

Summary:
Antibody-secreting plasma cells are a central component of short- and long-term adaptive immunity. Yet many fundamental questions about how activated B cells decide to yield functional plasma cells have yet to be answered. Likewise, the biochemical processes underpinning the ability of plasma cells to generate and secrete large numbers of antibodies, the capacity of some plasma cell to sustain antibody secretion, presumably without interruption, for decades, and the capacity of long-lived plasma cells to avoid apoptosis despite the high energy demands associated with sustained robust antibody synthesis and secretion each remain mysterious processes. Our objective here is to review what is currently known about these processes with an emphasis on the earliest phases of plasma cell genesis. Along the way we will work towards developing a model that ties the biochemistry of plasma cell function and survival. The chief idea imbedded in this model is that progress towards understanding plasma cell survival mechanisms may require increased focus on the unique cell autonomous processes inherent in plasma cell differentiation and function.
Introduction
To make a plasma cell from a naïve B cell is an especially involved process. Consider two pictures: a resting B cell characterized by a proportionately large nucleus and little cytoplasm, next to a plasma cell with a massively expanded endoplasmic reticulum (ER) and highly operational Golgi apparatus. It becomes obvious that the conversion of a B cell into a plasma cell requires a comprehensive re-working of cytoplasmic organelle structures, yielding what we view as cellular antibody secretion machines. Indeed, past studies suggest that the average plasma cell secretes some 10,000 antibody molecules every second1–3. The secretion of copious amounts of antibodies is thought to be necessary for achieving serum antibody concentrations that are protective. Indeed, serum antibodies play critical protective roles against numerous microbes, and the chief protective mechanism induced by most vaccines4,5. Hence high throughput antibody synthesis and secretion is inherent to plasma cell function. Remarkably however, little is known about how activated B cells implement and regulate the biochemical pathways that drive early plasma cell differentiation and full-blown plasma cell function.
Highly durable serum antibody titers are also a central feature of effective adaptive responses and considered one of several facets of immune memory. Because serum antibodies possess a half-life of only 0.5–8 days, depending on heavy chain class6,7, it is widely believed that long-term maintenance of serum antibody titers reflects the activity of equally long-lived plasma cells. Thus, while it is also generally believed that the majority of newborn plasma cells die days of their initial induction, others must survive for many decades while presumably maintaining high throughput antibody synthesis throughout this time frame. Importantly however, little is also known about how mature long-lived plasma cells receive, interpret, and integrate extrinsic and internal signals needed to avoid apoptosis while also maintaining robust antibody secretion.
How is the antibody secretion apparatus initiated and optimized beginning in activated B cells and sustained in long-lived plasma cells? The purpose of this review is to explore what is known about the biochemistry surrounding the first question with hopes of developing useful ideas about how to think why some plasma cells die while others do not.
Outside forces
We begin by considering the earliest phases of a primary antibody response. In adults newborn naïve B cells leave the bone marrow (BM) in a state of metabolic quiescence. This relatively inert metabolic state is disrupted when B cells are engaged by antigen together with signals delivered by T cells and/or ligands for certain toll-like receptors (TLRs). A chief outcome of successful B cell activation is several rounds of cell division. Additional outcomes include class switch recombination (CSR), plasma cell differentiation, migration into germinal centers (GCs), and for many cells death. CSR and plasma cell differentiation are each a common outcome available to responding B cells within the first 5–6 days of a typical response, before the onset of the venerable GC reaction. GC B cells and many memory B cells also retain the potential to undergo CSR or yield one or more plasma cells. Further, emerging data suggest that memory B cells can exist in a variety of distinct functional states defined by differences in their propensity to yield additional waves of plasma cells8,9. For T cell dependent responses, the classic view is that early antibody responses are induced upon ligation of surface immunoglobulin on naïve B cells together with additional cell surface receptors such as CD40, the latter of which binds to CD40-ligand (also known as CD154) on activated T cells. It should also be noted however that plasma cell differentiation can be induced by CD40 or TLR engagement without obvious surface Ig engagement, especially upon co-stimulation of a variety of cytokines, and these signals can be sufficient to generate large numbers of plasma cells10,11.
More mysterious are the oft overlooked responses to T cell independent antigens. The classic view is that there are two categories of T-independent immunogens. It should be emphasized that members of each category of T-independent immunogen possess the capacity to induce a full plasma cell program that including long-lived cells12–15, albeit typically with far fewer resulting plasma cells due to the absence of sustained clonal expansion within GCs. Antigens enriched for TLR ligands such as LPS are especially effective at inducing robust and rapid antibody responses and have been termed type 1 T-independent antigens. By contrast type 2 T-independent antigens possess dense arrays of a common epitope that lead to extensive surface BCR aggregation, which apparently causes many events associated with early B cell activation including plasma cell induction to occur without input from antigen-specific T cells. Thus, either TLR signaling or aggregation of surface BCRs are sufficient to drive induction of a complete plasma cell program.
When considering what it takes to induce differentiation, it should also be noted that subsets of naïve B cells are able to produce plasma cells faster and with fewer activation signals compared to the average peripheral B cell. This idea is consistent with the broader view that certain subpopulations of naïve B and T cells are poised for rapid effector function. It is likely that the development of such cells is highly advantageous for rapid short-term protection against a large array of microbes. Examples of naïve B cells that are poised to produce plasma cells include B1 and marginal zone (MZ) B cells, the latter of which localizes to the blood rich marginal sinus of the spleen where they are easily exposed to blood-borne bacterial antigens. As we will discuss in greater detail below, it turns out that the unique biochemical state of MZ B cells provides useful clues about the signals controlling the earliest phases of plasma cell induction.
Lost options
While it is clear that B cell blasts can experience several distinct outcomes within 6 days or so of initial activation, how proliferative daughter cells are partitioned into one or more outcomes is poorly understood. Importantly, each outcome requires distinct cell autonomous mechanisms16, raising questions about how B cell blasts coordinate the regulation of each process. In can be noted however that early class-switched B cells can yield either memory B cells or plasma cells, as can their non-switched IgM counterparts, and the same can be said for GC B cells. However, because CSR requires the enzyme Activation Induced Deaminase (AID), and AID expression is suppressed early in plasma cell differentiation17, plasma cells cannot undergo additional rounds of CSR. This specific idea extends to several additional aspects of B cell biology. Naïve and memory B cells use BCR, CD40, and/or TLR signals to proliferate, alter their migration patterns, and/or acquire different functions. During and after these events B cell retain the option of seeding additional GCs, undergoing additional rounds of CSR, and returning to quiescence as memory B cells. By contrast, plasma cell differentiation is accompanied by the loss of each of these options and cessation of proliferation, presumably to allow full focus on the energetic demands inherent in robust antibody synthesis and secretion. We will return to this issue after we first consider what is known, and what is unknown, about the biochemical coordination of the plasma cell fate.
