

Abstract
In recent years, several newly discovered viruses infecting free-living nematodes, sedentary plant-parasitic nematodes, and migratory root lesion nematodes have been described. However, to the best of our knowledge, no comprehensive research focusing exclusively on metagenomic analysis of the soil nematode community virome has thus far been carried out. In this work, we have attempted to bridge this gap by investigating viral communities that are associated with soil-inhabiting organisms, particularly nematodes. This study demonstrates a remarkable diversity of RNA viruses in the natural soil environment. Over 150 viruses were identified in different soil-inhabiting hosts, of which more than 139 are potentially new virus species. Many of these viruses belong to the nematode virome, thereby enriching our understanding of the diversity and evolution of this complex part of the natural ecosystem.
Keywords: meta-transcriptomics, virus discovery, soil nematodes, phylogeny
1. Introduction
Soil-inhabiting nematodes are a very heterogenous group of eukaryotic organisms, encompassing a complex and structured community. Based on their feeding strategies, soil nematodes can be grouped as herbivores or plant parasites, animal parasites that infect a wide range of invertebrates and vertebrates, bacterivores, fungivores, predators that feed on other nematodes, and omnivores that utilize several sources for food (McSorley 2009; Treonis et al. 2018). It is therefore presumed that the diversity of viruses concomitant with nematode populations would be high and include not only nematode-infecting pathogens but also viruses associated with their sources of food: plants, invertebrates, fungi, algae, and bacteria.
Not too many years ago, the interaction of viruses and nematodes was known only for a limited number of plant-parasitic nematodes (Longidoridae and Trichodoridae), implicated in the transmission of plant nepo- and tobraviruses (Brown et al. 1995). In recent years, several individual viruses have been identified to be associated with other nematodes, including free-living species (Bekal et al. 2011, 2014; Felix et al. 2011; Franz et al. 2012; Frézal et al. 2019), sedentary plant-parasitic nematodes (Bekal et al. 2011, 2014; Ruark et al. 2017; Lin et al. 2018), the migratory root lesion nematode (Vieira and Nemchinov 2019), and a parasite of several mammal species, Capillaria hepatica (Williams et al. 2019).
Currently, only a handful of metagenomics studies are available that relate, albeit indirectly, to the investigation of the nematode virome. These include a report on the soil virome in general (Starr et al. 2019) and an exploration of the invertebrate virome that contained a few nematodes along with 220 other species from nine animal phyla (Shi et al. 2016), mainly Arthropoda. The latter study resulted in the discovery of a large number of arthropod viruses along with a smaller number of viruses associated with animal-parasitic nematodes.
Here, we performed an extensive metatranscriptomic analysis of soil nematode samples collected in different regions of the USA. The purpose of this work was to specifically decipher viral communities associated with the complex network of soil-inhabiting nematodes and other soil-inhabiting microorganisms in order to (1) obtain a proof of concept and feasibility of a presence of a large viral community associated with soil-inhabiting nematodes and (2) fill the gaps in our limited knowledge of the diversity, structure, and evolutionary perspectives of this complex part of the natural ecosystem. This study demonstrates the occurrence of a remarkable diversity of RNA viruses and reveals a number of novel, previously undescribed viral species.
2. Results
2.1. Delineation of the viral hosts
To understand the diversity and proportion of the host species within each library and to reasonably suggest putative viral hosts, we used the widely adopted the taxonomic marker cytochrome c oxidase subunit I (COI; mitochondrial cytochrome C oxidase subunit I gene) (Damon et al. 2010). As anticipated, sequences belonging to the Nematoda constituted the prevalent group of COI contigs, reaching up to 89 per cent (60–89 per cent) of all COI sequences retrieved in each of the six pooled samples (Fig. 1). The remaining contigs were distributed among other Animal phyla (i.e. Annelida, Arthropoda, Mollusca, Rotifera, and Tardigrada) and the Kingdom Fungi (Fig. 1 and Supplementary Figure S1). Non-nematode species affiliated within these five phyla and the fungi were identified in the samples collected from all three geographic locations: California, Maryland, and Virginia (Fig. 2).
Figure 1.
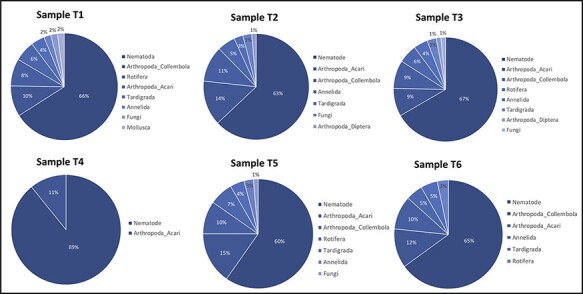
Proportions of the main (dominant) organisms in the soil samples, as revealed by using the mitochondrial cytochrome C oxidase subunit 1 (Cox1) gene as a marker. T1, pooled samples from California; T2–T4, samples from Virginia; T5 and T6, samples originated from Maryland.
