

Abstract
Sickle cell disease (SCD) is a worldwide distributed hereditary red cell disorder characterized by recurrent acute vaso-occlusive crises (VOCs and anemia). Gold standard treatments are hydroxycarbamide (HC) and/or different red blood cell (RBC) transfusion regimens to limit disease progression. Here, we report a retrospective study on 1,579 SCD patients (median age 23 years; 802 males/777 females), referring to 34 comprehensive Italian centers for hemoglobinopathies. Although we observed a similar proportion of Caucasian (47.9%) and African (48.7%) patients, Italian SCD patients clustered into two distinct overall groups: children of African descent and adults of Caucasian descent. We found a subset of SCD patients requiring more intensive therapy with a combination of HC plus chronic transfusion regimen, due to partial failure of HC treatment alone in preventing or reducing sickle cell-related acute manifestations. Notably, we observed a higher use of acute transfusion approaches for SCD patients of African descent when compared to Caucasian subjects. This might be related to (i) age of starting HC treatment; (ii) patients' low social status; (iii) patients' limited access to family practitioners; or (iv) discrimination. In our cohort, alloimmunization was documented in 135 patients (8.5%) and was more common in Caucasians (10.3%) than in Africans (6.6%). Alloimmunization was similar in male and female and more frequent in adults than in children. Our study reinforces the importance of donor-recipient exact matching for ABO, Rhesus, and Kell antigen systems for RBC compatibility as a winning strategy to avoid or limit alloimmunization events that negatively impact the clinical management of SCD-related severe complications.
Clinical Trial Registration
ClinicalTrials.gov, identifier: NCT03397017.
Keywords: hydroxycarbamide, multi-ethnicity, sickle cell disease, transfusion therapy, alloimmunization
Introduction
Sickle cell disease (SCD) is one of the most common inherited red cell disorders worldwide, recognized by the World Health Organization (WHO) and the United Nations as a “global health problem” (1, 2). About 300,000 infants are born each year with SCD; ~90,000 in Nigeria, 44,000 in India, 40,000 in the Democratic Republic of Congo, 12,000 in the United States (US), 2,000 in Europe, and 300 in the United Kingdom (UK) (3). Although SCD is part of the rare disease group, more than 10,000 SCD patients live in the UK and more than 25,000 in France, making SCD much more common than cystic fibrosis or hemophilia A (4, 5). The median life expectancy is now more than 60 years in Europe (6), but about 80% of patients with SCD in African regions die during childhood (7).
Treatment options for SCD are still limited, particularly in low-income settings. Hydroxycarbamide (HC) is the gold standard treatment for both children and adults with SCD (8). Hydroxycarbamide reduces painful vaso-occlusive events (VOCs) and acute chest syndrome as well as the need for red blood cell (RBC) transfusion. Until now, RBC transfusion has largely been used for clinical management of both acute and chronic sickle cell-related complications (9, 10). Despite the known indications for RBC transfusion and HC therapy (9, 11–16), treatment of patients with SCD in clinical practice is still not homogeneous.
Over the course of a lifetime, patients with SCD might be exposed to different RBC transfusion regimens, ranging from simple transfusion on-demand or manual/automatized RBC exchange to manage acute VOCs or to limit disease progression and organ damage (3, 17). Alloimmunization to RBC blood group antigens carries an increased risk of hemolytic transfusion reactions (DHTR), potentially delaying the identification of compatible RBC units (18, 19). In Europe, Italy presents a distinctive epidemiologic European niche for SCD, characterized by endemic SCD population mainly in Southern Italian regions and multi-ethnic SCD population localized in Central and Northern Italian regions (20–23). This affects the distribution of SCD genotypes, mainly represented by Sβ0/Sβ+ or SS genotype in patients of Caucasian descent, while SS or SC genotypes characterize patients of African descent. To better understand the scenario of transfusion regimens and alloimmunization of SCD patients in Italy, we carried out a national survey involving the National Comprehensive Reference Centers for SCD and the Italian Society of Thalassemia and Hemoglobinopathies (SITE), in collaboration with the Italian Society of Transfusion Medicine and Immunohematology (SIMTI) and the Italian Association of Hematology and Pediatric Oncology (AIEOP).
Materials and Methods
Study Design
This is a retrospective multicenter national study (ClinicalTrials.gov identifier NCT03397017). Data from the clinical records of eligible patients were collected from 2015 to 2018 through a standard web-based application (www.SITE-italia.org) encrypted by the Central Server. The recording of personal, therapy, and complications data of patients with sickle cell anemia included in the National Transfusion Treatment Survey was undertaken by the responsible investigator or sub-investigators selected by each center, following registration on the site. The operator can subsequently access the patient's clinical data and perform updates to follow clinical evolution over time. The study did not involve any additional tests compared to routine patient management.
The study aimed to identify the lifelong therapeutic approaches, focusing on transfusion regimens, in clinical management of a large multi-ethnic Italian cohort of patients with SCD. The study was coordinated by SITE, in collaboration with SIMTI and AIEOP. A total of 34 Italian reference centers from 15 Italian regions were involved. Data were collected through the National Comprehensive Reference Centers for SCD. The study was approved by the Ethics Committee of Fondazione IRCCS Ca' Granda, Ospedale Maggiore Policlinico of Milan, Italy, the coordinating center of the study.
The inclusion and exclusion criteria were, respectively, the presence or absence of a biochemical and/or genetic diagnosis of SCD and the capability or not to give informed consent. Written informed consent was obtained from all included patients after the study aims and protocol had been explained. When necessary, a Cultural Linguistic Mediator could be engaged to assist in the consent process. Besides demographic data, every center collected data concerning the type and sequence of therapy, acute transfusion therapy (ATR), chronic transfusion therapy (CTR, i.e., regular transfusion regimen for more than 1 year), or HC, the main indications for ACR and CTR, and details on alloimmunization (number and systems of antibodies) over the previous 4 years of treatment.
Statistical Analysis
Descriptive statistics of continuous variables were expressed using medians and interquartile range (IQR: 25th-75th percentile); dichotomous variables were summarized as counts and percentages. The Shapiro-Wilk test was used to test the normality of distribution. As no normal distribution was found, the differences between groups were analyzed with the Wilcoxon signed-rank test. Two- or multiple-proportion comparisons between variables were performed with the Z-test. All tests were two-sided, and p-values < 0.05 were considered as statistically significant. Data management and graphs were created with Excel (Microsoft, Seattle, WA, USA), and statistical analysis was performed using R version 3.5.1 for Windows (R Core Team, Vienna, Austria).
