

Abstract
The dopamine receptor 4 (D4R) is highly expressed in both motor, associative and limbic subdivisions of the cortico-basal ganglia network. Due to the distribution in the brain, there is mounting evidence pointing to a role for the D4R in the modulation of this network and its subsequent involvement in L-DOPA induced dyskinesias in Parkinson’s disease. As part of our continued effort in the discovery of novel D4R antagonists, we report the discovery and characterization of a new 3- or 4-benzyloxypiperidine scaffold as D4R antagonists. We report several D4R selective compounds (>30-fold vs. other dopamine receptor subtypes) with improved in vitro and in vivo stability over previously reported D4R antagonists.
Keywords: Dopamine 4 receptor, D4R, antagonists, benzyloxypiperidine, Parkinson’s disease
Graphical Abstract

Dopamine (DA) is a catecholamine neurotransmitter, first recognized in the 1950s, and is the major neurotransmitter in the central nervous system (CNS).1 The dopamine receptors (DR) were first characterized as members of the Class A G-protein-coupled receptors (GPCRs) superfamily in the 1980s and 1990s, and they are subdivided into five subtypes, D1-5.2, 3 The five subtypes are further divided into two distinct families (D1-like and D2-like) based on whether they activate or inhibit adenyl cyclase activity. The D1-like family consists of the adenyl cyclase activators D1 and D5, and the D2-like family consists of the adenyl cyclase inhibitors D2, D3, and D4.3 There is high homology between the dopamine receptors with ~80% homology between the D1-like receptors and ~75% homology within the D2-like family. Although there are several approved therapies targeting the DRs, this high level of homology has made it difficult to identify subtype selective dopamine ligands.
The approved medications that modulate DRs are for schizophrenia (SZ), Parkinson’s disease (PD) and others; however, very few are selective within the DRs, nor are they selective against the biogenic amine receptors.2, 4, 5 The early work on the discovery of DR modulators centered on the dopamine hypothesis of schizophrenia and most of the work was focused on the D1, D2, and D3 receptors.6, 7 The D4R has not been as extensively studied as the other DRs, although a few compounds were brought to clinical evaluation for SZ, these failed due to a lack of efficacy.8–10 Based on the localization of the D4R in the brain, we, and others, have a renewed interest in the identification of selective D4R antagonists as tool compounds, and potential treatments, for other CNS diseases such as addiction, cancer and PD L-DOPA-induced dyskinesias (LID).11 Herein we report additional work in the discovery of new piperidine based ligands as D4R antagonists.
Initial work from our laboratory established the morpholine scaffold as a potent D4R antagonist resulting in the discovery of 1, a potent and selective D4R antagonist (Figure 1).12 Further in vivo work utilizing a 6-OHDA mouse model showed that 1 produced a dose-dependent reduction in dyskinesia AIM (abnormal involuntary movement) scores.13 Although this was an important tool compound, 1 suffers from significant liabilities limiting the progression of this compound (high intrinsic clearance). In addition to the morpholine, our laboratory has published a 4,4-difluoropiperidine scaffold (not shown) which produced very potent compounds; however, the fluorine atoms attenuated the basicity of nitrogen which led to a drastic reduction in brain penetration (cLogP > 5).14 These compounds also suffered from high intrinsic clearance. We chose to modify the scaffold by moving the oxygen in the chain adjacent to the piperidine ring system as this would introduce a new scaffold and the docking scores indicated this could be a productive change. In addition, the 4-oxopiperidine scaffold would eliminate the stereocenter which would make the synthesis more versatile. Thus, we set out to explore additional scaffolds to probe the structure-activity relationship (SAR).
Figure 1.

The synthesis of the molecules in this study is shown in Scheme 1. Starting with the commercially available tert-butyl (S)-3-hydroxy or 4-hydroxypiperidine-1-carboxylate, 4 or 5, which were alkylated with NaH and the respective BnBr to form the benzyl ethers, followed by Boc deprotection to yield 6 or 7.12, 15 Next, the final targets, 8 or 9, were realized by either (a) N-alkylation (Cs2CO3 and BnBr) or (b) under reductive amination protocols (Et3N, ArCHO, NaHB(OAc)3).16 Lastly, the phenyl acetamides, 11, were synthesized by direct N-alkylation of the requisite 2-chloroacetamides, 10, under basic conditions.17
Scheme 1.

