

Abstract
Objective:
The aim of this study was to compare the hypermetabolic, and inflammatory trajectories in burned adults to gain insight into the pathophysiological alterations and outcomes after injury.
Summary of Background Data:
Burn injury leads to a complex response that is associated with hypermetabolism, morbidity, and mortality. The underlying pathophysiology and the correlations between humoral changes and organ function have not been well delineated in adult burn patients.
Methods:
Burned adult patients (n = 1288) admitted to our center from 2006 to 2016 were enrolled in this prospective study. Demographics, clinical data, metabolic and inflammatory markers, hypermetabolism, organ function, and clinical outcomes were obtained throughout acute hospitalization. We then stratified patients according to burn size (<20%, 20% to 40%, and >40% total body surface area [TBSA]) and compared biomedical profiles and clinical outcomes for these patients.
Results:
Burn patients were hypermetabolic with elevated resting energy expenditure (REE) associated with increased browning of white adipose tissue from weeks 2 to 4. Hyperglycemia and hyperinsulinemia peaked 7 to 14 days after injury. Oral glucose tolerance and insulin resistance (QUICKI, HOMA2) tests further confirmed these findings with similar areas under the curve for moderate (20% to 40% TBSA) and severe burn (>40% TBSA). Lipid metabolism in sera revealed elevated pro-inflammatory stearic and linoleic acid, with complementary increases in anti-inflammatory free fatty acids. Similar increases were observed for inflammatory cytokines, chemokines, and metabolic hormones. White adipose tissue from the site of injury had increased ER stress, mitochondrial damage, and inflammasome activity, which was exacerbated with increasing burn severity.
Conclusions:
In this large prospective trial, we delineated the complexity of the pathophysiologic responses postburn in adults and concluded that these profound responses are time and burn size dependent. Patients with medium-size (20% to 40% TBSA) burn demonstrated a very robust response that is similar to large burns.
Keywords: burn, hypermetabolism, inflammasome, inflammation, morbidity, mortality
Mortality of burn patients has significantly improved over the last decades with the establishment of critical bundles and protocolized burn care. Despite improvements in mortality, postburn morbidity is tremendous and remains a challenge for clinicians. Jeschke et al,1 and others have shown that after a severe thermal injury, patients are hyper-inflammatory and hypermetabolic, disabled, and debilitated over a period of at least 24 to 36 months.2 Thus, it is hypothesized that these long-term alterations are sequela from the pathophysiologic response that occurs during the acute phase after burn. The changes occurring during the acute phase have been delineated in the pediatric and recently in the elderly population,3–5 but not in the adult burn population. In addition, little data exist about the divergence in pathologic response that occurs according to burn size.
In pediatric and elderly patients, it has been shown that hypermetabolic responses induced by stress and inflammation start immediately after burn and persist for several months to years, impacting every aspect of the body. Furthermore, trajectories and alterations thereof determined outcomes and overall survival of burn. These results have been used to design novel treatment regimes and interventions to improve outcomes. For example, burn patients with poor outcomes have substantial alterations in their glucose profile encompassing hyperglycemia as well as hypoglycemia.6–8 Both hyperglycemia and hypoglycemia are detrimental to burn patients and can lead to infections, impaired wound healing, and mortality.9 In this case, novel treatment approaches are being investigated in order to control hyperglycemia without the competing risk of hypoglycemia susceptibility. Similarly, the impaired inflammatory and immune responses in elderly are now being studied to determine how to stimulate these dampened responses to improve and alter trajectories so when faced with complications, outcomes will shift from death to survival.
Despite the complex pathophysiological responses being extensively eluded in children and elderly, there is a lack of comprehensive oversight in adults. Therefore, the purpose of the present study was to characterize the pathophysiologic responses postburn in terms of clinical outcomes, hypermetabolism, inflammation, and organ function in a large prospective clinical trial in order to understand pathophysiologic mechanisms and allow burn care providers globally to develop new specific treatment options to improve outcome of severely burned adults.
PATIENTS AND METHODS
In this prospective study, we included patients admitted from 2006 to 2016 (n = 1288). In general, burn patients aged 18 to 65 years who were admitted to our burn center with thermal injuries were eligible for enrollment. Demographic data were collected on all patients. Patients who required surgery were additionally consented for blood and tissue collection. Procedures were approved by the Research Ethics Board of Sunnybrook Health Sciences Centre (Study #194-2010). All patients received standard of care according to our clinical protocols, including early excision and grafting, early nutrition, adequate ventilation, adequate antibiotic coverage, etc, as previously published.1,10–12 If needed, all patients received insulin for glucose control during their stay in the burn unit. Oral glucose control protocol targets a blood glucose range between 90 and 144 mg/dL (5 to 8 mmol/L).9 Insulin dosage was titrated on a sliding scale pursuant to the patient’s blood glucose levels and corresponding needs. Clinical data as well as white adipose tissue (WAT), serum, and plasma were obtained at various time points and processed according to established protocols.
Clinical Outcomes
Data were recorded prospectively and entered into our burn registry database. Demographic data included height, weight, burn size, length of stay, heart rate, blood pressure, nutritional intake, presence of inhalation injury, mechanical ventilation, number of surgeries, in-hospital complications, and mortality.1,10–12 In-hospital complications included infections, pneumonia, and septic episodes as defined by the American Burn Association guidelines.13,14 Organ function markers were troponin T, lactate, blood urea nitrogen, creatinine, amylase, lipase, aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), and bilirubin.
Metabolic Responses
Metabolic outcomes were compared for burn patients versus controls and among burn patients of different burn severities (<20%, 20% to 40, >40% total body surface area [TBSA]). We determined insulin sensitivity via oral glucose tolerance tests (OGTTs), conducted when a patient was 95% healed. Standard OGTT with intake of 75 g of glucose and subsequent glucose, insulin, and c-peptide measurements were conducted over a period of 2 hours15 and calculated according to well established protocols. We calculated insulin sensitivity indices: ISI Matsuda, QUICKI, HOMA2, and ISI quantitative insulin sensitivity check index, and homeostasis model assessment as previously described.15 Furthermore, resting energy expenditure (REE) was assessed weekly until discharge. The REE was measured with a Sensor Medics 2900 (SensorMedics corp, Yorba Linda, CA) metabolic measurement cart.16,17
Protein lysates from fat obtained from operation (OR) (greater than 10 days) were extracted using a RIPA lysis buffer and separated on SDS-PAGE gel before immunoblotting. Binding immunoglobulin protein (BiP), complex IV (COXIV), and AMP-activated protein kinase (AMPK) antibodies were from Cell Signaling Technology (MA) and β-actin antibody was from Thermo Fisher (MA).
Total RNA was extracted from fat tissue using TRIzol-chloro-form (Life Technologies, Carlsbad, CA) with subsequent purification using the RNeasy Kit (Qiagen, Germantown, MD) according to the manufacturer’s instructions. RNA (2 mg) was transcribed to cDNA using the high-capacity cDNA reverse transcription kit (Applied Biosystems, Waltham, MA). Real-time quantitative PCR was performed using the Applied Biosystems Step One Plus Real-Time PCR System. Primer sequences used are available upon request.
