

Abstract
Age-related cognitive decline is accompanied by an increase of neuronal apoptosis and a dysregulation of neuroplasticity-related molecules such as brain-derived neurotrophic factor and neurotoxic factors including beta amyloid (Aβ) peptide. Because it has been previously demonstrated that phosphodiesterase-5 inhibitors (PDE5-Is) protect against hippocampal synaptic dysfunction and memory deficits in mouse models of Alzheimer’s disease and physiological aging, we investigated the effect of a treatment with the PDE5-I, sildenafil, on cell death, pro- and antiapoptotic molecules, and Aβ production. We demonstrated that chronic intraperitoneal injection of sildenafil (3 mg/kg for 3 weeks) decreased terminal deoxyuridine triphosphate nick end labeling-positive cells in the CA1 hippocampal area of 26–30-month-old mice, downregulating the proapoptotic proteins, caspase-3 and B-cell lymphoma 2-associated X, and increasing antiapoptotic molecules such as B-cell lymphoma protein-2 and brain-derived neurotrophic factor. Also, sildenafil reverted the shifting of amyloid precursor protein processing toward Aβ42 production and the increase of the Aβ42:Aβ40 ratio in aged mice. Our data suggest that PDE5-I might be beneficial to treat age-related detrimental features in a physiological mouse model of aging.
Keywords: Aging, Sildenafil, Apoptosis, Caspase-3, Bax/Bcl-2 ratio, BDNF, APP processing, Beta-amyloid
1. Introduction
Cyclic guanosine monophosphate (cGMP) is a second messenger-regulating signal transduction in various biological systems, including the central nervous system (CNS). Its levels are maintained thanks to a balance between its production, catalyzed by guanylyl cyclase, and its degradation, carried out by phosphodiesterases (PDEs). Because cGMP has been recognized as a key molecule in synaptic plasticity and memory, an increasing number of studies have focused on strategies aimed to regulate its levels. Indeed, a decrease in cGMP-dependent signal transduction has been demonstrated in hippocampus during aging and Alzheimer’s disease (AD). The age-induced decrease in cGMP has been related to an increase in PDE expression and activity (Domek-Łopacińska and Strosznajder, 2010) and also to the accumulation of beta amyloid (Aβ) peptide, a key molecule in AD pathogenesis, which is capable of inhibiting the N-methyl-D-aspartate (NMDA) receptor-dependent activation of the nitric oxide (NO)–cGMP pathway (Chalimoniuk and Strosznajder, 1998) and the physiological increase of cGMP levels that occur during hippocampal synaptic plasticity (Puzzo et al., 2005).
Age-related decline of cognitive functions is thought to be associated with: (1) an increase of neuronal apoptosis (Elmore, 2007), a process of programmed cell death that might result in pathological processes such as degeneration (Pollack and Leeuwenburgh, 2001; Pollack et al., 2002; Reix et al., 2007); (2) a dysregulation of neuroplasticity-related molecules such as brain-derived neurotrophic factor (BDNF) (Assunção et al., 2010; Kim et al., 2010; Li et al., 2009; Liu et al., 2006; Zhao et al., 2009); and (3) an increase of neurotoxic factors including Aβ that could also exert a pathogenic role in cognitive decline of elderly nondemented individuals (Rodrigue et al., 2012). Interestingly, sildenafil (Viagra), a specific PDE5 inhibitor (PDE5-I) widely used as the elective drug to treat erectile dysfunction and pulmonary hypertension, has recently been proposed as a molecule to treat a variety of disorders including AD and aging.
In this study, we focused on normal aging, which is associated with substantial structural and functional changes in the CNS (Ziv and Melamed, 2010). However, it is not yet clear whether PDE5-Is can counteract neuronal apoptosis, BDNF dysregulation, and increased Aβ levels.
Apoptosis is regulated by a subtle balance between pro- and antiapoptotic molecules, like those of the proto oncogene B-cell lymphoma protein-2 (Bcl-2) family. Among the Bcl-2 family, the protein B-cell lymphoma 2-associated X (Bax) is activated in the initiation of apoptosis, which is followed by downstream events including caspase activation. In particular, caspase-3 is a widely studied apoptosis executioner (Cohen, 1997) implicated in synapse degeneration and dysfunction during AD and aging (D’Amelio et al., 2011; Louneva et al., 2008; Lynch and Lynch, 2002). On the other hand, the Bcl-2 protein plays an antiapoptotic role, which is, however, counteracted by caspase-3, leading to an enhancement of cell death (Yang et al., 2002).
The Bax/Bcl-2 system is regulated by the cyclic adenosine monophosphate-responsive element binding protein (CREB). This protein is known to play a critical role in neuronal survival (Dawson and Ginty, 2002; Walton and Dragunow, 2000) and exerts an antiapoptotic role in several cell types. The suppression of CREB by functional antagonists (Jaworski et al., 2003), or the postnatal disruption of CREB and cAMP responsive element modulator (Mantamadiotis et al., 2002) results in a progressive neuronal apoptosis and neurodegeneration. Moreover, CREB-null mice display an excess of apoptosis and degeneration in the peripheral nervous system (Lonze et al., 2002), and the age-related decline of CREB activity (Asanuma et al., 1996; Chung et al., 2002; Kudo et al., 2005; Monti et al., 2005) might be associated with an increase of neuronal apoptosis. CREB induces also the transcription of BDNF (Zieg et al., 2008), a neurotrophic factor involved in several neuronal functions during development and adult life, that has been recently widely studied for its role in the pathophysiology of neurodegenerative and psychiatric disorders. BDNF stimulates synaptic activity directly or by exerting a positive feedback on synaptic activity itself; in turn, BDNF levels are modulated by synaptic activity (Arancio and Chao, 2007). Furthermore, BDNF is capable of protecting neurons against apoptosis by stimulating the production of Bcl-2 antioxidant molecules (Mattson, 2000; Tamatani et al., 1998). Thus, the impairment of CREB phosphorylation might affect synaptic plasticity and apoptosis through a reduction of BDNF.
A suppression of CREB and BDNF activity can also be caused by an increase in Aβ levels such as those detected in AD or aging (Puzzo et al., 2005, 2009; Tong et al., 2001). In contrast, Aβ injections in hippocampal rat cortex have been recently demonstrated to stimulate the increase of apoptotic markers such as inflammatory cytokines, caspase-3, and terminal deoxynucleotidyl transferase (TdT)-mediated deoxyuridine triphosphate (dUTP)-biotin nick end labeling (TUNEL)-positive cells (Abdi et al., 2011). Additionally, caspase-3 is capable of stimulating the formation of Aβ by acting on amyloid precursor protein (APP), a transmembrane protein processed by a series of secretases to produce Aβ. Indeed, APP full-length might be initially cut by α-secretases (nonamyloidogenic pathway) generating the soluble (s) fragment, sAPPα and the carboxy-terminal fragment (CTF) 83; or by the β-secretases (amyloidogenic pathway) generating sAPPβ and CTF99. Then, γ-secretase generates a p3 fragment and APP intracellular domain fragment from C83 and Aβ40–42 and APP intracellular domain from C99.
In previous works, we have demonstrated that a 3-week treatment with the PDE5-I sildenafil counteracted the decrease of CREB phosphorylation observed in a mouse model of AD (Puzzo et al., 2009), and in a physiological mouse model of aging (Palmeri et al., 2013). In both models, the drug also rescued the decline of cognitive performance. In addition, the increase in Aβ levels of AD mice was rescued (Puzzo et al., 2009). Considering that CREB might regulate proapoptotic and antiapoptotic pathways, and that PDE5-Is modify CREB activity (Matsushita et al., 2012; Palmeri et al., 2013; Puerta et al., 2010; Puzzo et al., 2009), in this study we investigated whether sildenafil treatment might act on neuronal apoptosis, and interfere with the Bax/caspase-3 and Bcl-2 pathways in aged mice. We also examined the neurotrophic factor BDNF. Finally, we assessed whether sildenafil might act on APP processing and the formation of Aβ in aged mice.
2. Methods
2.1. Animals
We used C57Bl/6J WT mice from a colony kept in the animal facility of the Department of Bio-Medical Sciences - Section of Physiology (University of Catania). The animals were maintained on a 12-hour light/dark cycle (lights on at 6:00 AM) in temperature- and humidity-controlled rooms; food and water were available ad libitum. The protocol was approved by the University Institutional Animal Care and Use Committee.
2.2. Drug administration and experimental groups
Sildenafil was administered based on previous findings (Palmeri et al., 2013; Puzzo et al., 2009) indicating that the minimum effective concentration was 3 mg/kg/day and the minimum effective period was 3 weeks. Animals were first treated with intraperitoneal sildenafil or vehicle for 3 weeks. Then, brains were removed and stored according to the specific experimental procedure. All experiments were performed in parallel using 4 groups of mice: (1) young mice treated with vehicle; (2) young mice treated with sildenafil; (3) old mice treated with vehicle; and (4) old mice treated with sildenafil. Young mice were 3–6 months old, and old mice were 26–30 months old. Mice were sex-balanced for each group.
2.3. In situ detection and measurement of apoptotic cells (TUNEL)
For in situ detection of apoptosis at the level of single cells we used TUNEL (In Situ Cell Death Detection Kit, POD, Roche Applied Science) as previously reported (Musumeci et al., 2011). The method involves the addition of dUTP labeled with fluorescein to the ends of the DNA fragments by the catalytic action of TdT. All end-labeling experiments were performed in triplicate so that the results for various tissue samples could be standardized. Paraffin-embedded brain sections 1.5 μm in thickness were dewaxed as previously described. Slides were rinsed twice in 0.01 M phosphate-buffered saline (PBS) (pH 7.4), transferred to 0.07 M citrate buffer (pH 6.0), and subjected to 750 W microwave irradiation for 1 minute for permeabilization. Sections were then immersed in Tris-HCl 0.1 M, pH 7.5, containing 3% bovine serum albumin (both from Roche Diagnostics, Milano, Italy) and 20% normal bovine serum (Sigma-Aldrich) for 30 minutes at 20 °C, rinsed twice in PBS, and immersed in TdT buffer (Roche Applied Science). Sections were then covered with TdT and fluorescein-labeled dUTP in TdT buffer and incubated in a dark humid chamber at 37 °C for 60 minutes. All sections were incubated with an antibody specific for fluorescein conjugated to peroxidase with 30-minute incubation at 37 °C. Staining was visualized with diaminobenzidine (DAB), which stained nuclei with DNA fragmentation the color brown. Sections were counterstained with Mayer’s Hematoxylin (Histolab Products AB, Goteborg, Sweden). For negative control samples, TdT was omitted from the reaction. Ten fields from randomly selected slides were observed using a light microscope. Each field was photographed with a digital camera (Canon) at magnification ×20 and ×40. For each photomicrograph, 3 observers blinded to sample identity counted the number of cells exhibiting a positive TUNEL reaction. The proportion of positive cells was calculated for each photomicrograph and a mean value was obtained for each sample.