Results
SCD Italian Patients Cluster Into Two Overall Distinct Groups: Children of African Descent and Adults of Caucasian Descent
Our study shows that Italian SCD patients overall cluster into two distinct groups: children of African and adults of Caucasian descent (Figure 1A). We collected data from 1,579 patients, of whom 802 were male (51%) and 777 were female (49%) with a median age of 23 (IQR 10–41) years (Supplementary Figure S1). No significant difference in age was found between males and females (p = 0.18); the median age was 21.5 (IQR 10–40) years in males and 25.3 (IQR 10–42) years in females. Ethnicity was equally distributed between Caucasians (47.9%) and Africans (48.7%), with a minority of African American (2.8%) and Asian (0.4%) SCD patients. Ethnicity was not defined for 0.2% of patients (Table 1). In African patients, the most common genotype was homozygosity for HbS (SS; 71.3%). Compound heterozygosity with β-thalassemia mutations was 2.3% for S/β0-thalassemia, 3.9% for S/β+-thalassemia, and 20.8% for SC disease. Otherwise, the most prevalent genotype in Caucasians was S/β-thalassemia (74.5%; in particular, S/β0-thalassemia 34.8% and S/β+-thalassemia 39.7%). SS genotype was present in 21.4% and SC in 0.5% of SCD patients of Caucasian descent. The median age of African SCD patients was 12 (IQR 7–22) years compared with 38 (IQR 25–49) years in Caucasian patients (p < 0.001).
Figure 1.

(A) Distribution of sickle cell disease (SCD) patients by age and ethnicity. Data are shown as a column chart that reports, for each age group, the number of Africans and Caucasians patients of the cohort. (B) Distribution of SCD patients by genotype and therapy. Data are shown as a column chart that reports, for each genotype, the number of patients that received HC/ATR/CTR/NONE as their therapy regimen. ATR, acute transfusion; CTR, chronic transfusion; HC, hydroxycarbamide.
Table 1.
Demographic characteristics of patients with sickle cell disease (SCD).
| Ethnicity | Genotype | Counts (%) | Males (%) | Median age (IQR), yr |
|---|---|---|---|---|
| African (n = 769) | HbSS | 548 (71.3) | 280 (51.2) | 11 (6.9–19.8) |
| HbS/β°-thalassemia | 18 (2.3) | 12 (66.7) | 14.5 (9.5–19.8) | |
| HbS/β+-thalassemia | 30 (3.9) | 16 (53.3) | 26.5 (15.6–37.5) | |
| HbSC | 160 (20.8) | 78 (48.8) | 16.9 (7.8–27.0) | |
| Not defined | 13 (1.7) | 3 (23.1) | 40 (12–46) | |
| Caucasian (n = 756) | HbSS | 162 (21.4) | 76 (46.9) | 35.4 (18–48) |
| HbS/β°-thalassemia | 263 (34.8) | 138 (52.5) | 40 (27–50) | |
| HbS/β+-thalassemia | 300 (39.7) | 161 (53.7) | 39 (25–51) | |
| HbSC | 4 (0.5) | 3 (75.0) | 31.5 (20–40.8) | |
| Not defined | 27 (3.6) | 14 (51.9) | 31.9 (22–45) | |
| African-American (n = 45) | HbSS | 24 (53.3) | 9 (37.5) | 29 (22.6–36.2) |
| HbS/β°-thalassemia | 4 (8.9) | 2 (50.0) | 12.5 (6.8–23.2) | |
| HbS/β+-thalassemia | 4 (8.9) | 1 (25.0) | 25 (9.5–41.5) | |
| HbSC | 13 (28.9) | 4 (30.8) | 27.5 (21–35) |
Asian patients comprised 0.4%, and people of another ethnicity made up the other 0.2%.
Hb, hemoglobin; IQR, interquartile range; yr, years.
Combined Transfusion Approaches With Hydroxycarbamide Were Used to Limit HC-Resistant Sickle Cell-Related Clinical Manifestations
Full data on therapeutic management of SCD patients were collected for 1,364 out of the 1,579 patients (Table 2; Figure 1B; Supplementary Figure S1). The following therapeutic regimens were identified: (i) HC: chronic hydroxycarbamide therapy for more than 1 year; (ii) CTR: chronic transfusion regimen; (iii) ATR: acute transfusion approach; and (iv) no treatment (Figure 2A). When we considered therapeutic strategies in function of age, we found that blood transfusion approaches were used throughout patient's journey (Figure 2B). A total of 292 patients (21.4%) were on CTR, 196 (14.4%) on ATR, and 497 on HC (36.4%), of whom 100 needed transfusions for acute VOCs. Three hundred and seventy-nine (27.8%) patients never received long-term therapy. In both ATR and CTR clinical setting, donor-recipient exact matching for serological ABO, Rhesus and Kell antigen for RBC compatibility was carried out according to international and Italian guidelines, released in 2014 (9, 13, 24–27).
Table 2.
Treatments of SCD patients (n = 1,364) and distribution according to ethnicity and genotype.
| Therapya | Pts (%) | M vs. F | African vs. Caucasian vs. African-American | SS, No. (%) | S/β°, No. (%) | S/β+, No. (%) | SC, No. (%) | p-value |
|---|---|---|---|---|---|---|---|---|
| CTR | 292 (21.4) | 152/140 | 94/181/14 | 139 (49.5) | 64 (22.8) | 69 (24.6) | 9 (3.2) | <0.001 |
| ATR | 196 (14.4) | 92/104 | 116/69/10 | 103 (53.1) | 26 (13.4) | 32 (16.5) | 33 (17) | <0.001 |
| HC | 497 (36.4) | 273/224 | 238/246/11 | 268 (54.7) | 110 (22.4) | 96 (19.6) | 16 (3.3) | <0.001 |
| None | 379 (27.8) | 196/183 | 280/91/7 | 155 (42.6) | 30 (8.2) | 64 (17.6) | 115 (31.6) | <0.001 |
| bATR/CTR → HC | 236 (17.3) | 129/107 | 120/109/7 | 138 | 53 | 37 | 3 | – |
| bHC → CTR | 14 (1.0) | 8/6 | 6/7/1 | 9 | 2 | 2 | 1 | – |
The sum of partial counts may not correspond to the total in the case of different and/or not defined ethnicity/genotype.
Subset of 250 patients for whom it was possible to follow the timing of therapy.
A total of 100 patients under HC were transfused for the clinical management of acute vaso-occlusive events (VOCs).
