

Abstract
The application of genomic approaches is impacting all areas of laboratory testing including transfusion medicine. Use of DNA-based test methods is particularly applicable for red cell and platelet antigen typing as the majority of genes encoding the carrier proteins and carbohydrates are now known and were cloned in the 1990′s. Many of the antigenic polymorphisms are due to single nucleotide changes (SNP's) in the respective genes and DNA-arrays that target these changes have been validated by comparison with conventional serologic typing. Here we review the advances in the last decade in the application of DNA-based genotyping to transfusion therapy, specifically in sickle cell anemia (SCA), and discuss the practical integration and the value of this approach to improve outcomes and prevent complications in this patient population. The ability to test for antigens for which there are no serologic reagents is a major medical advance that promises to mitigate transfusion related morbidity and mortality due to alloimmunization.
Keywords: Red cell alloimmunization, Sickle cell anemia, Transfusion, Rh antigens, Antigen-matched, Red blood cell genotyping
1. Background
1.1. Transfusion therapy for sickle cell disease
Red cell transfusion remains a key intervention in the management of patients with sickle cell anemia (SCA). Red cell transfusions are often used in the management of acute complications including stroke, acute chest syndrome (ACS), aplastic crisis, and splenic sequestration, as well as prior to surgical procedures involving general anesthesia and lasting greater than 1 h [1]. Individuals with acute hepatic sequestration, sickle cell intrahepatic cholestasis, or multi-organ system failure may also benefit from acute red cell transfusion provided by simple or exchange transfusion (reviewed in [2]).
Primary and secondary stroke prevention have been the major indications for chronic RBC therapy, affecting approximately 10% of children with SCA [3-5]. Several studies in the past decade will impact the number of patients who will remain on, or require, chronic transfusions. The number of individuals receiving chronic transfusions is anticipated to decline following the Transfusions Changing to Hydroxyurea (TWiTCH) trial that demonstrated that hydroxyurea was not inferior to chronic red cell transfusion in lowering transcranial doppler (TCD) velocities in children with an abnormal TCD and absence of severe vasculopathy who had received a minimum of one year of transfusion therapy [6]. In contrast, indefinite chronic transfusion therapy is recommended for secondary stroke prevention as The Stroke With Transfusions Changing to Hydroxyurea (SWiTCH) trial addressed transition to hydroxyurea but was closed due to statistical futility on the composite endpoint of iron overload resolution and stroke prevention [7]. The Silent Infarct Transfusion (SIT) trial demonstrated that chronic transfusion therapy reduced the incidence of recurrent cerebral infarcts in patients with silent cerebral infarcts (SCIs) at baseline, no history of overt stroke, and normal TCD velocities [8]. These randomized, prospective trials show that red cell transfusion remains a major intervention for managing and preventing neurologic complications in individuals with SCA. Management and prevention of alloimmunization is a critical aspect of chronic red cell therapy. DNA-based genomic approaches for extended red cell antigen profiling of both patients and blood donors now offer substantial improvements to mitigate alloimmunization complications inherent with transfusion therapy.
1.2. Antibody specificities and prevalence
Alloimmunization to red cell antigens is a major complication associated with transfusions in patients with SCA, and can be associated with acute or delayed hemolytic transfusion reactions (DHTR), worsening anemia, or be asymptomatic termed “delayed serologic transfusion reactions” (DSTR) that result in compromised cell survival in the absence of overt signs or symptoms. If the total hemoglobin or hemoglobin A level following transfusion or at the next transfusion visit is lower than expected, a DHTR or DSTR should be considered and an antibody evaluation is warranted. The cause of increased alloimmunization is probably multifactorial, but disparity in blood group antigens expressed on donor versus patient red cells, which differ depending on ethnic group, is a major contributing factor, as is the large number of transfusion exposures (reviewed in [9,10]). Antibodies to the Rh and Kell (K antigen) systems comprise over two-thirds of antibodies in this patient population [11-13]. Hence, “prophylactic antigen matching” for C, E and K antigens, which is defined as transfusion of donor units lacking C, E, or K if the patient is negative for the antigen, is recommended. This approach however has not been universally adopted [14,15], primarily due to the high cost of antigen-negative donor units [16,17] and concerns for sufficient numbers of antigen-matched donors in some regions of the country, although this has not yet been sufficiently explored by large-scale donor testing.
Antigen matching for C, E, and K reduces the prevalence of alloantibody production from between 18%–75% (incidence rate: 1.7 to 3.9 antibodies/100 units transfused) [12,13,18-21] to approximately 5–29% (incidence rate: 0.26 to 0.50 antibodies/100 units transfused) [8,20-23]. Small, single institution studies demonstrate a further reduction to 0–7% (incidence rate: ≤ 0.10 antibodies/100 units transfused) with extended antigen matching beyond C, E, and K [24,25]. In our single institution study, prophylactic C, E, K-matched red cells from African-American donors resulted in no anti-K and a reduction in other specificities common in this patient population such as anti-Jkb, -Fyb and -S, but Rh immunization persisted. The most frequent antibodies encountered in patients receiving D, C, E and K antigen-matched units from African-American donors had Rh specificities [26]. While we observed a high prevalence of alloimmunization in patients who were chronically transfused, the incidence rate was comparable to multicenter studies that provided prophylactic C, E, and K matched units from general donor inventories (Table 1) [8,20]. The high prevalence of alloimmunization in our study is explained by the long observation period (15 years) and the very high number of red cell unit exposures (median 230 units) for patients chronically transfused.
Table 1.