2.4. Western blot analyses
Western blot analyses was performed as previously described (Podda et al., 2013; Puzzo et al., 2013). Crude extracts were prepared by homogenizing whole mouse brains in a buffer containing T-Per buffer (Thermoscientific, Rockford, IL, USA) and a protease inhibitor cocktail (Roche Diagnostics) in a Teflon-glass homogenizer followed by sonication. Protein concentrations were determined using Bradford’s method (Bradford, 1976) using bovine serum albumin as a standard. Sample proteins (50 μg) were diluted in sodium dodecyl sulphate protein gel loading solution (Invitrogen, Monza, Italy), boiled for 5 minutes, separated on 4%–12% Bis-tris gel (Invitrogen) and electroblotted onto nitrocellulose membranes (Invitrogen). We used the following antibodies: caspase-3 (Abcam, Cambridge, UK; 1:5000), Bax (Abcam; 1:1000), Bcl-2 (Abcam; 1:100), BDNF (Millipore, Billerica, MA, USA; 1:500), APP full-length (22C11; Millipore; 1:4000), soluble APPα (IBL, Gunma, Japan; 1:50), soluble APPβ (Covance, Emeryville, CA, USA; 1:500), CTFs (Invitrogen, 1:100), and tubulin (Santa Cruz Biotechnology Inc, Bergheimer, Germany; 1:20,000), which was used as loading control. Secondary antibodies (Amersham Biosciences, Milano, Italy) were diluted 1:10,000 and 1:4000. Nonspecific binding was blocked for 2 hours at 37 °C with 5% nonfat dry milk in Tween-Tris-buffered saline. All antibodies were prepared in 5% nonfat dry milk solution in Tween-Tris-buffered saline. Blots were developed using the enhanced chemiluminescence (ECL) technique (Amersham Biosciences) and relative band densities were quantified using ImageQuantTL 7.0 software. No signal was detected when the primary antibody was omitted (data not shown). Results were normalized with tubulin.
2.5. Immunohistochemistry
After overnight washing, specimens were dehydrated in graded ethanol, cleared in xylene, and paraffin-embedded, with their anatomical orientation preserved. Sections were processed as previously described (Musumeci et al., 2012; Palmeri et al., 2013). Briefly, they were incubated for 30 minutes in 0.3% H2O2/methanol to quench endogenous peroxidase activity then rinsed for 20 minutes with PBS (Bio-Optica, Milano, Italy). The sections were heated (5 minutes, 3 times) in capped polypropylene slide-holders with citrate buffer (10 mM citric acid, 0.05% Tween 20, pH 6.0; Bio-Optica), using a microwave oven (750 W) to unmask antigenic sites. The blocking step was performed before application of the primary antibody with goat serum (Vector Laboratories, Burlingame, CA, USA), 1:20 work dilution in PBS-Tween, for 1 hour in a moist chamber. Then, the sections were incubated overnight at 4 °C with the following rabbit antibodies: caspase-3 (1:25), Bax (1:50), Bcl-2 (1:100), and BDNF (1:2000). The secondary antibody, biotinylated anti-rabbit antibody was applied for 30 minutes at room temperature, followed by the avidin-biotin-peroxidase complex (Vector Laboratories) for an additional 30 minutes at room temperature. The immunoreaction was visualized by incubating the sections for 4 minutes in a 0.1% 3,3′-DAB and 0.02% hydrogen peroxide solution (DAB substrate kit, Vector Laboratories). The sections were lightly counterstained with Mayer’s Hematoxylin (Histolab Products AB) mounted in glycerol vinyl alcohol (GVA) mount (Zymed Laboratories, San Francisco, CA, USA) and observed using a Zeiss Axioplan light microscope (Carl Zeiss, Oberkochen, Germany).
The caspase-3, Bax, Bcl-2, and BDNF staining status was identified as either negative or positive. Immunohistochemistry-positive staining was defined as the presence of brown chromogen detection on the edge of the hematoxylin-stained cell nucleus, distributed within the cytoplasm or in the membrane via evaluation using light microscopy. Stain intensity and the proportion of immunopositive cells were also assessed using light microscopy. Intensity of staining was graded on a scale of 0 to 4, according to the following assessment: no detectable staining = 0, weak staining = 1, moderate staining = 2, strong staining = 3, and very strong staining = 4. The percentage of caspase-3, Bax, Bcl-2, and BDNF immunopositive cells (extent score) was independently evaluated by 4 investigators (2 anatomical morphologists and 2 histologists) and scored as a percentage of the final number of 100 cells in 5 categories: <5% (0); 5%–30% (+); 31%–50% (++); 51%–75% (+++), and >75% (++++). Counting was performed at magnification ×200. Negative control sections were processed like the experimental slides, except that they were incubated with PBS instead of the primary antibody.
To quantify immunohistochemical staining, 10 sections per animal were analyzed in stepwise fashion as a series of consecutive fields with a 40× objective and the stained area was expressed as pixels per field. Fields, randomly selected from each section, were analyzed and the percent area stained with caspase-3, Bax, Bcl-2, and BDNF antibodies was calculated using an image analyzer (Image-Pro Plus 4.5.1, Immagini & Computer, Milan, Italy), which quantifies the level of positive immunolabeling in each field. Values from all consecutive images of each biopsy were averaged. Negative control sections were processed like the experimental slides, except that they were incubated with PBS instead of the primary antibody. Digital pictures were taken using the Zeiss Axioplan light microscope and photographed with a Canon digital camera; evaluations were made by 3 blinded investigators, whose evaluations were assumed to be correct if values were not significantly different. In case of disputes concerning the interpretation, the case was revised to reach a unanimous agreement.
2.6. Congo red
For Congo red, hydrated sections were incubated in a freshly prepared alkaline alcoholic saturated sodium chloride solution (2.5 mM NaOH in 80% alcohol) for 20 minutes, then incubated in 0.2% Congo red solution (Bio-optica) for 30 minutes. The sections were lightly counterstained with Mayer’s Hematoxylin (Bio-optica), dehydrated in graded ethanol, and cleared in xylene. Finally, sections were coverslipped with GVA mount (Zymed Laboratories). Slice preparation and analyses were performed as described for immunohistochemistry.
2.7. Determination of Aβ levels
For analyses of Aβ42 and Aβ40 burden, frozen hemibrains were weighed and homogenized with an extraction buffer containing 100 mM NaCl, 0.4% diethylamine (Schmidt et al., 2005) and complete protease inhibitor cocktail (Roche). Human/Rat β(42)-Amyloid ELISA kit, High-Sensitive and Human/Rat β(40)-Amyloid ELISA kit (Wako Chemicals) were used. In brief, samples were centrifuged for 1 hour at 13,000 rpm, and supernatant diluted 1:1 before adding to ELISA plates coated with anti-human Aβ11–28 antibody and then incubated at 4 °C overnight. After washing with a wash solution, the sample was incubated for 1 hour in 100 μL of horseradish peroxide conjugated anti-human Aβ35–43 antibody (for Aβ42) or anti-human Aβ1–40 antibody (for Aβ40). The sandwiched Aβ1–42 and Aβ1–40 were visualized using a 3,3′,5,5′-tetramethylbenzidine solution and the absorbance was measured at a wavelength of 450 nm using a microtiter plate reader after the addition of a stop solution. Aβ concentrations were expressed as pmol/g tissue.
2.8. Statistical analysis
All experiments were blind with respect to treatment. Data were expressed as mean ± standard error of the mean. Statistical analysis was performed using dedicated software (Systat, Chicago, IL, USA). Comparisons were tested using analysis of variance and Bonferroni test. p Values < 0.05 were considered statistically significant.
3. Results
3.1. Sildenafil decreases apoptosis in aged mice
We first analyzed whether treatment with sildenafil might influence neuronal death at the hippocampal level. TUNEL, a widely used method for detecting DNA fragmentation caused by apoptosis (Gavrieli et al., 1992), showed an increase of apoptosis in the CA1 hippocampal area of old mice (young, 7.75 ± 0.62 cells per field vs. old, 59.75 ± 1.70 cells per field; F(1,6) = 821.468; p < 0.0001; Fig. 1). TUNEL-positive cells were dramatically reduced in old mice by sildenafil treatment (29.25 ± 1.03 cells per field; F(1,6) = 235.011; p < 0.0001; Fig. 1).
Fig. 1.
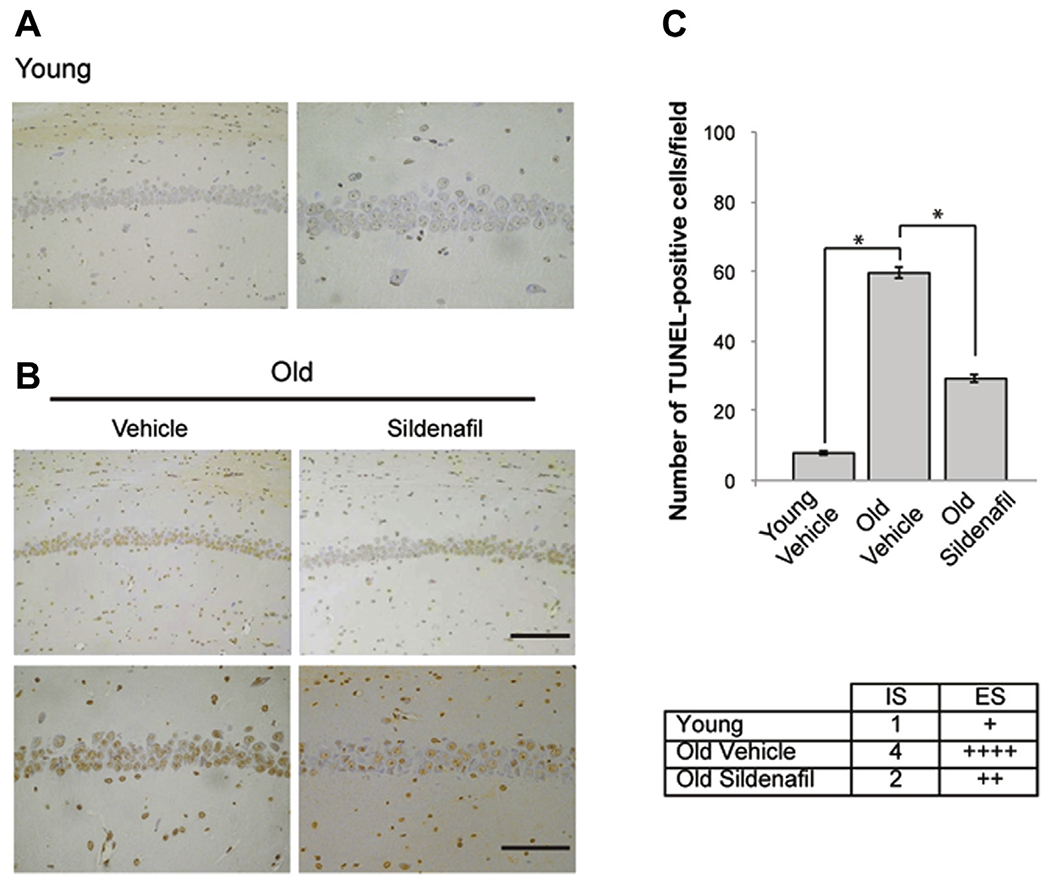
Sildenafil treatment reduces the age-induced neuronal apoptosis in the CA1 hippocampal area. (A) Few TUNEL-positive cells (brown) are present in the CA1 area of young animals (left panel, magnification ×20; right panel, magnification ×40). (B) The increase in DNA fragmentation present in old mice (left panels) is significantly reduced after treatment (right panels). Upper panels, magnification ×20, scale bar, 100 μM; bottom panels, magnification ×40, scale bar, 50 μM. (C) Bar graph showing the differences in the number of TUNEL-positive cells per field and table indicating a weak staining in young mice, a very strong staining in old mice, and a moderate staining in old mice treated with sildenafil. n = 4 mice for each group. * p < 0.0001. Abbreviations: ES, extent score; IS, intensity of staining; TUNEL, terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate-biotin nick end labeling.