ATR, acute transfusion regimen; CTR, chronic transfusion regimen; F, female; HC, Hydroxycarbamide; M, male; Pts, patients.
Figure 2.

(A) Distribution of sickle cell disease (SCD) patients by age and therapy. Data are shown as a column chart that reports, for each age group, the number of patients that received HC/ATR/CTR/NONE as their therapy regimen. (B) Distribution of the indications for transfusion therapy. Data are shown as counts of the indications for ATR and CTR. ACS, acute chest syndrome; ATR, acute transfusion; CTR, chronic transfusion; HC, hydroxycarbamide; MOFs, multi-organ failures; VOCs, vaso-occlusive events.
The main indications for CTR were recurrent severe VOCs (371 events; 37.1%), chronic anemia (306 events; 30.6%), primary stroke prevention (78 events, 7.8%) and secondary stroke prevention (55 events, 5.5%), acute chest syndrome (ACS; 107 events; 10.7%), splenic sequestration (31 events, 3.1%), HC-resistant pain (39 events; 3.9%) and leg ulcers (12 events; 1.2%) (Figure 2B). The median value of hemoglobin (Hb) before CTR was 9.2 g/dL (IQR: 8.5–10, min-max: 5.2–14.9 g/dL, data available for 85% of patients) and the median value of HbS was 50% of total Hb (IQR: 39–55, min-max: 8.5–89%, data available for 67% of patients).
The main indications for ATR were acute anemia (384 events; 35%), VOCs (352 events; 32.1%), ACS (170 events; 15.5%), surgery (82 events; 7.5%), pregnancy (64 events; 5.8%), splenic sequestration (26 events; 2.4%), stroke (9 events; 0.8%), multi-organ failure (6 events; 0.5%) and priapism (5 events; 0.5%) (Figure 2B). Before ATR, the median value of Hb level was 8.0 g/dL (IQR: 6.8–9.0 g/dL, min-max 2.7–15.2 g/dL, data available for 72% of patients), with 67% of HbS (IQR: 56–78%, min-max: 30–93, data available for 43% of patients). Patients who were not treated had a median Hb value of 10.7 g/dL (IQR: 9.3–12 g/dL). When we stratified SCD population according to age: children (<18 years) and adults (>18 years), the proportion of different indications for ATR was similar within the two groups except for pregnancy and priapism, which were present in adults (Supplementary Figure S2A). Considering CTR, we found CTR to be more frequently introduced in adults than in children with SCD (Supplementary Figure S2A). CTR protocols were more frequently applied in adults than in children to prevent ACS and to treat chronic pain resistant to HC therapy (Supplementary Figure S2A). We found a higher proportion of Caucasian adults (older than 20 years) under CTR (135/181, 75%) when compared to African adults (41/94, 44%). In children and adolescents (younger than 20 years), the proportion of Caucasians treated with CTR (46/181, 25%) was lower when compared with Africans (53/94, 56%). The ratio between patients in ATR and CTR decreases with during aging (p < 0.05, Figure 2A).
HC was generally started after 5 years of age in SCD patients of both Caucasian (6%) and African (94%) descent. Two hundred and fourteen of 246 Caucasian patients were adults (17% SS and 79% Sβ genotypes). As expected, SCD patients with SC genotype showed less intensive medical treatment than SCD patients with either SS or Sβ0/+ genotypes (Figure 1B).
For 250 out of 1,364 patients (18.3%), it was possible to follow the temporal sequence of change in therapy (Table 2). Fourteen patients (1.0% of the 1,364) switched from HC to CTR, and 236 patients (17.3%) switched from CTR to HC. A subset of 60 patients (4.3% of the 1,364 with treatment data available) received a simultaneous blood transfusion and HC for a median period of 4.6 years (IQR 2.2–11.1 years). This was due to the severity of sickle cell-related clinical presentation, which was only partially controlled by HC therapy. The main indications for CTR in these 60 patients were, respectively, VOCs (number of events: 33), anemia (n = 25), ACS (n = 13). HC-resistant pain (n = 6), splenic sequestration (n = 4), primary (n = 2) or secondary (n = 3) stroke prevention. Combined therapy (CTR+HC) was started at a median age of 20.5 (IQR 10.7–30.8) years. In 14/250 patients, HC was started at a median age of 30.7 (IQR 28.1–41.4) years, and CTR needed to be added to HC after a mean period of 3.8 (IQR 2.4–5.3) years at a median age of 34.4 (IQR 30.3–48.3) years. The proportion of SCD patients without any treatment decreased with aging (Figure 2A). Two out of 1,364 patients with SCD displayed hyperhemolysis reaction after RBC transfusion (1 African SS child: 5 years of age; 1 African SS adult: 23 years of age).
Collectively, our data indicate that acute and chronic transfusion approaches were chosen to limit disease progression in patients of both Caucasian and African descent. RBCs from Caucasian donors were used. The donor-recipient exact matching for ABO, Rhesus and Kell antigen for RBC compatibility strategy limits alloimmunization, which might further complicate clinical management of patients with SCD (9, 18, 19, 28).
Low Immunization Events Characterize the Italian Multi-Ethnic Cohort of SCD Patients
In our study, alloimmunization was documented in 135 (8.5%) out of 1,579 patients with SCD. Sixty-one of these 135 patients (45.2%) were already alloimmunized at their arrival at the comprehensive SCD center. Four of them (6.6%) developed alloimmunization against new RBC antigen (Supplementary Figure S3A) 36 of these 135 patients (27%) developed alloantibodies after they arrived at the Italian referral center (Supplementary Figure S3A). Data on alloantibody status in relation to the timing of arrival at the SCD center were not available for the other 38 alloimmunized SCD patients.
Among the alloimmunized SCD patients, 33 (24.4%) were younger than 18 years, 53 (39.3%) were aged 18–40 years, and 49 (36.3%) were older than 40 years (Supplementary Figure S3B). Fifty-one alloimmunized patients were African [median age 20 years (IQR 11–28 years)], corresponding to 6.6% of the African group; 78 patients were Caucasian [median age 41 years (IQR 31–51 years)], representing 10.3% of the Caucasian group; five patients were African American [median age 35 years (IQR 30–37 years)] and for one patient (age 29 years) ethnicity was not reported. Thus, alloimmunization was more frequent in adults than in children with SCD (p < 0.001). The frequency of alloimmunization was similar in female and male patients with SCD.