Similar alloimmunization rates in patients with SCA receiving chronic red cell transfusions with general versus minority donor inventories.
| Study | Donor inventory | Number of patients |
Number of red cell unit exposures in study |
Overall antibody rate (events/100 units transfused) |
Rh antibody rate (events/ 100 units transfused) |
Overall allo-immunization prevalence (%) |
|---|---|---|---|---|---|---|
| STOP trial | General | 63 | 1830 | 0.49 | 0.21 | 14% |
| SIT trial | General | 99 | 3235 | 0.28 | 0.16 | 6% |
| Philadelphia single center | Minority donor program | 123 | 44,210 | 0.30 | 0.21 | 58% |
1.3. Antibody persistence
Once an antibody is identified, the patient must receive donor units tested and found to be negative for that antigen for every subsequent transfusion life-long. This results in increased costs of both pre-transfusion testing and of the donor unit, and delays patient care. More importantly, patients who have made one antibody are at an increased risk of developing additional RBC antibodies. A principal concern is that the reactivity of most antibodies fall below the level of detection over time [26,27]. Many patients present for care at multiple institutions and in the absence of antibody history are at risk for HTRs associated with a robust secondary immune response. For referrals to another hospital for surgery or hematopoietic stem cell transplant, communication regarding the patient's alloimmunization history is critical. Some clinicians provide patients with a document summarizing the alloantibody history and direct communication between blood banks is key for transfusion safety. Five to 8 patient fatalities due to HTRs are reported to the FDA each year, with a majority in the last decade occurring in patients with SCA, and most due to the inability to detect pre-existing antibodies or the need for emergency transfusion [28].
2. RH variation
The Rh antigens defined by serology are D, C/c and E/e; the latter are allelic and co-dominantly expressed (Fig. 1A). The RHD gene encodes D antigen which represents the presence (“Rh positive”) or absence (“Rh negative”) of RhD protein in the RBC membrane, and RHCE encodes C/c and E/e antigens on the RhCE protein present in all but rare individuals (Fig. 1B). The genes arose by duplication, are linked, and are highly similar in overall sequence encoding 32–35 amino acid differences. Genetic exchange between them created hybrid proteins encoding novel Rh antigens (numbering > 50), identified in individuals who mounted an immune response to the foreign Rh protein.
Fig. 1.

Rh proteins and RH genes. A. RhD and RhCE proteins with 35 amino acid differences depicted by circles on RhD. Positions 103 and 225 in RhCE determine C or c and E or e expression, respectively. The four possible RhCE proteins are ce, cE, Ce, and CE. B. The RHD and RHCE genes are closely linked on chromosome 1, with ten highly homologous exons (boxes), and their inverted gene orientation promotes gene conversion with one gene acting as a donor template during replication. C. RHD*DIIIa-CE(4-7)-D. which arose by genetic exchange of RHCE exons 4 to 7 into the RHD*DIIIa locus. No RhD antigen but partial C antigen is encoded by this allele; red cells type strongly C+ with monoclonal anti-C but patients are at risk for allo-anti-C when exposed to conventional C+ red cells.
Routine serologic typing reagents are only available for the five common D, C, c, E and e antigens found in all ethnic groups. These RH gene exchanges or conversions can involve single or multiple exons and are more often found in African ethnic groups, and hence patients with SCA (reviewed in [29]). One primary RHD gene sequence is found in all populations, but > 500 allelic variations have now been reported to cause weak and/or altered D antigen expression. Three primary RHCE alleles, denoted RHCE*ce, RHCE*Ce, and RHCE*cE, are found in all populations but vary in frequency between different ethnic groups. More than 140 RHCE allelic variations have been reported to date [30,31]. Variant or altered RH alleles encode partial and/or weak expression of D, C, e and less often c and E antigens. The term “partial” describes red cells that lack some common epitopes associated with expression of the antigen. As a result, patients with SCA with variant RH can produce antibodies to Rh epitope(s) they lack, as well as to Rh epitopes that differ from those expressed on their own cells. In contrast, RH variants resulting in weak antigen expression result in lower red cell surface expression density but there is no loss of epitopes.
Approximately 90% of individuals with SCA carry at least one variant RH allele [26]. Equivalent RH gene diversity is also present in healthy African-American blood donors [32]. RH variation in both patients and donors likely contributes to the prevalence of Rh antibodies encountered in patients with SCA receiving donor units antigen-matched by serology for common D, C, E antigens and chosen specifically from the African-American donor pool [26]. In our single center retrospective study, antibodies were identified with apparent Rh specificities that were initially difficult to explain [26]. These included anti-D, -C, and/or -e in the plasma of patients whose red cells type as positive for the antigen, or anti-D, -C, and/or -E in the plasma of patients who had not been exposed to red cells positive for that Rh antigen. These Rh antibodies challenged conventions in that patients whose red cells are positive for an antigen, or type negative but were never exposed to the antigen, were not expected to have the corresponding antibody identified in their plasma. RH genotyping in these individuals demonstrated that inheritance of variant RH alleles resulting in partial antigen expression explained many of these Rh antibodies.
Blood group genotyping can be very useful in scenarios where serologic testing has limitations. In the Rh system, RhD and RhCE proteins have multiple, complex, potentially overlapping, and cross-reactive epitopes. Indeed, studies with monoclonal antibodies defined > 30 antigenic epitopes on RhD alone [33]. Hence, the fine specificity of the Rh antibodies made by patients with altered Rh proteins AND exposed to other altered Rh proteins of the donor are difficult to identify. These antibodies are cross-reactive with epitopes on conventional Rh proteins. In most cases, it is not possible to definitively identify the true or actual immunizing Rh epitope(s) because routine laboratory red cell reagents are primarily from Caucasians, and panels of RH genotyped red cells are not readily available. Individuals can become immunized when exposed to Rh epitopes on the donor red cells that differ, or are absent, from their own red cells [26,34]. Patients almost always present with a positive DAT, and hence these Rh antibodies can be confused with autoantibodies. Instead, these are often alloantibodies in patients homozygous for altered RH alleles. In some cases, the antibodies show anti-E or anti-C specificity by laboratory testing in the absence of stimulation by E or C positive donor red cells. These are termed “mimicking” antibodies, in that the antibody cross-reacts with Rh epitopes shared with E+ or C+ red cells, so appear in laboratory testing to be specific for these antigens. Approximately 30% of these complex Rh antibodies in patients with SCA receiving chronic or episodic transfusions caused signs of decrease red cell survival [26].