To further confirm the antiapoptotic effect of sildenafil, we analyzed proapoptotic molecules such as Bax and caspase-3. Western blot analyses on brain extracts showed that old animals presented an increase of proapoptotic molecules compared with young animals (Bax: 1.11 ± 0.01 vs. 0.27 ± 0.01; F(1,6) = 1483.403; p < 0.0001; caspase-3: 1.23 ± 0.01 vs. 0.32 ± 0.01; F(1,6) = 1939.653; p < 0.0001; Fig. 2A–C) and that a 3-week treatment with sildenafil induced a significant decrease of Bax (0.72 ± 0.02; F(1,6) = 162.715; p < 0.0001; Fig. 2A and B) and caspase-3 levels (0.69 ± 0.03; F(1,6) = 221.339; p < 0.0001; Fig. 2A and C) suggesting an antiapoptotic effect of the PDE5-I. Analyzing young animals, no differences were detected in Bax (0.26 ± 0.01; F(1,6) = 0.093; p = 0.770; Fig. 2A and B) and caspase-3 (0.34 ± 0.01; F(1,6) = 0.780; p = 0.411; Fig. 2A and B) expression in control or sildenafil-treated mice. We then focused on the CA1 hippocampal area where cells are particularly susceptible to age-related damage and where we have already demonstrated a decrease of cognitive function and CREB phosphorylation (Palmeri et al., 2013). CA1 immunohistochemical examination showed that old mice treated with sildenafil presented a decrease of Bax-positive cells (34.6% decrease compared with vehicle-treated brains; F(1,6) = 42,181.640; p < 0.0001; Fig. 2D) and caspase-3–positive cells (45.4% decrease compared with vehicle-treated brains; F(1,6) = 528,836.259; p < 0.0001; Fig. 2E).
Fig. 2.
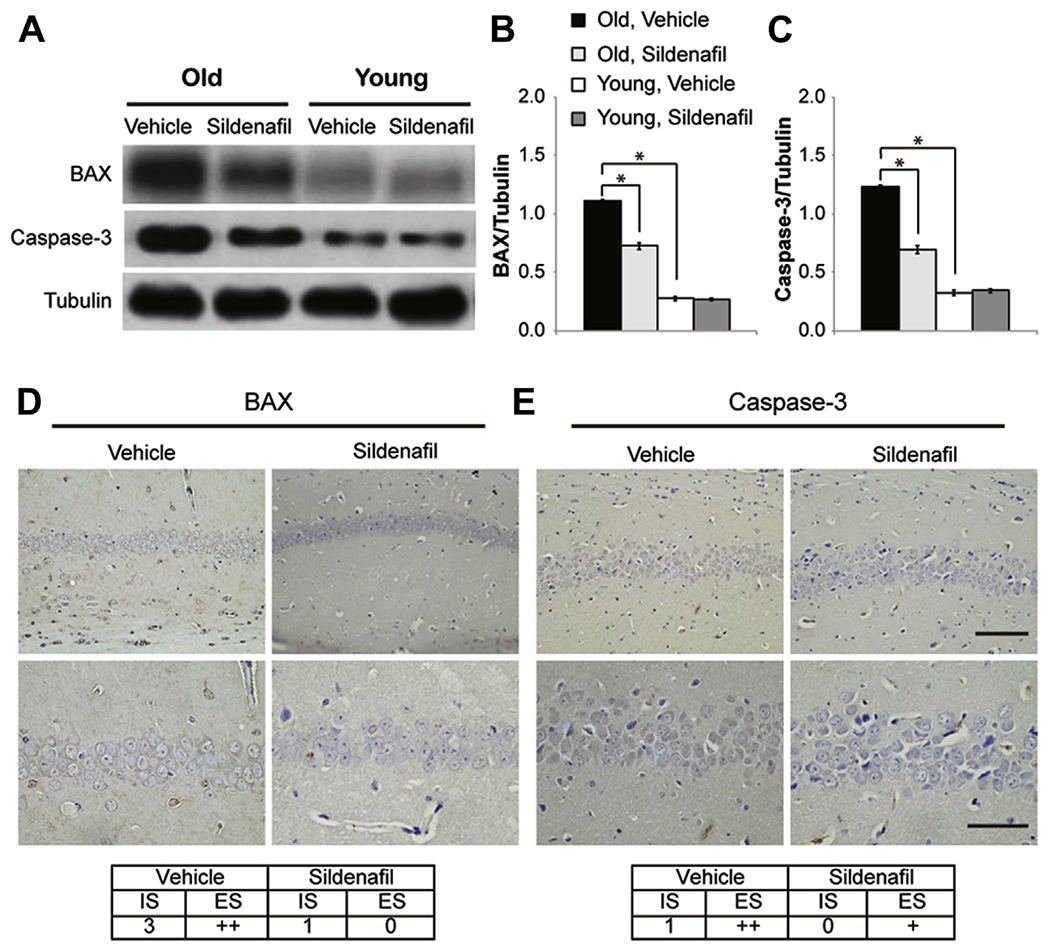
Sildenafil treatment reduces proapoptotic molecules in the CA1 hippocampal area of old mice. (A) Western blot analysis comparing the expression of Bax and caspase-3 from old and young mice treated with vehicle or sildenafil. (B and C) Bar graphs showing the results of the densitometric scan of the Western blots reported in (A) showing that sildenafil treatment decreases Bax and caspase-3 expression in old mice. (D) Bax strong immunoexpression in the CA1 area of old mice is reduced by sildenafil. (E) The weak staining for caspase-3 is further reduced after treatment. (D and E) Upper panels, magnification × 20, scale bar, 100 μM; bottom panels, magnification ×40, scale bar, 50 μM. n = 4 mice for each group. * p < 0.0001. Abbreviations: Bax, B-cell lymphoma 2-associated X protein; ES, extent score; IS, intensity of staining.
The effects of sildenafil on the levels of the antiapoptotic molecule, Bcl-2, and the Bax/Bcl-2 ratio were also examined. A strong decrease of Bcl-2 was found in the brains of old mice compared with young animals (0.31 ± 0.03 vs. 0.97 ± 0.04; F(1,6) = 139.531; p < 0.0001; Fig. 3A), that was partially rescued by sildenafil (0.51 ± 0.01; F(1,6) = 33.887; p = 0.001, comparing old mice treated with vehicle or sildenafil). The treatment did not completely restore Bcl-2 (F(1,6) = 95.668; p < 0.0001 comparing old treated mice with young mice), however, it modified the Bax/Bcl-2 ratio (3.7 ± 0.377 vs. 1.414 ± 0.070).
Fig. 3.
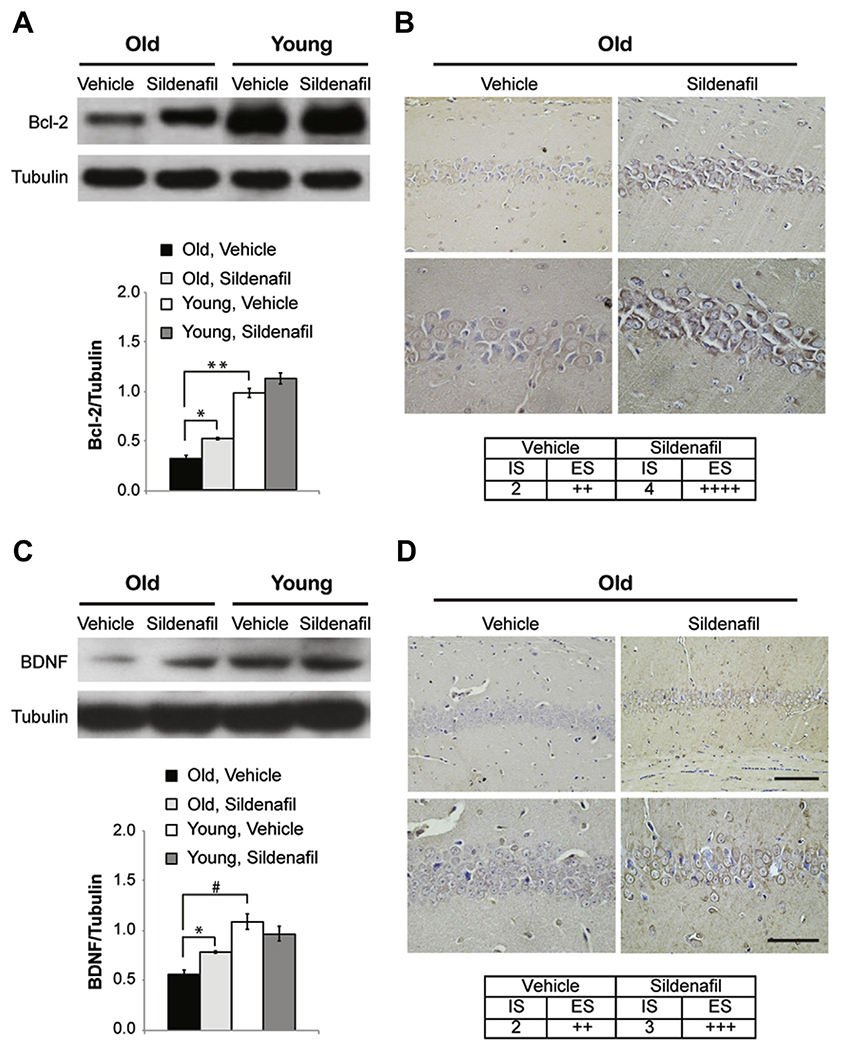
Sildenafil treatment increases antiapoptotic molecules in old mice. (A) Western blot analysis comparing the expression of Bcl-2 from old and young mice treated with vehicle or sildenafil and bar graph showing that the decrease of Bcl-2 in old mice is rescued by treatment with sildenafil. (B) Sildenafil increases Bcl-2 immunoexpression from moderate to very strong. (C) The expression of BDNF increases in old mice after a 3-week treatment with the PDE5-I. (D) The moderate BDNF staining observed in the CA1 of old mice changed to strong after treatment. (B and D) Upper panels, magnification ×20, scale bar, 100 μM; bottom panels, magnification ×40, scale bar, 50 μM. n = 4 mice for each group. ** p < 0.0001; * p < 0.001; # p < 0.05. Abbreviations: Bcl-2, B-cell lymphoma protein-2; BDNF, brain-derived neurotrophic factor; ES, extent score; IS, intensity of staining; PDE5-I, phosphodiesterase-5 inhibitor.