Immunohistochemistry
Tissues collected were immediately fixed in 10% formalin before paraffin embedding. Subsequently, tissues were sectioned and stained with hematoxylin and eosin (H&E) or incubated with uncoupling protein (UCP)-1 (Sigma-Aldrich, MO) antibody followed by DAB staining. Imaging was performed on a LSM confocal microscope (Zeiss, Zeiss, Germany).
Plasma Fatty Acids Composition
Plasma fatty acids composition from patients were analyzed by gas chromatography–mass spectrometry (GC-MS) performed by the Analytical Facility for Bioactive Molecules (AFBM) platform from the hospital for Sick Children, Toronto, Ontario, Canada. The respective concentrations of each fattyacid were added to determine the total fatty acid concentration and the percentage for each fatty acid.
Inflammatory and Immunological Responses
Inflammatory responses were determined in plasma and adipose tissue. Plasma cytokine profiling was conducted twice weekly until time of discharge. Adipose tissue inflammatory markers, cytokines, and NLRP3 inflammasome activity (IL-1β) were determined in WAT that was collected within the first week after injury as discarded tissue at surgeries. Inflammation from both the site of injury and systemically was determined by standard biochemical techniques (western blotting; Bio-Rad, Hercules, CA), by the Bio-Plex Suspension Array System (Millipore, MA) measuring 17 different cytokines and by quantitative RT-PCR (refer to above section).10,18–22
Statistical Analysis
Data are presented as mean ± standard deviation (SD), mean ± standard error of the mean (SEM), or median (IQR) for continuous variables and as number (%) for categorical variables. Statistical analysis was conducted using Student t test, analysis of variance (ANOVA) with Welch correction, Wilcoxon rank-sum test, and the χ2 test where appropriate. Statistical comparisons were conducted using SPSS 20 (IBM corp, New York, NY) and figures were generated using GraphPad Prism 6.0 (San Diego, CA) software or Microsoft Excel. Significance was accepted at a P value less than 0.05.
RESULTS
Clinical Outcomes
Clinical outcomes were assessed over a 10-year period, 2006 to 2016; during this time, 1288 patients met inclusion criteria (Supplemental Figure 1, http://links.lww.com/SLA/B152). Demographics, clinical markers, and incidence of various morbidities are summarized in Table 1. Five healthy controls were used to compare burn patients to normal tissue, of whom 2 were females and the mean age was 28 years. In terms of clinical outcomes, 34% of patients had burn wound infections, 14% pneumonia, 12% bacteremia, with 8% incidence of sepsis. Morbidities ranged from 2% for renal failure, to 1% for abdominal compartment syndrome. Crude mortality was 1.7% when futile patients were excluded.
TABLE 1.
Demographics and Outcomes of Patients by Injury Severity Group
| Characteristic | All | TBSA <20% | TBSA 20–40% | TBSA ≥40% | P * |
|---|---|---|---|---|---|
| No. of patients | 1288 | 1058 | 157 | 73 | |
| Demographics | |||||
| Age, y, mean ± SD | 40 ± 13 | 40 ± 13 | 43 ± 13 | 42 ± 12 | 0.014 |
| Male, no. (%) | 942 (73%) | 765 (72%) | 118 (75%) | 59 (81%) | 0.235 |
| Injury characteristics | |||||
| TBSA, %, mean ± SD | 12 ± 13 | 7 ± 5 | 27 ± 6 | 53 ± 10 | <0.0001 |
| 3rd TBSA, %, mean ± SD | 5 ± 11 | 2 ± 3 | 12 ± 12 | 38 ± 20 | <0.0001 |
| Inhalation injury, no. (%) | 181 (14%) | 92 (9%) | 52 (33%) | 37 (51%) | <0.0001 |
| Baux score, mean ± SD | 55 ± 22 | 48 ± 15 | 76 ± 17 | 104 ± 15 | <0.0001 |
| Etiology | |||||
| Flame, no. (%) | 723 (56%) | 540 (51%) | 118 (75%) | 65 (89%) | <0.0001 |
| Scald, no. (%) | 382 (30%) | 349 (33%) | 29 (19%) | 4 (6%) | <0.0001 |
| Electrical, no. (%) | 104 (8%) | 93 (9%) | 8 (5%) | 3 (4%) | 0.050 |
| Other, no. (%)† | 79 (6%) | 76 (7%) | 2 (1%) | 1 (1%) | 0.001 |
| Outcomes | |||||
| Mechanical ventilation, no. (%) | 375 (29%) | 199 (22%) | 111 (73%) | 65 (92%) | <0.0001 |
| OR procedure required, no. (%) | 855 (66%) | 640 (61%) | 144 (92%) | 71 (97%) | <0.0001 |
| OR visits, mean ± SD‡ | 2 ± 2 | 2 ± 1 | 3 ± 2 | 7 ± 4 | <0.0001 |
| Complications | |||||
| ACS, no. (%) | 1 (1%) | 0 | 0 | 1 (1%) | <0.001 |
| ARDS, no. (%) | 53 (4%) | 18 (2%) | 17 (11%) | 18 (25%) | <0.0001 |
| Pneumonia, no. (%) | 184 (14%) | 64 (6%) | 74 (47%) | 46 (63%) | <0.0001 |
| Bacteremia, no. (%) | 152 (12%) | 28 (3%) | 64 (41%) | 60 (82%) | <0.0001 |
| Sepsis, no. (%) | 106 (8%) | 21 (2%) | 41 (26%) | 44 (60%) | <0.0001 |
| Burn wound infections, no. (%) | 442 (34%) | 317 (30%) | 84 (54%) | 41 (56%) | <0.0001 |
| Renal failure, no. (%) | 32 (2%) | 9 (1%) | 7 (5%) | 16 (22%) | <0.0001 |
| LOS, days, mean ± SD§ | 18 ± 23 | 12 ± 10 | 33 ± 23 | 83 ± 54 | <0.0001 |
| LOS/TBSA, days/%, mean ± SD¶ | 2.6 ± 4.7 | 2.9 ± 5.1 | 1.2 ± 0.9 | 1.5 ± 1 | <0.0001 |
| Mortality, no. (%)§ | 22 (1.7%) | 5 (1%) | 6 (4%) | 11 (15%) | <0.0001 |
ACS indicates abdominal compartment syndrome; ARDS, acute respiratory distress syndrome; LOS, length of stay; OR, operating room; TBSA, total body surface area.
Significant differences among injury severity groups (P < 0.05).
Other etiology includes chemical, radiation, and contact burn.
Analysis restricted to patients with ≥1 OR visit.
Analysis excludes mortality ≤72 hours postadmission.
Analysis restricted to patients alive until discharge.