3. Extended RBC blood group antigen profiling
Although there are > 350 serologically distinct RBC antigens recognized to date [35], approximately fourteen “extended” antigens are considered for routine transfusion practice. This is based on the frequency of antigen expression on red cells, the degree of variation between patients and blood donors which impacts the incidence of antibody production and incompatibility, and historically, the ability to test for the antigen by serology. The National Heart, Lung, and Blood Institute (NHLBI) recommends extended red cell antigen typing for patients with SCA aged > 6 months and transfusion with prophylactic C, E, K matched red cells [1]. The extended antigen profile determined by serologic methods includes the Rh system antigens (CcEe), Kell (Kk), Duffy (Fya/b), Kidd (Jka/b), Glycophorin A (MN) and Glycophorin B (Ss). Some include Lewis (Lea/Leb) and P1, which are not usually clinically significant but this information can aid antibody identification. Although extending antigen-matching to minimize alloimmunization against Kidd, Duffy, and S antigens (in addition to C, E, and K) would be preferable, finding sufficient red cell units for chronically transfused patients is challenging for donor centers, and transfusion cost increases with higher levels of antigen-matching making it prohibitive at present for most providers. In addition to guiding antigen matching for C, E, and K, the patient extended red cell antigen profile facilitates antibody evaluation and identification when patients present with a newly formed antibody.
4. Genotyping
4.1. RBC antigen profile by genotype
Determining an individual's extended blood group antigen profile by DNA-methods (genotyping) is indirect, in contrast to direct detection by serology using a specific antibody (phenotyping). Because most red cell antigens result from single nucleotide differences (SNPs), typing by DNA methods is relatively straightforward and has become an important part of the practice of transfusion medicine. The results are highly correlated with serologic phenotyping, and are superior in situations of recent transfusion or the presence of a positive direct antiglobulin test (DAT). The ability to determine the patient or donor antigen profile in one assay increases accuracy, provides information for antigens for which no typing reagents are available [36,37] and for some institutions, can replace serologic typing at lower cost. Routine ABO and RhD typing is not done by DNA methods as the sole method due to the large number of different mutations that result in a Group O or a RhD negative phenotype.
A major benefit of genotyping is the ability to determine many blood group antigenic polymorphisms previously not possible due to a lack of specific and sensitive antibody reagents. This is potentially life-saving for alloimmunized patients. In cases where antibody specificity is not readily identified by routine antibody detection, which include Dombrock (Doa/b, Hy, Joa), other Kell system antigens (Kpa/b, Jsa/b), other Rh system (V/VS), Colton (Coa/b), Yt (a/b), Lutheran (Lua/b), Diego (Dia/b), and Scianna (Sc1/2) antibody specificities, incompatibility can be clinically significant and life-threatening, but no serologic reagents are available to aid identification, confirm the specificity, or to type donors to find compatible units. In the treatment of patients with SCA experiencing compromised survival of transfused cells, incompatibilities due to altered Rh antigens and Dombrock (Doa/b) should always be ruled out.
Case: anti-Dombrock
An 11-year old boy with SCA (Group O, RhD+ and C+c+E−e+, K−) presented with back pain and jaundice 5 days after transfusion of 1 unit of red cells “antigen-matched”, i.e. negative for E and K antigens. His hemoglobin was 6.7 gm/dL prior to the transfusion, and now 6.2 gm/dL on admission. A delayed hemolytic transfusion reaction (DHTR) was suspected, but his antibody screen using two different methods was negative and the direct antiglobulin test (DAT) was also negative. Transfusion with 2 cross-match compatible E−, K− RBC units increased his hemoglobin to 8.1 gm/dL, but his hemoglobin declined to 6.5 gm/dL three days later. His DAT remained negative but the antibody screen had become positive. However, no clear specificity was apparent on antibody identification panels. Fourteen units were cross-matched and all were incompatible. The sample was referred to a immunohematology reference laboratory (IRL). Anti-Knops was identified, but these are usually not clinically significant. DNA array extended antigen testing was performed and indicated the patient was positive for the majority of clinically significant antigens but lacked Fya and Dob.
Dombrock antibodies are very difficult and often impossible to detect in the serum/plasma despite causing severe hemolysis in vivo. Then antibody has poor avidity and is often lost from the test red cells during the indirect antiglobulin test (IAT) in vitro. The antibody can cause significant red cell destruction and be a silent killer. They are most often present with other antibody specificities, which further complicates laboratory identification and the clinical picture. There are no antibody reagents to type patients or donor red cells. DNA testing of the three red cell units associated with hemolysis revealed all were Fy(a−) but Do(b+). Subsequent transfusions with Do(b−) units have resulted in the expected hemoglobin A rise and the patient has been maintained on Do(b−) donor units. DHTRs caused by anti-Doa or -Dob are likely under-recognized since the antibody is weakly reactive or often undetectable and the DAT is often negative.
4.2. Methodology, availability, performance
A FDA-licensed commercial DNA-array is available for genotyping 35 blood group antigens, designated PreciseType human erythrocyte antigen (HEA) array (BioArray/Immucor), and at least one other is expected to be licensed soon. Commercial DNA-array platforms provide automated interpretation which has allowed more widespread use of genotyping. These commercial tests determine conventional Rh C, c, E, and e antigen status and some include a limited number of markers for RH variant alleles. Genotyping is primarily available in a number of blood donor centers and reference laboratories for typing blood donors and the hospital patients they serve, and also in some larger hospitals that have sufficient economy of scale. However, high-resolution RH genotyping and targeted RH gene sequencing is needed to comprehensively identify RH variants and to expertly interpret allelic associations in patients with SCA. Testing is performed in reference laboratories as research use only (RUO) and laboratory developed tests (LDTs).