RBC-specific antibodies were detected in 124 patients, of whom 70 (41 males, 29 females) had a single antibody and 43 (16 males and 27 females) had multiple antibodies (59.5%, 28.6, 9.5, and 2.4%, respectively, with two, three, four, and six antibodies). Specific antibodies were not identified in 11 patients (3 males and 8 females) (Supplementary Figure S3C). A higher proportion of Caucasian patients had multiple antibodies compared with patients of other ethnicities. In the Caucasian group, a single antibody was prevalently found in males (24/40, 60%), while there were no significant differences between genders in the African group. When genotypes were considered, we observed alloimmunization to be more frequently associated with SS genotype in both African and African American patients, whereas SS and Sβ0/+ genotypes were equally associated with alloimmunization in SCD patients of Caucasian descent (Supplementary Figure S3B).
As shown in Figure 3, alloimmunization predominantly involves RhCDE and Kell systems, with Rhesus (45%) the most represented antibody system, followed by Kell (15%), MNS (12%), Duffy (6.2%), Lewis (4.8%), Kidd (4.3%) and Lutheran (2.7%). In addition, antibodies against minor antigens (0.8%) and antibodies not better identified (8.8%) were also detected. Of interest, anti-e antibodies were found in two e-negative African SCD patients. When we considered alloimmunization profile and patients' age, a higher rate (26 vs. 6%) of alloantibodies with non-identified specificity in children's group was observed (Figure 3, right pie-charts).
Figure 3.

Number of children/adults in SCD patients by antibody system. The pie charts report the overall distribution of the specific antibodies for children and adults. Data are shown as column chart that reports, for each system and differentiating for children/adults, the number of African, Caucasian and African-American patients with the specific antibody.
Eleven patients (8.1%) with alloimmunization developed a delayed hemolytic transfusion reaction (DHTR) (8 females and 3 males) with a median age of 39 years (IQR: 18.5–49). Six of these patients had SS genotype (4 Africans, 2 Caucasians), and five were Caucasian with Sβ genotype. In 4 out of the 11 patients, a single antibody (D, C, Cw, Jsb) was detected, and in another 4 of the 11 (all females, 3 Caucasians, and 1 African) multiple antibodies (2–4 different specificities) were detected. In 2 patients, antibodies were not clearly identified. Taken together, these data indicate that low alloimmunization events recorded in both African and Caucasian patients might most likely be related to the donor-recipient exact matching for ABO, Rhesus and Kell antigen for RBC compatibility.
Discussion
Our study highlights the complexity of the clinical management of SCD manifestations throughout the patients' journey. The peculiar multi-ethnic profile of the Italian SCD population, with a roughly equal proportion of Caucasian and African patients, is an added value to the analysis of real-life data. In our series, Caucasian SCD patients were generally older than African patients. This is most likely related to the efficacy of the prenatal program for hemoglobinopathies that started in Italy in the 1980s (29, 30). In contrast, African SCD patients were mainly clustered in pediatric and young adult populations, reflecting the voluntary migration fluxes of the last two decades (22, 23, 31). Indeed, the presence of elderly Caucasian SCD patients represents a unique SCD cohort, providing important information on the combinatory effects of aging and SCD on disease natural history (32, 33).
In our study, the transfusion approach was similar in SS and S/β patients in terms of ATR and CTR regimens, and the percentage of patients with S/β+- and S/β° who were treated with ATR and CTR was similar. Both CTR and ATR were used more frequently in SS patients compared with either S/β+ or S/β°- patients (34). The main reasons for ATR were similar to those described by Rees et al. (35).
Notably, we found that SS African patients were treated with ATR more than Caucasian patients with S/β genotypes. This might be related to (i) patients' low social status; (ii) patients' limited access to family practitioners, generally related to troubles in speaking Italian. All these factors might contribute to the higher access to an emergency department for African SCD patients during acute events when compared to Caucasian SCD subjects (10).
Simple transfusions were the main transfusion strategy used during CTR, and single top-up transfusions were the main one in the acute setting. Manual and automatic exchange transfusions were used more in CTR than in ATR. This might be related to: (i) difficulties in vein access during VOCs; (ii) absence of expertise; (iii) overnight acute events (36, 37).
Of note, we found a subset of patients still symptomatic for acute SCD-related clinical manifestations under treatment with HC, highlighting the biocomplexity of SCD that lies beneath red cell sickling and dehydration (38–44). Indeed, HC partially acts on sickle cell-related inflammatory vasculopathy and chronic inflammation (4, 45–47). Thus, SCD patients considered partial responders to HC require a more intensive treatment, which has until now been represented by the combination of HC and chronic transfusion programs. In our cohort, combination therapy of CTR and HC was administered in a similar percentage of African and Caucasian SS patients. The percentage of SS patients who required long-term combination treatment was higher than the proportion with S/β°-S/β+- genotypes. Considering chronic therapies (CTR and HC), we confirmed that SC patients needed less intensive treatment than other SCD genotypes (14). Thus, our data highlight the presence of a subset of SCD patients with severe clinical manifestations resistant to HC treatment. These might now be eligible for new US Food and Drug Administration and European Medicines Agency therapeutic options such as crizanlizumab, a humanized antibody against P-selectin, or voxelotor, a novel oral anti-sickling agent, alone or in combination with HC (41, 42, 48–52).
In our cohort, alloimmunization was identified in SCD patients aged between 18 and 40 years; those aged <18 years had a lower alloimmunization events than those aged ≥18 years, most likely because they had a shorter transfusion history. A higher proportion of Caucasian patients (10.3%) than African patients (6.6%) had alloimmunization, and among these, Caucasian patients again were older than African patients (median 41 vs. 20 years).
Although the alloimmunization rate was not available due to the study's retrospective design, the low alloimmunization events in both African and Caucasian patients, might reflect the satisfactory level of donor-recipient exact matching for ABO Rhesus and Kell antigen for RBC compatibility, consistent with the Italian guidelines for children and adults released on 2014 (11, 15). Antigen mismatch between donor and recipient is the basis for antibody formation, as the recipient recognizes those antigens as non-self and, thereby, an immune response might be elicited (53, 54). Transfused SCD patients have the highest rates of RBC alloimmunization over their lifetimes (up to 40–50%), compared to fewer than 5% of other transfused individuals with either thalassemia major or myelodysplasia (55, 56). Detection of alloantibodies may make locating compatible RBC units for future transfusion difficult, delayed, and sometimes impossible (54). Potential reasons for alloimmunization include (i) number of donor exposures; (ii) antigenic mismatches between donor RBCs and recipient RBCs related to ethnicity (mainly between Caucasian donors and African descent recipients); (iii) the inflammatory state of recipients (57). SCD is characterized by a chronic inflammatory state, worsening during acute VOCs (57, 58). Thus, SCD patients transfused in their baseline state of health are thought to be less likely to become alloimmunized than patients transfused in a state of inflammation, such as during acute VOCs (58). Studies in SCD patients have shown that having an acute chest syndrome at the time of transfusion is a significant risk factor for becoming alloimmunized (53, 58). In our cohort, the antibodies identified were mainly of RhCDE and Kell systems, as reported in studies of transfused SCD patients from the United States and Brazil (9). Different antibodies systems (i.e., MNS and Kidd) and different frequencies of alloimmunization have been recently described in SCD patients in French Guiana when compared to our cohort or those from the United States or Brazil (18). These might reflect the heterogeneity of donor and recipient populations in different geographical settings, which increases the complexity of data analysis and comparison.
