

ABSTRACT
Activation of the stimulator of interferon gene (STING)-mediated innate immune response has been suggested as a promising therapeutic strategy for cancers. However, the effects of STING agonist on natural killer (NK) cell-mediated anti-tumor responses in pancreatic cancer remains unknown. Herein, we evaluated the effects of a classical STING agonist cyclic GMP-AMP (cGAMP) on NK cells in pancreatic cancer. We found that cGAMP could directly activate NK cells and enhance the sensitivity of pancreatic cancer cells to NK cell cytotoxicity, suggesting that cGAMP may become a potential adjuvant for NK cell therapy. In addition, combination of CAR-NK-92 cells targeting mesothelin and cGAMP displayed greater antitumor efficacy by inhibiting tumor growth and prolonging survival of the mouse model of pancreatic cancer. These results suggest that the combination of a STING agonist and NK cells may become a novel immunotherapy strategy for pancreatic cancer.
KEYWORDS: STING, STING agonist, cGAMP, pancreatic cancer, CAR-NK-92 cells
Introduction
Pancreatic cancer is characterized by a poor prognosis and the highest mortality, since the majority of patients presented advanced disease and developed metastases in the early stage.1 Current therapeutic strategies for pancreatic cancer are still limited to traditional methods such as surgical resection, radiation therapy, and chemotherapy, and the 5-year survival rate of patients with pancreatic cancer is less than 7%.2,3 Many clinical trials investigating new drugs or combinations of approved drugs have not shown benefits. Therefore, more effective therapeutic strategies for pancreatic cancer are urgently needed.
The stimulator of interferon genes (STING) has been suggested to play a critical role in the activation of the innate immune system, sensing of tumor cells, and initiating the immune responses of the immune responses of DC-CD8+ T cells.4,5 The DNA receptor cGAS recognizes exogeneous DNA viruses, bacteria, and DNA from damaged tumor cells and catalyzes the synthesis of endogenous cyclic GMP-AMP (cGAMP).6 As a second messenger, cGAMP can bind to the STING protein anchoring on the endoplasmic reticulum, which may subsequently recruit and activate TANK-binding kinase 1 (TBK1), NF-κB, and interferon regulatory factor 3 (IRF3), and further induce the production of type I interferon (IFN) and inflammatory cytokines.7,8 Owing to their important role in the tumor sensing process, STING agonists have emerged as promising immunotherapy agents to increase immunogenicity and inhibit tumor growth.9–11 STING agonists have been shown to have significant therapeutic effects on some tumors, such as B16 melanoma, 4T1 breast cancer, and CT26 colon cancer.12,13 However, most of these studies focused on the antitumor mechanisms induced by STING agonists involved in DC-CD8+ T cell-dependent antitumor efficacy, little attention has been paid to the effect of activation of the STING pathway on NK cells. A growing body of evidence indicates that STING may also participate in NK cell-mediated antitumor responses.14,15 Furthermore, tumor-derived cGAMP may trigger the STING-mediated interferon response in non-tumor cells, further activating NK cells and mediating the elimination of CD8+ T cell resistant tumors.15 However, it remains unknown whether STING agonists have direct effects on NK cells and are potentially used for tumor immunotherapy.
In this study, we conducted a series of experiments to examine whether STING agonist stimulation could directly modulate NK cell function and investigate the antitumor efficacy of STING agonist in combination with CAR-NK-92 cells targeting mesothelin in a preclinical mouse model of pancreatic cancer. We found that cGAMP could directly activate NK cells and enhance the sensitivity of pancreatic cancer cells to the cytotoxicity of NK cells. Furthermore, CAR-NK-92 cells targeting mesothelin combined with cGAMP showed greater antitumor efficacy. Our data suggested that the combination of STING agonist and NK cells may become a novel immunotherapy strategy for pancreatic cancer.
Materials and methods
Cell culture
The NK-92 and pancreatic cancer cell lines AsPC-1 and Capan-2 were obtained from the American Type Culture Collection (ATCC). NK-92 cells were cultured in α-MEM medium (GIBICO, USA) containing 0.1 mM β-mercaptoethanol (Thermo Fisher, USA), 12.5% horse serum (GIBICO, USA), 100 U/mL rhIL-2 (Changsheng, China), 12.5% fetal bovine serum (Changsheng, China), and 100 μg/mL of penicillin/streptomycin. AsPC-1 and Capan-2 cells were maintained in RPMI-1640 medium (GIBICO, USA) containing 10% fetal bovine serum and 100 μg/mL of penicillin/streptomycin. All cell lines have been authenticated with short tandem repeats (STR) analysis and maintained in incubators at 37°C under 5% CO2.
Cell proliferation assay
For the cell proliferation assay, AsPC-1 and Capan-2 cells (1 × 104 cells/well) were seeded in 96-well plates and treated with various concentration of STING agonist 2’-3’-cGAMP (Invitrogen, USA) followed by the addition of 20 μL/well MTT solution (10 mg/mL, Sigma, USA), and incubated at 37°C for 4 h. After centrifugation (2000 g, 25 mins), the supernatant (MTT solution) was removed. To solubilize the formazan crystals, 200 μL dimethyl-sulfoxide (Sigma, USA) was added to each well. Finally, absorbance was measured using a multifunctional microplate reader (BioTeK, USA) at a wavelength of 490 and 570 nm.
Apoptosis assay
The Annexin Ⅴ/PI apoptosis kit was used to detect cell apoptosis of AsPC-1 and Capan-2 cells according to the manufacturer’s instructions (LiankeBio, China). In brief, 3 × 105 cells/well were seeded in a 12-well plate and treated with various concentrations of cGAMP or control for 24 h. The cells were then washed with 1× PBS buffer twice and carefully collected. The cells were resuspended in 500 μL binding buffer and stained with 5 μL of Annexin Ⅴ at room temperature for 15 mins. Subsequently, the cells were washed with 1× binding buffer once and then stained with of 2.5 μL propidium iodide at room temperature for 5 mins. Finally, the cells were analyzed by flow cytometry (BD, USA) using FlowJo software (TreeStart, Inc., USA).
