

ABSTRACT
γδ T lymphocytes represent an emerging class of cellular immunotherapy with preclinical promise to treat cancer, notably neuroblastoma. The innate-like immune cell subset demonstrates inherent cytoxicity toward tumor cells independent of MHC recognition, enabling allogeneic administration of healthy donor-derived γδ T cell therapies. A current limitation is the substantial interindividual γδ T cell expansion variation among leukocyte collections. Overcoming this limitation will enable realization of the full potential of allogeneic γδ T-based cellular therapy. Here, we characterize γδ T cell expansions from healthy adult donors and observe that highly potent natural killer (NK) lymphocytes expand with γδ T cells under zoledronate and IL-2 stimulation. The presence of NK cells correlates with both the expansion potential of γδ T cells and the overall potency of the γδ T cell therapy. However, the potency of the cell therapy in combination with an antibody-based immunotherapeutic, dinutuximab, appears to be independent of γδ T/NK cell content both in vitro and in vivo, which minimizes the implication of interindividual expansion differences toward efficacy. Collectively, these studies highlight the utility of maintaining the NK cell population within expanded γδ T cell therapies and suggest a synergistic action of combined innate cell immunotherapy toward neuroblastoma.
KEYWORDS: γδ T cells, natural killer (NK) cells, neuroblastoma, dinutuximab, ADCC, adoptive cell therapy, allogeneic
Introduction
Adoptive cellular therapies (ACT) for cancer are commonly manufactured by genetically engineering αβ T lymphocytes to express a synthetic chimeric antigen receptor (CAR) for enhanced tumor recognition and cytoxic activity.1 CAR-engineered αβ T cells have shown clinical success for hematological malignancies,2 yet have been less effective as a solid tumor ACT, due to the overall paucity of solid tumor-specific antigens as well as the immune hostile solid tumor microenvironment (TME).3,4 Ex vivo expanded γδ T cells are an emerging class of cellular immunotherapy with tractable preclinical promise as an ACT approach to treat cancer.5,6 Notably, of all tumor infiltrating leukocytes, the genetic signature of γδ T cells was found to be most significantly correlated with favorable prognosis in solid tumors.7
γδ T cells express a variety of cell surface receptors (i.e. γδ-TCR, NKG2D, DNAM-1) that promote detection of and activation toward malignant cells via recognition of buterophylin on tumor cells with intracellular phosphoantigen accumulation or recognition of stress ligands preferentially expressed on the tumor cell surface.8 Therefore, γδ T cells detect and kill tumor cells independently of major histocompatibility complex (MHC) antigen recognition and without the requirement for co-stimulatory signals.9 Supporting their potential to counteract TME-mediated immune suppression often observed in solid tumors, γδ T cells produce inflammatory cytokines like TNF-α and IFN-γ,10 professionally present antigens to endogenous αβ T cells,11,12 and induce dendritic cell maturation.13 Subsets of expanded γδ T cells additionally express the FcγRIII receptor (CD16), required to mediate antibody dependent cellular cytotoxicity (ADCC), providing the possibility to synergize with monoclonal antibody treatments.14,15
We have developed a GMP compliant protocol for serum-free ex vivo expansion of the Vγ9Vδ2 subpopulation of γδ T cells from peripheral blood mononuclear cells (PBMCs) using zoledronate and IL-2.16 γδ T cells expanded in this fashion are bioactive against a variety of cancers in preclinical models including the pediatric solid tumor neuroblastoma, an aggressive extracranial solid tumor where more than half of high-risk patients relapse with incurable disease.17 Specifically, γδ T cells expanded from neuroblastoma patient apheresis products are directly cytotoxic toward neuroblastoma in vitro.18 Furthermore, the co-administration of the anti-GD2 monoclonal antibody dinutuximab or ch14.18/CHO with ex vivo expanded γδ T cells augmented cytotoxicity against neuroblastoma cells.18,19 The addition of temozolomide further enhanced anti-neuroblastoma cytotoxicity in combination with dinutuximab and γδ T cells, leading to complete tumor regression in aggressive mouse models of neuroblastoma.18
Despite the preclinical promise of γδ T cell ACT in neuroblastoma, the feasibility of sourcing an autologous patient-derived cell therapy is limited, due to high demand for apheresis products throughout the course of standard high-risk neuroblastoma therapy.20,21 High-risk neuroblastoma patients also demonstrate naïve T cell deficits following a single cycle of chemotherapy, suggesting not all patient-derived autologous ACTs will be potent.22 Healthy donor-derived γδ T cell expansions are a potentially superior “off-the-shelf” source as they are likely more potent and offer the opportunity for serial dosing to overcome any issues with persistence.23 We have recently adapted our ex vivo expansion protocol to include depletion of αβ T cells from allogeneic γδ T cell expansions. This modification yields a final αβ T lymphocyte contamination of <1% to minimize the risk for graft verses host disease.24
Significant healthy donor variability exists as it relates to the ex vivo expansion potential of γδ T cells.14,24,25 Specifically, γδ T cell expansion from healthy donor leukocytes stimulated with IL-2 and zoledronate varies donor-to-donor, with the final γδ T cell content of the expansion ranging from 20% to 80% of the total culture volume.24 The goal of the current study was to investigate the source of donor variability, the impact of the final γδ T cell percentage on cytotoxic potential, and the role of alternative immune cell subsets in the expanded cell therapy toward efficacy against neuroblastoma, both as monotherapy and in combination with dinutuximab. Additionally, characterization of the cellular constituents following expansion was performed to identify potential biomarkers of the the most potent cellular therapy and prospectively select the most promising healthy donors for γδ T cell expansions in future clinical trials.
Materials and methods
Neuroblastoma cell lines
Neuroblastoma cell lines, IMR5 and NLF, were obtained from the Children’s Oncology Group Childhood Cancer Cell Line Repository and cultured using RPMI 1640 (Sigma) medium completed with 10% Fetal Bovine Serum (Gemini) and 1% penicillin-streptomycin (Gemini) at 37°C in a humidified atmosphere with 5% CO2. Each cell line was STR genotyped (Texas Tech University Health Sciences Center) and the resulting identity was confirmed to match the COG cell line database (cccells.org). Cell lines were routinely verified to be free of mycoplasma contamination using the MycoAlert contamination kit (Lonza).
Healthy blood donors
PBMCs were obtained through whole blood collections from consenting volunteers through the Children’s Clinical and Translational Discovery Core (CTDC) at Emory University under the core’s IRB approved protocol (IRB00101797). To isolate PBMCs, approximately 40 mLs of whole blood was layered on Ficoll-Paque Plus (GE Healthcare Life Sciences) and subjected to density centrifugation. All donors that participated in this study were under age 50 and self-reported to be in good physical health.
Cell therapy expansion
PBMCs were expanded under our GMP-certified, serum-free protocol (Figure 1a) using zoledronate (Sigma) and IL-2 (PeproTech) in CTS™ OpTmizer™ T Cell Expansion medium (Fisher Scientific). On days 0, 3, 6, 9 and 12, PBMC/expansion cell number was determined using a hemocytometer count with trypan blue exclusion to determine viability. On day 6, expansions were depleted of αβ T cells as previously described.24 Briefly, αβ T cells were labeled with a biotinylated anti-TCR α/β antibody (Miltenyi Biotec) and then incubated with anti-biotin microbeads (Miltenyi Biotec) prior to capture using a LS column (Miltenyi Biotec). On days 0, 6 and 12, expansions were immunophenotyped for γδ T cell (CD3+γδ‐TCR+) and natural killer (NK) cell content (CD3−CD56+) using a Cytek Aurora 4 L (V, B, YG, R) cytometer. Unmixed data were exported and analyzed in FlowJo (v10.8). All information regarding antibodies for immunophenotyping is provided in Supplemental Table S1. Day 12 expanded cells were either used for analysis immediately or viably frozen as previously described.18 Frozen expanded cells were thawed into OpTmizer medium and allowed to rest in the presence of IL-2 (15.2 ng/mL) at 37°C for a minimum of 1 hour prior to experimentation.
Figure 1.
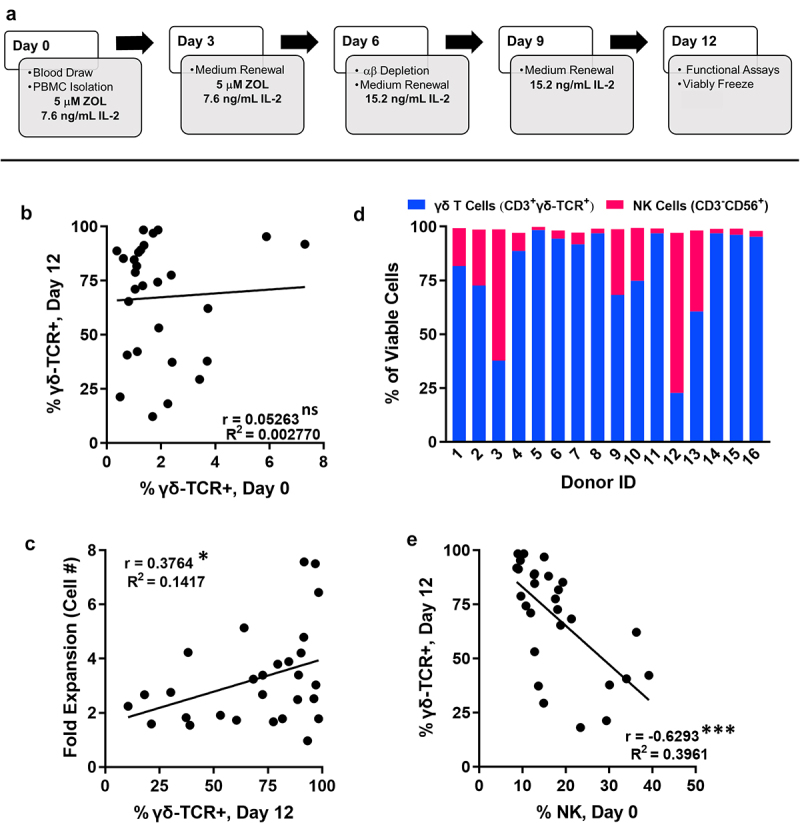
γδ T lymphocytes demonstrate variable interdonor expansion with NK cells identified as an additional cellular subset following zoledronate/IL-2 stimulation. A, Timeline depicting our GMP-compliant γδ T cell expansion protocol from healthy donor leukocytes. B, γδ T lymphocyte (viable CD3+γδ-TCR+ events as percent of total viable population) content in PBMCs on day 0 (used to seed expansion) vs. the final γδ T lymphocyte content of matched day 12 expansion. Data represent n = 28 independent expansions; not significant (ns), p = .7946. C, γδ T lymphocyte content of day 12 expanded cell therapy vs. matched fold expansion of cellular content (total cell number day 12/total cell number day 0). Data represent n = 28 independent expansions where fold expansion was tracked; *, p = .0484. D, Percentages of γδ T cells (blue) and NK cells (magenta) found within viable cellular expansions from 16 unique individual donors. E, NK lymphocyte (CD3−CD56+ events as percent of total viable population) content in PBMCs on day 0 vs. the final γδ T lymphocyte content of matched day 12 expansion. Data represent n = 28 independent expansions; ***, p = .0003).