Figure 2.
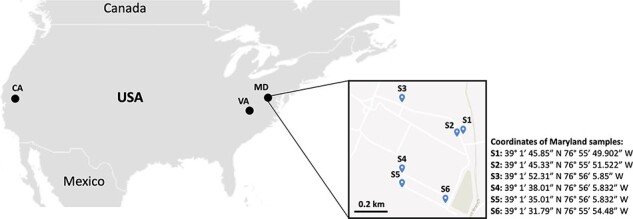
Geographic distribution of the soil samples collected in this study. Nematodes extracted from soil samples were combined into a total of six bulk nematode samples from California, and four bulk nematode samples from Virginia, resulting in a final set of four pooled RNA samples (T1 = California and T2–T4: Virginia). In Maryland, six bulked soil samples (S1–S6) were collected at the campus of Beltsville Agricultural Research Center, United States Department of Agriculture, Agricultural Research Service, resulting in two pooled RNA samples (T5 and T6).
2.2. Diversity of viruses
2.2.1. Known viruses
After removal of the low-quality reads, a total of ∼870 million clean reads were obtained (Supplementary Table S1). Mapping of the clean reads against the reference viral database (www.ncbi.nlm.nih.gov/genome/viruses) resulted in the detection of a group of known viruses (n = 18) that originated from nematodes, plants, insects, fungal plant pathogens, or the feces of birds (Supplementary Table S2). The only known nematode virus present in this group was the root lesion nematode virus 1 (Vieira and Nemchinov 2019), identified in two samples collected from Virginia, where it had not been reported prior to this study. Plant viruses included species transmitted by nematodes (Tobacco ringspot virus), eriophyid mites (Ryegrass mosaic virus), seeds (Festuca pratensis amalgavirus), vegetative propagation and grafting (Grapevine rupestris stem pitting-associated virus), and mechanical contact (Potato virus X). Among known insect viruses were those infecting honeybees and bumblebees, presumably nesting underground. Macrophomina phaseolina tobamo-like virus 1 and mycovirus Fusarium graminearum alternavirus 1, naturally infect phytopathogenic soil-borne fungi Macrophomina phaseolina and Fusarium graminearum, respectively, suggesting the presence of these pathogens in the collected soil samples.
One of the libraries contained Plasmopara viticola lesion associated narnavirus 5 that was originally reported from the causal agent of downy mildew, the oomycete Plasmopara viticola (Chiapello et al. 2020). The sources of the unclassified Hubei picorna-like virus 51 and goose dicistrovirus could presumably be bird feces. Characteristically, known viruses were often represented by a low number of reads (Supplementary Table S2), potentially indicating a share of their respective hosts among other soil-inhibiting organisms in the samples.
2.2.2. Novel viruses
To assess the diversity of novel viral sequences, a total of ∼1,650,588 contigs generated by de novo assembly (Supplementary Table S1) were searched for putative domains of viral RNA-dependent RNA polymerase (RdRp). In this manner, 139 presumably novel viruses belonging to the groups IV (positive sense single-stranded RNA viruses) and group III (double-stranded RNA (dsRNA) viruses) (Baltimore 1971) have been identified, reflecting a considerable biodiversity of viral communities associated with the different soil-inhabiting hosts (Fig. 3 and Supplementary Table S3). An extensive phylogenetic analysis of the universally conserved RdRps confirmed viral heterogeneity, delineated by three established phyla: Kitrinoviricota, Pisuviricota, and Lenarviricota (Fig. 3).
Figure 3.

Taxonomic distribution of viral sequences associated with soil nematodes samples. A. Distribution of viral sequences identified per sample and phylum. T1, pooled samples from California; T2–T4, samples from Virginia; T5 and T6, samples originated from Maryland. B. Generalized phylogenetic diversity of all viruses identified in the present study. Trees were deduced based on the alignments of viral RdRPs. New tentative viruses are marked in red color, known viruses in gray, and reference viruses are shown in black.
Novel viruses had on average 40.58 per cent identity with their best matches from National Center for Biotechnology Information, accompanying, in most cases, by a low query coverage. Interestingly, most of these novel viral sequences were specific to the sampled geographic regions. The different geographic distribution is likely due to the species of soil-inhabiting organisms characteristic for each natural ecosystem. Novel viruses identified in this research have been designated applying the following guidelines: (1) geographic location; (2) proposed taxonomic rank; (3) possible host, if proposed; and (4) successive number (Supplementary Table S3).
2.3. Phylogenetic assignments of novel viral contigs bearing RdRp motifs
To place the newly discovered viral sequences within the context of each phylum, phylogenetic distributions were tentatively allocated into seven clades at the level of the taxonomic rank ‘order’: Hepevirales, Tymovirales, Martellivirales, Tolivirales, Picornavirales, Durnaviralles and Wolframvirales (Supplementary Figures S2–S8).