Synthesis of the 3-(S)- or 4-ether piperidines, 8, 9, or 11.
Based on our previous work, we concentrated our SAR on a two-pronged approach by modifying both the nitrogen and oxygen substituents of the molecules (Table 1).12, 14 The oxygen group modification centered around aryl and heteroaryl groups containing both electron-donating and electron-withdrawing groups. The nitrogen substituent moieties consisted of the best groups that we have identified previously. Starting with the N-3-fluoro-4-methoxybenzyl group, we explored the O-alkylated area utilizing the benzyl groups that had previously provided active compounds. The 3-fluorobenzyl, 8a, showed good activity (Ki = 205.9 nM), and gratifyingly was selective against the other dopamine receptors (D1-3,5). Adding an additional fluoro (3,4-difluorophenyl, 8b, Ki = 169 nM), methyl group (4-fluoro-3-methyl, 8c, Ki = 135 nM) or having a 4-methylbenzyl (8e, Ki = 241 nM) or 2-methylbenzyl (8f, Ki = 343 nM) all produced active compounds, although there was a slight loss of activity with the 2-methyl group. As we have seen previously, the activity for these compounds resides in the (S)-enantiomer as the (R)-enantiomer is 15-fold less active (8d, Ki = 1980 nM). The 6-methyl-2-pyridine, 8g, lost significant activity (Ki = 1,040 nM). We have not previously looked at electron rich pyridines; however, compounds with an electron deficient pyridine (6-fluoro-2-pyridine) were active analogs in the morpholine scaffold. As with 8a, all the compounds were selective against the other dopamine receptors. Moving to the 2-methylimidazo[1,2-a]pyridine we saw activity divergence compared to the 3-fluoro-4-methoxybenzyl group. The 3-fluorobenzyl compound, 8h, was significantly less active (Ki = 1,939 nM) than the corresponding 8a. However, the potency could be regained by an additional fluorine (8i, Ki = 375 nM). The 4-fluoro-3-methylbenzyl was also active (8j, Ki = 188 nM); however, the 2-methylbenzyl lost activity and the 6-methyl-2-pyridine was again less active. The 3-trifluoromethoxybenzyl, 8m, was modestly active (Ki = 646 nM). The next analogs, 1-methyl and 3-methylimidazo[1,5-a]pyridine, showed a similar SAR pattern with the 3,4-difluorobenzyl (8o, Ki = 447 nM) and the 4-fluoro-3-methylbenzyl (8p, Ki = 166 nM) being the most active and the 3-methylbenzyl, 8n, 3-trifluoromethoxybenzyl, 8q, and 6-methyl-2-pyridine, 8r, being much less active or inactive. Interestingly, the 6-methyl-2-pyridine substituent was active when coupled with the 6-chloro-2-indole moiety, 8s (Ki = 319 nM). We then analyzed a variety of different N-benzyl substituents (8t-8ab) and only found a single compound that retained activity (8w, Ki = 165 nM). Lastly, we looked at direct phenyl substitution at the nitrogen; however, these compounds were not active. It is of note that all the 3-O-benzyl derivatives, regardless of the N-substitution were selective against the other dopamine receptors. In addition, as these compounds are targeting a central nervous system (CNS) target, we evaluated the multiparameter optimization (CNS MPO) score as a tool to assess the druglike attributes.18, 19 In this metric, compounds with scores >4 are considered to have a higher probability of success as CNS candidates. Compound 1 had a high (4.7) score, but the initial compounds, 8a-f, had lower scores that were predominantly driven by higher cLogP and cLogD scores due to higher lipophilicity. The next set of compounds (8g-s) where more polarity was introduced either through the pyridine or the imidazopyridine moieties all delivered higher CNS MPO scores (>4.5). Finally, all compounds in the last set had lower scores, again due to the higher cLogP and cLog D scores (8t-ab).
Table 1.
Modification of the 3-O-piperidine analogs

|

|

|

|
Ki values determined by competitive inhibition of [3H]N-methylspiperone (D2R, D3R, D4R) or [3H] SCH23390 (D1R, D5R) binding in membranes harvested from HEK293 cells stably expressing hD1-5R. All Ki are run in triplicate.