Cytotoxicity assay
The LDH assay kit (Biyuntian, China) was used to detect cytotoxicity of NK-92 cells. Briefly, 1 × 104 AsPC-1 or Capan-2 cells/well were seeded in a 96-well plate. NK-92 cells were added in an effector-to-target ratio of 5:1, 2.5:1, and 1.25:1, respectively, followed by the addition of 20 µL (5 mg/mL) of the maximum release agent of LDH and incubated at 37°C for 4 h. For the combination cytotoxicity assay, AsPC-1 or Capan-2 cells were stimulated by cGAMP for 20 h in a 96-well plate, and NK-92 cells or CAR-NK-92 cells (cGAMP-stimulated or not) were added and incubated for 4 h. A control group of cGAMP-treated tumor cells alone was added as a baseline group. The absorbance was measured using a microplate reader (BioTeK, USA) at 490/630 nm. Finally, the percentage of cytotoxicity was calculated using the following formula: Cytotoxicity = (experimental − target spontaneous)/(maximum – spontaneous) × 100%.
Reverse transcriptase-polymerase chain reaction assay
The cells were washed with 1× PBS and collected in the 1.5 mL tubes. Total cellular RNA was extracted using 500 uL of Trizol reagent (Invitrogen, USA). The cDNA was synthesized using the M-MLV Reverse Transcriptase Kit (Invitrogen, USA) and amplified by polymerase chain reaction (PCR) instrument (Bio-Rad, USA). The PCR products were detected by electrophoresis with 2% agarose gel and analyzed using a Gel imaging system. Real-time PCR (RT-PCR) experiments were performed with the LightCycler96 (Roche, Switzerland) using the TransStart SYBR qPCR Kit (TransGen, China). The primers for PCR and RT-PCR were synthesized by the Beijing Genomics Institute (Beijing, China) (Supplementary Table 1).
Western blotting analysis
Cells were washed with pre-cooled 1× PBS and collected in 100 µL lysis buffer containing 1% PMSF (Biyuntian, China) at 4°C for 15 mins. The protein concentration in the cell lysate was measured using the BCA concentration detection kit (Biyuntian, China). Equal amounts of total proteins were resolved by SDS-PAGE electrophoresis and transferred to PVDF membranes. The immunoblot was blocked with 5% nonfat milk in Tris-buffered saline containing 0.1% Tween 20 (TBST), incubated with the primary antibody at 4°C overnight. After washing in TBST three times, blots were incubated with the corresponding HRP-conjugated secondary antibodies at room temperature for 1 h, followed by three washes with TBST. Antibodies for p-IRF3 (4947S), IRF3 (1190S), Bax (2772S) and GAPDH (5174S) were purchased from Cell Signaling Technology and antibodies for Bcl2 (ab32124) were obtained from Abcam company. Protein bands were detected by Immobilon Western Chemiluminescent HRP Substrate (Millipore Corporation, USA) and visualized using the Chemi DocTM Touch Imaging System (Bio-Rad).
Flow cytometry analysis
For surface or intracellular staining analysis, tumor cells or NK-92 cells were treated with 4 μg/mL cGAMP for 12 h or 24 h and collected in the round bottom tube, respectively. Cells were washed with 1× PBS and then incubated with the corresponding antibody. AsPC-1 cells and Capan-2 cells were incubated with an ULBP2/5/6 fusion antibody (CST, USA), while NK-92 cells were incubated with antibodies of CCR5, CD107a, CD69, IFN-γ, TNF-α, granzyme B, and perforin (R&D, USA). NK cells were stimulated by PMA and ionomycin (LiankeBio, China), which were used as a positive control and isotype control, respectively. The stained cells were detected by FACS Calibur system (BD, USA) and analyzed with FlowJo 7.6.1 software (TreeStart, Inc., USA).
Detection of cytokines by ELISA
AsPC-1, Capan-2, and NK-92 cells (2 × 105 per well) were seeded in 24-well plates, respectively, and the culture supernatants were collected 24 h after stimulation with cGAMP. The levels of IFN-β, CCL5, and CXCL10 in the supernatants were detected using ELISA kits (LiankeBio, China) in accordance with the manufacturer’s instructions.
Chemotaxis assay
To measure NK-92 cell migration by chemotaxis assay, 2 × 105 NK-92 cells were seeded in the upper chamber of a 24-well Transwell plate with 100 μL serum free-medium, and 600 μL of conditioned media collected from the supernatant of AsPC-1 cells with or without cGAMP stimulation was placed in the lower chamber of the 24-well Transwell plate (5 μm pore size, Corning, USA). The cells were cultured in the incubator at 37°C under 5% CO2 for 8 h. Furthermore, NK-92 cells were pre-incubated with 1 μM anti-CCL5 antibody (R&D, USA) to block CCL5-mediated cell migration. Finally, the number of NK-92 cells that migrated to the lower chamber was counted and the data was presented as a percentage of migration based on total cell input.
Tumor growth in the mouse model of pancreatic cancer
The animal experiments and protocols involved in this study were approved by the Shandong University Animal Experiment Ethics Committee. For the animal study, 6 to 8-week-old female NOD-SCID mice were obtained from Hua Fukang Biological Technology Co. Ltd. (Beijing, China) and maintained in a specific-pathogen-free (SPF) environment at the Laboratory Animal Center of Shandong University. To establish a mouse model for pancreatic cancer, 5 × 106 AsPC-1 cells per mouse were injected into the mice subcutaneously (sc), and tumor growth was monitored and recorded daily. When the tumor volume reached approximately 100 mm3, all mice were randomly divided into six groups: (1) the untreated group, (2) the cGAMP treatment alone group, (3) NK-92 cells treatment alone group, (4) cGAMP combined with NK-92 cells treatment group, (5) anti-MSLN CAR-NK-92 cell treatment group, and (6) the cGAMP combined with anti-MSLN CAR-NK-92 cell treatment group. The mice in the NK cell treatment group were injected with 100 μL of 5 × 106 NK-92 cells or anti-MSLN CAR-NK-92 cells through the tail vein and 5000 IU of IL-2 intraperitoneally every three days to maintain the survival of NK-92 cells and CAR-NK-92 cells. Mice in the cGAMP treatment group were intratumorally injected with 0.5 mg/kg cGAMP three times a week. The tumor burden was measured with Vernier calipers every three days since the beginning of treatment. Tumor volume was calculated using the following formula: Tumor volume (cm3) = (long diameter) × (short diameter)2 × 0.5.
