

Abstract
Muscarinic receptors (MRs) in the G protein-coupled receptor superfamily are recipients and mediators of parasympathetic neural transmission within the central and enteric nervous systems. MR subtypes, M1R–M5R, encoded by CHRM1-CHRM5, expressed widely throughout the gastrointestinal (GI) tract, modulate a range of critical, highly regulated activities in healthy tissue, including secretion, motility, and cellular renewal. CHRM3/M3R overexpression in colon cancer is associated with increased cell proliferation, metastasis, and a worse outcome, but little is known about the role of the other four muscarinic receptor subtypes. To address this gap in knowledge, we queried the NCI Genomic Data Commons for publicly available TCGA-COAD samples collected from colon tissue. RNA-seq data were collected and processed for all available primary adenocarcinomas paired with adjacent normal colon. In this unbiased analysis, 78 paired samples were assessed using correlation coefficients and univariate linear regressions; gene ontologies were performed on a subset of correlated genes. We detected a consistent pattern of CHRM1 downregulation across colorectal adenocarcinomas. CHRM1 expression levels were positively associated with those for APC and SMAD4, and negatively associated with CTNNB1, the gene for β-catenin, and with coordinate changes in the expression of β-catenin target genes. These findings implicating CHRM1/M1R as an important deterrent of colon cancer development and progression warrant further exploration.
Keywords: colon (C26) carcinoma, muscarinic receptor (subtypes), acetylcholine, G protein-coupled receptors, Wnt/β-catenin signaling pathway
Introduction
Despite meaningful advances in colon cancer prevention and treatment in the United States, metastatic disease remains a highly prevalent cause of morbidity and mortality (Siegel et al., 2021). To address this concern, more must be learned about the molecular mechanisms that modulate progression from primary colon cancer to locoregional and distant metastases. An emerging area, and the focus of our current research, is the role muscarinic receptors (MRs) and alterations in post-MR signaling play in cancer progression (Ali et al., 2021; Schledwitz et al., 2021a; Tolaymat et al., 2021). The five MR subtypes, M1R–M5R, respectively, encoded by CHRM1-CHRM5, are G protein-coupled receptors comprised of seven transmembrane helix domains with an extracellular acetylcholine (ACh)-binding region, and extracellular allosteric binding regions for a variety of non-ACh ligands (Maeda et al., 2019; Tolaymat et al., 2021). Of these subtypes, M1R and M3R, expressed in healthy and neoplastic colon epithelial cells, appear to play prominent roles in disease initiation and progression (Harrington et al., 2010; Cheng et al., 2017; Schledwitz et al., 2021b; Tolaymat et al., 2021). M3R activation results in pro-proliferative downstream signaling, and its activity is most clearly associated with colon cancer progression. In mice, functional M3R deficiency, whether achieved by ablating Chrm3 or more recently by selective M3R antagonism, reduces tumor number and attenuates cancer cell invasion (Raufman et al., 2008, 2011; Hering et al., 2021). In contrast, M1R activation may protect against colon cancer, although its actions are understudied relative to M3R (Tolaymat et al., 2021). In mouse models of colon cancer, Chrm1/M1R deficiency does not reduce tumor size or number. Notably, in mice with combined Chrm1/M1R and Chrm3/M3R deficiency, M1R deficiency outweighs the effects of M3R deficiency; that is mice with combined M1R and M3R deficiency have as many colon tumors as control mice (Cheng et al., 2014). The molecular biology underlying the differential actions of M1R and M3R in colon cancer remains obscure.
β-catenin signaling is a critical player in colon cancer pathogenesis; over 90% of sporadic human colon cancers exhibit mutations in at least one protein in the β-catenin pathway - ∼10% exhibit activating mutations in β-catenin and ∼80% have inactivating mutations in APC, a facilitator of cytoplasmic β-catenin degradation (Muzny et al., 2012; Cheng et al., 2019). Our previous work supported the presence of functional interactions between MR and β-catenin signaling (Raufman et al., 2011). The ApcMin/+ mouse model generally recapitulates the effects of human β-catenin pathway mutations, although tumors predominate in the small intestine rather than colon (Boivin et al., 2003). Compared to ApcMin/+ littermate control mice, we observed that Chrm3/M3R-deficient ApcMin/+ mice had ∼70% fewer small intestinal adenomas (Raufman et al., 2011). Moreover, nuclear staining for β-catenin was significantly reduced in adenomas from Chrm3/M3R-deficient ApcMin/+ mice (Raufman et al., 2011). The mechanistic underpinnings of this interaction between MR and β-catenin signaling were unclear.
To shed light on the role of MR gene expression in colon cancer, we explored a subset of publicly available TCGA RNA-seq data for colon adenocarcinoma and matched normal tissue samples. We found CHRM1 expression was downregulated in adenocarcinomas and CHRM1 expression levels correlated with changes in the expression of β-catenin signaling pathway genes and their downstream targets. Gene Ontology analysis applied across all genes associated with CHRM1 suggested the effects of CHRM1 downregulation in colon cancer are not subtle, supporting a conceptual framework in which CHRM1 plays a protective role against colon cancer development.
Materials and Methods
Animals
M3R-Deficient ApcMin/+ Mice
As described previously (Raufman et al., 2011), male C57BL/6 ApcMin/+ mice (Jackson Laboratory, Bar Harbor, ME, United States) were crossed with female Chrm3–/– mice on a C57BL/6 genetic background (Taconic Farms). Our breeding schemes yielded ApcMin/+Chrm3+/+, ApcMin/+Chrm3+/–, and ApcMin/+Chrm3–/– littermates identified by genotyping using polymerase chain reaction amplification of tail DNA. Chrm3 primers were M3-A3 (5′-aagaccacagtagcagtg-3′), M3-B (5′ctctctacatccatagtccc-3′), and M3-NEO9 (5′-tggatgtggaatgtgtgcgagg-3′). To identify ApcMin/+ mice, we used common primer MAPC15 (5′-ttccactttggcataagg-3′), WT primer MAPC9 (5′-gccatcccttcacgttag-3′), and Min primer (5′-ttctgagaaagacagaagtta-3′) mixed in a single polymerase chain reaction (Jackson Labs protocol).
All experiments were approved by the Institutional Animal Care and Use Committee of the University of Maryland, Baltimore, and by the Research and Development Committee at the VA Maryland Health Care System.