Moving to the 4-oxopiperidine scaffold we kept the same strategy and looked at varying both the 4-oxygen and the piperidine nitrogen in a systematic fashion that utilizes key moieties that were shown to impart activity in previous scaffolds (Table 2). We started with the 2-imidazo[1,2-a]pyridine southern portion and evaluated halogenated benzyl groups, 9a-9d. The 3-fluorobenzyl, 9a (Ki = 167 nM), activity was vastly improved from the 3-O-derivative, 8g, by 10-fold. Adding an additional fluorine (9b, Ki = 338 nM) or 3-trifluoromethyl (9c, Ki = 166 nM) produced similarly active compounds, as did the addition of the 4-chloro (9d, Ki = 134 nM). Moving to the 3-imidazo[1,5-a]pyridine or the 1-imidazo[1,5-a]pyridines with the same O-benzyl groups produced active compounds, 9e-h. Addition of a 3-methyl group to the 1-imidazo[1,5-a]pyridine (9i-k) also generated active analogs, with 9j (Ki = 96 nM) being the most potent compound in the series. Moving to the 5-NH-benzimidazole compounds we saw a dramatic loss of activity (9l-n) which was also seen in the N-methylated benzimidazoles (9o-q). Moving the nitrogen one place to the indazole also produced less active compounds (9r-t); however, this activity could be regained by methylating the nitrogen of the indazole (9u, Ki = 276 nM; 9v, Ki = 170 nM; 9w, Ki = 201 nM). As we had found the 2-indole compound displayed surprising activity on the pyridine moiety in the above series, we incorporated it into this scaffold. The compounds were active against D4 (9x-z); however, they did have varying degrees of activity against the other dopamine receptors. Lastly, based on a recent report on the discovery of acetamide based D4 agonists, we incorporated this moiety into our southern portion to evaluate the effect on our scaffold.17 Interestingly, the acetamide (11a-d) produced varying activity against D4 as well as dopamine receptor selectivity. We did, however, identify active compounds (11a, Ki = 299 nM; 11d, Ki = 121 nM) that was selective for D4. The same trends for the CNS MPO scores held as previously described for the 3-oxopiperidine analogs, with our most potent compound, 9j, having a high score (5.0). Additionally, the 4-oxopiperidine scaffold eliminates the chiral center and thus allows an easier synthetic scheme and diversification.
Table 2.
4-O-piperidine

|

|

|

|
Ki values determined by competitive inhibition of [3H]N-methylspiperone (D2R, D3R, D4R) or [3H] SCH23390 (D1R, D5R) binding in membranes harvested from HEK293 cells stably expressing hD1-5R. All Ki are run in triplicate.
Having evaluated several new compounds around two new scaffolds, we next wanted to take advantage of the recently published X-ray crystal structure of the D4 receptor.20, 21 Using the published structure (PDB: 5WIU) we were able to dock our compounds using the Schrödinger Drug Discovery Suite (Maestro, Release 2021-3) and then generate ligand interaction poses using Glide (Figure 2).22–24 The compounds share similar interactions, namely an Asp115 interaction with the morpholine or piperidine nitrogen, and a π-π stacking interaction with Phe410 and the benzyl group off the nitrogen. The acetamide function of 11d appears to make an additional H-bonding interaction with Asp115 (Figure 2D); however, this does not appear to provide a favorable interaction as shown by the potency. Interestingly, the morpholine oxygen does not show any interactions within the receptor, nor does the side chain oxygen, as can be seen in compounds where we replaced the oxygen with a methylene linker. The presence of the side chain oxygen does play a couple of key roles however: 1. it reduces the cLogP, and 2. provides a synthetic handle to gain access to more diverse analogs.
Figure 2.