We then divided these patients on the basis of burn size: <20%, 20% to 40%, and >40% TBSA. We found that there was a slight but significant difference between these groups for age distribution. Burn size, inhalation injury, and burn severity were all significantly different between groups, P < 0.0001. In terms of clinical outcomes, we found that the amount of surgeries and escharotomies, number of ORs, length of stay (LOS), and all morbidities were significantly different, P < 0.001. In addition, there were significant differences between groups for clinical complications (eg, acute respiratory distress syndrome, pneumonia, sepsis, burn wound infection, and mortality) with burns over 40% TBSA being the worst in terms of adverse outcomes, P < 0.001 (Table 1). Mortality was also profoundly different between the severity groups with adjusted proportions, which includes the removal of futile patients, manifesting as 1.7% mortality for all burns, 1% for <20% TBSA, 4% for 20% to 40% TBSA, and 15% for >40% TBSA, P < 0.0001 (Table 1). This is represented in the Kaplan-Meier survival curve (Supplemental Figure 2, http://links.lww.com/SLA/B152).
Metabolic Responses
Glucose Metabolism
We measured daily average glucose, 6 am, minimum (Min), maximum (Max), and insulin levels. For patients with complete glucose measurements throughout hospital stay, we found that average and 6 am glucose levels are around 120 to 130 mg/dL, which is within our target range set by our glucose control protocols (Fig. 1A, B). Minimum and maximum glucose are tightly controlled, but become more variable at around 40 days when patients sample size decreases and patients at this time are sicker and require prospective glucose control (Fig. 1C, D). We confirmed a hyperglycemic and hyperinsulinemic response that was supported by the substantial amounts of insulin required to control elevated glucose, which peaked during the first 7 to 14 days (Fig. 1E).
FIGURE 1.
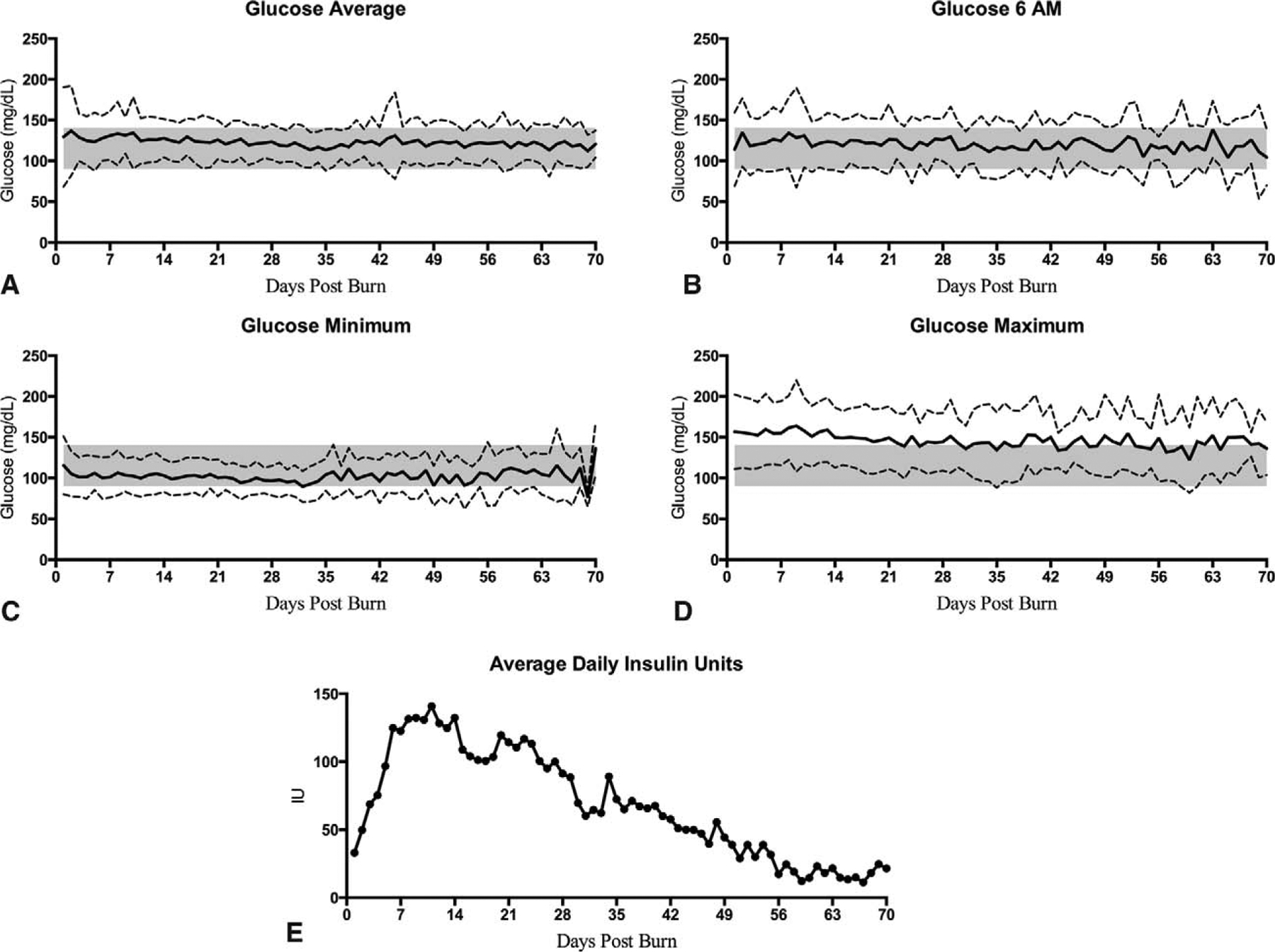
Patients’ glucose levels in burn adults (n = 600). Mean daily average glucose (A), 6AM glucose (B), minimum glucose (C), maximum glucose (D), and insulin units administered (E) during the course of hospital stay. Dashed lines indicate SD. Shaded area represents glucose control protocol target range of 90 to 144 mg/dL (5 to 8 mmol/L).
When looking at glucose and insulin with regard to burn size, it appeared that patients with smaller burns express hyperglycemia requiring high doses of insulin; however, it was not as long as burns over 20% TBSA (Supplemental Figure 3, http://links.lww.com/SLA/B152). Similarly, burns between 20% and 40% TBSA and over 40% TBSA have similar glucose patterns with the difference in the amount of insulin given to control hyperglycemia (Supplemental Figure 3, http://links.lww.com/SLA/B152), suggesting more severe and prolonged insulin resistance.
To determine whether increased hyperglycemia in burn patients was due to increased peripheral or central insulin resistance or lack of insulin production, we conducted OGTTs. As we only conducted OGTTs in patients with ≥20% TBSA burn, we compared patients with burns 20% and 40% and >40% TBSA. The OGTT profile indicated hyperglycemia and elevated fasting insulin (Supplemental Figure 4A–F, http://links.lww.com/SLA/B153). Glucose, insulin, and C-peptide had similar curves and area under the curves for both injury severities (Supplemental Figure 4G–L, http://links.lww.com/SLA/B153), indicating profound peripheral insulin resistance. Peripheral insulin resistance was confirmed when surrogate measures of insulin resistance indices were calculated (Supplemental Table 1, http://links.lww.com/SLA/B152). Both injury severity groups had abnormal QUICKI and HOMA2 estimates of insulin resistance. There was no significant differences between 20% and 40% and >40% TBSA burn in the OGTT profile or insulin indices.