Photomicrographs of Bcl-2–positive cells in the CA1 area (Fig. 3B) showed that sildenafil rescued the age-related decrease of Bcl-2 expression (65.5% increase compared with vehicle-treated brains; F(1,6) = 2410.775; p < 0.0001; Fig. 3B).
3.2. Sildenafil increases BDNF levels in aged mice
Western blot analyses showed that treatment with sildenafil ameliorated BDNF levels in old mice (0.50 ± 0.08 vs. 0.88 ± 0.01; F(1,6) = 32.632; p = 0.001), even if they were not equal to levels found in young animals (1.05 ± 0.09; F(1,6) = 15.459; p = 0.008 comparing young mice with sildenafil-treated old mice; Fig. 3C and D). Also in this case, immunohistochemistry focused on the CA1 hippocampal area confirmed the beneficial effect of the drug that allowed old mice to rescue BDNF levels (88.2% increase compared with vehicle-treated brains; F(1,6) = 70826.080; p < 0.0001; Fig. 3D).
3.3. Sildenafil modifies APP processing and Aβ levels
We analyzed whether sildenafil might influence APP processing and Aβ formation. First, we focused on old mice to understand whether APP and its fragments were changed with aging. We found an increase in full-length APP (1.16 ± 0.05 vs. 0.90 ± 0.05; F(1,14) = 11.385; p = 0.005 comparing old and young mice treated with vehicle; Fig. 4A and B) and sAPPβ (1.59 ± 0.04 vs. 0.95 ± 0.03; F(1,14) = 134.893; p < 0.0001; Fig. 4A and C) and a decrease of CTF83 (0.55 ± 0.06 vs. 0.97 ± 0.01; F(1,14) = 30.480; p < 0.0001; Fig. 4A and F). Treatment with sildenafil normalized these values (APP: 0.78 ± 0.05; F(1,14) = 27.701; p < 0.0001; sAPPβ: 0.89 ± 0.08; F(1,14) = 52.170; p < 0.0001; CTF83: 0.89 ± 0.08; F(1,14) = 10.595; p = 0.006 comparing treated and untreated old mice). Other components of the APP pathway were unchanged (sAPPα: 0.92 ± 0.01 vs. 0.81 ± 0.03; F(3,28) = 2.164; p = 0.115; CTF99: 0.49 ± 0.1 vs. 0.60 ± 0.13; F(3,28) = 3.197; p = 0.095 comparing old and young mice; Fig. 4A, D, and E).
Fig. 4.
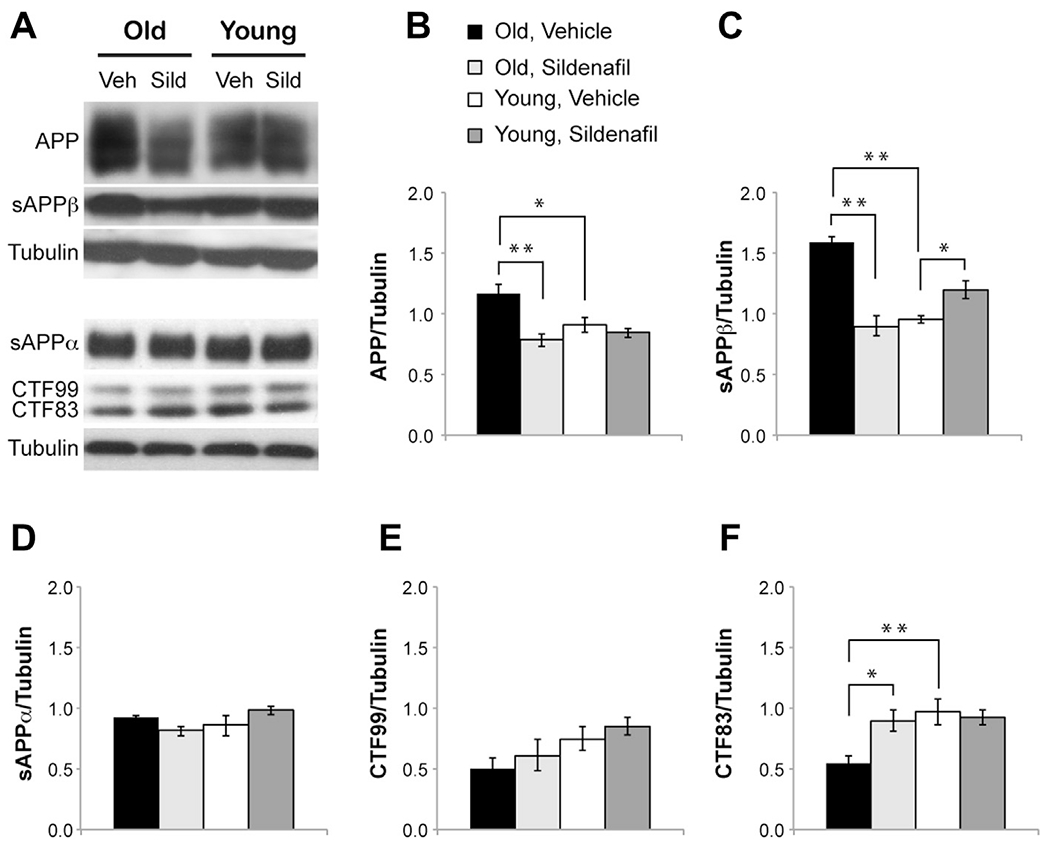
Sildenafil treatment modifies APP processing in young and old mice. (A) Western blot representative bands of APP and its fragments in young and old mice after treatment with vehicle or sildenafil. (B) Bar graph showing that chronic treatment with sildenafil decreases full-length APP in aged mice. (C) The age-induced increase in sAPPβ is rescued by sildenafil that, in contrast, produces a slight increase of sAPPβ in young mice. (D and E) No changes are detected in sAPPα and CTF99. (F) The decrease of CTF83 is rescued by sildenafil. n = 8 mice for each group. ** p < 0.0001; * p < 0.01. Abbreviations: APP, amyloid precursor protein; CTF, carboxy-terminal fragment; sAPP, soluble amyloid precursor protein; Sild, sildenafil; Veh, vehicle.
When we examined Aβ levels in aged mice, we found an increase of Aβ42 (54.49 ± 3.79 pmol/g of tissue vs. 37.83 ± 2.06, F(1,26) = 15.951; p = 0.001; Fig. 5A) and a decrease of Aβ40 (111.45 ± 6.68 vs. 158.31 ± 3.90 comparing old and young mice treated with vehicle, F(1,22) = 33.836; p < 0.0001; Fig. 5B) with a consequent increase of the Aβ42:Aβ40 ratio (0.51 ± 0.04 vs. 0.24 ± 0.01; F(1,26) = 31.157; p < 0.0001; Fig. 5C). 3-weeks treatment with sildenafil reported Aβ levels to values equal to those of young healthy mice (Aβ42: 40.23 ± 1.43, F(1,26) = 0.909; p = 0.349; Aβ40: 140.46 ± 7.66, F(1,22) = 3.662; p = 0.069; Fig. 5A, B) and normalized Aβ42:Aβ40 ratio (0.28 ± 0.02; F(1,26) = 3.230; p = 0.084; Fig. 5C). Conversely, young mice treated with sildenafil showed an increase of sAPPβ (1.19 ± 0.07, F(1,14) = 9.619; p = 0.008; Fig. 4A, C) resulting in a slight increase of both Aβ42 (45.47 ± 1.65, F(1,26) = 4.375; p = 0.048; Fig. 5A) and Aβ40 levels (171.59 ± 5.01, F(1,26) = 5.011; p = 0.036; Fig. 5B), Aβ42:Aβ40 ratio remaining unchanged (0.26 ± 0.01; F(1,26) = 1.095; p = 0.305; Fig. 5C). However, sAPPβ and Aβ42 levels were lower than those in old mice (sAPPβ: F(1,14) = 22.526; p < 0.0001; Aβ42: F(1,26) = 4.85; p = 0.037). Finally, young and old mice did not present senile plaques in the hippocampal CA1 area (Fig. 5D).
Fig. 5.
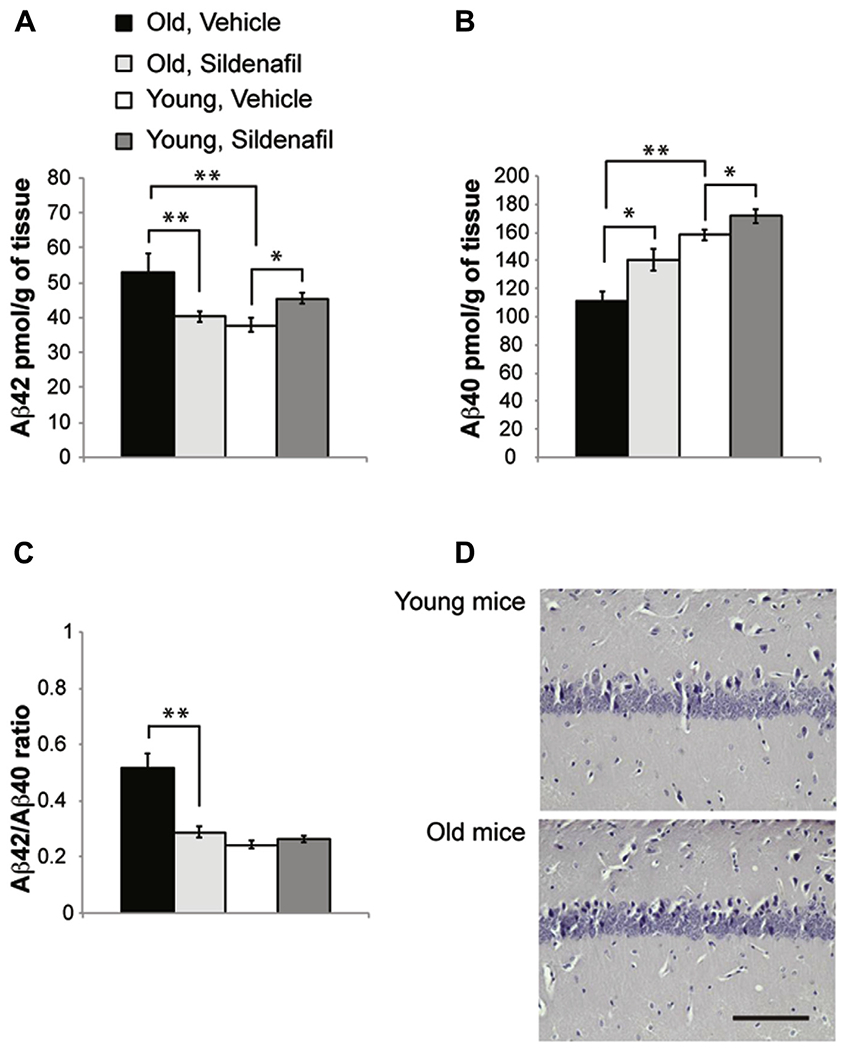
Sildenafil treatment modifies Aβ levels in young and old mice. (A) Old mice show an increase of Aβ42 levels that are reduced by sildenafil. The same treatment induces the opposite effect in young mice leading to a slight increase of Aβ42. (B) Old mice show a decrease of Aβ40 that is rescued by sildenafil. This sildenafil-induced increase of Aβ40 is also observed in young animals. (C) Sildenafil normalized Aβ42:Aβ40 ratio in old mice. (D) No plaques are detected using Congo red staining in young and old animals. Scale bar, 50 μM; n = 12 mice for Aβ40 and n = 14 mice for Aβ42. ** p < 0.01; * p < 0.05. Abbreviation: Aβ, beta amyloid.