For patients with SCA, DNA based testing has been shown to improve accuracy and greatly expand information on RBC antigens compared to serologic typing [37]. In 494 patients for whom previous serological typing results for 13 antigens were available, 71 discrepancies were identified among 6360 antigens when compared with results by genotyping (1.1%). All but one discrepancy was resolved in support of the genotype on repeat serologic typing. One Jk(b−) serologic phenotype, predicted to be Jk(b+) by genotype, was found by sequencing of the JK gene to be a novel silenced allele. Of note, it is recognized that rare silenced alleles will not be detected by using DNA-arrays that target known allelic polymorphisms. The concordance rate between genotyping and serologic confirmation was 0.9997. Importantly, red cell genotyping provided greater accuracy and additional antigen information not possible with serologic typing, including the clinically relevant partial/altered RhC, Doa, Dob, Hy, Joa, Jsb, Uvar, V/VS and hrB antigens, as well as less commonly encountered polymorphisms including Dia/b, Coa/b, Lua/b, Sc1/2, and LWa/b. DNA arrays also target the FY gene GATA erythroid promoter mutation that results in RBC-specific loss of Fyb antigen expression. Patients whose red cells type Fy(b−) by serology, but who have the FY erythroid GATA mutation can receive Fy(b+) red cells without risk for anti-Fyb as tissue expression is intact. When extended phenotype matching is practiced, this increases the number of donors available as antigen-matched for these patients.
5. Specific considerations for sickle cell anemia
5.1. Absence of high prevalence antigens in patients with sickle cell anemia
Approximately 2% of people from African backgrounds lack glycophorin B on their red cells and are U− or U variant, and approximately 1% are Js(b−), or Jo(a−) or Hy- and are at risk for clinically significant anti-U, -Js(b), -Jo(a), or -Hy, respectively. Antibodies to these antigens react with all routine reagent red cells used for antibody screening and identification, and can be confused with warm autoantibodies because the patient often also has a positive DAT. Among 494 patients with SCA in our study, 66 lacked high prevalence antigens: five U-, three U variant, two Js(b−), three Jo(a−), one Hy−, and 54 hrB− in the Rh system [37]. Although providing antigen negative units as a prophylactic prevention strategy for these patients is not possible at this time, this information is key to guiding interpretation of pre-transfusion antibody evaluations over the entire course of the patient's medical care.
Individuals homozygous for variant RH alleles can also lack common high prevalence Rh antigens (for example: hrB, hrS, HrB) or express Rh antigens exclusive to African ethnic groups (for example: V, VS, DAK, Rh32, Go(a)) [38]. The plasma of patients who develop antibodies to high prevalence Rh antigens show the same panagglutination pattern in routine laboratory testing as that seen with warm autoantibodies. RH genotyping can identify individuals who lack high prevalence Rh antigens and facilitate antibody evaluation and donor selection.
5.2. Precise C antigen determination
Approximately 20% of SCA patients with C+ red cells express variant C and do not have a conventional RhCe protein (Fig. 2). In these patients, the C antigen is encoded by a hybrid RHD*DIIIa-CE(4-7)-D gene that is actually located in the RHD locus but does not encode D antigen, but encodes a C+ serologic phenotype (Fig. 1C). Thirty percent of patients with this hybrid gene (in the absence of a conventional RHCE*Ce allele in trans) made anti-C when repeatedly exposed to conventional C+ red cells [39]. These anti-C represent alloantibodies and not autoantibodies, since the antibody developed in response to “foreign” C epitopes. Because of the risk for allo-anti-C, patients who are C+ due to this hybrid allele are better served on C− rather than C+ transfusion when prophylactic antigen matching. In our cohort of 494 patients with SCA, RH genotyping confirmed RHD*DIIIa-CE(4-7)-D alleles in 27 of 138 C+ individuals (20%) [37]. These patients receive C− donor units despite testing C+ by serology.
Fig. 2.
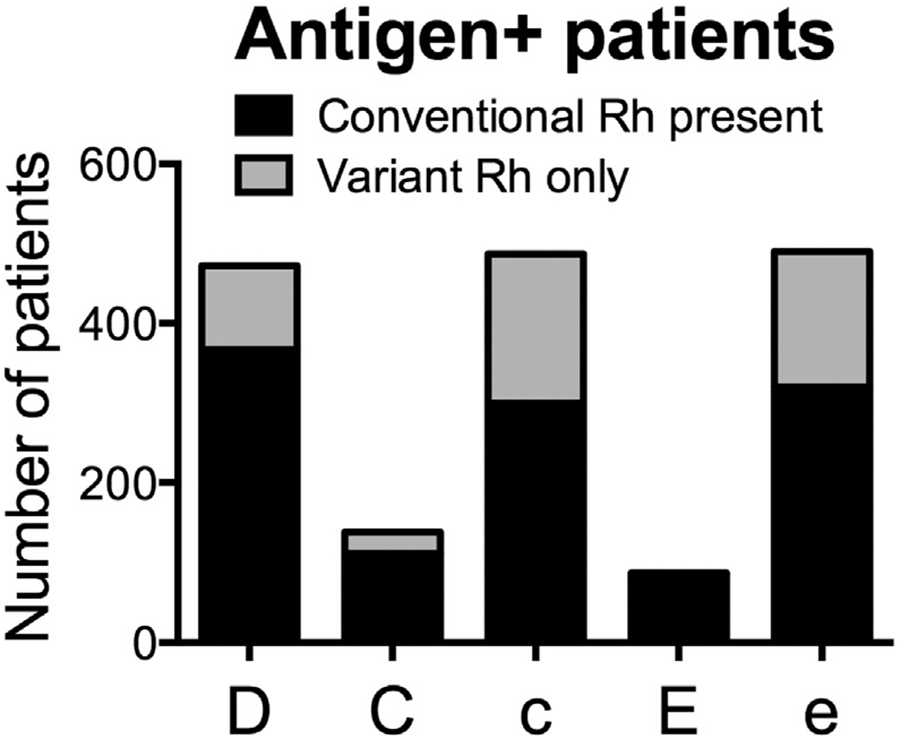
RH genotype predicted antigen expression for the five principal Rh antigens in patients with SCA. For each Rh antigen, the number of antigen positive individuals whose red cells express conventional protein or variant antigen only is shown for 494 individuals with SCA as determined by high resolution RH genotyping using DNA-arrays combined with AS-PCR assays and exon-specific sequencing.