Statistical analysis
All statistical analyses were performed using SPSS version 23.0 software (SPSS Inc) and GraphPad Prism 8.0 software (GraphPad Software Inc. USA). For two groups, a two-tailed Student’s t test of parametric statistical analysis was performed when data had normal distribution and homogeneity of variance. Otherwise, the Mann-Whitney U test was used. For multiple groups, One-way ANOVA analysis was performed for data meeting homogeneity of variance, whereas Bonferroni post hoc analysis was used when data with skewed distribution and not meeting homogeneity of variance. Multi-way ANOVA was also used in this study. All data were presented as mean ± Standard error of the mean (SEM). A P-value <0.05 was considered statistically significant.
Results
The STING agonist cGAMP activated NK cells directly and promoted the killing activity of NK cells
To explore whether the STING agonist can directly activate NK cells, we first observed spontaneous STING mRNA expression (TMEM173) in NK cells using the HPA database (http://www.proteinatlas.org/) (Supplementary Figure 1A). We also detected the expression of the STING gene in the NK-92 cell line, and found that NK-92 cells expressed the STING gene (Supplementary Figure 1B). NK-92 cells were then stimulated by 4 μg/mL cGAMP, and the levels of IRF3 phosphorylation, which leads to the production of type I interferons16 were determined. We found that IRF3 phosphorylation was induced in a time-dependent manner (Figure 1a). Additionally, stimulation with cGAMP promoted gradual increase in IFN- mRNA expression of IFN-β gradually in a dose-dependent manner (Figure 1b), and the IFN- protein level of IFN-β in the supernatant of NK-92 cells was markedly increased after stimulation with cGAMP (Figure 1c). These findings indicate that the STING agonist cGAMP could stimulate the activation of the STING signaling pathway in NK-92 cells, resulting in an increase in IFN-β production.
Figure 1.
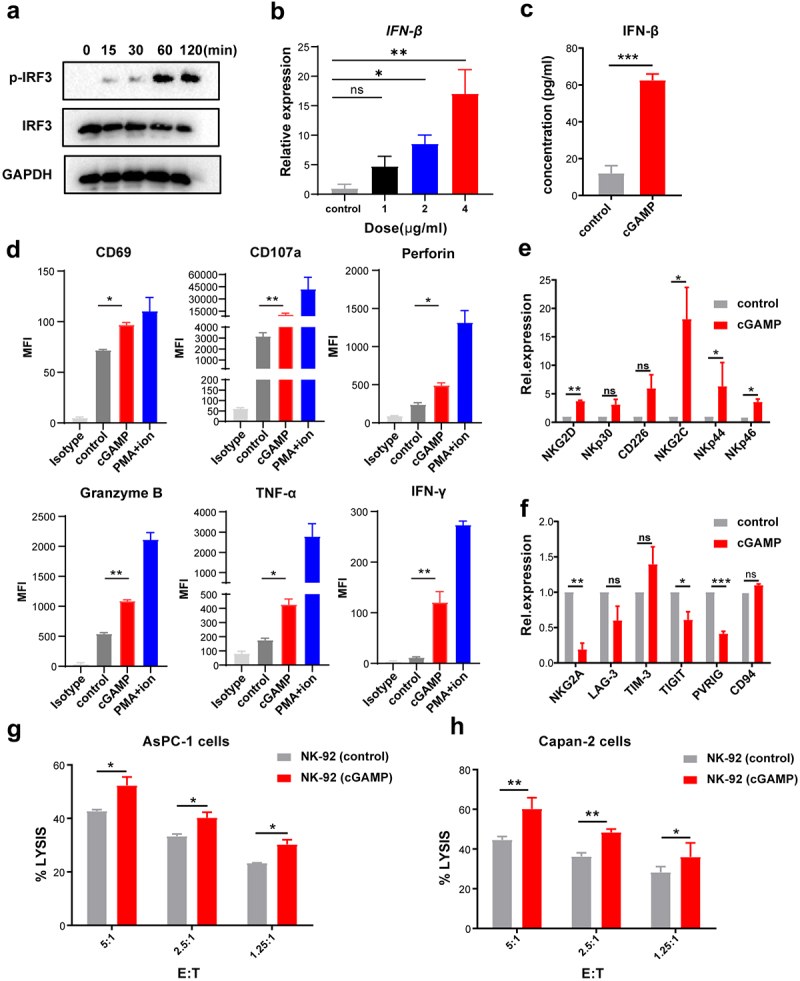
STING agonist cGAMP activated NK cells directly and promotes the killing capability of NK cells. (a) The time-dependent phosphorylation of IRF3 in NK-92 cells is shown after 4 μg/mL cGAMP stimulation. The data shown are representative of three independent experiments. (b) Up-regulation of IFN-β in NK-92 cells was detected by RT-PCR after stimulation with 1, 2, and 4 μg/mL cGAMP, respectively, for 24 h. (c) The levels of IFN-β in the supernatant of NK-92 cells measured by ELISA after stimulation with 4 μg/mL cGAMP. (d) Flow cytometry analysis of CD69, CD107a, perforin, granzyme B, IFN-γ, and TNF-α in NK-92 cells following stimulation with 4 μg/mL cGAMP for 24 h. The isotype group was used as a negative control to exclude background differences; PMA and ionomycin were used as positive controls. MFI, the mean fluorescence intensity. (e) The expression of activating receptors on the surface of NK-92 cells detected by RT-PCR after stimulation with 4 μg/mL cGAMP for 24 h and normalized to GAPDH. (f) The expression of inhibitory receptors on the surface of NK-92 cells detected by RT-PCR after stimulation with 4 μg/mL cGAMP for 24 h and normalized to GAPDH. (g, h) NK-92 cells were stimulated with 4 μg/mL cGAMP for 20 h, and co-cultured with AsPC-1 cells (g) and Capan-2 cells (h) for 4 h, then the cytolytic activity of NK cells was detected using the LDH assay. Data are shown as means ± SEM, and statistical significance was determined as ns: not significant, ***p < .001, **p < .01 and *p < .05.