RNA-Seq Data Accession and Processing
Data publicly available on the National Cancer Institute (NCI) Genomic Data Commons were downloaded from the TCGA-COAD analysis of tissue samples of colorectal cancer (Grossman et al., 2016). A subset of these data comprised of paired adenocarcinoma and adjacent normal colon samples were selected for further analysis. Fragments per kilobase of transcript per million mapped reads (FPKM) normalized RNA-seq count data and metadata were collected for 78 primary adenocarcinoma samples with an adjacent normal control (Supplementary Figure 1A). Count tables and metadata were pooled into an expression set and the following processing steps performed using the “hpgltools” package in R (Belew and Hughitt, 2018). Expression data were log2 transformed, quantile normalized, and underwent filtering for low count genes (using the default cbcb filter) prior to SVAseq batch correction (Supplementary Figure 1B). Principal component analysis (PCA) was used to assess appropriateness of the pipeline, and clustering based on condition was present following the pipeline described above (Supplementary Figure 1C).
Differential Expression and Model Analysis
Differential expression profiles were generated for the processed adenocarcinoma and matched normal tissue counts using basic log2 fold change calculations, edgeR, and limma-voom analyses for each set of tissues via the “hpgltools” package (McCarthy et al., 2012; Ritchie et al., 2015; Belew and Hughitt, 2018). An adjusted p-value using Benjamini Hochberg FDR was applied (Benjamini and Yosef, 1995). Univariate mixed linear models comparing the final processed counts of various genes and CHRM1, controlling for participant ID, were generated using the nlme package in R (Pinheiro et al., 2021). R2-values and residual standard error (RSE) were assessed for all models to ensure generation of a reliable model; residuals were assessed to ensure they were homoscedastic and independent and identically distributed (i.i.d.). The “MuMIn” package was used to generate R2-values for the models (Bartoń, 2020). For the initial models described in Figure 1, the conditional R2s ranged from 0.00926 to 0.2330, where a conditional R2 indicates the variation captured by the model (i.e., CTNNB1 and CHRM1 levels, accounting for variation among samples), while the marginal R2 describes only the variance explained by the fixed effects (i.e., CTNNB1 and CHRM1 levels only). Three of the six models produced conditional R2s suggesting small relationships despite the statistical significance: conditional R2s of 0.00926, 0.0317, and 0.115 (Axin1, GSK3b, and FZD1, respectively), resulting in no further analysis, and thus in three final models for APC, SMAD4, and CTNNB1 (Equations 1–3):
FIGURE 1.

Changes in CHRM1 mRNA expression levels are associated with changes in the expression levels of APC, CTNNB1, and SMAD4. We used univariate mixed models with participant ID as a random effect to model processed (low count filtered, quantile normalized, log2 transformed, and batch corrected) CHRM1 expression data as a function of key genes in colorectal cancer. CHRM1 expression levels was significantly associated with APC (A), CTNNB1 (encoding β-catenin) (B), and SMAD4 (C) expression levels, which showed differential clustering of values based on cancer status.
| (1) |
| (2) |
| (3) |
The RSEs of these ranged from 35.74 to 36.62%, where an RSE describes the typical variance from a model, altogether suggesting Equations 1–3 are predicative but imperfect models (Table 1). “Heatmap.2” was used to generate the heatmap in Figure 2A (Warnes et al., 2020) while the “ggplot2” package was used for all other portions of Figures 1–4 with the exception of Figures 4B,C (Wickham, 2016). The “stats” package in R was used for t-tests and to generate Pearson correlation coefficients (R Core Team, 2021). Gene Ontologies were performed using the “gprofiler2” package in R (Kolberg et al., 2020).
TABLE 1.
Model evaluation of equations described in Table 3.
| RSE | Marginal R2 | Conditional R2 | Pearson correlation coefficient | Residuals iid | |
| CHRM1-APC model | 35.74% | 0.1420 | 0.2330 | 0.3729 (p-value 7.716 E-04) | √ |
| CHRM1- CTNNB1 model | 36.72% | 0.1223 | 0.1895 | 0.3466 (p-value 1.878E-03) | √ |
| CHRM1- SMAD4 model | 37.02% | 0.09763 | 0.1811 | 0.3007 (p-value 7.468E-03) | √ |
Residuals, RSE, and R2 of the models described in Table 3 were probed to assess model fit. Residuals were found to be independent, normally distributed, and homoscedastic. Individual Pearson correlation coefficients were all ≥ 0.30, and all models had R2 within a reasonable range given the complex interplay and network of genes that is interacting at any given time. Although RSEs were not low, the associative (vs. predictive) intent of our models and the relatively small denominators altogether with the other results suggests the models are reasonable.
FIGURE 2.

Association of CHRM1 expression with multiple β-catenin signaling genes predictive of cancer status. (A) A heatmap of expression profiles of 72 β-catenin downstream genes demonstrated unsupervised clustering of samples into adenocarcinoma (red) or normal (black) tissue. (B) Individual gene expression levels were assessed in each sample, and levels in adenocarcinoma samples were found to be, on average, more varied across all samples.
FIGURE 4.

Gene pathways differentially expressed in colon adenocarcinomas associated with changes in CHRM1 expression levels. (A) We used a series of univariate mixed models (participant ID as random effect) to assess the relationship between expression levels of CHRM1 and other genes in the dataset, with 4,724 genes demonstrating statistically significant (p-value ≤ 0.01) negative associations and 4,463 demonstrating significant positive associations with CHRM1. (B) We detected 3,921 genes that had a model with a surrogate effect magnitude ≥ 0.25 with KEGG data available. Seventy-seven GO terms were upregulated in adenocarcinoma samples—the top 10 are displayed. (C) Twenty-five GO terms were downregulated in adenocarcinoma samples.
Statistical Analysis for Animal Data
Animal data are presented as means ± SE and were analyzed by two-tailed unpaired Student’s t-test. P < 0.05 were considered statistically significant.
