75 Mean age (range), years: bud/form 64 (42 to 86), bud 64 (41 to 85), form 63 (41 to 84), placebo 65 (43 to 85) Smoking history (mean pack‐years): bud/form 33, bud 39, form 36, placebo 30 Mean % predicted FEV1 (SD): bud/form 36 (10), bud 36 (10), form 36 (10), placebo 36 (10) Inclusion criteria: GOLD‐defined COPD (stages III and IV); ≥ 40 years; COPD symptoms > 2 years; smoking history ≥ 10 pack‐years; FEV1/FVC ≤ 70% pre‐bronchodilator; FEV1 ≤ 50% predicted; use of short‐acting beta2‐agonists as reliever medication; ≥ 1 COPD exacerbation requiring oral corticosteroids/antibiotics 2 to 12 months before first clinic visit Exclusion criteria: history of asthma/rhinitis before 40 years of age; any relevant cardiovascular disorders; exacerbation of COPD requiring medical intervention within 4 weeks of run‐in/during run‐in phase; non‐allowed medications: oxygen therapy; ICS (aside from study medication), disodium cromoglycate, leukotriene‐antagonists, 5‐lipoxygenase inhibitors, bronchodilators (other than study medication and terbutaline 0.5 mg as needed), antihistamines, medication containing ephedrine, beta‐blocking agents |
|
| Interventions |
Run‐in: All participants received 30 mg oral prednisolone twice daily and 2 × 4.5 mg formoterol twice daily (2 weeks) Treatments: 1. Budesonide/formoterol 320/9 mcg twice daily 2. Budesonide 400 mcg twice daily 3. Formoterol 9 mcg twice daily 4. Placebo twice daily Inhaler device: Turbuhaler Co‐treatment: terbutaline 0.5 mg as needed, courses of oral corticosteroids (maximum 3 weeks per course) and antibiotics in the event of exacerbations, parenteral steroids and/or nebulised treatment (single injections/inhalations) at emergency visits |
|
| Outcomes |
Primary: time to first exacerbation and change in postmedication FEV1 Secondary: number of exacerbations, time to and number of oral corticosteroid–treated episodes, change in postdose FEV1, slow VC, morning and evening PEF, quality of life (SGRQ), symptoms, use of reliever medication and adverse events |
|
| Notes |
Funding: GSK Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised trial (no other details—funded by AstraZeneca and presumed to adhere to usual methods) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind (assume participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | An intention‐to‐treat analysis was used but dropout was high in all groups (ranging from 29% to 44%) |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the industry document were reported in full. Checked with study authors |
Calverley 2007 TORCH.
| Methods |
Design: multi‐centre, double‐blind, placebo‐controlled, randomised, parallel‐group study Three‐year treatment period Conducted at 444 centres in 42 countries |
|
| Participants |
Participants: 6184 people were randomly assigned to fluticasone/salmeterol combination (1546), fluticasone (1551), salmeterol (1542) and placebo (1545)
Baseline characteristics: Male %: flut/salm 75, flut 75, salm 76, placebo 76 Mean age (SD), years: flut/salm 65.0 (8.3), flut 65.0 (8.4), salm 65.1 (8.2), placebo 65.0 (8.2) Smoking history (mean (SD) pack‐years): flut/salm 47.0 (26.5), flut 49.2 (28.6), salm 49.3 (27.7), placebo 48.6 (26.9) Mean % predicted FEV1 (SD): flut/salm 44.3 (12.3), flut 44.1 (12.3), salm 43.6 (12.6), placebo 44.1 (12.3) Inclusion criteria: current or former smokers with at least a 10‐pack‐year history. Eligible individuals were 40 to 80 years of age and had received a diagnosis of COPD, with a pre‐bronchodilator forced expiratory volume in one second (FEV1) of less than 60% of predicted value, an increase in FEV1 with the use of 400 mcg of albuterol of less than 10% of the predicted value for that individual and a ratio of pre‐bronchodilator FEV1 to forced vital capacity (FVC) equal to or less than 0.70 Exclusion criteria: diagnosis of asthma, current respiratory disorders other than COPD (e.g. lung cancer, sarcoidosis, tuberculosis, lung fibrosis); chest x‐ray indicating diagnosis other than COPD that might interfere with the study (chest x‐ray to be taken up to six months before entry to the treatment period); lung volume reduction surgery and/or lung transplant, requirement for long‐term oxygen therapy (LTOT is defined as oxygen therapy prescribed for 12 or more hours per day) at start of study; receiving long‐term oral corticosteroid therapy defined as continuous use for longer than 6 weeks; serious, uncontrolled disease (including serious psychological disorders) likely to interfere with the study and/or likely to cause death within the 3‐year study duration; received any other investigational drugs in the last 4 weeks before entry to visit one; evidence of alcohol, drug or solvent abuse; known or suspected hypersensitivity to inhaled corticosteroids, bronchodilators or lactose; known deficiency of α1‐antitrypsin |
|
| Interventions |
Run‐in: Before the 2 week run‐in period, all use of corticosteroids and inhaled long‐acting bronchodilators was stopped, but participants could continue other medications for COPD Treatments: 1. Fluticasone/salmeterol 500/50 mcg twice daily 2. Fluticasone 500 mcg twice daily 3. Salmeterol 50 mcg twice daily 4. Placebo twice daily Inhaler device: multi‐dose dry powder inhaler (Diskus or Accuhaler) Co‐treatment: all COPD medications except corticosteroids and inhaled long‐acting bronchodilators |
|
| Outcomes |
Primary: time to death from any cause by three years Secondary: frequency of exacerbations, health status (SGRQ), postbronchodilator spirometry and adverse events |
|
| Notes |
Funding: GSK Clinicaltrials.gov ID: NCT00268216 Definition of pneumonia: Study authors state there was no prospective definition because the finding was unexpected NOTE: Participants previously enrolled into TRISTAN (SFCB3024) may be recruited to this trial 4 weeks after stopping their previous study medication |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random assignment with permuted blocks with stratification |
| Allocation concealment (selection bias) | Low risk | Medication was allocated with the use of three numbers as follows.
A specialist IVR system company, ClinPhone, managed this system. At the randomisation visit (visit two), the principal investigator or designee contacted the IVR system through an automated 24‐hour telephone number; upon provision of a unique personal identification number (PIN) and answers to a series of questions, the site was provided with the participant's treatment number as well as a pack number |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Neither the subject nor the investigator knew to which treatment arm a participant had been allocated. At each treatment visit, each participant was issued with a treatment pack containing DISKUS/ACCUHALER inhalers. The inhalers contained one of the four treatments (salmeterol/fluticasone propionate combination product, fluticasone propionate, salmeterol or placebo) in accordance with the randomisation schedule. The inhalers were labelled in accordance with all applicable regulatory requirements. Each treatment pack and study treatment inhaler was labelled with the protocol number; storage and dosing instructions were provided by GW Research and Development |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The investigator is responsible for the detection and documentation of events meeting the definition of an AE or SAE as provided in this protocol. However, no prospective definition of pneumonia is provided in the study protocol |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk |
Outcome all‐cause mortality: Participants who prematurely discontinue study drug will be followed up for 156 weeks from randomisation for assessment of survival. The risk of bias due to incomplete outcome data for mortality is low General Withdrawal rates were high but fairly even (with the exception of the placebo group, which had an even higher withdrawal rate; placebo 44%, LABA 36%, ICS 38% and ICS/LABA 34%). However, for participants who withdrew from the study prematurely, all data on exacerbations, health status and lung function available at the time of a participant's withdrawal from the study were included in the analysis. All efficacy analyses were performed according to the intention‐to‐treat principle |
| Selective reporting (reporting bias) | Low risk | All collected data reported. Checked with study authors |
Calverley 2010.
| Methods |
Design: double‐blind, double‐dummy, randomised, active‐controlled, parallel‐group study 11‐Month treatment period (48 weeks) Conducted at 76 centres in 8 countries across Europe |
|
| Participants |
Participants: 481 people were randomly assigned to budesonide/formoterol combination (242) and formoterol (239)
Baseline characteristics: Male %: bud/form 81.5, form 81.1 Mean age (SD), years: bud/form 64.1 (9.1), form 63.7 (8.8) Smoking history (mean (SD) pack‐years): bud/form 37.8 (14.6), form 39.7 (19.1) Mean % predicted FEV1 (SD): bud/form 42.3 (6.0), form 42.5 (5.9) Inclusion criteria: hospital outpatients with severe stable COPD according to the GOLD guidelines; 40 years of age with a diagnosis of symptomatic COPD for > 2 years, at least a 20‐pack‐year smoking history, a postbronchodilator FEV1 between 30% and 50% of predicted normal and at least 0.7 L absolute value and a predose FEV1/forced vital capacity (FVC) of 0.7; at least one exacerbation requiring medical intervention (oral corticosteroid and/or antibiotic treatment and/or need for a visit to an emergency department and/or hospitalisation) within 2 to 12 months before the screening visit and the need to be clinically stable for the 2 months before study entry; change in FEV1 < 12% of predicted normal value 30 minutes following inhalation of 200 mg of salbutamol pMDI Exclusion criteria: history of asthma, allergic rhinitis or other atopic disease, variability of symptoms from day to day and frequent symptoms at night and early morning (suggestive of asthma); receiving long‐term oxygen therapy or having a lower respiratory tract infection or having been hospitalised for an acute COPD exacerbation within two months before screening or during the run‐in period. Treatments with oral, injectable or depot corticosteroids and antibiotics, long‐acting antihistamines and changes in the dose of an oral modified‐release theophylline in the two months before screening and during the run‐in period were excluded |
|
| Interventions |
Run‐in: During the 4‐week run‐in period, all non‐permitted COPD treatments were discontinued and eligible participants were treated with combination ipratropium/salbutamol (20/100 mg, two inhalations three times daily) Treatments: 1. Budesonide/formoterol 400/12 mcg twice daily 2. Formoterol 12 mcg twice daily Inhaler device: dry powder inhaler Co‐treatment: not described |
|
| Outcomes |
Primary: change in predose morning FEV1 and mean rate of COPD exacerbations per participant per year Secondary: FVC, PEF, SGRQ total score, six‐minute walking test, BMI, BODE index, safety evaluations including ECG |
|
| Notes |
Funding: Chiesi Farmaceutici Clinicaltrials.gov ID: NCT00476099 Definition of pneumonia: not defined |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The randomisation scheme followed a balanced‐block centre‐stratified design and was prepared via a computerised system |
| Allocation concealment (selection bias) | Low risk | Participants were centrally assigned, in each centre, to one of the three treatment arms at the end of the run‐in period through an Interactive voice/web response System (IXRS) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | On each study day, participants took both active medications and matched placebo twice daily, to maintain blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | On each study day, participants took both active medications and matched placebo twice daily, to maintain blinding. In cases of emergency, unbinding of the treatment code was done through IXRS |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 12.3% withdrew from the combination group and 14.2% from the formoterol group. Judged to be relatively low and even between groups, and for the intention‐to‐treat population, last observation carried forward was used |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the prospectively registered protocol were reported in full |
Choudhury 2005.
| Methods |
Design: randomised, double‐blind, placebo‐controlled trial 12‐Month treatment period in total, but participants/clinicians could stop study inhalers and return to usual (prerandomisation) steroid inhalers at any point during the study. These participants remained in the study, continued completing their diary cards and were followed up Conducted at 31 general practices in East London and Essex, UK |
|
| Participants |
Participants: 260 people were randomly assigned to fluticasone (128) and placebo (132)
Baseline characteristics: Male %: flut 48, placebo 56 Mean age (SD), years: flut 67.6 (8.9), placebo 67.3 (9.0) Smoking history (mean (SD) pack‐years): flut 40.0 (24.2), placebo 38.8 (22.3) Mean % predicted FEV1 (SD): flut 53.2 (18.2), placebo 55.0 (17.1) Inclusion criteria: Investigators searched the medical record database at each practise to identify people 40 years of age and older, with a history of smoking, who had been prescribed ICS for a minimum of six months. We invited people who fulfilled these criteria to attend a recruitment interview. Individuals with lung function consistent with international guidelines for the diagnosis of COPD were invited to join the study: postbronchodilator FEV1 less than 80% predicted, FEV1/FVC ratio less than 70% and a pre‐bronchodilator to postbronchodilator change in FEV1 of less than 15%. Participants with an FEV1 greater than 15% but with a volume change of less than 200 mL were also included Exclusion criteria: At interview, we excluded people if they were on long‐term oral corticosteroids, were not taking their prescribed ICS for at least four days a week or had other chronic active lung disease or lung cancer |
|
| Interventions |
Run‐in: two‐week run‐in period before randomisation, when participants stopped their regular ICS Treatments: 1. Fluticasone propionate 500 mcg twice daily 2. Placebo twice daily Inhaler device: Accuhaler Co‐treatment: General practitioners were advised to manage exacerbations according to usual guidance with antibiotics and/or oral steroids. Decisions about stopping study inhalers and returning to usual (prerandomisation) steroid inhalers were made by the general practitioner and the participant. Participants who did return to their usual steroid inhaler after randomisation remained in the study, continued completing their diary cards and were followed up for one year |
|
| Outcomes |
Primary: COPD exacerbation frequency Secondary: time to first exacerbation (from diary cards and medical records), reported symptoms, peak expiratory flow rate and reliever inhaler use (from diary cards) and lung function and health=related quality of life (at follow‐up visits) |
|
| Notes |
Funding: The British Lung Foundation and Newham National Health Service Trust Research and Development funded the study. GlaxoSmithKline provided the study inhalers free of charge but was not involved in study design, data collection or analysis or interpretation of results Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were allocated with minimisation to intervention or control using the programme MINIM v1.3. Minimisation factors were age, smoking status, pretrial weekly dose of ICS, self reported COPD exacerbation frequency and percentage predicted FEV1 |
| Allocation concealment (selection bias) | Low risk | Inhalers were given an alphanumerical code to conceal allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study nurses and regular clinicians were blind to allocation throughout the study. Inhalers were given an alphanumerical code to conceal allocation |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Study nurses performed the measurements and were blind to allocation |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Decisions about stopping study inhalers and returning to usual (prerandomisation) steroid inhalers were made by the general practitioner and the participant. Participants who did return to their usual steroid inhaler after randomisation remained in the study, continued completing their diary cards and were followed up for one year |
| Selective reporting (reporting bias) | High risk | Key outcomes were not reported (e.g. total mortality, breakdown of all and serious adverse events). No reply from study authors |
Dal Negro 2003.
| Methods |
Design: randomised, double‐blind, parallel‐group, pilot study 12‐Month treatment period Conducted at a single centre in Italy |
|
| Participants |
Participants: 12 people were randomly assigned to fluticasone/salmeterol combination (6) and salmeterol alone (6)
Baseline characteristics: Male %: flut/salm 83.3, salm 100 Age range (mean not reported), years: flut/salm 53 to 77, salm 55 to 78 Smoking history (mean (SD) pack‐years): flut/salm 40.1 (6.3), salm 43.1 (5.3) Mean % predicted FEV1 (SD): flut/salm 50 (2.0), salm 48 (5.6) Inclusion criteria: basal FEV1 < 80% predicted normal value, but > 800 mL; FEV1/FVC ratio < 70% predicted; FEV1 change of < 12% as a percentage of predicted normal value following salbutamol 400 mg; regular treatment with oral theophylline 200 mg bid and short‐acting beta2‐adrenergics prn for a period of at least six months; current smokers or ex‐smokers with a smoking history of at least 10 pack‐years Exclusion criteria: current evidence of asthma or other pulmonary diseases; regular treatment with ICS; unstable respiratory disease requiring oral/parenteral corticosteroids within four weeks before the beginning of the study; changes in COPD medication in the last four weeks before entering the run‐in period; upper or lower respiratory tract infection within four weeks before the screening visit; unstable angina or unstable arrhythmias; recent myocardial infarction or hearth failure; insulin‐dependent diabetes mellitus; neuropsychiatric disorders; concurrent use of medications that affected COPD (e.g. beta‐blockers) or interacted with methylxanthine products such as macrolides or fluoroquinolones; known or suspected hypersensitivity to ICS, beta2‐agonist or lactose; evidence of alcohol abuse |
|
| Interventions |
Run‐in: Patients entered a two‐week run‐in period during which they assumed their regular treatment with theophylline and salbutamol as required Treatments: 1. Fluticasone/salmeterol 250/50 mcg twice daily 2. Salmeterol 50 mcg twice daily Participants were also randomly assigned to placebo (6), but this group was not included in the current review because no group received fluticasone monotherapy Inhaler device: Diskus Co‐treatment: theophylline and salbutamol as required |
|
| Outcomes | Unclear which outcome was primary Exacerbations per year, FEV1, morning PEF, daily symptom scores, use of rescue medication and adverse events |
|
| Notes |
Funding: unclear Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | At the end of the run‐in, eligible participants will be randomly assigned to receive one of the three double‐blind treatments (no other details and does not appear to be industry funded) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Assigned to receive one of the three double‐blind treatments, all via Diskus |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropout |
| Selective reporting (reporting bias) | High risk | Several key outcomes not reported (mortality, adverse events). Difficulty contacting study authors |
Dransfield 2013.
| Methods |
Design: two replicate double‐blind parallel‐group trials 12‐Month treatment period Study one was conducted at 167 centres in 15 countries (Argentina, Australia, Canada, Chile, Estonia, Germany, Italy, Mexico, Netherlands, Peru, Phillipines, South Africa, Sweden, the United Kingdom and the United States). Study two was conducted at 183 centres in 15 countries |
|
| Participants |
Participants: 1622 people were randomly assigned in study one and 1633 in study two to vilanterol 25 (818), fluticasone/vilanterol combination 50/25 (820), 100/25 (806) and 200/25 (811)
Baseline characteristics: Male %: Vil 58.0, F/Vil 50/25 58.0, F/Vil 100/25 56.2, F/Vil 200/25 57.6 Mean age, years: Vil 63.6, F/Vil 50/25 63.7, F/Vil 100/25 63.8, F/Vil 200/25 63.7 Smoking history (mean (SD) pack‐years): not reported Mean % predicted FEV1: Vil 45.2, F/Vil 50/25 45.4, F/Vil 100/25 46.1, F/Vil 200/25 45.2 Inclusion criteria: Eligible patients were 40 years of age or older and had a history of COPD, a smoking history of 10 or more pack‐years, a ratio of forced expiratory volume in one second (FEV1) to forced vital capacity of 0.70 or less after bronchodilators (and an FEV1 of 70% or less of predicted) and a documented history of one or more moderate or severe disease exacerbations in the year before screening Exclusion criteria: Appendix listing the exclusion criteria and the drugs that were and were not permissible for the study duration could not be located |
|
| Interventions |
Run‐in: four weeks during which participants received open‐label combination fluticasone propionate (250) and salmeterol (50) twice daily to establish adherence to treatment and a stable baseline Treatments: 1. Fluticasone furoate/vilanterol 50/25 mcg daily 2. Fluticasone furoate/vilanterol 100/25 mcg daily 3. Fluticasone furoate/vilanterol 200/25 mcg daily 4. Vilanterol 25 daily Inhaler device: dry powder inhaler Co‐treatment: Appendix listing the exclusion criteria and the drugs that were and were not permissible for the study duration could not be located |
|
| Outcomes |
Primary: annual rate of moderate and severe exacerbations Secondary: time to first moderate or severe exacerbation; annual rate of exacerbations requiring oral corticosteroids; predose AM FEV1 |
|
| Notes |
Funding: GSK Clinicaltrials.gov ID: NCT01009463 and NCT01017952 Definition of pneumonia: confirmed by chest x‐ray |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The central randomisation schedule was generated by the GSK statistics group, which used a validated computerised system (RandAll; GSK, London, UK) |
| Allocation concealment (selection bias) | Unclear risk | The statistician entered the parameters for the randomisation but was masked to treatment assignment and did not have access to the master randomisation schedule until the study was unmasked at database lock. The Registration and Medication Ordering System (RAMOS; GSK, London, UK) was used to register and randomly assign participants and assign drugs |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and investigators were masked to allocation, and the Ellipta dry powder inhalers were identical in appearance. Every effort was made to ensure that the statistics and programming department of GSK was not unmasked to any treatment allocations ahead of the database freeze |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Participants and investigators were masked to allocation, and the Ellipta dry powder inhalers were identical in appearance. Every effort was made to ensure that the statistics and programming department of GSK was not unmasked to any treatment allocations ahead of the database freeze |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropout quite high but even between groups (ranging from 23% to 31%). A generalised linear model was used for the intention‐to‐treat analyses of each study, including all randomly assigned participants receiving at least one dose of study drug (representing 100% of those randomly assigned) |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the prospectively registered protocol were reported in the published paper |
Ferguson 2008.
| Methods |
Design: randomised, double‐blind, parallel‐group study 12‐Month study period Conducted at 94 research sites in the United States and Canada |
|
| Participants |
Participants: 782 people were randomly assigned to fluticasone/salmeterol combination (394) and salmeterol alone (388)
Baseline characteristics: Male %: flut/salm 58, salm 52 Mean age (SD), years: flut/salm 64.9 (9.0), salm 65.0 (9.1) Smoking history (mean (SD) pack‐years): flut/salm 58.5 (30.6), salm 54.4 (25.7) Mean % predicted FEV1 (SD): flut/salm 32.8 (11.0), salm 32.8 (10.1) Inclusion criteria: 40 years of age or older with a diagnosis of COPD, a cigarette smoking history of greater than or equal to 10 pack‐years, a prealbuterol FEV1/FVC of 0.70 or less, an FEV1 of 50% of predicted normal or less and a history of 1 or more exacerbations of COPD in the year before the study, which required treatment with oral corticosteroids or antibiotics, or hospitalisation Exclusion criteria: diagnosis of asthma, a significant lung disease other than COPD, a clinically significant and uncontrolled medical disorder including but not limited to cardiovascular, endocrine or metabolic, neurological, psychiatric, hepatic, renal, gastric and neuromuscular diseases, or had a COPD exacerbation that was not resolved at screening |
|
| Interventions |
Run‐in: four‐week run‐in period during which participants received open‐label fluticasone/salmeterol 250/50 via Diskus twice daily Treatments: 1. Fluticasone/salmeterol 250/50 mcg twice daily 2. Salmterol 50 mcg twice daily Inhaler device: Diskus Co‐treatment: As‐needed albuterol was provided for use throughout the study. Concurrent inhaled long‐acting bronchodilators (beta2‐agonist and anticholinergic), ipratropium/albuterol combination products, oral beta‐agonists, inhaled corticosteroids and theophylline preparations were not allowed during the treatment period. Oral corticosteroids and antibiotics were allowed for the short‐term treatment of COPD exacerbations |
|
| Outcomes |
Primary: annual rate of moderate to severe exacerbations Secondary: time to first moderate to severe exacerbation, annual rate of exacerbations requiring oral corticosteroids, predose FEV1, diary records of dyspnoea, night‐time awakenings due to COPD and use of supplemental albuterol |
|
| Notes |
Funding: GSK (ID SCO40043) Clinicaltrials.gov ID: NCT00144911 Definition of pneumonia: no objective definition given |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Centre‐based randomisation schedule |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Dropout high and fairly even (30% vs 38%). Method of imputation described only for the primary outcome (‘Endpoint was defined as the last scheduled measurement of predose AM FEV1 during the 52‐week treatment period’) |
| Selective reporting (reporting bias) | Low risk | Checked GSK documents—all stated and expected outcomes are reported |
Fukuchi 2013.