The plasma cell transcriptional program
Much of the focus on the early plasma cell differentiation program has centered on its unique transcriptional networks. It is now well appreciated that the generation of fully functional plasma cells requires the activity of at least three transcription factors: Blimp1, IRF4, and Xbp1. These proteins are components of a unique network of transcriptional factors that also include at least two regulators that oppose plasma cell differentiation by repressing one or more plasma cell requisite regulator. Because the transcriptional regulation of the plasma cell fate is the chief focus of several other reviews in this issue (see Patterson et al and Trezise and Nutt, this issue), we will keep our comments on transcriptional networks in plasma cell differentiation brief.
Blimp1 functions as both a transcriptional activator and repressor, up-regulating genes needed for robust antibody expression and other aspects of plasma cell function, but also suppressing transcription for genes associated with various aspects of B cell identity18. Notably, Blimp1 expression increases in a step wise manner as early B220+ plasma cells in the spleen and BM give way to more mature long-lived B220− plasma cells, and these changes occur in concert with progressive increases in Ig protein production (see19,20 and Figure 1). These data are consistent with the notion that Blimp1 acts in part to increase Ig gene transcription, and thus plays a central albeit indirect role in the biochemical processes whereby plasma cells adapt to increased protein production rates. We will elaborate on this point below.
Figure 1. Coordinated increases in intracellular Blimp1 and Igkappa levels.
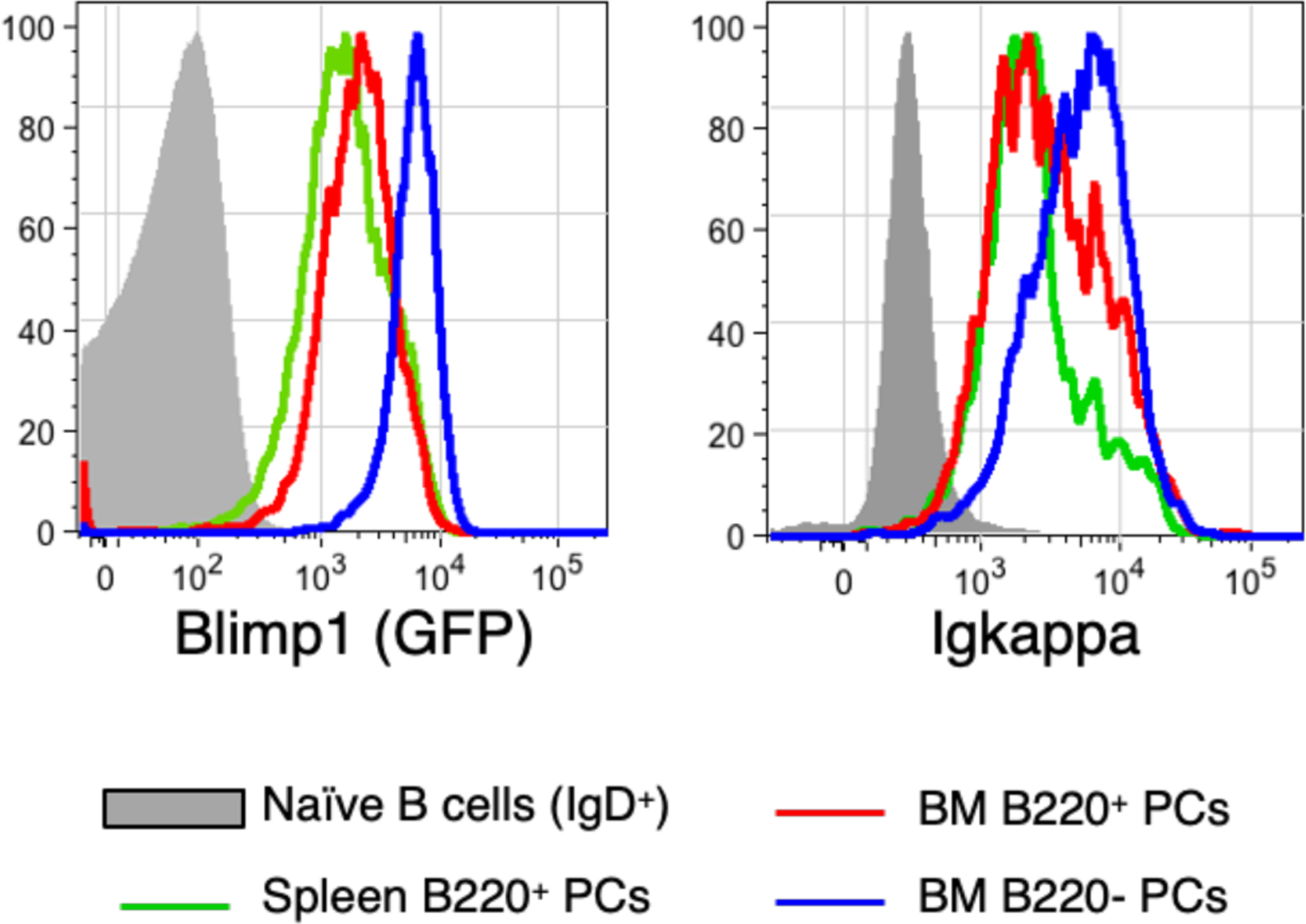
Cells from an adult B6.Blimp1+/GFP mouse were stained to resolve newborn (B220+) and mature long-lived (B220−) plasma cells (CD138+ Sca1+) in the spleen (for B220+ cells alone) or BM.
The role of IRF4 in plasma cell differentiation has been more challenging to study, owing to its role in other B-lineage cells including in GC B cells, and its capacity to evoke district transcriptional programs based on differences in intracellular IRF4 concentration21. Nonetheless three observations indicate that IRF4 is uniquely and intimately associated with plasma cell survival mechanisms. First, induced inhibition of IRF4 expression in myeloma cell lines in vitro causes their rapid demise. Second, induced mutation of IRF4 in mature plasma cells in vivo also caused rapid loss in BM plasma cell numbers, in contrast to the impact of mutating Blimp1. Importantly, how IRF4 promotes plasma cell survival has not been uncovered. Because normal mature plasma cell survival requires the BAFF family cytokine receptor BCMA, it is tempting at first glance to consider the possibility that BCMA expression and/or function requires IRF4 activity. However, BCMA mutation in myeloma cell lines does not appear to cause their profound demise in a manner that parallels the impact of eliminating IRF4. Therefore, IRF4 may promote survival through an alternative pathway that is central to the cell autonomous mechanisms employed by mature plasma cells to avoid apoptosis. And finally, data from Boss and colleagues suggests that IRF4 orchestrates relevant transcriptional changes before Blmp1 induction22. It is clear based on these observations that improved understanding on the mechanistic role played by IRF4 at different stages of B cell and plasma cell differentiation should be a priority.
Too many proteins?