Results
Chrm3 Gene Ablation-Induced Attenuation of β-Catenin Signaling and Intestinal Neoplasia in Mice Identifies Crosstalk Between Muscarinic Receptor and β-Catenin Signaling
Previous evidence supporting a functional interaction between MRs and β-catenin signaling consisted of finding fewer and smaller adenomas in the small intestines of ApcMin/+ mice deficient in Chrm3/M3R (Raufman et al., 2011). Moreover, we observed diminished nuclear staining for β-catenin in these small intestinal adenomas (Raufman et al., 2011). Although ApcMin/+ mice are generally considered an animal model of genetic colon cancer, β-catenin signaling is almost equally important in sporadic colon cancer, where approximately 90% of cancers harbor mutations in genes involved in β-catenin signaling. Collectively, these previous findings suggested an important role for MR signaling in the translocation of β-catenin, a key transcription co-factor in colon cancer, to its site of activity in the cell nucleus. Activated β-catenin signaling is known to upregulate the transcription of numerous genes that promote colon neoplasia (Cheng et al., 2019; Chen et al., 2021).
To determine if M3R deficiency also attenuated neoplasia in ApcMin/+ mouse colons, we analyzed colon tumor data recorded during our previous study (Raufman et al., 2011). This previously unpublished analysis revealed that besides impacting the production of small intestinal adenomas, in ApcMin/+ mice M3R deficiency also robustly attenuated colon neoplasia; we detected colon tumors in all six ApcMin/+ mice expressing M3R but in only one of eight mice with homozygous M3R deficiency (p = 0.005; Figure 5). These findings provide strong support for the concept that functional crosstalk between MR and β-catenin signaling robustly impacts the progression of colon neoplasia.
FIGURE 5.
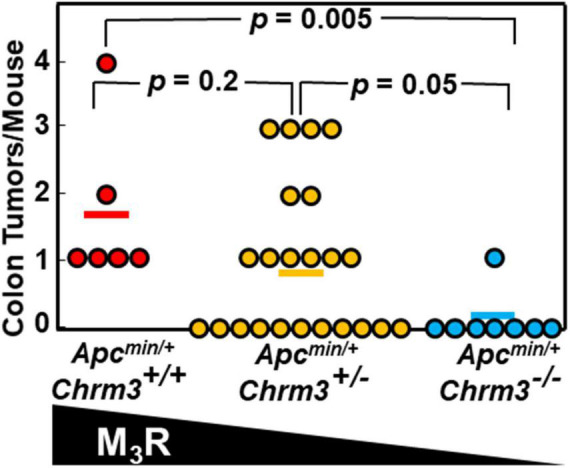
M3R deficiency attenuates colon tumor formation in ApcMin/+ mice with aberrant β-catenin signaling. In ApcMin/+ mice with aberrant β-catenin signaling resulting in exuberant intestinal polyposis, M3R deficiency attenuates colon tumor formation. Apcmin/+ mice with varying M3R expression were created as described in Methods. We euthanized mice at 14 weeks of age, harvested colons, and counted and analyzed tumors. Each symbol represents one Apcmin/+ mouse; bars show means.
CHRM1 Expression Is Attenuated in Human Colorectal Adenocarcinomas
Comparing muscarinic receptor subtype expression levels in adenocarcinoma vs. normal colon samples, we consistently observed reduced CHRM1, CHRM2, and CHRM4 and increased CHRM3 RNA levels (log2fc normal relative to cancer tissue; Figure 6). Following low count filtration, CHRM5 expression data were removed from our analysis. Only CHRM2 and CHRM4 levels were found to have an adjusted p ≤ 0.01 for log2fc across all three analytical methods, while CHRM1 had an adjusted p ≤ 0.01 for all methods except edgeR (Table 2). Since normal tissue samples were derived from full thickness colon tissue samples and M2 and M4 muscarinic receptor subtypes are predominantly localized in intestinal smooth muscle, we were not surprised to observe reduced CHRM2 and CHRM4 expression levels in samples of adenocarcinomas which derive from intestinal epithelial cells; in fact, these findings provided an internal validation of our methodology. Since M1R and M3R are expressed by both normal and neoplastic intestinal epithelial cells, we considered CHRM1 and CHRM3 more disease-relevant candidates for further exploration. Based on these considerations, we explored the relationship between reduced expression of CHRM1, increased expression of CHRM3, and the expression levels of genes commonly mutated in colorectal cancer.
FIGURE 6.

Downregulated expression of CHRM1, CHRM2, and CHRM4 and upregulated expression of CHRM3 mRNA levels in colon cancer. Log2fc was assessed for CHRM1–4 levels in adenocarcinoma compared to normal colon tissues using basic fold change, limma-voom, and edgeR normalization. Changes in CHRM2 and CHRM4 expression were statistically significant (adjusted p ≤ 0.05) across all three assessment methods, while changes in CHRM1 were significant when assessed by basic fold change and limma models; changes in CHRM3 were only significant using basic log2fc. ***p ≤ 0.001, *p ≤ 0.05.
TABLE 2.
Log2 fold changes in CHRM1–4.
| Basic log2 fold change | Adjusted p-value | Limma-voom fold change | Adjusted p-value | edgeR fold change | Adjusted p-value | |
| CHRM1 | −0.2067 | 8.5897E-04 | −0.3310 | 6.044E-05 | −0.8446 | 6.791E-02 |
| CHRM2 | −0.7694 | 1.9037E-17 | −1.1010 | 4.462E-21 | −2.3380 | 3.025E-07 |
| CHRM3 | 0.1477 | 4.2514E-02 | 0.1046 | 1.902E-01 | 0.09757 | 1.000 |
| CHRM4 | −0.4018 | 1.0158E-07 | −0.6219 | 7.594E-11 | −1.8890 | 1.426E-04 |
Log2fc was assessed for CHRM1–4 levels in adenocarcinoma vs. solid normal colorectal tissue samples using basic fold changes, limma-voom, and edgeR normalization, respectively, employing the processing scheme illustrated in Supplementary Figure 1B.
CHRM1 Expression Levels Are Associated With APC, SMAD4, and Inversely Associated With CTNNB1 Expression Levels
We first explored the relationship between CHRM1 levels and APC, KRAS, TP53, and SMAD4 RNA levels by generating a series of univariate linear mixed models with participant ID as a random factor. The relationship describing CHRM1 expression values as a function of KRAS changes was not statistically significant. Whereas the model of CHRM1 expression as a function of TP53 technically showed a significant association, the scatterplot revealed a bimodal distribution of cancer samples along the axis for TP53, suggesting the relationship between CHRM1 and TP53 expression is inconsistent in colorectal cancer. Thus, even if a relationship exists, it appears to be independent of cancer status.