We then investigated the effect of cGAMP on the activation of NK-92 cells. The results of flow cytometry showed that the expression of activation markers (such as CD69), cytotoxic molecules (such as CD107a, perforin, and granzyme B), and cytokines (such as TNF-α and IFN-γ), were up-regulated in NK-92 cells stimulated with cGAMP compared to the control group (Figure 1d). All these markers are known as indicators of NK cell activation and cytotoxicity.17 Furthermore, we detected activating receptors and inhibitory receptors of NK cells using RT-PCR, and found that inhibitory receptors such as NKG2A, TIGIT, and PVRIG decreased in cGAMP-stimulated NK-92 cells, while activating receptors such as NKG2D, NKG2C, NKp44, and NKp46 increased (Figure 1e and f). Furthermore, we found that cGAMP stimulation could significantly enhance the cytotoxicity of NK-92 cells against AsPC-1 and Capan-2 cells compared to untreated NK-92 cells (Figure 1g and h). Our data suggest that cGAMP may directly activate NK-92 cells and enhance their cytolytic activity against pancreatic cancer cells by stimulating the STING signaling pathway.
The STING agonist cGAMP induced the apoptosis of pancreatic cancer cells
To better understand the role of STING in tumor cells, we detected its expression in pancreatic cancer cell lines AsPC-1 and Capan-2 cells (Supplementary Figure 2A-2C). We initially found that STING could be detected in pancreatic cancer cells (Supplementary Figures 2A,2B), and we verified this finding by HPA analysis of the TCGA data set (http://www.proteinatlas.org/) (Supplementary Figure 2C). Based on these observations, we hypothesized that pancreatic cancer cells were likely to respond to stimulation of GAMP. The phosphorylation of IRF3 was up-regulated in both AsPC-1 (Figure 2a) and Capan-2 cells (Figure 2b) in a time-dependent manner on exposure to cGAMP in the pancreatic cancer cells. Furthermore, IFN-β mRNA and protein levels increased in a dose-dependent manner in AsPC-1 cells (Figure 2c,d) and Capan-2 cells (Figure 2e,f) after cGAMP stimulation. These results indicated that the STING agonist cGAMP may activate the STING signaling pathway in pancreatic cancer cell lines and induce the production of type I IFN.
Figure 2.
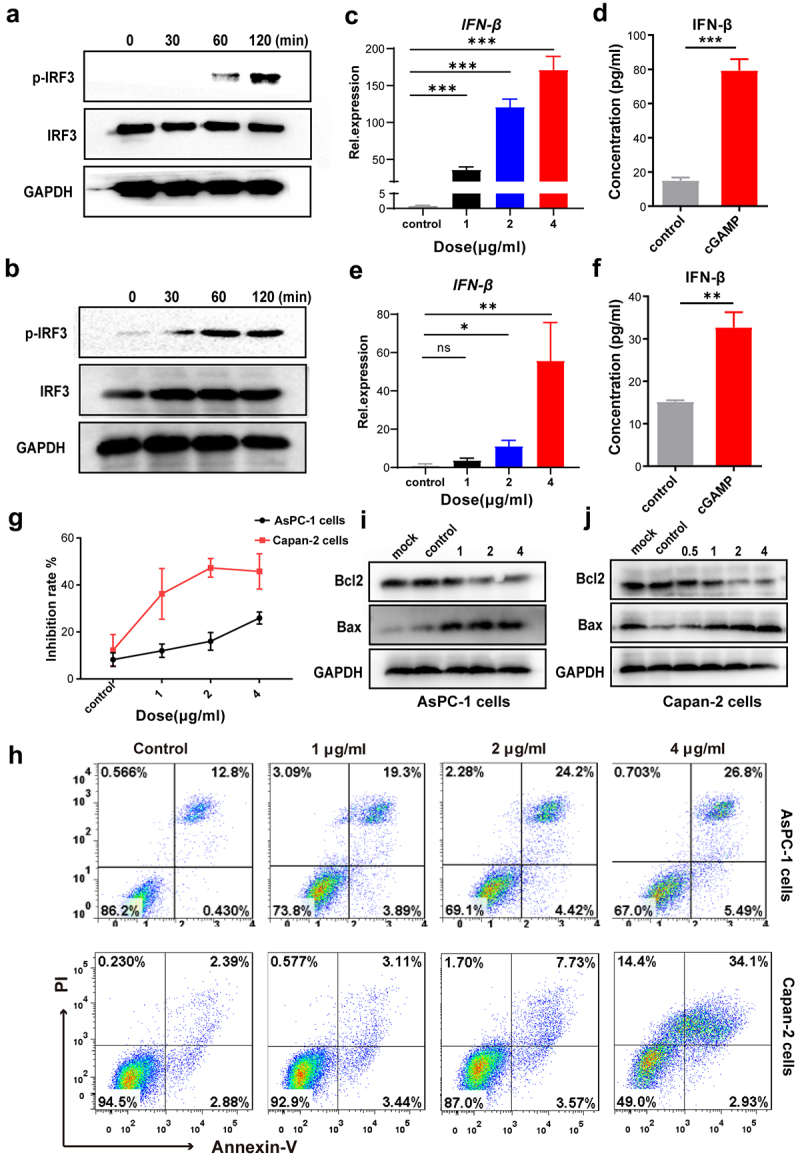
STING agonist cGAMP induced the apoptosis of pancreatic cancer cells. (a) AsPC-1 cells were stimulated with 4 μg/mL cGAMP for the indicated time points, and the expression of p-IRF3, total IRF3 and GAPDH was measured by western blotting. (b) Capan-2 cells were stimulated with 4 μg/mL cGAMP for the indicated time points, and the expression of p-IRF3, total IRF3 and GAPDH was measured by western blotting. The data shown are representative of three independent experiments. (c) AsPC-1 cells were stimulated with cGAMP at indicated dosage for 24 h, and mRNA expression of IFN-β was detected by RT-PCR. (d) The protein expression of IFN-β in the supernatant of AsPC-1 cells was measured by ELISA. (e) Capan-2 cells were stimulated with cGAMP at the indicated dosage for 24 h, and mRNA expression of IFN-β was detected by RT-PCR. (f) The protein expression of IFN-β in the supernatant of Capan-2 cells was measured by ELISA. (g) AsPC-1 and Capan-2 cells were stimulated with cGAMP at indicated dosages for 24 h, cell viability was detected by the MTT assay. (h) Apoptosis of AsPC-1 and Capan-2 cells were analyzed by flow cytometry and Annexin V and propidium iodide double staining assays. (i) AsPC-1 cells were stimulated with cGAMP for 24 h at the indicated dose, and the amount of Bcl2, Bax and GAPDH was measured by western blotting. (j) Capan-2 cells were stimulated with cGAMP for 24 h at the indicated dose, and the level of Bcl2, Bax, and GAPDH was evaluated by western blotting, the data shown are representative of three independent experiments. Data are shown as means ± SEM, statistical significance was determined as ns: not significant, ***p < .001, **p < .01 and *p < .05.