Resting Energy Expenditure and Adipose Tissue Browning
REE expressed as percent predicted indicated that adults are hypermetabolic after burn injury (Fig. 2A). We found that over time, REE slightly decreases but remains elevated after 4 weeks. When stratified by burn size, it appears that even smaller TBSA burn patients are somewhat hypermetabolic during the first 2 weeks after burn. Larger burn patients (>40% TBSA) remain hypermetabolic after 4 weeks postinjury and an ANOVA test revealed it was significantly greater than smaller burns (20% to 40% TBSA) that normalize, P < 0.05 (Fig. 2B). Recently, the phenomena of WAT browning, in which subcutaneous white adipose converts to a more brown-like adipose, has been implicated in facilitating persistent hypermetabolism in burns.23,24 Furthermore, these studies have supported that browning of WAT occurs over time and is most apparent at later time points (>10 days). Currently, we measured UCP-1 expression, a widely accepted indicator of browning, and found that burn induces browning of the WAT beyond 10 days after injury with a greater signal occurring in the severe group (>40% TBSA, Fig. 2C). Using RT-PCR, we showed that browning is higher in burn patients relative to controls and also confirmed our aforementioned findings by showing an exacerbated effect in the >40% TBSA burn group (Fig. 2D, E).
FIGURE 2.
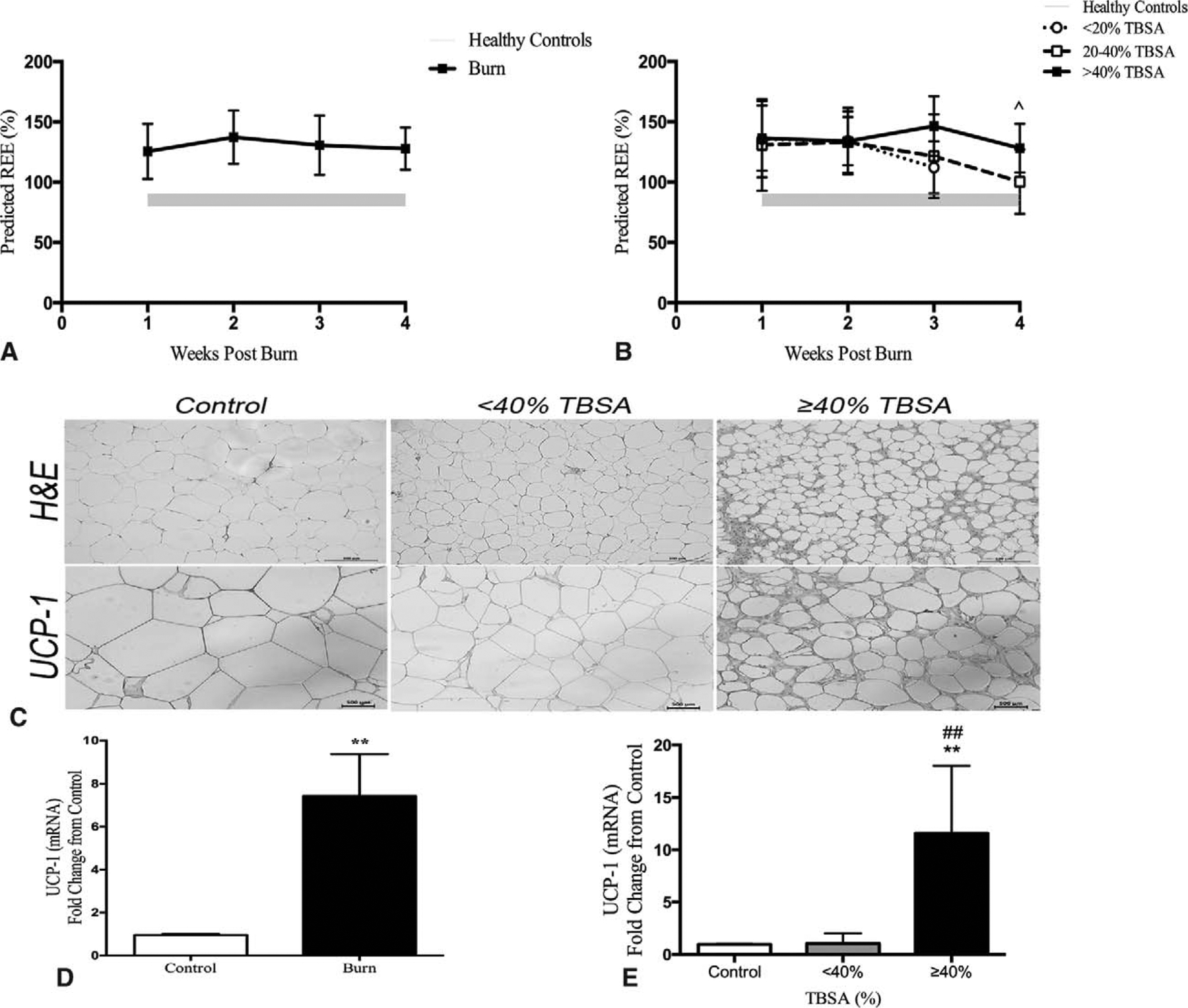
Hypermetabolic response and WAT browning. Burn adults (n = 92) had increased resting energy expenditure expressed as a percentage throughout the course of hospital stay (A). When stratified by burn severity (B), all burn groups (<20%, 20–40%, and >40% TBSA) demonstrated hypermetabolic response with the severe burn persisting beyond 4 weeks postinjury. Data represented as mean ± SEM, *significant difference between 20–40% TBSA and >40% TBSA burned adults, P < 0.05. Morphological and immunohistochemical evidence of browning in white adipose tissue postburn. H&E and UCP-1 staining of representative cross sectional area of paraffin sections of sWAT obtained from burned patients and healthy controls (C). Quantitative RT-PCR gene expression comparing controls to burn patients (>10 days postburn) revealed several fold increased expression in browning marker UCP-1 at the site of injury (D). This increased expression of UCP-1 was further demonstrated when stratified based on injury severity (<40 TBSA vs ≥40% TBSA). Data represented as mean ± SEM. ** = significant difference between controls (n = 5) and burned adults (n = 10); ## = significant difference between < 40% TBSA and ≥40% TBSA burned adults, P < 0.01.
Lipid Metabolism
We found that burn causes an elevation in pro-inflammatory fatty acids stearic acid and linoleic acid (Fig. 3A, B). Similarly, augmented responses were also observed for oleic, vaccenic, and eicosapentaenoic fatty acids (Fig. 3C, D). The collective effects were most profound and significant within the first 14 days, relative to healthy controls. When stratifying by burn size, it appears that >40% TBSA had the most alterations after burn (Fig. 3E to H).
FIGURE 3.