| Methods |
Design: randomised, double‐blind, parallel‐group study Three‐month study period Conducted at 163 centres in 9 countries (India, Japan, Republic of Korea, Phillipines, Poland, Russian Federation, Taiwan, Ukraine and Vietnam) |
|
| Participants |
Participants: 1293 people were randomly assigned to budesonide/formoterol combination (636) and formoterol alone (657)
Baseline characteristics: Male %: bud/form 87.6, form 90.3 Mean age (range), years: bud/form 64.5 (40 to 89), form 65.6 (40 to 87) Smoking history (mean (range) pack‐years): bud/form 43.4 (10 to 160), form 44.7 (0 to 300) Mean % predicted FEV1 (range): bud/form 40.9 (12 to 79), form 40.8 (8‐84) Inclusion criteria: male and female individuals, 40 years of age or older with a diagnosis of moderate to severe COPD for at least 2 years (pre‐bronchodilator forced expiratory volume in one second (FEV1) 50% of predicted normal or less, postbronchodilator FEV1/forced vital capacity (FVC) < 70%), current or previous smoking history of 10 or more pack‐years and having at least one COPD exacerbation in 12 months Exclusion criteria: history or current clinical diagnosis of asthma or atopic disease such as allergic rhinitis; significant or unstable ischaemic heart disease, arrhythmia, cardiomyopathy, heart failure, uncontrolled hypertension or any other relevant cardiovascular disorder; experiencing a COPD exacerbation during the run‐in period or within 4 weeks before randomisation that required hospitalisation and/or a course of oral or parenteral steroids; and requiring regular oxygen therapy |
|
| Interventions |
Run‐in: 1‐ to 2‐week run‐in period during which participants received open‐label formoterol 4.5 mg two inhalations twice daily and all other COPD medications were discontinued, with the exception of salbutamol 100 mg/actuation via pMDI Treatments: 1. Budesonide/formoterol combination 320/9 mcg twice daily 2. Formoterol 9 mcg twice daily Inhaler device: Turbuhaler Co‐treatment: Salbutamol 100 mg/actuation was available as reliever medication throughout the treatment period. During the randomised treatment period, participants were not permitted to take any other medication for their COPD, including beta2‐agonists, anticholinergics, leukotriene receptor antagonists, medications containing ephedrine or xanthine‐containing derivatives or inhaled corticosteroids |
|
| Outcomes |
Primary: change in predose FEV1 from baseline to treatment period Secondary: 1 hour postdose FEV1, predose and one hour postdose FVC, COPD symptoms, time to first COPD exacerbation, number of COPD exacerbations, health‐related quality of life on the SGRQ, morning and evening PEF, adverse events and vital signs |
|
| Notes |
Funding: AstraZeneca (ID: D589DC00007) Clinicaltrials.gov ID: NCT01069289 Definition of pneumonia: pneumonia, brochopneumonia and bacterial pneumonia counted as serious adverse events. Diagnosis not given |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomly assigned 1:1 to study treatment. No details of sequence generation methods, but presumed to adhere to usual AstraZeneca methods |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | From clinicaltrials.gov: masking: double‐blind (participant, caregiver, investigator, outcomes assessor) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | From clinicaltrials.gov: masking: double‐blind (participant, caregiver, investigator, outcomes assessor) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropout low and even between groups |
| Selective reporting (reporting bias) | Low risk | All outcomes were fully reported on clinical.trials.gov and in the published report |
GSK FCO30002 2005.
| Methods |
Design: multi‐centre, randomised, placebo‐controlled, double‐blind trial Three‐month treatment period Conducted at 32 centres in Germany |
|
| Participants |
Participants: 140 people were randomly assigned to salmeterol plus fluticasone (68) and salmeterol plus placebo (69)
Baseline characteristics: Male %: flut+salm 60.6, salm+placebo 71.2 Mean age (SD), years: flut+salm 61 (8), salm+placebo 63 (10) Smoking history (mean (SD) pack‐years): not reported Mean % predicted FEV1 (SD): not reported Inclusion criteria: documented history of COPD; male and female subjects 40 to 79 years of age; smokers and ex‐smokers with a smoking history of ≥ 10 pack‐years; FEV1 40% to 80% of predicted, FEV1/FVC < 70% at visit 1 or 2; low reversibility of airway obstruction at visit 1 or 2: increase in FEV1 (normal value) < 10% at 30 minutes after inhalation of 200 mcg salbutamol; symptomatic COPD during run‐in as documented in the participant diary: on ≥ 5 days, symptom score was > 5 and/or salbutamol inhalation; ability to correctly use the Mini‐Wright Peak‐Flow‐Meter and the Diskus™ inhaler Exclusion criteria: long‐term oxygen therapy; use of inhaled or systemic corticosteroids during the 8 weeks before study entry; acute exacerbation, antibiotic treatment or hospital stay within 4 weeks before study entry; use of beta blockers within two weeks before study entry |
|
| Interventions |
Run‐in: 2 weeks during which all participants received salmeterol 50 mcg twice daily as bronchodilator treatment and salbutamol as rescue medication Treatments: 1. Salmeterol 50 mcg plus fluticasone 500 mcg twice daily (and placebo tablets for 2 weeks) 2. Salmeterol 50 mcg plus placebo inhaler twice daily (and placebo tablets for 2 weeks) A third group receiving oral prednisolone in combination with salmeterol was not included in this review Inhaler device: Diskus Co‐treatment: salbutamol as rescue medication |
|
| Outcomes |
Primary: change in FEV1 Secondary: Participants' self assessment of exercise capacity and morning serum cortisol concentrations |
|
| Notes |
Funding: GSK Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Described as randomised (presumed to adhere to usual GSK methodology) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropout relatively low and even between groups (17.6% vs 14.5%). Full analysis set (FAS) and per‐protocol analyses were reported, but only the FAS was extracted |
| Selective reporting (reporting bias) | Unclear risk | All outcomes stated were reported. Pneumonia‐related outcomes were not reported. GSK data request was not successful at the time of publication |
GSK FLTA3025 2005.
| Methods |
Design: randomised, double‐blind, parallel‐group, comparative trial Six‐month treatment period Conducted at 55 centres in the United States |
|
| Participants |
Participants: 640 people were randomly assigned to fluticasone 250 (216), fluticasone 500 (218) and placebo (206)
Baseline characteristics: Male %: flut250 72.2, flut500 66.1, placebo 68.0 Mean age (SD), years: flut250 65.2 (8.7), flut500 63.3 (10.0), placebo 64.8 (9.5) Smoking history (mean (SD) pack‐years): not reported Mean % predicted FEV1 (SD): not reported Inclusion criteria: Male or female individuals were eligible if they were diagnosed with COPD; were at least 40 years of age; had a current or prior history of at least 20 pack‐years of cigarette smoking; had a history of cough productive of sputum on most days for at least 3 months of the year, for at least 2 years that was not attributable to another disease process; had a baseline FEV1 < 65% of predicted normal but > 0.70 L or FEV1 ≤ 0.70 L and > 40% of predicted normal and FEV1/forced vital capacity (FVC) ratio of < 0.70; had a score of ≥ 2 on the Modified Medical Research Council (MMRC) Dyspnea Scale at screening and a score of ≥ 4 on the CBSQ at randomisation and had not received systemic corticosteroids or high‐dose inhaled corticosteroid therapy for at least 6 months before screening Exclusion criteria: current diagnosis of asthma, concurrent participation in a pulmonary rehabilitation programme, respiratory disease other than COPD or other significant concurrent disease, an abnormal and clinically significant ECG at screening and the occurrence of a moderate or severe COPD exacerbation during the run‐in period |
|
| Interventions |
Run‐in: 2‐week placebo run‐in period Treatments: 1. Fluticasone propionate 250 mcg twice daily 2. Fluticasone propionate 500 mcg twice daily 3. Placebo twice daily Inhaler device: Diskus Co‐treatment: Concurrent use of the following respiratory medications was not allowed: beta‐agonists (other than salbutamol), cromolyns, corticosteroids (oral, inhaled and intranasal), anti‐leukotrienes and ipratropium. Concurrent use of theophylline was allowed. Use of antibiotics for the treatment of up to three COPD exacerbations was allowed |
|
| Outcomes |
Primary: morning predose FEV1 Secondary: Chronic Bronchitis Symptoms Questionnaire (CBSQ), Transition Dyspnoea Index (TDI, exacerbations of COPD, participant‐recorded daily morning PEF rate, supplemental salbutamol use, night‐time awakenings and quality of life (CDRQ) |
|
| Notes |
Funding: GSK Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised trial (GSK funded, likely to be computerised randomisation schedule) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded trial (presumed participant and personnel/investigator) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropout high but even across groups. The intent‐to‐treat (ITT) population consisted of all randomly assigned participants who received at least 1 dose of study medication. The ITT population was the primary population for all efficacy and safety analyses |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported in the results summary |
GSK SCO100470 2006.
| Methods |
Design: multi‐centre, randomised, double‐blind, double‐dummy, parallel‐group design 6 month treatment period Conducted at 135 centres in 20 countries (Australia (10), Bulgaria (5), Croatia (1), Czech Republic (8), France (14), Germany (18), Greece (4), Italy (16), Latvia (5), Lithuania (2), Netherlands (12), Philippines (3), Poland (5), Romania (3), Russian Federation (8), Slovakia (4), Slovenia (4), Sweden (4), Thailand (4) and United Kingdom (5)) |
|
| Participants |
Participants: 1050 people were randomly assigned to fluticasone/salmeterol combination (518) and salmeterol alone (532)
Baseline characteristics: Male %: flut/salm 78.4, salm 77.3 Mean age (SD), years: flut/salm 63.5 (9.4), salm 63.7 (9.0) Smoking history (mean pack‐years): not reported Mean % predicted FEV1 (SD): not reported Inclusion criteria: male or female, 40 to 80 years of age with an established history of GOLD stage II COPD; poor reversibility of airflow obstruction (defined as ≤ 10% increase in FEV1 as a percentage of normal predicted value); minimum score of ≥ 2 on the Modified Medical Research Council Dyspnoea Scale and a smoking history of at least 10 pack‐years. In addition, participants had to achieve a composite symptom score of ≥ 120 (out of 400 maximum score, measured using visual analogue scales) on at least 4 of the last 7 days of the run‐in period, and to have a Baseline Dyspnoea Index (BDI) score of ≤ 7 units at visit 2 Exclusion criteria: Participants would be excluded if they had asthma or atopic disease, had a lung disease likely to confound the drug response other than COPD, had a recent exacerbation (within 4 weeks of screening or during run‐in); were receiving long‐term oxygen therapy or pulmonary rehabilitation or had taken tiotropium bromide, inhaled corticosteroids or anti‐leukotriene medication within 14 days of visit one |
|
| Interventions |
Run‐in: run‐in mentioned, unclear duration Treatments: 1. Salmeterol/fluticasone propionate 50/250 mcg twice daily 2. Salmeterol 50 mcg twice daily Inhaler device: Diskus accuhaler Co‐treatment: not reported |
|
| Outcomes |
Primary: FEV1, Transitional Dyspnoea Index (TDI) Secondary: change from baseline in trough FEV1, change from baseline in trough FVC and FVC/FEV1 ratio, TDI focal score, change from baseline in postdose FEV1, FVC and FVC/FEV1 ratio, change from baseline in mean morning PEF, change from baseline in St George's Respiratory Questionnaire |
|
| Notes |
Funding: GSK Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Described as randomised (assumed to adhere to usual GSK methodology) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described—only results summary available |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropout low and even between groups (11.4% vs 13.9%). The ITT (intent‐to‐treat) population (all participants randomly assigned and confirmed as having received at least one dose of double‐blind study medication) was the primary population for analysis of all efficacy and health outcome variables; the safety population (identical to the ITT population) was used for analysis of all safety variables |
| Selective reporting (reporting bias) | Low risk | All stated outcomes were reported and no expected outcomes were missing |
GSK SCO104925 2008.
| Methods |
Design: multi‐centre, randomised, double‐blind, placebo‐controlled, parallel‐group study Three‐month treatment period Conducted at 11 centres (4 centres in the Russian Federation, 4 centres in the United States, 2 centres in Chile and 1 centre in Estonia) |
|
| Participants |
Participants: 161 people were randomly assigned to fluticasone (42), placebo (42), fluticasone/salmeterol combination (39) and salmeterol (38)
Baseline characteristics: Male %: flut 69.0, placebo 76.2, flut/salm 82.1, salm 78.9 Mean age (SD), years: flut 64.2 (11.2), placebo 65.2 (8.6), flut/salm 63.6 (7.8), salm 64.0 (9.3) Smoking history (mean (SD) pack‐years): not reported Mean % predicted FEV1 (SD): not reported Inclusion criteria: Males or females of non‐childbearing potential 40 years of age or older were eligible to participate if they had an established clinical history of COPD, evidence of bronchitis as a component of the COPD disease and a current or prior history of at least 10 pack‐years of cigarette smoking. Participants had a measured postalbuterol FEV1/FVC ≤ 70% at visit 1 (screening) and a measured postalbuterol FEV1 ≥ 30% and ≤ 70% of predicted normal Exclusion criteria: no other criteria reported |
|
| Interventions |
Run‐in: not reported Treatments: 1. Fluticasone propionate 500 mcg twice daily 2. Placebo twice daily 3. Fluticasone/salmeterol combination 500/50 mcg twice daily 4. Salmeterol 50 mcg twice daily Inhaler device: not reported Co‐treatment: not reported |
|
| Outcomes |
Primary: predose resistance difference between 5 Hz and 15 Hz (R5 to R15) as measured by IOS Secondary: predose‐ and 2 hours postdose low‐frequency reactance area (AX); 2 hours postdose R5 to R15; postalbuterol computed tomography (CT) parameters of area of airway wall and area of airway lumen |
|
| Notes |
Funding: GSK Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomly assigned to treatment (assumed to adhere to GSK protocol) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Described as double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Withdrawal uneven between groups but all less than 20%. ITT analysis used |
| Selective reporting (reporting bias) | Low risk | All outcomes reported in detail |
GSK SCO30002 2005.
| Methods |
Design: multi‐centre, randomised, double‐blind, parallel‐group, placebo‐controlled study 12‐Month treatment period Conducted at 49 centres in Italy and 7 in Poland |
|
| Participants |
Participants: 256 people were randomly assigned to fluticasone (131) and placebo (125)
Baseline characteristics: Male %: flut 83, placebo 80 Mean age (SD), years: flut 64.6 (8.7), placebo 65.7 (9.0) Smoking history (mean (SD) pack‐years): not reported Mean % predicted FEV1 (SD): not reported Inclusion criteria: male or female individuals aged > 40 years with an established clinical history of COPD; participants who demonstrated at visit 1 a pre‐bronchodilator baseline FEV1/VC < 88% for men and < 89% for women of predicted normal values and FEV1 ≤ 70% of predicted normal value, but > 800 mL; participants who demonstrated at visit 1, poor reversibility of airflow obstruction, defined as an increase in FEV1 < 10% of the normal predicted FEV1 value (or < 200 mL from baseline), 30 minutes after inhalation of 400 mcg salbutamol via MDI; current smokers or ex‐smokers with a smoking history of at least 10 pack‐years Exclusion criteria: as above |
|
| Interventions |
Run‐in: two‐week run‐in during which all inhaled corticosteroids and long‐acting beta2‐agonists were discontinued Treatments: 1. Fluticasone propionate 500 mcg twice daily 2. Placebo twice daily Inhaler device: metered‐dose inhaler Co‐treatment: not reported |
|
| Outcomes |
Primary: time to first moderate or severe exacerbation Secondary: number and severity of exacerbations, withdrawals due to exacerbations, clinic FEV1, VC, FEV1/VC, daily record card symptoms, PEFR, distance walked in the six‐minute walk test (SWT), perceived breathlessness before and after SWT, quality of life (SGRQ), use of relief medication, adverse events, SAEs on therapy |
|
| Notes |
Funding: GSK Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomly assigned to treatment. No details given but assumed to adhere to GSK methodology |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropout high at 26% and 32% for ICS and placebo, respectively. The safety population/Intent‐to‐treat (ITT) population consisted of all randomly assigned participants who took study medication (all of those randomly assigned) |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the GSK summary were reported in detail |
GSK SCO40041 2008.
| Methods |
Design: randomised, double‐blind, parallel‐group trial Three‐year treatment period Conducted at 31 centres in the United States |
|
| Participants |
Participants: 186 people were randomly assigned to fluticasone/salmeterol combination (92) and salmeterol alone (94)
Baseline characteristics: Male %: flut/salm 59.8, salm 62.8 Mean age (SD), years: flut/salm 65.4 (8.4), salm 65.9 (9.5) Smoking history (mean (SD) pack‐years): not reported Mean % predicted FEV1 (SD): not reported Inclusion criteria: male/female participants with an established clinical history of COPD (including a history of exacerbations), a baseline (pre‐bronchodilator) FEV1 < 70% of the predicted normal value, a baseline (pre‐bronchodilator) FEV1/FVC ratio of 70%, at least one evaluable native hip and a smoking history of ≥ 10 pack‐years Exclusion criteria: no information |
|
| Interventions |
Run‐in: not reported Treatments: 1. Fluticasone propionate/salmeterol 250/50 mcg twice daily 2. Salmeterol 50 mcg twice daily Inhaler device: Diskus Co‐treatment: 'permitted COPD therapy' unclear |
|
| Outcomes |
Primary: change in bone mineral density at the lumbar spine Secondary: change in bone mineral density at the hip, adverse events, serious adverse events, fatal SAEs |
|
| Notes |
Funding: GSK Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomly assigned to treatment (no specific information but assumed to adhere to GSK methods) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind (presumed participant and personnel/investigator) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawal very high in both groups (39% and 41%) |
| Selective reporting (reporting bias) | Low risk | All outcomes described in the GSK summary were reported |
Hanania 2003.
| Methods |
Design: randomised, double‐blind, multi‐centre, placebo‐controlled study Six‐month treatment period Conducted at 76 investigative sites in the United States |
|
| Participants |
Participants: 723 people were randomly assigned to fluticasone/salmeterol combination (178), fluticasone (183), salmeterol (177) and placebo (185)
Baseline characteristics: Male %: flut/salm 61, flut 66, salm 58, placebo 68 Mean age (range), years: flut/salm 63 (40 to 87), flut 63 (40 to 84), salm 64 (42 to 87), placebo 65 (40 to 81) Smoking history (median (range) pack‐years): flut/salm 53 (20 to 220), flut 60 (20 to 162), salm 57 (20 to 224), placebo 56 (20 to 165) Mean % predicted FEV1 (SD): flut/salm 41 (11), flut 42 (11), salm 42 (12), placebo 42 (12) Inclusion criteria: Participants were > 40 years of age, were current or former smokers with a > 20 pack‐year history and had received a diagnosis of COPD, as defined by the American Thoracic Society. Inclusion criteria required a baseline FEV1/FVC ratio of < 70% and a baseline FEV1 of < 65% of predicted normal, but > 0.70 L (or if < 0.70 L, then > 40% of predicted normal). Participants were required to have symptoms of chronic bronchitis and moderate dyspnoea Exclusion criteria: current diagnosis of asthma; use of oral corticosteroids within the past six weeks; abnormal clinically significant ECG; long‐term oxygen therapy; moderate or severe exacerbation during the run‐in period; and any significant medical disorder that would place the individual at risk, interfere with evaluations or influence study participation |
|
| Interventions |
Run‐in: two‐week, single‐blind run‐in period during which participants received placebo and albuterol and discontinued use of corticosteroids and bronchodilators with the exception of stable regimens of theophylline Treatments: 1. Fluticasone propionate 250 mcg twice daily 2. Salmeterol 50 mcg twice daily 3. Fluticasone/salmeterol 250/50 mcg twice daily 4. Placebo twice daily Inhaler device: Diskus Co‐treatment: Participants were given as‐needed albuterol and were not allowed corticosteroids or bronchodilators, with the exception of stable regimens of theophylline |
|
| Outcomes |
Primary: predose and 2 hours postdose FEV1 Secondary: morning PEF, dyspnoea (TDI), supplemental albuterol use, health status (CRDQ), Chronic Bronchitis Symptom Questionnaire, exacerbations, adverse events, ECG, vital signs and clinical laboratory evaluations |
|
| Notes |
Funding: GSK (ID: SFCA3007) Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was stratified by reversibility (no other info but GSK sponsored—likely to adhere to GSK methods) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind trial (presumed participant and personnel/investigator) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropout relatively high but even across groups. To account for participant withdrawals, endpoint was used as the primary time point and was defined as the last on‐treatment post‐baseline assessment excluding any data from the discontinuation visit |
| Selective reporting (reporting bias) | Low risk | Compared with GSK result summary and protocol—no evidence of publication bias |
Hattotuwa 2002.
| Methods |
Design: double‐blind, placebo‐controlled study Three‐month study period Conducted at the London Chest Hospital |
|
| Participants |
Participants: 36 people were randomly assigned to fluticasone (17) and placebo (19)
Baseline characteristics: Male %: flut 81.3, placebo 92.9 Mean age (SD), years: flut 64.7 (6.2), placebo 64.7 (6.5) Smoking history (mean (SD) pack‐years): flut 64.9 (50.3), placebo 60.3 (46.6) Mean % predicted FEV1 (SD): flut 46.2 (13.6), placebo 45.5 (16.1) Inclusion criteria: male or female, 40 to 75 years of age, current smokers or ex‐smokers with more than 20 pack‐years of smoking, non‐atopic and with an FEV1 25% to 80% of predicted, which improved by less than 15% over baseline and 200 mL after 200 mcg inhaled salbutamol Exclusion criteria: Individuals with severe concurrent medical problems, psychological impairment on immunosuppressive treatment or with a chest infection within 8 weeks were excluded. Participants who were already taking inhaled steroids had the drug withdrawn and had to be stable for at least 8 weeks before the first biopsy |
|
| Interventions |
Run‐in: After recruitment, participants had a run‐in period of 8 weeks to ensure that they were stable before the first bronchoscopy Treatments: 1. Fluticasone 500 mcg twice daily 2. Placebo twice daily Inhaler device: multi‐dose dry powder Accuhaler Co‐treatment: All reliever medications (inhaled beta2‐agonist, anticholinergics and theophylline) were continued as before |
|
| Outcomes |
Primary: bronchoscopy Secondary: spirometry (FEV1 and VC), biopsy processing and counts, symptom scores |
|
| Notes |
Funding: Glaxo‐Wellcome and Departmental Funds Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomly assigned to FP or P in a double‐blind manner using a random numbers table |
| Allocation concealment (selection bias) | Low risk | Randomly assigned in a double‐blind manner |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Seven of the randomly assigned participants did not appear in the analyses (18.9%) but were counted in the adverse event data relevant for this review |
| Selective reporting (reporting bias) | High risk | Serious adverse events not reported. No reply from study authors at time of publication |
Kardos 2007.
| Methods |
Design: randomised, double‐blind, parallel‐group study 10‐Month study period Conducted at 95 respiratory centres in Germany |
|
| Participants |
Participants: 994 people were randomly assigned to fluticasone/salmeterol combination (507) and salmeterol alone (487)
Baseline characteristics: Male %: flut/salm 74.0, salm 77.6 Mean age (SD), years: flut/salm 63.8 (8.3), salm 64.0 (8.2) Smoking history (mean pack‐years): flut/salm 36.8, salm 37.0 Mean % predicted FEV1 (SD): flut/salm 40.4 (8.9), salm 40.3 (8.5) Inclusion criteria: outpatients with postbronchodilator FEV1 < 50% predicted, FEV1/FVC of 70% predicted or less, age 40 years or older, smoking history of 10 or more pack‐years and a documented history of two or more moderate to severe exacerbations during the year before the study Exclusion criteria: Individuals with COPD exacerbations, hospital admissions or change in COPD therapy during the 4 weeks before visit one or during the 4‐week run‐in period were excluded. Those with asthma, significant lung diseases other than COPD and need for long‐term oxygen therapy or long‐term systemic steroid use were also excluded |
|
| Interventions |
Run‐in: four weeks Treatments: 1. Salmeterol/fluticasone 50/500 mcg twice daily 2. Salmeterol 50 mcg twice daily Inhaler device: Diskus Co‐treatment: Inhaled salbutamol was used as reliever medication, and regular treatment with short‐acting bronchodilators, antioxidants/mucolytics, short‐acting oral beta2‐agonists and theophylline was permitted |
|
| Outcomes |
Primary: number of exacerbations Secondary: Time to first exacerbation, pre‐bronchodilator PEF, postbronchodilator FEV1, SGRQ, symptoms and breathlessness, reliever medication use and use of other COPD medications were recorded on diary cards |
|
| Notes |
Funding: GSK Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Consecutive numbers assigned to participants determined the blinded treatment based on a centrally generated list with blocks of six |
| Allocation concealment (selection bias) | Low risk | Randomisation list was centrally generated |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind treatment (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar withdrawal rates in each group. ITT included 99.6% of the randomly assigned population (4 participants were excluded as the result of a randomisation error) |
| Selective reporting (reporting bias) | Unclear risk | Unable to locate prospective trial registration to check that all outcomes were reported. Study author contacted and forwarded request to GSK—no data were provided in time for publication |
Kerwin 2013.