Expansion immunophenotyping
γδ T cells and NK cells were characterized for markers of activation, inhibition, senescence, and exhaustion using flow cytometry. Expanded cells were washed with FACs buffer (1x PBS with 10% FBS, 0.1% sodium azide, 5 mM EDTA) and stained with ZombieNiR viability dye (BioLegend) for 30 min at room temperature according to the manufacturer’s protocol. Cells were washed and resuspended in FACs buffer containing FC receptor block (BD Biosciences) and incubated for 10 min at room temperature. Immediately following, cells were stained with the antibody panel (Supplemental Table S2) for 15 min at room temperature. After staining, cells were washed twice with FACs buffer and immediately analyzed for cell surface marker expression on a Cytek Aurora 5 L (UV, V, B, YG, R) instrument. Data analysis was performed using the Cytek SpectroFlo software.
Enrichment and depletion studies
Depletions of CD56+ or γδ-TCR+ cells were performed following manufacturer’s guidelines. Briefly, day 0 PBMCs or day 11–12 expanded cells were rinsed with 1X PBS containing 0.5% BSA and EDTA prior to incubation with anti-human γδ-TCR or CD56 biotinylated antibodies (Miltenyi Biotec) for 10 min at 4°C. Antibody-labeled cell suspensions were washed and incubated with anti-biotin magnetic microbeads (Miltenyi Biotec) for 15 min at 4°C. Cell suspensions were again washed and applied to a LS magnetic separation column (Miltenyi Biotec). The column flow through was collected and seeded for standard expansion if depletion was performed on day 0 or allowed to rest overnight in OpTmizer medium with added IL-2 (15.2 ng/mL) prior to immunophenotyping and cytotoxicity assay if depletion was performed on day 11–12.
Cytotoxicity assays
Flow cytometry-based assay. The in vitro cytotoxic potential of expanded γδ T and NK cells was measured against the neuroblastoma cell lines IMR5 and NLF. To distinguish between target and effector populations, neuroblastoma cells were labeled with Violet Proliferation Dye 450 (VPD450, BD Biosciences) following the manufacturer’s provided protocol. Labeled neuroblastoma cells were seeded in 24-well plates at 200,000 cells per well in 500 μL of HIcRPMI (RPMI 1640 completed with 10% heat-inactivated FBS and 1% penicillin-streptomycin). Cells were allowed to adhere overnight. On the following morning, expanded day 12 effector cells (expanded cells) were added to wells containing neuroblastoma target cells at designated effector:target ratios (i.e., 0:1, 1:1, 5:1, 10:1) in the presence and absence of dinutuximab to determine ADCC. Cells were co-incubated for a 4 h cytotox assay and then harvested. To harvest, supernatant was collected from each well. Remaining adherent target cells were detached using accutase (BioLegend) and pooled with the previously collected supernatant. Cells were washed and resuspended in annexin-binding buffer (BioLegend) containing annexin-V-APC antibody (BioLegend). Immediately pror to analysis, BD-ViaProbe (7-AAD, BD Biosciences) was to discriminate live vs. dead cells. Cytotoxicity was calculated by subtracting background cell death of each target cell line from the experimental sample.
Bioluminescence imaging (BLI)-based assay. Alternatively, the cytotoxic potential of expanded γδ T and NK cells against adherent neuroblastoma cells was also determined by BLI through measurement of light emitted from target cells expressing luciferase.26 Luciferase expressing IMR5 cells (IMR5lucGFP; courtesy of Dr. Robert Schnepp), were generated through lentivirus infection with GFP/luciferase plasmid.27 Prior to experimentation, it was confirmed that the amount of light (RLU) produced upon the addition of substrate luciferin (75 μg/mL, PerkinElmer) correlated with IMR5lucGFP cell number (Supplemental Fig. S1). IMR5lucGFP cells were seeded at 34,000 cells/well in 100 μL HIcRPMI and allowed to adhere overnight. On the following morning, expanded day 12 effector cells were added to wells containing neuroblastoma target cells at designated effector:target ratios (i.e., 0:1, 1:1, 5:1, 10:1) in the presence and absence of dinutuximab, 5 μg/mL. After 4 hours of co-incubation, luciferin was added to each well and incubated for 10 min prior to detecting luminescence on Synergy™ Mx Microplate reader (BioTek). Killing was determined by calculating specific lysis using the equation % specific lysis = 100*(spontaneous death RLU – test RLU)/(spontaneous death RLU – maximal killing RLU). Maximal killing was induced by lysing cells with 0.1% Triton solution. Effector-induced target cell death was also visually assessed and imaged by detecting GFP signal using a Lionheart FX microscope (BioTek).
Activation assays
The activation status of γδ T cells and NK cells was determined following combination with neuroblastoma cells in the presence and absence of dinutuximab, 5 μg/mL. IMR5 cells were labeled with VPD450 and seeded as described above for the flow cytometry-based cytotoxicity assay. On the following morning, expanded γδ T/NK cells were added to the plated neuroblastoma cells at a 1:1 effector:target ratio and activation/degranulation assay was executed as previously described.28 Briefly, cells were co-incubated for 1 hour prior to the addition of anti-human CD107a antibody and BD GolgiPlug (BD Biosciences) to capture degranulation and then for an additional 3 hours prior to collection of non-adherent cells. Cells were washed and stained with designated antibodies to characterize activation of γδ and NK lymphocytes (Supplemental Table S3). After staining, cells were washed twice with FACs buffer and immediately analyzed for cell surface marker expression on a Cytek Aurora 5 L (UV, V, B, YG, R) instrument. Unmixed data were exported and analyzed in FlowJo (v10.8.0).
Mouse xenograft studies
All animal studies were conducted in accordance with policies set forth by the Emory University Institutional Animal Care and Use Committee (IACUC) under an approved animal use protocol (PROTO201700089). Tumors were established in six-to-eight-week-old male and female NOD-scid IL2Rgammanull (NSG) mice (Jackson Laboratory) by subcutaneously injecting the right flank with a 1:1 mixture of Matrigel (Corning) and single cell tumor suspension containing 2 × 106 IMR5 cells. After 2 weeks, palpable tumors were detectable and tumor dimensions were measured every 2–3 days. Tumor volume was calculated using the formula L x W x H x π/6. When tumor volume reached 300–350 mm3, mice were evenly distributed between experimental arms and treatments were initiated. All mice received the chemoimmunotherapy backbone of dinutuximab (100 μg/ mouse, intravenous (IV), days 1, 7, and 13) and temozolomide (20 mg/kg, intraperitoneal (IP), days 2, 5, 8, 11, and 14). Between experimental arms mice received vehicle control (HIcRPMI, 100 μL), γδ T dominant cell therapy (2.5 x 106 cells, 100 μL HIcRPMI), or NK-dominant cell therapy (2.5 x 106 cells, 100 μL HIcRPMI) administered intravenously (IV) or intratumorally (IT) on days 2, 5, 8, 11, 14, and 17 of treatment schedule for a total of six cell therapy injections. When temozolomide and cell therapy were administered on the same day, temozolomide was dosed in the morning, at least 4 hours prior to injection of cell therapy in the afternoon. Originally, 4–5 mice were distributed across experimental groups to detect 50% differences in the group means, assuming sample standard deviations < 50% of the mean (α = 0.05, power = 0.8). A second confirmatory study was performed for intratumoral injections; the data were combined for presentation bringing the total enrolled to n = 7 for this arm of the study. Once tumor volume exceeded 2000 mm3 or reached ulceration, humane euthanasia was perfomed by CO2 asphyxiation followed by cervical dislocation.
Cell therapy trafficking studies: For preliminary investigations, mice with subcutaneous xenografts established to 300–350 mm3 received either a bolus dose of cells (20 x 106 cells, 100 μL HIcRPMI) from a day 12 γδ T cell expansion product (approximately 70% γδ T cell and 30% NK cell content) or vehicle control (HI cRPMI) injected intravenously. Following 8 hours, mice were sacrificed, and tumors were excised from flank and analyzed for immune cell content by flow cytometry (detailed below). For paired trafficking studies corresponding to complete treatment regimen, mice were treated with dinutuximab (days 1 and 7), temozolomide and cell therapy (days 2,5,8). Mice were given an additional dose of IV cell therapy on day 9. Four hours post day 9 administration, animals were sacrificed and tumors extracted for immune cell conent by flow cytometry. To do so, tumor tissue was weighed and dissociated into a viable single-cell suspension under mechanical pressure using GentleMacs platform (Miltenyi). The resulting cell suspension was collected and washed prior to lysing contaminating red blood cells with red blood cell lysis buffer (Abcam) under manufacturer’s protocol. The remaining cells were washed twice with FACs buffer and stained with anti-human antibodies for characterization of tumor-infiltrating immune cells (Supplemental Table S4). Cell suspensions were stained for 15 min at room temperature and then washed twice with FACs buffer prior to analysis on a Cytek Aurora 5 L (UV, V, B, YG, R) instrument. Unmixed data were exported and analyzed using Cytek SpectroFlo software.
Statistical analysis
GraphPad Prism (v9.0) was used for all statistical analysis. Where necessary, a student’s t test or analysis of variance (ANOVA) with additional post hoc test was used to assess statistical significance. A log rank (Mantel-Cox) test was performed on the Kaplan-Meier survival plots to determine statistically distinct curves. In all experiments, statisitcal significance was defined as p < .05.
Results
Peripheral blood mononuclear cells (PBMCs) stimulated with zoledronate and IL-2 produce phenotypically diverse γδ T cell expansions among healthy adult donors.
PBMCs were isolated from whole blood of healthy adults and expanded with zoledronate and IL-2, using our previously established GMP-compliant protocol (Figure 1a,).[16] On day 6 of ex vivo expansion, alloreactive αβ T cells were depleted to enhance the safety margin for allogeneic administration. Similar to previous investigations, day 6 αβ-depletion consistently resulted in <1% αβ T cell contamination.24
To evaluate for healthy donor γδ T cell expansion differences, 47 expansions representing 16 independent donors were characterized. The resulting cell therapy expansions demonstrated significant proliferation of γδ T cells compared to the γδ T cell content of baseline PBMCs (p < .0001); however, the final γδ T cell (CD3+ γδ-TCR+) concentration on the twelfth day of expansion was highly variable and ranged from 12.2% to 98.4% of the total expanded population (Supplemental Fig. S2). It was hypothesized that the starting γδ T cell content of PBMCs may contribute to the final γδ T cell content of expanded cells. However, no correlation between the starting γδ T cell content of PBMCs (day 0) and the expanded day 12 γδ T cell concentration was observed (p = .7946, Figure 1b). The percentage of γδ T cells within the expansion on day 12 weakly correlated with the overall volume of cell therapy (defined as total number of cells) produced (p = .0484, Figure 1c).