Increased synthesis of Ig heavy and light chains and associated proteins is thought to cause a stress response centered on the ER, which in turn changes transcription rates for numerous genes, many of which encode proteins that allow activated B cells or early plasma cells to adapt by increasing the mass and function of the ER, the Golgi, and other pathways and structures needed to increase protein secretion. This process is called the Unfolded Protein Response (UPR), the induction of which is a common feature of cells whose function is intricately linked to high throughput protein synthesis. Notably, homologs for many UPR key regulators have been characterized in eukaryotic cells dating back to yeast23,24. The UPR in mammalian cells is regulated by three ER sensors: IRE1, ATF6, and PERK. Among these, only null mutation of IRE1 has an obvious negative impact on induction of plasma cells25–27, which we have to say is a bit surprising. IRE1 serves to both sense ER protein overload and regulate the function of the Xbp1 transcription factor. In turn, IRE1-driven activation allows Xbp1 to amplify transcription of a wide array of targets associated with the UPR26,28.
How IRE1 regulates Xbp1 is both highly conserved and quite remarkable. To generate a functional Xbp1 transcriptional activator, full length Xbp1 transcripts must undergo IRE1-mediated splicing29. Aggregation of IRE1 proteins in the ER cause its conversion into an RNA splicing endoribonuclease, which then acts to convert full-length Xbp1 transcripts into mRNAs that encode a functional transcriptional regulator. Although there have been hints of a few others, the chief target for IRE1 is Xbp1. Hence, accumulation of poorly folded proteins within the ER due to increased antibody synthesis causes activation of IRE1, which then plays an essential role in sustaining plasma cell function by generating a functional Xbp1 transcription factor. This process cements UPR function in established plasma cells.
UPR onset in waves
Since it is thought that Xbp1 is the true workhorse in building the ER of the nascent plasma cell, the field has termed this Xbp1-dominated process the physiological UPR of plasma cell differentiation to differentiate from the stress response UPR that can occur in all eukaryotic cells30. It’s becoming clear that this specialized UPR does not occur solely as reaction to increased immunoglobulin sensed by IRE1 and transmitted through Xbp1. Instead, activated B cells enter a phase of early Xbp1-independent ER remodeling that in effect sets the stage for an Xbp1-centric response to massive immunoglobulin translation (see Figure 2). Initially and quite rapidly the cell upregulates expression of UPR-associated genes required for sustaining the processes of mRNA transcription and protein translation31. The upregulation of this gene cohort achieves maximal levels early in the differentiation process, is also observed in activated B cell blasts that have not experienced plasma cell inductive stimuli, and includes genes that encode amino acid transporters, tRNA synthetases, ribosomal subunits, and other regulators of the processes necessary for transcription and translation (Figure 3). Additionally, transcription of genes necessary for the regulation of these processes as well as the UPR itself is increased. Over a period of time, which for most B cell blasts can be days, a second cohort of UPR-associated genes is activated as B cells move closer to the plasma cell fate. These include genes encoding a large number of both regulatory and protein folding chaperones, genes required for ER-Golgi transit and function, protein isomerases and regulators of ER-associated protein degradation (ERAD). Importantly, these changes in gene expression changes occur before induced transcription of the Prdm1 locus, which encodes Blimp1, and also prior to bona fide activation of the Xbp1-dependent UPR. Surprisingly, transcription of the entire plasma cell specific UPR geneset is increased before Xbp1 splicing. Once these initial changes prepare the differentiating cell, Prdm1 transcription is increased to levels required for full plasma cell function. Full implementation of Blimp1 function causes amplification of a wide variety of plasma cell affiliated genes including the Ig loci. Increased Ig gene transcription leads to increases in Ig translation, which in turn causes IRE1-mediated Xbp1 activation and full implementation of the UPR (Figure 2).
Figure 2. Stepwise UPR implementation across B cell differentiation.
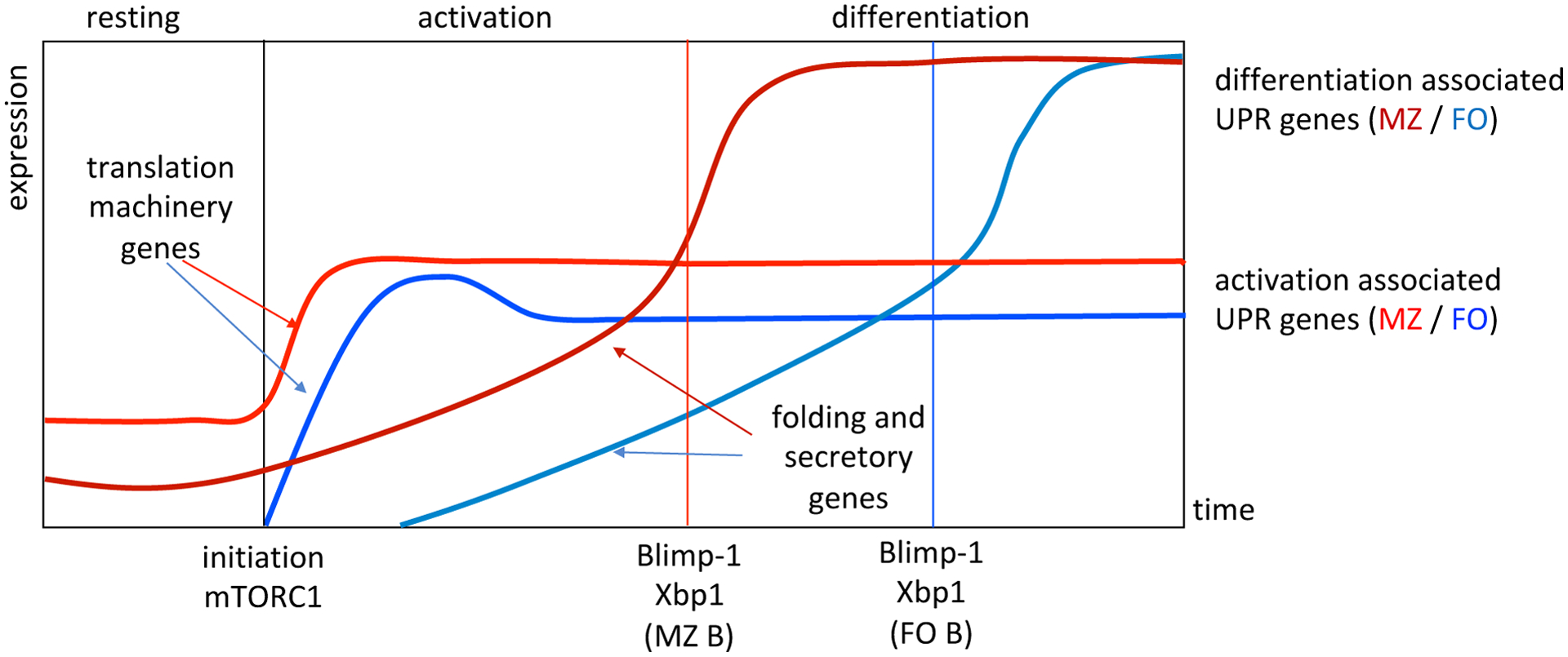
This schematic illustrates the distinct segments of gene expression changes that occur in the earliest phases of plasma cell differentiation. Initiation marks the start of stimulus and the rapid induction of genes required for building the translation machinery, which is followed by the more gradual onset of genes required for building the secretory apparatus. Indicated are the relative times to full Blimp1 induction and Xbp1 splicing for both follicular and MZ B cells. Note that basal expression of these genes occurs along with basal mTORC1 activity in MZ B cells yielding faster differentiation kinetics.