| Methods |
Design: randomised, multi‐centre, placebo‐controlled, parallel‐group trial Six‐month treatment period Conducted at 221 centres in nine countries (Chile, Estonia, Germany, Japan, Korea, Phillipines, Poland, Russian Federation and the United States) |
|
| Participants |
Participants: 1030 people were randomly assigned to placebo (207), fluticasone furoate 100 mcg (206), vilanterol 25 mcg (205), fluticasone furoate 50 mcg and vilanterol combination (206) and fluticasone furoate 100 mcg and vilanterol combination (206)
Baseline characteristics: Male %: placebo 68, flut100 64, vil 68, flut50/vil 66, flut100/vil 67 Mean age (SD), years: placebo 62.1, flut100 62.7, vil 63.4, flut50/vil 62.8, flut100/vil 62.3 Smoking history (mean (SD) pack‐years): placebo 45.6, flut100 46.2, vil 47.6, flut50/vil 44.2, flut100/vil 46.6 Mean % predicted FEV1 (SD): placebo 42.4, flut10041.5, vil 44.5, flut50/vil 42.5, flut100/vil 42.3 Inclusion criteria: over 40 years of age and have a clinical diagnosis of COPD, a smoking history of 10 pack‐years, a postbronchodilator FEV1/forced vital capacity (FVC) ratio of less than or equal to 0.70, a postbronchodilator FEV1 less than or equal to 70% predicted (NHANES III) and a score of greater than or equal to 2 on the Modified Medical Research Council Dyspnoea Scale (mMRC). A history of COPD exacerbations was not required for individuals to be eligible to enter the study. Albuterol reversibility was assessed at the screening visit, and both reversible and non‐reversible individuals were eligible to enter the study Exclusion criteria: current diagnosis of asthma or other non‐COPD respiratory disorders; lung volume reduction surgery within 12 months of visit one; poorly controlled COPD, defined as acute worsening of COPD requiring patient‐managed therapy with corticosteroids or antibiotics or treatment prescribed by a physician within 6 weeks before visit 1; hospitalisation due to poorly controlled COPD within 12 weeks before visit 1; lower respiratory tract infection that required the use of antibiotics within 6 weeks before visit 1; the need for long‐term oxygen therapy or nocturnal oxygen therapy (greater than or equal to 12 hours/d) |
|
| Interventions |
Run‐in: 2‐week, single‐blind run‐in period during which participants received placebo once daily in the morning via a dry powder inhaler that contains two strips Treatments: 1. Fluticasone furoate 100 mcg daily 2. Vilanterol 25 mcg daily 3. Fluticasone 50 mcg/ vilanterol 25 mcg daily 4. Fluticasone 100 mcg/ vilanterol 25 mcg daily 5. Placebo Inhaler device: dry powder inhaler Co‐treatment: Albuterol, ipratropium (provided that the person was on a stable dose from the screening visit throughout the study), mucolytics, antibiotics for short‐term treatment, cough suppressants for short‐term treatment, intranasal decongestants and corticosteroids, flu and pneumonia vaccines, MAOIs, medications for other disorders as long as the dose remained constant whenever possible and their use were not expected to affect lung function |
|
| Outcomes |
Primary: weighted mean FEV1 (0 to 4 hours postdose) on day 168 to assess bronchodilation by FF/VI and VI (vs placebo) and FF/VI vs FF; and the change from baseline in trough (23 to 24 hours postdose) FEV1 on day 169 to assess the 24‐hour effect of VI and to determine the contribution of FF to lung function (i.e. FF/VI vs VI) Secondary: CRQ self administered standardised dyspnoea domain on day 168, various FEV1 parameters, PEF, symptom measures, adverse events, exacerbations and pneumonia events |
|
| Notes |
Funding: GSK (ID: HZC112206) Clinicaltrials.gov ID: NCT01053988 Definition of pneumonia: presumptive diagnosis or radiographically confirmed |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation schedule was generated using a validated computerised system (RandAll; GlaxoSmithKline, London, UK) |
| Allocation concealment (selection bias) | Low risk | Participants were randomly assigned using the Registration and Medication Ordering System (RAMOS; GlaxoSmithKline, London UK) to register and randomly assign the participant and to receive medication assignment information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Protocol stated that the study medication was double‐blind for participant and investigator |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated in protocol. Some details in supplementary material about outcome assessors but unclear who was blind |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropout was quite high but even across groups (26.7% to 33.3%). ITT population used |
| Selective reporting (reporting bias) | Low risk | All stated outcomes were available in full in the published report and on clinicaltrials.gov |
Lapperre 2009.
| Methods |
Design: double‐blind, parallel, 4‐group, placebo‐controlled, randomised design 2.5‐Year treatment period Conducted at 2 centres in the Netherlands |
|
| Participants |
Participants: 55 people were randomly assigned to fluticasone (26) and placebo (29)
Baseline characteristics: Male %: flut 88.5, placebo 83.3 Mean age (SD), years: flut 62 (8), placebo 59 (8) Smoking history (mean (range) pack‐years): flut 44 (31 to 55), placebo 42 (34 to 54) Mean % predicted FEV1 (SD): flut 57 (9.9), placebo 54 (8.3) Inclusion criteria: 45 to 75 years of age, were current or former smokers, had smoked for 10 or more pack‐years and had lung function levels compatible with Global Initiative for Chronic Obstructive Lung Disease (GOLD) stages II and III Exclusion criteria: asthma and receipt of ICS within 6 months before random assignment. Important co‐morbid conditions |
|
| Interventions |
Run‐in: not reported Treatments: 1. Fluticasone 500 mcg twice daily 2. Placebo twice daily Inhaler device: Diskus dry powder inhaler Co‐treatment: short‐acting bronchodilators |
|
| Outcomes |
Primary: inflammatory cell counts in bronchial biopsies and induced sputum Secondary: postbronchodilator spirometry, hyperresponsiveness to methacholine PC20, dyspnoea score by the MRC scale, health status by the SGRQ and the Clinical COPD Questionnaire |
|
| Notes |
Funding: Netherlands Asthma Foundation, both centres and GSK Clinicaltrials.gov ID: NCT00158847 Definition of pneumonia: confirmed by chest x‐ray |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | At entry, an independent randomisation centre provided participant and medication numbers by using a minimisation procedure that balanced treatment groups for centre, sex, smoking status, FEV1/IVC 60% and methacholine PC20 (the provocative concentration of methacholine that causes a 20% decrease in FEV1) 2 mg/mL) |
| Allocation concealment (selection bias) | Low risk | An independent randomisation centre provided participant and medication numbers |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study medications were individually numbered, and we used Diskus dry powder inhalers (GlaxoSmithKline, Zeist, The Netherlands) with 60 doses per inhaler; all active treatment medications and placebo were identical in appearance |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawal rates were very high compared with the numbers of events for the different outcomes. Per‐protocol analysis was used |
| Selective reporting (reporting bias) | Low risk | Data for several outcomes not available from the published report, but study authors provided data upon request |
Laptseva 2002.
| Methods |
Design: randomised, double‐blind, parallel‐group study Six‐month treatment period |
|
| Participants |
Participants: 49 people were randomly assigned to budesonide (25) and placebo (24)
Baseline characteristics: none reported—abstract only Inclusion criteria: individuals between 40 and 65 years of age, FEV1 40% to 60% of predicted normal, FEV1/VC < 55%, bronchodilator reversibility < 15% Exclusion criteria: not reported |
|
| Interventions |
Run‐in: not reported Treatments: 1. Budesonide 400 mcg twice daily 2. Placebo Inhaler device: not reported Co‐treatment: All participants received anticholinergic drug and methylxanthine or short‐acting beta2 agent |
|
| Outcomes | Number and severity of exacerbations, FEV1, FVC, diary card symptoms, PEFR | |
| Notes |
Abstract only Funding: not reported Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No details provided |
| Selective reporting (reporting bias) | High risk | Only abstract available. Outcomes could not be used. Could not find contact information for study authors |
Mahler 2002.
| Methods |
Design: randomised, double‐blind, placebo‐controlled, parallel‐group, multi‐centre trial 6 ‐month treatment period Conducted at 65 centres |
|
| Participants |
Participants: 674 people were randomly assigned to fluticasone (168), placebo (181), fluticasone/salmeterol combination (165) and salmeterol alone (160)
Baseline characteristics: Male %: flut 61, placebo 75, flut/salm 62, salm 64 Mean age (range), years: flut 64.4 (42 to 82), placebo 64 (44 to 90), flut/salm 61.9 (40 to 86), salm 63.5 (40 to 84) Smoking history (median (range) pack‐years): flut 54 (20 to 200), placebo 60 (20 to 165), flut/salm 55 (15 to 150), salm 52.5 (20 to 193) Mean % predicted FEV1 (no SD reported): flut 41, placebo 41, flut/salm 41, salm 40 Inclusion criteria: baseline FEV1/FVC of 70% or less and baseline FEV1 of less than 65% of predicted but more than 0.70 L. Participants were required to have daily cough productive of sputum for 3 months of the year for 2 consecutive years and dyspnoea Exclusion criteria: Specific exclusion criteria were current diagnosis of asthma, oral corticosteroid use within the past 6 weeks, abnormal clinically significant electrocardiogram, long‐term oxygen therapy, moderate or severe exacerbation during the run‐in and any clinically significant medical disorder |
|
| Interventions |
Run‐in: 2‐week, single‐blind, run‐in period during which participants received placebo via Diskus on an as‐needed basis and discontinued use of corticosteroids and bronchodilators, with the exception of stable regimens of theophylline Treatments: 1. Fluticasone 500 mcg twice daily 2. Placebo twice daily 3. Fluticasone/Salmeterol combination 500/50 mcg twice daily 4. Salmeterol 50 mcg twice daily Inhaler device: Diskus Co‐treatment: as needed albuterol and stable regimens of theophylline |
|
| Outcomes |
Primary: change in predose FEV1 values and change in 2‐hour postdose FEV1 values Secondary: morning and evening PEF, supplemental albuterol use, dyspnoea as assessed by the TDI, Chronic Bronchitis Symptom Questionnaire, exacerbations defined by treatment, health status on the Chronic Respiratory Disease Questionnaire, adverse events, 24‐hour Holter monitoring and vital signs |
|
| Notes |
Funding: GSK (protocol number SFCA3006) Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was stratified by reversibility and investigative site to ensure balance between treatment groups at each site and in terms of the number of reversible participants (no other details, industry‐sponsored) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind (presumed participant and personnel/investigator) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 17 participants at one investigative site were not evaluable because of poor study practices, and of the remaining 674 participants, 645 had an evaluable baseline assessment. A total of 234 participants were discontinued from the study (between 28% and 40% across groups) |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were reported in detail. Only one secondary outcome was not available |
Martinez 2013.
| Methods |
Design: multi‐centre, randomised, stratified (by smoking status), placebo‐controlled, double‐blind, parallel‐group study 6 month treatment period Conducted at 145 study centres in 8 countries (Czech Republic, Germany, Japan, Poland, Romania, Russian Federation, Ukraine and the United States) |
|
| Participants |
Participants: 1224 people were randomly assigned to fluticasone furoate 100 mcg (204), fluticasone furoate 200 mcg (203), placebo (205), fluticasone furoate 100 mcg and vilanterol combination (204), fluticasone furoate 100 mcg and vilanterol combination (205) and vilanterol (203)
Baseline characteristics: Male %: flut100 74, flut200 74, placebo 74, flut100/vil 71, flut200/vil 67, vil 74 Mean age (SD), years: flut100 61.8 (8.3), flut200 61.8 (9.0), placebo 61.9 (8.1), flut100/vil 61.9 (8.8), flut200/vil 61.1 (8.6), vil 61.2 (8.6) Smoking history (mean (SD) pack‐years): flut100 39.8 (21.3), flut200 43.5 (22.5), placebo 45.7 (25.8), flut100/vil 42.8 (23.9), flut200/vil 41.5 (23.4), vil 42.0 (23.3) Mean % predicted FEV1 (SD): flut100 48.4 (12.2), flut200 47.1 (12.0), placebo 48.3 (12.7), flut100/vil 48.1 (12.9), flut200/vil 47.1 (12.8), vil 48.5 (12.9) Inclusion criteria: clinical diagnosis of COPD, 40 years of age or older, smoking history of 10 or more pack‐years, postbronchodilator FEV1/ forced vital capacity (FVC) ratio of 0.70 or less, postbronchodilator FEV1 70% predicted or less (NHANES III) and score of 2 or higher on the Modified Medical Research Council Dyspnoea Scale (mMRC). No prior history of COPD exacerbations was required for individuals to be eligible to enter the study. Reversibility to albuterol was assessed at the screening visit; both reversible and non‐reversible individuals were eligible to enter the study Exclusion criteria: any respiratory disorder other than COPD; lung volume reduction surgery within 12 months of screening; acute worsening (participant‐managed corticosteroid or antibiotic treatment or physician prescription) of COPD within six weeks of screening, hospitalisation for COPD over 12 weeks or lower respiratory tract infection that required the use of antibiotics in the 6 weeks before screening; the need for long‐term oxygen therapy or nocturnal oxygen therapy (12 or more hours/d) |
|
| Interventions |
Run‐in: 2‐week, single‐blind run‐in period during which participants received placebo once daily in the morning via a dry powder inhaler (DPI) that contained two strips Treatments: 1. Fluticasone furoate 100 mcg daily 2. Fluticasone furoate 200 mcg daily 3. Placebo 4. Fluticasone furoate 100 mcg/vilanterol 25 mcg daily 5. Fluticasone furoate 200 mcg/vilanterol 25 mcg daily 6. Vilanterol 25 mcg Inhaler device: dry powder inhaler Co‐treatment: albuterol, ipratropium (provided that the person was on a stable dose from the screening visit throughout the study), mucolytics, antibiotics for short‐term treatment, cough suppressants for short‐term treatment, intranasal decongestants and corticosteroids, flu and pneumonia vaccines, MAOIs, medications for other disorders as long as the dose remained constant whenever possible and when their use was not expected to affect lung function |
|
| Outcomes |
Primary: weighted mean FEV1 (zero to four hours postdose) on day 168 to assess bronchodilation by FF/VI and VI (vs placebo) and FF/VI versus FF; change from baseline in trough (23 to 24 hours postdose) FEV1 on day 169 to assess the 24‐hour effect of VI and to determine the contribution of FF to lung function (i.e. FF/VI vs VI) Secondary: CRQ self administered standardised dyspnoea domain on day 168, various FEV parameters, PEF, symptom measures, adverse events, exacerbations and pneumonia events |
|
| Notes |
Funding: GSK (ID: HZC112207) Clinicaltrials.gov ID: NCT01054885 Definition of pneumonia: presumptive diagnosis or radiographically confirmed |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation schedule was generated using a validated computerised system (RandAll; GlaxoSmithKline, London, UK) |
| Allocation concealment (selection bias) | Low risk | Participants were randomly assigned using the Registration and Medication Ordering System (RAMOS; GlaxoSmithKline, London, UK) to register and randomly assign the participant and to receive medication assignment information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Protocol stated that the study medication was double‐blind for participant and investigator |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated in protocol. Some details in supplementary material about outcome assessors but unclear who was blind |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropout in all groups was less than 30% (range 20.1% to 29.4%) and an ITT analysis was used |
| Selective reporting (reporting bias) | Low risk | All stated outcomes were available in full in the published report and in supplementary tables |
Mirici 2001.
| Methods |
Design: randomised, placebo‐controlled, double‐blind, parallel‐group study Three‐month treatment period Conducted at a single centre in Turkey |
|
| Participants |
Participants: 50 people were randomly assigned to budesonide (25) and placebo (25)
Baseline characteristics: Male %: bud 70, placebo 80 Mean age (SD), years: bud 51.8 (9.5), placebo 54.5 (10.3) Smoking history (mean (SD) pack‐years): bud 21.7 (12.5), placebo 31.3 (19.1) Mean % predicted FEV1 (SD): bud 64.1 (6.5), placebo 59.9 (8.2) Inclusion criteria: FEV1 < 70%, FEV1 reversibility after inhalation of terbutaline from a Turbuhaler of less than 15% of pre‐bronchodilator FEV1. All participants were smokers who refused or failed a programme to quit smoking Exclusion criteria: long‐term treatment with oral or inhaled corticosteroids within 6 months of study entry, respiratory tract infection in the previous 3 months, pregnancy or lactation and the presence of other serious systemic disease |
|
| Interventions |
Run‐in: not reported Treatments: 1. Budesonide 400 mcg twice daily 2. Placebo twice daily Inhaler device: Turbuhaler Co‐treatment: Beta2‐agonists of all kinds, theophylline and mucolytics were allowed. Inhaled corticosteroids other than study medication and oral or parenteral corticosteroids were not allowed |
|
| Outcomes | Spirometry and sputum cell analysis | |
| Notes |
Funding: unclear Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation sequence was computer‐generated at the Research Centre of the Faculty of Medicine |
| Allocation concealment (selection bias) | Low risk | Randomisation was masked and case numbers were allocated in consecutive order |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind. All inhalers had the same appearance and did not have drug labels |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | All spirometric indices and sputum cell analyses were performed at baseline and after treatment, blind to the clinical details |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Equal and fairly low dropout per group, values not imputed |
| Selective reporting (reporting bias) | High risk | Key expected outcomes were not reported (mortality and adverse events). Named outcomes were well reported. No reply from authors at the time of publication |
Ozol 2005.
| Methods |
Design: randomised, double‐blind, placebo‐controlled design Six‐month treatment period Conducted at a single centre in Turkey |
|
| Participants |
Participants: 26 people were randomly assigned to budesonide (13) and placebo (13)
Baseline characteristics: Male %: bud 84.6, placebo 53.8 Mean age (SD), years: bud 64.9 (6.1), placebo 65.9 (8.1) Smoking history (mean (SD) pack‐years): bud 45.6 (22.2), placebo 44.4 (23.0) Mean % predicted FEV1 (SD): bud 61.1 (9.7), placebo 57.3 (11.2) Inclusion criteria: (1) FEV1/FVC <70% and FEV1 50% of predicted value, (2) reversibility with inhaled beta2‐agonists (400 mg salbutamol) of less than 200 mL or less than 12% of predicted FEV1, (3) stable COPD defined as no acute exacerbation within preceding 3 months, (4) no history of systemic disease or other pulmonary disease, (5) no therapy with inhaled or systemic corticosteroids within 3 months before entry into the study and (6) no history of asthma or atopy Exclusion criteria: no additional information |
|
| Interventions |
Run‐in: not reported Treatments: 1. Budesonide 400 mcg twice daily 2. Placebo twice daily Inhaler device: dry powder inhaler Co‐treatment: All participants were receiving therapy with inhaled salbutamol and ipratropium bromide. For 9 participants, sustained‐released theophyline was also given |
|
| Outcomes |
Primary: unclear Secondary: FVC, FEV1, diary card data, inflammatory measures |
|
| Notes |
Funding: unclear Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomly assigned by a computer‐generated, blinded randomisation list |
| Allocation concealment (selection bias) | Low risk | ‘Blinded’ randomisation list |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Cells were counted by our pathologist, who was also blinded. Not clear for other outcomes |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | ‘The results presented are an analysis of 22 subjects (12 budesonide‐treated subjects and 10 placebo‐treated subjects) who completed the study’. Both dropout rates were low but uneven between groups (two were excluded from the placebo group and one from the budesonide group. One extra, presumed to have been randomly assigned to placebo (assuming equal group size at randomisation) was excluded for failure to take the medication consistently) |
| Selective reporting (reporting bias) | High risk | Key expected outcomes missing (mortality, serious adverse events). No reply from study authors by time of publication |
Paggiaro 1998.
| Methods |
Design: multi‐centre, randomised, placebo‐controlled trial Six‐month treatment period Conducted in 13 European countries, New Zealand and South Africa |
|
| Participants |
Participants: 281 people were randomly assigned to fluticasone (142) and placebo (139)
Baseline characteristics: Male %: flut 70, placebo 78 Mean age (no SD reported), years: flut 62, placebo 64 Smoking history (mean (SD) pack‐years): not reported Mean % predicted FEV1 (SD): flut 59 (18), placebo 55 (17) Inclusion criteria: current or ex‐smokers, 50 to 75 years of age with a history of smoking equivalent to at least 10 pack‐years and chronic bronchitis (a cough with excess sputum production for at least 3 months in at least 2 consecutive years with no other pathology). Participants also had to have a history of at least one exacerbation per year for the previous 3 years that required a visit to their doctor or hospital; high expectation, according to the investigator, of experiencing an exacerbation during the 6‐month treatment period; regular productive cough; predicted FEV1 of 35% to 90%, ratio of FEV1 to forced vital capacity of 70% or less and reversibility in FEV1 of less than 15% after inhalation of 400 mcg or 800 mcg salbutamol via a metered‐dose inhaler or Diskhaler Exclusion criteria: Individuals with abnormal chest radiographs or who had received oral or depot steroids, inhaled steroids of more than 500 mcg daily or antibiotic therapy; had been admitted to hospital in the 4 weeks before the study; or were currently taking fluticasone propionate were excluded |
|
| Interventions |
Run‐in: 2 week run‐in period during which usual inhaled steroids were stopped and participants received salbutamol as required Treatments: 1. Fluticasone propionate 500 mcg twice daily 2. Placebo twice daily Inhaler device: metered‐dose inhalers, with a spacer if desired Co‐treatment: Participants could take short‐acting beta2‐agonists for relief of symptoms as required throughout the study. Other COPD medications, such as anticholinergics and xanthine derivatives, could be continued throughout the study without dose changes |
|
| Outcomes |
Primary: COPD exacerbations Secondary: FEV1, morning PEF, FVC, 6‐minute walk test, Borg score, diary card symptom scores, daily sputum volume, total adverse events, serum cortisol concentration |
|
| Notes |
Funding: unclear (‘code was held by the sponsor company’s statisticians’) Definition of pneumonia:Not provided |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random numbers were computer‐generated on PACT (version 2.7) |
| Allocation concealment (selection bias) | Low risk | All investigators were given a set of four or more sealed envelopes containing assignment codes, from which they assigned treatment, starting with the lowest number |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind (presumed participant and personnel/investigator) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 'We did analysis by intention to treat of all patients who took at least one dose of study medication’ ‘Only available data was analysed’. Dropout uneven |
| Selective reporting (reporting bias) | High risk | Key expected outcomes not reported (mortality and serious adverse events). No reply from study authors by time of publication |
Pauwels 1999.