The other substantial cellular constituent identified within the expansions was phenotyped as CD3−CD56+ natural killer (NK) lymphocytes (Figure 1d). When characterizing interdonor (between donors) γδ T cell expansion amongst 16 unique donors, we found that the majority of donors expanded predominant (> 50%) γδ T cell content, with only 2/16 expansions yielding > 50% NK cell content (Figure 1d). Unlike γδT cells, the NK cell content of the PBMCs used to seed the expansion on day 0 inversely correlated with the final γδT cell content post-expansion (p = .0003), such that greater NK cell percentage in day 0 PBMCs resulted in expansions with lower overall γδ T cell content (Figure 1e).
Intradonor expansion consistency and cellular stability through cryopreservation support reliable, multi-dose administration. The ideal healthy donor for γδ T cell ACT is one who can repeatedly donate a potent expansion over multiple PBMC harvests to support a consistent and reliable cell therapy. Furthermore, expanded cells should remain viable with substantive antitumor potency both before and after cryopreservation. For the majority of donors, intradonor (from a single donor) expansions were found to be fairly consistent in γδ T cell content over time (Figure 2a). For example, Donor 1, a predominant γδ T cell expander, and Donor 3, whose PBMCs produced expansions with robust NK cell content, yielded similar intradonor expansions over the course of 5 months and 12 months, respectively (Figure 2a). However, in the case of Donor 2, the γδ T cell to NK cell composition was variable, with phenotypically different cellular content in the expansions, even occurring within the same month (Figure 2a). Evaluation of the Donor 2 PBMC samples used to seed the November 13, 2019 and November 26, 2019 expansions revealed a lower abundance of T cells (both αβ and γδ) and greater NK cell content in the Nov. 26, 2019 pre-expansion PBMCs that produced an NK dominant expansion (Figure 2b). Importantly, one freeze and thaw cycle of expanded cells did not dramatically shift their γδ T cell to NK cell make-up, although a small proportion of NK cells was lost after one freeze/thaw (Figure 2c).
Figure 2.
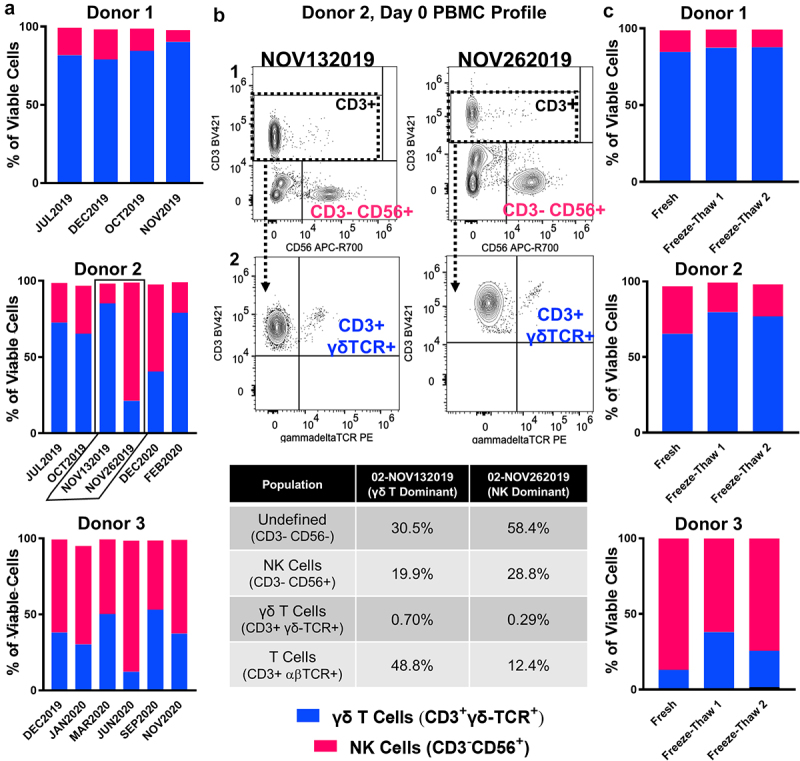
Intradonor expansion consistency and stability following cryopreservation support clinical utility of γδ T cell therapy. A, Percentages of γδ T cells (blue) and NK cells (magenta) found within expansions from three individual donors (Donor 1, 2, 3) across independent blood collections performed over the course of 6–12 months. Donors selected are representative of n = 16 individual donors expanded over the course of these investigations and depict expansions with high (Donor 1), moderate (Donor 2), and low γδ T cell content (Donor 3) on day 12. B, Representative immunoprofiling strategy used to characterize PBMCs seeded for γδ T cell expansion. Plots in row 1 (CD56 vs. CD3) were used to define NK content (CD3−CD56+, magenta). CD3+ events were selected (dashed box) and carried forward to gate γδ T cells (CD3+γδ-TCR+, blue) in row 2 (γδ-TCR vs CD3). The specific plots provided depict back-to-back expansions from Donor 2 (NOV132019, NOV262019), highlighted by black outline in A. Populations of interest are quantifed directly below in table. C, Percentages of γδ T cells (blue) and NK cells (magenta) found within expansions from three donors (Donor 1, 2, 3) prior to (Fresh) and after (Freeze-Thaw 1 and 2) cryopreservation. Two aliquots from the same freeze point were independently thawed and characterized on different days for replicate comparison (Freeze-Thaw 1 vs. Freeze-Thaw 2).
Immunoprofiling reveals potent cytotoxic phenotypes for both expanded γδ T and NK lymphocytes. Day 12 expansions were immunophenotyped to characterize both the γδ T cells (CD3+γδ-TCR+) and NK cells (CD3−CD56+) present (Figure 3a, Supplemental Fig. S3). For comparison, expansions with high γδ T cell content (>50% CD3+γδ-TCR+) were defined as “γδ T dominant” while those with high NK content (>50% CD3−CD56+) were represented as “NK dominant.” In line with our previous observations, the bulk γδ T cells within expansions represented an effector memory phenotype (CD45RA−CCR7−).18,24 Here, we also phenotypically characterized NK cells within expansions and found the predominant NK phenotype to be CD56brightCD16+, which was also independent of an expansion’s γδ T:NK content (Figure 3b). The expanded NK phenotype differed from the standard CD56dimCD16+ and CD56brightCD16− NK phenotypes observed pre-expansion (Figure 3b).
Figure 3.

Expanded γδ T cells and NK cells demonstrate potent cytotoxic effector phenotypes. A, Schematic depicting technique used to immunoprofile individual γδ T cells and NK cells within complete expansions. Throughout Figure 3, characterization of the γδ T population is provided in blue and characterization of the NK population is provided in magenta. B, Representative immunoprofiling and gating strategy characterizing effector status (CD16−/+ vs CD56dim/bright) of NK cells on day 0 (PRE-EXPANSION) and day 12 (POST-EXPANSION). Percent of expanded γδ T lymphocytes (left, blue) or NK lymphocytes (right, magenta) that express (c) activating receptors NKG2D and DNAM, (d) inhibitory receptors KIR2DL1 and NKG2A, (e) exhaustion markers PD-1, TIM3, CTLA-4 and TIGIT, or (f) the apoptotic receptor FAS and its activating ligand (FasL) following 12 days of expansion and cryopreservation. For C-F, Donor symbols are provided so marker expression can be compared across individual expansions and between γδ T and NK cellular components. Data represent n = 8 individual expansions from different donors.
Both expanded γδ T cells and NK cells expressed uniformly high levels (>90% positive) of activating receptors that may promote tumor recognition, including CD314 (NKG2D) and CD226 (DNAM-1) (Figure 3c). Although both were identified post-expansion, γδ T cells and NK cells expressed lower levels of inhibitory receptors and exhaustion markers compared to activating receptors (Figure 3d and 3e). Of note, CD159a (NKG2A), a classical inhibitory member of the NKG2 family, was identified on both expanded γδ T cells and NK cells (Figure 3d). CD279 (PD-1), CD366 (TIM3) and TIGIT were the most highly expressed exhaustion markers identified post-expansion; increased levels of TIGIT were observed on the expanded NK cell population compared to expanded γδ T cells (Figure 3e). Additionally, both expanded γδ T cells and NK cells expressed CD95 (Fas), with γδ T cells at a higher frequency than NK cells, and lower CD178 (FasL), both markers involved in apoptotic signaling. (Figure 3f).
Anti-neuroblastoma potency of expanded immune cells correlates with overall NK cell content in vitro. γδ T cells expanded from neuroblastoma patient apheresis products are potent toward human-derived neuroblastoma cell lines both in vitro and in vivo when combined with dinutuximab and low-dose temozolomide.18 However, expanded immune cells used in those prior investigations were not depleted of αβ T cells and did not account for the impact of NK cells toward overall potency. Ex vivo γδ T cell expansions depleted of αβ T cells successfully target and kill neuroblastoma cell lines in vitro. NK dominant expansions (NK cells ≥50% of the total expanded cells), induced significantly higher levels of cell death against both IMR5 and NLF neuroblastoma cell lines at 1:1 and 5:1 effector:target ratios, respectively (Figure 4a, Supplemental Fig. S4A and S4B). A significant positive correlation was observed between anti-neuroblastoma cytotoxic activity and overall NK content in the expanded cells (p = .0033, Figure 4b). NK cells preferentially activated in the presence of neuroblastoma, marked by significantly increased NK cell surface levels of CD107a (degranulation, p < .0001), compared to no observed increase in γδ T cells when complete expansions where co-cultured with IMR5 cells (p > .9999, Figure 4c).
Figure 4.