Case: anti-C in patients whose red cells type as C+
Anti-C was detected on a routine antibody screen for a 12-year old female with SCA receiving chronic red cell transfusions every 3–4 weeks. Her serologic phenotype was RhD+ and C+c+E−e+, K− and she had been transfused with E− K− units according to standard protocol for prophylactic antigen matching. The identification of anti-C in a patient whose red cells type as C+ prompted referral for RH genotyping. Genotyping confirmed the presence of the hybrid RHD*DIIIa-CE(4-7)-D which encodes a partial C antigen with risk for anti-C, despite the fact that her red cells type C+ with monoclonal anti-C typing reagents. RH genotype confirmed absence of conventional C antigen, and therefore, her anti-C represents an alloantibody made in response to exposure to conventional C antigen. These antibodies are clinically significant. Prevention of allo-anti-C in patients with SCA requires RH genotyping for accurate C antigen matching.
5.3. Precise e antigen determination
The red cells of nearly all patients with SCA of African descent will be e+, but one-third is homozygous for partial or altered e antigen (Fig. 2). Anti-e in these e+ patients is alloantibodies (rather than autoantibodies) and are more problematic to manage. The clinical significance is often associated with the specific RHCE*ce allele present. For example, those alleles that also encode lack of the high prevalence Rh antigen, hrs, can be associated with significant destruction of transfused red cells [40,41]. If the patient is also E+, then transfusion with E+e− red cells is straightforward. Although only 2% of donors are E+e−, donor units are usually readily available and only become challenging if patients have multiple additional antibodies. For patients who are not E+, e− donor units will be E+ with significant risk for clinically significant anti-E production. Transfusion is best guided by the clinical presentation. If a e+ patient has anti-e, (and/or -hrB or -hrS) and signs of RBC destruction are observed with e+ RBC transfusion, RH genotype-matched red cells should be considered.
5.4. Precise D antigen determination
In patients with SCA, one-third of RHD alleles are altered and 20% of D+ individuals exclusively express altered RhD resulting in partial D (missing D epitopes) and in a minority of cases, weak partial D (red cell surface antigen density decreased and missing D epitopes) (Fig. 2). Anti-D in patients who are RhD positive with SCA is associated with alleles encoding partial D antigen [26,34]. Routine serologic RhD typing cannot distinguish red cells with altered D antigen and RHD genotyping is required.
Case: anti-D in patients whose red cells type RhD+
A 5-year old boy with SCA (Group O, RhD+ and C−c+E−e+, K−) presented to the emergency department with pain, fatigue, and dark urine 10 days post-transfusion. He had received 1 red cell unit negative for C, E, and K antigens. His hemoglobin was 6.5 gm/dL prior to transfusion, but was now 5.6 gm/dL at presentation, and nadired at 4.2 gm/dL during hospital admission. The antibody screen was weakly positive in a gel-based assay but negative with the tube method. The DAT was negative. A selected red cell panel detected 1 + reactivity to all cells tested except one, suggesting an autoantibody given the near pan-reactivity. However, the laboratory subsequently identified an unexpected anti-D given the patient was confirmed RhD+. RHD genotyping found the patient was homozygous for RHD*DAU4 which encodes a partial D antigen [42], confirming the anti-D is an alloantibody (not an autoantibody) formed after transfusion with RhD+ red cells that express D epitopes the patient lacks.
Six months later, he received 1 unit cross-match compatible RhD− red cells lacking C, E, and K antigens in preparation for a tonsillectomy and adenoidectomy with general anesthesia. He returned 6 days later with a DHTR with a nadir hemoglobin of 4.5 gm/dL (pre-transfusion hemoglobin 7.0 gm/dL). Anti-Jkb was identified and he was treated with steroids, IVIg and transfused with red cells also lacking Jkb. Extended antigen profile by genotyping was performed and for all future transfusions extended matching was recommended with D−, C−, E−, K−, Jk(b−) as well as Fy(a−) and S− red cells since he lacked those antigens as well. Red cell units antigen-matched for his extended profile are found in ~1 in 200 units from the general donor pool and 1 in 50 African-American donors [38].
Avoiding risk for anti-D in patients whose RHD genotype indicates they have partial D phenotype and no conventional D expression would require transfusion with RhD negative donor units which are primarily from Caucasian donors (17% of Caucasians are RhD− but only 5% of African Americans). Therefore, providing RhD negative red cells to patients with partial D antigen may increase alloimmunization risk to other antigens (Fya, Jkb, and S) that are more common in Europeans compared to Africans. Alternatively, these patients can receive RhD positive donor units that have the same RHD genotype, which requires RH genotyping of large numbers of African American donors.
5.5. Transfusion support for bone marrow transplantation for sickle cell disease
Patients who have multiple antibodies and for whom it is difficult to find compatible donors are increasingly being offered the option of hematopoietic stem cell (HSC) transplantation. Although most antibodies to red cells including ABO are not a barrier to engraftment, red cell alloantibodies can cause complications for highly alloimmunized patients. Pre-transplant exchange transfusion is often performed to achieve a hemoglobin S level of 30% or less and it can be challenging to find compatible donor units for both pre-transplant exchange and post-transplant support prior to engraftment. Importantly, non-myeloablative conditioning results in mixed chimerism with both recipient and donor-derived lymphocytes and persistent recipient plasma cells. Pre-existing non-ABO blood group antibodies can cause overt hemolysis of incompatible donor red cells or delayed erythropoiesis, and new red cell antibodies whether donor or patient derived, can complicate transfusion support and recovery. Genotyping of the recipient peripheral blood sample and buccal cells can be done in an attempt to determine the origin of new antibodies post-transplant, i.e. donor-derived or recipient-derived, to inform selection of units for transfusion. Genotyping both the donor and recipient for extended blood group antigens prior to transplantation would proactively inform transfusion management in patients with existing alloantibodies.