Figure 3. Gene expression data for early plasma cell UPR-associated gene expression.
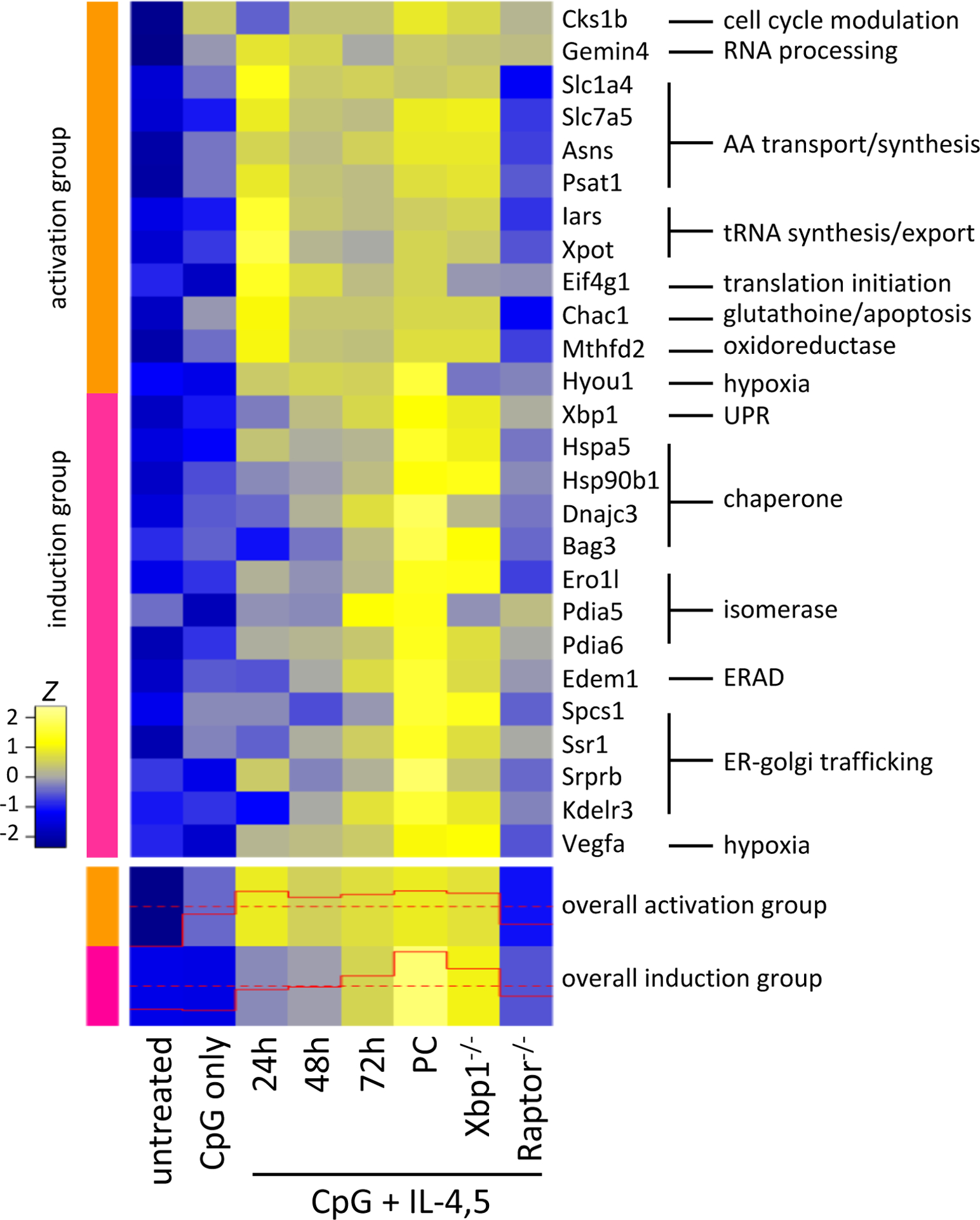
(top panel) Selected genes from activation associated UPR hallmark genes (orange) and plasma cell associated UPR hallmark genes (magenta) are displayed as the z score across each row. 3–5 individual mice are averaged for each group shown. In all groups, follicular B cells were sorted from either Cre expressing control mice or mice with the indicated genes floxed out. General gene functional categories are indicated at right. (bottom panel) Gene expression for all genes included in the activation and plasma cell induction UPR genesets are summarized for the general trend of expression with the levels illustrated in the red trace. From31.
Plasma cell wannabes
The majority of peripheral B cells are termed follicular B cells due to their propensity to recirculate through follicular structures in peripheral lymphoid organs32. Follicular B cells are thought to dominate responses to proteinaceous antigens by generating GCs as well as short- and long-lived plasma cells33. By contrast, relatively small numbers of naïve B cells possess properties reminiscent of innate immune function. These B cells yield plasma cells efficiently in response to TLR signaling and with increased kinetics relative to follicular B cells34. There is also a propensity for innate-like B cells to home to unique sites in the body where they may play unique protective roles. Perhaps most notably, MZ B cells are actively retained in the blood-rich marginal sinus of the spleen35. Thus, MZ B cells play a unique role in protection against blood-born microbes, due to both their propensity to lodge in the marginal sinus of the spleen and the efficiency with which they generate plasma cells36.
How MZ B cells become poised to produce plasma cells and how microbial signals are translated into rapid plasma cell induction is largely unknown. However, in this regard, we were intrigued to learn that resting MZ B cells possess a modestly expanded ER37. Due to this and other observations we took on a comprehensive comparison of gene expression in freshly isolated follicular and MZ B cells. Our results revealed that resting MZ B cells possess increased transcript abundance for numerous genes associated with antibody secretion and the UPR31,37. Importantly however, at baseline MZ B cells do not secrete antibody and have no apparent change in unprocessed/processed Xbp1 mRNA nor Xbp1s protein compared with naïve follicular B cells31,37. Indeed, induced deletion of Xbp1 in MZ B cells failed to change their enrichment of UPR-associated gene expression, with only 5 total genes effected31. These results indicate that at steady state the canonical Xbp1-driven UPR program is not engaged in MZ B cells. Consistent with this view, MZ B cells do in fact retain the capacity to yield non-plasmacytic B-lineage cells including GC B cells38. Nonetheless, it seems fair to conclude that during development MZ B cells acquire a unique biochemical state that confers a propensity for rapid induction of effector function. Furthermore, because both the development, unique gene expression patterns, and unique functions of MZ B cells remain intact in germ free mice, it is highly likely that their unique biochemical state is not the result of chronic TLR stimulation by microbes or microbial products.