Previous studies have suggested that activation of the STING signaling pathway may promote tumor cell apoptosis,18,19 so we examined the effect of cGAMP on the proliferation and apoptosis of pancreatic cancer cells. Cell viability of AsPC-1 and Capan-2 cells decreased in a dose-dependent manner after cGAMP stimulation (Figure 2g). We then detected AsPC-1 and Capan-2 cell apoptosis using the Annexin Ⅴ-PI assay and found that the proportion of AsPC-1 and Capan-2 cells with early and late apoptosis gradually increased as the concentration of cGAMP increased (Figure 2h). To delineate the mechanism of apoptosis, we found that the expression of Bcl2 was down-regulated while Bax was up-regulated in AsPC-1 cells (Figure 2i) and Capan-2 cells (Figure 2j), indicating that cGAMP may induce apoptosis by an intrinsic apoptosis pathway. In accordance with our results, other studies have reported that the STING signaling pathway induces apoptosis of T cells and tumor cells.20,21 In addition, it has been reported that IFN-β can inhibit proliferation and induce the apoptosis of pancreatic cancer cells,22,23 thus we considered that IFN-β induced by cGAMP might play a critical role in this process.
The STING agonist cGAMP enhanced the sensitivity of pancreatic cancer cells to NK cell cytotoxicity
To evaluate whether cGAMP could enhance the sensitivity of pancreatic cancer cells to NK cell cytotoxicity, AsPC-1 and Capan-2 cells were stimulated by cGAMP and then co-cultured with NK-92 cells. The results revealed that the sensitivity of cancer cells was significantly enhanced to NK-92 cell cytotoxicity (Figure 3a,b).
Figure 3.
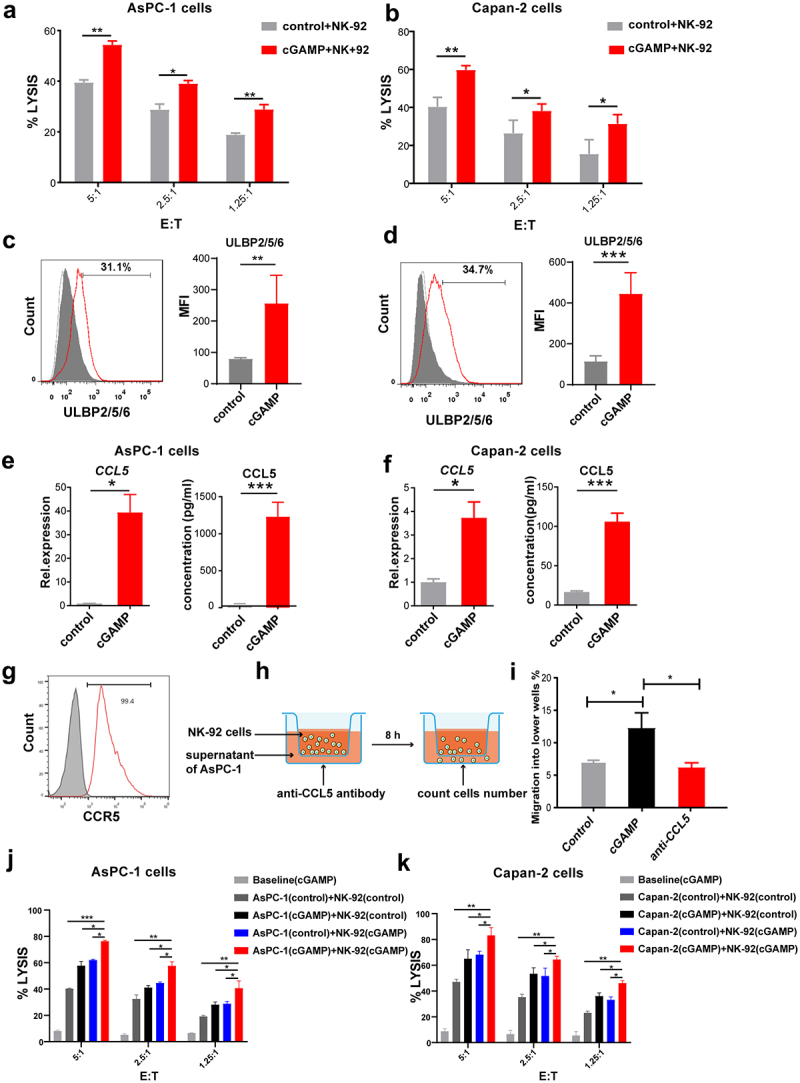
STING agonist cGAMP enhanced NK cell cytotoxicity to pancreatic cancer cells. (a) AsPC-1 cells were stimulated with 4 μg/mL cGAMP for 20 h and co-cultured with NK-92 cells for 4 h. Cell lysis was detected by the LDH assay. (b) Capan-2 cells were stimulated with 4 μg/mL cGAMP for 20 h and co-cultured with NK-92 cells. Cell lysis was detected by the LDH assay. (c) Flow cytometry analysis of the fusion protein expression of ULBP2/5/6 in AsPC-1 cells after stimulation with 4 μg/mL cGAMP. (d) Flow cytometry analysis of the fusion protein expression of ULBP2/5/6 in Capan-2 cells after stimulation with 4 μg/mL cGAMP. (e) The mRNA and protein levels of CCL5 were determined respectively using RT-PCR and ELISA assays in AsPC-1 cells after stimulation with 4 μg/mL cGAMP. (f) The mRNA and protein levels of CCL5 were determined respectively using RT-PCR and ELISA assays in Capan-2 cells after stimulation with 4 μg/mL cGAMP. (g) Flow cytometry analysis of CCR5 expression in NK-92 cells. (h) A schematic diagram of the Transwell migration assay of NK-92 cells. (i) Migration of NK-92 cells into the lower chamber of the Transwell containing the conditioned media from the AsPC-1 cell cultures with or without the stimulation with 4 μg/mL cGAMP, or anti-CCL5 antibody. (j) Both AsPC-1 cells and NK-92 cells were pre-stimulated with 4 μg/mL cGAMP for 20 h and then co-cultured for 4 h. Cell lysis was detected by LDH assay. (k) Both Capan-2 cells and NK-92 cells were pre-stimulated with 4 μg/mL cGAMP for 20 h and then co-cultured for 4 h. Cell lysis was detected by LDH assay. A control group of the cGAMP-treated tumor cells alone (no NK cells) were used as a baseline group. Data are shown as means ± SEM, and statistical significance was determined as ns: not significant, ***p < .001, **p < .01 and *p < .05.