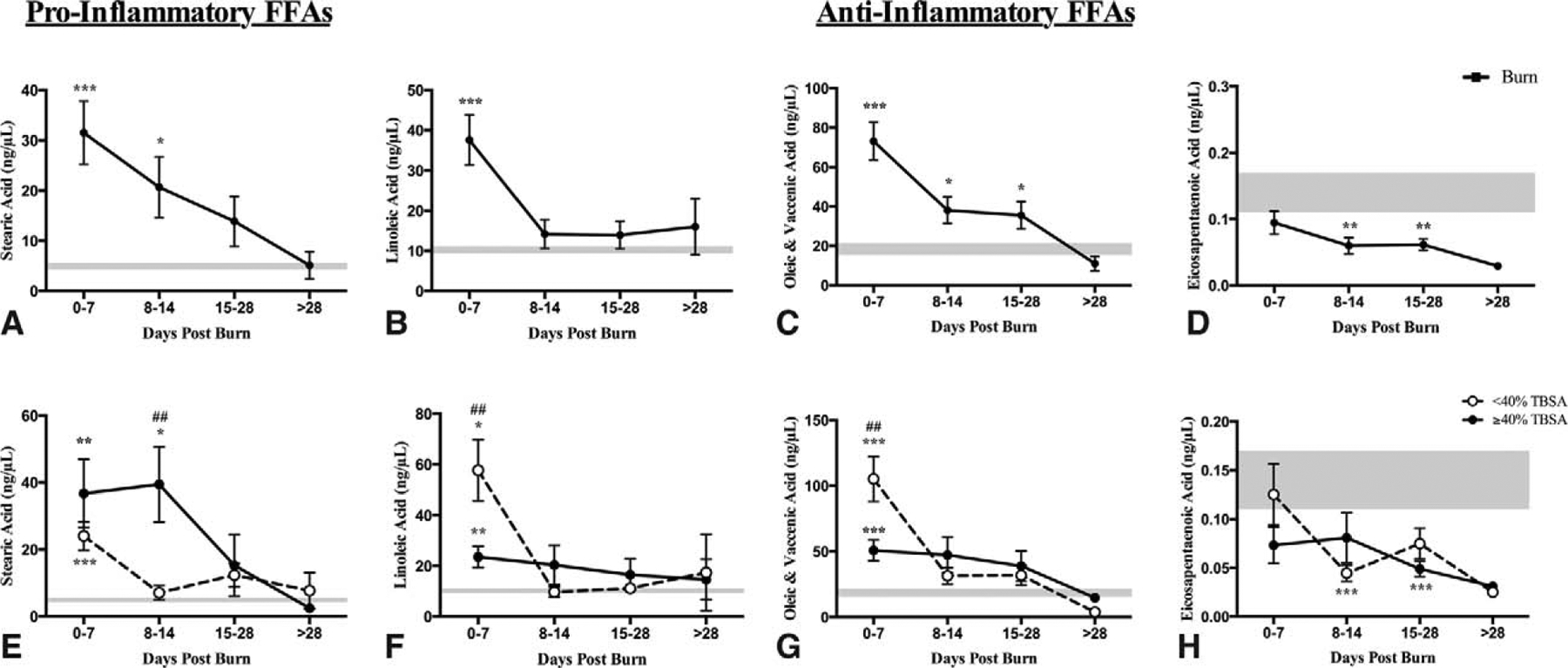
Lipid FFA alterations in burn patients over time. Pro-inflammatory FFA species (stearic and linoleic acids) were all significantly increased within the first 14 days after injury relative to controls (A, B). Anti-inflammatory oleic and vaccenic acid and eicosapentaenoic acid also showed augmented profiles throughout sampling period (C, D). When stratified based on burn size (<40% TBSA vs ≥40% TBSA), both groups had similar profiles with steric acid revealing the most divergent trajectory in the severe groups (E to H). Data expressed as mean ± SEM.*, ** & *** = significant difference between burn (n = 46) and healthy controls (n=5); #, ## & ### = significant difference between < 40% TBSA and ≥40% TBSA burn groups; P < 0.05, P < 0.01, and P < 0.001, respectively.
Metabolic Alterations and Inflammasome Activity
As an acute phase sentinel of stress and inflammation, the byproduct of NLRP3 inflammasome assembly results in the production of IL-1β,25 both of which have been shown to be upregulated after burn injury.3,26 In addition, the NLRP3 inflammasome assembly has been shown to be activated by a number of sources, including ER stress27 and mitochondrial dysfunction.28 Currently, we hypothesized that increasing burn severity causes cellular alterations that are associated with metabolic changes and hence result in representative inflammasome activity. All burn sizes caused marked upregulation of genetic and protein expression of ER stress marker BiP relative to controls, with elevated proportions in the >40% TBSA group (Fig. 4A). Similarly, there was also a downregulation of COXIV, a well-established mitochondrial marker, indicating mitochondrial damage and concurrently AMPK activation supporting a compensatory response to stimulate mitochondrial biogenesis (Fig. 4B, C). As a byproduct of NLRP3 inflammasome activation, IL-1β RNA expression was upregulated in WAT that increased with burn severity (Fig. 4D). Using a multiplex platform, we found that pro-inflammatory biomarkers are substantially increased after burn, with a steady decrease over time, while anti-inflammatory IL-10 was elevated acutely after burn and normalized over time (Fig. 5). Collectively, the exacerbated and uncoordinated response may be contributing to the susceptibility of infection and sepsis.29,30 This notion was further supported when we compared burn size with substantial changes occurring in the first 2 weeks; while smaller burns usually recover and approach normal levels, big burns remain augmented (Supplemental Figure 5, http://links.lww.com/SLA/B153). Collectively, these data suggest a consort of augmented immunometabolic response that worsens with increasing burn severity.
FIGURE 4.