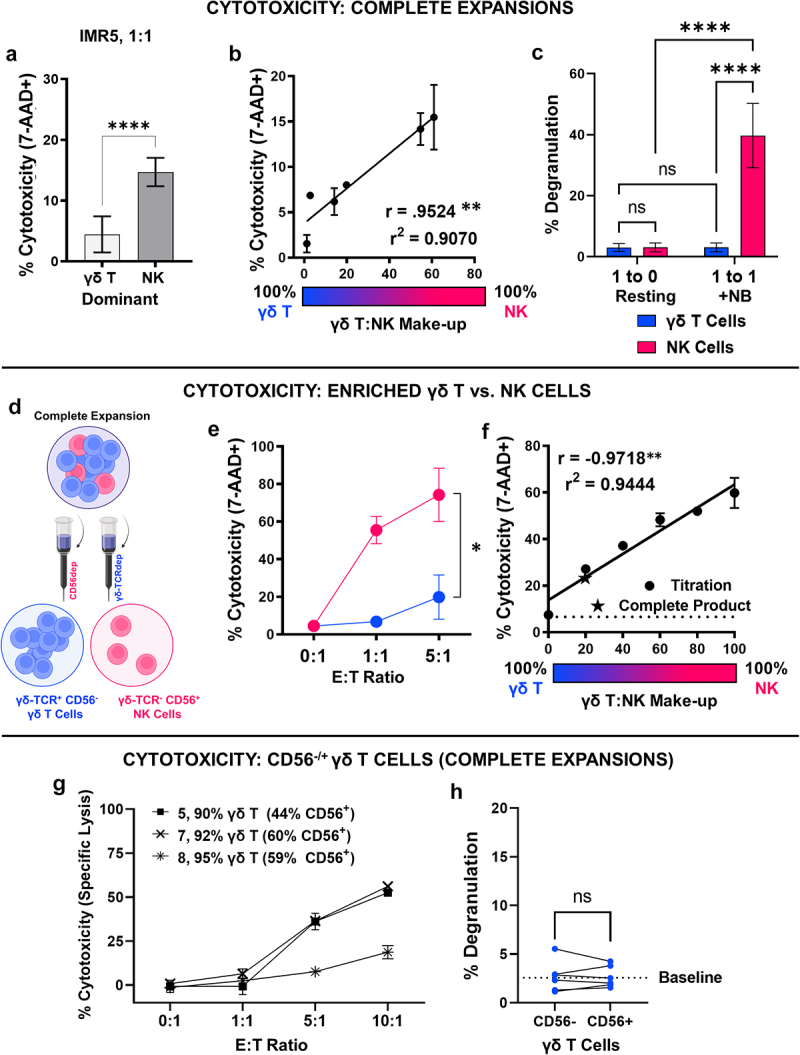
NK cell content predicts in vitro cytotoxic potential of γδ T cell expansions against neuroblastoma. A, Effector-induced cell death represented as percent of neuroblastoma cells positive for 7-AAD following a 4-hour cytotoxicity assay with either γδ T cell dominant or NK cell dominant expansion. IMR5 cells were co-cultured at 1:1 effector:target ratios with expanded immune cells. Data represent mean ± SD with n = 3 expansions per condition; ****, p < .0001. B, Effector-induced cell death following a 4-hour cytotoxicity assay with expansions defined by varying γδ T:NK cell content. Each expansions’s percent NK cell content is mapped against the percent of IMR5 (target) cells found to be positive for 7-AAD. Data represent mean ± SD from n = 6 independent donors/expansions; **, p = .0033. C, Degranulation of γδ T cells and NK cells within complete expansions following co-culture with IMR5 cells at 1:1 effector:target ratio for 4 hours compared to resting controls (effector only, 1:0 effector:target). Data represent mean ± SD of n = 5 independent expansions/donors; ****, p < .0001, ns, p > .9999. D, Schematic depicting the negative selection strategy used to enrich either γδ T cells or NK cells from a complete expansion containing both populations. E, Effector-induced cell death of IMR5 cells following a 4-hour cytotox with either γδ T cells (CD56−) or NK cells enriched from a complete day 12 expansion. Data represent mean ± SD from n = 3 independent expansion enrichments; *, p = .02. F, Effector-induced cell death of IMR5 cells following a 4-hour cytotoxicity assay with enriched γδ T cells and NK cells admixed together at set ratios (i.e. 20% NK, 80% γδ) to yield a final 1:1 effector:target ratio. Each titration’s percent NK cell content is mapped against the percent of IMR5 (target) cells found to be positive for 7-AAD. Data represent mean ± SD of experimental duplicates; **, p = .0012. Experiment was repeated with a second donor and analysis is provided in Supplemental Fig. S4D G, Effector-induced cell death represented as specific lysis of neuroblastoma cells measured using BLI-based cytotox assay following 4 hours of effector/target co-culture. Data represent mean ± SD from n = 3 donors (5, 7, 8) with expansionss containing >90% γδ T cell content but varying CD56 expression. H, Degranulation of CD56− or CD56+ γδ T cells from same expansion following co-culture with IMR5 cells at 1:1 effector:target ratio for 4 hours. A baseline average for resting CD107a expression found in γδ T cells in the absence of neuroblastoma targets is provided as dashed line for reference. Data represent mean ± SD of n = 5 independent expansions/donors. Cells from the same expansion are connected for direct comparison; ns, p = .9670.
To confirm the differing cytotoxic potentials of co-expanded γδ T and NK cells toward neuroblastoma, NK cells were enriched by depleting the cellular expansion of γδ‐TCR+ cells, and separately, γδ T cells were enriched by depleting the complete cell expansion of CD56+ cells. Negative selection following cellular expansion, yielded isolated populations of CD56− γδ T cells and NK cells for comparison (Figure 4d, Supplemental Fig. S4C). Isolated NK cells induced significantly more neuroblastoma cell death at both 1:1 and 5:1 effector:target ratios when compared to isolated CD56− γδ T cells from the same expansion (p = .02, Figure 4e). Enriched NK and γδ T cell populations were then admixed at various ratios, and target cell death was enumerated by 7-AAD positivity. In two separate donor experiments, significant linear relationships were found between the ratio of CD56− γδ T cells to NK cells present and target cell death induced, with greater NK content yielding greater neuroblastoma cytotoxicity (p = .0012, Figure 4f, Supplemental Fig. S4D).
Since CD56 was used as the selection marker to deplete NK cells from the cellular expansions, CD56+ γδ T cells were likely simultaneously removed (Figure 4d), which may in turn underestimate enriched γδ T cell potency through depletion of potent CD56+ γδ T cell subsets.29 To investigate the role of CD56+ γδ T cells, the cytotoxicity of expanded cells predominantly populated by γδ T cells (>90%) with varying levels of CD56 expression was compared. Two donors (7, 8) with similar levels of CD56+ γδ T cells (around 60%) had different cytotoxicity curves, with Donor 7 being more potent than Donor 8. A third donor (5) with lower levels of CD56+ γδ T cells (44%) showed increased tumor cell killing compared to Donor 8 that contained roughly 15% higher CD56+ γδ T cell content (Figure 4g). No degranulation was observed in either CD56+ or CD56− γδ T cells compared to background when co-incubated with neuroblastoma cells (p = .9670, Figure 4h).
NK cells do not expand independently of γδ T cells using stimulation with zoledronate and IL-2. Lymphocytes expressing either the γδ-TCR (γδ T cells) or CD56 (NK cells) were depleted from starting PBMC populations using negative selection to investigate the expansion potential of each population independent of one another (Figure 5a, Supplemental Fig. S5). PBMCs depleted of CD56+ cells on day 0 proliferated similarly to the complete PBMC control, while depletion of γδ-TCR+ cells on day 0 caused a sharp decrease in overall expansion potential (p = .0543, Figure 5b). The final composition of all expansions depleted of CD56+ cells on day 0 neared 100% γδ T cells on day 12 with poor anti-neuroblastoma cytotoxicity (Figure 5c). While day 0 γδ-TCR+ depleted PBMCs consistently failed to expand in number, the cells present on day 12 mainly represented NK cells that maintained strong anti-neuroblastoma cytotoxicity (Figure 5c).
Figure 5.
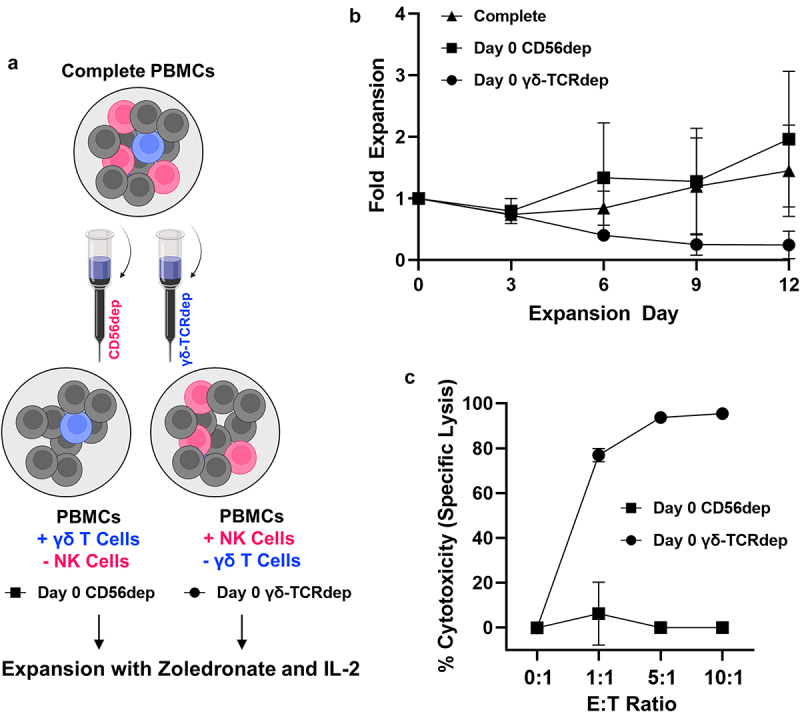
The presence of γδ T lymphocytes facilitates NK cell expansion under zoledronate and IL-2 stimulation. A, Schematic depicting negative selection strategy used to deplete CD56+ or γδ-TCR+ cells from PBMCs prior to expansion. B, Fold expansion of cellular content (total cell number day 0, 3, 6, 9, or 12/total cell number day 0) following CD56 (■) or γδ-TCR depletion (●) of PBMCs on day 0 compared to complete PBMCs (▲). Data represent mean ± SD from n = 3 independent depletion expansions with multiple donors. C, Effector-induced cell death represented as specific lysis of neuroblastoma cells measured using BLI-based cytotoxicity assay following 4 hours of effector and target co-culture. Effector cells were expanded from either day 0 CD56 depleted PBMCs yielding a predominant γδ T cell population (■) or day 0 γδ-TCR depleted PBMCs yielding a predominant NK cell population (●). While NK cells did not “expand” independently of γδ T cells, marked by decrease in total fold change (b), enough viable cells were present after 12 days to complete cytotoxicity assay for one donor. Data represent mean ± SD from n = 1 donor.