Diverse roles for mTORC1
Emerging data suggest unique roles for the mTOR kinase complex known as mTORC1 in several phases of plasma cell induction and long-term function. mTOR (mechanistic target of rapamycin) is a highly conserved serine/threonine kinase that exists in two distinct complexes: mTORC1, which is inhibited by rapamycin, and mTORC2, which is not. mTORC1 plays an important role in promoting translation in response to external cell growth inducing signals, whereas mTORC2 is often associated with cell survival mechanisms via the additional kinase Akt (also called protein kinase B)39. mTORC1 has been connected to each wave of UPR induction including in activated B cells and fully functional mature plasma cells. Interestingly, mTORC1 has also been connected via poorly understood mechanisms to changes in gene expression required for early UPR and plasma cell induction.
It appears that fine tuning of mTORC1 activity to different levels at different stages of activation and differentiation is an essential aspect of B cell lifestyle. For starters, many of the UPR-related genes expressed in MZ B cells are also canonical indicators of mTORC1 activation. Targets of the mTORC1 signaling program increased in MZ B cells include genes required for synthesis and import of biomolecules required for transcription and protein translation. Unlike the full UPR, basal levels of mTORC1 activation are apparent in steady state MZ B cells and is independent of PI3K signaling and tied to extracellular nutrient availability31,40. Sintes et al41 showed recently a connection between TACI signaling through MyD88 and mTOR activation in MZ B cells which certainly seems to amplify TLR signaling through mTOR. However, as alluded to above we have observed that both germ free and MyD88−/− mice have similar numbers of MZ B cells with unchanged mTOR activity, suggesting that increased basal mTOR activation in MZ B may require another mechanism (our unpublished data). Consistent with MZ B cells using mTORC1 to drive expression of genes needed for building of the secretory apparatus, relief of inhibition of mTOR via mutation Tsc1, a conical negative regulator of mTORC1, can complement Xbp1 mutation to allow for plasma cell induction42. Furthermore, upon induced mutation of Tsc1 naive follicular B cells acquire gene expression patterns that mirror WT MZ B cells and upon stimulation produce plasma cells with increased kinetics (our unpublished data). Alternatively, and somewhat mysteriously, chronic increases in mTORC1 activity due to long-term Tsc1 deficiency causes the specific loss of MZ B cells43.
Other data solidify the notion that innate-like B cells including those in the MZ B cell pool possess a distinct metabolic state. For MZ B cells one aspect of this unique state centers on heightened lipid uptake mediated by the scavenger receptor CD36, a process which appears important for importing extracellular low-density lipid. Expression of several members of the aryl hydrocarbon receptor pathway that regulate the metabolism of exogenous sterols are also significantly increased in MZ B cells (Ahr, Cyp39a1, Cyp8b1, Fdx1, Pomc, Fdxr, Cyp11a131. Further evidence that this pathway is engaged in resting MZ B cells centers on the loss of Glutathione peroxidase 4 (Gpx4), which for MZ B cells causes lipid peroxidation and ferroptosis44. Furthermore, CD36 transcript abundance is quickly increased upon activation of follicular B cells to levels observed in naive MZ B cells and loss of CD36 impairs both T-dependent and T–independent humoral responses 31,45. Together these studies suggest that differentiation-poised MZ B cells utilize alternative mTORC1-regulated pathways for nutrient uptake to initiate the building of organelles necessary to initiate rapid expansion of the secretory apparatus and plasma cell function.
The mTORC1/UPR axis in GC B cells
In activated follicular B cells mTORC1 activity fosters the transcription of many genes that are considered UPR targets in the context of ER stress, yet apparently with minimal ER stress. In this context mTORC1 acts before Blimp1 induction and increased Ig translation to facilitate the expression of numerous UPR-related genes, thus establishing a primed state that allows for rapid and comprehensive UPR induction by Xbp1. This is very similar to how basal mTORC1 signaling in resting MZ B cells activates a variety of UPR-associated genes, priming cells for rapid ER expansion and plasma cell differentiation as discussed above.
mTORC1 plays additional unique roles for GC B cells such as in promoting the expression of the GC-affiliated transcription factor BCL646. Further understanding of mTORC1 function in GC B cell maturation and selection was provided recently by Ersching and colleagues47. The latter work showed that mTORC1 signaling initiated from T cell mediated CD40L-CD40 interactions drives anabolic pathways needed to prepare for rapid cell division as GC light zone B cells prepare to enter or re-enter the GC dark zone. Within the dark zone B cells are likely to experience further rounds of mitosis and somatic hypermutation. For light zone B cells increased mTORC1 activity coincided with increased albeit transient Myc expression, and mTORC1 signaling appeared to be dispensable once cells initiate cell division. Thus, it is attractive to consider that relatively strong Myc and mTORC1 signaling, due to prolonged BCR-Ag and/or CD40-CD40L signaling for GC B cells bearing high affinity BCR, cause increased UPR-related gene expression in cohorts of cells destined to yield plasma cells. Interestingly however, it should also be noted that unrestrained mTORC1 activity can abbreviate the effects of T cell-mediated selection, causing the emergence of greater numbers of plasma cells characterized by the secretion of low affinity antibodies. Thus, within the GC mTORC1 activity must also be tuned to appropriate levels.
The mTORC1-UPR pathway in long-lived plasma cells?
We probed for evidence of ongoing mTORC1 activity in long-lived plasma cells by inoculating immunized wild type or lupus-prone mice with rapamycin. We were surprised to learn that long-term drug-mediated inhibition of mTORC1 arrests antibody synthesis in long-lived plasma cells without causing their overt death. Instead, mTORC1-inhibited plasma cells appeared to adapt to interrupted protein synthesis by increasing autophagy. Remarkably, surviving plasma cells rapidly resumed antibody synthesis upon cessation of rapamycin treatment, again highlighting the resilience of these cells. Furthermore, it has been shown that newly formed plasmablasts and long-lived plasma cells utilize autophagy at different rates and that this appears to be tied to more efficient inhibition of autophagy through mTORC1 signaling. This would further a continuum of mTORC1 establishing the secretory program gradually giving way to Blimp1-Xbp1 amplified and sustained program even after differentiation into maturity48–50. These data also suggest these changes in metabolism feed into the ability to continue high levels of protein production and maximize adaptability and survival. Nonetheless, these results show that mTORC1-regulated protein translation is as expected integral to Ig synthesis and secretion, which in turn would be expected to drive continuous UPR activation.
It should be noted however that these studies do not test directly the role of the UPR in long-term plasma cell survival or function. In fact, surprisingly Xbp1 mutation in mature plasma cells does not appear to cause their rapid demise, and this result allows for the separation of the mechanisms controlling differentiation from processes dedicated to the antibody secretion machinery51. These are additional areas where more work is needed. Given that the rate of Ig protein synthesis is maximal in mature plasma cells (see Figure 1), one must consider the possibility that mature long-lived plasma cells employ alternative mechanisms to maintain engagement of the UPR. In this regard, it is tempting to speculate that the alternative ER sensors PERK and ATF6 play a more dominant role in mature plasma cell function relative to their apparent minimal role managing the acute ER stress inherent to early plasma cell induction. It is also important to consider that, though PERK-deficient animals have comparable numbers of plasma cells, there is evidence that the mTORC1-regulated gene chaperone P58IPK is a direct inhibitor of PERK. This provides another mechanism whereby mTOR is important for enforcing an IRE1/Xbp1 focused UPR and also a potential for PERK activity affecting plasma cell survival and function in times of dysregulated mTOR activity25,52.