4. Discussion
The present study showed that a 3-week treatment with the PDE5-I sildenafil inhibits brain apoptosis in aged mice. We came to this conclusion through the analyses of TUNEL assay and the quantification of proapototic and antiapoptotic molecules such as caspase-3, Bax, and Bcl-2. We particularly focused on the hippocampal CA1 region, because of its particular vulnerability to neuronal cell death during aging and neurodegeneration (Schmidt-Kastner and Freund, 1991) and the correlation between CA1 apoptotic signs with the impairment of hippocampal-dependent forms of learning and memory during aging (Bennett et al., 1998; Uysal et al., 2012).
The decrease of TUNEL-positive neurons after treatment is consistent with several studies indicating that the stimulation of the NO/cGMP pathway exerts an antiapoptotic effect. The NO donor S-nitroso-N-acetyl-d,l-penicillamine and the membrane-permeable cGMP analogue 8-Br-cGMP (Podda et al., 2008, 2012) inhibited serum deprivation-induced apoptosis (Kim et al., 1999) and 6-hydroxydopamine—induced apoptosis (Ha et al., 2003) in PC12 cells by inhibiting mitochondrial cytochrome c release, caspase-3 activation, and DNA fragmentation. Other NO-generating compounds, cGMP analogues, soluble guanylyl cyclase activators, and cGMP-specific phosphodiesterase inhibitors have been demonstrated to protect: (1) PC12 cells and rat sympathetic neurons from death after withdrawal of trophic support (Farinelli et al., 1996); (2) PC12 cells against glutamate-induced apoptosis (Yang et al., 2001); and (3) astrocytes from H2O2-induced apoptosis (Takuma et al., 2001). In PC12 cells, NO-induced cGMP elevations also exerted an antiapoptotic effect that counter-balances the proapoptotic actions of high doses of NO (Fiscus et al., 2002), emphasizing how NO might have a proapoptotic or antiapoptotic effect based on the doses and the cells on which it acts (Choi et al., 2002; for a review see Kang et al., 2004) consistently with the phenomenon of hormesis (see later in text), according to which several physiological molecules exhibit opposite effects at high or low concentrations (Calabrese et al., 2010; Puzzo et al., 2012).
The NO antiapoptotic effect was caused by the inhibition of caspase-3 via a cGMP-dependent mechanism and by direct inhibition of caspase-3–like activity through protein S-nitrosylation (Kim et al., 1997). Sildenafil has also been shown to reverse the hypoxia-induced neuronal apoptosis (Caretti et al., 2008) and tadalafil to counteract ischemia-induced neuronal cell death, thus facilitating recovery after ischemic cerebral injury (Ko et al., 2009). Besides in the CNS, sildenafil exerts its antiapoptotic effect in different types of cells such as skeletal muscle (Armstrong et al., 2013), cardiomyocytes (Das et al., 2005; Ebrahimi et al., 2009; Fisher et al., 2005; Westermann et al., 2012), endothelial cells (Gokce et al., 2010), tissue samples of rats with colitis (Karakoyun et al., 2011), cavernous tissues (Mostafa et al., 2010), and renal tubular cells (Akgül et al., 2011).
Aging-induced apoptosis was associated with an increase of caspase-3 and Bax activity, a decrease of Bcl-2, and an increase of the Bax/Bcl-2 ratio (Dorszewska et al., 2004; Kaufmann et al., 2001; Marcotte et al., 2004; Pollack et al., 2002). This is in contrast with other studies showing no changes in proapoptotic proteins, TUNEL-positive cells, and antiapoptotic proteins in the hippocampus with age (Shimohama et al., 1998; Wu et al., 2006). However, these studies were performed on different animal models and at different ages (24-month-old rats and 16-month-old senescence-accelerated mice, respectively), and our investigation was conducted on a physiological mouse model of aging at 26–30 months of age. In our conditions, the observed apoptotic profile was reverted by sildenafil treatment. The reduction of caspase-3 by sildenafil has also been observed in ischemia models on skeletal muscle (Armstrong et al., 2013), on myocardium (Das et al., 2005; Fisher et al., 2005; Milano et al., 2011), and in hyperammonia (Arafa and Atteia, 2013), as the reduction of Bax, the increase of Bcl-2, or the modification of the Bax/Bcl-2 ratio toward an antiapoptotic pathway in different tissues (Bae et al., 2012; Barros-Miñones et al., 2013; Caretti et al., 2008; Mostafa et al., 2010). In this regard, previous studies on the antiapoptotic effect of NO underlined how NO could inhibit Bcl-2 cleavage that occurred in association with an inhibition of apoptosis and caspase-3–like activation, blocking the vicious cycle of caspase—Bcl-2 (Kim et al., 1998).
We then focused on BDNF, a neurotrophin with a crucial role in synaptic plasticity and memory which has been involved in hippocampal age-related changes (for a review, see Tapia-Arancibia et al., 2008 and von Bohlen und Halbach, 2010). It is still a matter of discussion whether BDNF levels decrease or increase with aging, depending on the species, the cerebral region investigated, and the concomitant presence of neurodegenerative diseases. Several studies have demonstrated an age-related decrease of BDNF in the hippocampus of aged rats and mice (Assunção et al., 2010; Kim et al., 2010; Li et al., 2009; Liu et al., 2006; Zhao et al., 2009), but it has also been reported that the alteration of synaptic plasticity occurring in aged rats (Lapchak et al., 1993) or gerbils (Hwang et al., 2006) was not related to a modification of BDNF content. Some reports, on the contrary, indicated that the concentration of BDNF increased with normal aging in the rat and mouse hippocampus and it decreased in the cortex, and no differences in BDNF levels were found in the hippocampus of senescence-accelerated mice with memory impairment (Katoh-Semba et al., 1998). BDNF decrease has been found to be determinant during aging in primates (Hayashi et al., 1997, 2001) and in humans, where a decrease in BDNF serum levels has been detected (Erickson et al., 2010; Lommatzsch et al., 2005) related to memory dysfunction, even if hippocampal BDNF messenger RNA seems not to be modified by age (Webster et al., 2006). In neurodegenerative diseases, such as AD, almost all studies consistently showed that there is a decrease in BDNF levels related to cognitive impairment (for a review, see Tapia-Arancibia et al., 2008). In conclusion, these findings indicate that BDNF has an important role during aging. In this study, we demonstrated that the decrease of BDNF levels found in old mice, concomitant with the apoptotic condition and the impairment of synaptic plasticity and memory (Palmeri et al., 2013), was rescued by sildenafil treatment. This is consistent with previous studies indicating that PDE5 inhibitors increase BDNF levels in a mouse model of AD (Cuadrado-Tejedor et al., 2011), in a model of neurotoxicity resembling some of the neuropathological features of Huntington’s disease (Puerta et al., 2010), and in a mouse model of type 2 diabetes (Wang et al., 2011). Moreover, because BDNF is thought to activate the mitogen-activated protein kinases / extracellular-regulated kinase cascade (Chiou et al., 2006; Peng et al., 2008) leading to an increase of CREB and Bcl-2, and in turn, CREB regulates BDNF and Bcl-2 transcription (Perianayagam et al., 2006; Zieg et al., 2008); the positive effect of sildenafil on age-related features might be because of the regulation of these neuroplasticity-related proteins.
Another finding of this study was the modification of APP processing and Aβ levels by sildenafil. We have previously demonstrated that PDE5 inhibition decreased Aβ load in APP/PS1 transgenic models of AD (Puzzo et al., 2009), and it did not exert the same effect in Tg2576 mice (Cuadrado-Tejedor et al., 2011). However, because in this work we did not use mice overexpressing human Aβ but physiological models of aging, we first analyzed whether murine Aβ and the relative APP pathway were modified in aged wild type mice. We found an intensification of the amyloidogenic pathway of APP cleavage (increase of full-length APP and sAPPβ but a decrease of CTF83) toward the formation of Aβ42 whereas Aβ40 levels were decreased. Thus, consistent with other studies (Placanica et al., 2009), aged mice presented an increase of the Aβ42:Aβ40 ratio considered an index of neurotoxicity (Kuperstein et al., 2010; Pauwels et al., 2012). As expected, we did not find senile plaques in normal old mice, consistent with the more recent view indicating that soluble Aβ produces more severe synaptic dysfunction and neuronal damage than do plaques (Selkoe, 2002). This shifting in the APP processing was counteracted by sildenafil that normalized APP, sAPPβ, CTF83, and the Aβ42:Aβ40 ratio in old mice.
Surprisingly, young mice treated with sildenafil showed a slight increase of sAPPβ and Aβ, whose levels, much lower than those found in old animals, should not exert toxic effects. This could be in line with recent evidence suggesting that the PDE4 inhibitor rolipram increases APP protein expression and Aβ in neuronal cells (Canepa et al., 2013). Moreover, considering that: (1) the cyclic adenosine monophosphate/protein kinase A pathway acts in parallel with the cGMP/protein kinase G (PKG) pathway leading to CREB phosphorylation (Lu et al., 1999); (2) the NO/cGMP/PKG pathway might represent a crucial target through which Aβ acts when at high doses (Puzzo et al., 2005); and (3) a moderate Aβ increase might exert positive effects on hippocampal long-term potentiation and memory in normal mice (Puzzo et al., 2008, 2012), it is feasible that the low increase in Aβ levels induced by sildenafil might mediate some cGMP-induced beneficial effect on cognition in young mice, even if further investigations are needed to address this issue.