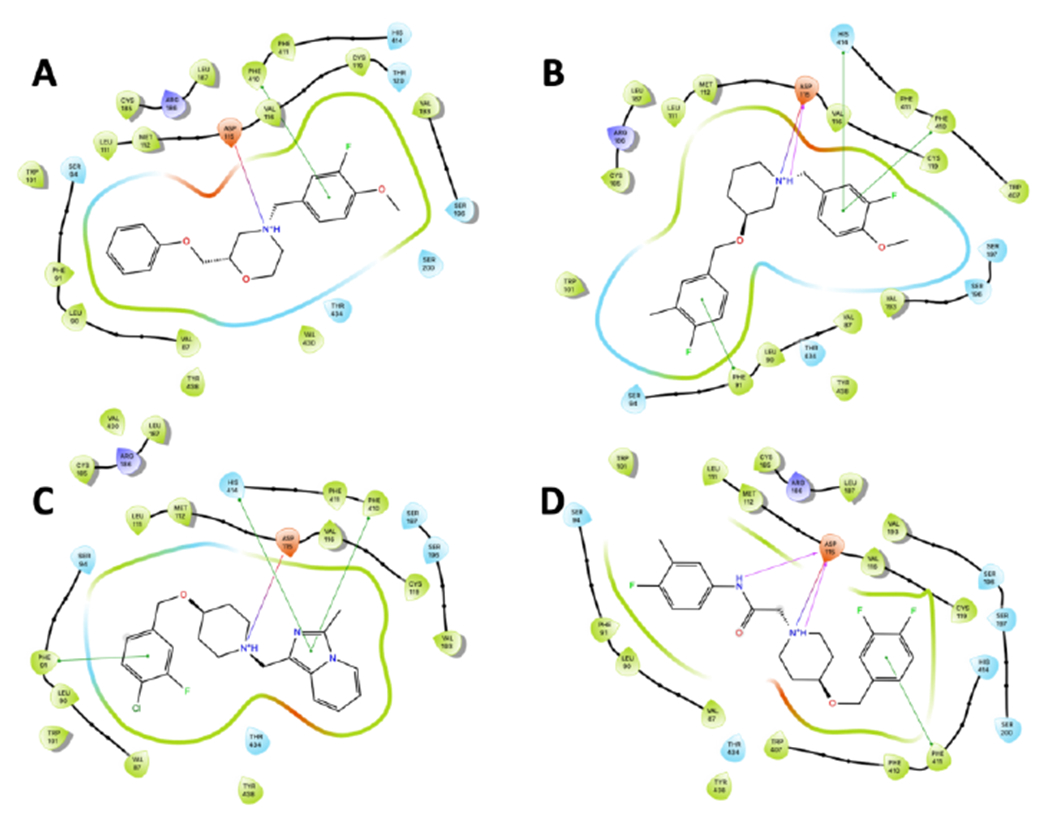
Ligand interaction poses generated using Glide docking software (PDB: 5WIU). A. Compound 1, B. Compound 8c, C. Compound 9j, D. Compound 11d.
Next, we further evaluated selected compounds based on liver microsome stability and plasma protein binding (Table 3).25–27 We again used 1 as our comparator compound because despite being highly unstable in liver microsomes (human and mouse) it did show good free fraction in both human and mouse. A metabolite ID study on 1 indicated that N-dealkylation was the major metabolic liability, followed by further O-phenyl oxidation of the unsubstituted moiety. Only three of the 3-oxypiperidine analogs were tested (8b, 8i, 8o); however, these represented compounds with varying N-alkylated moieties from the substituted N-benzyl to the N-methylimidazopyridine. Moving to the 4-oxopiperidine scaffold reversed this trend. Evaluating the N-methylimidazo[1,2-a]pyridine showed these compounds to have significantly improved liver microsome stability in both human and mouse. In fact, 9a and 9b were tested as stable in human liver microsomes but were modestly stable in mouse liver microsomes. The imidazo[1,5-a]pyridine moieties were not as stable (9e, 9h) and addition of a methyl group to block potential oxidation at the carbon adjacent to the two nitrogen atoms did not improve the stability (9i, 9j). The NH-benzimidazole analogs also displayed improved stability; however, these compounds were not as active (Ki > 1,000 nM). The addition of a methyl group to the nitrogen did not impact their stability negatively but did improve the potency by 10-fold. Lastly, the acetamide analog, 11d, was moderately stable in both human and mouse liver microsomes. As seen in 1, most of the compounds tested displayed good free fraction (> 3%) in both human and mice; although there were some species differences which trended to higher free fraction in mice. Most of the compounds that showed this species difference contained an imidazopyridine southern fragment. We were concerned that there may be plasma instability in mice which would contribute to this; however, aside from 11d which was not stable in mouse plasma, the other compounds were stable in both human and mice plasma when tested.
Table 3.