γδ T cells demonstrate enhanced potency in combination with anti-GD2 immunotherapy. γδ T cells and NK cells within all expansions expressed the antibody-recognizing cell surface receptor CD16, which is necessary for antibody dependent cellular cytotoxicity (ADCC). Therefore, the potency of the expanded cells when given in combination with dinutuximab, the humanized monoclonal anti-GD2 antibody used to treat patients with high risk neuroblastoma, was evaluated against two neuroblastoma cells lines, one with high expression of GD2 (IMR5) and one with low GD2 (NLF) (Supplemental Fig. S6A). Dinutuximab enhanced the potency of γδ T cell expansions against GD2high IMR5 with no increased cytotoxicity observed against GD2low NLF cells (Supplemental Fig. S6B). Expansions with varying γδ T:NK lymphocyte ratios from different donors were tested in combination with dinutuximab against IMR5 cells. Results showed that the potency of expanded immune cells when combined with dinutuximab is agnostic of γδ T cell or NK cell content (Figure 6a, Supplemental Fig. S6C). To understand the lack of variation in cytotoxic potential due to γδ T:NK content in the presence of dinutuximab, CD16 expression was queried specifically on γδ T cells from both γδ T dominant and NK dominant expansions. In all cases, γδ T cells demonstrated expression of CD16, with percent positivity ranging from 31% to 86% of the expanded population (Figure 6b). Considering CD16 expression among γδ T cells may contribute to enhanced cytotoxicity in combination with anti-GD2 immunotherapy, complete expansions were profiled for activation against neuroblastoma in the presence of antibody. In combination with dinutuximab compared to administration as a single agent, γδ T cells within a complete expansion demonstrated preferential activation marked by enhanced CD107a cell surface expression as a surrogate for degranulation (p = .0063, Figure 6c). In contrast, the activation of NK cells within complete cellular expansions was not further enhanced by the addition of dinutuximab (p = .3701, Figure 6c).
Figure 6.
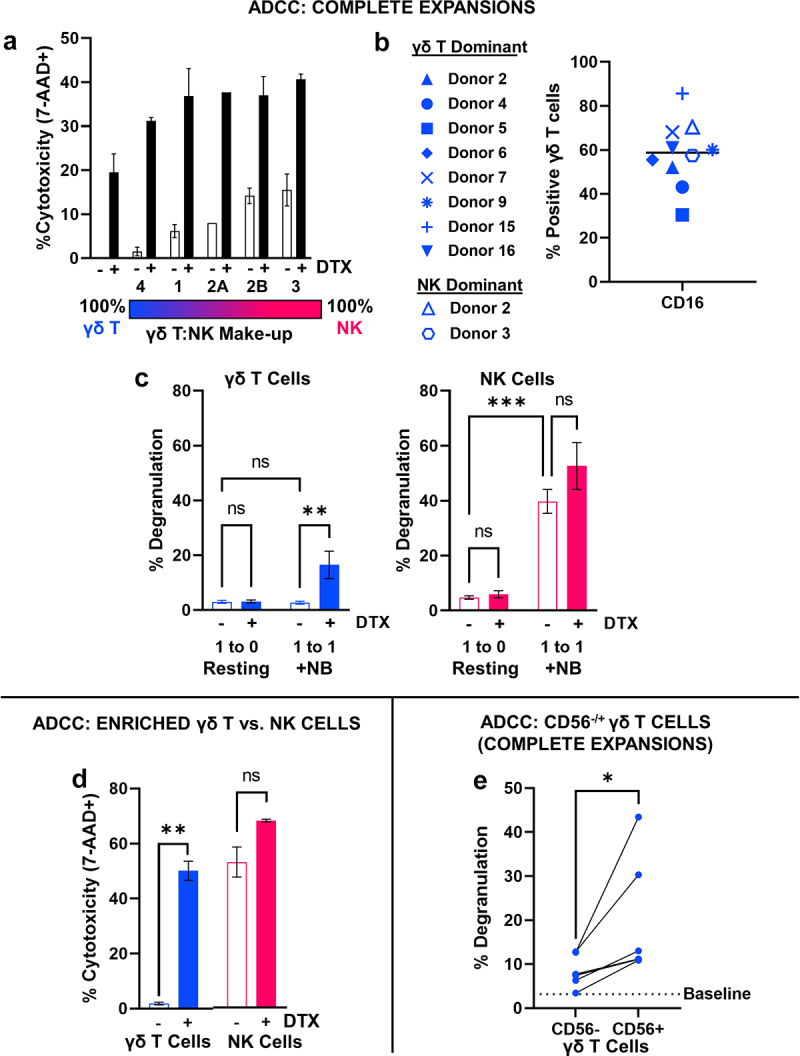
Combination with dinutuximab promotes in vitro anti-neuroblastoma activity of γδ T cells. A, Effector-induced cell death (percent 7-AAD+) of IMR5 cells following a 4-hour cytotoxicity assay with γδ T cell expansions defined by varying NK cell content in the absence (open) and presence (closed) of dinutuximab, 5 μg/mL. Data represent mean ± SD of n = 5 independent expansions from 4 different healthy donors. Donor 2 provides comparison of same donor but low vs. high NK content post-expansion. B, CD16 (FCγIII) expression on γδ T cells following 12 days of expansion. γδ T cells within NK cell dominant expansions are characterized by an open symbol. Donor symbols are provided so marker expression can be compared between donors. C, Degranulation of γδ T cells (left, blue) or NK cells (right, magenta) within complete expansions following co-culture with IMR5 cells at 1:1 effector:target ratio for 4 hours in the absence (open) and presence (closed) of dinutuximab, 5 μg/mL. Data represent mean ± SD of n = 5 independent expansions/donors; **, p = .0063, ***, p = .0003, ns, p = .3701. D, Effector-induced cell death of IMR5 cells represented by 7-AAD positivity following a 4-hour cytotoxicity assay with either γδ T cells (CD56−) or NK cells isolated from a complete day 12 expansion. Data represent mean ± SD from n = 2 independent expansion depletions; **, p = .003, ns, p = .060. E, Degranulation of CD56− or CD56+ γδ T cells within same expansion following co-culture with IMR5 cells at 1:1 effector:target ratio for 4 hours in the presence of dinutuximab, 5 μg/mL. A baseline average for resting CD107a expression in γδ T cells incubated with dinutuximab but absent of target neuroblastoma cells is provided for reference. Data represent mean ± SD of n = 5 independent expansions/donors. Cells from the same expansion are connected by line for direct comparison; *, p = .0453.
To confirm the differing cytotoxic potentials of co-expanded γδ T and NK cells toward neuroblastoma in combination with dinutuximab, neuroblastoma cells were cocultured with either γδT cells (CD56−) or NK cells enriched from a single expansion (Figure 4d) in combination with dinutuximab. While dinutuximab modestly elevated the cytotoxic potential of already potent NK cells, it caused an approximate 50% increase in neuroblastoma cell death in vitro when combined with isolated expanded γδT cells (CD56−) (p = .003, Figure 6d). Specifically considering the role of CD56+ γδ T cells, which are disguised by the enrichment strategy, the degranulation of CD56+ and CD56− γδ T cells was evaluated in complete expansions in combination with dinutuximab against IMR5 cells. Compared to resting levels of CD107a expression, both CD56+ γδ T cells and CD56− γδ T cells demonstrated increased levels of cell surface degranulation after co-culture with neuroblastoma in the presence of dinutuximab, and CD56+ γδ T cells had significantly higher levels of activation compared to their CD56− counterparts from the same expansion (p = .0453, Figure 6e).
Expanded γδ T/NK cells impart sustained tumor regression of neuroblastoma xenografts in vivo agnostic to γδ T:NK composition when administered intratumorally in combination with IV chemoimmunotherapy. In vitro assays lack the complexity required to account for in vivo persistence and tumor trafficking of expanded γδ T cell therapies. To evaluate the antitumor efficacy of the different cellular expansions, IMR5 cells were subcutaneously xenografted in the flank of NOD-scid IL2Rgammanull (NSG) mice. The ability of γδ T cells and NK cells to traffic to the tumor microenvironment following intravenous (IV) administration of a complete cell therapy expansion (70% γδ, 30% NK) was confirmed using a bolus cell therapy (20 x 10^6 cells) dose. Eight hours post administration, both γδ T cells and NK cells were found within the tumor but in limited numbers (Supplemental Fig. S7).
To define how cellular content contributes to potency in vivo, a γδ T cell dominant expansion (>90% γδ T cell composition, “γδ T”) and a dominant NK cell expansion (>75% NK cell composition,) were combined in separate arms of an in vivo preclinical investigation in combination with temozolomide and dinutuximab (“chemoimmunotherapy”) found to be most effective in previous preclinical investigations using patient-derived γδ T cell expansions (Figure 7a and 7b).[18] Of note, to manufacture sufficient cells to support the demands of the present animal trial, starting with less than 40 mL of whole blood, three donor expansions (from Donor 3) were required to generate the NK cell expansions where two donor expansions (from Donor 6) were required to generate sufficient γδ T cell dominant expansions. Consistent with prior in vitro results, NK dominant expansions outperformed γδ T cell dominant expansions in the absence of dinutuximab in vitro (Figure 7c, -DTX). However, both expansion types were equally effective at a 5:1 effector:target ratio in combination with dinutuximab in vitro prior to cryopreservation for subsequent in vivo use (Figure 7c, +DTX).
Figure 7.