Our study presents some limitations: (i) the retrospective design of the study, which did not allow us to evaluate the alloimmunization rate; (ii) lacking data on patients' transfusion history before their arrival to the comprehensive centers for hemoglobinopathies; (iii) no record on patients' mortality.
In conclusion, our study highlights that transfusion regimens are still crucial as intensive treatments for both acute and chronic SCD-related complications. In our cohort, HC was offered to symptomatic SCD children in agreement with national recommendations (10, 15). This clearly favors a more extensive use of transfusion approaches in the pediatric setting in our cohort in case of acute complications. Since alloimmunizations remains a barrier for safe and effective transfusion of patients with SCD, a careful evaluation of RBC transfusion guidelines transfusion becomes a key factor, along with the implementation of transfusion protocols (such as phenotypically-matched RBCs: RhCDE and Kell), genotyping of patients and donors to identify RBC antigenic variants and maintenance of records on patient transfusion history (59, 60). In addition, dissemination of the knowledge on management of DHTR in patients with SCD, might positively impact patients' outcome (61). These measures are expected to reduce the occurrence of alloimmunization as well as the incidence of delayed hemolytic transfusion reactions, optimizing the management of SCD subjects of different descent throughout the patients' journey.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of Fondazione IRCCS Ca' Granda, Ospedale Maggiore Policlinico of Milan, Italy. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
GG, LDF, and GLF contributed to the conceptualization and design of the study, acquisition and curation of the data, contributed to data analysis and interpretation, writing, critical appraisal and comments, and reviewing and editing. LS, DV, NM, PB, AV, MC, GL, and VV contributed to the conceptualization and design of the study, acquisition and curation of the data, and critical appraisal and comments. PR, VP, AQ, LN, GR, MA, RR, DD'A, EF, SMar, FA, FB, ER, AS, SC, GC, FG, RL, PG, GB, AF, SMac, PM, MM, RO, FL, MB, RC, RD, AP, PC, CF, GP, and LB contributed to acquisition and curation of the data. BG and FP contributed to data analysis and interpretation. All authors have read and agreed to the published version of the manuscript.
Funding
Financial support for Medical Editorial Assistance was provided by Novartis Farma SpA. Novartis Farma SpA was not involved in the study design, collection, analysis, interpretation of data, and the writing of this article or the decision to submit it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Ray Hill, an Independent Medical Writer who provided medical writing assistance with this manuscript on behalf of Springer Healthcare, which was funded by Novartis Farma SpA, Italia in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3). Authors had full control of the content and made the final decision for all aspects of this article.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2022.832154/full#supplementary-material
Flow-chart of study population. Pts, patients; SCD, sickle cell disease.
{kind=link}
Distribution of the indications for transfusion therapy regimen for children and adults, (A) Indications for acute transfusion regimen (ATR). (B) Indications for chronic transfusion regimen (CTR). Data are shown as counts of the indications for ATR and CTR. ACS, acute chest syndrome; ATR, acute transfusion; CTR, chronic transfusion; HC, hydroxycarbamide; MOFs, multi-organ failures; VOCs, vaso-occlusive events.
{kind=link}
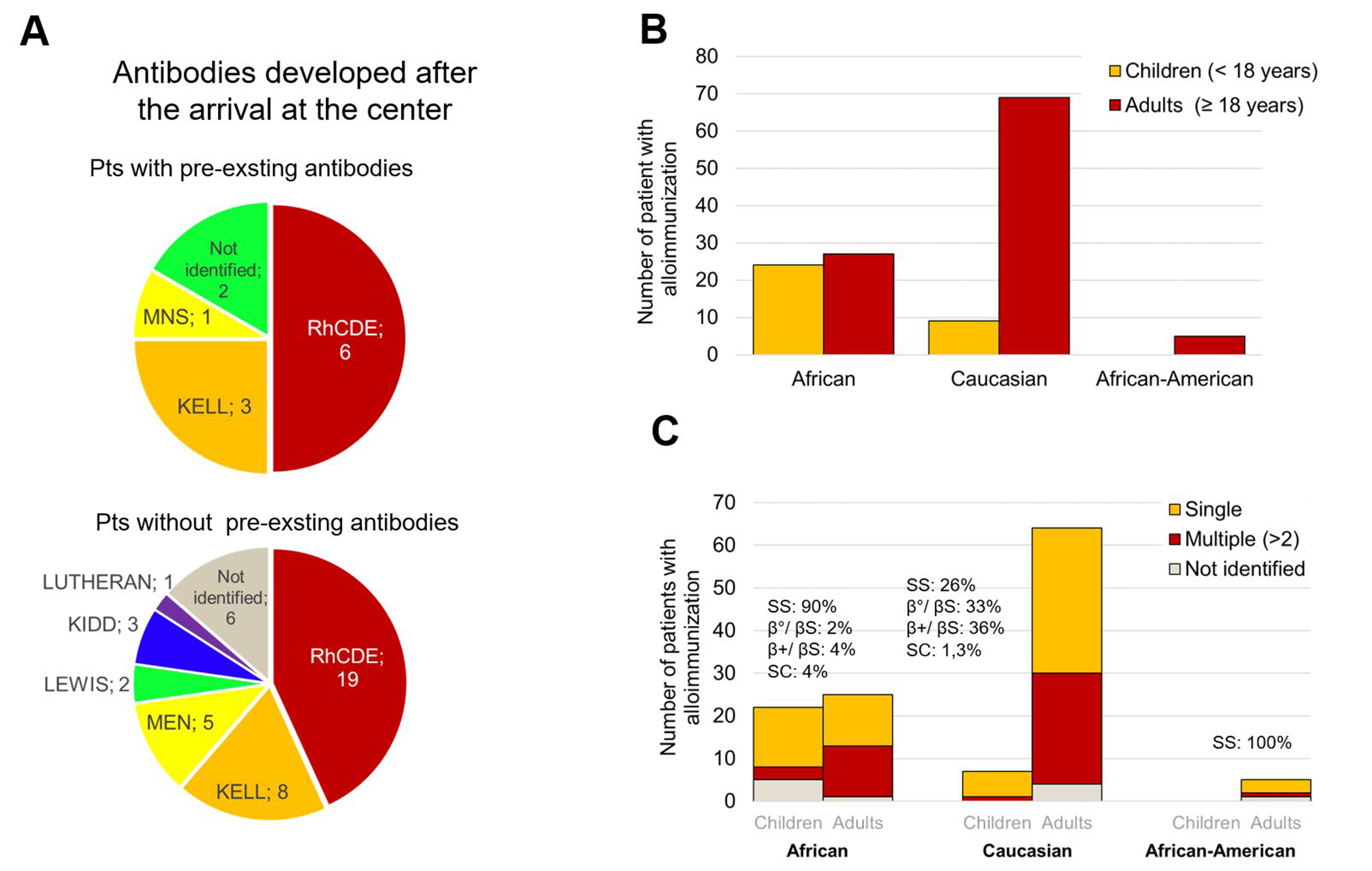
(A) Distribution of new antibodies developed in after the arrival at the center for sickle cell disease (SCD) patients with pre-existing (up) and without (low) pre-existing antibodies. (B) Number of SCD children/adults patients with alloimmunization by ethnicity. (C) Number of SCD children/adults patients with alloimmunization presenting single, multiple or unidentified antibody/ies. Data are shown as pie and column charts with the number of patients with alloimmunization.