| Methods |
Design: parallel‐group, double‐blind, placebo‐controlled, randomised, multi‐centre study 3 year treatment period Conducted at 39 study centres in nine European countries (Belgium, Denmark, Finland, Italy, the Netherlands, Norway, Spain, Sweden and the United Kingdom) |
|
| Participants |
Participants: 1277 people were randomly assigned to budesonide (634) and placebo (643)
Baseline characteristics: Male %: bud 73.5, placebo 72.2 Mean age (SD), years: bud 52.5 (7.5), placebo 52.4 (7.7) Smoking history (mean (SD) pack‐years): bud 39.4 (20.1), placebo 39.2 (20.2) Mean % predicted FEV1 (SD): bud 76.8 (12.4), placebo 76.9 (13.2) Inclusion criteria: Persons 30 to 65 years of age were eligible if they were currently smoking at least five cigarettes per day and had smoked cigarettes for at least 10 years or had a smoking history of at least 5 pack‐years. FEV1 after use of a bronchodilator had to be between 50% and 100% of predicted normal value, and ratio of pre‐bronchodilator FEV1 to slow vital capacity had to be less than 70%. Increase in FEV1 after inhalation of 1 mg of terbutaline from a dry powder inhaler had to be less than 10% of predicted normal value. Change in FEV1 between the end of the first three‐month period of the run‐in phase and the end of the second had to be less than 15% Exclusion criteria: Participants with history of asthma, allergic rhinitis or allergic eczema and those who had used oral glucocorticoids for longer than four weeks during the preceding six months were excluded. Use of inhaled glucocorticoids other than the study medication, beta‐blockers, cromones or long‐acting inhaled beta2‐adrenergic agonists was not allowed |
|
| Interventions |
Run‐in: three‐month smoking cessation programme. For participants who did not stop smoking, this phase was followed by a three‐month period during which compliance with inhaled medication was assessed with the use of a placebo containing dry powder inhaler with a hidden mechanical counter. Participants who continued smoking and were at least 75% compliant with the recommended treatment regimen were randomly assigned Treatments: 1. Budesonide 400 mcg twice daily 2. Placebo twice daily Inhaler device: 1, Pulmicort; 2, dry powder turbuhaler Co‐treatment: Use of inhaled glucocorticoids other than the study medication, beta‐blockers, cromones or long‐acting inhaled beta2‐adrenergic agonists was not allowed |
|
| Outcomes |
Primary: change over time in postdose FEV1 Secondary: serious adverse events, mortality, glucocorticoid‐related adverse effects, bone density, non‐serious adverse events |
|
| Notes |
Funding: funded by a grant from Astra Draco, Lund, Sweden Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | ‘Randomly assigned’. No specific details given but industry‐sponsored |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind (presumed participant and personnel/investigator) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Central evaluator who was unaware of the treatment received and was analysed according to a standardised computerised protocol |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data on randomly assigned participants were analysed on an intention‐to‐treat basis. Withdrawal rates under 30% and even in both groups |
| Selective reporting (reporting bias) | High risk | Several missing outcomes. Could not locate protocol to check that all prospectively registered outcomes were reported. Contacted second study author; no reply by time of publication |
Renkema 1996.
| Methods |
Design: parallel‐group, double‐blind, randomised, placebo‐controlled study 2‐year study period Conducted at a single centre in the Netherlands |
|
| Participants |
Participants: 39 people were randomly assigned to budesonide (21) and placebo (18)
Baseline characteristics: Male %: bud 100, placebo 100 Mean age (SD), years: bud 56 (8), placebo 54 (10) Smoking history, cigarettes/year (SD): bud 635 (530), placebo 729 (495) Mean % predicted FEV1 (SD): bud 67 (15), placebo 60 (18) Inclusion criteria: clinical diagnosis of COPD based on history (persistent dyspnoea, mainly on exertion, without sudden attacks of dyspnoea); FEV1 less than 80% of predicted value; residual volume (RV) greater than 100% of predicted value; specific compliance expressed as percentage of predicted value greater than 100% after bronchodilation; when, however, air trapping (calculated as thoracic gas volume measured by body plethysmography minus functional residual capacity measured with an indicator gas) was greater than 1.5 L Csp was allowed to be less than 100% of predicted; no signs of allergy (negative skin test results, total serum IgE < 200 IU/mL, eosinophils in peripheral blood < 250 × l03/mL); and stable phase of the disease Exclusion criteria: Excluded were participants older than 70 years at entry, participants receiving continuous corticosteroid therapy and participants with severe concomitant disease, likely to interfere with the purpose of the study. All participants had alpha‐1 antitrypsin serum levels within the normal range. All participants were smokers or ex‐smokers. Smoking history was expressed as cigarette‐years |
|
| Interventions |
Run‐in: three months with no corticosteroid medication Treatments: 1. Budesonide 800 mcg twice daily (plus placebo tablet once daily) 2. Placebo twice daily (plus placebo tablet once daily) Inhaler device: metered‐dose inhaler (MDI) through a 750‐mL spacer (Nebuhaler; ASTRA, Ryswylc, The Netherlands) Co‐treatment: Throughout the study, participants were maintained on regimens of their usual bronchodilator medication, consisting of anticholinergics. beta‐agonists, theophylline or a combination of these drugs |
|
| Outcomes |
Primary: FEV1 Secondary: compliance, symptom scores, fasting morning plasma cortisol levels |
|
| Notes |
Funding: grants from the Netherlands Asthma Foundation, ASTRA BV Holland and AB DRACO Sweden Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | By computerised randomisation, stratified for smoking |
| Allocation concealment (selection bias) | Low risk | Allocated blindly |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Uneven withdrawal rates, no description of imputation to account for dropout |
| Selective reporting (reporting bias) | High risk | Key expected outcomes not reported (mortality and adverse events). No reply from study authors by time of publication |
Rennard 2009.
| Methods |
Design: Randomised, double‐blind, double‐dummy, parallel‐group, active‐ and placebo‐controlled, multi‐centre study 12‐Month treatment period 237 sites in the US, Europe and Mexico |
|
| Participants |
Participants: 1483 people were randomly assigned to budesonide/formoterol at high (494) and low dose (494), and to formoterol alone (495)
Baseline characteristics: Male %: bud/form high 62.3, bud/form low 62.8, form 65.3 Mean age (SD), years: bud/form high 63.2 (8.9), bud/form low 63.6 (9.2), form 62.9 (9.1) Smoking history (median pack‐years): bud/form high 40, bud/form low 40, form 40 Mean % predicted FEV1 (SD): bud/form high 38.6 (11.4), bud/form low 39.6 (10.9), form 39.3 (11.9) Inclusion criteria: current smokers or ex‐smokers aged > 40 years with clinical diagnosis of COPD and symptoms for > 2 years were eligible for this study. Participants were required to have a history of at least one COPD exacerbation treated with a course of oral corticosteroids and/or antibacterials, with 1 to 12 months before screening and documented use of an inhaled short‐acting bronchodilator as rescue medication. Prebronchodilator FEV1 of < 50% of predicted normal and pre‐bronchodilator FEV1/FVC of < 70% were required at screening. Smoking history of at least 10 pack‐years, score ≥ 2 on the Modified Medical Research Council dyspnoea scale at the time of screening and breathlessness, cough and sputum scale score ≥ 2 per day for at least half of the 2‐week run‐in period Exclusion criteria: Individuals were excluded if they had any of the following conditions: history of asthma, history of allergic rhinitis before 40 years of age, significant/unstable CV disorder, clinically significant respiratory tract disorder other than COPD and homozygous alpha1‐antitrypsin deficiency or any other clinically significant co‐morbidities. Individuals were also excluded if they needed additions or alterations to their usual COPD maintenance therapy or an increment in rescue therapy because of worsening symptoms within 30 days before screening. Oral or ophthalmic non‐cardioselective beta‐adrenoceptor antagonists, oral corticosteroids, pregnancy and breast‐feeding also were exclusionary |
|
| Interventions |
Run‐in: 2 weeks during which previous inhaled corticosteroids were discontinued Treatments: 1. Budesonide/formoterol 320/9 mcg twice daily 2. Budesonide/formoterol 160/9 mcg twice daily 3. Formoterol 9 mcg twice daily Inhaler device: 1 and 2, pressurised metered‐dose inhaler. 3, dry powder inhaler Co‐treatment: salbutamol allowed as reliever medication |
|
| Outcomes |
Primary: predose FEV1 and one hour postdose FEV1 Secondary: morning and evening PEF, COPD exacerbations, quality of life (SGRQ), symptom scores, percentage of awakening‐free nights, any adverse event (AE), pneumonia‐related AEs, serious AEs, mortality, vital signs and cortisol levels |
|
| Notes |
Funding: AstraZeneca Clinicaltrials.gov ID: NCT00206167 Definition of pneumonia: reported by physicians based on the Medical Dictionary for Regulatory Activities (version 10.0) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Not described. Industry sponsored, presumed to follow usual AZ methods |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Described as double‐blind [presumed participants and personnel/investigators] |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Safety was assessed by adverse event (AE) reporting. Pneumonia events were reported by physicians based on the Medical Dictionary for Regulatory Activities (version 10.0) pneumonia‐related preferred terms (pneumonia, bronchopneumonia, lobar pneumonia or pneumonia staphylococcal). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The withdrawal rates were very high compared to the number of events for the different outcomes |
| Selective reporting (reporting bias) | Low risk | All outcomes reported [checked against protocol]. Only one secondary outcome not reported. |
Schermer 2009.
| Methods |
Design: randomised, double‐blind, double‐dummy, placebo‐controlled phase IV trial Three‐year treatment period Conducted at 44 general practices in the Netherlands |
|
| Participants |
Participants: 190 people were randomly assigned to fluticasone (94) and placebo (96)
Baseline characteristics: Male %: flut 73, placebo 68 Mean age (SD), years: flut 58.4 (9.9), placebo 59.6 (10.1) Smoking history (mean (SD) pack‐years): flut 30.2 (18.2), placebo 26.5 (16.7) Mean % predicted FEV1 (SD): flut 63.2 (17.1), placebo 65.7 (17.7) Inclusion criteria: age 35 to 75 years; current or former smoker; chronic dyspnoea, sputum production and cough for at least three consecutive months per year during the previous two years; postbronchodilator forced expiratory volume in one second (FEV1) < 90% of predicted value and/or postbronchodilator FEV1/FVC (forced vital capacity) of predicted value < 88% for men and < 89% for women Exclusion criteria: postbronchodilator FEV1 < 40% of predicted and/or a history of asthma, allergic rhinitis or allergic eczema |
|
| Interventions |
Run‐in: Trial was preceded by the following (in chronological order): an optional smoking cessation attempt supported by the GP in trial candidates who were current smokers; a three‐month washout phase to eliminate possible carry‐over effects of a successful smoking cessation attempt or withdrawal of prior treatment with N‐acetylcysteine and/or inhaled corticosteroids; and a 14‐day pretreatment phase (30 mg oral prednisolone) to attain the highest possible baseline condition Treatments: 1. Fluticasone propionate 500 mcg twice daily 2. Placebo twice daily Inhaler device: Diskus dry powder inhaler. Unclear from trial report whether placebo was administered to match the fluticasone inhaler or the other active treatment, which was delivered as effervescent tablets dissolved in a glass of tap Co‐treatment: not reported |
|
| Outcomes | Primary: rate of exacerbation and quality of life as measured by the interviewer‐administered version of the Chronic Respiratory Questionnaire (CRQ) | |
| Notes |
Funding: Dutch Council for Health Insurances, with complementary funding by the Netherlands Asthma Foundation (authors had received various GSK and other pharma research grants) Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | An independent statistician generated a randomisation list based on a block size of three for treatment allocation to balance the three treatment arms by study centre. |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Neither investigators nor patients were aware of the group assignment. Placebo described as ‘matching’ |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Neither investigators nor patients were aware of the group assignment [presuming the investigators were those doing the outcome assessments] |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Dropout was high in both groups. The primary analyses were done on an intention‐to‐treat basis. Additional per protocol analyses were done on patients with a trial medication compliance rate >80%. Unclear how data were imputed or who was included in the ITT population |
| Selective reporting (reporting bias) | High risk | All outcomes stated in the protocol were reported but some key expected outcomes were missing (serious adverse events and pneumonia). No reply from author by time of publication. |
Senderovitz 1999.
| Methods |
Design: placebo‐controlled, randomised, double‐blind, multi‐centre trial Six‐month treatment period Five centres in Denmark |
|
| Participants |
Participants: unclear how many people were randomly assigned. 26 were evaluable in the budesonide (14) and placebo (12) groups
Baseline characteristics: Male %: bud 57, placebo 50 Median age (range), years: bud 58.5 (51 to 74), placebo 62.5 (57 to 74) Smoking history (mean (SD) pack‐years): not reported Mean % predicted FEV1 (SD): not reported Inclusion criteria: outpatients 18 to 75 years of age with stable COPD were included. FEV1, forced vital capacity (FVC) < 0.7, postbronchodilator FEV1 < 70% of predicted, FEV1 > 40% of predicted and increase in FEV1 < 15% after inhalation of 0 to 5 mg terbutaline Exclusion criteria: clinical evidence of asthma (e.g. pollen season–related symptoms, exercise‐induced symptoms only and significantly elevated levels of eosinophils and IgE), history of atopy (hay fever and/or atopic dermatitis), treatment with inhaled corticosteroids within the past 6 months, treatment with oral corticosteroids, cromoglycate or nedocromil within the past 4 weeks, other systemic disease making compliance and participation in the study difficult, pregnancy and breast‐feeding and an increase in FEV1 > 30% of baseline after 2 weeks of prednisolone treatment |
|
| Interventions |
Run‐in: All participants received 2 weeks of treatment with oral prednisolone 37.5 mg daily. Reversible participants with 15% < AFEV1 < 30% of baseline and irreversible participants with AFEV1 < 15% were separately randomly assigned to inhaled budesonide 400 pg bid or placebo Treatments: 1. Budesonide 400 mcg twice daily 2. Placebo twice daily Inhaler device: Spirocort Turbuhaler Co‐treatment: not reported |
|
| Outcomes |
Primary: FEV1 Secondary: exacerbations, adverse events and symptom scores |
|
| Notes |
Funding: not reported Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Reversible participants with 15% < AFEV1 < 30% of baseline and irreversible participants with AFEV1 < 15% were separately randomly assigned |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 26 of 37 were evaluable (12 in placebo group, 14 in active group—30% dropout overall). Number randomly assigned and number of dropouts not provided for each group. ITT not adopted |
| Selective reporting (reporting bias) | High risk | Several key expected outcomes not reported (mortality, adverse events, withdrawal per group). Study author contacted but not able to provide data |
Shaker 2009.
| Methods |
Design: randomised, double‐blinded, placebo‐controlled, parallel‐group, single‐centre study Two‐ to four‐year treatment period Conducted at a single centre in Denmark |
|
| Participants |
Participants: 254 people were randomly assigned to budesonide (127) and placebo (127)
Baseline characteristics: Male %: bud 62.2, placebo 54.3 Mean age (SD), years: bud 63.6 (7.5), placebo (63.6 (7.2) Smoking history (mean (SD) pack‐years): bud 56 (23), placebo 56 (24) Mean % predicted FEV1 (SD): bud 51 (11), placebo 53 (11) Inclusion criteria: Individuals 50 to 80 years of age were eligible if they were current smokers with a clinical diagnosis of COPD for not less than 2 years. All participants should have a significant smoking history of at least 10 cigarettes per day during the past 6 months and a previous history of at least 20 pack‐years. Ex‐smokers were excluded. Baseline lung function criteria were as follows: FEV1 between 35% and 70% of predicted (pre‐bronchodilator), and FEV1/forced vital capacity (FEV1/FVC) ≤ 60% Exclusion criteria: Reversibility of ≥ 12% and 200 mL in FEV1 from baseline values, 15 minutes after inhalation of 1 mg terbutaline or ≥ 15% and 300 mL after 2 weeks on oral prednisolone (25 mg), was an exclusion criterion. Individuals were also excluded if they had any severe concomitant disease; had an exacerbation within 30 days before the first visit; received oral steroids for longer than four weeks within six months of the first visit; or were on long‐term oxygen therapy |
|
| Interventions |
Run‐in: two‐week run‐in period on oral prednisolone Treatments: 1. Budesonide 400 mcg twice daily 2. Placebo twice daily Inhaler device: Pulmicort Turbuhaler Co‐treatment: Bronchodilators, mucolytics and short courses of oral corticosteroids (maximum 3 courses of 4 weeks' duration per year) and antibiotics were allowed during the study |
|
| Outcomes |
Primary: 15th percentile density (PD15) Secondary: change over time in the relative area of emphysema at a threshold of –910 Hounsfield units (RA‐910), FEV1 and diffusion capacity (DLCO) and in number of exacerbations, which was defined as a combination of 2 of the 3 following criteria: increased dyspnoea, increased sputum production and change in sputum colour |
|
| Notes |
Funding: AstraZeneca Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were allocated to either group in a proportion of 1:1 by block randomisation using a random sequence generated by a computer programme at AstraZeneca |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind. To maintain blinding, all Turbuhalers were of identical appearance |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | High proportion of dropouts in both groups (43% intervention and 49% placebo) |
| Selective reporting (reporting bias) | Low risk | Could not locate protocol to check that all prospectively registered outcomes were reported, but study authors provided all relevant outcomes upon request |
Sharafkhaneh 2012.
| Methods |
Design: randomised, double‐blind, double‐dummy, parallel‐group, multi‐centre study 12‐Month treatment period Conducted at 180 study sites in the United States (106 sites), Central and South America (53 sites) and South Africa (21 sites) |
|
| Participants |
Participants: 1219 people were randomly assigned to high‐dose budesonide/formoterol (407), low‐dose budesonide/formoterol (408) and formoterol alone (404)
Baseline characteristics: Male %: bud/form high 64.4, bud/form low 64.7, form 56.8 Mean age (SD), years: bud/form high 63.8 (9.4), bud/form low 62.8 (9.2), form 62.5 (9.4) Smoking history (mean pack‐years): bud/form high 46, bud/form low 44, form 43 Mean % predicted FEV1 (SD): bud/form high 37.9 (11.8), bud/form low 37.6 (11.6), form 37.5 (12.4) Inclusion criteria: Individuals were current smokers or ex‐smokers with a smoking history of 10 pack‐years, 40 years of age, with a clinical diagnosis of COPD with symptoms for >2 years. Participants were required to have a history of 1 COPD exacerbation requiring treatment with a course of systemic corticosteroids, antibiotics or both, within one to 12 months before screening (visit 1) and documented use of an inhaled short‐acting bronchodilator as rescue medication. At screening, a pre‐bronchodilator forced expiratory volume in one second (FEV1) of 50% of predicted normal and a pre‐bronchodilator FEV1/forced vital capacity (FVC) of < 70% also were required Exclusion criteria: Exclusion criteria included current, previous (within past 60 days) or planned enrolment in a COPD pulmonary rehabilitation programme, treatment with oral corticosteroids and incidence of a COPD exacerbation or any other significant medical diagnosis between screening and randomisation visits |
|
| Interventions |
Run‐in: 2‐week run‐in period Treatments: 1. Budesonide/formoterol 320/9 mcg twice daily 2. Budesonide/formoterol 160/9 mcg twice daily 3. Formoterol DPI 9 mcg twice daily Inhaler device: 1 and 2, pressurised metered‐dose inhaler. 3, dry powder inhaler Co‐treatment: Rescue medication (albuterol pMDI 90 mg 2 inhalations) was provided for as needed use during screening and run‐in, and throughout the study |
|
| Outcomes |
Primary: COPD exacerbations Secondary: FEV1, FVC, morning and evening PEF, diary card symptoms, rescue medication use, BODE index, exercise capacity, health‐related quality of life (SGRQ), adverse events. Unclear which was primary |
|
| Notes |
Funding: AstraZeneca ID: D589CC00003 Clinicaltrials.gov ID: NCT00419744 Definition of pneumonia: based on clinical judgement, not on radiological or microbial assessments |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Assignments were made sequentially by interactive voice response system in a a computer generated allocation schedule produced in advance. |
| Allocation concealment (selection bias) | Low risk | Assignments were made sequentially by interactive voice response system following a computer generated allocation schedule produced in advance. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | To maintain patient and investigator blinding, all active treatments were provided in blinded treatment kits. Patients in the budesonide/formoterol pMDI groups received a placebo DPI and those in the formoterol DPI group received a placebo pMDI. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The withdrawal rates were relatively even but high, especially compared to the low event rates for the outcomes of interest |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were reported in detail |
Szafranski 2003.
| Methods |
Design: Randomised, double‐blind, placebo‐controlled, parallel‐group, multi‐centre study 12 month treatment period Conducted in 89 centres from 11 countries |
|
| Participants |
Participants: 812 people were randomly assigned to budesonide (198), placebo (205), budesonide/formoterol (208) and formoterol (201)
Baseline characteristics: Male %: bud 80, placebo 83, bud/form 76, form 76 Mean age (range), years: bud 64 (40 to 90), placebo 65 (47 to 92), bud/form 64 (41 to 82), form 63 (40 to 90) Smoking history (mean pack‐years (SD not reported)): bud 44, placebo 45, bud/form 44, form 45 Mean % predicted FEV1 (SD not reported): bud 37, placebo 36, bud/form 36, form 36 Inclusion criteria: Adults with moderate to severe asthma were included and were selected according to the following criteria: outpatients aged 0 to 40 years; COPD symptoms for 2 years; > 10 pack‐year smoking history; FEV1/FVC 70%; FEV1 50% predicted normal (stages IIB and III according to the GOLD classification); total symptom score of two per day during at least 7 days of the run‐in period; documented use of short‐acting inhaled bronchodilators for reliever medication; > 1 severe COPD exacerbation within 2 to 12 months before the first clinic visit Exclusion criteria: History of asthma and/or seasonal allergic rhinitis before the age of 40; any relevant cardiovascular disorders as judged by the investigator; using beta‐blocking agents; current respiratory tract disorders other than COPD or any other significant diseases or disorders that may have put them at risk or that may have influenced the results of the study; requirement for regular use of oxygen therapy or an exacerbation during run‐in. Individuals for whom it would have been considered unethical to withdraw inhaled steroids were also excluded |
|
| Interventions |
Run‐in: 2‐week run‐in period Treatments: 1. Budesonide 400 mcg twice daily (Pulmicort) 2. Placebo twice daily 3. Budesonide/formoterol 320/9 mcg twice daily (Symbicort) 4. Formoterol 9 mcg twice daily (Oxis) Inhaler device: as above Co‐treatment: Only study medication was allowed during the treatment period, plus terbutaline 0.5 mg when needed as reliever medication |
|
| Outcomes |
Primary: severe exacerbations and FEV1 Secondary: VC and PEF, health‐related quality of life, diary card data, reliever medication use, mild exacerbations, adverse events and clinical chemistry |
|
| Notes |
Funding: AstraZeneca Definition of pneumonia: Not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Total of 812 participants were randomly assigned (no other details, industry‐sponsored) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind (presumed participants and personnel/investigator) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawal high and uneven between groups (formoterol 31.8%, placebo 43.9%, 31% ICS and 28% LABA/ICS). An intention‐to‐treat analysis was used, but imputation methods were unclear for some outcomes |
| Selective reporting (reporting bias) | Unclear risk | Could not locate trial registration to check protocol adherence—pneumonia outcomes not reported. Difficulty contacting study author to clarify |
Tashkin 2008 SHINE.