In vitro PK parameters of selected compounds.
| Cmpd | D4, Ki (nM) | Intrinsic clearance (mL/min/kg)a,d | Plasma Protein Binding (%fu)b,d | ||||
|---|---|---|---|---|---|---|---|
| hCLINT | hCLHEP | mCLINT | mCLHEP | hPPB | mPPB | ||
| 1 | 5.8 | 71.9 | 16.3 | 2128 | 67.8 | 3.1 | 3.7 |
| 8b | 169.3 | 272.5 | 18.7 | 1261.3 | 84.1 | 1.1 | 2.6 |
| 8i | 187.5 | 689.3 | 19.5 | 1402.8 | 84.7 | 0.2 | 8.7 |
| 8o | 165.5 | 621.7 | 19.5 | 5368.8 | 88.6 | 3.9 | 18.1 |
| 9a | 167.0 | <23.1 | 10.7 | 380.2 | 72.8 | 7.0 | 35.5 |
| 9b | 337.5 | <20 | <10 | 158.3 | 57.4 | 6.2 | 42.8 |
| 9c | 166.1 | 44.6 | 13.9 | 309.7 | 69.8 | 1.8 | 17.1 |
| 9d | 133.9 | 44.1 | 13.8 | 96.8 | 46.7 | 1.8 | 22.4 |
| 9e | 157.6 | 212.5 | 18.4 | 916.9 | 82.0 | 1.0 | 1.7 |
| 9h | 165.6 | 340.0 | 19.0 | 1261.3 | 84.1 | 2.3 | 40.5 |
| 9i | 309.1 | 85.3 | 16.3 | 272.0 | 67.7 | 5.2 | 3.6 |
| 9j | 95.5 | 114.5 | 17.1 | 140.3 | 54.9 | 7.1 | 13.2 |
| 9l | 1,517.8 | 48.3 | 14.2 | 90.4 | 45.1 | 7.7 | 17.1 |
| 9m | 1,803.4 | 32.9 | 12.5 | 76.4 | 41.3 | 2.5 | 16.1 |
| 9n | 4,279.6 | <20 | <10 | 93.9 | 46.0 | 22.9 | 40.1 |
| 9p | 640 | 63.3 | 15.3 | 85.2 | 43.8 | 1.0 | 5.7 |
| 9u | 276.1 | 32.8 | 12.5 | 103.7 | 48.2 | 3.6 | 16.6 |
| 9v | 170.0 | 42.7 | 13.7 | 85.3 | 43.8 | 0.9 | 5.2 |
| 9w | 200.5 | 63.3 | 15.3 | 85.2 | 43.8 | 1.0 | 5.7 |
| 9y | 149.4 | 23.5 | 10.8 | 83.2 | 43.3 | <0.3 | 0.4 |
| 9z | 211.4 | 26.9 | 11.5 | 71.5 | 39.9 | 0.1 | 0.1 |
| 11a | 299.4 | 144.7 | 17.6 | 287.1 | 68.6 | 0.3 | 1.5 |
| 11d | 120.9 | 49.5 | 14.3 | 306.2 | 69.6 | 0.5 | c |
Predicted hepatic clearance based on intrinsic clearance in mouse and human liver microsomes using the well-stirred organ CL model (binding terms excluded).
fu = fraction unbound.
Unstable in human or mouse plasma.
In vitro DMPK studies performed at Q2 Solutions, Indianapolis, IN
Lastly, we evaluated a few key compounds in an in vivo cassette experiment utilizing a 4-in-1 experiment design.28, 29 In order to conserve resources as well as animals, we chose to do the in vivo PK experiments in rats so that we could serially sample as well as utilize the cassette design. We chose a range of compounds to evaluate to get a better idea around the SAR of the PK parameters. As can be seen in Table 3, all of the heteroaryl southern containing compounds displayed very high clearance with short half-lives (CL > 100 mL/min/kg; t1/2 < 2 hr). The compounds also had high Vss (5 – 19 L/kg); although all the compounds were highly brain penetrant (Kp > 6). In contrast, the acetamide derivative, 11a, displayed very favorable in vivo PK parameters (CL = 22 mL/min/kg and t1/2 = 4.4 hr) and was brain penetrant (Kp = 2.9). The brain penetration is lower than for the other compounds, which may be attributed to the addition of the free NH of the amide; however, the compound had appreciable levels in the brain.