Inside forces: plasma cell induction and survival
A fair amount of work has been devoted to understanding how cell extrinsic ligands that induce antibody responses or promote long-term plasma cell survival. By contrast, far less is known about the cell intrinsic processes controlling the number of plasma cells produced during early phases of antibody responses. Ultimately, this number is likely to be influenced by several processes including early B cell (and T cell) clonal burst size, the timing and efficiency with which proliferative B cells enact a plasma cell differentiation program, and the number of activated B cells and newborn plasma cells that die. Thus, it appears worthwhile to consider how different cell autonomous processed affect each outcome during the earliest phases of T-dependent and T-independent antibody responses. Such information may also ultimately shed light on the processes whereby mature plasma cells persist as long-lived cells. This idea gains support from work revealing different types of antigen/adjuvant combinations influence the degree to which the transcription factor Zbtb20 influences the longevity of mature BM plasma cells53.
Inside forces: clonal burst size
One poorly understood aspect of early plasma cell differentiation centers on how daughter cells derived from each activated B cell blast are partitioned into distinct fates. Emerging data lend to the notion that proliferation, CSR, and plasma cell differentiation are each controlled by separate cell autonomous processes, often operating in parallel in individual cells. Consequently, the timing with which activated B cells complete CSR versus plasma cell differentiation is likely to vary from clone to clone and even cell to cell within a responding clone. It has been argued that clone-to-clone variability in outcome and outcome timing is advantageous because it ensures the functional diversity of immune responses: hence for B-lineage cells some cells will yield plasma cells, while others seed a GC and/or exit activation programs to become antigen experienced memory cells.
The degree to which cell extrinsic factors influence the number of plasma cells produced is an open question. It has been proposed that the number of B cell blasts partitioned to the plasma cell fate is influenced by the duration of the interaction between B cell and antigen, with sustained high-affinity Ag-BCR interactions driving the genesis of greater numbers of plasma cells. However, this remains a cloudy area, in part due to the low frequency of antigen responsive B cells during the early phases of a response and the lack of experimental systems to define and vary BCR-Ag affinities across polyclonal naïve B cells populations. Indeed, earlier experimental data centered on monoclonal B cell populations first interpretated to reflect a direct connection between BCR-Ag affinities and numbers of early plasma cells produced have been reinterpreted to suggest that sustained BCR aggregation is more likely to increase B cell and plasma cell clonal expansion, without directly increasing the proportion of daughter B cells that yield plasma cells54,55.
It has also been proposed that the length of time required to complete each round of mitosis naturally varies for clone to clone. If so, then the number of cell divisions experienced within each clone during a specified time frame would also vary, increasing with faster division rates. This idea stems from careful study of the length of time individual cells can be found in the G0/G1 versus S/G2M phases of the cell cycle after in vitro stimulation56,57, and from work suggesting that subtle increases in the availability of the MYC proto-oncogene cause increased division numbers per unit time58. Although these ideas would be enforced by experimentation in vivo, the impact of distinct intraclonal division rates on the number of plasma cells is worth considering. Specifically, if the duration needed to complete mitosis differs for each clone, and the probability that each daughter cell will experience the plasma cell fate is identical for each clone59,60, the number of early plasma cells produced would be expected to increase in proportion to the number of cell divisions experienced by each respective clone per unit time. Further assuming that the forces controlling long-term survival apply equally to all newborn plasma cells, naturally proliferative clones might also contribute to a greater degree to long-lived plasma cell pools.
Outside and inside forces controlling plasma cell survival
It is widely believed that the majority of newborn plasma cells die within days of their generation. Why so many nascent plasma cells die and the relationship of this process to survival mechanisms for mature long-lived plasma cells is unknown. Early plasma cells are believed to proliferate for an undetermined but relatively abbreviated time frame before dying or integrating as non-dividing cells into unique microniches in the BM. Mature in the BM plasma cells must gain access to the pro-survival cytokines APRIL or BAFF, each of which stimulates mitochondrial pro-survival pathways overseen by the global anti-apoptotic protein Mcl1. Mature plasma cells sense external APRIL and BAFF availability via the plasma cell restricted BAFF-family receptor BCMA61,62. Access to APRIL and perhaps BAFF appears to be enforced through the tethering of mature plasma cells to specialized stromal cells in the BM via the integrins LFA-1 (lymphocyte-function-associated antigen, αLβ2, CD11a/CD18) and VLA-4 (very late antigen, CD49d/CD29). Recent results suggest that plasma cell interactions with BM stromal cells augments both PI-3-kinase and BCMA signaling to allow mature plasma cells to suppress apoptotic pathways associated with chronic ER stress63. This result is important because it solidifies the notion that mature plasma cells must sidestep UPR-associated death pathways and because it provides insights into how long-lived plasma cells achieve this end game.
It is tempting, perhaps even fashionable, to imagine that the overwhelming death rate ascribed to newborn plasma cells, both outside and inside the BM, is due to the failure of these cells to gain access to BCMA ligands and pro-survival interactions with key BM stromal elements. However, there are other possibilities. We propose an alternative model wherein immature plasma cell death reflects the failure of many cells to adapt to acute ER stress. In this regard, hyperacute or unresolved chronic ER stress can cause apoptosis by increasing availability of Bim, a pro-apoptotic Bcl2 family member. ER stress activates the UPR transcription factor CHOP, which up-regulates Bim expression to prime cells for apoptosis. In viable cells Bim is sequestered by Bcl2 and other anti-apoptotic proteins including Mcl1, but upon increased abundance, Bim is unleased to activate the death effector molecules Bak and Bax. Thus, we propose that newborn and mature plasma cells avoid apoptosis via two highly distinct mechanisms: Many newborn plasma cells die because they fail to adapt to the hyperacute stress associated with the initial upregulation of Ig heavy and light chain translation. In this scenario, which cells avoid apoptosis may be a stochastic process. Hence, while differentiation is triggered initially by antigen and other ligands, the resulting signals may set forth a cell autonomous ER-based death pathway for many newborn plasma cells. In contrast, mature plasma cells in the BM and in potentially other sites such as the spleen64 avoid apoptosis by utilizing BCMA signaling, influenced heavily by availability of APRIL and/or BAFF. This also provides mechanism by which circulating plasma blasts appear to be universally short lived, the increased pro-apoptotic signaling that comes with massive UPR signaling must be met with increases in survival sequencing from extrinsic stromal cues that are not met in the circulation. Therefore, while ultimately both pathways feed into processes based in the mitochondria, the cell intrinsic and cell extrinsic events regulating these processes may be quite distinct. Therefore, while ultimately both pathways feed into processes based in the mitochondria, the cell intrinsic and cell extrinsic events regulating these processes may be quite distinct.