| Methods |
Design: randomised, double‐blind, double‐dummy, placebo‐controlled, parallel‐group, multi‐centre study Six‐month treatment period Conducted at 194 centres in the US, Czech Republic, the Netherlands, Poland and South Africa |
|
| Participants |
Participants: 1704 people were randomly assigned to six groups, but the three ICS/LABA groups were merged for analysis. Participant numbers in the four categories were as follows: budesonide (275), placebo (300), budesonide/formoterol (845) and formoterol (284)
Baseline characteristics: Male %: bud 67.6, placebo 69.0, bud/form 68.9, form 65.5 Mean age (SD), years: bud 63.4 (8.8), placebo 63.2 (9.6), bud/form 63.5, form 63.5 (9.5) Smoking history (mean pack‐years (SD not reported)): bud 41, placebo 40, bud/form 41, form 40 Mean % predicted FEV1 (SD): bud 39.7 (12.0), placebo 41.3 (12.1), bud/form 39.4, form 39.6 (12.8) Inclusion criteria: Current smokers or ex‐smokers older than 40 years of age with a clinical diagnosis of COPD and symptoms for longer than two years were eligible for this study. Participants were required to have a history of at least one COPD exacerbation treated with a course of oral corticosteroids and/or antibacterials with documented use of an inhaled short‐acting bronchodilator as rescue medication one to 12 months before screening. Prebronchodilator FEV1 < 50% of predicted normal and pre‐bronchodilator FEV1/FVC < 70% were required at screening. Smoking history of at least 10 pack‐years, score two or higher on the Modified Medical Research Council dyspnoea scale at the time of screening and a breathlessness, cough and sputum scale score of 2 or higher per day for at least half of the 2‐week run‐in period Exclusion criteria: Individuals were excluded if they had any of the following conditions: history of asthma, history of allergic rhinitis before 40 years of age, significant/unstable CV disorder, clinically significant respiratory tract disorder other than COPD and homozygous alpha1‐antitrypsin deficiency or any other clinically significant co‐morbidities. Individuals were excluded if they needed additions or alterations to their usual COPD maintenance therapy or an increment in rescue therapy because of worsening symptoms within 30 days before screening. Oral or ophthalmic non‐cardioselective beta‐adrenoceptor antagonists, oral corticosteroids, pregnancy and breast‐feeding also were exclusionary |
|
| Interventions |
Run‐in: 2‐week run‐in period Treatments: 1. Budesonide 160 mcg × 2 inhalations (320 mcg) twice daily 2. Placebo twice daily 3. Budesonide/formoterol 160/4.5 mcg × 2 inhalations (320/9 mcg) twice daily OR Budesonide/formoterol 80/4.5 mcg × 2 inhalations (160/9 mcg) twice daily OR Budesonide 160 mcg × 2 inhalations (320 mcg) twice daily plus formoterol DPI 4.5 mcg × 2 inhalations (9 mcg) twice daily 4. Formoterol DPI 4.5 mcg × 2 inhalations (9 mcg) twice daily Inhaler device: 1 and 3, pressurised metered‐dose inhaler. 4, dry powder inhaler. 2, unclear Co‐treatment: Participants were allowed the following concomitant medications during the study period: ephedrine‐free antitussives and mucolytics, nasal corticosteroids, stable‐dose non‐nebulised ipratropium bromide, oral or ophthalmic cardioselective beta‐adrenoceptor antagonists or study‐provided salbutamol as rescue medication. The following medications were allowed for exacerbations after randomisation: oral and parenteral corticosteroids, short‐term use of xanthines, increased use of inhaled beta2‐adrenoceptor agonists and ipratropium bromide, nebulised beta2‐adrenoceptor agonists and ipratropium bromide |
|
| Outcomes |
Primary: predose FEV1 and 1 hour postdose FEV1 Secondary: 12‐hour spirometry, predose and 1 hour postdose morning and evening PEF, dyspnoea, health‐related quality of life, COPD exacerbations, breathlessness diary and symptom scores, use of rescue medication, adverse events, serious adverse events and mortality |
|
| Notes |
Funding: AstraZeneca Clinicantrials.gov identifier: NCT00206154 Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Eligible participants were randomly assigned in balanced blocks according to a computer‐generated randomisation scheme at each site |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind. To maintain blinding, participants received both a pressurised metered‐dose inhaler (pMDI) and a dry powder inhaler (DPI) containing active treatment or placebo (PL), or combinations of active treatment and placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | ECG results were evaluated by a cardiologist in a blinded fashion through an independent ECG service provider. Unclear for other outcomes |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Withdrawal rates lower in combination groups (14.8% combined) than in other groups (22.9%, 25.7% and 21.5% for ICS, placebo and LABA, respectively). ITT analysis used with last observation carried forward for missing values |
| Selective reporting (reporting bias) | Low risk | Checked against protocol and contacted study authors. All stated outcomes reported in full |
van Grunsven 2003.
| Methods |
Design: randomised, placebo‐controlled, double‐blind trial 24‐Month treatment period Conducted at 10 general practices in Holland |
|
| Participants |
Participants: 48 people were randomly assigned to fluticasone (24) and placebo (24)
Baseline characteristics: Male %: flut 50, placebo 54 Mean age (SD), years: flut 46 (10), placebo 47 (11) Smoking history (mean (SD) pack‐years): flut 11.9 (9.5), placebo 5.8 (8.4) Mean % predicted FEV1 (SD): flut 95 (18), placebo 98 (17) Inclusion criteria: chronic cough and/or sputum production for at least three consecutive months and showed an annual decline in pre‐bronchodilator FEV1 of 40 to 80 mL Exclusion criteria: previous diagnosis of a pulmonary condition; presence of a co‐morbid condition with reduced life expectancy; intolerance for inhaled beta2‐agonists; use of beta‐blocking agents; inability to use inhalation devices or peak flow meters |
|
| Interventions |
Run‐in: general population screened, followed by 2‐year monitoring of participants with respiratory symptoms, then randomisation Treatments: 1. Fluticasone propionate 250 mcg twice daily 2. Placebo twice daily Inhaler device: Rotadisk dry powder inhaler Co‐treatment: Apart from short‐acting ("rescue") bronchodilators in case of acute dyspnoea, participants were not allowed to use other pulmonary medication |
|
| Outcomes |
Primary outcome: FEV1 Secondary outcomes: PC20, exacerbations, COOP/WONCA |
|
| Notes |
Funding: GSK Definition of pneumonia: not given |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised. No details but assumed to adhere to usual GSK methods |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Described as double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropout high but even between groups (25%). All participants with at least one follow‐up measurement for the primary outcome (postbronchodilator FEV1) were included in an intention to‐treat analysis |
| Selective reporting (reporting bias) | High risk | Key outcomes not reported (e.g. mortality). SAEs not given per arm. No reply from study authors by time of publication |
Verhoeven 2002.
| Methods |
Design: randomised, double‐blind, placebo‐controlled study Six‐month treatment period Single study centre in the Netherlands |
|
| Participants |
Participants: 23 people were randomly assigned to fluticasone (10) and placebo (13)
Baseline characteristics: Male %: flut 80, placebo 84.6 Mean age (range), years: flut 54 (42 to 65), placebo 56 (42 to 67) Smoking history (mean (range) pack‐years): flut 25 (five to 50), placebo 26 (11 to 50) Mean % predicted FEV1 (range): flut 66 (55 to 93), placebo 61 (34 to 72) Inclusion criteria: chronic productive cough, FEV1 < 70% of predicted normal value, FEV1 reversibility of < 10% predicted after 750 mg terbutaline administered by metered‐dose inhalation, negative serological examination (Phadiatop test) and negative skin prick tests for standard inhaled allergens. Individuals with an FEV1/inspiratory vital capacity (IVC) ratio of < 0.70 were also included, provided their total lung capacity (TLC) was greater than the predicted value + 1.64 SD. Participants had to be current and persistent smokers 40 to 70 years of age Exclusion criteria: history of asthma characterised by attacks of dyspnoea, chest tightness or wheezing; respiratory tract infection in the 4 weeks preceding the first visit; or suffering from serious or unstable concomitant disease |
|
| Interventions |
Run‐in: two weeks Treatments: 1. Fluticasone propionate 500 mcg twice daily 2. Placebo Inhaler device: Diskhaler Co‐treatment: Eligible participants using anti‐inflammatory treatment including non‐steroidal anti‐inflammatory drugs were asked to refrain from oral prescriptions for at least three months and from inhaled corticosteroids, sodium cromoglycate or nedocromil sodium for at least 6 weeks before the start of the study. Long‐acting beta2‐agonists, xanthine derivatives and antihistamine drugs also had to be stopped at least 6 weeks before the start of the study |
|
| Outcomes | Primary outcome unclear. Outcomes reported were use of secondary medication, compliance, FEV1, PC20, FEV1/FVC, cortisol levels and inflammatory markers | |
| Notes |
Funding: GlaxoWellcome (FLIL44/FMS40060) Definition of pneumonia: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomly allocated (no details, but industry‐funded) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts in either group |
| Selective reporting (reporting bias) | Low risk | Adverse event data not adequately reported but information supplied by the study author |
Vestbo 1999.
| Methods |
Design: double‐blind, parallel‐group, randomised clinical trial nested in a continuing epidemiological study, the Copenhagen City Heart Study (CCHS) 36‐Month treatment period Conducted at a single centre in Denmark |
|
| Participants |
Participants: 290 people were randomly assigned to budesonide (145) and placebo (145)
Baseline characteristics: Male %: bud 85, placebo 90 Mean age (SD), years: bud 59 (8.3), placebo 59.1 (9.7) Smoking history (mean (SD) pack‐years): not reported Mean % predicted FEV1 (SD): bud 86.2 (20.6), placebo 86.9 (21.1) Inclusion criteria: CCHS participant; 30 to 70 years of age; FEV1/vital capacity ratio 0.7 or less; FEV1 reversibility after inhalation of 1·0 mg terbutaline from Turbuhaler (Bricanyl, Lund, Sweden) of less than 15% of pre‐bronchodilator FEV1; FEV1 reversibility after 10 days of treatment with oral prednisolone 37.5 mg daily of less than 15% of pre‐bronchodilator FEV1; and informed consent. Pack‐years and other measures of cigarette smoking were not part of inclusion criteria Exclusion criteria: Long‐term treatment (more than two episodes of longer than 4 weeks) with oral or inhaled steroids within 6 months of study entry. Other exclusion criteria were pregnancy or lactation, intention to become pregnant, other serious systemic disease that could influence the results of this study (investigators’ judgement), chronic alcohol or drug use and participation in other clinical studies of COPD within 1 month of inclusion |
|
| Interventions |
Run‐in: not described Treatments: 1. First six months: budesonide 800 mcg am and 400 mcg pm; following 30 months: 400 mcg twice daily 2. Placebo twice daily Inhaler device: Turbuhaler Co‐treatment: Continuous use of inhaled corticosteroids other than study medication was not allowed. Oral, inhaled or parenteral steroids could be used during exacerbations for up to three periods of four weeks each year. Treatment with beta2‐agonists of all kinds, theophylline, disodium cromoglycate and mucolytics was allowed but kept constant. Concomitant use of beta‐blockers during the study was not allowed |
|
| Outcomes |
Primary: spirometric indices (FEV1, VC, FVC) Secondary: respiratory symptoms (e.g. wheeze, wheeze without a cold, breathlessness at rest and at different grades of exertion, cough night and day, phlegm night and day, chest tightness), exacerbations, chronic mucus hypersecretion, adverse events |
|
| Notes |
Funding: AstraZeneca Definition of pneumonia: not given |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was masked and the randomisation sequence generated by computer at Astra. Study numbers were allocated in a consecutive order |
| Allocation concealment (selection bias) | Low risk | The randomisation code was held by Astra and was not available to the researchers until the study had been completed. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind. All study inhalers (budesonide and placebo) had the same appearance. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Dropout higher in placebo group (35% vs 25% in budesonide group). ITT used. |
| Selective reporting (reporting bias) | Unclear risk | All stated outcomes reported but unable to check against trial registration. Difficulty contacting authors. |
Yildiz 2004.
| Methods |
Design: randomised, double‐blind, placebo‐controlled design Three‐month treatment period Conducted at a single centre in Turkey |
|
| Participants |
Participants: 38 people randomly assigned to budesonide plus existing bronchodilator therapy (20) and placebo (18)
Baseline characteristics: Male %: bud 100, placebo 100 Mean age (SD), years: bud 70 (7), placebo 64 (9) Smoking history (mean (SD) pack‐years): bud 55 (31), placebo 47.5 (18) Mean % predicted FEV1 (SD): bud 51 (22), placebo 40 (14) Inclusion criteria: pre‐bronchodilator FEV1 between 30% and 80% of predicted and FEV1/FVC < 70% (stage II according to the GOLD classification), irreversible airway obstruction suggested by < 10% improvement in FEV1 after inhalation of 200 mg salbutamol, smoking history of more than 20 pack‐years and no exacerbation or respiratory tract infection in the previous four weeks Exclusion criteria: history suggestive of asthma, clinical signs of right heart failure, recent hospitalisation or admission to the emergency department because of exacerbation, requirement for regular use of oxygen therapy or used inhaled or oral ICS in the past six weeks |
|
| Interventions |
Run‐in: no information Treatments: 1. 800 mcg budesonide twice daily plus existing bronchodilator therapy 2. Placebo twice daily plus existing bronchodilator therapy Inhaler device: Miflonide inhaler, Novartis Co‐treatment: All participants were receiving combined bronchodilator therapy consisting of inhaled long‐acting beta2‐agonist (Formoterol, Foradil Aerolizer, Novartis) plus inhaled anticholinergic (ipratropium bromide, Atrovent inhaler, Boehringer Ingelheim) |
|
| Outcomes |
Primary: unclear which outcome was primary Secondary: St. George's Respiratory Questionnaire, FEV1, arterial blood gas analysis, serious adverse events, exacerbations |
|
| Notes |
Funding: unclear Definition of pneumonia: not given |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | 'Randomised'. No other details, funding unclear |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Withdrawal rates were low (2/18 placebo, 0/20 ICS) |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported except one secondary outcome—not deemed to reflect bias. Difficulty finding correct contact details for study author |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| GSK FLIP63 2005 | Treatment period less than 12 weeks |
| GSK SAM30022 2005 | Comparison not of interest (beclomethasone vs salmeterol/fluticasone combination) |
| GSK SAM40004 2006 | Asthma, not COPD |
| GSK SAS40015 2007 | Comparison not of interest (fluticasone plus oral montelukast vs salmeterol/fluticasone combination) |
| GSK SCO100540 2006 | Comparison not of interest (salmeterol/fluticasone combination vs placebo) |
| GSK SCO100646 2008 | Comparison not of interest (participants received salmeterol and salmeterol/fluticasone combination for varying amounts of time during the study) |
| GSK SCO30005 2006 | Comparison not of interest (salmeterol/fluticasone combination vs placebo) |
| GSK SCO40030 2005 | Treatment period less than 12 weeks |
| GSK SCO40034 2009 | Comparison not of interest (salmeterol/fluticasone combination vs tiotropium) |
| GSK SCO40036 2009 | Comparison not of interest (salmeterol/fluticasone combination vs tiotropium) |
| GSK SFCB3019 2004 | Asthma, not COPD |
| Lung Health Study 2000 | Comparison not of interest (triamcinolone acetonide used as inhaled steroid) |
| van der Valk 2002 | ICS discontinuation study; all participants received treatment for three months before randomisation |
| Weir 1999 | Comparison not of interest (beclomethasone used as inhaled steroid) |
| Wouters 2005 | ICS discontinuation study; all participants received treatment for three months before randomisation |
Characteristics of studies awaiting assessment [ordered by study ID]
Ohar 2013.
| Methods | Six‐month randomised controlled trial |
| Participants | Participants were > 40 years of age with a historical FEV1/FVC < 0.7. Six‐month history of hospitalisation attributed to AECOPD was also required |
| Interventions | Fluticasone/salmeterol combination or salmeterol alone within 14 days of an exacerbation event: < 10‐day hospitalisation for AECOPD, or AECOPD requiring treatment with OCS or OCS + antibiotics in an emergency department, or during a physician’s office visit (if the index event was office‐based) |
| Outcomes | Exacerbation rates |
| Notes | Abstract only |
Characteristics of ongoing studies [ordered by study ID]
Vestbo 2013.
| Trial name or title | The Study to Understand Mortality and Morbidity in COPD (SUMMIT) study protocol (NCT01313676) |
| Methods |
Design: multi‐centre, placebo‐controlled, double‐blind, randomised, parallel‐group trial 15‐ to 44‐month treatment period (duration of treatment phase depended on mortality rate in the study; the study will last until 1000 deaths have been recorded) |
| Participants | Inclusion criteria: male or female, 40 to 80 years of age Current smokers or ex‐smokers with a smoking history of at least 10 pack‐years Established history of COPD with FEV1/FVC ratio 0.70 and FEV1 greater than 50 and less than 70% of predicted normal History of CVD or at increased risk for CVD For participants older than 40 years of age, this is defined as any one of the following: established coronary artery disease, established peripheral vascular disease, previous stroke, previous myocardial infarction or diabetes mellitus with target organ disease For participants older than 60 years of age, any one of the above or two of the following: treated for hypercholesterolaemia, treated for hypertension, treated for diabetes mellitus or treated for peripheral vascular disease Exclusion criteria: current diagnosis of asthma or respiratory disorders other than COPD Chest radiograph indicating diagnosis other than COPD Undergone lung volume reduction surgery and/or lung transplant Requirement for long‐term oxygen therapy at start of study (12 hours per day) Receiving long‐term oral corticosteroid therapy Current severe heart failure (NYHA class IV); individuals will also be excluded if they have a known ejection fraction of 30% or if they have an implantable cardioverter‐defibrillator Any life‐threatening condition with life expectancy of three years, other than vascular disease or COPD, that might prevent the individual from completing the study End‐stage chronic renal disease |
| Interventions |
Run‐in: 4 to 10 days Treatments: 1. Placebo; 2. Fluticasone furoate (100 mcg once daily) 3. Vilanterol (25 mcg) 4. Fluticasone furoate/vilanterol combination (100/25 mcg once daily) Inhaler device: novel dry powder inhaler Co‐treatment: All prior use of ICS and inhaled long‐acting bronchodilators will be discontinued at entry to the run‐in period |
| Outcomes |
Primary: mortality Secondary: decline in FEV1 and effect on a composite cardiovascular endpoint |
| Starting date | Recruitment commenced in March 2011 and was ongoing in March 2013 |
| Contact information | J. Vestbo, Department of Respiratory Medicine J, Odense University Hospital, Sdr Ringvej 29, 5000 Odense C, Denmark E‐mail: jvestbo@dadlnet.dk |
| Notes | None |
Differences between protocol and review
The subgroup analysis stated in the protocol based on diagnostic criteria for pneumonia could not be conducted, as most trials did not state the definition. We included studies using fluticasone furoate, alone or in combination with the LABA vilanterol; these new formulations were not anticipated during the writing of the protocol. For reasons explained in the review, we chose not to calculate an indirect comparison of fluticasone/LABA versus budesonide LABA.
We added the following additional comparisons: fluticasone furoate/vilanterol versus vilanterol; fluticasone/vilanterol versus budesonide/formoterol.
We added to the methods a section on assessing transitivity.
Contributions of authors
Alieksei Seniukowich and Kayleigh Kew extracted data. Kayleigh conducted the analysis, interpreted the data and wrote up the results, with clinical input from Alieksei.
Charlotta Karner, authored the protocol, sifted the search and contributed to data extraction.
Sources of support
Internal sources
-
NIHR, UK.
This review is supported by a programme grant from NIHR.
External sources
No sources of support supplied
Declarations of interest
None known.
Edited (no change to conclusions)
References
References to studies included in this review
Anzueto 2009 {published and unpublished data}
- Anzueto A, Ferguson GT, Feldman G, Chinsky K, Seibert A, Emmett A, et al. Effect of fluticasone propionate/salmeterol (250/50) on COPD exacerbations and impact on patient outcomes. Journal of Chronic Obstructive Pulmonary Disease 2009;6(5):320‐9. [DOI] [PubMed] [Google Scholar]
- Dalal AA, Blanchette CM, Petersen H, St. Charles M. Cost‐effectiveness of fluticasone propionate/salmeterol (250/50mcg) compared to salmeterol (50mcg) in patients with chronic obstructive pulmonary disease: economic evaluation of study SCO100250 [Abstract]. American Thoracic Society International Conference; 2009 May 15‐20; San Diego. 2009:A1502 [Poster K54].
- DiSantostefano RL, Li H, Rubin DB, Stempel DA. Which patients with chronic obstructive pulmonary disease benefit from the addition of an inhaled corticosteroid to their bronchodilator? A cluster analysis. BMJ Open 2013;3(4):e001838. [DOI: 10.1136/bmjopen-2012-001838] [DOI] [PMC free article] [PubMed] [Google Scholar]
- GlaxoSmithKline. A Randomized, Double‐Blind, Parallel‐Group, 52‐Week Study to Compare the Effect of Fluticasone Propionate/Salmeterol DISKUS 250/50mcg BID with Salmeterol DISKUS 50mcg BID on the Annual Rate of Moderate/Severe Exacerbations in Subjects with Chronic Obstructive Pulmonary Disease (COPD). SCO100250. www.gsk‐clinicalstudyregister.com/ (accessed 25 May 2013).
- GlaxoSmithKline. A randomized, double‐blind, parallel‐group, 52‐week study to compare the effect of fluticasone propionate/salmeterol DISKUS 250/50mcg bid with salmeterol DISKUS 50mcg bid on the annual rate of moderate/severe exacerbations in subjects with chronic obstructive pulmonary disease (COPD). www.gsk‐clinicalstudyregister.com/ (accessed 8 April 2013).
Bourbeau 1998 {published data only}
- Bourbeau J, Rouleau MY, Boucher S. Randomised controlled trial of inhaled corticosteroids in patients with chronic obstructive pulmonary disease. Thorax 1998;53(6):477‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bourbeau 2007 {published data only}
- Bourbeau J, Christodoulopoulos P, Maltais F, Yamauchi Y, Olivenstein R, Hamid Q. Effect of salmeterol/fluticasone propionate on airway inflammation in COPD: a randomised controlled trial. Thorax 2007;62(11):938‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Burge 2000 {published and unpublished data}
- Bale G, Martinez‐Camblor P, Burge PS, Soriano JB. Long‐term mortality follow‐up of the ISOLDE participants: causes of death during 13 years after trial completion. Respiratory Medicine 2008;102(10):1468‐72. [DOI] [PubMed] [Google Scholar]
- Bale GA, Burge PS, Burge C, Moore VC. The ISOLDE Study 11‐13 years on: what were the causes of death? [Abstract]. American Thoracic Society International Conference; 2007 May 18‐23; San Francisco. 2007:Poster H42.
- Briggs AH, Lozano‐Ortega G, Spencer S, Bale G, Spencer MD, Burge PS. Estimating the cost‐effectiveness of fluticasone propionate for treating chronic obstructive pulmonary disease in the presence of missing data. Value In Health 2006;9(4):227‐35. [DOI] [PubMed] [Google Scholar]
- Burge PS, Calverley PM, Jones PW, Spencer S, Anderson JA. Prednisolone response in patients with chronic obstructive pulmonary disease: results from the ISOLDE study. Thorax 2003;58(8):654‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge PS, Calverley PM, Jones PW, Spencer S, Anderson JA, Maslen TK. Randomised, double blind, placebo controlled study of fluticasone propionate in patients with moderate to severe chronic obstructive pulmonary disease: the ISOLDE trial. British Medical Journal 2000;320(7245):1297‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge S, Sondhi S, Williams MK, Wilson K, Efthimiou J. An economic evaluation of fluticasone propionate in patients with moderate to severe chronic obstructive pulmonary disease (COPD) from the isolde trial. American Journal of Respiratory and Critical Care Medicine 2000;161 Suppl 3:A491. [Google Scholar]
- Calverley PM, Burge PS, Spencer S, Anderson JA, Jones PW. Bronchodilator reversibility testing in chronic obstructive pulmonary disease. Thorax 2003;58(8):659‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calverley PM, Spencer S, Willits L, Burge PS, Jones PW. Withdrawal from treatment as an outcome in the ISOLDE study of COPD. Chest 2003;124(4):1350. [DOI] [PubMed] [Google Scholar]
- D'Urzo AD, Calverley PMA. Withdrawal of treatment in the ISOLDE study [letter]. Chest 2004;125(6):2368. [DOI] [PubMed] [Google Scholar]
- GlaxoSmithKline F. A multi‐centre, double‐blind, placebo‐controlled, parallel group study of the efficacy and tolerability of long‐term inhaled fluticasone propionate 500 mcg twice daily via a Volumatic Spacer device in patients with non‐asthmatic chronic obstructive pulmonary disease, including an acute oral corticosteroid trial. FLTB3054. www.gsk‐clinicalstudyregister.com/ (accessed 20 May 2013).
- Jarad NA, Wedzicha JA, Burge PS, Calverley PM. An observational study of inhaled corticosteroid withdrawal in stable chronic obstructive pulmonary disease. ISOLDE Study Group. Respiratory Medicine 1999;93(3):161‐6. [DOI] [PubMed] [Google Scholar]
- Jebrak G. Corticosteroid therapy and chronic obstructive broncho‐pneumopathies: the ISOLDE study. Presse Medicale 2000;29(16):1461‐2. [PubMed] [Google Scholar]
- Jones PW, Willits LR, Burge PS, Calverley PMA. Disease severity and the effect of fluticasone propionate on chronic obstructive pulmonary disease exacerbations. European Respiratory Journal 2003;21(1):68‐73. [DOI] [PubMed] [Google Scholar]
- Marchand E. Withdrawal from and study design of the ISOLDE trial [comment]. Chest 2004;126(0012‐3692):659. [DOI] [PubMed] [Google Scholar]
- Spencer S, Calverley PM, Burge PS, Jones PW. Health status deterioration in patients with chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine 2001;163(1):122‐8. [DOI] [PubMed] [Google Scholar]
- Spencer S, Calverley PM, Burge PS, Jones PW. Impact of preventing exacerbations on deterioration of health status in COPD. European Respiratory Journal 2004;23(5):698‐702. [DOI] [PubMed] [Google Scholar]
Calverley 2003b {published and unpublished data}
- Borgstrom L, Asking L, Olsson H, Peterson S. Lack of interaction between disease severity and therapeutic response with budesonide/formoterol in a single inhaler [Abstract]. American Thoracic Society 100th International Conference; 2004 May 21‐26; Orlando. 2004:C22 Poster 505.