In the order Hepelivirales, to which many known plant-, invertebrate-, and vertebrate-infecting viruses belong, sequences depicting novel viruses formed several distinct clusters, broadly associated with the families Benyviridae and Alphatetraviridae (Supplementary Fig. S2). According to Basic Local Alignment Search Tool (BLAST) searches, percentage identity of all new sequences grouped into the order Hepelivirales to known viruses available at NCBI varied between ∼26 and 56 per cent, signifying potentially new viral species (Supplementary Table S3). This assumption was also supported by their genome organization (Supplementary Fig. S2).
Many novel viral sequences placed in the order Tymovirales formed well-defined clusters, some possibly at the level of new families (Supplementary Fig S3). Notably, these groups included viral sequences unveiled from samples collected in different locations (i.e. Maryland and Virginia), thus accentuating the occurrence of their respective host(s) in the corresponding geographic areas. Among the twenty-four putative new viral contigs in this order, five comprised coding-complete or substantially complete genomes (Supplementary Table S3).
Thirty-four of the new viral sequences belonged to the order Martellivirales. The majority of these appeared to be novel viruses that form distinct clades within this order (Supplementary Fig. S4). At least three out of the thirty-four sequences appeared to incorporate coding-complete or nearly complete viral genomes (Supplementary Table S3).
Novel viral sequences (n = 21) identified in the order Durnavirales for the most part were associated with partitiviruses, dsRNA viruses with bisegmented genomes (Supplementary Fig. S5). Notwithstanding, a major portion of them assembled into a separate cluster, sharing less than 45 per cent identity with their best BLAST match (Supplementary Table S3).
A total of forty new sequences detected in this study were associated with the order Picornavirales. Among them, six most likely infected nematodes as their primary host (as described in more detail below). Their phylogenetic placements, low similarity, and coverage values suggested that these contigs represented novel viruses. Except for few sequences, many were distributed into distinct phylogenetic RdRp clades linked to the established taxa (Supplementary Fig. S6). More than ten novel sequences contained coding-complete or nearly complete genomes, ranging between 7,117 and 10,641 nt. Overall, these substantially complete sequences displayed genome organization characteristic for representative members of the order Picornavirales, therefore supporting the results of our phylogenetic analysis (Supplementary Fig. S6).
Among other novel sequences, two were identified that belonged to the orders Tolivirales and Wolframvirales. The former originated from the sample collected in Maryland and the latter from California (Supplementary Table S3). The Maryland sequence had the best low-coverage (6 per cent) hit against the Dipteran tombus-related virus distantly related to Tombusviridae, a plant-infecting virus family (Paraskevopoulou et al. 2021). Phylogenetic analysis placed this sequence together with other tombus-like viruses in the order Tolivirales (Supplementary Fig. S7). Judging from the low identity/coverage percentage, it can potentially represent a segment of a new virus that was provisionally named Maryland toli-like virus 1 (MTLV-1). The California sequence was tentatively assigned to the order Wolframvirales (Supplementary Fig. S8) and provisionally named California wolfra-like virus 1 (CWLV-1).
In total, there were thirty-four coding-complete or essentially complete novel genomes identified in this study (Supplementary Table S3). Two were tentatively placed in the order Durnavirales, four in the Martellivirales, five in the Tymovirales, nine in the Hepelivirales, and fourteen in the order Picornavirales. Genome structures of these longest contigs exhibited a wide range of patterns, not always related to the established architecture of the representative species (see Supplementary Figs S2–S8 for each of the orders).
2.4. Novel nematode-associated viral sequences
Among the 139 potentially novel virus species, there were at least seven that most likely naturally infected nematodes, consistent with their phylogenetic distribution: California martelli-like virus 8 (CMLV-8), Virginia picorna-like virus 10 (VPLV-10), Virginia picorna-like virus 11 (VPLV-11), Virginia picorna-like virus 17 (VPLV-17), Maryland picorna-like virus 7 (MPLV-7), California picorna-like virus 12 (CPLV-12), and Virginia picorna-like virus 1 (VPLV-1).
Phylogenetic analysis of the RdRp sequences of VPLV-10, -11, and -17 and MPLV-7 placed these viruses in a separate monophyletic group of the order Picornavirales (Supplementary Fig. S6). This clade is evolutionarily related to the known plant-parasitic nematode-infecting viruses, including RLNV1 (Vieira and Nemchinov 2019), Soybean cyst nematode virus 1 (Bekal et al. 2011), and potato cyst nematode picorna-like virus (Ruark et al. 2017). CPLV-12 clustered with Wuhan spirurian nematode virus 1 (APG78447) and Xinzhou nematode virus 2 (YP_009345045), both previously associated with animal-parasitic nematodes (Supplementary Fig. 6). VPLV-1 grouped with Hubei picorna-like virus 12 (YP_009336568) and Hubei picorna-like virus 11 (YP_009336580), both of which were found to be associated with the large pig roundworm (parasitic nematode Ascaris suum) (Shi et al. 2016).