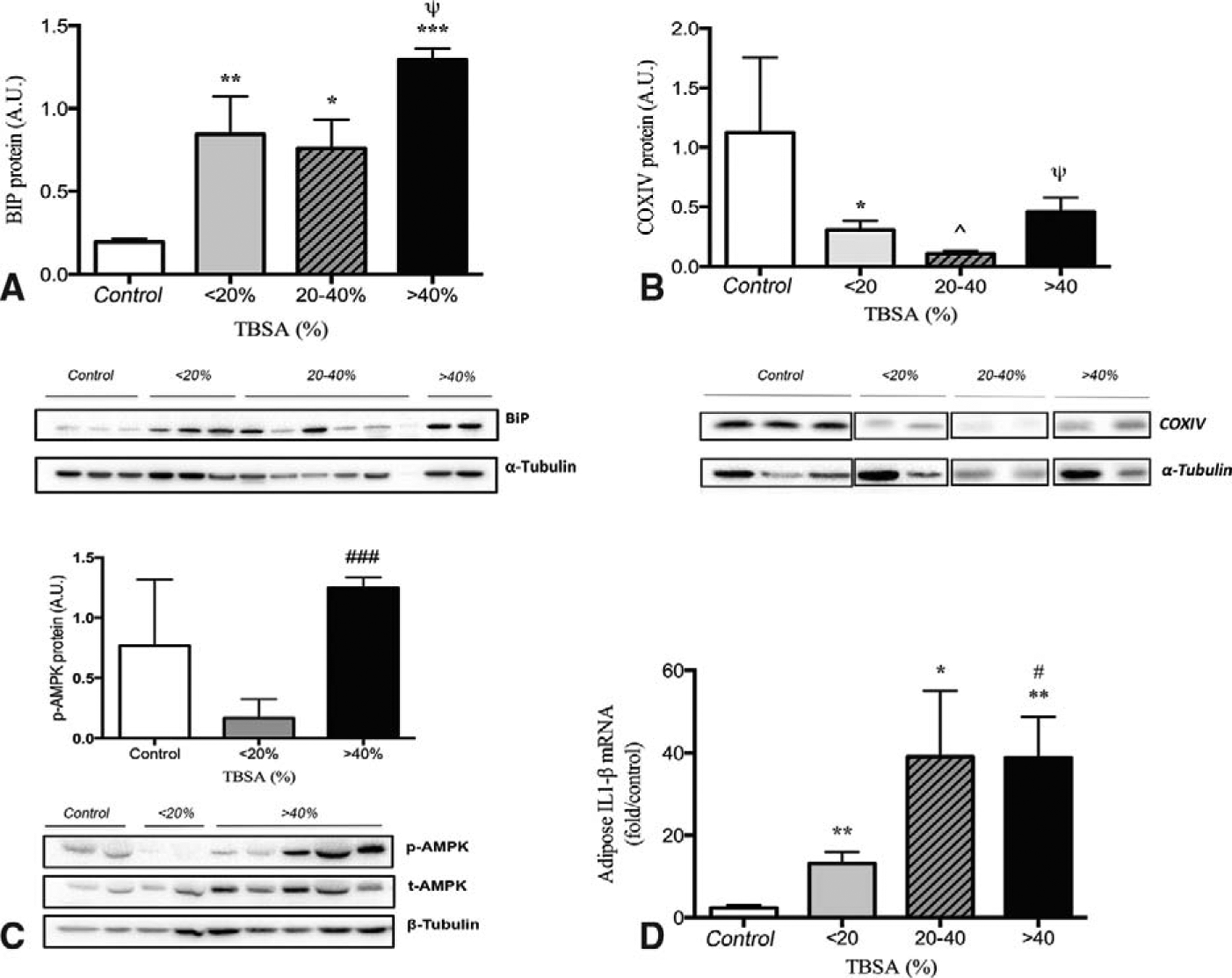
Metabolic markers of augmented response in WAT. ER stress marker BiP was upregulated in all burn groups relative to controls with highest proportion in >40 TBSA (A). Mitochondrial dysfunction was present and manifested by decreased expression of COXIV (B) and increased AMPK (C). As a measure of NLRP3 inflammasome activity, adipose tissue IL-1β was increased in all burn groups and increased with increasing burn severity (D). All tissues were taken within 7 days postburn. Data are represented as mean ± SEM.*, ** & *** = significant difference between controls (n = 5) and burned adults (n = 34); ^, ^^ & ^^^ = significant difference between < 20% TBSA and 20% to 40% TBSA; #, ## & ### = significant difference between <20% TBSA and > 40%TBSA; Ψ, Ψ Ψ & Ψ Ψ Ψ = significant difference between 20–40% TBSA and > 40% TBSA; P < 0.05, P < 0.01, and P < 0.001, respectively.
FIGURE 5.
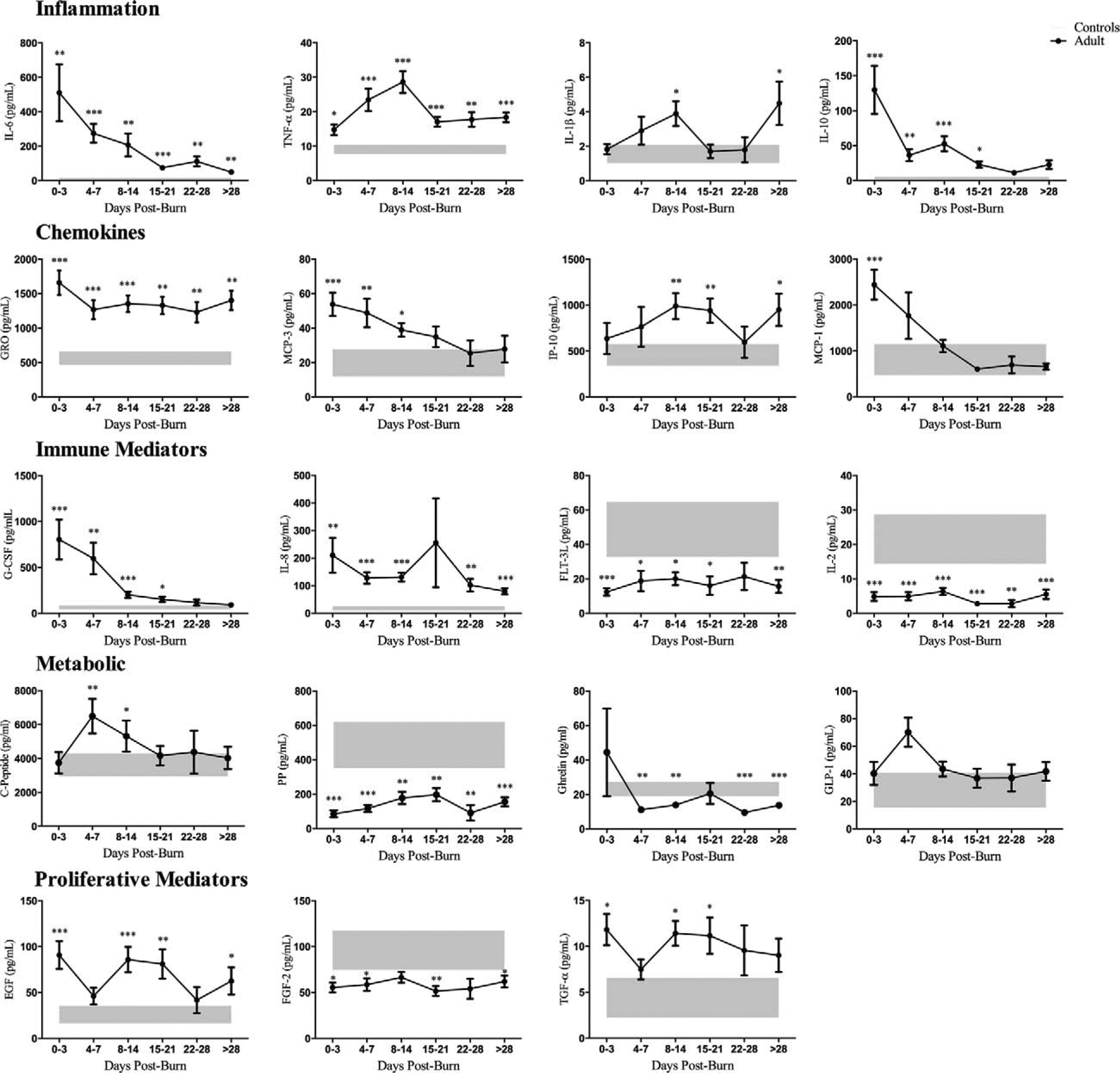
Plasma cytokine profiling of burn (n = 128) and healthy controls (n = 10) during the course of hospital stay (0 to 3, 4 to 7, 8 to 14, 15 to 21, 22 to 28, >28 days postburn). A significantly augmented inflammatory, chemokine, and metabolic response was present in burn patients and persisted throughout hospital course, with greatest alterations during the acute phase (within 14 days postburn). Data are represented as mean ± SEM.*, ** &*** = significant difference between controls (grey boxes) vs. burned adults; P < 0.05, P < 0.01 and P < 0.001, respectively.