- Calverley PM, Bonsawat W, Cseke Z, Zhong N, Peterson S, Olsson H. Maintenance therapy with budesonide and formoterol in chronic obstructive pulmonary disease. European Respiratory Journal 2003;22(6):912‐9. [DOI] [PubMed] [Google Scholar]
- Calverley PMA. Effect of budesonide/formoterol on severe exacerbations and lung function in moderate to severe COPD. Thorax 2002;57 Suppl III:iii44. [Google Scholar]
- Calverley PMA, Cseke Z, Peterson S. Budesonide/formoterol reduces the use of oral corticosteroids in the treatment of COPD [Abstract]. European Respiratory Journal 2003;22 Suppl 45:P436. [DOI] [PubMed] [Google Scholar]
- Calverley PMA, Kuna P, Olsson H. COPD exacerbations are reduced by budesonide/formoterol in a single inhaler [Abstract]. European Respiratory Journal 2003;22 Suppl 45:P1587. [Google Scholar]
- Calverley PMA, Olsson H, Symbicort International COPD Study Group. Budesonide/formoterol in a single inhaler sustains improvements in lung function over 12 months compared with mono‐components and placebo in patients with COPD [abstract]. American Thoracic Society 99th International Conference; 2003 May 16‐21; Seattle. 2003:B024 Poster 418.
- Calverley PMA, Peterson S. Combining budesonide/formoterol in a single inhaler reduces exacerbation frequency in COPD [abstract]. American Thoracic Society 99th International Conference; 2003 May 16‐21; Seattle. 2003:D092 Poster 211.
- Calverley PMA, Stahl E, Jones PW. Budesonide/formoterol improves the general health status of patients with COPD [Abstract]. American Thoracic Society International Conference; 2005 May 20‐25; San Diego. 2005:B93 Poster 303.
- Calverley PMA, Szafranski W, Andersson A. Budesonide/formoterol is a well‐tolerated long term maintenance therapy for COPD. European Respiratory Journal 2005;26 Suppl 49:Poster 1917. [Google Scholar]
- Calverley PMA, Thompson NC, Olsson H. Budesonide/formoterol in a single inhaler sustains lung function improvements in COPD [Abstract]. European Respiratory Journal 2003;22 Suppl 45:P435. [Google Scholar]
- Halpin D, Ståhl E, Lundback B, Anderson F, Peterson S. Treatment costs and number needed to treat (NNT) with budesonide/formoterol to avoid one exacerbation of COPD [Abstract]. American Thoracic Society 100th International Conference; 2004 May 21‐26; Orlando. 2004:D22 Poster 525.
- Halpin DMG, Larsson T, Calverley PMA. How many patients with COPD must be treated with budesonide/formoterol compared with formoterol alone to avoid 1 day of oral steroid use? [Abstract]. American Thoracic Society International Conference; 2005 May 20‐25; San Diego. 2005:B93 Poster 314.
- Jones PW, Ståhl E. Budesonide/formoterol in a single inhaler improves health status in patients with COPD [abstract]. American Thoracic Society 99th International Conference; 2003 May 16‐21; Seattle. 2003:B024 Poster 419.
- Jones PW, Ståhl E. Budesonide/formoterol sustains clinically relevant improvements in health status in COPD [Abstract]. European Respiratory Journal 2005;26(Suppl 49):Abstract No. 1352. [Google Scholar]
- Jones PW, Ståhl E. Reducing exacerbations leads to a better health‐related quality of life in patients with COPD. European Respiratory Society 13th Annual Congress; 2003 Sep 28‐29; Vienna. 2003:P1586.
- Lofdahl CG. Reducing the impact of COPD exacerbations: clinical efficacy of budesonide/formoterol. European Respiratory Review 2004;13(88):14‐21. [Google Scholar]
- Lofdahl CG, Andreasson E, Svensson K, Ericsson A. Budesonide/formoterol in a single inhaler improves health status in patients with COPD without increasing healthcare costs [Abstract]. European Respiratory Journal 2003;22(Suppl 45):P433. [Google Scholar]
- Lofdahl CG, Ericsson A, Svensson K, Andreasson E. Cost effectiveness of budesonide/formoterol in a single inhaler for COPD compared with each mono‐component used alone. Pharmacoeconomics 2005;23(4):365‐75. [DOI] [PubMed] [Google Scholar]
Calverley 2003 TRISTAN {published and unpublished data}
- Calverley P, Pauwels R, Vestbo J, Jones P, Pride N, Gulsvik A, et al. Combined salmeterol and fluticasone in the treatment of chronic obstructive pulmonary disease: a randomised controlled trial. Lancet 2003;361(9356):449‐56. [DOI] [PubMed] [Google Scholar]
- Calverley PMA, Pauwels R, Vestbo J, Jones P, Pride N, Gulsvik A, et al. Safety of salmeterol/fluticasone propionate combination in the treatment of chronic obstructive pulmonary disease. European Respiratory Journal 2002;20(Suppl 38):242s [P1572]. [Google Scholar]
- Calverley PMA, Pauwels RA, Vestbo J, Jones PW, Pride NB, Gulsvik A, et al. Clinical improvements with salmeterol / fluticasone propionate combination in differing severities of COPD [A035] [Poster D50]. http://www.abstracts2view.com (accessed 20 April 2012).
- Calverley PMA, Pauwels RA, Vestbo J, Jones PW, Pride NB, Gulsvik A, et al. Salmeterol/fluticasone propionate combination for one year provides greater clinical benefit than its individual components [A98] [Poster 306]. http://www.abstracts‐on‐line.com/abstracts/ATS (accessed 20 April 2012).
- Hunjan MK, Chandler F. Numbers needed to treat (NNT) to avoid an exacerbation in patients with chronic obstructive pulmonary disease (COPD) using salmeterol/fluticasone propionate combination (SFC) and associated costs [Abstract]. American Thoracic Society 100th International Conference; 2004 May 21‐26; Orlando. 2004:D22 Poster 503.
- Hunjan MK, Williams DT. Costs of avoiding exacerbations in patients with chronic obstructive pulmonary disease (COPD) treated with salmeterol/fluticasone propionate combination (Seretide) and salmeterol. European Respiratory Journal 2004;24 Suppl 48:291s. [Google Scholar]
- Hunjan MK, Williams DT. Salmeterol/fluticasone propionate combination is clinically effective in avoiding exacerbations in patients with moderate/severe COPD. European Respiratory Journal 2004;24 Suppl 48:513s. [Google Scholar]
- Jones PW, Edin HM, Anderson J. Salmeterol/fluticasone propionate combination improves health status in COPD patients.. http://www.abstracts‐on‐line.com/abstracts/ATS (accessed 21 April 2012).
- Jones PW, Ståhl E. Budesonide/formoterol sustains clinically relevant improvements in health status in COPD [Abstract]. European Respiratory Journal 2005;26 Suppl 49:Abstract No. 1352. [Google Scholar]
- Jones PW, Vestbo J, Pauwels RA, Calverley PMA, Anderson JA, Spencer MD. Informative drop out in COPD studies. Investigation of health status of withdrawals in the TRISTAN study. European Respiratory Society 13th Annual Congress; 2003 Sep 28‐29; Vienna. 2003:P1593.
- Nitschmann S. Inhalational combination therapy in chronic obstructive lung disease. TRISTAN study. German Internist 2004;45(6):727‐8. [DOI] [PubMed] [Google Scholar]
- Pauwels RA, Calverly PMA, Vestbo J, Jones PW, Pride N, Gulsvik A, et al. Reduction of exacerbations with salmeterol/fluticasone combination 50/500 mcg bd in the treatment of chronic obstructive pulmonary disease. European Respiratory Journal 2002;20 Suppl 38:240 [P1569]. [Google Scholar]
- Pauwels RA, Vestbo J, Calverley PMA, Jones PW, Pride NB, Gulsvik A. Characterization of exacerbations in the TRISTAN study of salmeterol / fluticasone propionate (SFC) combination in moderate to severe COPD. http://www.abstracts2view.com (accessed 20 April 2012).
- SFCB3024. A multi‐centre, randomised, double‐blind, parallel group, placebo‐controlled study to compare the efficacy and safety of the salmeterol/FP combination product at a strength of 50/500mcg bd with salmeterol 50mcg bd alone and FP 500mcg bd alone, delivered via the DISKUS™/ACCUHALER™, in the treatment of subjects with chronic obstructive pulmonary disease (COPD) for 12 months. http://ctr.gsk.co.uk (accessed 21 April 2012).
- Spencer M, Briggs AH, Grossman RF, Rance L. Development of an economic model to assess the cost effectiveness of treatment interventions for chronic obstructive pulmonary disease. Pharmacoeconomics 2005;23(6):619‐37. [DOI] [PubMed] [Google Scholar]
- Spencer MD, Karia N, Anderson J. The clinical significance of treatment benefits with the salmeterol/fluticasone propionate 50/500mcg combination in COPD. European Respiratory Journal 2004;24 Suppl 48:290s. [Google Scholar]
- Vestbo J, Calverley PMA, Pauwels R, Jones P, Pride N, Gulsvik A, et al. Absence of gender susceptibility to the combination of salmeterol and fluticasone in the treatment of chronic obstructive pulmonary disease. European Respiratory Journal 2002;20 Suppl 38:240 [P1570]. [Google Scholar]
- Vestbo J, Pauwels R, Anderson JA, Jones P, Calverley P. Early onset of effect of salmeterol and fluticasone propionate in chronic obstructive pulmonary disease. Thorax 2005;60(4):301‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestbo J, Pauwels RA, Calverley PMA, Jones PW, Pride NB, Gulsvik A. Salmeterol/fluticasone propionate combination produces improvement in lung function detectable within 24 hours in moderate to severe COPD. http://www.abstracts2view.com (accessed 20 April 2012).
- Vestbo J, Soriano JB, Anderson JA, Calverley P, Pauwels R, Jones P. Gender does not influence the response to the combination of salmeterol and fluticasone propionate in COPD. Respiratory Medicine 2004;98(11):1045‐50. [DOI] [PubMed] [Google Scholar]
Calverley 2007 TORCH {published and unpublished data}
- Allegra L. TORCH study: An invitation to clinical considerations. GIMT ‐ Giornale Italiano delle Malattie del Torace 2007;61(1):15‐23. [Google Scholar]
- Briggs AH, Glick HA, Lozano‐Ortega G, Spencer M, Calverley PM, Jones PW, et al. Is treatment with ICS and LABA cost‐effective for COPD? Multinational economic analysis of the TORCH study. European Respiratory Journal 2010;35(3):532‐9. [DOI] [PubMed] [Google Scholar]
- Briggs AH, Lozano‐Ortega G, Spencer S, Bale G, Spencer MD, Burge PS. Estimating the cost‐effectiveness of fluticasone propionate for treating chronic obstructive pulmonary disease in the presence of missing data. Value in Health 2006;9(4):227‐35. [DOI] [PubMed] [Google Scholar]
- Calverley P, Celli B, Anderson J, Ferguson G, Jenkins C, Jones P, et al. Salmeterol/fluticasone propionate combination (SFC) improves survival in COPD over three years: on‐treatment analysis from the TORCH study [Abstract]. American Thoracic Society International Conference; 2009 May 15‐20; San Diego. 2009:A6191[Poster #219].
- Calverley P, Celli B, Anderson JA, Ferguson GT, Jenkins C, Jones PW, et al. The TOwards a Revolution in COPD Health (TORCH) study: salmeterol/fluticasone propionate improves survival in COPD over three years [Abstract]. Respirology 2006;11 Suppl 5:A149 [PS‐3‐8]. [Google Scholar]
- Calverley PM, Anderson JA, Celli B, Ferguson GT, Jenkins C, Jones PW, et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. New England Journal of Medicine 2007;356(8):775‐89. [DOI] [PubMed] [Google Scholar]
- Calverley PM, Anderson JA, Celli B, Ferguson GT, Jenkins C, Jones PW et al and TORCH Investigators. Cardiovascular events in patients with COPD: TORCH study results. Thorax 2010;65(8):719‐25. [DOI] [PubMed] [Google Scholar]
- Calverley PM, Celli B, Anderson JA, Ferguson GT, Jenkins C, Jones PW, et al. The TOwards a Revolution in COPD Health (TORCH) study: fluticasone propionate /salmeterol improves survival in COPD over three years. Chest 2006;130(4):122s. [Google Scholar]
- Calverley PMA, Celli B, Andersen JA, Ferguson GT, Jenkins C, Jones PW, et al. The TORCH (TOwards a Revolution in COPD Health) study salmeterol/fluticasone propionate (SFC) improves survival in COPD over three years. European Respiratory Journal 2006;28 Suppl 50:34. [Google Scholar]
- Celli B, Calverley PM, Anderson JA, Ferguson GT, Jenkins C, Jones P, et al. The TOwards a Revolution in COPD HEalth (TORCH) Study: salmeterol/fluticasone propionate reduces the rate of exacerbations over three years. Respirology 2006;11 Suppl 5:A140. [Google Scholar]
- Celli B, Calverley PM, Anderson JA, Ferguson GT, Jenkins C, Jones PW, et al. The TOwards a Revolution in COPD Health (TORCH) study: fluticasone propionate/salmeterol reduces the rate of exacerbations over 3 years [Abstract]. Chest 2006;130(4):177s. [Google Scholar]
- Celli B, Calverley PMA, Anderson JA, Ferguson GT, Jenkins C, Jones PW, et al. The TORCH (TOwards a Revolution in COPD Health) study salmeterol/fluticasone propionate (SFC) improves health status reduces exacerbations and improves lung function over three years [Abstract]. European Respiratory Journal 2006;28 Suppl 50:34s [E312]. [Google Scholar]
- Celli B, Emmett A, Crater G, Kalberg C. Fluticasone propionate/salmeterol (FSC) improves the inspiratory to total lung capacity ratio (IC/TLC) and exercise endurance time in patients with COPD. European Respiratory Society 16th Annual Congress; 2006 Sep 2‐6; Munich. 2006:A320 (Poster 323).
- Celli B, Ferguson GT, Anderson JA, Jenkins CR, Jones PW, Vestbo J, et al. Salmeterol/fluticasone propionate (SFC) improves lung function and reduces the rate of decline over three years in the TORCH survival study [Abstract]. American Thoracic Society International Conference; 2007 May 18‐23; San Francisco. 2007:A763.
- Celli B, Vestbo J, Jenkins CR, Jones PW, Ferguson GT, Calverley PMA, et al. Sex differences in mortality and clinical expressions of patients with chronic obstructive pulmonary disease: the TORCH experience. American Journal of Respiratory and Critical Care Medicine 2011;183(3):317‐22. [DOI] [PubMed] [Google Scholar]
- Celli BR, Thomas NE, Anderson JA, Ferguson GT, Jenkins CR, Jones PW, et al. Effect of pharmacotherapy on rate of decline of lung function in chronic obstructive pulmonary disease: results from the TORCH study. American Journal of Respiratory and Critical Care Medicine 2008;178(4):332‐8. [DOI] [PubMed] [Google Scholar]
- Celli D, Emmett A, Crater G, Kalberg C. Salmeterol/fluticasone propionate (SFC) improves the inspiratory to total lung capacity ratio (IC/TLC) and exercise endurance time in patients with COPD. European Respiratory Journal 2006;28 Suppl 50:764 [4395]. [Google Scholar]
- Corhay JL, Louis R. Clinical study of the month: the TORCH study (TOwards a Revolution in COPD Health) [L'étude clinique du mois. L'étude TORCH (TOwards a Revolution in COPD Health): vers une revolution de la santé des patients souffrant de BPCO]. Revue Medicale de Liege 2007;62(4):230‐4. [PubMed] [Google Scholar]
- Ferguson GT, Calverley PM, Anderson JA, Celli B, Jenkins C, Jones PW, et al. The TOwards a Revolution in COPD Health (TORCH) study: fluticasone propionate/salmeterol is well tolerated in patients with COPD over 3 years [Abstract]. Chest 2006;130(4):178s. [Google Scholar]
- Ferguson GT, Calverley PM, Anderson JA, Celli B, Jenkins CR, Jones PW, et al. Effect of salmeterol/fluticasone propionate (SFC) on bone mineral density (BMD) and eye disorders over three years in the TORCH trial [Abstract]. American Thoracic Society International Conference; 2007 May 18‐23; San Francisco. 2007:A763.
- Ferguson GT, Calverley PM, Anderson JA, Jenkins CR, Celli B, et al. Prevalence and progression of osteoporosis in patients with COPD results from the towards a revolution in COPD health study. Chest 2009;136(6):1456‐65. [DOI] [PubMed] [Google Scholar]
- Ferguson GT, Calverley PMA, Anderson JA, et al. The TORCH (TOwards a Revolution in COPD Health) study: salmeterol/fluticasone propionate (SFC) improves survival in COPD over three years. European Respiratory Journal 2006;28 Suppl 50:34s. [Google Scholar]
- Glick H, Briggs A, Lozano‐Ortega G, Spencer M, Vestbo J, Calverley P. Is treatment with ICS/LABA combination good value for money in COPD? Evidence from the TORCH study [Abstract]. American Thoracic Society International Conference; 2007 May 18‐23; San Francisco. 2007:[C97].
- Jenkins C, Celli B, Anderson J, Ferguson G, Calverley P, Jones P, et al. Salmeterol/fluticasone propionate (SFC) is efficacious in GOLD stage II COPD patients: analysis from the TORCH study [Abstract]. American Thoracic Society International Conference; May 15‐20; San Diego. 2009:A6186 [Poster #213].
- Jenkins CR, Celli B, Anderson JA, Ferguson GT, Jones PW, Vestbo J, et al. Seasonality and determinants of moderate and severe COPD exacerbations in the TORCH study. European Respiratory Journal 2012;39:38‐45. [DOI] [PubMed] [Google Scholar]
- Jenkins CR, Celli B, Anderson JA, Ferguson GT, Jones PW, Vestbo J, et al. The TORCH survival study: consistent efficacy results seen in geographic regions in a multi‐national study [Abstract]. American Thoracic Society International Conference; May 18‐23; San Francisco. 2007:[C97].
- Jenkins CR, Jones PW, Calverley PM, Celli B, Anderson JA, Ferguson GT, et al. Efficacy of salmeterol/fluticasone propionate by GOLD stage of chronic obstructive pulmonary disease: analysis from the randomised, placebo‐controlled TORCH study. Respiratory Research 2009;10:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PW, Anderson JA, Calverley PM, Celli BR, Ferguson GT, Jenkins C, et al. Health status in the TORCH study of COPD: treatment efficacy and other determinants of change. Respiratory Research 2011;12:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PW, Calverley PM, Celli B, Anderson JA, Ferguson GT, Jenkins C, et al. The TOwards a Revolution in COPD Health (TORCH) study: fluticasone propionate/salmeterol improves and sustains health status in COPD over 3 years. Chest 2006;130(4):177s. [Google Scholar]
- Jones PW, Vestbo J, Anderson JA, Celli B, Ferguson GT, Jenkins CR, et al. Informative withdrawal in a COPD study. An example from the TORCH Study [Abstract]. American Thoracic Society International Conference; 2007 May 18‐23; San Francisco. 2007:[C97].
- Keene ON, Vestbo J, Anderson JA, Calverley PM, Celli B, Ferguson GT, et al. Methods for therapeutic trials in COPD: Lessons from the TORCH trial. European Respiratory Journal 2009;34(5):1018‐23. [DOI] [PubMed] [Google Scholar]
- McDonough C, Blanchard AR. TORCH study results: pharmacotherapy reduces lung function decline in patients with chronic obstructive pulmonary disease. Hospital Practice 2010;38(2):92‐3. [DOI] [PubMed] [Google Scholar]
- McGarvey LP, John M, Anderson JA, Zvarich M, Wise RA, Committee TCE et al. Ascertainment of cause‐specific mortality in COPD: operations of the TORCH Clinical Endpoint Committee. Thorax 2007;62(5):411‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta RS, Kathman SJ, Daley‐Yates PT, Cahn T, Beerahee M, Kunka RL, et al. Pharmacokinetics and pharmacodynamics in COPD patients following long‐term twice‐daily treatment with salmeterol/fluticasone propionate (SFC) 50/500mg and the individual components [Abstract]. American Thoracic Society International Conference; 2007 May 18‐23; San Francisco. 2007:Poster #A41.
- SCO30003. A multi‐centre, randomised, double‐blind, parallel group, placebo‐controlled study to investigate the long‐term effects of salmeterol/fluticasone propionate (SERETIDE®/VIANI®/ADVAIR®) 50/500mcg bd, salmeterol 50mcg bd and fluticasone propionate 500mcg bd, all delivered via the DISKUS®/ACCUHALER® inhaler, on the survival of subjects with chronic obstructive pulmonary disease (COPD) over 3 years of treatment. www.ctr.gsk.co.uk (accessed 21 April 2012).
- Sacchetta A. Long term therapy and outcome of chronic obstructive pulmonary disease with or without co‐morbidity: The TORCH Study. Italian Journal of Medicine 2008;2(3):11‐5. [Google Scholar]
- Vestbo J, Anderson JA, Calverley PM, Celli B, Ferguson GT, Jenkins C, et al. Bias due to withdrawal in long‐term randomised trials in COPD: Evidence from the TORCH study. Clinical Respiratory Journal 2011;5(1):44‐9. [DOI] [PubMed] [Google Scholar]
- Vestbo J, TORCH Study Group. The TORCH (TOwards a Revolution in COPD Health) survival study protocol. European Respiratory Journal 2004;24(2):206‐10. [DOI] [PubMed] [Google Scholar]
Calverley 2010 {published data only}
- Calverley PMA, Kuna P, Monso E, Costantini M, Petruzzelli Sd, Sergio F, et al. Beclomethasone/formoterol in the management of COPD: a randomised controlled trial. Respiratory Medicine 2010;104:1858‐68. [DOI] [PubMed] [Google Scholar]
Choudhury 2005 {published data only}
- Choudhury AB, Dawson CM, Kilvington HE, Eldridge HE, James WY, Wedzicha JA, et al. Withdrawal of inhaled corticosteroids in people with chronic obstructive pulmonary disease (COPD) in primary care ‐ a randomised controlled trial [Abstract]. European Respiratory Journal 2005;26 Suppl 49:Abstract No. 1328. [Google Scholar]
- Choudhury AB, Dawson CM, Kilvington HE, Eldridge S, James WY, Wedzicha JA, et al. Withdrawal of inhaled corticosteroids in people with COPD in primary care: a randomised controlled trial. Respiratory Research 2007;8:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Dal Negro 2003 {published data only}
- Dal Negro R, Micheletto C, Trevsian F, Tognella S. Salmeterol and fluticasone 50ug/250ug BiD versus salmeterol 50ug bid and versus placebo in the long term treatment of COPD. American Thoracic Society 98th International Conference; May 17‐22; Georgia. 2002.