It is interesting to notice that Aβ (Puzzo et al., 2012) and the NO/cGMP pathway (Calabrese et al., 2010) behave in a hormetic fashion, stimulating or inhibiting cellular functions based on the applied dose (i.e., low doses of Aβ or NO stimulate synaptic plasticity and memory, and high doses inhibit these processes). This is in line with an increasing number of studies pointing to the importance of hormesis in aging and neurodegenerative diseases (for a review, see Calabrese et al., 2013). During aging, the body’s ability to regulate the homeostatic balance between oxidant and antioxidant systems decreases leading to an increased production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) (Calabrese et al., 2011, 2012) responsible for the tissue damage. Although the continuous presence of a low concentration of ROS—RNS is able to stimulate the production of antioxidant enzymes and other defense mechanisms, overcoming a sustainable threshold determines cell damage. The CNS is particularly vulnerable to oxidative stress and the increased production of ROS—RNS causes an impairment of neuronal plasticity (Gilgun-Sherki et al., 2002) and neuroplasticity-related molecules (i.e., BDNF; Wu et al., 2004). Moreover, oxidative stress leads to accumulation of unfolded or misfolded proteins, such as Aβ, which is known to play a fundamental role in AD pathogenesis and during aging (Calabrese et al., 2008). Thus, longevity results on the ability of cells to activate adaptation phenomena in response to oxidative insults, and, depending on the severity of this insult and the individual genetic predisposition, homeostasis can be restored, making the body less vulnerable to disease, or adaptation can fail, paving the way to neurodegenerative disease. In this context, modulation of endogenous cellular defense mechanisms represents an interesting and innovative therapeutic approach. Recently, the vitagene system, encoding for heat shock proteins, heme oxygenase-1, thioredoxin reductase, and sirtuins, has been identified as a potential novel target for cytoprotection acting through a hormetic mechanism (Calabrese et al., 2008, 2010, 2011, 2012, 2013) and the NO/cGMP system might be involved in this neuroprotective network as a prosurvival pathway.
In conclusion, our findings strongly suggest that inhibition of PDE5 can counteract apoptosis during aging by modulating pro- and antiapoptotic molecules and the APP pathway. This, together with the modification of CREB phosphorylation (Palmeri et al., 2013; Puzzo et al., 2009) might explain the improvement of cognitive function in AD and aged mice treated with sildenafil (see Fig. 6 for a summary of the effects of sildenafil in aged mice). However, as previously emphasized, none of the existing commercially available PDE5-Is are optimized for the CNS, thus, further studies are needed to develop new safe and effective drugs for human use in chronic diseases such as aging and AD.
Fig. 6.

Overview diagram of sildenafil effects in aged mice. Red arrows indicate inhibition, and green arrows indicate stimulation. Abbreviations: Aβ, beta amyloid; APP, amyloid precursor protein; Bax, B-cell lymphoma 2-associated X protein; Bcl2, B-cell lymphoma protein-2; BDNF, brain-derived neurotrophic factor; CTF, carboxy-terminal fragment; i.p., intraperitoneal; p-CREB, Phosphorylated cAMP Response Element Binding protein; sAPP, soluble amyloid precursor protein.
Acknowledgements
The authors thank Lucia Privitera for help with animal treatment. This work was supported by the “Progetto di Ricerca d’Ateneo” from the University of Catania to A.P. and the NIH grant U01-AG032973 to O.A.
Footnotes
Disclosure statement
The authors declare that they have no actual or potential conflicts of interest.
This project was approved by the I.A.C.U.C. Committee (12/17/2010 project #137, University of Catania).
References
- Abdi A, Sadraie H, Dargahi L, Khalaj L, Ahmadiani A, 2011. Apoptosis inhibition can be threatening in Aβ-induced neuroinflammation, through promoting cell proliferation. Neurochem. Res 36, 39–48. [DOI] [PubMed] [Google Scholar]
- Akgül T, Huri E, Yagmurdur H, Ayyıldız A, Ustün H, Germiyanoğlu C, 2011. Phosphodiesterase 5 inhibitors attenuate renal tubular apoptosis after partial unilateral ureteral obstruction: an experimental study. Kaohsiung J. Med. Sci 27, 15–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arafa MH, Atteia HH, 2013. Sildenafil citrate attenuates the deleterious effects of elevated ammonia. Toxicol. Mech. Methods 23, 402–411. [DOI] [PubMed] [Google Scholar]
- Arancio O, Chao MV, 2007. Neurotrophins, synaptic plasticity and dementia. Curr. Opin. Neurobiol 17, 325–330. [DOI] [PubMed] [Google Scholar]
- Armstrong DM, Armstrong Ada C, Figueiredo RC, Florentino JE, Saad PF, Fox-Talbot K, Halushka MK, Berkowitz DE, Taha MO, Fagundes DJ, 2013. Sildenafil citrate protects skeletal muscle of ischemia-reperfusion injury: immunohistochemical study in rat model. Acta Cir. Bras 28, 282–287. [DOI] [PubMed] [Google Scholar]
- Asanuma M, Nishibayashi S, Iwata E, Kondo Y, Nakanishi T, Vargas MG, Ogawa N, 1996. Alterations of cAMP response element-binding activity in the aged rat brain in response to administration of rolipram, a cAMP-specific phosphodiesterase inhibitor. Brain Res. Mol. Brain Res 41, 210–215. [DOI] [PubMed] [Google Scholar]
- Assunção M, Santos-Marques MJ, Carvalho F, Andrade JP, 2010. Green tea averts age-dependent decline of hippocampal signaling systems related to antioxidant defenses and survival. Free Radic. Biol. Med 48, 831–838. [DOI] [PubMed] [Google Scholar]
- Bae EH, Kim IJ, Joo SY, Kim EY, Kim CS, Choi JS, Ma SK, Kim SH, Lee JU, Kim SW, 2012. Renoprotective effects of sildenafil in DOCA-salt hypertensive rats. Kidney Blood Press. Res 36, 248–257. [DOI] [PubMed] [Google Scholar]
- Barros-Miñones L, Martín-de-Saavedra D, Perez-Alvarez S, Orejana L, Suquía V, Goñi-Allo B, Hervias I, López MG, Jordan J, Aguirre N, Puerta E, 2013. Inhibition of calpain-regulated p35/cdk5 plays a central role in sildenafil-induced protection against chemical hypoxia produced by malonate. Biochim. Biophys. Acta 1832, 705–717. [DOI] [PubMed] [Google Scholar]
- Bennett SA, Tenniswood M, Chen JH, Davidson CM, Keyes MT, Fortin T, Pappas BA, 1998. Chronic cerebral hypoperfusion elicits neuronal apoptosis and behavioral impairment. Neuroreport 9, 161–166. [DOI] [PubMed] [Google Scholar]
- Bradford MM, 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Calabrese EJ, Iavicoli I, Calabrese V, 2013. Hormesis: its impact on medicine and health. Hum. Exp. Toxicol 32, 120–152. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Cornelius C, Cuzzocrea S, Iavicoli I, Rizzarelli E, Calabrese EJ, 2011. Hormesis, cellular stress response and vitagenes as critical determinants in aging and longevity. Mol. Aspects Med 32, 279–304. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Cornelius C, Dinkova-Kostova AT, Calabrese EJ, Mattson MP, 2010. Cellular stress responses, the hormesis paradigm, and vitagenes: novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid. Redox Signal 13, 1763–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese V, Cornelius C, Dinkova-Kostova AT, Iavicoli I, Di Paola R, Koverech A, Cuzzocrea S, Rizzarelli E, Calabrese EJ, 2012. Cellular stress responses, hormetic phytochemicals and vitagenes in aging and longevity. Biochim. Biophys. Acta 1822, 753–783. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Cornelius C, Mancuso C, Pennisi G, Calafato S, Bellia F, Bates TE, Giuffrida Stella AM, Schapira T, Dinkova-Kostova AT, Rizzarelli E, 2008. Cellular stress response: a novel target for chemoprevention and nutritional neuroprotection in aging, neurodegenerative disorders and longevity. Neurochem. Res 33, 2444–2471. [DOI] [PubMed] [Google Scholar]
- Canepa E, Domenicotti C, Marengo B, Passalacqua M, Marinari UM, Pronzato MA, Fedele E, Ricciarelli R, 2013. Cyclic adenosine monophosphate as an endogenous modulator of the amyloid-β precursor protein metabolism. IUBMB Life 65, 127–133. [DOI] [PubMed] [Google Scholar]
- Caretti A, Bianciardi P, Ronchi R, Fantacci M, Guazzi M, Samaja M, 2008. Phosphodiesterase-5 inhibition abolishes neuron apoptosis induced by chronic hypoxia independently of hypoxia-inducible factor-1alpha signaling. Exp. Biol. Med 233, 1222–1230. [DOI] [PubMed] [Google Scholar]
- Chalimoniuk M, Strosznajder JB, 1998. Aging modulates nitric oxide synthesis and cGMP levels in hippocampus and cerebellum. Effects of amyloid beta peptide. Mol. Chem. Neuropathol 35, 77–95. [DOI] [PubMed] [Google Scholar]
- Chiou SH, Ku HH, Tsai TH, 2006. Moclobemide (MB) up-regulated Bcl-2 expression and induced neural stem cell differentiation into serotoninergic neuron via extracellular-regulated kinase (ERK) pathway. Br. J. Pharmacol 148, 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BM, Pae HO, Jang SI, Kim YM, Chung HT, 2002. Nitric oxide as a proapoptotic as well as anti-apoptotic modulator. J. Biochem. Mol. Biol 35, 116–126. [DOI] [PubMed] [Google Scholar]
- Chung YH, Kim EJ, Shin CM, Joo KM, Kim MJ, Woo HW, Cha CI, 2002. Age-related changes in CREB binding protein immunoreactivity in the cerebral cortex and hippocampus of rats. Brain Res. 956, 312–318. [DOI] [PubMed] [Google Scholar]
- Cohen GM, 1997. Caspases: the executioners of apoptosis. Biochem. J 326, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado-Tejedor M, Hervias I, Ricobaraza A, Puerta E, Pérez-Roldán JM, García-Barroso C, Franco R, Aguirre N, García-Osta A, 2011. Sildenafil restores cognitive function without affecting β-amyloid burden in a mouse model of Alzheimer’s disease. Br. J. Pharmacol 164, 2029–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Amelio M, Cavallucci V, Middei S, Marchetti C, Pacioni S, Ferri A, Diamantini A, De Zio D, Carrara P, Battistini L, Moreno S, Bacci A, Ammassari-Teule M, Marie H, Cecconi F, 2011. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat. Neurosci 14, 69–76. [DOI] [PubMed] [Google Scholar]