In contrast, the model using APC expression levels yielded a statistically significant relationship, as did the model using SMAD4 (p ≤ 0.01) (Table 3, Equations 1 and 2, Figures 1A,C), prompting us to investigate the relationship between CHRM1 levels and other genes relevant to β-catenin signaling, particularly since SMAD4 activates this pathway (Hussein et al., 2003; Romero et al., 2008; Freeman et al., 2012; Du et al., 2020). We detected a statistically significant linear relationship between CHRM1 and Axin1, CTNNB1 (gene encoding β-catenin; Figure 1B), FZD1 (gene encoding the Wnt receptor), and GSK3b RNA levels (Table 3) using the stringent p-value of effect size (beta coefficient) but did not detect statistically significant relationships between CHRM1 and WNT, GSK3a, CK1α, DVL1, LRP5, or LRP6. A combination of R2-values, RSE, and assessment of residuals as well as simple Pearson correlation used to assess the models, identified three genes of interest—APC, SMAD4, and CTNNB1 (Equations 1–3).
TABLE 3.
Linear regression results for the expression levels of CHRM1 and genes commonly mutated in colon cancer.
| Beta coefficient | Standard error | Degrees of freedom | t-value | p-value | |
| Intercept | 0.1752 | 0.1303 | 38 | 1.3444 | 0.1868 |
| APC levels | 0.2714 | 0.7273E-01 | 38 | 3.7313 | 6.2110E-04 |
| Intercept | 1.9058 | 0.3745 | 38 | 5.0886 | 1.0062E-05 |
| CTNNB1 levels | −0.1985 | 0.5898E-01 | 38 | −3.3660 | 1.7553E-03 |
| Intercept | 0.70369E-01 | 0.1978 | 38 | 0.3558 | 0.7239 |
| SMAD4 levels | 0.2415 | 0.8159E-01 | 38 | 2.9597 | 5.2787E-03 |
Univariate linear mixed models with participant id incorporated a random factor generated to model the relationship between CHRM1 and APC, CTNNB1, and SMAD4 levels were statistically significant. FPKM normalized and processed (log2 transformed, low count filtered, quantile normalized, and SVAseq batch corrected) expression levels were used for model generation.
Performing the same analysis using CHRM3, we detected statistically significant linear relationships between increasing LRP5 and Axin1 RNA levels, decreasing GSK3b RNA levels, and upregulated CHRM3 expression. However, none of the models were i.i.d. with conditional R2s ≥ 0.15 and correlation coefficients of ≥ 0.30. As a result of this analysis, moving forward we focused on gene associations with CHRM1 expression levels.
Downstream Genes in the β-Catenin Signaling Pathway Predict Cancer Status
To clarify the relationship between CHRM1 and components of the β-catenin pathway and gauge the potential for functional MR-induced effects on β-catenin signaling, a subset of 72 genes previously identified to be downstream of β-catenin signaling and curated in our dataset were analyzed in normal colon vs. primary colon adenocarcinoma tissues (Herbst et al., 2014). Statistically significant fold changes (adjusted p ≤ 0.01) were observed for 58 genes; 23 were downregulated and 35 were upregulated in cancer (Supplementary Table 1). We generated a heatmap of expression levels for those β-catenin-related genes which demonstrated clustering by cancer status (normal vs. adenocarcinoma tissue) using unsupervised clustering, confirming this gene set predicted cancer status (Figure 2A). To examine the potential variation across each individual gene, we generated a plot of the normalized counts for each sample and each gene (Figure 2B). Whereas the variation in expression of some genes was greater in normal compared to cancer samples, most showed a wider range of variation in expression in the cancer samples, suggesting some genes were upregulated and playing a role in a subset of the cancers, but there is variability across samples. We used the paired Student’s t-test to analyze these findings, comparing the ranges of normalized values; this revealed a mean increase in variation of 0.7348 in adenocarcinoma (95% CI = 0.4672–1.0023).
Association of CHRM1 Expression Levels With Those of Genes in the β-Catenin Signaling Pathway
Using a series of univariate regressions, we explored the relationship between expression levels for the subset of the aforementioned 72 genes and CHRM1. Statistically significant (adjusted p-value ≤ 0.01) increased RNA expression for 10 (AC2, AC9, ITF2, AC6, CASL, ALEX1, TIAM-1, AC5, FAAR, and FLVI2/BMI1) and decreased RNA expression for 16 genes (BMP4, FOSL1, BCL1, B2T, CMD1, AUTS9, ADCADN, CD87, API4, CCL28, VEGF, LEF-1, FAN1, NMA, CFND, and SFRS3) were associated with increasing CHRM1 levels, adjusting for participant ID with a regression coefficient of ≥ 0.1. Regression coefficients were used as surrogate effect sizes given the univariate nature of the mixed model which only controlled for participant ID. Expression levels of an additional seven genes (MRTL, ASH2, DKK-4, CLD1, MMP-7, ATF, and FEX) were more modest decreased in association with increasing CHRM1 levels (Figure 3 and Supplementary Table 2).
FIGURE 3.

CHRM1 expression is associated with multiple β-catenin signaling target genes. We modeled CHRM1 processed expression data as a function of the relevant β-catenin signaling and target genes using mixed (univariate) models, with participant ID as a random effect. β-catenin target genes previously associated with colon cancer and having a significant positive (blue symbols) or negative (red symbols) association with CHRM1 are shown.
After expanding the univariate regression analysis to all 36,987 genes, we found 9,187 genes were significantly related to CHRM1expression levels—of these, 5,776 had a surrogate effect size with a magnitude ≥ 0.25 (Figure 4A and Supplementary Table 3). Several are pseudogenes, or long noncoding (lnc)RNAs. To identify pathways associated with downregulated CHRM1 expression in colon cancer we performed a Gene Ontology (GO) analysis on the 5,776 gene subset which also had an adjusted p-value of ≤ 0.01 and had available KEGG data (3,921 genes). Of 25 GO terms downregulated in cancer, the following were prominent: phospholipid binding, S/T kinase activity, nucleoside triphosphatase regulator activity, GTPase regulator and activator activity, guanyl nucleotide exchange factor activity, Ras guanyl nucleotide exchange factor activity, and PI and PIP binding (Figure 4C). Of 77 GO terms upregulated in cancer, the following were prominent: catalytic activity on DNA/RNA, histone binding, nuclease and methyltransferase activity, DNA/helicase activity, and single stranded DNA and RNA as well as damaged DNA binding (Figure 4B).