Conclusions
In this review, we have put forth ideas and described experiments centered on the earliest phases of plasma cell differentiation. Our view that this is a critical yet understudied phase of the antibody response, and that the processes inherent in plasma cell differentiation and triggered by T-independent or T-dependent antigens will one day be understood well enough to allow manipulation of short- and long-term antibody titers. Such information may pave the way not only for arresting the production of pathogenic and otherwise problematic antibodies in autoimmunity and transplant rejection, but also for intentional amplification of protective antibody titers.
Acknowledgments:
We thank all members of our laboratory for insightful discussions on all aspects of plasma cell biology. Our work in this area is supported by National Institutes of Health (NIH) grants National Institutes of Health (NIH) grants T32-CA009140 to BG and R01-AI097590 and R01-AI154932 to DA.
Footnotes
The authors declare that there is no conflict of interest.
References
- 1.Helmreich E, Kern M, Eisen HN. The secretion of antibody by isolated lymph node cells. J Biol Chem. 1961;236:464–473. [PubMed] [Google Scholar]
- 2.Hibi T, Dosch HM. Limiting dilution analysis of the B cell compartment in human bone marrow. Eur J Immunol. 1986;16(2):139–145. [DOI] [PubMed] [Google Scholar]
- 3.Wiest DL, Burkhardt JK, Hester S, Hortsch M, Meyer DI, Argon Y. Membrane biogenesis during B cell differentiation: most endoplasmic reticulum proteins are expressed coordinately. The Journal of cell biology. 1990;110(5):1501–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amanna IJ, Carlson NE, Slifka MK. Duration of humoral immunity to common viral and vaccine antigens. N Engl J Med. 2007;357(19):1903–1915. [DOI] [PubMed] [Google Scholar]
- 5.Amanna IJ, Slifka MK. Mechanisms that determine plasma cell lifespan and the duration of humoral immunity. Immunol Rev. 2010;236:125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vieira P, Rajewsky K. The bulk of endogenously produced IgG2a is eliminated from the serum of adult C57BL/6 mice with a half-life of 6–8 days. Eur J Immunol. 1986;16(7):871–874. [DOI] [PubMed] [Google Scholar]
- 7.Vieira P, Rajewsky K. The half-lives of serum immunoglobulins in adult mice. Eur J Immunol. 1988;18(2):313–316. [DOI] [PubMed] [Google Scholar]
- 8.Zuccarino-Catania GV, Sadanand S, Weisel FJ, et al. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat Immunol. 2014;15(7):631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson JL, Rosenthal RL, Knox JJ, et al. The Transcription Factor T-bet Resolves Memory B Cell Subsets with Distinct Tissue Distributions and Antibody Specificities in Mice and Humans. Immunity. 2020;52(5):842–855 e846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pone EJ, Lou Z, Lam T, et al. B cell TLR1/2, TLR4, TLR7 and TLR9 interact in induction of class switch DNA recombination: modulation by BCR and CD40, and relevance to T-independent antibody responses. Autoimmunity. 2015;48(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boeglin E, Smulski CR, Brun S, Milosevic S, Schneider P, Fournel S. Toll-like receptor agonists synergize with CD40L to induce either proliferation or plasma cell differentiation of mouse B cells. PLoS One. 2011;6(10):e25542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bortnick A, Chernova I, Quinn WJ 3rd, Mugnier M, Cancro MP, Allman D. Long-lived bone marrow plasma cells are induced early in response to T cell-independent or T cell-dependent antigens. J Immunol. 2012;188(11):5389–5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foote JB, Mahmoud TI, Vale AM, Kearney JF. Long-term maintenance of polysaccharide-specific antibodies by IgM-secreting cells. J Immunol. 2012;188(1):57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taillardet M, Haffar G, Mondiere P, et al. The thymus-independent immunity conferred by a pneumococcal polysaccharide is mediated by long-lived plasma cells. Blood. 2009;114(20):4432–4440. [DOI] [PubMed] [Google Scholar]
- 15.Garcia de Vinuesa C, O’Leary P, Sze DM, Toellner KM, MacLennan IC. T-independent type 2 antigens induce B cell proliferation in multiple splenic sites, but exponential growth is confined to extrafollicular foci. Eur J Immunol. 1999;29(4):1314–1323. [DOI] [PubMed] [Google Scholar]
- 16.Hasbold J, Corcoran LM, Tarlinton DM, Tangye SG, Hodgkin PD. Evidence from the generation of immunoglobulin G-secreting cells that stochastic mechanisms regulate lymphocyte differentiation. Nat Immunol. 2004;5(1):55–63. [DOI] [PubMed] [Google Scholar]
- 17.Xu Z, Pone EJ, Al-Qahtani A, Park SR, Zan H, Casali P. Regulation of aicda expression and AID activity: relevance to somatic hypermutation and class switch DNA recombination. Crit Rev Immunol. 2007;27(4):367–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaffer AL, Lin KI, Kuo TC, et al. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity. 2002;17(1):51–62. [DOI] [PubMed] [Google Scholar]
- 19.Chernova I, Jones DD, Wilmore JR, et al. Lasting antibody responses are mediated by a combination of newly formed and established bone marrow plasma cells drawn from clonally distinct precursors. J Immunol. 2014;193(10):4971–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kallies A, Hasbold J, Tarlinton DM, et al. Plasma cell ontogeny defined by quantitative changes in blimp-1 expression. J Exp Med. 2004;200(8):967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity. 2006;25(2):225–236. [DOI] [PubMed] [Google Scholar]
- 22.Scharer CD, Patterson DG, Mi T, Price MJ, Hicks SL, Boss JM. Antibody-secreting cell destiny emerges during the initial stages of B-cell activation. Nat Commun. 2020;11(1):3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–1086. [DOI] [PubMed] [Google Scholar]
- 24.Korennykh AV, Egea PF, Korostelev AA, et al. The unfolded protein response signals through high-order assembly of Ire1. Nature. 2009;457(7230):687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gass JN, Jiang HY, Wek RC, Brewer JW. The unfolded protein response of B-lymphocytes: PERK-independent development of antibody-secreting cells. Molecular immunology. 2008;45(4):1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4(4):321–329. [DOI] [PubMed] [Google Scholar]
- 27.Reimold AM, Iwakoshi NN, Manis J, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412(6844):300–307. [DOI] [PubMed] [Google Scholar]
- 28.Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21(1):81–93. [DOI] [PubMed] [Google Scholar]
- 29.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–891. [DOI] [PubMed] [Google Scholar]
- 30.Bettigole SE, Glimcher LH. Endoplasmic reticulum stress in immunity. Annual review of immunology. 2015;33:107–138. [DOI] [PubMed] [Google Scholar]