- Das A, Xi L, Kukreja RC, 2005. Phosphodiesterase-5 inhibitor sildenafil pre-conditions adult cardiac myocytes against necrosis and apoptosis. Essential role of nitric oxide signaling. J. Biol. Chem 280, 12944–12955. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Ginty DD, 2002. CREB family transcription factors inhibit neuronal suicide. Nat. Med 8, 450–451. [DOI] [PubMed] [Google Scholar]
- Domek-Łopacińska KU, Strosznajder JB, 2010. Cyclic GMP and nitric oxide synthase in aging and Alzheimer’s disease. Mol. Neurobiol 41, 129–137. [DOI] [PubMed] [Google Scholar]
- Dorszewska J, Adamczewska-Goncerzewicz Z, Szczech J, 2004. Apoptotic proteins in the course of aging of central nervous system in the rat. Respir. Physiol. Neurobiol 139, 145–155. [DOI] [PubMed] [Google Scholar]
- Ebrahimi F, Shafaroodi H, Asadi S, Nezami BG, Ghasemi M, Rahimpour S, Hashemi M, Doostar Y, Dehpour AR, 2009. Sildenafil decreased cardiac cell apoptosis in diabetic mice: reduction of oxidative stress as a possible mechanism. Can. J. Physiol. Pharmacol 87, 556–564. [DOI] [PubMed] [Google Scholar]
- Elmore S, 2007. Apoptosis: a review of programmed cell death. Toxicol. Pathol 35, 495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson KI, Prakash RS, Voss MW, Chaddock L, Heo S, McLaren M, Pence BD, Martin SA, Vieira VJ, Woods JA, McAuley E, Kramer AF, 2010. Brain-derived neurotrophic factor is associated with age-related decline in hippocampal volume. J. Neurosci 30, 5368–5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinelli SE, Park DS, Greene LA, 1996. Nitric oxide delays the death of trophic factor-deprived PC12 cells and sympathetic neurons by a cGMP-mediated mechanism. J. Neurosci 16, 2325–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiscus RR, Yuen JP, Chan SL, Kwong JH, Chew SB, 2002. Nitric oxide and cyclic GMP as pro- and anti-apoptotic agents. J. Card. Surg 17, 336–339. [DOI] [PubMed] [Google Scholar]
- Fisher PW, Salloum F, Das A, Hyder H, Kukreja RC, 2005. Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiotoxicity. Circulation 111, 1601–1610. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson SA, 1992. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol 119, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilgun-Sherki Y, Rosenbaum Z, Melamed E, Offen D, 2002. Antioxidant therapy in acute central nervous system injury: current state. Pharmacol. Rev 54, 271–284. [DOI] [PubMed] [Google Scholar]
- Gokce C, Gulsen S, Yilmaz C, Guven G, Caner H, Altinors N, 2010. The effect of the sildenafil citrate on cerebral vasospasm and apoptosis following experimental subarachnoid hemorrhage in rats. J. Neurosurg. Sci 54, 29–37. [PubMed] [Google Scholar]
- Ha KS, Kim KM, Kwon YG, Bai SK, Nam WD, Yoo YM, Kim PK, Chung HT, Billiar TR, Kim YM, 2003. Nitric oxide prevents 6-hydroxydopamine-induced apoptosis in PC12 cells through cGMP-dependent PI3 kinase/Akt activation. FASEB J. 17, 1036–1047. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Mistunaga F, Ohira K, Shimizu K, 2001. Changes in BDNF- immunoreactive structures in the hippocampal formation of the aged macaque monkey. Brain Res. 918, 191–196. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Yamashita A, Shimizu K, 1997. Somatostatin and brain-derived neurotrophic factor mRNA expression in the primate brain: decreased levels of mRNAs during aging. Brain Res. 749, 283–289. [DOI] [PubMed] [Google Scholar]
- Hwang IK, Yoo KY, Jung BK, Cho JH, Kim DH, Kang TC, Kwon YG, Kim YS, Won MH, 2006. Correlations between neuronal loss, decrease of memory, and decrease expression of brain-derived neurotrophic factor in the gerbil hippocampus during normal aging. Exp. Neurol 201, 75–83. [DOI] [PubMed] [Google Scholar]
- Jaworski J, Mioduszewska B, Sánchez-Capelo A, Figiel I, Habas A, Gozdz A, Proszynski T, Hetman M, Mallet J, Kaczmarek L, 2003. Inducible cAMP early repressor, an endogenous antagonist of cAMP responsive element-binding protein, evokes neuronal apoptosis in vitro. J. Neurosci 23, 4519–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YC, Kim PK, Choi BM, Chung HT, Ha KS, Kwon YG, Kim YM, 2004. Regulation of programmed cell death in neuronal cells by nitric oxide. In Vivo 18, 367–376. [PubMed] [Google Scholar]
- Karakoyun B, Uslu U, Ercan F, Aydin MS, Yuksel M, Ogunc AV, Alican I, 2011. The effect of phosphodiesterase-5 inhibition by sildenafil citrate on inflammation and apoptosis in rat experimental colitis. Life Sci. 89, 402–407. [DOI] [PubMed] [Google Scholar]
- Katoh-Semba R, Semba R, Takeuchi IK, Kato K, 1998. Age-related changes in levels of brain-derived neurotrophic factor in selected brain regions of rats, normal mice and senescence-accelerated mice: a comparison to those of nerve growth factor and neurotrophin-3. Neurosci. Res 31, 227–234. [DOI] [PubMed] [Google Scholar]
- Kaufmann JA, Bickford PC, Taglialatela G, 2001. Oxidative-stress-dependent up-regulation of Bcl-2 expression in the central nervous system of aged Fisher-344 rats. J. Neurochem 76, 1099–1108. [DOI] [PubMed] [Google Scholar]
- Kim SE, Ko IG, Kim BK, Shin MS, Cho S, Kim CJ, Kim SH, Baek SS, Lee EK, Jee YS, 2010. Treadmill exercise prevents aging-induced failure of memory through an increase in neurogenesis and suppression of apoptosis in rat hippocampus. Exp. Gerontol 45, 357–365. [DOI] [PubMed] [Google Scholar]
- Kim YM, Chung HT, Kim SS, Han JA, Yoo YM, Kim KM, Lee GH, Yun HY, Green A, Li J, Simmons RL, Billiar TR, 1999. Nitric oxide protects PC12 cells from serum deprivation-induced apoptosis by cGMP-dependent inhibition of caspase signaling. J. Neurosci 19, 6740–6747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YM, Kim TH, Seol DW, Talanian RV, Billiar TR, 1998. Nitric oxide suppression of apoptosis occurs in association with an inhibition of Bcl-2 cleavage and cytochrome c release. J. Biol. Chem 273, 31437–31441. [DOI] [PubMed] [Google Scholar]
- Kim YM, Talanian RV, Billiar TR, 1997. Nitric oxide inhibits apoptosis by preventing increases in Caspase-3-like activity via two distinct mechanisms. J. Biol. Chem 272, 31138–31148. [DOI] [PubMed] [Google Scholar]
- Ko IG, Shin MS, Kim BK, Kim SE, Sung YH, Kim TS, Shin MC, Cho HJ, Kim SC, Kim SH, Kim KH, Shin DH, Kim CJ, 2009. Tadalafil improves short-term memory by suppressing ischemia-induced apoptosis of hippocampal neuronal cells in gerbils. Pharmacol. Biochem. Behav 91, 629–635. [DOI] [PubMed] [Google Scholar]
- Kudo K, Wati H, Qiao C, Arita J, Kanba S, 2005. Age-related disturbance of memory and CREB phosphorylation in CA1 area of hippocampus of rats. Brain Res. 1054, 30–37. [DOI] [PubMed] [Google Scholar]
- Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M, Vandersteen A, Segers-Nolten I, Van Der Werf K, Subramaniam V, Braeken D, Callewaert G, Bartic C, D’Hooge R, Martins IC, Rousseau F, Schymkowitz J, De Strooper B, 2010. Neurotoxicity of Alzheimer’s disease Aβ peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J. 29, 3408–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapchak PA, Araujo DM, Beck KD, Finch CE, Johnson SA, Hefti F, 1993. BDNF and trkB mRNA expression in the hippocampal formation of aging rats. Neurobiol. Aging 14, 121–126. [DOI] [PubMed] [Google Scholar]
- Li Q, Zhao HF, Zhang ZF, Liu ZG, Pei XR, Wang JB, Cai MY, Li Y, 2009. Long-term administration of green tea catechins prevents age-related spatial learning and memory decline in C57BL/6 J mice by regulating hippocampal cyclic amp-response element binding protein signaling cascade. Neuroscience 159, 1208–1215. [DOI] [PubMed] [Google Scholar]
- Liu J, He QJ, Zou W, Wang HX, Bao YM, Liu YX, An LJ, 2006. Catalpol increases hippocampal neuroplasticity and up-regulates PKC and BDNF in the aged rats. Brain Res. 1123, 68–79. [DOI] [PubMed] [Google Scholar]
- Lommatzsch M, Zingler D, Schuhbaeck K, Schloetcke K, Zingler C, Schuff-Werner P, Virchow JC, 2005. The impact of age, weight and gender on BDNF levels in human platelets and plasma. Neurobiol. Aging 26, 115–123. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Riccio A, Cohen S, Ginty DD, 2002. Apoptosis, axonal growth defects, and degeneration of peripheral neurons in mice lacking CREB. Neuron 34, 371–385. [DOI] [PubMed] [Google Scholar]
- Louneva N, Cohen JW, Han LY, Talbot K, Wilson RS, Bennett DA, Trojanowski JQ, Arnold SE, 2008. Caspase-3 is enriched in postsynaptic densities and increased in Alzheimer’s disease. Am. J. Pathol 173, 1488–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YF, Kandel ER, Hawkins RD, 1999. Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J. Neurosci 19, 10250–10261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch AM, Lynch MA, 2002. The age-related increase in IL-1 type I receptor in rat hippocampus is coupled with an increase in Caspase-3 activation. Eur. J. Neurosci 15, 1779–1788. [DOI] [PubMed] [Google Scholar]
- Mantamadiotis T, Lemberger T, Bleckmann SC, Kern H, Kretz O, Martin Villalba A, Tronche F, Kellendonk C, Gau D, Kapfhammer J, Otto C, Schmid W, Schütz G, 2002. Disruption of CREB function in brain leads to neurodegeneration. Nat. Genet 31, 47–54. [DOI] [PubMed] [Google Scholar]
- Marcotte R, Lacelle C, Wang E, 2004. Senescent fibroblasts resist apoptosis by downregulating caspase-3. Mech. Ageing Dev 125, 777–783. [DOI] [PubMed] [Google Scholar]
- Matsushita H, Matsuzaki M, Han XJ, Nishiki TI, Ohmori I, Michiue H, Matsui H, Tomizawa K, 2012. Antidepressant-like effect of sildenafil through oxytocin-dependent cyclic AMP response element-binding protein phosphorylation. Neuroscience 200, 13–18. [DOI] [PubMed] [Google Scholar]
- Mattson MP, 2000. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol 1, 120–129. [DOI] [PubMed] [Google Scholar]