{kind=link}
References
- 1.World Health Organization. Sickle-Cell Anaemia: Report by the Secretariat. Geneva: World Health Organization (2006). Available online at: https://apps.who.int/gb/archive/pdf_files/WHA59/A59_9-en.pdf (accessed June 23, 2021).
- 2.United Nations Secretary-General,. Secretary-General's Message on Sickle-Cell Anaemia (2009). Available online at: https://www.un.org/sg/en/content/sg/statement/2009-06-19/secretary-generals-message-sickle-cell-anaemia (accessed June 18, 2021).
- 3.Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of sickle cell anaemia in children under five, 2010-2050: modelling based on demographics, excess mortality, and interventions. PLoS Med. (2013) 10:e1001484. 10.1371/journal.pmed.1001484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brousse V, Makani J, Rees DC. Management of sickle cell disease in the community. BMJ. (2014) 348:g1765. 10.1136/bmj.g1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leleu H, Arlet JB, Habibi A, Etienne-Julan M, Khellaf M, Adjibi Y, et al. Epidemiology and disease burden of sickle cell disease in France: a descriptive study based on a French nationwide claim database. PLoS ONE. (2021) 16:e0253986. 10.1371/journal.pone.0253986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gardner K, Bell C, Bartram JL, Allman M, Awogbade M, Rees DC, et al. Outcome of adults with sickle cell disease admitted to critical care - experience of a single institution in the UK. Br J Haematol. (2010) 150:610–3. 10.1111/j.1365-2141.2010.08271.x [DOI] [PubMed] [Google Scholar]
- 7.Rees DC. To begin at the beginning: sickle cell disease in Africa. Lancet Haematol. (2014) 1:e50–51. 10.1016/S2352-3026(14)00006-4 [DOI] [PubMed] [Google Scholar]
- 8.Colombatti R, Palazzi G, Masera N, Notarangelo LD, Bonetti E, Samperi P, et al. Hydroxyurea prescription, availability and use for children with sickle cell disease in Italy: Results of a National Multicenter survey. Pediatr Blood Cancer. (2018) 65:26774. 10.1002/pbc.26774 [DOI] [PubMed] [Google Scholar]
- 9.Chou ST, Alsawas M, Fasano RM, Field JJ, Hendrickson JE, Howard J, et al. American Society of Hematology 2020 guidelines for sickle cell disease: transfusion support. Blood Adv. (2020) 4:327–55. 10.1182/bloodadvances.2019001143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forni GL, Finco G, Graziadei G, Balocco M, Rigano P, Perrotta S, et al. Development of interactive algorithm for clinical management of acute events related to sickle cell disease in emergency department. Orphanet J Rare Dis. (2014) 9:91. 10.1186/1750-1172-9-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Società Italiana Talassemie ed Emoglobinopatie – Società Italiana di Medicina Trasfusionale. Raccomandazioni per le strategie trasfusionali nelle Emoglobinopatie n.3 [Italian] . Bonomo P, Carta MP, Forni GL, Prati D, Rigano P, Vassanelli A, editors. Bologna: SITE (2014). Available online at: www.SITE.italia.org (accessed June 16, 2021).
- 12.Lottenberg R, Hassell KL. An evidence-based approach to the treatment of adults with sickle cell disease. Hematology Am Soc Hematol Educ Program. (2005) 58–65. 10.1182/asheducation-2005.1.58 [DOI] [PubMed] [Google Scholar]
- 13.Davis BA, Allard S, Qureshi A, Porter JB, Pancham S, Win N, et al. Guidelines on red cell transfusion in sickle cell disease. Part I: principles and laboratory aspects. Br J Haematol. (2017) 176:179–91. 10.1111/bjh.14346 [DOI] [PubMed] [Google Scholar]
- 14.Davis BA, Allard S, Qureshi A, Porter JB, Pancham S, Win N, et al. Guidelines on red cell transfusion in sickle cell disease Part II: indications for transfusion. Br J Haematol. (2017) 176:192–209. 10.1111/bjh.14383 [DOI] [PubMed] [Google Scholar]
- 15.Casale M, Casciana ML, Ciliberti A, Colombatti R, Del Vecchio GC, Fasoli S, et al. Raccomandazioni per la gestione della Malattia Drepanocitica in Età Pediatrica in Italia [Italian]. (2018). Available online at: www.aieop.org (accessed June 16, 2021).