Discussion
Previous studies implicated MRs in the progression and spread of many cancer types, including colon cancer (Shah et al., 2009). To date, no studies attempted to discern the relationship between MR subtype expression and key players in colon cancer. We sought to fill this gap in knowledge using an unbiased approach to explore expression of MR subtypes in publicly available RNA-seq data. We found CHRM1 levels were downregulated in colon adenocarcinoma, while, as expected, CHRM3 was upregulated. Further, we identified an imperfect but linear relationship between CHRM1 and APC and CTNNB1 mRNA levels, supporting a relationship between CHRM1/M1R and β-catenin signaling in colon cancer. APC plays a key role in the β-catenin destruction complex to ensure that, in the absence of activation by Wnt ligands, cytosolic β-catenin is ubiquitinated and degraded (Parker and Neufeld, 2020). Notably, Ras degradation is also regulated by GSK3β, a member of the β-catenin destruction complex (Lee et al., 2018). As M1R activation results in downstream Ras/MAPK activation (Qian et al., 1995), this illuminates a potential node of intersection between M1R and β-catenin signaling.
Although M3R activation results in similar downstream cascades to those modulated by M1R activation, M3R expression is upregulated in colorectal adenocarcinoma and promotes cancer progression. The divergent effects of M1R vs. M3R activation on colon cancer were described previously (Tolaymat et al., 2021) and remain poorly understood. Notably, other differences between the expression and actions of these MR subtypes are reported. For example, M1R and M3R differ in their propensity to engage in cAMP signaling via non-canonical pathways. Following treatment with a muscarinic agonist, Chinese hamster ovary (CHO) cells expressing only M1R maximally accumulate four times as much cAMP as CHO cells expressing only M3R, despite similar receptor density, pharmacokinetics, and efficiency of signaling via phospholipase C, inositol triphosphate, and calcium. Investigation of the underlying mechanism suggested post-M1R stimulation of cAMP accumulation involves activation of Gs (Burford et al., 1995; Burford and Nahorski, 1996). As β-catenin is a target of phosphorylation by PKA, which is activated by cAMP (Katoh and Katoh, 2017), reduced expression of CHRM1/M1R in colon cancer would attenuate β-catenin signaling mediated by this non-canonical M1R/cAMP/PKA/β-catenin axis. This differential impact of M1R and M3R activation on cAMP signaling adds additional complexity and potential fine tuning to the impact of MR expression and activation on β-catenin signaling.
It is possible that differential localization of MRs within cells comprising the tumor microenvironment also plays a role in discrepant effects; M3R is more consistently expressed throughout multiple tissue layers, including circular and longitudinal muscle, myenteric nerve cell bodies, and mucosal epithelial tissue, while M1R expression is largely restricted to myenteric and submucosal nerve cells and epithelial cells (Harrington et al., 2010). In support of this, the wide variation in identified expression levels in CHRM3 in colon cancer, specifically across a range of experimental methods and cell lines, suggests tissue localization may play a role in the observed changes (Felton et al., 2018; Ali et al., 2021). Further, unlike the other muscarinic receptor subtypes, both M1R and M3R undergo pre-coupling with Gi/o G-proteins, that are their non-preferential G proteins. Differential pre-coupling in different tissues could result in variable downstream effects following MR activation (Jakubík et al., 2011).
Our exploration of the relationship between CHRM1 and genes known to be differentially expressed downstream of β-catenin (Figure 3) was similarly thought-provoking. Despite great variation across expression levels of those genes, particularly in cancer tissue, they remained predictive of cancer status. By exploring the relationship between those genes and CHRM1 levels, we identified a subset of genes with a statistically significant association. Univariate analyses identifying concurrent changes in gene expression suggested functional interactions. Of particular interest were positive correlations with ITF2 and AC2, which had the highest surrogate effect sizes. ITF2 is implicated as both a tumor suppressor and proto-oncogene in colon cancer (Davidsen et al., 2018); relevant to the current analysis, in human colon cancer cells and tissue, ITF2 is reported to prevent activation of the β-catenin-TCF4 complex and transcription of β-catenin gene targets (Shin et al., 2014). Decreased levels of AC2 and the general class of adenylyl cyclases was previously demonstrated in human colon cancer cell lines (Nelson and Holian, 1988; Yi et al., 2018; Fan et al., 2019). These changes in AC2 expression may also be relevant to the non-canonical M1R/cAMP/PKA/β-catenin axis described above.
Additionally, we identified a negative correlation with SFRS3, implicated previously in colon cancer (Kuranaga et al., 2018; Zhou et al., 2020). We detected more modest inverse correlations with BMP4 and FOSL1 (but with higher conditional R2-values and high p-values); these genes are also implicated in colon cancer cell proliferation and metastasis (Diesch et al., 2014; Yokoyama et al., 2017; Liu et al., 2021). Notably, MMP7, has a well-documented role in colon cancer progression and metastasis (Kitamura et al., 2009; Koskensalo et al., 2011; Polistena et al., 2014), as does VEGF via vasculature remodeling in tumor-adjacent tissues (Ellis et al., 2000; Liu et al., 2011; Ahluwalia et al., 2014; Yang et al., 2015). Notably, our group previously observed that M3R activation induces MMP7 expression in human colon cancer cells (Xie et al., 2009). Hence, the inverse relationship between CHRM1 and MMP7 expression levels is of particular interest. While we anticipated detecting genes previously implicated in colon cancer, the relative relationship to changes in CHRM1 expression suggests a common relationship between the concurrent changes observed in CHRM1 and APC/CTNNB1, and these downstream β-catenin target genes.