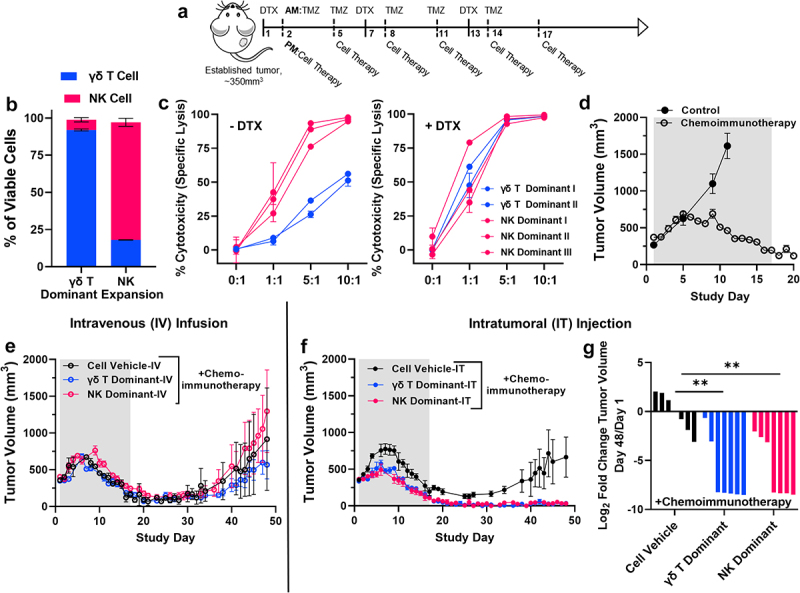
γδ T cell therapy in combination with dinutuximab in vivo supports utility agnostic of γδ T/NK lymphocyte content. A, Schematic depicting therapy protocol followed for in vivo investigations; dinutuximab (DTX, 100 μg) was dosed intravenous, temozolomide (TMZ, 20 mg/kg) was dosed intraperitoneal, cell therapy (γδ T dominant or NK dominant) was dosed intravenous (IV) or intratumoral (IT) depending on experimental conditions defined below. B, Phenotypes, defined by γδ T cells and NK cells, of expansions used as cellular therapy in subsequent in vivo investigations. Data represent mean ± SD of n = 2 independent expansions for γδ T dominat therapy and n = 3 independent expansions for NK dominant therapy. C, Effector-induced cell death represented as specific lysis of IMR5 cells measured using BLI-based cytotoxicity assay following 4 hours of co-culture. Effector cells were from either a γδ T cell dominant expansion (blue) or NK cell dominant expansion (magenta) defined in B. Left panel represents cytotoxicity assay performed without dinutuximab, while right panel represents the addition of dinutuximab at 5 μg/mL. Data represent mean ± SD for n = 2 expansions used for γδ T cell therapy and n = 3 expansions used for NK cell therapy. D, Average tumor volume curve demonstrating effect of dinutuximab and temozolomide (chemoimmunotherapy) treatment on IMR5 tumor growth compared to control tumors over time. Data represent mean ± SEM for n = 4 mice for control group and n = 12 mice for chemoimmunotherapy group. Average tumor volume curve demonstrating tumor growth for mice treated with either γδ T cell dominant or NK cell dominant cell therapy or cellular vehicle (eE) intravenously (IV) or (fF) intratumorally (IT) plus chemoimmunotherapy. Data represent mean ± SEM for n = 4 mice per arm for intravenous study and n = 7 mice per arm for intratumoral study. Shaded area in both graphs represents portion of study timeline while mice received treatment (days 1 through 17). G, Log2 fold change in tumor volume comparing day 48 measurements with those from day 1 of study schedule. Day 48 was chosen for statistical analysis as it was the last time point recorded before a mouse was removed from study due to excessive tumor burden (≥ 2000 mm3) in the cellular vehicle arm. Each bar represents a tumor-bearing mouse with n = 7 mice per arm comparing mice treated with cellular vehicle compared to γδ T cell dominant or NK cell dominant cell therapy; **, p = .0013 γδ T, **, p =.0032 NK.
All mice with established IMR5 subcutaneous tumors that received the chemoimmunotherapy tumor priming regimen of dinutuximab and temozolomide demonstrated tumor regression by day 12 compared to unhindered growth of control IMR5 tumors (Figure 7d). Intravenous infusion of a γδ T cell or an NK cell expansion in combination with the chemoimmunotherapy conditioning showed transient tumor regression followed by tumor regrowth similar to chemoimmunotherapy alone treated mice (Figure 7e). A limited accumulation/persistence of both cell types was found at the tumor site after 3 rounds of cell therapy injections (Supplemental Fig. S8). We hypothesized that insufficient tumor accumulation of immune cells limited the efficacy of our γδ T/NK cell therapy and performed intratumoral injections of the different NK and γδ T cell dominant expansions with the same chemoimmunotherapy backbone from above to assess the potency of each type in the in situ tumor environment. While intratumoral γδ T or NK cellular therapy alone or in combination with temozolomide or dinutuximab were ineffective at achieving complete regression compared to vehicle treated controls (Supplemental Fig. S9), both intratumoral γδ T cell or NK dominant cell therapy in combination with systemic dinutuximab (IV) and temozolomide (IP) induced complete and sustained tumor regression compared to intratumoral vehicle plus chemoimmunotherapy treated mice (Figure 7f). Tumor regression was significantly greater in cell therapy treated mice compared to those treated with the cellular vehicle as control (p = .0013 γδ T, p = .0032 NK; Figure 7g), and an absence of tumors persisted out to 50 days after therapy enrollment and at least 30 days after therapy completion. Ultimately, a significant prolongation of survival was obserbed for mice that received either cell therapy compared to those that did not (p = .0072 γδ T Dominant, p = .0229 NK Dominant; Supplemental Fig. S10).
Discussion
We have previously demonstrated the utility of patient-dervied, ex-vivo expanded γδ T cells to treat preclinical models of neuroblastoma in combination with immunotherapy (dinutuximab) and chemotherapy (temozolomide),18 which provided the framework to translate ex vivo expanded allogeneic γδ T cell therapy in combination with chemoimmunotherapy for children with refractory or relapsed neuroblastoma. Although our published preclinical data defines that γδ T cells expanded from neuroblastoma patients can effectively kill neuroblastoma cell lines in mouse models, others have shown that naïve T cells from therapy-treated neuroblastoma patients have significant defects that may limit their potency as a cell therapy.22 Given γδ T cells make up only 1–5% of peripheral blood mononuclear cells (PBMCs),30 it is necessary to procure sufficient cells for expansion through apheresis. Newly diagnosed high-risk neuroblastoma patients undergo apheresis during induction therapy, after receiving two cycles of chemotherapy,21 stressing that a patient-derived γδ T cell therapy would be expanded from apheresis products isolated after chemotherapy and may therefore yield sub-optimal therapeutic efficacy.22 There is also an increased demand for stem cell rescues in upfront high-risk neuroblastoma therapy (for standard of care tandem autologous stem cell transplants and sometimes 131I-MIBG therapy), potentially limiting residual apheresis units to manufacutre patient-derived ACTs. This limitation can be overcome by capitalizing on the MHC-independent nature of γδ T cells and substituting an allogeneic, healthy donor-derived cell therapy in place of the autologous patient-derived product.31,32 In this light, the overarching goal of the present study was to define the feasibility and utility of sourcing an allogeneic γδ T cellular therapy from healthy adult donors, while identifying potential biomarkers of robust and potent anti-neuroblastoma activity.
As previously noted, ex vivo γδ T cell expansion from healthy donor PBMCs tends to be highly variable at the interindividual level.12,14,24 The present investigations confirmed this finding, and now demonstrate that the composition of immune cell therapy expanded from healthy donor leukocytes with zoledronate and IL-2 can be defined by two predominant populations: CD3+γδTCR+CD56−/+ γδ T cells and CD3−CD56+ NK lymphocytes. NK cells, an innate lymphocyte population with similar cytotoxic characteristics to γδ T cells,33,34 are also not MHC-restricted and are actively under preclinical and clinical investigation as allogeneic ACTs for multiple cancers, including neuroblastoma.35 Therefore, the presence of NK cells within γδ T cell expansions is intriguing but does not warrant safety concerns. Co-expansion of NK cells alongside γδ T cells is also logical considering the role of IL-2 in ex vivo expansion protocols used to specifically enrich and expand NK cells.36 Notably, across all expansions, only those from 4/16 donors reached a threshold of >50% NK cells, suggesting that donor PBMCs more commonly yield γδ T cell-dominant expansions with our serum-free expansion protocol using zoledronate and IL-2, which also tend to yield higher overall cellular content. Ou et al.’s recent work corroborates this finding, demonstrating that of 43 healthy donors expanded using zoledronate and IL-2, approximately 70% of expansions produced a cellular product with >50% γδ T cell content.37
Previously, the starting γδ T cell number and Vδ2 percentage have been identified as adequate predictors of γδ T cell expansion.16,37 Here no significant correlation was found between the starting γδ-TCR+ percentage of PBMCs, when measured using a pan-γδ-TCR antibody, and the final γδ-TCR+ content of expanded cells, which is agreeable with findings from another recent investigation.24 Contrastingly, the most significant predictor of γδ T cell content within expansions was identified to be the starting content of NK cells within the leukocytes used to seed expansion, where greater NK cell content on day 0 yields lower γδ T cell content on day 12. Ultimately, it is likely that both γδ T cells and NK cells present in PBMCs used to expand the cell therapy impact its final consistency, and the ratio of both populations should be considered when selecting healthy donors to establish a more uniform interdonor expansion consistency.
Interestingly, NK cells required the presence of γδ T cells for proliferation, failing to expand to significant numbers in IL-2 and zoledronate when γδ T cells were removed from the seeded leukocytes. The few NK cells that did expand remained potent against neuroblastoma cells in vitro. This suggests that γδ T cells likely secrete NK sustaining and proliferating factors that are more commonly given exogenously or through feeder cells in established NK cell expansion protocols. γδ T cells, however, expanded quite well under IL-2 and zoledronate stimulation despite NK cell depletion prior to expansion. Independent of the percentage of NK cells post-expansion, the majority of day 12 NK cells were identified as CD3−CD56brightCD16+. This distinct phenotype is uncharacteristic from the NK populations (CD56dimCD16+ and CD56brightCD16−/dim) typically identified within circulating PBMCs.38 In line with other adoptive NK cell therapies, including those used against neuroblastoma, this distinct effector population of NK cells displays an activated phenotype marked by high expression of CD56, CD16, NKG2D, and DNAM-1.39 Expanded γδ T cells within the cell therapy also expressed high levels of the same activation markers, supporting a potent γδ T cell phenotype. In contrast, both expanded γδ T cells and NK cells expressed relatively lower levels of inhibitory (excluding NKG2A) and exhaustion markers (TIGIT≥ PD-1≥ TIM3> CTLA-4). CD159α (NKG2A), which was found on expanded γδ T and NK cells across all donors, is classically considered an inhibitory receptor for NK, NK T, and CD8+ αβ T cells;40,41 however, recently, constitutive expression of NKG2A on Vδ2 T lymphocytes has been associated with a subset of γδ T cells licensed with potent cytotoxic effector function and hyper-responsive anti-tumor activity.42 High expression of CD95 (FAS receptor) was also observed among expanded cells (γδ T > NK). While FAS expression on expanded γδ T/NK cells could be a setup for autocrine activation and fratricide given expanded γδ T cells and NK cells also expressed its activating ligand, FasL, our current findings do not support that this limits expansion, persistence, or cytotoxic potential.
Importantly, all expansions effectively killed neuroblastoma cells above background levels in vitro, with NK dominant expansions demonstrating enhanced anti-tumor killing compared to those with less NK cell content when administered as a single agent in vitro. Depletion of γδ-TCR+ cells from expansions revealed the predominant potency of the NK cell population, while depletion of CD56+ cells resulted in a γδ T cell population with reduced killing potential compared to the complete expansion. Our findings for the variation in killing potential in vitro between γδ T cells and NK cells is consistent with previous reports that found NK lymphocytes to be more potent than γδ T cells and αβ T cells against other cancer models.43,44 The cytotoxicity of NK dominant and γδ T dominant cell expansions equalized when combined with immunotherapy or chemoimmunotherapy, having similar and potent antitumor effects in vitro and in vivo against neuroblastoma models, respectively. Enhanced tumor killing of either an NK dominant or γδ dominant expansion observed in combination with dinutuximab is presumed to be specific to the CD16 receptor on the immune cells, mediating ADCC. Indeed, when expanded γδ T cells are combined with a murine version of anti-GD2 antibody, 14G2a, due to its weakened ability to activate human CD16,45,46 antitumor cytotoxicity was significantly reduced, supporting CD16 as the γδ T/NK cell receptor involved in dinutuximab response (Supplemental Fig. S11). Therefore, a cytotoxicity assay in combination with dinutuximab (or any targeted monoclonal antibody recognized by human CD16) should be performed as release criteria to predict an expanded γδ T cell therapy’s overall efficacy for a clinical trial employing antibody therapy that requires ADCC for efficacy. In combination with monoclonal antibody in vitro (and chemoimmunotherapy in vivo), an optimal biomarker of a potent ex vivo expanded γδ T cell therapy is high expression of CD16+ on all immune cells in the expansion, which synergize with dinutuximab to achieve potent neuroblastoma killing. Additionally, a therapy with high overall CD56 (neural cell adhesion molecule, NCAM-1) content among both NK cells and γδ T cells shows particular promise, for CD56+ γδ T cells demonstrate stronger activation than CD56− γδ T cells against neuroblastoma when combined with dinutuximab.