2.5. In situ hybridization assays to verify origin of the selected viruses
In an attempt to evaluate the association of the identified viruses with nematodes, we performed in situ hybridization (ISH) assays with selected viruses that were represented in the Maryland dataset by an abundant number of reads. Using the antisense probes (Supplementary Table S4), transcripts of three distinct viruses (MHLV-10, MMLV-5, and T5_contig_6437) were detected in different parts of the nematode body sections (Fig. 4). While MHLV-10 and MMLV-5 contained annotated RdRp domains, T5_contig_6437 did not and thus has not been included in the phylogenetic analysis. The latter virus was provisionally designated as Maryland picorna-like virus 9 (MPLV-9). The fragment of MPLV-9 genome encoded a single open reading frame (ORF), 743-aa-long, with 33 per cent identity to the hypothetical protein of Picornavirales spp. (QJI53518.1). The detection of MHLV-10 and MMLV-5 by ISH reinforced the idea that other viral sequences in the dataset are potentially affiliated with nematodes.
Figure 4.
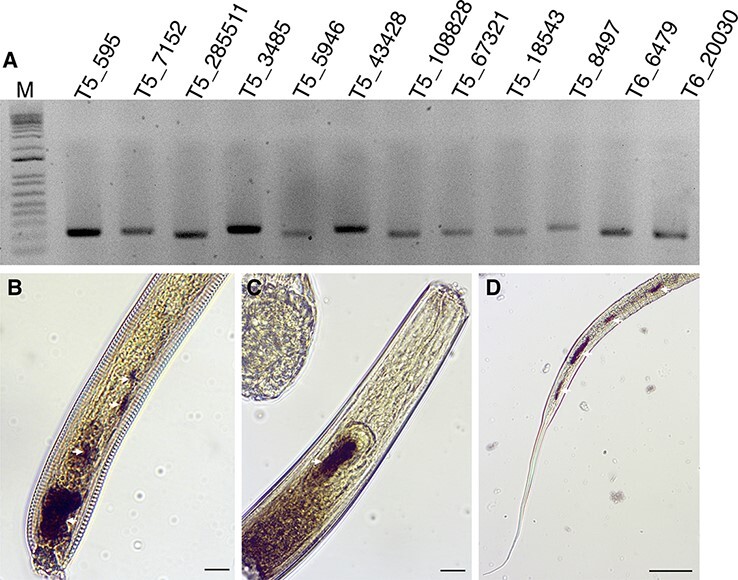
Detection of viral transcripts within nematodes. A. PCR detection of randomly selected viral sequences in different soil samples from Maryland. M, 1 Kb Plus DNA Ladder (Thermo Fisher Scientific, USA); 1, T5_contig_43428 (Maryland martelli-like virus 6); 2, T5_contig_7152 (Maryland picorna-like virus 5); 3, T5_contig_108828 (Maryland tymo-like virus 2); 4, T5_contig_5946 (Maryland martelli-like virus 5); 5, T5_contig_18543 (Maryland hepe-like virus 7); 6, T5_contig_67321(Maryland hepe-like virus 10); 7, T6_contig_6479 (Maryland martelli-like virus 4); T6_contig_20030 (Maryland hepe-like virus 11). B–D. Detection of the viral transcripts within the nematode body sections by ISH with antisense DIG-labeled DNA probes. Dark, brown-colored regions (white arrows) represent a positive detection of three viral sequences in different nematode species. (B) T5_contig_5946 (Martellivirales; Maryland martelli-like virus 5); (C) T5_contig_67321 (Hepelivirales; Maryland hepe-like virus 10); (D) T5_contig_6437 (Maryland picorna-like virus 9). Scale bars represent 20 μm.
Due to the complexity of the samples (i.e. sectioned and pooled nematodes), nematode species associated with ISH-detected viruses were not identified. Hybridization with the sense probes used as negative controls revealed no signal for these three viruses, as well as for the remaining viral species (data not shown). The experiment had demonstrated that some of the viral sequences without any guiding BLAST hits or phylogenetic implications with known nematode-infecting viruses may indeed correspond to novel viral genomes associated with nematodes as their hosts.
3. Discussion
Nematodes are among the most abundant animals on Earth. Following arthropods, they are considered the second most diverse animal phylum. While the virome of many arthropods is becoming available (Shi et al. 2016; Calisher and Higgs, 2018), the virome associated with nematodes remains mostly unexplored. Therefore, the main objective of this research was to investigate the biological diversity of viral communities associated with soil nematodes. To the best of our knowledge, this task has not been previously undertaken on a large-scale metagenomic approach. Given that separating nematodes from a heterogenous variety of species present in soil samples is a difficult if not unfeasible endeavor, the scope of this study has broadened in the process to include viruses of other soil-inhabiting organisms as well.