- Dal Negro RW, Pomari C, Tognella S, Micheletto C. Salmeterol & fluticasone 50 mcg/250 mcg bid in combination provides a better long‐term control than salmeterol 50 microg bid alone and placebo in COPD patients already treated with theophylline. Pulmonary Pharmacology and Therapeutics 2003;16(4):241‐6. [DOI] [PubMed] [Google Scholar]
Dransfield 2013 {published data only}
- Dransfield MT, Bourbeau J, Jones PW, Hanania NA, Mahler DA, Vestbo J, et al. Once‐daily inhaled fluticasone furoate and vilanterol versus vilanterol only for prevention of exacerbation of COPD: two replicate double‐blind, parallel‐group, randomised controlled trials. The Lancet: Respiratory Medicine 2013;1(3):210‐23. [DOI] [PubMed] [Google Scholar]
Ferguson 2008 {published data only}
- Dalal AA, Blanchette CM, Petersen H, Manavi K, St. Charles M. Cost‐effectiveness of fluticasone propionate/salmeterol (250/50 mcg) compared to salmeterol (50 mcg) in patients with COPD: economic evaluation of a randomized, double‐blind, parallel‐group, multi‐centre trial (Study SCO40043) [Abstract]. Chest 2008;134(4):106003s. [Google Scholar]
- Ferguson G, Anzueto A, Fei R, Emmett A, Crater G, Knobil K, et al. A randomized, double‐blind trial comparing the effect of fluticasone/salmeterol 250/50 to salmeterol on COPD exacerbations in patients with COPD. Chest 2007;132(4):530b‐531. [Google Scholar]
- Ferguson GT, Anzueto A, Fei R, Emmett A, Knobil K, Kalberg C. Effect of fluticasone propionate/salmeterol (250/50 mcg) or salmeterol (50 mcg) on COPD exacerbations. Respiratory Medicine 2008;102:1099‐108. [DOI] [PubMed] [Google Scholar]
- SCO40043. A randomized, double‐blind, parallel‐group, 52‐week study to compare the effect of fluticasone propionate/salmeterol DISKUS® 250/50mcg bid with salmeterol DISKUS® 50mcg bid on the annual rate of moderate/severe exacerbations in subjects with chronic obstructive pulmonary disease (COPD). http://ctr.gsk.co.uk (accessed 8 April 2008).
Fukuchi 2013 {published data only}
- Fukuchi Y, Samoro R, Fassakhov R, Taniguchi H, Ekelund J, Carlsson LG, et al. Budesonide/formoterol via Turbuhaler® versus formoterol via Turbuhaler® in patients with moderate to severe chronic obstructive pulmonary disease: Phase III multinational study results. Respirology 2013; Vol. 18:866‐73. [DOI] [PubMed]
- Ichinose M, Samoro R, Fassakhov R, Oguri M, Ekelund J, Carlsson LG, et al. Budesonide/formoterol vs formoterol, both via Turbuhaler®, in patients with moderate to severe COPD: Phase III study results [Abstract]. European Respiratory Society Annual Congress; 2012 Sep 1‐5; Vienna. 2012; Vol. 40, issue Suppl 56:374s [P2108].
GSK FCO30002 2005 {unpublished data only}
- FCO30002. A multi‐centre, randomised, placebo‐controlled, double‐blind comparison with 3 parallel groups to investigate the efficacy and safety of inhaled glucocorticoid fluticasone (500 μg bd via Diskus™) vs. oral glucocorticoid therapy vs. placebo in subjects with chronic obstructive airway disease (COPD) under therapy with Salmeterol (50 μg bd). GlaxoSmithKline Clinical Trial Register (accessed 15 April 2012).
GSK FLTA3025 2005 {unpublished data only}
- FLTA3025. A randomized, double‐blind, parallel‐group, comparative trial of inhaled fluticasone propionate 250mcg BID, 500mcg BID, and placebo BID via the DISKUS in subjects with chronic obstructive pulmonary disease (COPD). GlaxoSmithKline Clinical Trial Register (accessed 18 April 2012).
GSK SCO100470 2006 {unpublished data only}
- SCO100470. A multi‐centre, randomised, double‐blind, parallel group, 24‐week study to compare the effect of the salmeterol/fluticasone propionate combination product 50/250mcg, with salmeterol 50mcg both delivered twice daily via the DISKUS/ACCUHALER inhaler on lung function and dyspnoea in subjects with Chronic Obstructive Pulmonary Disease (COPD). http:ctr.gsk.co.uk (accessed 20 April 2012).
GSK SCO104925 2008 {unpublished data only}
- Evaluation of Novel Endpoints in Subjects with Chronic Obstructive Pulmonary Disease(COPD) in a Randomized, Double‐Blind, Placebo‐Controlled Study of Treatment with FluticasonePropionate/Salmeterol 500/50mcg combination and its individual components, FluticasonePropionate 500mcg and Salmeterol 50mcg. http://www.gsk‐clinicalstudyregister.com/ 2008 (accessed 19 April 2012).
- SCO104925. Evaluation of Novel Endpoints in Subjects with Chronic Obstructive Pulmonary Disease(COPD) in a Randomized, Double‐Blind, Placebo‐Controlled Study of Treatment with FluticasonePropionate/Salmeterol 500/50mcg combination and its individual components, FluticasonePropionate 500mcg and Salmeterol 50mcg. http:ctr.gsk.co.uk (accessed 19 April 2012).
GSK SCO30002 2005 {unpublished data only}
- SCO30002. A multi‐centre, randomised, double‐blind, parallel group, placebo‐controlled study to compare the efficacy and safety of inhaled salmeterol/fluticasone propionate combination product 25/250 µg two puffs bd and fluticasone propionate 250µg two puffs bd alone, all administered via metered dose inhalers (MDI), in the treatment of subjects with chronic obstructive pulmonary disease (COPD) for 52 weeks. GlaxoSmithKline Clinical Trial Register (accessed 21 April 2012).
GSK SCO40041 2008 {unpublished data only}
- NCT00355342. A Randomized, Double‐Blind, Parallel‐Group Clinical Trial Evaluating the Effect of the Fluticasone Propionate/Salmeterol Combination Product 250/50mcg BID Via DISKUS Versus Salmeterol 50mcg BID Via DISKUS on Bone Mineral Density in Subjects With Chronic Obstructive Pulmonary Disease (COPD). http://clinicaltrials.gov/show/NCT00355342 (accessed 19 April 2012).
Hanania 2003 {published data only}
- Hanania NA, Darken P, Horstman D, Reisner C, Lee B, Davis S, et al. The efficacy and safety of fluticasone propionate (250 mcg)/salmeterol (50 mcg) combined in the Diskus inhaler for the treatment of COPD. Chest 2003;124(3):834‐43. [DOI] [PubMed] [Google Scholar]
- Hanania NA, Knobil K, Watkins M, Wire P, Yates J, Darken P. Salmeterol and fluticasone propionate therapy administered by a single Diskus in patients with COPD. Chest 2002:S129. [Google Scholar]
- Hanania NA, Ramsdell J, Payne K, Davis S, Horstman D, Lee B, et al. Improvements in airflow and dyspnoea in COPD patients following 24 weeks treatment with salmeterol 50mcg and fluticasone propionate 250mcg alone or in combination via the Diskus. American Journal of Respiratory and Critical Care Medicine 2001;163 Suppl 5:A279. [Google Scholar]
- Horstman D, Darken P, Davis S, Lee B. Improvements in FEV1 and symptoms in poorly reversible COPD patients following treatment with salmeterol 50mcg/fluticasone propionate 250mcg combination [Abstract]. European Respiratory Journal 2003;22 Suppl 45:P434. [Google Scholar]
- Mahler DA, Darken P, Brown CP, Knobil K. Predicting lung function responses to combination therapy in chronic obstructive pulmonary disease (COPD) [Abstract]. National COPD Conference; 2003 Nov 14‐15; Arlington. 2003:1081.
- Mahler DA, Darken P, Brown CP, Knobil K. Predicting lung function responses to salmeterol/fluticasone propionate combination therapy in COPD [Abstract]. European Respiratory Journal 2003;22 Suppl 45:P429. [Google Scholar]
- SFCA3007. A randomized, double‐blind, placebo‐controlled, parallel‐group trial evaluating the safety and efficacy of the DISKUS formulations of salmeterol (SAL) 50mcg BID and fluticasone propionate (FP) 250mcg BID individually and in combination as salmeterol 50mcg/fluticasone propionate 250mcg BID (SFC 50/250) compared to placebo in COPD subjects. http://ctr.gsk.co.uk (accessed 20 April 2012).
- Spencer M, Wire P, Lee B, Chang CN, Darken P, Horstman D. Patients with COPD using salmeterol/fluticasone propionate combination therapy experience improved quality of life. European Respiratory Journal 2003;22 Suppl 45:51s. [Google Scholar]
- Spencer MD, Karia N, Anderson J. The clinical significance of treatment benefits with the salmeterol/fluticasone propionate 50/500mcg combination in COPD. European Respiratory Journal 2004;24 Suppl 48:290s. [Google Scholar]
Hattotuwa 2002 {published data only}
- Gizycki MJ, Hattotuwa KL, Barnes N, Jeffery PK. Effects of fluticasone propionate on inflammatory cells in COPD: an ultrastructural examination of endobronchial biopsy tissue. Thorax 2002;57(9):799‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattotuwa KL, Gizycki MJ, Ansari TW, Jeffery PK, Barnes NC. The effects of inhaled fluticasone on airway inflammation in chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine 2002;165(12):1592‐9. [DOI] [PubMed] [Google Scholar]
Kardos 2007 {published and unpublished data}
- Kardos P, Wencker M. Combination therapy with salmeterol and fluticasone propionate (SFC) is more effective than salmeterol (SAL) alone in reducing exacerbations of COPD. European Respiratory Journal 2005;26 Suppl 49:Abstract 1944. [Google Scholar]
- Kardos P, Wencker M, Glaab T, Vogelmeier C. Impact of salmeterol/fluticasone propionate versus salmeterol on exacerbations in severe chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine 2007;175(2):144‐9. [DOI] [PubMed] [Google Scholar]
- SCO30006. A randomised, double‐blind, parallel‐group study to investigate the protective effect of the combination of fluticasone and salmeterol (500/50 µg bid via the DISKUS) compared with salmeterol (50 µg bid via the DISKUS) on the incidence of moderate to severe exacerbations in patients with severe chronic obstructive pulmonary disease (COPD) (GOLD III/IV). http:ctr.gsk.co.uk (accessed 21 April 2012).
- Vogelmeier C. Combination therapy with salmeterol and fluticasone propionate (SFC) improves quality of life (QoL) more than salmeterol (SAL) alone in COPD. 42nd Nordic Lung Conference; 2005 Jun 9‐11; Trondheim. 2005; Vol. 13 Suppl 22.
- Vogelmeier CF, Wencker M, Glaab TH, Kardos P. Number needed to treat (NNT) to reduce exacerbations in severe COPD comparing salmeterol/fluticasone propionate (SFC) with salmeterol (SAL) treatment.. American Thoracic Society International Conference; May 19‐21; San Diego. 2006:A110.
Kerwin 2013 {published data only}
- Kerwin EM, Scott‐Wilson C, Sanford L, Rennard S, Agusti A, Barnes N, et al. A randomised trial of fluticasone furoate/vilanterol (50/25 mug; 100/25 mug) on lung function in COPD. Respiratory Medicine 2013; Vol. 107, issue 4:560‐9. [0954‐6111] [DOI] [PubMed]
- Kerwin EM, Scott‐Wilson C, Sanford L, Rennard SI, Agusti A, Barnes N, et al. Lung function effects and safety of fluticasone furoate (FF)/vilanterol (VI) in patients with COPD: Low‐mid dose assessment [Abstract]. European Respiratory Society Annual Congress; Sep 1‐5; Vienna. 2012; Vol. 40, issue Suppl 56:545s [3082].
Lapperre 2009 {published and unpublished data}
- Kunz LIZ, Strebus J, Budulac SE, Lapperre TS, Sterk PJ, Postma DS, et al. Inhaled Steroids Modulate Extracellular Matrix Composition in Bronchial Biopsies of COPD Patients: A Randomized, Controlled Trial. PloS one 2013; Vol. 8, issue 5:e63430. [DOI] [PMC free article] [PubMed]
- Lapperre TS, Snoeck‐Stroband JB, Gosman MM, Jansen DF, Schadewijk A, Thiadens HA, et al. Effect of fluticasone with and without salmeterol on pulmonary outcomes in chronic obstructive pulmonary disease: a randomized trial. Annals of Internal Medicine 2009;151(8):517‐27. [DOI] [PubMed] [Google Scholar]
Laptseva 2002 {published data only}
- Laptseva IM, Laptseva EA, Borshchevsky VV, Gurevich G, Kalechits O. Inhaled budesonide in the management of chronic obstructive pulmonary disease. European Respiratory Journal 2002;20 Suppl 38:244s. [Google Scholar]
Mahler 2002 {published and unpublished data}
- Mahler DA, Darken P, Brown CP, Knobil K. Predicting lung function responses to combination therapy in chronic obstructive pulmonary disease (COPD). http://www.abstracts2view.com (accessed 15 April 2012).
- Mahler DA, Wire P, Horstman D, Chang CN, Yates J, Fischer T, et al. Effectiveness of fluticasone propionate and salmeterol combination delivered via the Diskus device in the treatment of chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine 2002;166(8):1084‐91. [DOI] [PubMed] [Google Scholar]
- SFCA3006. A randomized, double‐blind, placebo‐controlled, parallel‐group trial evaluating the safety and efficacy of the DISKUS formulations of salmeterol (SAL) 50mcg BID and fluticasone propionate (FP) 500mcg BID individually and in combination as salmeterol 50mcg/fluticasone propionate 500mcg BID (SFC 50/500) compared to placebo in COPD subjects. http://ctr.gsk.co.uk (accessed 17 April 2012).
- Spencer M, Wire P, Lee B, Chang CN, Darken P, Horstman D. Patients with COPD using salmeterol/fluticasone propionate combination therapy experience improved quality of life. European Respiratory Journal 2003;22 Suppl 45:51. [Google Scholar]
- Spencer MD, Anderson JA. Salmeterol/fluticasone combination produces clinically important benefits in dyspnoea and fatigue [Abstract]. American Thoracic Society 2005 International Conference; 2005 May 20‐25; San Diego. 2005:B93 [Poster 308].
- Spencer MD, Karia N, Anderson J. The clinical significance of treatment benefits with the salmeterol/fluticasone propionate 50/500mcg combination in COPD. European Respiratory Journal 2004;24 Suppl 48:290. [Google Scholar]
Martinez 2013 {published data only}
- Martinez F, Boscia J, Feldman G, Scott‐Wilson C, Kilbride S, Fabbri L, et al. Lung function effects and safety of fluticasone furoate (FF)/vilanterol (VI) in patients with COPD: mid‐high dose assessment [Abstract]. European Respiratory Society Annual Congress, Vienna, Austria, September 1‐5. 2012; Vol. 40, issue Suppl 56:527s [P2887].
- Martinez FJ, Boscia J, Feldman G, Scott‐Wilson C, Kilbride S, Fabbri L, et al. Fluticasone furoate/vilanterol (100/25; 200/25 mug) improves lung function in COPD: a randomised trial. Respiratory Medicine 2013;107(4):550‐9. [0954‐6111] [DOI] [PubMed] [Google Scholar]
Mirici 2001 {published data only}
- Mirici A, Bektas Y, Ozbakis G, Erman Z. Effect of inhaled corticosteroids on respiratory function tests and airway inflammation in stable chronic obstructive pulmonary disease. Clinical Drug Investigation 2001;21(12):835‐42. [Google Scholar]
Ozol 2005 {published data only}
- Ozol D, Aysan T, Solak ZA, Mogulkoc N, Veral A, Sebik F. The effect of inhaled corticosteroids on bronchoalveolar lavage cells and IL‐8 levels in stable COPD patients. Respiratory Medicine 2005;99(12):1494‐500. [DOI] [PubMed] [Google Scholar]
Paggiaro 1998 {published data only}
- FLIT97. A multi‐centre, randomised, double‐blind, parallel group study of the efficacy and safety of inhaled fluticasone propionate 1000 µg daily with placebo in chronic obstructive pulmonary disease. http://ctr.gsk.co.uk (accessed 20 April 2012).
- Paggiaro PL, Dahle R, Bakran I, Frith L, Hollingworth K, Efthimiou J. Multicentre randomised placebo‐controlled trial of inhaled fluticasone propionate in patients with chronic obstructive pulmonary disease. International COPD Study Group. Lancet 1998;351(9105):773‐80. [DOI] [PubMed] [Google Scholar]
Pauwels 1999 {published data only}
- Johnell O, Pauwels R, Löfdahl C‐G, Laitinen LA, Postma DS, Pride NB, et al. Bone mineral density in patients with chronic obstructive pulmonary disease treated with budesonide Turbuhaler. European Respiratory Journal 2002;19(6):1058‐63. [DOI] [PubMed] [Google Scholar]
- Lofdahl CG, Postma DS, Laitinen LA, Ohlsson SV, Pauwels RA, Pride NB. The European Respiratory Society study on chronic obstructive pulmonary disease (EUROSCOP): recruitment methods and strategies. Respiratory Medicine 1998;92(3):467‐72. [DOI] [PubMed] [Google Scholar]
- Pauwels RA, Lofdahl CG, Laitinen LA, Schouten JP, Postma DS, Pride NB, et al. Long‐term treatment with inhaled budesonide in persons with mild chronic obstructive pulmonary disease who continue smoking. European Respiratory Society Study on Chronic Obstructive Pulmonary Disease. New England Journal of Medicine 1999;340(25):1948‐53. [DOI] [PubMed] [Google Scholar]
- Pauwels RA, Lofdahl CG, Pride NB, Postma DS, Laitinen LA, Ohlsson SV. European Respiratory Society study on chronic obstructive pulmonary disease (EUROSCOP): hypothesis and design. European Respiratory Journal 1992;5(10):1254‐61. [PubMed] [Google Scholar]
Renkema 1996 {published data only}
- Renkema TE, Schouten JP, Koeter GH, Postma DS. Effects of long‐term treatment with corticosteroids in COPD. Chest 1996;109(5):1156‐62. [DOI] [PubMed] [Google Scholar]
- Grunsven PM, Schayck CP, Derenne JP, Kerstjens HA, Renkema TE, Postma DS, et al. Long term effects of inhaled corticosteroids in chronic obstructive pulmonary disease: a meta‐analysis. Thorax 1999;54(1):7‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rennard 2009 {published data only}
- Bleecker ER, Meyers DA, Bailey WC, Sims AM, Bujac SR, Goldman M, et al. ADRB2 polymorphisms and budesonide/formoterol responses in COPD. Chest 2012; Vol. 142, issue 2:320‐8. [DOI] [PMC free article] [PubMed]
- Rennard SI, Tashkin DP, McElhattan J, Goldman M, Ramachandran S, Martin UJ, et al. Efficacy and tolerability of budesonide/formoterol in one hydrofluoroalkane pressurized metered‐dose inhaler in patients with chronic obstructive pulmonary disease. Results from a 1‐year randomized controlled clinical trial. Drugs 2009;69(5):549‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schermer 2009 {published data only}
- Chavannes NH, Schermer TR, Wouters EF, Akkermans RP, Dekhuijzen RP, Muris JW, et al. Predictive value and utility of oral steroid testing for treatment of COPD in primary care: the COOPT study. International Journal of COPD 2009;4:431‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schermer T, Chavannes N, Dekhuijzen R, Wouters E, Muris J, Akkermans R, et al. Fluticasone and N‐acetylcysteine in primary care patients with COPD or chronic bronchitis. Respiratory Medicine 2009;103(4):542‐51. [DOI] [PubMed] [Google Scholar]
Senderovitz 1999 {published data only}
- Senderovitz T, Vestbo J, Frandsen J, Maltbaek N, Norgaard M, Nielsen C, et al. Steroid reversibility test followed by inhaled budesonide or placebo in outpatients with stable chronic obstructive pulmonary disease. Respiratory Medicine 1999;93(10):715‐8. [DOI] [PubMed] [Google Scholar]
Shaker 2009 {published and unpublished data}
- Shaker SB, Dirksen A, Ulrik CS, Hestad M, Stavngaard T, Laursen LC, et al. The effect of inhaled corticosteroids on the development of emphysema in smokers assessed by annual computed tomography. COPD: Journal of Chronic Obstructive Pulmonary Disease 2009;6(2):104‐11. [DOI] [PubMed] [Google Scholar]
- Shaker SB, Stavngaard T, Laursen LC, Stoel BC, Dirksen A. Rapid fall in lung density following smoking cessation in COPD. COPD: Journal of Chronic Obstructive Pulmonary Disease 2011;8(1):2‐7. [DOI] [PubMed] [Google Scholar]
Sharafkhaneh 2012 {published data only}
- Sharafkhaneh A, Southard JG, Goldman M, Uryniak T, Martin UJ. Effect of budesonide/formoterol pMDI on COPD exacerbations: a double‐blind, randomized study. Respiratory Medicine 2012;106(2):257‐68. [DOI] [PubMed] [Google Scholar]
- Sharafkhaneh A, Uryniak T, Martin U. Long‐term effects of budesonide/formoterol pressurized metered‐dose inhaler on Chronic Obstructive Pulmonary Disease (COPD) symptoms and health status in patients with COPD [Abstract]. Chest 2011;140(4):528A. [Google Scholar]
Szafranski 2003 {published data only}
- Anderson P. Budesonide/formoterol in a single inhaler (Symbicort) provides early and sustained improvement in lung function in moderate to severe COPD. Thorax 2002;57 Suppl 3:iii43. [Google Scholar]
- Borgstrom L, Asking L, Olsson H, Peterson S. Lack of interaction between disease severity and therapeutic response with budesonide/formoterol in a single inhaler [Abstract].. American Thoracic Society 100th International Conference; May 21‐26; Orlando. 2004:C22 [Poster 505].
- Calverley P, Pauwels Dagger R, Löfdahl CG, Svensson K, Higenbottam T, Carlsson LG, et al. Relationship between respiratory symptoms and medical treatment in exacerbations of COPD. European Respiratory Journal 2005;26(3):406‐13. [DOI] [PubMed] [Google Scholar]
- Calverley PMA. Effect of budesonide/formoterol on severe exacerbations and lung function in moderate to severe COPD. Thorax 2002;BTS Winter Meeting 2002:S145. [Google Scholar]
- Calverley PMA, Szafranski W, Andersson A. Budesonide/formoterol is a well‐tolerated long term maintenance therapy for COPD. European Respiratory Journal 2005;26 Suppl 49:Poster 1917. [Google Scholar]
- Calverley PMA, Thompson NC, Olsson H. Budesonide/formoterol in a single inhaler sustains lung function improvements in COPD [Abstract]. European Respiratory Journal 2003;22 Suppl 45:P435. [Google Scholar]
- Campbell LM, Szafranski W. Budesonide/formoterol in a single inhaler (Symbicort) provides sustained relief from symptoms in moderate to severe COPD. Thorax. 2002; Vol. BTS Winter Meeting 2002:S143.
- Campbell LW, Szafranski W. Budesonide/formoterol in a single inhaler (Symbicort) reduces severe exacerbations in patients with moderate‐severe COPD. Thorax. 2002; Vol. BTS Winter Meeting 2002:S141.
- Dahl R, Cukier A, Olsson H. Budesonide/formoterol in a single inhaler reduces severe and mild exacerbations in patients with moderate to severe COPD. European Respiratory Journal 2002;20 Suppl 38:242 [P1575]. [Google Scholar]
- Egede F, Menga G. Budesonide/formoterol in a single inhaler provides sustained relief from symptoms and night‐time awakenings in moderate‐severe COPD: results from symptoms and night‐time awakenings in moderate to severe COPD: results from a 1‐year study. European Respiratory Journal 2002;20 Suppl 38:242 [P1574]. [Google Scholar]
- Halpin D, Stahl E, Lundback B, Anderson F, Peterson S. Treatment costs and number needed to treat (NNT) with budesonide/formoterol to avoid one exacerbation of COPD [Abstract]. American Thoracic Society 100th International Conference; May 21‐26; Orlando. 2004:D22 Poster 525.