To be effective in the clinical setting, both γδ T cells and NK cells should traffic to the tumor, which presented as a primary limitation of the therapy in this study. Only modest tumor localization for both cell types was observed within subcutaneous neuroblastoma xenografts following intravenous administration, and in this context the cell therapy offered no additional control of tumor burden compared to the combination of dinutuximab and temozolomide. Alternatively, when chemoimmunotherapy was administered IV with either cell therapy directly injected into tumors, complete tumor regression occurred in tumors injected with either the γδ T or NK cell therapy. Together, these findings support the additional benefit of expanded γδ T/NK cell therapy in combination with chemoimmunotherapy for aggressive neuroblastoma tumors, but highlight that the overall tumor resident cell therapy content was too low for efficacy following intravenous injection. This is in contrast to our earlier studies showing that IV injected patient-derived γδ T expansions synergized with chemoimmunotherapy to reduce neuroblastoma xenografts in vivo (although the neuroblastomas and the cell therapy were not derived from the same patient, so they too were allogeneic, but patient-derived).18 The exact reason as to why healthy-donor derived γδ T cells appear to be less effective than patient-derived γδ T cells remains unclear, but given αβ T lymphocytes were not depleted from patient-derived γδ T expansions in our earlier studies (Zoine et al.) this may suggest that αβ lymphocytes play a synergistic role with γδ T cells to induce a more potent antitumor response against neuroblastoma when delivered IV. Supporting this hypothesis, it has been shown that γδ T cells can act as professional antigen presenting cells that assist with αβ T cell activation and cytotoxicity.11,12
Overall our recent studies continue to support the relevance and necessity for an expanded γδ- T/NK cell therapy in combination with tumor-targeting antibody as it contains two immune cell subsets that are necessary for ADCC. Furthermore, this work now demonstrates that novel methods to increase γδ T cell localization and sustainability are critical and will significantly strengthen the potency of the γδ T cell therapy when delivered intravenously. The off-the-shelf utility of the allogeneic cell therapy allows for more frequent dosing to increase the amount of circulating therapeutic, ideally leading to an increase in tumor localized γδ T/NK cells based on their natural trafficking propensity in response to chemokines.47 Additional methods to localize cell therapy to its tumor target are readily available. Tumors can be conditioned for γδ T cell recognition through agents that upregulate tumor phosphoantigens19,48,49 or stress antigens.50–54 Alternatively, there is ample opportunity to engineer γδ T lymphocytes to traffic toward solid tumors through CAR expression.55–58
In conclusion, highly potent NK cells are found within γδ T cell expansions and directly contribute to anti-neuroblastoma activity. When used in combination with dinutuximab, the cell therapy’s potency is independent of its overall γδ T:NK cell content, minimizing the implication of interindividual expansion differences. In light of freeze-thaw stability and expansion potential, the ideal therapy candidate appears to be one predominantly populated by γδ T cells, but subpopulations of NK cells do not appear to harm and instead promote anti-neuroblastoma potency.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health under R21 Grant [5R21CA223300] awarded to KCG and HTS; Curing Kids Cancer awarded to HTS; Rally Foundation for Childhood Cancer Research under Postdoctoral Research Fellow Grant [20FN06] awarded to HCJ; and Batcole Foundation under Postdoctoral Research Fellow Grant [22FC05] awarded to HCJ. We thank the Children’s Healthcare of Atlanta and Emory University’s Pediatric Flow Cytometry Core for support with flow cytometry experiments. Diagrams in some figures were created with BioRender.com
Funding Statement
This work was supported by the National Institutes of Health [5R21CA223300] and Rally Foundation [20FN06, 22FC05].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Harris DT, Kranz DM.. Adoptive T cell therapies: a comparison of T cell receptors and chimeric antigen receptors. Trends Pharmacol Sci. 2016;37(3):220–16. doi: 10.1016/j.tips.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao Z, Chen Y, Francisco NM, Zhang Y, Wu M. The application of CAR-T cell therapy in hematological malignancies: advantages and challenges. Acta Pharm Sin B. 2018;8(4):539–551. doi: 10.1016/j.apsb.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeRenzo C, Krenciute G, Gottschalk S. The landscape of CAR T cells beyond acute lymphoblastic leukemia for pediatric solid tumors. Am Soc Clin Oncol Educ Book. 2018;38(38):830–837. doi: 10.1200/EDBK_200773. [DOI] [PubMed] [Google Scholar]
- 4.Richards RM, Sotillo E, Majzner RG. CAR T cell therapy for neuroblastoma. Front Immunol. 2018;9:2380. doi: 10.3389/fimmu.2018.02380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lawand M, Dechanet-Merville J, Dieu-Nosjean MC. Key features of gamma-delta t-cell subsets in human diseases and their immunotherapeutic implications. Front Immunol. 2017;8:761. doi: 10.3389/fimmu.2017.00761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu YL, Ding Y-P, Tanaka Y, Shen L-W, Wei C-H, Minato N, Zhang W. gammadelta T cells and their potential for immunotherapy. Int J Biol Sci. 2014;10(2):119–135. doi: 10.7150/ijbs.7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21(8):938–945. doi: 10.1038/nm.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sebestyen Z, Prinz I, Déchanet-Merville J, Silva-Santos B, Kuball J. Translating gammadelta (gammadelta) T cells and their receptors into cancer cell therapies. Nat Rev Drug Discov. 2020;19(3):169–184. doi: 10.1038/s41573-019-0038-z. [DOI] [PubMed] [Google Scholar]
- 9.Vantourout P, Hayday A. Six-of-the-best: unique contributions of gammadelta T cells to immunology. Nat Rev Immunol. 2013;13(2):88–100. doi: 10.1038/nri3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beetz S, Wesch D, Marischen L, Welte S, Oberg -H-H, Kabelitz D. Innate immune functions of human gammadelta T cells. Immunobiology. 2008;213(3–4):173–182. doi: 10.1016/j.imbio.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 11.Brandes M, Willimann K, Bioley G, Lévy N, Eberl M, Luo M, Tampé R, Lévy F, Romero P, Moser B, et al. Cross-presenting human γδ T cells induce robust CD8 + αβ T cell responses. Proc Natl Acad Sci U S A. 2009;106(7):2307–2312. doi: 10.1073/pnas.0810059106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Himoudi N, Morgenstern DA, Yan M, Vernay B, Saraiva L, Wu Y, Cohen CJ, Gustafsson K, Anderson J. Human gammadelta T lymphocytes are licensed for professional antigen presentation by interaction with opsonized target cells. J Immunol. 2012;188(4):1708–1716. doi: 10.4049/jimmunol.1102654. [DOI] [PubMed] [Google Scholar]
- 13.Conti L, Casetti R, Cardone M, Varano B, Martino A, Belardelli F, Poccia F, Gessani S. Reciprocal activating interaction between dendritic cells and pamidronate-stimulated gammadelta T cells: role of CD86 and inflammatory cytokines. J Immunol. 2005;174(1):252–260. doi: 10.4049/jimmunol.174.1.252. [DOI] [PubMed] [Google Scholar]
- 14.Ryan PL, Sumaria N, Holland CJ, Bradford CM, Izotova N, Grandjean CL, Jawad AS, Bergmeier LA, Pennington DJ. Heterogeneous yet stable Vδ2 (+) T-cell profiles define distinct cytotoxic effector potentials in healthy human individuals. Proc Natl Acad Sci U S A. 2016;113(50):14378–14383. doi: 10.1073/pnas.1611098113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gogoi D, Chiplunkar SV. Targeting gamma delta T cells for cancer immunotherapy: bench to bedside. Indian J Med Res. 2013;138:755–761. [PMC free article] [PubMed] [Google Scholar]
- 16.Sutton KS, Dasgupta A, McCarty D, Doering CB, Spencer HT. Bioengineering and serum free expansion of blood-derived gammadelta T cells. Cytotherapy. 2016;18(7):881–892. doi: 10.1016/j.jcyt.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 17.Whittle SB, Smith V, Doherty E, Zhao S, McCarty S, Zage PE. Overview and recent advances in the treatment of neuroblastoma. Expert Rev Anticancer Ther. 2017;17(4):369–386. doi: 10.1080/14737140.2017.1285230. [DOI] [PubMed] [Google Scholar]