Unlike other metagenomic studies performed on environmental soil specimens, our research focused specifically on soil nematodes associated with agricultural systems. The adapted methodology led to impactful results: 157 viruses have been identified in different soil-inhabiting hosts including more than 139 novel, previously undescribed viruses. These new sequences not only contribute to the existing dataset of viruses found in soil-inhabiting microorganisms, but also reveal with some level of confidence at least seven new viruses that most likely naturally infect soil nematodes. ISH assays conducted with selected viruses, including those that presumably infect nematodes, confirmed the presence of three different viruses within the nematode bodies, supporting the results of an in silico analysis and our overall virus discovery strategy in nematodes.
Phylogenetic analysis of the RdRp domains enabled a broad classification of all identified viruses into seven orders. Although viruses belonging to each of these taxa have some common hosts, these associations are not strict and cannot be employed to predict the virus-host infectivity. Based on the COI profiling, the potential range of hosts from which the viruses have originated spread throughout at least seven different phyla and three kingdoms. Nevertheless, the phylum Nematoda represented on average 68 per cent of all detected organisms. Even though this ratio cannot be mechanically applied toward the attribution of potential hosts of all identified viruses, it implies that nematode RNAs and related transcripts were the most abundant constituents in each sample, presumably contributing more to the diversity and origin of the novel viral sequences.
Apart from nematodes, the identified viruses could stem from other sources including different life forms found in the soil, decaying plant material (shrub leaves, grasses, seeds, and roots), remains of animal corpses, decomposed insect residues, and the excretory products of all levels. It is also important to emphasize that many of the soil-inhabiting organisms could be part of the nematodes’ diet, as they feed on plants, algae, bacteria, arthropods, and other nematodes, and thus carry all viruses infecting their food source. In this regard, it is expected that a large proportion of the novel sequences positioned in the order Tymovirales are plant-like viruses and/or may have originated from plants through nematode feeding, animal ingestion and soil-inhabiting zoosporic organisms, phytopathogenic fungi, plant-feeding insects, groundwater contamination, and other means (Roossinck 2013; Andika et al. 2017).
Taken together, these findings provide new insights into the manifold diversity of viruses associated with such micro-scale soil environment. In addition to the expansion of our knowledge of the nematode virome, this study also points to the important role of viruses in the structural and evolutionary aspects of soil communities. For example, distinct phylogenetic grouping of a considerable number of the identified viruses could potentially lead to the establishment of new species and genera, particularly within the orders Hepelivirales, Tymovirales, Martellivirales, Durnaviralles, and Picornavirales.
In spite of the fact that soil samples used in this study were collected from agricultural fields, they often contained many unknown nematode species, including new plant-parasitic nematodes. This large representation of unexplored nematode taxa may, in part, explain the great diversity of new viruses associated with these samples, suggesting that nematodes are more prone to viral infections than previously assumed.
Except for those identified in free-living nematodes of the genus Caenorhabditis (Caenorhabditis elegans and C. briggsae), most of the nematode-associated viruses described so far are restricted to animal- and plant-parasitic nematodes. Therefore, the diversity of virus sequences identified in this study presents a more comprehensive picture of the viral communities in the network of soil nematode fauna.
Discovery of new viruses in nematodes could also have biotechnological applications related to their use for control of plant-parasitic nematodes. Recently, it was demonstrated that C. elegans infected with Orsay virus had reduced fitness and modulated sexual behavior in comparison with non-infected specimens (Frézal et al. 2019; van Sluijs et al. 2021). Moreover, nematode-infecting viruses can potentially be used as tools for genetic modification of plant-parasitic nematodes, as it was recently shown for animal-parasitic nematodes (Hagen et al. 2021). This type of methodology has a great promise to increase our knowledge on the biology of these resilient plant pathogens. As of today, genetic modification of plant-parasitic nematodes is still in its infancy due to the large technical restrains (Kranse et al. 2021).
In conclusion, the findings reported here demonstrate that viruses associated with nematodes and other soil-inhabiting communities appear to be vastly heterogenous, encompassing many novel, emerging and known species across different orders. They also enrich our understanding of the diversity and evolution of this complex part of natural ecosystem and provide the foundation for further discoveries.
4. Methods
4.1. Sample collection and processing
In the states of California and Virginia, soil samples randomly collected by growers from agricultural land were received for processing in the respective diagnostics labs (the Plant Pest Diagnostics Branch in California and the Virginia Tech Nematode Assay lab in Virginia). Specific details and coordinates of the collection sites in these two states were intentionally omitted in order to protect growers’ privacy. Nematodes extracted from soil samples were combined into a total of six bulk nematode samples from California and four bulk nematode samples from Virginia, representing a large-scale geographic distribution of nematodes within each state.