- 31.Gaudette BT, Jones DD, Bortnick A, Argon Y, Allman D. mTORC1 coordinates an immediate unfolded protein response-related transcriptome in activated B cells preceding antibody secretion. Nat Commun. 2020;11(1):723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allman D, Pillai S. Peripheral B cell subsets. Current opinion in immunology. 2008;20(2):149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goodnow CC, Vinuesa CG, Randall KL, Mackay F, Brink R. Control systems and decision making for antibody production. Nat Immunol. 2010;11(8):681–688. [DOI] [PubMed] [Google Scholar]
- 34.Oliver AM, Martin F, Gartland GL, Carter RH, Kearney JF. Marginal zone B cells exhibit unique activation, proliferative and immunoglobulin secretory responses. Eur J Immunol. 1997;27(9):2366–2374. [DOI] [PubMed] [Google Scholar]
- 35.Lu TT, Cyster JG. Integrin-mediated long-term B cell retention in the splenic marginal zone. Science. 2002;297(5580):409–412. [DOI] [PubMed] [Google Scholar]
- 36.Cerutti A, Cols M, Puga I. Marginal zone B cells: virtues of innate-like antibody-producing lymphocytes. Nat Rev Immunol. 2013;13(2):118–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gunn KE, Brewer JW. Evidence that marginal zone B cells possess an enhanced secretory apparatus and exhibit superior secretory activity. J Immunol. 2006;177(6):3791–3798. [DOI] [PubMed] [Google Scholar]
- 38.Song H, Cerny J. Functional heterogeneity of marginal zone B cells revealed by their ability to generate both early antibody-forming cells and germinal centers with hypermutation and memory in response to a T-dependent antigen. J Exp Med. 2003;198(12):1923–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annual review of immunology. 2012;30:39–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Donahue AC, Fruman DA. Distinct signaling mechanisms activate the target of rapamycin in response to different B-cell stimuli. Eur J Immunol. 2007;37(10):2923–2936. [DOI] [PubMed] [Google Scholar]
- 41.Sintes J, Gentile M, Zhang S, et al. mTOR intersects antibody-inducing signals from TACI in marginal zone B cells. Nat Commun. 2017;8(1):1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benhamron S, Pattanayak SP, Berger M, Tirosh B. mTOR activation promotes plasma cell differentiation and bypasses XBP-1 for immunoglobulin secretion. Mol Cell Biol. 2015;35(1):153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Benhamron S, Tirosh B. Direct activation of mTOR in B lymphocytes confers impairment in B-cell maturation and loss of marginal zone B cells. Eur J Immunol. 2011;41(8):2390–2396. [DOI] [PubMed] [Google Scholar]
- 44.Muri J, Thut H, Bornkamm GW, Kopf M. B1 and Marginal Zone B Cells but Not Follicular B2 Cells Require Gpx4 to Prevent Lipid Peroxidation and Ferroptosis. Cell reports. 2019;29(9):2731–2744 e2734. [DOI] [PubMed] [Google Scholar]
- 45.He C, Wang S, Zhou C, et al. CD36 and LC3B initiated autophagy in B cells regulates the humoral immune response. Autophagy. 2021:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raybuck AL, Cho SH, Li J, et al. B Cell-Intrinsic mTORC1 Promotes Germinal Center-Defining Transcription Factor Gene Expression, Somatic Hypermutation, and Memory B Cell Generation in Humoral Immunity. J Immunol. 2018;200(8):2627–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ersching J, Efeyan A, Mesin L, et al. Germinal Center Selection and Affinity Maturation Require Dynamic Regulation of mTORC1 Kinase. Immunity. 2017;46(6):1045–1058 e1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dossou AS, Basu A. The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers. 2019;11(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arnold J, Murera D, Arbogast F, Fauny JD, Muller S, Gros F. Autophagy is dispensable for B-cell development but essential for humoral autoimmune responses. Cell death and differentiation. 2016;23(5):853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pengo N, Scolari M, Oliva L, et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol. 2013;14(3):298–305. [DOI] [PubMed] [Google Scholar]
- 51.Taubenheim N, Tarlinton DM, Crawford S, Corcoran LM, Hodgkin PD, Nutt SL. High rate of antibody secretion is not integral to plasma cell differentiation as revealed by XBP-1 deficiency. J Immunol. 2012;189(7):3328–3338. [DOI] [PubMed] [Google Scholar]
- 52.Ma Y, Shimizu Y, Mann MJ, Jin Y, Hendershot LM. Plasma cell differentiation initiates a limited ER stress response by specifically suppressing the PERK-dependent branch of the unfolded protein response. Cell stress & chaperones. 2010;15(3):281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y, Bhattacharya D. Adjuvant-specific regulation of long-term antibody responses by ZBTB20. J Exp Med. 2014;211(5):841–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chan TD, Gardam S, Gatto D, Turner VM, Silke J, Brink R. In vivo control of B-cell survival and antigen-specific B-cell responses. Immunol Rev. 2010;237(1):90–103. [DOI] [PubMed] [Google Scholar]
- 55.Chan TD, Gatto D, Wood K, Camidge T, Basten A, Brink R. Antigen affinity controls rapid T-dependent antibody production by driving the expansion rather than the differentiation or extrafollicular migration of early plasmablasts. J Immunol. 2009;183(5):3139–3149. [DOI] [PubMed] [Google Scholar]
- 56.Duffy KR, Wellard CJ, Markham JF, et al. Activation-induced B cell fates are selected by intracellular stochastic competition. Science. 2012;335(6066):338–341. [DOI] [PubMed] [Google Scholar]
- 57.Dowling MR, Kan A, Heinzel S, et al. Stretched cell cycle model for proliferating lymphocytes. Proc Natl Acad Sci U S A. 2014;111(17):6377–6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heinzel S, Binh Giang T, Kan A, et al. A Myc-dependent division timer complements a cell-death timer to regulate T cell and B cell responses. Nat Immunol. 2017;18(1):96–103. [DOI] [PubMed] [Google Scholar]
- 59.Duffy KR, Hodgkin PD. Intracellular competition for fates in the immune system. Trends Cell Biol. 2012;22(9):457–464. [DOI] [PubMed] [Google Scholar]
- 60.Kuchina A, Espinar L, Cagatay T, et al. Temporal competition between differentiation programs determines cell fate choice. Mol Syst Biol. 2011;7:557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Benson MJ, Dillon SR, Castigli E, et al. Cutting edge: the dependence of plasma cells and independence of memory B cells on BAFF and APRIL. J Immunol. 2008;180(6):3655–3659. [DOI] [PubMed] [Google Scholar]
- 62.O’Connor BP, Raman VS, Erickson LD, et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med. 2004;199(1):91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cornelis R, Hahne S, Taddeo A, et al. Stromal Cell-Contact Dependent PI3K and APRIL Induced NF-kappaB Signaling Prevent Mitochondrial- and ER Stress Induced Death of Memory Plasma Cells. Cell reports. 2020;32(5):107982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Erickson LD, Lin LL, Duan B, Morel L, Noelle RJ. A genetic lesion that arrests plasma cell homing to the bone marrow. Proc Natl Acad Sci U S A. 2003;100(22):12905–12910. [DOI] [PMC free article] [PubMed] [Google Scholar]