NKG2D is one of the main activating receptors for NK cells and interaction of NKG2D with its ligands can promote NK cell activation and contribute to NK cell-mediated cytolytic activity and tumor immune surveillance.24 Herein, we determined the expression of NKG2D ligands on the surface of the tumor cells, which can bind to the membrane NKG2D receptor on NK cells, thereby enhancing the killing ability of NK cells against tumor cells. NKG2D ligands include activating ligands MHC-I chain-related molecules A and B (MICA/B) and unique long 16 (UL16)-binding proteins.25 The results showed that the protein expression of ULBP-2/5/6 was up-regulated in AsPC-1 cells (Figure 3c) and Capan-2 cells (Figure 3d) after stimulation. Furthermore, the expressions of CCL5 (Figure 3e,f) and CXCL10 mRNA and protein (Supplementary Figure 3A,3B) increased significantly in AsPC-1 and Capan-2 cells treated with cGAMP. It should be noted that CCL5 and CXCL10 were involved in NK cell activation and infiltration into tumor tissues.26 Furthermore, CCR5 (the ligand of CCL5) was detected in NK-92 cells (Figure 3g), indicating that cGAMP may play a crucial role in recruiting NK cells. To verify this notion, we performed a microscopy-based chemotaxis assay (Figure 3h). Our results showed that 15% of NK-92 cells migrated from the top chamber of the Transwell to the lower chamber within 8 h of coculture with supernatants from cGAMP-stimulated AsPC-1 cells, while less than 7% of NK-92 cells migrated to the lower chamber in the presence of the control supernatant (Figure 3i). Finally, the migration of NK-92 cells was reduced to background levels in the presence of anti-CCL5 antibody (Figure 3i). These findings suggest that the STING agonist cGAMP may promote the recruitment of NK cells to tumor cells and enhance the sensitivity of pancreatic cancer cells to NK cytotoxicity. Finally, to assess whether there is an additional effect when both pancreatic cancer cells and NK-92 cells were stimulated with cGAMP, we pre-treated AsPC-1 cells and NK-92 cells with cGAMP for 20 h separately, and then detected the cytotoxicity of NK-92 cells (Figure 3j,k). We found an significantly increased cytolytic capacity of NK-92 cells when both tumor cells and NK-92 cells were pre-stimulated with cGAMP than alone stimulated group. Taken together, all these results indicate that cGAMP not only directly increase the anti-tumor activity of NK cells but also enhance the sensitivity of pancreatic cancer cells to NK cell cytotoxicity.
The STING agonist cGAMP enhanced the sensitivity of pancreatic cancer cells to CAR-NK-92 cell cytotoxicity
The results described above showed that the STING signaling pathway was activated in both pancreatic cancer cells and NK cells, and the cytotoxicity of NK cells against pancreatic cancer cells was enhanced, suggesting that STING agonists may be used as adjuvants for NK cell therapy. Chimeric antigen receptor (CAR)-modified NK cell–based immunotherapy is revolutionizing the field of cancer treatment.27,28 Compared to NK cells, CAR-NK cells can specifically target tumors and have stronger antitumor capabilities.29–31 Mesothelin (MSLN) has been reported to be highly expressed on the surface of some solid tumor cells, including pancreatic cancer cells, but little or no expression in normal cells, thus becoming a promising target in cancer immunotherapy.32–35 We first verified the positive expression of MSLN in AsPC-1 cells (Figure 4a) and Capan-2 cells (Figure 4b) by flow cytometry and further confirmed its expression in AsPC-1 cells and pancreatic cancer tissues by immunohistochemistry (Figure 4c). These results were consistent with other studies,33,36 which suggested MSLN was a potential target of CAR for pancreatic cancer. Herein, we developed a second-generation MSLN-targeting CAR-modified NK-92 cells (anti-MSLN CAR-NK-92) with the 4–1 BB co-stimulatory module by the piggyBac transposon system (Figure 4d). Approximately 87.5% of anti-MSLN CAR-NK-92 cells expressed MSLN after-transfection (Figure 4e), and these cells exerted increased cytotoxicity against AsPC-1 cells (Figure 4f) and Capan-2 cells (Figure 4g) compared to NK-92 cells. These results suggested that we successfully established that anti-MSLN CAR-NK-92 cells displayed more efficient killing activity against pancreatic cancer cells.