In conclusion, we have identified new 3- and 4-oxopiperidine scaffolds as potent and selective dopamine 4 antagonists. This work builds upon our previous disclosures on chiral morpholines and 4,4-difluoropiperidine compounds. Although this scaffold does not impart the same level of activity, chemical optimization has improved human and mouse liver microsome stability. We have also evaluated the scaffolds using the published X-ray crystal structure and have identified the key Asp115 and Phe410 interactions that are present in all the compounds. This analysis will enable further scaffold diversification and chemical optimization. In vivo PK studies identified the acetamide containing compound, 11a, as lead molecule with low clearance and good brain penetration. Further optimization and in vivo animal studies are on-going and will be reported in due course.
Supplementary Material
Table 4.
In vivo cassette for selected compounds.
| 9b | 9d | 9l | 9n | 9p | 9u | 9y | 11a | |
|---|---|---|---|---|---|---|---|---|
| Rat cassette (0.25 mpk) a,b,c | ||||||||
| CL (ml/min/kg) | 116 | 134 | 199 | 235 | 107 | 140 | 127 | 22.0 |
| T1/2 (h) | 0.9 | 0.6 | 1.1 | 1.2 | 1.1 | 1.4 | 1.8 | 4.4 |
| C0 (ng/mL) | 49.4 | 52.2 | 23.5 | 22.9 | 60.2 | 32.5 | 36.9 | 91 |
| Vss (L/kg) | 6.4 | 5.1 | 13.6 | 19.2 | 6.3 | 12.9 | 12.6 | 5.5 |
| AUC (h*ng/mL) | 142 | 125 | 79.9 | 65.6 | 149 | 112 | 126 | 747 |
| Rat cassette (Day 2) d | ||||||||
| Plasma (ng/mL) | 34.9 | 29.9 | 14.1 | 7.0 | 30.1 | 30.6 | 19.4 | 45.8 |
| Brain (ng/g) | 232 | 453 | 139 | 112 | 214 | 355 | 562 | 134 |
| Kp (B:P) | 6.6 | 15.5 | 9.9 | 16.1 | 7.2 | 11.6 | 28.9 | 2.9 |
In vivo cassette performed at Pharmaron, Inc. Louisville, KY.
Vehicle: DMSO:PEG400:EtOH:Saline (5:48:10:37).
IV blood sampling at 0.0833, 0.25, 0.5, 1, 3, 4, 8 and 24 hrs post dose.
Brain sampling on Day 2 at 0.25 hr post dose.
Acknowledgements
This work was generously supported by a grant from the US National Institutes of Health (NINDS: R01NS119266) to C.R.H. The authors would like to thank Q2 Solutions (Indianapolis, IN USA) and Pharmaron, Inc. (Louisville, KY) for the in vitro and in vivo DMPK experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jaber M, Robinson SW, Missale C, Caron MG. Dopamine receptors and brain function. Neuropharmacol. 1996;35: 1503–1519. [DOI] [PubMed] [Google Scholar]
- 2.Zhou Q-Y, Grandy DK, Thambi L, et al. Cloning and expression of human and rat D1 dopamine receptors. Nature. 1990;347: 76–80. [DOI] [PubMed] [Google Scholar]
- 3.Beaulieu J-M, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63: 182–217. [DOI] [PubMed] [Google Scholar]
- 4.Boyd KN, Mailman RB. Dopamine receptor signaling and current and future antipsychotic drugs. Handb Exp Pharmacol. 2012;212: 53–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hurley MJ, Jenner P. What has been learnt from study of dopamine receptors in Parkinson’s disease? Pharm Therap. 2006; 111: 715–728. [DOI] [PubMed] [Google Scholar]
- 6.Brisch R, Saniotis A, Wolf R, et al. The role of dopamine in schizophrenia from a neurobiological and evolutionary perspective: old fashioned, but still in vogue. Front Psychiatry. 2014;5: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nikam SSA AK Evaluation of schizophrenia drugs: a focus on dopaminergic systems. Curr Opin Investig Drugs. 2008;9: 37–46. [PubMed] [Google Scholar]
- 8.Tarazi FI, Baldessarini RJ. Dopamine D4 receptors: significance for molecular psychiatry at the millenium. Mol Psychiatry. 1999;4: 529–538. [DOI] [PubMed] [Google Scholar]
- 9.Tarazi FI, Zhang K, Baldessarini RJ. Dopamine D4 receptors: beyond schizophrenia. J Recept Signal Transduct Res. 2004;24: 131–147. [DOI] [PubMed] [Google Scholar]
- 10.Bristow LJ, Kramer MS, Kulagowski J, Patel S, Ragan CI, Seabrook GR. Schizophrenia and L-745,870, a novel dopamine D4 receptor antagonist. Trends Pharm Sci. 1997;18: 186–188. [DOI] [PubMed] [Google Scholar]