Lastly, we used the relationship between expression levels of genes with a significant relationship to CHRM1 expression to identify gene ontology (GO) terms and pathways of importance to cancer progression. Several GPCR regulation- and activation-related GO terms were downregulated, as were several potentially CHRM1 cascade-related GO terms such as PI and PIP binding and guanyl nucleotide exchange factor activity. Although these genes were specifically stratified based on altered levels correlating with changes in CHRM1 expression, it remains interesting that changes in CHRM1 and related genes had sufficient impact to associate with downregulated pathways. While the sheer number of genes with changing expression levels paralleling CHRM1 was high, the fact that the most concurrently downregulated pathways were CHRM1-related while the most concurrently upregulated GO terms were growth- and proliferation-related, implies the strong correlations were unlikely due to chance and reflect the impact of CHRM1 downregulation in colon cancer. This further validates that CHRM1/M1R downregulation observed in colon cancer is not only non-random, but very likely involved in important aspects of development and/or progression of disease.
Our findings suggest an important relationship exists between CHRM1/M1R and β-catenin signaling in colorectal cancer. Nonetheless, we acknowledge important limitations. Our analytical approach cannot identify a specific mechanism (s) that underlies directionality of gene changes nor a specific cause and effect relationship. Moreover, our results do not exclude the possibility that additional players are involved; additional studies are required to elucidate the processes driving these changes. For example, RNA-seq experiments in Chrm1/M1R knockout mice may elucidate the temporal dynamics and confirm the directionality of such changes in gene and protein expression, thus better delineating the mechanisms whereby these pathways intersect.
Collectively, these patterns of changes in gene expression levels and cancer-related pathways support a conceptual framework wherein CHRM1/M1R expression contributes to protection against the development and progression of colorectal adenocarcinoma. Further elucidation of these novel mechanistic insights regarding intersection of CHRM1/M1R and β-catenin signaling, beyond the scope of the current project, warrant additional experimentation.
Data Availability Statement
The datasets analyzed for this study can be found in the Genomic Data Commons, at https://portal.gdc.cancer.gov/, under the TCGA-COAD project.
Author Contributions
MA and J-PR conceptualized the project. KC performed the experiments and analyzed the results. MA, AS, and J-PR wrote the initial draft, proofread, edited, contributed additional material, and completed the final draft. MA, AS, KC, and J-PR approved the final manuscript. All authors contributed to the article and approved the submitted version.
Author Disclaimer
The contents do not represent the views of the U.S. Department of Veterans Affairs or the U.S. Government.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Funding
This work was funded by the United States (U.S.) Department of Veterans Affairs Biomedical Laboratory Research and Development Program, VA Merit Award grant no. BX004890. MA was supported by the U.S. National Institutes of Health (grant no. T32 DK067872).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2022.857563/full#supplementary-material
References
- Ahluwalia A., Jones M. K., Matysiak-Budnik T., Tarnawski A. S. (2014). VEGF and colon cancer growth beyond angiogenesis does VEGF directly mediate colon cancer growth via a non-angiogenic mechanism? Curr. Pharm. Des. 20 1041–1044. 10.2174/1381612819999131218175905 [DOI] [PubMed] [Google Scholar]
- Ali O., Tolaymat M., Hu S., Xie G., Raufman J. P. (2021). Overcoming obstacles to targeting muscarinic receptor signaling in colorectal cancer. Int. J. Mol. Sci. 22:716. 10.3390/ijms22020716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belew A., Hughitt K. (2018). “hpgltools, A Pile of (hopefully) Useful R Functions.” R package version 2018.03. Available online at: https://rdrr.io/github/elsayed-lab/hpgltools/man/hpgltools.html [Google Scholar]
- Bartoń K. (2020). “MuMIn, Multi-Model Inference.” R Package Version 0.12.2/r18. Available at online at: https://r-forge.r-project.org/R/?group_id=346 (accessed December 23, 2021). [Google Scholar]
- Benjamini Y., Yosef H. (1995). Controlling the false discovery rate a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57 289–300. [Google Scholar]
- Boivin G. P., Washington K., Yang K., Ward J. M., Pretlow T. P., Russell R., et al. (2003). Pathology of mouse models of intestinal cancer consensus report and recommendations. Gastroenterology 124 762–777. 10.1053/gast.2003.50094 [DOI] [PubMed] [Google Scholar]
- Burford N. T., Nahorski S. R. (1996). Muscarinic m1 receptor-stimulated adenylate cyclase activity in Chinese hamster ovary cells is mediated by Gs alpha and is not a consequence of phosphoinositidase C activation. Biochem. J. 315 883–888. 10.1042/bj3150883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burford N. T., Tobin A. B., Nahorski S. R. (1995). Differential coupling of m1 m2 and m3 muscarinic receptor subtypes to inositol 1,4,5-trisphosphate and adenosine 3’,5’-cyclic monophosphate accumulation in Chinese hamster ovary cells. J. Pharmacol. Exp. Ther. 274 134–142. [PubMed] [Google Scholar]
- Chen G. T., Tifrea D. F., Murad R., Habowski A. N., Lyou Y., Duong M. R., et al. (2021). Disruption of β-Catenin-Dependent wnt signaling in colon cancer cells remodels the microenvironment to promote tumor invasion. Mol. Cancer Res. 10.1158/1541-7786.MCR-21-0349 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K., Shang A. C., Drachenberg C. B., Zhan M., Raufman J. P. (2017). Differential expression of M3 muscarinic receptors in progressive colon neoplasia and metastasis. Oncotarget 8 21106–21114. 10.18632/oncotarget.15500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K., Xie G., Khurana S., Heath J., Drachenberg C. B., Timmons J., et al. (2014). Divergent effects of muscarinic receptor subtype gene ablation on murine colon tumorigenesis reveals association of M3R and zinc finger protein 277 expression in colon neoplasia. Mol. Cancer 13:77. 10.1186/1476-4598-13-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X., Xu X., Chen D., Zhao F., Wang W. (2019). Therapeutic potential of targeting the Wnt/β-catenin signaling pathway in colorectal cancer. Biomed. Pharmacother. 110 473–481. [DOI] [PubMed] [Google Scholar]