In Maryland, a total of six pooled soil samples were collected from experimental agricultural fields at the Beltsville Agricultural Research Center’s campus, representing a local geographic distribution (Fig. 2). Since our main goal was to validate the presence of viruses among different nematode communities as a concept proof, no additional parameters related to soil composition or potential host plants were registered.
Each pooled soil sample was composed of a minimum of five soil cores of 20-cm depth. Soil samples were mixed thoroughly, and nematodes were extracted from 100-g soil samples through a series of mesh screens (20-, 60-, and 400-µm mesh) following the sucrose-centrifugation method (Barker 1985). The recovered nematode solution together with any other extracted microorganisms (Supplementary Fig. S9) was placed in a modified Baermann funnel and incubated for 24 h. Nematodes were pelleted in 50–100 µl of distilled water, frozen in liquid nitrogen, and stored at −80ºC.
4.2. Total RNA extraction and Illumina RNA sequencing
Total RNA extraction was performed with the RNeasy Plant Mini kit (Qiagen) following the manufacturer’s instructions. Samples from each separate geographical location were pooled together to obtain the needed amount of mRNA for sequencing that resulted in a final set of six pooled RNA samples (California = T1; Virginia = T2-T4; and Maryland = T5 and T6). The purity and quantity of the samples were verified with a NanoDrop spectrophotometer (ThermoFisher Scientific) and a Bionalyser (Agilent Technologies). An average integrity value of 8.6 was observed for the six RNA samples. cDNA libraries were generated using a poly (A) selection method to obtain a higher coverage with better accuracy (Zhao et al. 2018) and to enrich RNAs of many viruses that generate polyadenylated transcripts. A minimum of 2 µg of total RNA per sample was used, following the manufacturer’s instructions (Illumina Inc., San Diego, CA). Paired-end reads (2 × 150 bp) were obtained on the Illumina HiSeq 2500 platform performed by Novogene Inc., USA.
4.3. Bioinformatic analysis
4.3.1. Quality reads assessment and assembly
After removal of low-quality reads and reads shorter than 50 nt, a total of ∼870 million clean reads were obtained. To identify known viral contigs, the clean reads (Supplementary Table S2) were initially aligned to the reference viral genome database available at NCBI (www.ncbi.nlm.nih.gov/genome/viruses). The reads that mapped to the viral genome sequences were extracted and saved to a file using a Python script. The reads were then assembled into contigs using CLC Workbench (QIAGEN CLC Genomics Workbench version 20.0) with default parameters.
4.3.2. Identification of viral contigs
The resulted contigs were then searched against known viral genome sequences using Blast2Go (https://www.blast2go.com/) with the default parameters (e-value < 10−5). Afterward, all unmapped reads were de novo assembled using CLC Workbench version 20.0. Two complementary steps were then performed for the identification of new putative viral sequences using the contigs assembled for each individual library. In the first step, all contigs were compared (using BLASTx) against the reference viral proteins available at NCBI to discriminate potential viral sequences based on identity percentage (e-value < 10−5), with specific emphasis on the detection of RdRp sequences. In the second step, viral contigs were re-identified using publicly available Hidden Markov models (HMM) profiles of different types of RdRp of eukaryotic RNA viruses following Starr et al. (2019). PFAM analysis was performed using the TransDecoder.LongOrfs (https://github.com/TransDecoder/TransDecoder/blob/master/TransDecoder.LongOrfs) on the assembled contigs to find all possible ORFs in each contig. Then, a Perl script (included with the TransDecoder package) was performed to obtain the longest ORF from each contig and translate them to proteins. Next, hmmscan was used to search the resulting proteins against the PFAM-A v33 dataset obtained locally, including those RdRps of eukaryotic RNA viruses, such as Mononeg_RNA_pol [PF00946], RdRP_5 [PF07925], Flavi_NS5 [PF00972], Bunya_RdRp [PF04196], Mitovir_RNA_pol [PF05919], RdRP_1 [PF00680], RdRP_2 [PF00978], RdRP_3 [PF00998], RdRP_4 [PF02123], RVT_1 [PF00078], RVT_2 [PF07727], Viral_RdRp_C [PF17501], and Birna_RdRp [PF04197]. Short RdRp sequences that did not overlap with the core RdRp domains were removed. All the remaining contigs with a positive hit against RdRp domain sequences were recovered and then used to extend viral contigs (where possible) using the Geneious Prime software v.2020.
4.3.3. Phylogenetic analysis
RdRp sequences were aligned using MAFFT version 7 employing the E-INS-i algorithm. All alignments were manually trimmed using Geneious to contain only viral RdRps and neighboring conserved domains. Ambiguously aligned regions were removed using the trimAI software with default parameters (Capella-Gutiérrez et al. 2009). Phylogenetic trees were built using a maximum likelihood (ML) approach on the IQ-TREE server with the in-built automated test to choose the best substitution model for each tree (Trifinopoulos et al. 2016). The robustness of clades was inferred using 1,000 bootstrap replicates. The resultant trees were visualized using FigTree version 1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/).