- Milano G, Bianciardi P, Rochemont V, Vassalli G, Segesser LK, Corno AF, Guazzi M, Samaja M, 2011. Phosphodiesterase-5 inhibition mimics intermittent reoxygenation and improves cardioprotection in the hypoxic myocardium. PLoS One 6, e27910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti B, Berteotti C, Contestabile A, 2005. Dysregulation of memory-related proteins in the hippocampus of aged rats and their relation with cognitive impairment. Hippocampus 15, 1041–1049. [DOI] [PubMed] [Google Scholar]
- Mostafa T, Rashed LA, Kotb K, 2010. Testosterone and chronic sildenafil/tadalafil anti-apoptotic role in aged diabetic rats. Int. J. Impot. Res 22, 255–261. [DOI] [PubMed] [Google Scholar]
- Musumeci G, Carnazza ML, Loreto C, Leonardi R, Loreto C, 2012. β-Defensin-4 (HBD-4) is expressed in chondrocytes derived from normal and osteoarthritic cartilage encapsulated in PEGDA scaffold. Acta Histochem. 114, 805–812. [DOI] [PubMed] [Google Scholar]
- Musumeci G, Loreto C, Carnazza ML, Strehin I, Elisseeff J, 2011. Oa cartilage derived chondrocytes encapsulated in poly(ethylene glycol) diacrylate (PEGDA) for the evaluation of cartilage restoration and apoptosis in an in vitro model. Histol. Histopathol 26, 1265–1278. [DOI] [PubMed] [Google Scholar]
- Palmeri A, Privitera L, Giunta S, Loreto C, Puzzo D, 2013. Inhibition of phosphodiesterase-5 rescues age-related impairment of synaptic plasticity and memory. Behav. Brain Res 240, 11–20. [DOI] [PubMed] [Google Scholar]
- Pauwels K, Williams TL, Morris KL, Jonckheere W, Vandersteen A, Kelly G, Schymkowitz J, Rousseau F, Pastore A, Serpell LC, Broersen K, 2012. Structural basis for increased toxicity of pathological aβ42:aβ40 ratios in Alzheimer disease. J. Biol. Chem 287, 5650–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng CH, Chiou SH, Chen SJ, Chou YC, 2008. Neuroprotection by imipramine against lipopolysaccharide-induced apoptosis in hippocampus-derived neural stem cells mediated by activation of BDNF and the MAPK pathway. Eur. Neuropsychopharmacol 18, 128–140. [DOI] [PubMed] [Google Scholar]
- Perianayagam MC, Madias NE, Pereira BJ, Jaber BL, 2006. CREB transcription factor modulates Bcl2 transcription in response to C5a in HL-60-derived neutrophils. Eur. J. Clin. Invest 36, 353–361. [DOI] [PubMed] [Google Scholar]
- Placanica L, Zhu L, Li YM, 2009. Gender- and age-dependent gamma-secretase activity in mouse brain and its implication in sporadic Alzheimer disease. PLoS One 4, e5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podda MV, D’Ascenzo M, Leone L, Piacentini R, Azzena GB, Grassi C, 2008. Functional role of cyclic nucleotide-gated channels in rat medial vestibular nucleus neurons. J. Physiol 586, 803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podda MV, Leone L, Piacentini R, Cocco S, Mezzogori D, D’Ascenzo M, Grassi C, 2012. Expression of olfactory-type cyclic nucleotide-gated channels in rat cortical astrocytes. Glia 60, 1391–1405. [DOI] [PubMed] [Google Scholar]
- Podda MV, Piacentini R, Barbati SA, Mastrodonato A, Puzzo D, D’Ascenzo M, Leone L, Grassi C, 2013. Role of cyclic nucleotide-gated channels in the modulation of mouse hippocampal neurogenesis. PLoS One 8, e73246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack M, Leeuwenburgh C, 2001. Apoptosis and aging: role of the mitochondria. J. Gerontol. A Biol. Sci. Med. Sci 56, B475–B482. [DOI] [PubMed] [Google Scholar]
- Pollack M, Phaneuf S, Dirks A, Leeuwenburgh C, 2002. The role of apoptosis in the normal aging brain, skeletal muscle, and heart. Ann. N. Y. Acad. Sci 959, 93–107. [DOI] [PubMed] [Google Scholar]
- Puerta E, Hervias I, Barros-Miñones L, Jordan J, Ricobaraza A, Cuadrado-Tejedor M, García-Osta A, Aguirre N, 2010. Sildenafil protects against 3-nitropropionic acid neurotoxicity through the modulation of calpain, CREB, and BDNF. Neurobiol. Dis 38, 237–245. [DOI] [PubMed] [Google Scholar]
- Puzzo D, Bizzoca A, Privitera L, Furnari D, Giunta S, Girolamo F, Pinto M, Gennarini G, Palmeri A, 2013. Aug 12. F3/contactin promotes hippocampal neurogenesis, synaptic plasticity and memory in adult mice. Hippocampus. 10.1002/hipo.22186. [DOI] [PubMed] [Google Scholar]
- Puzzo D, Privitera L, Leznik E, Fà M, Staniszewski A, Palmeri A, Arancio O, 2008. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci 28, 14537–14545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzzo D, Privitera L, Palmeri A, 2012. Hormetic effect of amyloid-beta peptide in synaptic plasticity and memory. Neurobiol. Aging 33, 1484.e15–1484.e24. [DOI] [PubMed] [Google Scholar]
- Puzzo D, Staniszewski A, Deng SX, Privitera L, Leznik E, Liu S, Zhang H, Feng Y, Palmeri A, Landry DW, Arancio O, 2009. Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-beta load in an Alzheimer’s disease mouse model. J. Neurosci 29, 8075–8086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzzo D, Vitolo O, Trinchese F, Jacob JP, Palmeri A, Arancio O, 2005. Amyloid-beta peptide inhibits activation of the nitric oxide/cGMP/cAMP-responsive element-binding protein pathway during hippocampal synaptic plasticity. J. Neurosci 25, 6887–6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reix S, Mechawar N, Susin SA, Quirion R, Krantic S, 2007. Expression of cortical and hippocampal apoptosis-inducing factor (AIF) in aging and Alzheimer’s disease. Neurobiol. Aging 28, 351–356. [DOI] [PubMed] [Google Scholar]
- Rodrigue KM, Kennedy KM, Devous MD Sr., Rieck JR, Hebrank AC, Diaz-Arrastia R, Mathews D, Park DC, 2012. β-Amyloid burden in healthy aging: regional distribution and cognitive consequences. Neurology 78, 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt SD, Nixon RA, Mathews PM, 2005. ELISA method for measurement of amylod-β levels. Methods Mol. Biol 299, 279–297. [DOI] [PubMed] [Google Scholar]
- Schmidt-Kastner R, Freund TF, 1991. Selective vulnerability of the hippocampus in brain ischemia. Neuroscience 40, 599–636. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, 2002. Alzheimer’s disease is a synaptic failure. Science 298, 789–791. [DOI] [PubMed] [Google Scholar]
- Shimohama S, Fujimoto S, Sumida Y, Tanino H, 1998. Differential expression of rat brain bcl-2 family proteins in development and aging. Biochem. Biophys. Res. Commun 252, 92–96. [DOI] [PubMed] [Google Scholar]
- Takuma K, Phuagphong P, Lee E, Mori K, Baba A, Matsuda T, 2001. Anti-apoptotic effect of cGMP in cultured astrocytes: inhibition by cGMP-dependent protein kinase of mitochondrial permeable transition pore. J. Biol. Chem 276, 48093–48099. [DOI] [PubMed] [Google Scholar]
- Tamatani M, Ogawa S, Nunez G, Tohyama M, 1998. Growth factors prevent changes in Bcl-2 and Bax expression and neuronal apoptosis induced by nitric oxide. Cell Death Differ. 5, 911–919. [DOI] [PubMed] [Google Scholar]
- Tapia-Arancibia L, Aliaga E, Silhol M, Arancibia S, 2008. New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Res. Rev 59, 201–220. [DOI] [PubMed] [Google Scholar]
- Tong L, Thornton PL, Balazs R, Cotman CW, 2001. Beta-amyloid-(1-42) impairs activity-dependent cAMP-response element-binding protein signaling in neurons at concentrations in which cell survival Is not compromised. J. Biol. Chem 276, 17301–17306. [DOI] [PubMed] [Google Scholar]
- Uysal N, Tugyan K, Aksu I, Ozbal S, Ozdemir D, Dayi A, Gönenç S, Açikgöz O, 2012. Age-related changes in apoptosis in rat hippocampus induced by oxidative stress. Biotech. Histochem 87, 98–104. [DOI] [PubMed] [Google Scholar]
- von Bohlen und Halbach O, 2010. Involvement of BDNF in age-dependent alterations in the hippocampus. Front. Aging Neurosci 2, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton MR, Dragunow I, 2000. Is CREB a key to neuronal survival? Trends Neurosci. 23, 48–53. [DOI] [PubMed] [Google Scholar]
- Wang L, Chopp M, Szalad A, Liu Z, Bolz M, Alvarez FM, Lu M, Zhang L, Cui Y, Zhang RL, Zhang ZG, 2011. Phosphodiesterase-5 is a therapeutic target for peripheral neuropathy in diabetic mice. Neuroscience 193, 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster MJ, Herman MM, Kleinman JE, Shannon Weickert C, 2006. BDNF and trkB mRNA expression in the hippocampus and temporal cortex during the human lifespan. Gene Expr. Patterns 6, 941–951. [DOI] [PubMed] [Google Scholar]
- Westermann D, Becher PM, Lindner D, Savvatis K, Xia Y, Fröhlich M, Hoffmann S, Schultheiss HP, Tschöpe C, 2012. Selective PDE5A inhibition with sildenafil rescues left ventricular dysfunction, inflammatory immune response and cardiac remodeling in angiotensin II-induced heart failure in vivo. Basic Res. Cardiol 107, 308. [DOI] [PubMed] [Google Scholar]
- Wu A, Ying Z, Gomez-Pinilla F, 2004. The interplay between oxidative stress and brain-derived neurotrophic factor modulates the outcome of a saturated fat diet on synaptic plasticity and cognition. Eur. J. Neurosci 19, 1699–1707. [DOI] [PubMed] [Google Scholar]
- Wu Y, Zhang AQ, Wai MS, Lai HW, Wu SX, Yew DT, 2006. Changes of apoptosis-related proteins in hippocampus of SAM mouse in development and aging. Neurobiol. Aging 27, 782.e1–782.e10. [DOI] [PubMed] [Google Scholar]
- Yang B, Johnson TS, Thomas GL, Watson PF, Wagner B, Furness PN, El Nahas AM, 2002. A shift in the Bax/Bcl-2 balance may activate Caspase-3 and modulate apoptosis in experimental glomerulonephritis. Kidney Int. 62, 1301–1313. [DOI] [PubMed] [Google Scholar]
- Yang X, Wang Y, Luo J, Liu S, Yang Z, 2001. Protective effects of YC-1 against glutamate induced PC12 cell apoptosis. Cell. Mol. Neurobiol 31, 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Li Q, Pei X, Zhang Z, Yang R, Wang J, Li Y, 2009. Long-term ginsenoside administration prevents memory impairment in aged C57BL/6J mice by up-regulating the synaptic plasticity-related proteins in hippocampus. Behav. Brain Res 201, 311–317. [DOI] [PubMed] [Google Scholar]
- Zieg J, Greer PL, Greenberg ME, 2008. SnapShot: Ca(2+)-dependent transcription in neurons. Cell 134, 1080–1080.e2. [DOI] [PubMed] [Google Scholar]
- Ziv I, Melamed E, 2010. Editorial: apoptosis in the aging brain. Apoptosis 15, 1285–1291. [DOI] [PubMed] [Google Scholar]