- 16.Russo G, De Franceschi L, Colombatti R, Rigano P, Perrotta S, Voi V, et al. Current challenges in the management of patients with sickle cell disease - a report of the Italian experience. Orphanet J Rare Dis. (2019) 14:120. 10.1186/s13023-019-1099-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carrara P, Balocco M, Pinto V, Olcese F, Sold Strada P, Forni GL. Manual erythroexchange for chronic transfusion therapy in patients with sickle cell syndromes unresponsive to hydroxyurea: A long-term follow-up. Am J Hematol. (2010) 85:974. 10.1002/ajh.21895 [DOI] [PubMed] [Google Scholar]
- 18.Conrath S, Vantilcke V, Parisot M, Maire F, Selles P, Elenga N. Increased prevalence of alloimmunization in sickle cell disease? Should we restore blood donation in French Guiana? Front Med. (2021) 8:681549. 10.3389/fmed.2021.681549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Linder GE, Chou ST. Red cell transfusion and alloimmunization in sickle cell disease. Haematologica. (2021) 106:1805–15. 10.3324/haematol.2020.270546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Russo-Mancuso G, Sciotto A, Munda SE, Romano V, Schilirò G. Alloimmunization and autoimmunity in Caucasian patients with sickle cell disease. Int J Pediatr Hematol Oncol. (1998) 5:443–7. [Google Scholar]
- 21.Russo-Mancuso G, La Spina M, Schiliro G. The changing profile of sickle cell disease in Italy. Eur J Epidemiol. (2003) 18:923–4. 10.1023/A:1025635118305 [DOI] [PubMed] [Google Scholar]
- 22.Rigano P, De Franceschi L, Sainati L, Piga A, Piel FB, Cappellini MD, et al. Real-life experience with hydroxyurea in sickle cell disease: a multicenter study in a cohort of patients with heterogeneous descent. Blood Cells Mol Dis. (2018) 69:82–9. 10.1016/j.bcmd.2017.08.017 [DOI] [PubMed] [Google Scholar]
- 23.De Franceschi L, Lux C, Piel FB, Gianesin B, Bonetti F, Casale M, et al. Access to emergency departments for acute events and identification of sickle cell disease in refugees. Blood. (2019) 133:2100–3. 10.1182/blood-2018-09-876508 [DOI] [PubMed] [Google Scholar]
- 24.Alli NA, Patel M, Alli HD, Bassa F, Coetzee MJ, Davidson A, et al. Recommendations for the management of sickle cell disease in South Africa. S Afr Med J. (2014) 104:743–51. 10.7196/SAMJ.8470 [DOI] [PubMed] [Google Scholar]
- 25.Società Italiana Talassemie ed Emoglobinopatie,. Raccomandazioni per la gestione del paziente adulto affetto da anemia falciforme [Italian] . In: De Franceschi L, Graziadei G, Rigano P, Cianciulli P, Forni GL, editors. Bologna: SITE: (2014). Available online at: www.SITE.italia.org (accessed June 16, 2021). [Google Scholar]
- 26.Yawn BP, Buchanan GR, Afenyi-Annan AN, Ballas SK, Hassell KL, James AH, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. (2014) 312:1033–48. 10.1001/jama.2014.10517 [DOI] [PubMed] [Google Scholar]
- 27.Qureshi A, Kaya B, Pancham S, Keenan R, Anderson J, Akanni M, et al. Guidelines for the use of hydroxycarbamide in children and adults with sickle cell disease: a British Society for Haematology Guideline. Br J Haematol. (2018) 181:460–75. 10.1111/bjh.15235 [DOI] [PubMed] [Google Scholar]
- 28.Sins JW, Biemond BJ, Van Den Bersselaar SM, Heijboer H, Rijneveld AW, Cnossen MH, et al. Early occurrence of red blood cell alloimmunization in patients with sickle cell disease. Am J Hematol. (2016) 91:763–9. 10.1002/ajh.24397 [DOI] [PubMed] [Google Scholar]
- 29.Giambona A, Damiani G, Vinciguerra M, Jakil C, Cannata M, Cassara F, et al. Incidence of haemoglobinopathies in Sicily: the impact of screening and prenatal diagnosis. Int J Clin Pract. (2015) 69:1129–38. 10.1111/ijcp.12628 [DOI] [PubMed] [Google Scholar]
- 30.Società Italiana Talassemie ed Emoglobinopatie,. Raccomandazioni per lo screening neonatale nelle sindromi falciformi [Italian] . In: De Franceschi L, Russo G, Sainati L, editors. Bologna: SITE: (2017). Available online at: www.SITE.italia.org (accessed June 16, 2021). [Google Scholar]
- 31.Piel FB, Tatem AJ, Huang Z, Gupta S, Williams TN, Weatherall DJ. Global migration and the changing distribution of sickle haemoglobin: a quantitative study of temporal trends between 1960 and 2000. Lancet Glob Health. (2014) 2:e80–89. 10.1016/S2214-109X(13)70150-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shet AS, Thein SL. A growing population of older adults with sickle cell disease. Clin Geriatr Med. (2019) 35:349–67. 10.1016/j.cger.2019.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tumburu L, Ghosh-Choudhary S, Seifuddin FT, Barbu EA, Yang S, Ahmad MM, et al. Circulating mitochondrial DNA is a proinflammatory DAMP in sickle cell disease. Blood. (2021) 137:3116–26. 10.1182/blood.2020009063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ware RE, De Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet. (2017) 390:311–23. 10.1016/S0140-6736(17)30193-9 [DOI] [PubMed] [Google Scholar]
- 35.Rees DC, Robinson S, Howard J. How I manage red cell transfusions in patients with sickle cell disease. Br J Haematol. (2018) 180:607–17. 10.1111/bjh.15115 [DOI] [PubMed] [Google Scholar]
- 36.Swerdlow PS. Red cell exchange in sickle cell disease. Hematology Am Soc Hematol Educ Program. (2006) 48–53. 10.1182/asheducation-2006.1.48 [DOI] [PubMed] [Google Scholar]
- 37.Biller E, Zhao Y, Berg M, Boggio L, Capocelli KE, Fang DC, et al. Red blood cell exchange in patients with sickle cell disease-indications and management: a review and consensus report by the therapeutic apheresis subsection of the AABB. Transfusion. (2018) 58:1965–72. 10.1111/trf.14806 [DOI] [PubMed] [Google Scholar]
- 38.Brugnara C, De Franceschi L. Effect of cell age and phenylhydrazine on the cation transport properties of rabbit erythrocytes. J Cell Physiol. (1993) 154:271–80. 10.1002/jcp.1041540209 [DOI] [PubMed] [Google Scholar]
- 39.De Franceschi L, Corrocher R. Established and experimental treatments for sickle cell disease. Haematologica. (2004) 89:348–56. [PubMed] [Google Scholar]