- 11.Lindsley CW, Hopkins CR. Return of D4 dopamine receptor antagonists in drug discovery. J Med Chem. 2017;60: 7233–7243. [DOI] [PubMed] [Google Scholar]
- 12.Witt JO, McCollum AL, Hurtado MA, et al. Synthesis and characterization of a series of chiral alkoxymethyl morpholine analogs as dopamine receptor 4 (D4R) antagonists. Bioorg Med Chem Lett. 2016;26: 2481–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sebastianutto I, Maslava N, Hopkins CR, Cenci MA. Validation of an improved scale for rating L-DOPA-induced dyskinesia in the mouse and effects of specific dopamine receptor antagonists. Neurobiol Dis. 2016;96: 156–170. [DOI] [PubMed] [Google Scholar]
- 14.Jeffries DE, Witt JO, McCollum AL, et al. Discovery, characterization and biological evaluation of a novel (R)-4,4-difluoropiperidine scaffold as dopamine receptor 4 (D4R) antagonists. Bioorg Med Chem Lett. 2016;26: 5757–5764. [DOI] [PubMed] [Google Scholar]
- 15.Berry CB, Bubser M, Jones CK, et al. Discovery and characterization of ML398, a potent and selective antagonist of the D4 receptor with in vivo activity. ACS Med Chem Lett. 2014;5: 1060–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abdel-Magid AF, Carson KG, Harris BD, Maryanoff CA, Shah RD. Reductive amination of aldehydes and ketones with sodium triacetoxyborohydride. Studies on direct and indirect reductive amination procedures. J Org Chem. 1996;61: 3849–3862. [DOI] [PubMed] [Google Scholar]
- 17.Keck TM, Free RB, Day MM, et al. Dopamine D4 receptor-selective compounds reveal structure-activity relationships that engender agonist efficacy. J Med Chem. 2019;62: 3722–3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wager TT, Hou X, Verhoest PR, Villalobos A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem Neurosci. 2010;1: 435–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wager TT, Chandrasekaran RY, Hou X, et al. Defining desirable central nervous system drug space through the alignment of molecular properties, in vitro ADME, and safety attributes. ACS Chem Neurosci. 2010;1: 420–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang S, Wacker D, Levit A, et al. D4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science. 2017;358: 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lyu J, Wang S, Balius TE, et al. Ultra-large library docking for discovering new chemotypes. Nature. 2019;566: 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Small-Molecule Drug Discovery Suite 2021-3, Schrödinger, LLC, New York, NY, 2021. [Google Scholar]
- 23.Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47: 1739–1749. [DOI] [PubMed] [Google Scholar]
- 24.Halgren TA, Murphy RB, Friesner RA, et al. Glide: a new approach for rapide, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47: 1750–1759. [DOI] [PubMed] [Google Scholar]
- 25.Obach RS. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Disp. 1999;27: 1350–1359. [PubMed] [Google Scholar]
- 26.Obach RS, Baxter JG, Liston TE, et al. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J Pharmacol Exp Ther. 1997;283: 46–58. [PubMed] [Google Scholar]
- 27.Chang G, Steyn SJ, Umland JP, Scott DO. Strategic Use of Plasma and Microsome Binding To Exploit in Vitro Clearance in Early Drug Discovery. ACS Med Chem Lett. 2010;1: 50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bridges TM, Morrison RD, Byers FW, Luo S, Daniels JS. Use of a novel rapid and resource-efficient cassette dosing approach to determine the pharmacokinetics and CNS distribution of small molecule 7-transmembrane receptor allosteric modulators in rat. Pharmacol Res Perspect. 2014;2: e00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith NF, Raynaud FI, Workman P. The application of cassette dosing for pharmacokinetic screening in small-molecule cancer drug discovery. Mol Cancer Ther. 2007;6: 428–440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.