- Davidsen J., Larsen S., Coskun M., Gögenur I., Dahlgaard K., Bennett E. P., et al. (2018). The VTI1A-TCF4 colon cancer fusion protein is a dominant negative regulator of Wnt signaling and is transcriptionally regulated by intestinal homeodomain factor CDX2. PLoS One 13:e0200215. 10.1371/journal.pone.0200215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diesch J., Sanij E., Gilan O., Love C., Tran H., Fleming N. I., et al. (2014). Widespread FRA1-dependent control of mesenchymal transdifferentiation programs in colorectal cancer cells. PLoS One 9:e88950. 10.1371/journal.pone.0088950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X., Li Q., Yang L., Liu L., Cao Q., Li Q. (2020). SMAD4 activates Wnt signaling pathway to inhibit granulosa cell apoptosis. Cell Death Dis. 11:373. 10.1038/s41419-020-2578-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis L. M., Takahashi Y., Liu W., Shaheen R. M. (2000). Vascular endothelial growth factor in human colon cancer, biology and therapeutic implications. Oncologist 5 (Suppl. 1), 11–15. 10.1634/theoncologist.5-suppl_1-11 [DOI] [PubMed] [Google Scholar]
- Fan Y., Mu J., Huang M., Imani S., Wang Y., Lin S., et al. (2019). Epigenetic identification of ADCY4 as a biomarker for breast cancer an integrated analysis of adenylate cyclases. Epigenomics 11 1561–1579. 10.2217/epi-2019-0207 [DOI] [PubMed] [Google Scholar]
- Felton J., Hu S., Raufman J. P. (2018). Targeting M3 muscarinic receptors for colon cancer therapy. Curr. Mol. Pharmacol. 11 184–190. 10.2174/1874467211666180119115828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman T. J., Smith J. J., Chen X., Washington M. K., Roland J. T., Means A. L., et al. (2012). Smad4-mediated signaling inhibits intestinal neoplasia by inhibiting expression of β-catenin. Gastroenterology 142 562–571.e2. 10.1053/j.gastro.2011.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman R. L., Heath A. P., Ferretti V., Varmus H. E., Lowy D. R., Kibbe W. A., et al. (2016). Toward a shared vision for cancer genomic data. N. Engl. J. Med. 375 1109–1112. 10.1056/NEJMp1607591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington A. M., Peck C. J., Liu L., Burcher E., Hutson J. M., Southwell B. R. (2010). Localization of muscarinic receptors M1R M2R and M3R in the human colon. Neurogastroenterol. Motil. 22 999–1008. 10.1111/j.1365-2982.2009.01456.x [DOI] [PubMed] [Google Scholar]
- Herbst A., Jurinovic V., Krebs S., Thieme S. E., Blum H., Göke B., et al. (2014). Comprehensive analysis of β-catenin target genes in colorectal carcinoma cell lines with deregulated Wnt/β-catenin signaling. BMC Genomics 15:74. 10.1186/1471-2164-15-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering N. A., Verena L., Rayoung K., Benjamin W., Raoul D. A., Marco A., et al. (2021). Blockage of cholinergic signaling via muscarinic acetylcholine receptor 3 inhibits tumor growth in human colorectal adenocarcinoma. Cancers 13:3220. 10.3390/cancers13133220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein S. M., Duff E. K., Sirard C. (2003). Smad4 and beta-catenin co-activators functionally interact with lymphoid-enhancing factor to regulate graded expression of Msx2. J. Biol. Chem. 278 48805–48814. 10.1074/jbc.M305472200 [DOI] [PubMed] [Google Scholar]
- Jakubík J., Janíčková H., Randáková A., El-Fakahany E. E., Doležal V. (2011). Subtype differences in pre-coupling of muscarinic acetylcholine receptors. PLoS One 6:e27732. 10.1371/journal.pone.0027732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh M., Katoh M. (2017). Molecular genetics and targeted therapy of WNT-related human diseases (Review). Int. J. Mol. Med. 40 587–606. 10.3892/ijmm.2017.3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T., Biyajima K., Aoki M., Oshima M., Taketo M. M. (2009). Matrix metalloproteinase 7 is required for tumor formation, but dispensable for invasion and fibrosis in SMAD4-deficient intestinal adenocarcinomas. Lab. Invest. 89 98–105. 10.1038/labinvest.2008.107 [DOI] [PubMed] [Google Scholar]
- Kolberg L., Raudvere U., Kuzmin I., Vilo J., Peterson H. (2020). gprofiler2- an R package for gene list functional enrichment analysis and namespace conversion toolset g:Profiler. F1000Res. 9:ELIXIR-709. 10.12688/f1000research.24956.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskensalo S., Louhimo J., Nordling S., Hagström J., Haglund C. (2011). MMP-7 as a prognostic marker in colorectal cancer. Tumour Biol. 32 259–264. 10.1007/s13277-010-0080-2 [DOI] [PubMed] [Google Scholar]
- Kuranaga Y., Sugito N., Shinohara H., Tsujino T., Taniguchi K., Komura K., et al. (2018). SRSF3 a Splicer of the PKM Gene, regulates cell growth and maintenance of cancer-specific energy metabolism in colon cancer cells. Int. J. Mol. Sci. 19:3012. 10.3390/ijms19103012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. K., Jeong W. J., Cho Y. H., Cha P. H., Yoon J. S., Ro E. J., et al. (2018). β-Catenin-RAS interaction serves as a molecular switch for RAS degradation via GSK3β. EMBO Rep. 19:e46060. 10.15252/embr.201846060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Xu J., Wang M., Wang Q., Bi Y., Han M. (2011). Tumor-derived vascular endothelial growth factor (VEGF)-a facilitates tumor metastasis through the VEGF-VEGFR1 signaling pathway. Int. J. Oncol. 39 1213–1220. 10.3892/ijo.2011.1138 [DOI] [PubMed] [Google Scholar]
- Liu Y., Yue M., Li Z. (2021). FOSL1 promotes tumorigenesis in colorectal carcinoma by mediating the FBXL2/Wnt/β-catenin axis via Smurf1. Pharmacol. Res. 165:105405. 10.1016/j.phrs.2020.105405 [DOI] [PubMed] [Google Scholar]