4.4. Host species identification
To investigate the diversity of putative host species present in each library, we used the COI as a barcode. For each sample, local mega BLASTn searches were performed against all CO1 sequences downloaded from NCBI (e-value < 10−5). The BLAST results were then filtered for matches of a minimum length of 500 nt, with a nucleotide identity of at least 98 per cent. The corresponding predicted COI proteins were then classified by BLAST analyses against the non-redundant database at NCBI and annotated using PFAM domain searches against the PFAM-A v33 dataset obtained at https://pfam.xfam.org (Mistry et al. 2021) and run through CLC Main Workbench v.20. The COI protein sequences were dereplicated and clustered at 98 per cent amino acid identity, following the methodology previously described (Starr et al. 2019), and used to generate phylogenetic trees using known COI sequences from NCBI. Proteins were aligned using MAFFT version 7 employing the E-INS-i algorithm. The resulting alignments were trimmed for the conserved domain manually on CLC Main Workbench v. 20. Phylogenetic trees were built using the ML approach using the IQ-TREE server (Trifinopoulos et al. 2016). The robustness of ML analysis was inferred using 1,000 bootstrap replicates. The resultant trees were visualized using CLC Main Workbench v. 20.
4.5. In situ hybridization assays
RNA was treated with RNase-free DNase (Qiagen) before reverse transcription. cDNA was synthesized using the iScript first-strand synthesis kit (Bio-Rad) following the manufacturer’s instructions. Whole-mount ISHs were performed following the protocol of de Boer et al. (1998). Specific primers were designed to amplify a range of gene products (104–305 bp) using the cDNA library produced from the mixed pool of nematodes. The resulting polymerase chain reaction (PCR) products were then used as a template for the generation of sense (negative control) and antisense digoxigenin (DIG)-labeled probes using a DIG-nucleotide labeling kit (Roche). Hybridized probes within the nematode tissues were detected using an anti-DIG antibody conjugated to alkaline phosphatase and its substrate. Nematode segments were observed using a Nikon Eclipse 5i light microscope.
Data availability
All relevant data are within the manuscript and its Supplementary Material files. The raw data will be submitted to NCBI after the acceptance of the manuscript.
Supplementary Material
Contributor Information
Paulo Vieira, USDA-ARS Mycology & Nematology Genetic Diversity & Biology Laboratory, Beltsville, MD 20705, USA.
Sergei A Subbotin, Plant Pest Diagnostics Branch, California Department of Food & Agriculture, Sacramento, CA 95832, USA; Center of Parasitology of A.N. Severtsov Institute of Ecology and Evolution of the Russian Academy of Sciences, Leninskii Prospect 33, Moscow 117071, Russia.
Nadim Alkharouf, Department of Computer & Information Sciences Faculty, Towson University, Towson, MD 21204, USA.
Jonathan Eisenback, School of Plant and Environmental Sciences, Virginia Tech, Blacksburg, VA 24061, USA.
Lev G Nemchinov, USDA-ARS Molecular Plant Pathology Laboratory, Beltsville, MD 20705, USA.
Supplementary data
Supplementary data is available at Virus Evolution online.
Funding
This project was supported by the United States Department of Agriculture Agricultural Research Service CRIS project number 8042-21000-300-00D.
Conflict of interest:
None declared.
References
- Andika I. B. et al. (2017) ‘Phytopathogenic Fungus Hosts A Plant Virus: A Naturally Occurring Cross-kingdom Viral Infection’, Proceedings of the National Academy of Sciences of the USA, 114: 12267–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltimore D. (1971) ‘Expression of Animal Virus Genomes’, Bacteriological Reviews, 35: 235–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker K. R. (1985) ‘Nematode Extraction and Bioassays’. In: Barker, K. R., Carter, C. C., and Sasser, J. N. (eds) An Advanced Treatise on Meloidogyne, Volume 2. Methodology, pp. 19–35. North Carolina State University. Raleigh, NC. [Google Scholar]
- Bekal S. et al. (2011) ‘Discovery and Initial Analysis of Novel Viral Genomes in the Soybean Cyst Nematode’, The Journal of General Virology, 92: 1870–9. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2014) ‘A Novel Flavivirus in the Soybean Cyst Nematode’, The Journal of General Virology, 95: 1272–80. [DOI] [PubMed] [Google Scholar]
- Brown D. J. F. et al. (1995) ‘Transmission of Viruses by Plant Nematodes’, Annual Review of Phytopathology, 33: 223–49. [DOI] [PubMed] [Google Scholar]
- Calisher C.H. et al. (2018) ‘The discovery of arthropod-specific viruses in hematophagous arthropods: an open door to understanding the mechanisms of arbovirus and arthropod evolution?’, Annual Review Entomology, 63: 87–103. [DOI] [PubMed] [Google Scholar]