- 40.Matte A, Recchiuti A, Federti E, Koehl B, Mintz T, El Nemer W, et al. Resolution of sickle cell disease-associated inflammation and tissue damage with 17R-resolvin D1. Blood. (2019) 133:252–65. 10.1182/blood-2018-07-865378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matte A, Zorzi F, Mazzi F, Federti E, Olivieri O, De Franceschi L. New therapeutic options for the treatment of sickle cell disease. Mediterr J Hematol Infect Dis. (2019) 11:e2019002. 10.4084/mjhid.2019.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matte A, Cappellini MD, Iolascon A, Enrica F, De Franceschi L. Emerging drugs in randomized controlled trials for sickle cell disease: are we on the brink of a new era in research and treatment? Expert Opin Investig Drugs. (2020) 29:23–31. 10.1080/13543784.2020.1703947 [DOI] [PubMed] [Google Scholar]
- 43.Park SY, Matte A, Jung Y, Ryu J, Anand WB, Han EY, et al. Pathologic angiogenesis in the bone marrow of humanized sickle cell mice is reversed by blood transfusion. Blood. (2020) 135:2071–84. 10.1182/blood.2019002227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valenti MT, Mattè A, Federti E, Puder M, Anez-Bustillos L, Deiana M, et al. Dietary ω-3 fatty acid supplementation improves murine sickle cell bone disease and reprograms adipogenesis. Antioxidants. (2021) 10:799. 10.3390/antiox10050799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hebbel RP, Vercellotti G, Nath KA. A systems biology consideration of the vasculopathy of sickle cell anemia: the need for multi-modality chemo-prophylaxsis. Cardiovasc Hematol Disord Drug Targets. (2009) 9:271–92. 10.2174/1871529X10909040271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Franceschi L, Cappellini MD, Olivieri O. Thrombosis and sickle cell disease. Semin Thromb Hemost. (2011) 37:226–36. 10.1055/s-0031-1273087 [DOI] [PubMed] [Google Scholar]
- 47.Sparkenbaugh E, Pawlinski R. Interplay between coagulation and vascular inflammation in sickle cell disease. Br J Haematol. (2013) 162:3–14. 10.1111/bjh.12336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Telen MJ. Beyond hydroxyurea: new and old drugs in the pipeline for sickle cell disease. Blood. (2016) 127:810–9. 10.1182/blood-2015-09-618553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Telen MJ, Malik P, Vercellotti GM. Therapeutic strategies for sickle cell disease: towards a multi-agent approach. Nat Rev Drug Discov. (2019) 18:139–58. 10.1038/s41573-018-0003-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Delgado J, Voltz C, Stain M, Lapvetelainen T, Urach S, Lahteenvuo J, et al. The European Medicines Agency review of crizanlizumab for the prevention of recurrent vaso-occlusive crises in patients with sickle cell disease. Hemasphere. (2021) 5:e604. 10.1097/HS9.0000000000000604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karki NR, Kutlar A. P-selectin blockade in the treatment of painful vaso-occlusive crises in sickle cell disease: a spotlight on crizanlizumab. J Pain Res. (2021) 14:849–56. 10.2147/JPR.S278285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weaver SB, Rungkitwattanakul D, Singh D. Contemporary management and prevention of vaso-occlusive crises (VOCs) in adults with sickle cell disease. J Pharm Pract. (2021) 8971900211026644. 10.1177/08971900211026644. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 53.Tormey CA, Hendrickson JE. Transfusion-related red blood cell alloantibodies: induction and consequences. Blood. (2019) 133:1821–30. 10.1182/blood-2018-08-833962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hendrickson JE. Red blood cell alloimmunization and sickle cell disease: a narrative review on antibody induction. Ann Blood. (2020) 5:33. 10.21037/aob-2020-scd-01 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chou ST, Jackson T, Vege S, Smith-Whitley K, Friedman DF, Westhoff CM. High prevalence of red blood cell alloimmunization in sickle cell disease despite transfusion from Rh-matched minority donors. Blood. (2013) 122:1062–71. 10.1182/blood-2013-03-490623 [DOI] [PubMed] [Google Scholar]
- 56.Karafin MS, Westlake M, Hauser RG, Tormey CA, Norris PJ, Roubinian NH, et al. Risk factors for red blood cell alloimmunization in the Recipient Epidemiology and Donor Evaluation Study (REDS-III) database. Br J Haematol. (2018) 181:672–81. 10.1111/bjh.15182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yazdanbakhsh K, Ware RE, Noizat-Pirenne F. Red blood cell alloimmunization in sickle cell disease: pathophysiology, risk factors, and transfusion management. Blood. (2012) 120:528–37. 10.1182/blood-2011-11-327361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fasano RM, Booth GS, Miles M, Du L, Koyama T, Meier ER, et al. Red blood cell alloimmunization is influenced by recipient inflammatory state at time of transfusion in patients with sickle cell disease. Br J Haematol. (2015) 168:291–300. 10.1111/bjh.13123 [DOI] [PubMed] [Google Scholar]
- 59.Nickel RS, Horan JT, Fasano RM, Meyer E, Josephson CD, Winkler AM, et al. Immunophenotypic parameters and RBC alloimmunization in children with sickle cell disease on chronic transfusion. Am J Hematol. (2015) 90:1135–41. 10.1002/ajh.24188 [DOI] [PubMed] [Google Scholar]
- 60.Lin Y, Saskin A, Wells RA, Lenis M, Mamedov A, Callum J, et al. Prophylactic RhCE and Kell antigen matching: impact on alloimmunization in transfusion-dependent patients with myelodysplastic syndromes. Vox Sang. (2017) 112:79–86. 10.1111/vox.12455 [DOI] [PubMed] [Google Scholar]
- 61.Hendrickson JE, Fasano RM. Management of hemolytic transfusion reactions. Hematol Am Soc Hematol Educ Program. (2021) 704–09. 10.1182/hematology.2021000308 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Flow-chart of study population. Pts, patients; SCD, sickle cell disease.
Distribution of the indications for transfusion therapy regimen for children and adults, (A) Indications for acute transfusion regimen (ATR). (B) Indications for chronic transfusion regimen (CTR). Data are shown as counts of the indications for ATR and CTR. ACS, acute chest syndrome; ATR, acute transfusion; CTR, chronic transfusion; HC, hydroxycarbamide; MOFs, multi-organ failures; VOCs, vaso-occlusive events.
(A) Distribution of new antibodies developed in after the arrival at the center for sickle cell disease (SCD) patients with pre-existing (up) and without (low) pre-existing antibodies. (B) Number of SCD children/adults patients with alloimmunization by ethnicity. (C) Number of SCD children/adults patients with alloimmunization presenting single, multiple or unidentified antibody/ies. Data are shown as pie and column charts with the number of patients with alloimmunization.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.