Organ Biomarkers
We measured biomarkers to indicate integrity and function of the heart, kidney, pancreas, and liver. We found that burn causes alteration in cardiac biomarkers after 3 weeks postinjury and is associated with decreased organ perfusion. Initial lactate was increased, which is part of the initial response after burn (Supplemental Figure 6A, http://links.lww.com/SLA/B153). Over time with an increasing incidence of organ complications and sepsis, lactate rose and so did troponin (Supplemental Figure 6B, http://links.lww.com/SLA/B153). Renal biomarkers BUN and creatinine followed the aforementioned trajectory (Supplemental Figure 6C–D, http://links.lww.com/SLA/B153). A similar trend was observed for pancreatic amylase and lipase with early increases and peaks between days 15 and 21 (Supplemental Figure 6E–F, http://links.lww.com/SLA/B153). The liver profile revealed plateaus early and values decreasing over time (Supplemental Figure 6G–J, http://links.lww.com/SLA/B153).
When stratified into the 3 burn groups, patients with larger burns (>40% TBSA) had significantly higher values than smaller burns (<20% TBSA) for these markers indicating impaired organ function. These observations parallel the clinical incidence of organ complications and sepsis where increasing burn injury severity is reflected in cardiac, renal, and liver markers alike (Supplemental Figure 7, http://links.lww.com/SLA/B153).
DISCUSSION
In general, burn outcomes have significantly improved over the last decades and survival can be expected. However, mortality and substantial morbidities still occur, and lead to debilitation of severely burned patients. The responses after burn are complex, profoundly altered, and are very difficult to dissect. Despite the identification and delineation of the numerous components of the postburn response, no prospective large clinical study has ever fully characterized the acute phase postburn comprehensively in adult patients. The purpose of the present study was to determine the pathophysiologic response in terms of hypermetabolism, inflammatory stress responses, and organ function in a large prospective clinical trial to enable developments of future interventions and treatment options.
This trial included 1288 patients with the vast majority being burn under 20% TBSA. The overall incidence of morbidities and complications was low, but it is interesting to note that 30% of these patients under 20% TBSA burn had wound infections, 2% sepsis, and 5 patients deceased. When comparing patients suffering from 20% to 40% TBSA burns, it revealed that these patients have the shortest LOS and a mortality of 4%. The morbidities within this patient group are similar to the severe burn group (>40% TBSA), with smaller than expected differences, for example, pneumonia 47% versus 63%. However, profound differences in incidence proportions were supported when comparing the 20% to 40% and >40% TBSA groups for sepsis (26% vs 60%), renal failure (5% vs 22%), and mortality (4% vs 15%), respectively. These results indicate the profound and substantial impact of larger burns on systemic morbidities.
The present study yielded both expected and some unexpected results. The expected results included the difference in clinical outcomes, the augmented and prolonged hypermetabolic response, and the profound inflammatory and stress responses in larger burn patients. Energy requirements were significantly increased and prolonged in burn patients over 40% TBSA when compared with smaller burns. In addition, glucose and lipid profiles indicate profound alterations. Surprisingly, the glucose tolerance test did not reveal any major difference in term of insulin resistance and sensitivity between the 20% and 40% TBSA and >40% TBSA burn groups indicating that even “smaller” injuries undergo a hyperinsulinemic hyperglycemic response. However, the underlying signals and mechanisms are not entirely clear, as the majority of inflammatory stress markers such as cytokines are increased to a greater extent in the large burn group than the <40% TBSA burn groups. One possible explanation could be ER stress in adipose tissue. We showed that even in the adipose tissue from smaller burns, ER stress is similarly increased when compared with larger burns. The adipose tissue is an important “organ” and has recently gained attention as 2 groups independently showed that the adipose tissue undergoes a response called browning.23,24 Browning of the adipose tissue is a desired change in diabetic or obese patients, but in burns patients, others and we hypothesize that this mechanism contributes to catastrophic hypermetabolism and catabolism. We currently showed that browning occurs in burn patients and is even initiated at smaller burn sizes. Hence, regardless of the burn size, ER-associated stress was present. These results indicate that even smaller burns undergo hypermetabolic alterations, which can be detrimental. Agonists of the hypermetabolic response include catecholamines and stress hormones such as cortisol.31–34 In the present study, we did not determine catecholamines or cortisol, but Jeschke et al10 have recently shown that in burned children, serum and urine cortisol increased 5 to 8-fold and remained elevated throughout the entire acute hospital stay. Stress hormones such as glucocorticoids have been described as one of the major hormones responsible for proteolysis and catabolism.35–38 Glucocorticoid levels are markedly increased postburn, and therefore, a hypothetical approach to attenuate protein breakdown and hypermetabolism would be to block cortisol production. In addition, Jeschke et al1 have determined that catecholamines (epinephrine and norepinephrine) in serum are elevated during acute hospitalization about 10 to 20-fold and remain elevated for up to 3 to 5 years. As catecholamines are mediators of inflammation and adipose browning, it seems therefore logical for future studies to utilize catecholamine blockers and determine whether these agents can reduce hypermetabolism, stress, inflammation, and browning.
Of greater interest is the change in serum lipids, triglycerides, and free fatty acids, all of which are significantly increased throughout nearly the entire acute hospital stay. Fat transporter proteins are decreased postburn, while triglycerides and free fatty acids are increased, which could explain the fatty infiltration of the liver and other organs postburn. We have shown that hepatomegaly with fatty infiltration is associated with increased incidence of sepsis and mortality implying the importance of organ integrity and function.39 The exact lipid profile after burn has not been measured and it is of great importance for future studies to look at lipidomic profiles and correlate lipid profiles to outcomes after burn to hopefully gain insights into novel therapeutic approaches.
When considered as many parts working collectively and contributing to real physical manifestations, the impact of all these augmented biomedical markers is demonstrated by organ function, sepsis, and death. We found impairments in the heart, liver, and most importantly kidney, all of which are relative to burn size. Although organ function returned to normal in the majority of patients, subsets of patients remained impaired and had organ dysfunction, further supporting underlying mechanisms dividing these injury groups.
Another point worth emphasizing is that the present study cohort is heterogeneous. We did not eliminate patients on the basis of gender, inhalation injury, sepsis, multiple organ failure, or death to smoothen out the trajectory or variables measured. The inclusion criteria facilitated achieving a large adult patient population in order to be able to perform robust statistics. We propose that the development of trajectories or patterns of these detrimental outcomes will be the focus of future studies.
In summary, on the basis of our findings, we suggest that a burn injury involving more than 20% of the total body surface can cause hypermetabolic, inflammatory, and stress responses that are in some aspects as profound as in patients with over 40% TBSA burns. Despite being descriptive and retrospective, this study highlights the important elements for all global burn care providers alike to utilize in order to understand the complicated immune and metabolic responses in order to treat their patients. Collectively, treatments should focus on several aspects of the pathophysiologic events postburn, such as alleviate hyperglycemia, anti-inflammation, attenuate hypermetabolism, as well as browning.
Supplementary Material
ACKNOWLEDGMENT
We thank all the individuals who participated in this clinical trial. We also would like to thank Jason Bian and the clinical and research stuff at Ross Tilley Burn Centre.