- Capella-Gutiérrez S. et al. (2009) ‘trimAl: A Tool for Automated Alignment Trimming in Large-scale Phylogenetic Analyses’, Bioinformatics, 25: 1972–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiapello M. et al. (2020) ‘Analysis of the Virome Associated to Grapevine Downy Mildew Lesions Reveals New Mycovirus Lineages’, Virus Evolution, 6: veaa058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damon C. et al. (2010) ‘Performance of COX1 Gene as a Marker for the Study of Metabolically Active Pezizomycotina and Agaricomycetes Fungal Communities from the Analysis of Soil RNA’, FEMS Microbiology Ecology, 74: 693–705. [DOI] [PubMed] [Google Scholar]
- de Boer J. M. et al. (1998) ‘In-situ Hybridization to Messenger RNA in Heterodera Glycines’, Journal of Nematology, 30: 309–12. [PMC free article] [PubMed] [Google Scholar]
- Félix M.-A. et al. (2011) ‘Natural and Experimental Infection of Caenorhabditis Nematodes by Novel Viruses Related to Nodaviruses’, PLoS Biology, 9: e1000586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz C. J. et al. (2012) ‘Complete Genome Sequence of Le Blanc Virus, a Third Caenorhabditis Nematode-infecting Virus’, The Journal of Virology, 86: 11940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frézal L. et al. (2019) ‘Noda-Like RNA Viruses Infecting Caenorhabditis Nematodes: Sympatry, Diversity, and Reassortment’, The Journal of Virology, 93: e01170–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen J. et al. (2021) ‘Lentiviral Transduction Facilitates RNA Interference in the Nematode Parasite Nippostrongylus Brasiliensis’, PLoS Pathogens, 17: e1009286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranse O. et al. (2021) ‘Toward Genetic Modification of Plant-parasitic Nematodes: Delivery of Macromolecules to Adults and Expression of Exogenous mRNA in Second Stage Juveniles’, G3, 11: jkaa058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J. et al. (2018) ‘A Novel Picornavirus-like Genome from Transcriptome Sequencing of Sugar Beet Cyst Nematode Represents A New Putative Genus’, The Journal of General Virology, 99: 1418–24. [DOI] [PubMed] [Google Scholar]
- McSorley R. (2009) ‘Soil-inhabiting Nematodes’. In: Rhodes, E. (ed.) Featured Creatures. Gainesville, FL: pp. 1–3.University of Florida. Publication Number: EENY-12. [Google Scholar]
- Mistry J. et al. (2021) ‘Pfam: The Protein Families Database in 2021’, Nucleic Acids Research, 49: D412–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paraskevopoulou S. et al. (2021) ‘Viromics of Extant Insect Orders Unveil the Evolution of the Flavi-like Superfamily’, Virus Evolution, 7: veab030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roossinck M. J. (2013) ‘Plant Virus Ecology’, PLoS Pathogens, 9: e1003304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruark C. L. et al. (2017) ‘Soybean Cyst Nematode Culture Collections and Field Populations from North Carolina and Missouri Reveal High Incidences of Infection by Viruses’, PLoS One, 12: e0171514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M. et al. (2016) ‘Redefining the Invertebrate RNA Virosphere’, Nature, 540: 539–43. [DOI] [PubMed] [Google Scholar]
- Starr E. P. et al. (2019) ‘Metatranscriptomic Reconstruction Reveals RNA Viruses with the Potential to Shape Carbon Cycling in Soil’, Proceedings of the National Academy of Sciences of the USA, 116: 25900–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treonis A. M. et al. (2018) ‘Characterization of Soil Nematode Communities in Three Cropping Systems through Morphological and DNA Metabarcoding Approaches’, Science Report, 8: 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifinopoulos J. et al. (2016) ‘W-IQ-TREE: A Fast Online Phylogenetic Tool for Maximum Likelihood Analysis’, Nucleic Acids Research, 44: W232–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Sluijs L. et al. (2021) ‘Virus Infection Modulates Male Sexual Behavior in Caenorhabditis Elegans’, Molecular Ecology, 30: 6776–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira P., and Nemchinov L. G. (2019) ‘A Novel Species of RNA Virus Associated with Root Lesion Nematode Pratylenchus Penetrans’, The Journal of General Virology, 100: 704–8. [DOI] [PubMed] [Google Scholar]
- Williams S. H. et al. (2019) ‘Discovery of Two Highly Divergent Negative-sense RNA Viruses Associated with the Parasitic Nematode, Capillaria Hepatica, in Wild Mus Musculus from New York City’, The Journal of General Virology, 100: 1350–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S. et al. (2018) ‘Evaluation of Two Main RNA-seq Approaches for Gene Quantification in Clinical RNA Sequencing: PolyA+ Selection versus rRNA Depletion’, Science Report, 8: 4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data are within the manuscript and its Supplementary Material files. The raw data will be submitted to NCBI after the acceptance of the manuscript.