Case: post HSC transplant hemolysis
A 10-year old female with SCD (Group O, RhD+ and C−E−c+e+K−) with multiple antibodies including anti-C, -E, -K, -S, - Jsa, and -Ytb underwent allogenic stem cell transplantation. She received 24 units lacking the antigens corresponding to these antibodies during the first four weeks post-transplant, but she demonstrated overt hemolysis and prolonged reticulocytopenia. Her hemoglobin fell to 2.4 gm/dL and she had an increasing titer of anti-C and a newly formed anti-Jkb was identified. Extended blood group genotyping was performed on her pre-transplant sample and the HSC donor to determine origin of the new antibody production. The patient's extended genotype was Fy(a−b−), Jk(a+b+), MN+, S−s+, U+, V+VS+, Js(a−), Yt (a+b−), Di(a−b+), Co(a+b−), Do(a+b+). The donor was Group O, RhD+ and antigen compatible for all except C+ and was Jk(b−). The anti-Jk(b−) represents the donor response to Jk(b+) patient red cells. The anti-C was patient-derived and exacerbated by C+ donor red cells. The combination of multiple specificities in routine hospital laboratory workup had the pattern of a non-specific warm autoantibody, but anti-e like specificity was identified by extensive reference laboratory evaluation. RH genotyping demonstrated the patient was homozygous for altered RHCE*ce alleles, while the donor had conventional RHCE*ce. A search for RH genotyped, antigen-negative donor units was undertaken but severe anemia with overt hemolysis continued and the patient died 9 weeks post-transplant. The clinical course of transplant recipients who demonstrate both donor and/or patient-derived antibodies can be highly variable from prolonged erythrocytopenia, transient hemolysis, to this rare severe case with fatality (our unpublished observations).
5.6. Red cell genotyping of donors
Prophylactic antigen matching beyond C, E, and K has been proposed to further reduce alloimmunization in SCA [24,25], which would require extensive recruitment of ethnically-matched donors. To date, a few small feasibility studies have investigated whether DNA-typing of patients and donor inventories could provide extended matched red cells to patients with SCA (matched for ABO/RhD, C/c, E/e, K, Fya/b, Jka/b, S/s, and Doa/b and Dia/b for some) [43,44]. A small fraction of patients (7–8%) had no extended matched components available due to a predominantly Caucasian donor pool in one study [43] or infrequent or rare patient phenotypes [44]. The availability of high-throughput red cell genotyping for donor populations can identify donors with uncommon antigen-negative combinations and facilitate an extended red cell matching strategy. Further minimizing alloimmunization for patients with SCA will require recruitment and retention of repeat African-American donors with extended red cell typing including RH genotyping.
6. Summary
For patients with SCA, red cell genotyping provides information on numerous additional antigens not possible by serology and RH genotyping identifies altered Rh antigens not detected serologically. DNA-based RBC antigen typing can facilitate complex antibody evaluations and guide RBC selection by focusing on antibodies the patient is at risk for, or that may be mistaken as or masked by warm autoantibodies. This is particularly advantageous when routine testing cannot demonstrate red cell compatibility or when no specific antibody is identified despite evidence of red cell destruction. Genotyping can also be used to determine red cell antigen phenotypes in individuals who are recently transfused, have discrepant serologic typing, need transfusion support for bone marrow transplantation, or when no typing antisera is available.
It has been increasingly appreciated that variant RH genes encoding amino acid changes in the Rh proteins contribute to alloimmunization despite Rh serologic matching in patients with SCA [26,34,45]. There is an emerging role for molecular methods to improve red cell matching in the Rh system. At present, providing antigen negative red cells for individuals predicted by RH genotype to exclusively express partial Rh antigens may be performed on an individual case basis. Consideration of their extended red cell antigen profile, alloimmunization history and specificities, and the availability of appropriate donors will determine the feasibility.
7. Future
Blood group antigens are encoded by 45 genes with nearly 2000 alleles identified to date associated with differences in phenotype or with antibody production. Next generation sequencing (NGS) methods are rapidly replacing Sanger sequencing and targeted NGS panels to predict blood group antigens that include ABO and Rh are in development. Alignment of NGS sequence reads is more challenging for duplicated and homologous gene families including RHD/RHCE. The development of sequencing technology that allows longer read lengths and automated algorithms for interpretation will improve accuracy and availability for RH and other blood group genes. As entire genes can be analyzed, NGS may provide a solution for routine genetic determination of all key blood group antigens at a level of fidelity that cannot be achieved with current DNA-arrays [46]. NGS technology would overcome many of the limitations associated with DNA-array based assays and is successfully being applied to human leukocyte antigens (HLA) and human neutrophil antigens (HNA). In the future, improved access to cost-efficient red cell genotyping technology for both recipient and donor populations has the potential to be transformative for transfusion in patients with SCA.
8. Take away points.
For multiply transfused patients, especially those experiencing transfusion reactions or hemolysis, the additional information provided by DNA-based extended antigen typing is relevant for clinical decision making, antibody evaluation, and donor unit selection.
- The prevalence of altered RH alleles in patients with SCA suggests an important role for molecular methods to improve red cell matching in the Rh system.
- Determination of C status by genomics to identify altered C antigen in all SCA patients who type C+ by serology is recommended. For individuals predicted to express partial C antigen only, transfusion with C− units can reduce the risk of alloanti-C.
- RH genotyping is recommended when unexplained Rh antibodies are detected in patients despite Rh antigen matching.
- Donor unit selection for individuals with variant Rh antigens may be tailored to consider RH genotype matching on an individual case basis, particularly for those who are alloimmunized.
Reference blood group genomics laboratories use a combination of DNA-arrays and laboratory developed tests (LDTs) to identify relevant RH variants. RHD- and RHCE-specific DNA arrays targeting the more common variants are available, but additional PCR-based assays and exon-specific sequencing are needed for comprehensive RH analysis.