- 18.Zoine JT, Knight KA, Fleischer LC, Sutton KS, Goldsmith KC, Doering CB, Spencer HT. Ex vivo expanded patient-derived gammadelta T-cell immunotherapy enhances neuroblastoma tumor regression in a murine model. Oncoimmunology. 2019;8(8):1593804. doi: 10.1080/2162402X.2019.1593804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fisher JP, Flutter B, Wesemann F, Frosch J, Rossig C, Gustafsson K, Anderson J. Effective combination treatment of GD2-expressing neuroblastoma and Ewing’s sarcoma using anti-GD2 ch14.18/CHO antibody with Vgamma9Vdelta2+ gammadeltaT cells. Oncoimmunology. 2016;5(1):e1025194. doi: 10.1080/2162402X.2015.1025194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith V, Foster J. High-risk neuroblastoma treatment review. Children (Basel). 2018;5(9). doi: 10.3390/children5090114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pinto NR, Applebaum MA, Volchenboum SL, Matthay KK, London WB, Ambros PF, Nakagawara A, Berthold F, Schleiermacher G, Park JR, et al. Advances in risk classification and treatment strategies for neuroblastoma. J Clin Oncol. 2015;33(27):3008–3017. doi: 10.1200/JCO.2014.59.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Das RK, Vernau L, Grupp SA, Barrett DM. Naive T-cell deficits at diagnosis and after chemotherapy impair cell therapy potential in pediatric cancers. Cancer Discov. 2019;9(4):492–499. doi: 10.1158/2159-8290.CD-18-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tough DF, Sprent J. Lifespan of gamma/delta T cells. J Exp Med. 1998;187(3):357–365. doi: 10.1084/jem.187.3.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burnham RE, Zoine JT, Story JY, Garimalla SN, Gibson G, Rae A, Williams E, Bixby L, Archer D, Doering CB, et al. Characterization of donor variability for gammadelta T cell ex vivo expansion and development of an allogeneic gammadelta T cell immunotherapy. Front Med (Lausanne). 2020;7:588453. doi: 10.3389/fmed.2020.588453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khan MW, Curbishley SM, Chen H-C, Thomas AD, Pircher H, Mavilio D, Steven NM, Eberl M, Moser B. Expanded Human Blood-Derived gammadeltaT cells display potent antigen-presentation functions. Front Immunol. 2014;5:344. doi: 10.3389/fimmu.2014.00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kiesgen S, Messinger JC, Chintala NK, Tano Z, Adusumilli PS. Comparative analysis of assays to measure CAR T-cell-mediated cytotoxicity. Nat Protoc. 2021;16(3):1331–1342. doi: 10.1038/s41596-020-00467-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen D, Cox J, Annam J, Weingart M, Essien G, Rathi KS, Rokita JL, Khurana P, Cuya SM, Bosse KR, et al. LIN28B promotes neuroblastoma metastasis and regulates PDZ binding kinase. Neoplasia. 2020;22(6):231–241. doi: 10.1016/j.neo.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lorenzo-Herrero S, Sordo-Bahamonde C, Gonzalez S, López-Soto A.. CD107a degranulation assay to evaluate immune cell antitumor activity. In: López-Soto A, Folgueras A, editors. Cancer immunosurveillance. Methods in molecular biology, vol 1884. New York, NY: Humana Press; 2019. https://doi-org.proxy.library.emory.edu/10.1007/978-1-4939-8885-3_7 [DOI] [PubMed] [Google Scholar]
- 29.Van Acker HH, Capsomidis A, Smits EL, Van Tendeloo VF. CD56 in the immune system: more than a marker for cytotoxicity? Front Immunol. 2017;8:892. doi: 10.3389/fimmu.2017.00892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hviid L, Akanmori BD, Loizon S, Kurtzhals JAL, Ricke CH, Lim A, Koram KA, Nkrumah FK, Mercereau-Puijalon O, Behr C, et al. High frequency of circulating gamma delta T cells with dominance of the v(delta)1 subset in a healthy population. Int Immunol. 2000;12(6):797–805. doi: 10.1093/intimm/12.6.797. [DOI] [PubMed] [Google Scholar]
- 31.Lamb LS Jr., Musk P, Ye Z, van Rhee F, Geier SS, Tong -J-J, King KM, Henslee-Downey PJ. Human gammadelta(+) T lymphocytes have in vitro graft vs leukemia activity in the absence of an allogeneic response. Bone Marrow Transplant. 2001;27(6):601–606. doi: 10.1038/sj.bmt.1702830. [DOI] [PubMed] [Google Scholar]
- 32.Handgretinger R, Schilbach K. The potential role of gammadelta T cells after allogeneic HCT for leukemia. Blood. 2018;131(10):1063–1072. doi: 10.1182/blood-2017-08-752162. [DOI] [PubMed] [Google Scholar]
- 33.Morandi F, Yazdanifar M, Cocco C, Bertaina A, Airoldi I. Engineering the bridge between innate and adaptive immunity for cancer immunotherapy: focus on gammadelta T and NK cells. Cells. 2020;9(8):1757. doi: 10.3390/cells9081757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paul S, Lal G. The molecular mechanism of natural killer cells function and its importance in cancer immunotherapy. Front Immunol. 2017;8:1124. doi: 10.3389/fimmu.2017.01124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shin MH, Kim J, Lim SA, Kim J, Kim S-J, Lee K-M. NK cell-based immunotherapies in cancer. Immune Netw. 2020;20(2):e14. doi: 10.4110/in.2020.20.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Becker PS, Suck G, Nowakowska P, Ullrich E, Seifried E, Bader P, Tonn T, Seidl C. Selection and expansion of natural killer cells for NK cell-based immunotherapy. Cancer Immunol Immunother. 2016;65(4):477–484. doi: 10.1007/s00262-016-1792-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ou L, Wang H, Liu Q, Zhang J, Lu H, Luo L, Shi C, Lin S, Dong L, Guo Y, et al. Dichotomous and stable gamma delta T-cell number and function in healthy individuals. J Immunother Cancer. 2021;9(5):e002274. doi: 10.1136/jitc-2020-002274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity. 2017;47(5):820–833. doi: 10.1016/j.immuni.2017.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y, Wu H-W, Sheard MA, Sposto R, Somanchi SS, Cooper LJN, Lee DA, Seeger RC. Growth and activation of natural killer cells ex vivo from children with neuroblastoma for adoptive cell therapy. Clin Cancer Res. 2013;19(8):2132–2143. doi: 10.1158/1078-0432.CCR-12-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andre P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, Bléry M, Bonnafous C, Gauthier L, Morel A, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell. 2018;175(7):1731–1743 e13. doi: 10.1016/j.cell.2018.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Creelan BC, Antonia SJ. The NKG2A immune checkpoint - a new direction in cancer immunotherapy. Nat Rev Clin Oncol. 2019;16(5):277–278. doi: 10.1038/s41571-019-0182-8. [DOI] [PubMed] [Google Scholar]
- 42.Cazzetta V, Bruni E, Terzoli S, Carenza C, Franzese S, Piazza R, Marzano P, Donadon M, Torzilli G, Cimino M, et al. NKG2A expression identifies a subset of human Vdelta2 T cells exerting the highest antitumor effector functions. Cell Rep. 2021;37(3):109871. doi: 10.1016/j.celrep.2021.109871. [DOI] [PubMed] [Google Scholar]
- 43.Deng X, Terunuma H, Terunuma A, Takane T, Nieda M. Ex vivo-expanded natural killer cells kill cancer cells more effectively than ex vivo-expanded gammadelta T cells or alphabeta T cells. Int Immunopharmacol. 2014;22(2):486–491. doi: 10.1016/j.intimp.2014.07.036. [DOI] [PubMed] [Google Scholar]
- 44.Niu C, Jin H, Li M, Xu J, Xu D, Hu J, He H, Li W, Cui J. In vitro analysis of the proliferative capacity and cytotoxic effects of ex vivo induced natural killer cells, cytokine-induced killer cells, and gamma-delta T cells. BMC Immunol. 2015;16(1):61. doi: 10.1186/s12865-015-0124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barker E, Mueller BM, Handgretinger R, Herter M, Yu AL, Reisfeld RA. Effect of a chimeric anti-ganglioside GD2 antibody on cell-mediated lysis of human neuroblastoma cells. Cancer Res. 1991;51:144–149. [PubMed] [Google Scholar]
- 46.Dhillon S. Dinutuximab: first global approval. Drugs. 2015;75(8):923–927. doi: 10.1007/s40265-015-0399-5. [DOI] [PubMed] [Google Scholar]
- 47.Kabelitz D, Wesch D. Features and functions of gamma delta T lymphocytes: focus on chemokines and their receptors. Crit Rev Immunol. 2003;23(5–6):339–370. doi: 10.1615/CritRevImmunol.v23.i56.10. [DOI] [PubMed] [Google Scholar]
- 48.Mattarollo SR, Kenna T, Nieda M, Nicol AJ. Chemotherapy and zoledronate sensitize solid tumour cells to Vgamma9Vdelta2 T cell cytotoxicity. Cancer Immunol Immunother. 2007;56(8):1285–1297. doi: 10.1007/s00262-007-0279-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Varesano S, Zocchi MR, Poggi A. Zoledronate triggers vdelta2 T cells to destroy and kill spheroids of colon carcinoma: quantitative image analysis of three-dimensional cultures. Front Immunol. 2018;9:998. doi: 10.3389/fimmu.2018.00998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Story JY, Zoine JT, Burnham RE, Hamilton JAG, Spencer HT, Doering CB, Raikar SS. Bortezomib enhances cytotoxicity of ex vivo-expanded gamma delta T cells against acute myeloid leukemia and T-cell acute lymphoblastic leukemia. Cytotherapy. 2021;23(1):12–24. doi: 10.1016/j.jcyt.2020.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Poggi A, Catellani S, Garuti A, Pierri I, Gobbi M, Zocchi MR. Effective in vivo induction of NKG2D ligands in acute myeloid leukaemias by all-trans-retinoic acid or sodium valproate. Leukemia. 2009;23(4):641–648. doi: 10.1038/leu.2008.354. [DOI] [PubMed] [Google Scholar]
- 52.Satwani P, Bavishi S, Saha A, Zhao F, Ayello J, van de Ven C, Chu Y, Cairo MS. Upregulation of NKG2D ligands in acute lymphoblastic leukemia and non-Hodgkin lymphoma cells by romidepsin and enhanced in vitro and in vivo natural killer cell cytotoxicity. Cytotherapy. 2014;16(10):1431–1440. doi: 10.1016/j.jcyt.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 53.Morisaki T, Onishi H, Koya N, Kiyota A, Tanaka H, Umebayashi M, Ogino T, Nagamatsu I, Katano M. Combinatorial cytotoxicity of gemcitabine and cytokine-activated killer cells in hepatocellular carcinoma via the NKG2D-MICA/B system. Anticancer Res. 2011;31:2505–2510. [PubMed] [Google Scholar]
- 54.Hoeres T, Smetak M, Pretscher D, Wilhelm M. Improving the efficiency of vgamma9Vdelta2 T-cell immunotherapy in cancer. Front Immunol. 2018;9:800. doi: 10.3389/fimmu.2018.00800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Capsomidis A, Benthall G, Van Acker HH, Fisher J, Kramer AM, Abeln Z, Majani Y, Gileadi T, Wallace R, Gustafsson K, et al. Chimeric antigen receptor-engineered human gamma delta T cells: enhanced cytotoxicity with retention of cross presentation. Mol Ther. 2018;26(2):354–365. doi: 10.1016/j.ymthe.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fisher J, Anderson J. Engineering approaches in human gamma delta T cells for cancer immunotherapy. Front Immunol. 2018;9:1409. doi: 10.3389/fimmu.2018.01409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rischer M, Pscherer S, Duwe S, Vormoor J, Jürgens H, Rossig C. Human gammadelta T cells as mediators of chimaeric-receptor redirected anti-tumour immunity. Br J Haematol. 2004;126(4):583–592. doi: 10.1111/j.1365-2141.2004.05077.x. [DOI] [PubMed] [Google Scholar]
- 58.Fleischer LC, Spencer HT, Raikar SS. Targeting T cell malignancies using CAR-based immunotherapy: challenges and potential solutions. J Hematol Oncol. 2019;12(1):141. doi: 10.1186/s13045-019-0801-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.