- Maeda S., Qu Q., Robertson M. J., Skiniotis G., Kobilka B. K. (2019). Structures of the M1 and M2 muscarinic acetylcholine receptor/G-protein complexes. Science 364 552–557. 10.1126/science.aaw5188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy D. J., Chen Y., Smyth G. K. (2012). Differential expression analysis of multifactor, RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40 4288–4297. 10.1093/nar/gks042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzny D. M., Matthew N. B., Kyle C., Huyen H. D., Jennifer A. D., Gerald Fowler C. L., et al. (2012). Comprehensive molecular characterization of human colon and rectal cancer. Nature 487 330–337. 10.1038/nature11252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson R. L., Holian O. (1988). Adenylate cyclase activity and cyclic adenosine monophosphate levels in colon cancer lines and dermal fibroblasts and the effects of cholera toxin and epidermal growth factor. J. Surg. Oncol. 38 108–112. 10.1002/jso.2930380211 [DOI] [PubMed] [Google Scholar]
- Parker T. W., Neufeld K. L. (2020). APC controls Wnt-induced β-catenin destruction complex recruitment in human colonocytes. Sci. Rep. 10:2957. 10.1038/s41598-020-59899-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro J., Bates D., DebRoy S., Sarkar D. R Core Team (2021). _nlme, Linear and Nonlinear Mixed Effects Models_.”R Package Version 3.1-155, Available online at: https://CRAN.R-project.org/package=nlme (accessed December 23, 2021). [Google Scholar]
- Polistena A., Cucina A., Dinicola S., Stene C., Cavallaro G., Ciardi A., et al. (2014). MMP7 expression in colorectal tumours of different stages. In Vivo 28 105–110. [PubMed] [Google Scholar]
- Qian N. X., Russell M., Johnson G. L. (1995). Acetylcholine muscarinic receptor regulation of the Ras/Raf/MAP kinase pathway. Life Sci. 56 945–949. 10.1016/0024-3205(95)00032-2 [DOI] [PubMed] [Google Scholar]
- R Core Team (2021). R: A Language and Environment for Statistical Computing. (Vienna: R Foundation for Statistical Computing; ). [Google Scholar]
- Raufman J. P., Samimi R., Shah N., Khurana S., Shant J., Drachenberg C., et al. (2008). Genetic ablation of M3 muscarinic receptors attenuates murine colon epithelial cell proliferation and Neoplasia. Cancer Res. 68 3573–3578. 10.1158/0008-5472.CAN-07-6810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raufman J. P., Shant J., Xie G., Cheng K., Gao X. M., Shiu B., et al. (2011). Muscarinic receptor subtype-3 gene ablation and scopolamine butylbromide treatment attenuate small intestinal neoplasia in Apcmin/+ mice. Carcinogenesis 32 1396–1402. 10.1093/carcin/bgr118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., et al. (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43:e47. 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero D., Iglesias M., Vary C. P., Quintanilla M. (2008). Functional blockade of Smad4 leads to a decrease in beta-catenin levels and signaling activity in human pancreatic carcinoma cells. Carcinogenesis 29 1070–1076. 10.1093/carcin/bgn054 [DOI] [PubMed] [Google Scholar]
- Schledwitz A., Sundel M. H., Alizadeh M., Hu S., Xie G., Raufman J. P. (2021a). Differential actions of muscarinic receptor subtypes in gastric, pancreatic, and colon cancer. Int. J. Mol. Sci. 22:13153. 10.3390/ijms222313153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schledwitz A., Xie G., Raufman J. P. (2021b). Exploiting unique features of the gut-brain interface to combat gastrointestinal cancer. J. Clin. Invest. 131:e143776. 10.1172/JCI143776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N., Khurana S., Cheng K., Raufman J. P. (2009). Muscarinic receptors and ligands in cancer. Am. J. Physiol. Cell Physiol. 296 C221–C232. 10.1152/ajpcell.00514.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H. W., Choi H., So D., Kim Y. I., Cho K., Chung H. J., et al. (2014). ITF2 prevents activation of the β-catenin-TCF4 complex in colon cancer cells and levels decrease with tumor progression. Gastroenterology 147 430–442.e8. 10.1053/j.gastro.2014.04.047 [DOI] [PubMed] [Google Scholar]
- Siegel R. L., Miller K. D., Fuchs H. E., Jemal A. (2021). Cancer Statistics, 2021. CA Cancer J. Clin. 71 7–33. [DOI] [PubMed] [Google Scholar]
- Tolaymat M., Margaret S. H., Madeline A., Guofeng X., Jean-Pierre R. (2021). Potential role for combined subtype-selective targeting of M1 and M3 muscarinic receptors in gastrointestinal and liver diseases. Front. Pharmacol. 12:786105. 10.3389/fphar.2021.786105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnes G. R., Bolker B., Bonebakker L., Gentleman R., Huber W., Liaw A., Lumley T., Maechler M., et al. (2020). “gplots, Various R Programming Tools for Plotting Data. Available at online at: https://github.com/talgalili/gplots (accessed December 23, 2021). [Google Scholar]
- Wickham H. (2016). ggplot2, Elegant Graphics for Data Analysis. Berlin: Springer-Verlag. [Google Scholar]
- Xie G., Cheng K., Shant J., Raufman J. P. (2009). Acetylcholine-induced activation of M3 muscarinic receptors stimulates robust matrix metalloproteinase gene expression in human colon cancer cells. Am. J. Physiol. Gastrointest. Liver Physiol. 296 G755–G763. 10.1152/ajpgi.90519.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Zhang Y., Hosaka K., Andersson P., Wang J., Tholander F., et al. (2015). VEGF-B promotes cancer metastasis through a VEGF-A-independent mechanism and serves as a marker of poor prognosis for cancer patients. Proc. Natl. Acad. Sci. U.S.A. 112 E2900–E2909. 10.1073/pnas.1503500112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi H., Wang K., Jin J. F., Jin H., Yang L., Zou Y., et al. (2018). Elevated adenylyl cyclase 9 expression is a potential prognostic biomarker for patients with Colon Cancer. Med. Sci. Monit. 24 19–25. 10.12659/msm.906002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama Y., Watanabe T., Tamura Y., Hashizume Y., Miyazono K., Ehata S. (2017). Autocrine BMP-4 signaling is a therapeutic target in colorectal cancer. Cancer Res. 77 4026–4038. 10.1158/0008-5472.CAN-17-0112 [DOI] [PubMed] [Google Scholar]
- Zhou Z., Gong Q., Lin Z., Wang Y., Li M., Wang L., et al. (2020). Emerging Roles of SRSF3 as a therapeutic target for cancer. Front. Oncol. 10:577636. 10.3389/fonc.2020.577636 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets analyzed for this study can be found in the Genomic Data Commons, at https://portal.gdc.cancer.gov/, under the TCGA-COAD project.