Figure 4.
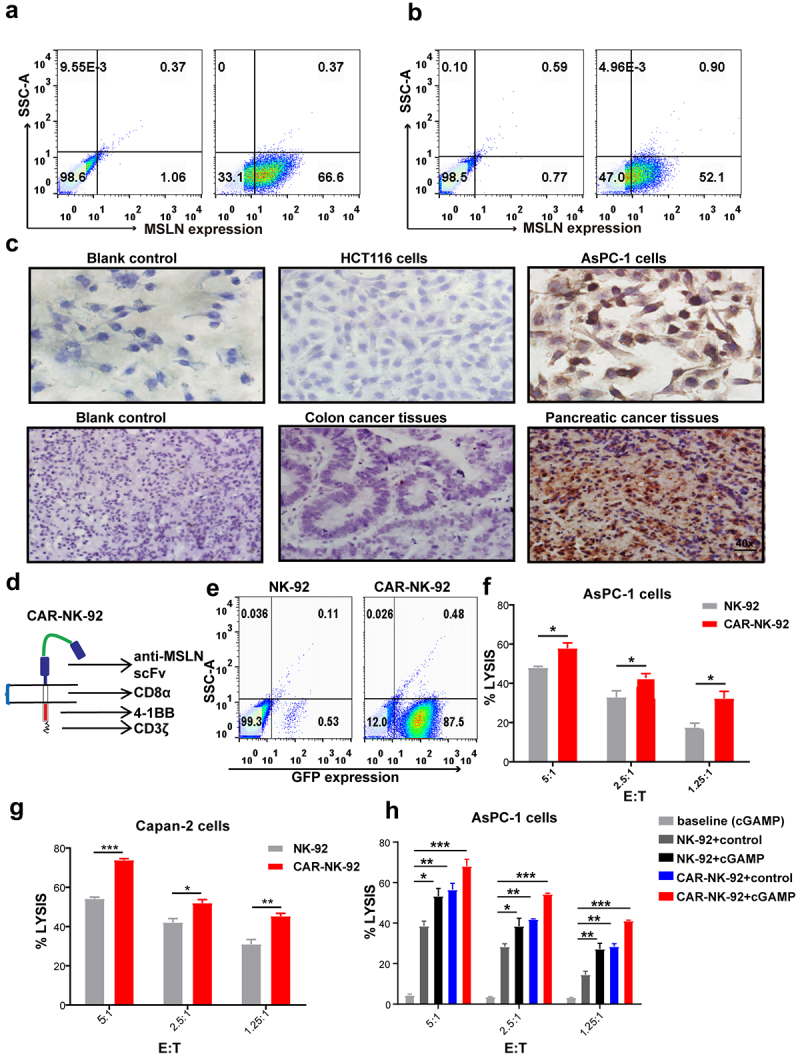
STING agonist cGAMP enhanced the sensitivity of pancreatic cancer cells to anti-MSLN CAR-NK-92 cells. (a) Flow cytometry analysis of MSLN expression in AsPC-1 cells. (b) Flow cytometry analysis of MSLN expression in Capan-2 cells. (c) The positive expression of MSLN was detected in AsPC-1 cells and pancreatic cancer tissues but not in the colon cancer cell line HCT116 or colon cancer tissues by immunohistochemical staining. 40x: Scale bar = 50 μm. The data shown are representative of three independent experiments. (d) The structure of MSLN CAR. (e) The expression of green fluorescence protein (GFP) in CAR-NK-92 cells. The data shown are representative of three independent experiments. (f) The cytolytic activity of anti-MSLN CAR-NK-92 cells against AsPC-1 cells determined by LDH assay. (g) The cytolytic activity of anti-MSLN CAR-NK-92 cells against Capan-2 cells determined by the LDH assay. (h) AsPC-1 cells were stimulated with 4 μg/mL cGAMP for 20 h, and co-cultured with NK-92 cells or anti-MSLN CAR-NK-92 cells for 4 h. cGAMP-treated tumor cells alone were added as a baseline group. The cytolytic activity was detected by the LDH assay. Data are shown as means ± SEM, and statistical significance was determined as ns: not significant, ***p < .001, **p < .01 and *p < .05.
To examine whether the STING agonist could enhance pancreatic cancer cell sensitivity to anti-MSLN CAR-NK-92 cell cytotoxicity in vitro, AsPC-1 cells were treated with 4 μg/mL cGAMP and co-cultured with anti-MSLN CAR-NK-92 cells. A control group of the cGAMP-treated tumor cells without NK cells were used as the baseline group. We found that cGAMP stimulation significantly improved AsPC-1 cell sensitivity to anti-MSLN CAR-NK-92 cell cytotoxicity (Figure 4h). These results also indicate that the STING agonist cGAMP could be used as an adjuvant for anti-MSLN CAR-NK-92 cell therapy.
The STING agonist cGAMP enhanced the therapeutic effects of CAR-NK-92 cells on pancreatic cancer cells in vivo
To further evaluate the anti-tumor effects of cGAMP and CAR-NK-92 cells in vivo, we established the pancreatic cancer mouse model bearing the AsPC-1 cells (Figure 5a). When the tumor volume reached approximately 100 mm3, the mice were randomly divided into six groups: (1) the control ttgroup, (2) the cGAMP treatment group, (3) the NK-92 cell treatment group, (4) the NK-92 cells combined with cGAMP treatment group, (5) the anti-MSLN CAR-NK-92 cell treatment group, and (6) the anti-MSLN CAR-NK-92 cells combined with cGAMP treatment group. Tumor burden was measured with vernier calipers every three days after treatment. Our results showed that compared to the control group, treatment with NK-92 cells, cGAMP, or anti-MSLN CAR-NK-92 cells alone inhibited xenograft growth (Figure 5b,c). Importantly, combination treatment of cGAMP and CAR-NK-92 cells resulted in a superior inhibitory effect on tumor growth (Figure 5b,c) and significantly prolonged the survival of mice bearing AsPC-1 cells (Figure 5d), suggesting that STING agonist cGAMP may enhance the anti-tumor effect of anti-MSLN CAR-NK-92 cells against pancreatic cancer cells.