This study was supported by Canadian Institutes of Health Research # 123336 and CFI Leader’s Opportunity Fund: Project #25407 NIH RO1 GM087285-01.
The authors declare no conflicts of interest.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s Web site (www.annalsofsurgery.com).
REFERENCES
- 1.Jeschke MG, Gauglitz GG, Kulp GA, et al. Long-term persistence of the pathophysiologic response to severe burn injury. PLoS One. 2011;6:e21245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams FN, Herndon DN, Jeschke MG. The hypermetabolic response to burn injury and interventions to modify this response. Clin Plast Surg. 2009;36:583–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jeschke MG, Patsouris D, Stanojcic M, et al. Pathophysiologic response to burns in the elderly. EBioMedicine. 2015;2:1536–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kraft R, Herndon DN, Al-Mousawi AM, et al. Burn size and survival probability in paediatric patients in modern burn care: a prospective observational cohort study. Lancet. 2012;379:1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stanojcic M, Chen P, Xiu F, et al. Impaired immune response in elderly burn patients: new insights into the immune-senescence phenotype. Ann Surg. 2016;264:195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finney SJ, Zekveld C, Elia A, et al. Glucose control and mortality in critically ill patients. JAMA. 2003;290:2041–2047. [DOI] [PubMed] [Google Scholar]
- 7.Jeschke MG, Pinto R, Herndon DN, et al. Hypoglycemia is associated with increased postburn morbidity and mortality in pediatric patients. Crit Care Med. 2014;42:1221–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mecott GA, Al-Mousawi AM, Gauglitz GG, et al. The role of hyperglycemia in burned patients: evidence-based studies. Shock. 2010;33:5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rehou S, Burnett M, Jeschke MG. Burned adults develop profound glucose intolerance. Crit Care Med. 2016;44:1059–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeschke MG, Chinkes DL, Finnerty CC, et al. Pathophysiologic response to severe burn injury. Ann Surg. 2008;248:387–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jeschke MG, Finnerty CC, Emdad F, et al. Mild obesity is protective after severe burn injury. Ann Surg. 2013;258:1119–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kraft R, Herndon DN, Finnerty CC, et al. Association of postburn fatty acids and triglycerides with clinical outcome in severely burned children. J Clin Endocrinol Metab. 2013;98:314–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kraft R, Herndon DN, Finnerty CC, et al. Occurrence of multiorgan dysfunction in pediatric burn patients: incidence and clinical outcome. Ann Surg. 2014;259:381–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greenhalgh DG, Saffle JR, Holmes JHt. et al. American Burn Association consensus conference to define sepsis and infection in burns. J Burn Care Res. 2007;28:776–790. [DOI] [PubMed] [Google Scholar]
- 15.Gauglitz GG, Herndon DN, Kulp GA, et al. Abnormal insulin sensitivity persists up to three years in pediatric patients post-burn. J Clin Endocrinol Metab. 2009;94:1656–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeschke MG, Mlcak RP, Finnerty CC, et al. Gender differences in pediatric burn patients: does it make a difference? Ann Surg. 2008;248:126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeschke MG, Norbury WB, Finnerty CC, et al. Propranolol does not increase inflammation, sepsis, or infectious episodes in severely burned children. J Trauma. 2007;62:676–681. [DOI] [PubMed] [Google Scholar]
- 18.Gauglitz GG, Halder S, Boehning DF, et al. Post-burn hepatic insulin resistance is associated with Er stress. Shock. 2010;33:299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gauglitz GG, Herndon DN, Jeschke MG. Insulin resistance postburn: underlying mechanisms and current therapeutic strategies. J Burn Care Res. 2008;29:683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gauglitz GG, Song J, Herndon DN, et al. Characterization of the inflammatory response during acute and post-acute phases after severe burn. Shock. 2008;30:503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gauglitz GG, Toliver-Kinsky TE, Williams FN, et al. Insulin increases resistance to burn wound infection-associated sepsis. Crit Care Med. 2010;38:202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jeschke MG, Mlcak RP, Finnerty CC, et al. Burn size determines the inflammatory and hypermetabolic response. Crit Care. 2007;11:R90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patsouris D, Qi P, Abdullahi A, et al. Burn induces browning of the subcutaneous white adipose tissue in mice and humans. Cell Rep. 2015;13:1538–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sidossis LS, Porter C, Saraf MK, et al. Browning of subcutaneous white adipose tissue in humans after severe adrenergic stress. Cell Metab. 2015;22:219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gross O, Thomas CJ, Guarda G, et al. The inflammasome: an integrated view. Immunol Rev. 2011;243:136–151. [DOI] [PubMed] [Google Scholar]
- 26.Stanojcic M, Chen P, Harrison RA, et al. Leukocyte infiltration and activation of the NLRP3 inflammasome in white adipose tissue following thermal injury. Crit Care Med. 2014;42:1357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Menu P, Mayor A, Zhou R, et al. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis. 2012;3:e261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iyer SS, He Q, Janczy JR, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39:311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lord JM, Midwinter MJ, Chen YF, et al. The systemic immune response to trauma: an overview of pathophysiology and treatment. Lancet. 2014; 384:1455–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shankar R, Melstrom KA Jr, Gamelli RL. Inflammation and sepsis: past, present, and the future. J Burn Care Res. 2007;28:566–571. [DOI] [PubMed] [Google Scholar]
- 31.Goodall M, Stone C, Haynes BW Jr. Urinary output of adrenaline and noradrenaline in severe thermal burns. Ann Surg. 1957;145:479–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goodall MG. Sympathetic nerve and adrenal medullary response to thermal burn. Clinical analysis of adrenaline and noradrenaline depletion. Am Surg. 1966;32:448–452. [PubMed] [Google Scholar]
- 33.Wilmore DW. Hormonal responses and their effect on metabolism. Surg Clin North Am. 1976;56:999–1018. [DOI] [PubMed] [Google Scholar]
- 34.Wilmore DW, Long JM, Mason AD Jr, et al. Catecholamines: mediator of the hypermetabolic response to thermal injury. Ann Surg. 1974;180:653–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hasselgren PO. Muscle protein metabolism during sepsis. Biochem Soc Trans. 1995;23:1019–1025. [DOI] [PubMed] [Google Scholar]
- 36.Hasselgren PO. Glucocorticoids and muscle catabolism. Curr Opin Clin Nutr Metab Care. 1999;2:201–205. [DOI] [PubMed] [Google Scholar]
- 37.Tiao G, Fagan J, Roegner V, et al. Energy-ubiquitin-dependent muscle proteolysis during sepsis in rats is regulated by glucocorticoids. J Clin Invest. 1996;97:339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang L, Luo GJ, Wang JJ, et al. Dexamethasone stimulates proteasome- and calcium-dependent proteolysis in cultured L6 myotubes. Shock. 1998;10:298–306. [DOI] [PubMed] [Google Scholar]
- 39.Barret JP, Jeschke MG, Herndon DN. Fatty infiltration of the liver in severely burned pediatric patients: autopsy findings and clinical implications. J Trauma. 2001;51:736–739. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.