Clinical interpretation of genotyping can be a challenge. Immunohematology reference laboratories doing this testing as American Association of Blood Banks (AABB) accredited institutions can provide guidance for donor red cell selection.
Occasional discrepancies seen between serologic phenotype and genotype are most often due to gene mutations that weaken the antigen below serologic detection levels, mutations that silence expression of the antigen (null phenotypes), or due to stem cell transplantation or natural chimeras who inherit a mixed population of cells.
Acknowledgments
This work was supported by the Innovations in Clinical Research Award from the Doris Duke Charitable Foundation Grants #2011097 and #2015133 to STC and CMW, and National Institutes of Health U01 HL134696-01 to STC and CMW.
Footnotes
Author disclosures
The authors have no conflicts of interest to disclose.
References
- [1].Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. , Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members, JAMA 312 (2014) 1033–1048. [DOI] [PubMed] [Google Scholar]
- [2].Chou ST, Fasano RM, Management of patients with sickle cell disease using transfusion therapy: guidelines and complications, Hematol. Oncol. Clin. North Am 30 (2016) 591–608. [DOI] [PubMed] [Google Scholar]
- [3].Adams RJ, McKie VC, Hsu L, et al. , Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography, N. Engl. J. Med 339 (1998) 5–11. [DOI] [PubMed] [Google Scholar]
- [4].Adams RJ, Brambilla D, Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease, N. Engl. J. Med 353 (2005) 2769–2778. [DOI] [PubMed] [Google Scholar]
- [5].Fasano RM, Meier ER, Hulbert ML, Cerebral vasculopathy in children with sickle cell anemia, Blood Cells Mol. Dis 54 (2015) 17–25. [DOI] [PubMed] [Google Scholar]
- [6].Ware RE, Davis BR, Schultz WH, et al. , Hydroxycarbamide versus chronic transfusion for maintenance of transcranial doppler flow velocities in children with sickle cell anaemia-TCD With Transfusions Changing to Hydroxyurea (TWiTCH): a multicentre, open-label, phase 3, non-inferiority trial, Lancet 387 (2016) 661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ware RE, Schultz WH, Yovetich N, et al. , Stroke With Transfusions Changing to Hydroxyurea (SWiTCH): a phase III randomized clinical trial for treatment of children with sickle cell anemia, stroke, and iron overload, Pediatr. Blood Cancer 57 (2011) 1011–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].DeBaun MR, Gordon M, McKinstry RC, et al. , Controlled trial of transfusions for silent cerebral infarcts in sickle cell anemia, N. Engl. J. Med 371 (2014) 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chou ST, Liem RI, Thompson AA, Challenges of alloimmunization in patients with haemoglobinopathies, Br. J. Haematol 159 (2012) 394–404. [DOI] [PubMed] [Google Scholar]
- [10].Yazdanbakhsh K, Ware RE, Noizat-Pirenne F, Red blood cell alloimmunization in sickle cell disease: pathophysiology, risk factors, and transfusion management, Blood 120 (2012) 528–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Vichinsky EP, Earles A, Johnson RA, et al. , Alloimmunization in sickle cell anemia and transfusion of racially unmatched blood, N. Engl. J. Med 322 (1990) 1617–1621. [DOI] [PubMed] [Google Scholar]
- [12].Aygun B, Padmanabhan S, Paley C, Chandrasekaran V, Clinical significance of RBC alloantibodies and autoantibodies in sickle cell patients who received transfusions, Transfusion 42 (2002) 37–43. [DOI] [PubMed] [Google Scholar]
- [13].Castro O, Sandler SG, Houston-Yu P, Rana S, Predicting the effect of transfusing only phenotype-matched RBCs to patients with sickle cell disease: theoretical and practical implications, Transfusion 42 (2002) 684–690. [DOI] [PubMed] [Google Scholar]
- [14].Osby M, Shulman IA, Phenotype matching of donor red blood cell units for nonalloimmunized sickle cell disease patients: a survey of 1182 North American laboratories, Arch. Pathol. Lab. Med 129 (2005) 190–193. [DOI] [PubMed] [Google Scholar]
- [15].Afenyi-Annan A, Willis MS, Konrad TR, Lottenberg R, Blood bank management of sickle cell patients at comprehensive sickle cell centers, Transfusion 47 (2007) 2089–2097. [DOI] [PubMed] [Google Scholar]
- [16].Winkler AM, Josephson CD, Transfusion practices for patients with sickle cell disease at major academic medical centers participating in the Atlanta Sickle Cell Consortium, Immunohematology 28 (2012) 24–26. [PubMed] [Google Scholar]
- [17].Westhoff C, Ness PM, The promise of extended donor antigen typing, Transfusion 55 (2015) 2541–2543. [DOI] [PubMed] [Google Scholar]
- [18].Ambruso DR, Githens JH, Alcorn R, et al. , Experience with donors matched for minor blood group antigens in patients with sickle cell anemia who are receiving chronic transfusion therapy, Transfusion 27 (1987) 94–98. [DOI] [PubMed] [Google Scholar]
- [19].Rosse WF, Gallagher D, Kinney TR, et al. , Transfusion and alloimmunization in sickle cell disease. The Cooperative Study of Sickle Cell Disease, Blood 76 (1990) 1431–1437. [PubMed] [Google Scholar]
- [20].Vichinsky EP, Luban NL, Wright E, et al. , Prospective RBC phenotype matching in a stroke-prevention trial in sickle cell anemia: a multicenter transfusion trial, Transfusion 41 (2001) 1086–1092. [DOI] [PubMed] [Google Scholar]
- [21].Sakhalkar VS, Roberts K, Hawthorne LM, et al. , Allosensitization in patients receiving multiple blood transfusions, Ann. N. Y. Acad. Sci 1054 (2005) 495–499. [DOI] [PubMed] [Google Scholar]