- Jones PW, Stahl E, Svensson K. Improvement in health status in patients with moderate to severe COPD after treatment with budesonide/formoterol in a single inhaler. European Respiratory Journal 2002;20 Suppl 38:250 [P1613]. [Google Scholar]
- Korsgaard J, Sansores R. Budesonide/formoterol (single inhaler) provides sustained relief from shortness of breath and chest tightness in a 1‐year study of patients with moderate to severe COPD. European Respiratory Journal 2002;20 Suppl 38:242 [P1577]. [Google Scholar]
- Lange P, Saenz C. Budesonide/formoterol in a single inhaler is well tolerated in patients with moderate to severe COPD: results of a 1 year study. European Respiratory Journal 2002;20 Suppl 38:242 [P1573]. [Google Scholar]
- Lofdahl CG. Reducing the impact of COPD exacerbations: clinical efficacy of budesonide/formoterol. European Respiratory Review 2004;13(88):14‐21. [Google Scholar]
- Milanowski J, Nahabedian S. Budesonide/formoterol in a single inhaler acts rapidly to improve lung function and relieve symptoms in patients with moderate to severe COPD. European Respiratory Journal 2002;20 Suppl 38:242 [P1576]. [Google Scholar]
- Szafranski W, Cukier A, Ramirez A, Menga G, Sansores R, Nahabedian S, et al. Efficacy and safety of budesonide/formoterol in the management of chronic obstructive pulmonary disease. European Respiratory Journal 2003;21:74‐81. [DOI] [PubMed] [Google Scholar]
- Szfranski W, Ramirez A, Peterson S. Budesonide/formoterol in single inhaler provides sustained improvements in lung function in patients with moderate to severe COPD [Abstract]. European Respiratory Society Annual Congress; Sep 14‐18; Stockholm. 2002.
Tashkin 2008 SHINE {published and unpublished data}
- AstraZeneca. A 6‐month double‐blind, double‐dummy, randomized, parallel group, multicenter efficacy & safety study of SYMBICORT® pMDI 2 x 160/4.5 μg & 80/4.5 μg bid compared to formoterol TBH, budesonide pMDI (& the combination) & placebo in COPD Patients (SHINE) (study number D589900002). www.astrazenecaclinicaltrials.com (accessed 18 April 2012).
- Bleecker ER, Meyers DA, Bailey WC, Sims AM, Bujac SR, Goldman M, et al. Effect of ß2‐Adrenergic Receptor Gene Polymorphism Gly16Arg on Response to Budesonide/Formoterol Pressurized Metered‐Dose Inhaler in Chronic Obstructive Pulmonary Disease [Abstract]. American Journal of Respiratory and Critical Care Medicine 2011;183:A4086. [Google Scholar]
- Tashkin DP, Rennard SI, Martin P, Ramachandran S, Martin UJ, Silkoff PE. Efficacy and safety of budesonide and formoterol in one pressurized metered‐dose inhaler in patients with moderate to very severe chronic obstructive pulmonary disease: results of a 6‐month randomized clinical trial. Drugs 2008;68(14):1975‐2000. [DOI] [PubMed] [Google Scholar]
van Grunsven 2003 {published data only}
- Grunsven P, Schermer T, Akkermans R, Albers M, Boom G, Schayck O, et al. Short‐ and long‐term efficacy of fluticasone propionate in subjects with early signs and symptoms of chronic obstructive pulmonary disease. Results of the DIMCA study. Respiratory Medicine 2003;97(12):1303‐12. [DOI] [PubMed] [Google Scholar]
Verhoeven 2002 {published and unpublished data}
- Verhoeven GT, Garrelds IM, Hoogsteden HC, Zijlstra FJ. Effects of fluticasone propionate inhalation on levels of arachidonic acid metabolites in patients with chronic obstructive pulmonary disease. Mediators of Inflammation 2001;10(1):21‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven GT, Hegmans JP, Mulder PG, Bogaard JM, Hoogsteden HC, Prins JB. Effects of fluticasone propionate in COPD patients with bronchial hyper‐responsiveness. Thorax 2002;57(8):694‐700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven GT, Wijkhuijs AJ, Hooijkaas H, Hoogsteden HC, Sluiter W. Effect of an inhaled glucocorticoid on reactive oxygen species production by bronchoalveolar lavage cells from smoking COPD patients. Mediators of Inflammation 2000;9(2):109‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Vestbo 1999 {published data only}
- Vestbo J, Sorensen T, Lange P, Brix A, Torre P, Viskum K. Long‐term effect of inhaled budesonide in mild and moderate chronic obstructive pulmonary disease: a randomised controlled trial. Lancet 1999;353(9167):1819‐23. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Yildiz 2004 {published data only}
- Yildiz F, Basyigit I, Yildirim E, Boyaci H, Ilgazli A. Does addition of inhaled steroids to combined bronchodilator therapy affect health status in patients with COPD?. Respirology 2004;9(3):352‐5. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
GSK FLIP63 2005 {published data only}
- GlaxoSmithKline F. A single‐centre, double‐blind study to investigate the effects of inhaled fluticasone propionate (FP) 1500mcgdaily on inflammatory processes in the lungs of patients with chronic obstructive airways disease [FLIP63]. www.gsk‐clinicalstudyregister.com (accessed 20 April 2012).
GSK SAM30022 2005 {published data only}
- GSK SAM30022. A phase IV, multi‐centre, double‐blind, double‐dummy, parallel group, randomised study comparing Seretide (25/50 2 puffs bd) via the Evohaler (MDI‐HFA) with Beclometasone dipropionate (200mcg 2 puffs bd) via the MDI‐CFC in adolescents and adults experiencing moderate symptoms of reversible airways obstruction. www.gsk‐clinicalstudyregister.com (accessed 20 April 2012).
GSK SAM40004 2006 {published data only}
- GSK SAM40004. A multi‐centre, randomised, double‐blind, placebo‐controlled parallel group study to compare the effect on airway inflammation and remodelling of treatment with salmeterol/fluticasone propionate combination product (50/100μg strength) bd via the Accuhaler inhaler, or fluticasone propionate 100μg bd via the Accuhaler inhaler or placebo via the Accuhaler inhaler for 16 weeks, followed by double‐blind treatment for 52 weeks with the salmeterol/fluticasone propionate combination product (50/100μg strength) bd via the Accuhaler inhaler or fluticasone propionate 100μg bd via the Accuhaler inhaler, in adults with reversible airways obstruction (SIRIAS ‐ Seretide in Inflammation and Remodelling In Asthma Study). www.gsk‐clinicalstudyregister.com (accessed 20 April 2012).
GSK SAS40015 2007 {published data only}
- GSK SAS40015. A multi‐centre, randomised, double‐blind, double‐dummy, parallel‐group, 12‐week, active control comparison of the salmeterol/fluticasone propionate combination product (50/100 mcg strength) bd via the DISKUS/ACCUHALER inhaler with fluticasone propionate (100 mcg strength) bd via the DISKUS/ACCUHALER inhaler plus oral montelukast 10 mg od in adolescents and adults with reversible airways obstruction [SAS40015]. www.gsk‐clinicalstudyregister.com (accessed 20 April 2012).
GSK SCO100540 2006 {published data only}
- GSK SCO100540. A multi‐centre, randomised, double‐blind, parallel group study to investigate the efficacy and safety of the Salmeterol/fluticasone propionate combination at a strength of 50/500µg BD, compared with placebo via Accuhaler, added to usual chronic obstructive pulmonary disease (COPD) therapy, in subjects with COPD for 24 weeks [SCO100540]. www.gsk‐clinicalstudyregister.com (accessed 20 April 2012).
GSK SCO100646 2008 {published data only}
- GSK SCO100646. Clinical Evaluation of SFC for Chronic Obstructive Pulmonary Disease (Chronic Bronchitis, Emphysema) ‐Assessment of the Effect of Addition of Fluticasone Propionate to Salmeterol Xinafoate 50 mcg after Switching under Double‐blinded Conditions to SFC 50/250 mcg in Chronic Obstructive Pulmonary Disease. Study number SCO100646. www.gsk‐clinicalstudyregister.com (accessed 20 April 2012).
GSK SCO30005 2006 {published data only}
- GSK SCO30005. A 13‐week, double‐blind, parallel‐group, multi‐centre study to compare the bronchial anti‐inflammatory activity of the combination of salmeterol/ fluticasone propionate (SERETIDE™/ADVAIR™/VIANI™) 50/500 mcg twice daily compared with placebo twice daily in patients with Chronic Obstructive Pulmonary Disease [SCO30005]. www.gsk‐clinicalstudyregister.com (accessed 20 April 2012).
GSK SCO40030 2005 {published data only}
- GSK SCO40030. A Randomized, Double‐Blind, Placebo‐Controlled, Parallel Group Clinical Trial Evaluating the Effect of the Fluticasone Propionate/Salmeterol Combination Product 250/50mcg BID via DISKUS and Salmeterol 50mcg BID via DISKUS on Lung Hyperinflation in Subjects with Chronic Obstructive Pulmonary Disease (COPD). [SCO40030]. www.gsk‐clinicalstudyregister.com (accessed 20 April 2012).
GSK SCO40034 2009 {published data only}
- GSK SCO40034. A multi‐centre, randomised, double‐blind, double dummy, parallel group 12‐week exploratory study to compare the effect of the fluticasone/salmeterol propionate combination product (SERETIDE™) 50/500mcg bd via the DISKUS™/ACCUHALER™ inhaler with tiotropium bromide 18 mcg od via the Handihaler inhalation device on efficacy and safety in patients with Chronic Obstructive Pulmonary Disease (COPD). www.gsk‐clinicalstudyregister.com (accessed 20 April 2012).
GSK SCO40036 2009 {published data only}
- GSK SCO40036. Multicentre, Randomised, Double‐Blind, Double Dummy, Parallel Group, 104‐week Study to Compare the Effect of the Salmeterol/Fluticasone Propionate Combination Product (SERETIDE*) 50/500mcg Delivered Twice Daily via the DISKUS*/ACCUHALER* Inhaler with Tiotropium Bromide 18 mcg Delivered Once Daily via the HandiHaler Inhalation Device on the Rate of Health Care Utilisation Exacerbations in Subjects with Severe Chronic Obstructive Pulmonary Disease (COPD). www.gsk‐clinicalstudyregister.com (accessed 20 April 2012).
- GSK SCO40036. Multicentre, randomised, double‐blind, double‐dummy, parallel group, 104‐week study to compare the effect of the salmeterol/fluticasone propionate combination product (SERETIDE*) 50/500mcg delivered twice daily via the DISKUS*/ACCUHALER* inhaler with tiotropium bromide 18 mcg delivered once daily via the HandiHaler inhalation device on the rate of health care utilisation exacerbations in subjects with severe chronic obstructive pulmonary disease (COPD). www.gsk‐clinicalstudyregister.com (accessed 8 April 2012).
GSK SFCB3019 2004 {published data only}
- GSK SFCB3019. A multi‐centre randomized, double‐blind, double‐dummy, parallel‐group comparison of the salmeterol/fluticasone propionate combination product (50/500mcg strength) BD via one DISKUS/Accuhaler inhaler with salmeterol 50mcg BD via one DISKUS/Accuhaler and fluticasone propionate 500mcg BD via another DISKUS/Accuhaler and with fluticasone propionate 500mcg BD via one DISKUS/Accuhaler in adolescents and adults with reversible airways obstruction. www.gsk‐clinicalstudyregister.com (accessed 20 April 2012).
Lung Health Study 2000 {published data only}
- Lung Health Study Research Group. Effect of inhaled triamcinolone on the decline in pulmonary function in chronic obstructive pulmonary disease. New England Journal of Medicine 2000;343(26):1902‐9. [DOI] [PubMed] [Google Scholar]
van der Valk 2002 {published data only}
- Monninkhof E, Valk P, Palen J, Zielhuis G, Hervaarden C, Herwaarden C. Effect of discontinuation of inhaled steroids on health‐related quality of life in patients with COPD. American Journal of Respiratory and Critical Care Medicine 2002;165 Suppl 8:A226. [DOI] [PubMed] [Google Scholar]
- Valk P, Monninkhof E, Palen J, Zielhuis G, Herwaarden C, Phalen J. Effect of discontinuation of inhaled corticosteroids in patients with chronic obstructive pulmonary disease: the COPE study. American Journal of Respiratory and Critical Care Medicine 2002;166(1073‐449X):1358‐63. [DOI] [PubMed] [Google Scholar]
- Valk P, Monninkhof EM, Palen JAM, Zielhuis GA, Herwaarden CLA. Effect of discontinuation of inhaled fluticasone propionate on exacerbations in patients with COPD. European Respiratory Journal 2001;18 Suppl 33:183s. [Google Scholar]
Weir 1999 {published data only}
- Weir DC, Bale GA, Bright P, Sherwood Burge P. A double‐blind placebo‐controlled study of the effect of inhaled beclomethasone dipropionate for 2 years in patients with nonasthmatic chronic obstructive pulmonary disease. Clinical and Experimental Allergy 1999;29(Suppl 2):125‐8. [DOI] [PubMed] [Google Scholar]
Wouters 2005 {published data only}
- Wouters EF. COPD and Seretide: a multi‐centre intervention and characterisation (COSMIC) study: a rationale and baseline characteristics [abstract]. European Respiratory Society Annual Congress; Sep 14‐18; Stockholm. 2002:P1571.
- Wouters EF, Postma DS, Fokkens B, Hop WC, Prins J, Kuipers AF, et al. Withdrawal of fluticasone propionate from combined salmeterol/fluticasone treatment in patients with COPD causes immediate and sustained disease deterioration: a randomised controlled trial. Thorax 2005;60:480‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters EF, Postma DS, Fokkens B, Hop WC, Prins J, Kuipers AFTCC, et al. [Withdrawal of fluticasone propionate from combined salmeterol/fluticasone treatment in patients with COPD causes immediate and sustained disease deterioration: a randomised controlled trial]. [Portuguese]. Revista Portuguesa de Pneumologia 2005;11(6):587‐9. [PubMed] [Google Scholar]
References to studies awaiting assessment
Ohar 2013 {published data only}
- Ohar JA, Crater G, Emmett A, Ferro T, Morris A, Raphiou I, et al. Effects of Fluticasone Propionate/Salmeterol Combination 250/50mcg BID (ADVAIR DISKUS™) vs. Salmeterol 50mcg BID (SEREVENT DISKUS™) on Chronic Obstructive Pulmonary Disease (COPD) Exacerbation Rate, Following Acute Exacerbation or Hospitalization. American Journal of Respiratory and Critical Care Medicine. 2013; Vol. 187(Meeting Abstracts):A2439.
References to ongoing studies
Vestbo 2013 {published data only}
- Vestbo J, Anderson J, Brook RD, Calverley PMA, Celli BR, Crim C, et al. The study to understand mortality and morbidity in COPD (SUMMIT) study protocol. European Respiratory Journal 2013;41(5):1017‐22. [0903‐1936] [DOI] [PubMed] [Google Scholar]
Additional references
Agarwal 2010
- Agarwal R, Aggarwal AN, Gupta D, Jindal SK. Inhaled corticosteroids vs placebo for preventing COPD exacerbations. Chest 2010;137(2):318‐25. [DOI] [PubMed] [Google Scholar]
Agusti 2013
- Agusti A, Teresa L, Backer W, Zvarich MT, Locantore N, Barnes N, et al. A comparison of the efficacy and safety of once‐daily fluticasone furoate/vilanterol with twice‐daily fluticasone propionate/salmeterol in moderate to very severe COPD. The European Respiratory Journal. 2013/10/12 2013 Oct 10 [Epub ahead of print]. [1399‐3003: (Electronic)]
ATS/ERS 2004
- American Thoracic Society / European Respiratory Society Task Force. Standards for the diagnosis and management of patients with COPD [Internet]. Version 1.2. http://www.thoracic.org/go/copd. New York: American Thoracic Society, (accessed 4 May 2012). [URL: http://www.thoracic.org/go/copd]
Blais 2010
- Blais L, Forget A, Ramachandran S. Relative effectiveness of budesonide/formoterol and fluticasone propionate/salmeterol in a 1‐year, population‐based, matched cohort study of patients with chronic obstructive pulmonary disease (COPD): effect on COPD‐related exacerbations, emergency department visits and hospitalizations, medication utilization, and treatment adherence. Clinical Therapeutics 2010;32(7):1320‐8. [DOI] [PubMed] [Google Scholar]
BTS 2009
- British Thoracic Society, Community Acquired Pneumonia in Adults Guideline Group. Guidelines for the management of community acquired pneumonia in adults: update 2009. Thorax 2009;64(Suppl III):iii1–iii55. [DOI] [PubMed] [Google Scholar]
Bucher 1997
Cates 2013
- Cates C. Inhaled corticosteroids in COPD: quantifying risks and benefits. Thorax 2013;68(6):499‐500. [DOI] [PubMed] [Google Scholar]
Cipriani 2013
- Cipriani A, Higgins JPT, Geddes JR, Salanti GS. Conceptual and technical challenges in network meta‐analysis. Annals of Internal Medicine 2013;159(2):130‐7. [DOI] [PubMed] [Google Scholar]
Drummond 2008
- Drummond MB, Dasenbrook EC, Pitz MW, Murphy DJ, Fan E. Inhaled corticosteroids in patients with stable chronic obstructive pulmonary disease: a systematic review and meta‐analysis. JAMA 2008;300(20):2407‐16. [1538‐3598: (Electronic)] [DOI] [PMC free article] [PubMed] [Google Scholar]
Effing 2007
- Effing T, Monninkhof EEM, Valk PPDLPM, Zielhuis GGA, Walters EH, Palen JJ, et al. Self‐management education for patients with chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2007, Issue 4. [DOI: 10.1002/14651858.CD002990.pub2] [DOI] [PubMed] [Google Scholar]
Ernst 2007
- Ernst P, Gonzalez AV, Brassard P, Suissa S. Inhaled corticosteroid use in chronic obstructive pulmonary disease and the risk of hospitalization for pneumonia. American Journal of Respiratory and Critical Care Medicine 2007;176:162‐6. [DOI] [PubMed] [Google Scholar]
Glenny 2005
- Glenny AM, Altman DG, Song F, Sakarovitch C, Deeks JJ, D'Amico R, et al. Indirect comparisons of competing interventions. Health Technology Assessment 2005;9(26):1‐134, iii‐iv. [1366‐5278: (Print)] [DOI] [PubMed] [Google Scholar]
GOLD 2013
- GOLD. Global Strategy for the Diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2013. http://www.goldcopd.com (accessed 22 November 13).
Halpin 2011
- Halpin DM, Gray J, Edwards SJ, Morais J, Singh D. Budesonide/formoterol vs. salmeterol/fluticasone in COPD: a systematic review and adjusted indirect comparison of pneumonia in randomised controlled trials. International Journal of Clinical Practice 2011;65(7):764‐74. [1742‐1241: (Electronic)] [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 [updated September 2009]. The Cochrane Collaboration, 2009. www.cochrane‐handbook.org.
Janson 2013 [PATHOS]
- Janson C, Larsson K, Lisspers KH, Stallberg B, Stratelis G, Goike H, et al. Pneumonia and pneumonia related mortality in patients with COPD treated with fixed combinations of inhaled corticosteroid and long acting beta2 agonist: observational matched cohort study (PATHOS). BMJ. 2013/05/31 2013; Vol. 346:f3306. [1756‐1833: (Electronic)] [DOI] [PMC free article] [PubMed]
Johnson 1998
- Johnson M. Development of fluticasone propionate and comparison with other inhaled corticosteroids. Journal of Allergy and Clinical Immunology 1998;101(4):S434‐9. [DOI] [PubMed] [Google Scholar]
Lacasse 2006
- Lacasse Y, Goldstein R, Lasserson TJ, Martin S. Pulmonary rehabilitation for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2006, Issue 4. [DOI: 10.1002/14651858.CD003793.pub2] [DOI] [PubMed] [Google Scholar]
Marzoratti 2013
Nannini 2012
- Nannini LJ, Lasserson TJ, Poole P. Combined corticosteroid and long‐acting beta2‐agonist in one inhaler versus long‐acting beta2‐agonists for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2012, Issue 9. [DOI: 10.1002/14651858.CD006829.pub2] [DOI] [Google Scholar]
Nannini 2013
- Nannini LJ, Poole P, Milan SJ, Holmes R, Normansell R. Combined corticosteroid and long‐acting beta2‐agonist in one inhaler versus placebo for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2013, Issue 11. [DOI: 10.1002/14651858.CD003794.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
NICE 2011
- National Institute for Health and Clinical Excellence. Chronic obstructive pulmonary disease, costing report, implementing NICE guidance. http://guidance.nice.org.uk/CG101/CostingReport/pdf/English (accessed 4 February 2012). [URL: http://guidance.nice.org.uk/CG101/CostingReport/pdf/English]
Partridge 2009
- Partridge MR, Schuermann W, Beckman O, Persson T, Polanowski T. Effect on lung function and morning activities of budesonide/formoterol versus salmeterol/fluticasone in patients with COPD. Therapeutic Advances in Respiratory Disease 2009;3(4):147‐57. [DOI] [PubMed] [Google Scholar]
Puhan 2011
- Puhan MA, Gimeno‐Santos E, Scharplatz M, Troosters T, Walters EH, Steurer J. Pulmonary rehabilitation following exacerbations of chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2011, Issue 10. [DOI: 10.1002/14651858.CD005305.pub3] [DOI] [PubMed] [Google Scholar]
Roberts 2011
- Roberts M, Mapel D, Petersen H, Blanchette C, Ramachandran S. Comparative effectiveness of budesonide/formoterol and fluticasone/salmeterol for COPD management. Journal of Medical Economics 2011;14(6):769‐76. [DOI] [PubMed] [Google Scholar]
Rossios 2011
- Rossios C, To Y, To M, Ito M, Barnes PJ, Adcock IM, Johnson M, Ito K. Long‐acting fluticasone furoate has a superior pharmacological profile to fluticasone propionate in human respiratory cells. European Journal of Pharmacology 2011;670(1):244‐51. [DOI] [PubMed] [Google Scholar]
Ruiz 2011
Sin 2009
- Sin DD, Tashkin D, Zhang X, Radner F, Sjobring U, Thoren A, et al. Budesonide and the risk of pneumonia: a meta‐analysis of individual patient data. Lancet 2009;374(9691):712‐9. [1474‐547X: (Electronic)] [DOI] [PubMed] [Google Scholar]
Singanayagam 2010
- Singanayagam A, Chalmers J, Hill A. Inhaled corticosteroids and risk of pneumonia: evidence for and against the proposed association. Quarterly Journal of Medicine 2010;103:379‐85. [DOI] [PubMed] [Google Scholar]
Singh 2009
- Singh S, Amin AV, Loke YK. Long‐term use of inhaled corticosteroids and the risk of pneumonia in chronic obstructive pulmonary disease: a meta‐analysis. Archives of Internal Medicine 2009;169(3):219‐29. [1538‐3679: (Electronic)] [DOI] [PubMed] [Google Scholar]
Song 2008
- Song F, Harvey I, Lilford R. Adjusted indirect comparison may be less biased than direct comparison for evaluating new pharmaceutical interventions. Journal of Clinical Epidemiology 2008;61(5):455‐63. [0895‐4356: (Print)] [DOI] [PubMed] [Google Scholar]
Suissa 2013
- Suissa S, Patenaude V, Lapi F, Ernst P. Inhaled corticosteroids in COPD and the risk of serious pneumonia. Thorax 2013;68:1029‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Van der Meer 2001
- Meer RM, Wagena E, Ostelo RWJG, Jacobs AJE, Schayck O. Smoking cessation for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2001, Issue 1. [DOI: 10.1002/14651858.CD002999] [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 2012
- World Health Organization. Chronic respiratory diseases. http://www.who.int/respiratory/en/ (accessed 7 February 2012).
Yang 2009
- Yang IA, Fong K, Sim EHA, Black PN, Lasserson TJ. Inhaled corticosteroids for stable chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD002991.pub2] [DOI] [PubMed] [Google Scholar]
Yang 2012
- Yang IA, Clarke MS, Sim EH, Fong KM. Inhaled corticosteroids for stable chronic obstructive pulmonary disease. The Cochrane Database of Systematic Reviews. 2012/07/13 2012; Vol. 7:CD002991. [1469‐493X: (Electronic)] [DOI] [PMC free article] [PubMed]
Yawn 2013
- Yawn BP, Li Y, Tian H, Zhang J, Arcona S, Kahler KH. Inhaled corticosteroid use in patients with chronic obstructive pulmonary disease and the risk of pneumonia: a retrospective claims data analysis. International Journal of Chronic Obstructive Pulmonary Disease. 2013/07/10 2013; Vol. 8:295‐304. [1178‐2005: (Electronic)] [DOI] [PMC free article] [PubMed]