- [22].O'Suoji C, Liem RI, Mack AK, et al. , Alloimmunization in sickle cell anemia in the era of extended red cell typing, Pediatr. Blood Cancer 60 (2013) 1487–1491. [DOI] [PubMed] [Google Scholar]
- [23].Nickel RS, Horan JT, Fasano RM, et al. , Immunophenotypic parameters and RBC alloimmunization in children with sickle cell disease on chronic transfusion, Am. J. Hematol 90 (2015) 1135–1141. [DOI] [PubMed] [Google Scholar]
- [24].Lasalle-Williams M, Nuss R, Le T, et al. , Extended red blood cell antigen matching for transfusions in sickle cell disease: a review of a 14-year experience from a single center, Transfusion 51 (2011) 1732–1739. [DOI] [PubMed] [Google Scholar]
- [25].Tahhan HR, Holbrook CT, Braddy LR, Brewer LD, Christie JD, Antigen-matched donor blood in the transfusion management of patients with sickle cell disease, Transfusion 34 (1994) 562–569. [DOI] [PubMed] [Google Scholar]
- [26].Chou ST, Jackson T, Vege S, et al. , High prevalence of red blood alloimmunization in sickle cell disease despite transfusion from Rh-matched minority donors, Blood 122 (2013) 1062–1071. [DOI] [PubMed] [Google Scholar]
- [27].Tormey CA, Stack G, The persistence and evanescence of blood group alloantibodies in men, Transfusion 49 (2009) 505–512. [DOI] [PubMed] [Google Scholar]
- [28].Food and Drug Administration, Transfusion/donation fatalities. http://www.fda.gov/BiologicsBloodVaccines/SafetyAvailability/ReportaProblem/TransfusionDonationFatalities/, (2016). [Google Scholar]
- [29].Chou ST, Westhoff CM, The role of molecular immunohematology in sickle cell disease, Transfus. Apher. Sci 44 (2011) 73–79. [DOI] [PubMed] [Google Scholar]
- [30].I.S.o.B.T. (ISBT), Red cell immunogenetics and blood group terminology. http://www.isbtweb.org/working-parties/red-cell-immunogenetics-and-blood-group-terminology/. [Google Scholar]
- [31].BGMUT, Blood Group Antigen Mutation Database (BGMUT). https://www.ncbi.nlm.nih.gov/projects/gv/rbc/xslcgi.cgi?cmd=bgmut%2Fhome&user_id=0&probe_id=0&source_id=0&locus_id=0&locus_group=3&proto_id=0&kit_id=0&dummy=0. [Google Scholar]
- [32].Reid ME, Hipsky CH, Hue-Roye K, Hoppe C, Genomic analyses of RH alleles to improve transfusion therapy in patients with sickle cell disease, Blood Cells Mol. Dis 52 (2014) 195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Scott ML, Voak D, Jones JW, et al. , A structural model for 30 Rh D epitopes based on serological and DNA sequence data from partial D phenotypes, Transfus. Clin. Biol 3 (1996) 391–396. [DOI] [PubMed] [Google Scholar]
- [34].Sippert E, Fujita CR, Machado D, et al. , Variant RH alleles and Rh immunisation in patients with sickle cell disease, Blood Transfus. 13 (2015) 72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Storry JR, Castilho L, Daniels G, et al. , International Society of Blood Transfusion Working Party on red cell immunogenetics and blood group terminology: Cancun report (2012), Vox Sang. 107 (2014) 90–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hashmi G, Shariff T, Seul M, et al. , A flexible array format for large-scale, rapid blood group DNA typing, Transfusion 45 (2005) 680–688. [DOI] [PubMed] [Google Scholar]
- [37].Casas J, Friedman DF, Jackson T, et al. , Changing practice: red blood cell typing by molecular methods for patients with sickle cell disease, Transfusion 55 (2015) 1388–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Reid ME, Lomas-Francis C, Olsson ML, The Blood Group Antigen Facts Book, Elsevier Academic Press, San Diego, CA, 2012. [Google Scholar]
- [39].Tournamille C, Meunier-Costes N, Costes B, et al. , Partial C antigen in sickle cell disease patients: clinical relevance and prevention of alloimmunization, Transfusion 50 (2010) 13–19. [DOI] [PubMed] [Google Scholar]
- [40].Westhoff CM, Vege S, Horn T, et al. , RHCE*ceMO is frequently in cis to RHD*DAU0 and encodes a hr(S) -, hr(B) -, RH:-61 phenotype in black persons: clinical significance, Transfusion 53 (2013) 2983–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Westhoff CM, Vege S, Halter-Hipsky C, et al. , DIIIa and DIII Type 5 are encoded by the same allele and are associated with altered RHCE*ce alleles: clinical implications, Transfusion 50 (2010) 1303–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ipe TS, Wilkes JJ, Hartung HD, et al. , Severe hemolytic transfusion reaction due to anti-D in a D+ patient with sickle cell disease, J. Pediatr. Hematol. Oncol 37 (2015) e1.35–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wilkinson K, Harris S, Gaur P, et al. , Molecular blood typing augments serologic testing and allows for enhanced matching of red blood cells for transfusion in patients with sickle cell disease, Transfusion 52 (2012) 381–388. [DOI] [PubMed] [Google Scholar]
- [44].Ribeiro KR, Guarnieri MH, da Costa DC, et al. , DNA array analysis for red blood cell antigens facilitates the transfusion support with antigen-matched blood in patients with sickle cell disease, Vox Sang. 97 (2009) 147–152. [DOI] [PubMed] [Google Scholar]
- [45].Noizat-Pirenne F, Tournamille C, Relevance of RH variants in transfusion of sickle cell patients, Transfus. Clin. Biol 18 (2011) 527–535. [DOI] [PubMed] [Google Scholar]
- [46].Lane WJ, Westhoff CM, Uy JM, et al. , Comprehensive red blood cell and platelet antigen prediction from whole genome sequencing: proof of principle, Transfusion 56 (2016) 743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]