Figure 5.
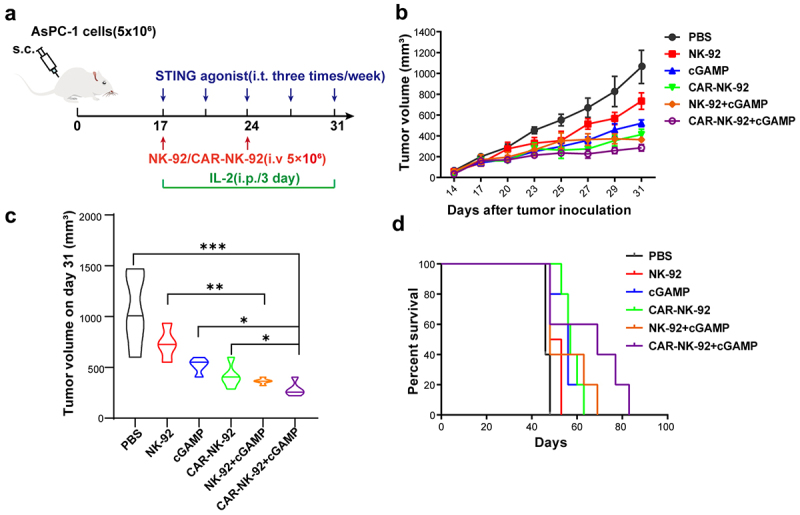
STING agonist cGAMP enhanced the therapeutic effect of anti-MSLN CAR-NK-92 cells on pancreatic cancer. (a) Diagram depicting the treatment schedule of STING agonist in combination with anti-MSLN CAR-NK cells in subcutaneous (s.c.) pancreatic tumors of NOD-SCID mice, blue arrows indicate cGAMP treatment, red arrow indicates NK-92 cells or CAR-NK-92 cells treatment, and green arrow indicates IL-2 treatment. (b) Tumor volume was measured with vernier calipers every two or three days. (c) The Violin diagram showing tumors volume measured on day 31. (d) Survival analyses of tumor-bearing mice treated with the indicated control and experimental groups, and were analyzed using Log-rank (Mantel-Cox) test. The statistical significance was determined as ***p < .001, **p < .01 and *p < .05.
Discussion
This study was carried out to investigate the activity of the STING agonist cGAMP in modulating NK cell activities and to evaluate the combination treatment of cGAMP and the anti-MSLN CAR-NK-92 cells for pancreatic cancer. To our knowledge, this is the first study to report the significant effect of cGAMP in combination with anti-MSLN CAR-NK-92 cells on the treatment of pancreatic cancer.
We observed the anti-tumor activity of cGAMP against pancreatic cancer in the AsPC-1-bearing mouse model. Since the STING agonist has been reported to induce anti-tumor responses in mice that were deficient in T cell functions,15 this suggested that cGAMP may exert antitumor effects by improving STING-mediated innate immune responses and/or directly inducing apoptosis of tumor cells. Unexpectedly, we found that cGAMP stimulation may increase the sensitivity of pancreatic cancer cells to NK cells and enhance the cytotoxicity of NK cells.
We found that STING was detected in both NK-92 cells and pancreatic cancer cell lines, which was consistent with a previous report that STING in host immune cells and tumor cells could work cooperatively in immune responses.37 IRF3 phosphorylation has been used as a sign of STING activation,38 which results in the production of type I IFNs.8,39 In our study, we found that IRF3 phosphorylation increased in both NK-92 cells and pancreatic cancer cells with cGAMP stimulation. We also observed increased expression of IFN-β in a time and dosage-dependent manner. Although combination treatment with the STING agonist and IL-15 could activate NK cells in prostate cancer,31 the role of STING agonist alone is still uncertain. In this study, our results indicated that the STING agonist cGAMP may activate the STING signaling pathway in both NK-92 cells and pancreatic cancer cells.
NK cells play a critical role in the killing of cancer cells. Although intraperitoneal injection of 2’-3’-cGAMP could activate NK cells,14,15 the direct effect of cGAMP on NK cells remains largely unclear. Here we stimulated NK-92 cells with cGAMP in vitro in the absence of other cells and observed the activation of the STING signaling pathway in NK-92 cells, as exemplified by the phosphorylation of IRF3 and synthesis of IFN-β (Figure 1a–c). NK cell activation was observed, as evidenced by upregulation of CD69, CD107a, perforin, granzyme B, TNF-α, and IFN-γ in cGAMP-stimulated NK-92 cells compared with the control group (Figure 1d). These findings suggested that the STING agonist cGAMP could directly stimulate the STING signaling pathway in NK cells and further promote NK cell activation. Interestingly, Marcus et al. reported that NK cell-intrinsic STING signaling is independent of NK activation in mice models of RMA lymphoma and B16F10 melanoma.15 Monocytes (macrophages and DCs) are potentially responsible for the cGAMP-induced activation of NK cells in vivo, and the role of NK cells is likely to be neglected. In our study, we found that the NK cell-intrinsic STING signaling is equally important for NK activation when the role of other cell populations is ruled out. We proposed that the STING agonist could not only prime NK cell activation by directly inducing NK cell-intrinsic STING signaling, but it may also promote NK cell activation indirectly via the production of cytokines (e.g., IFN-β) by other cells such as monocytes, B cells or tumor cells actively stimulated by the STING agonist.
It has been reported that the STING agonist may induce apoptosis in breast cancer cells and malignant B lymphoma cells.40,41 However, STING agonists were not effective against B16F10 melanoma, SCCFV II upper gastrointestinal squamous cell carcinoma, CT26 colon cancer, or Hepa1-6 hepatoma,19 suggesting that STING-mediated apoptosis could be tumor type and/or cell dependent due to differential expression of STING between different cell types.42 Another interesting finding of this study is that significant apoptosis was observed in pancreatic cancer cells after cGAMP stimulation. To decipher the underlying mechanism, we detected the expression of Bcl2 and Bax, and found that Bcl2 expression was down-regulated, whereas Bax levels were up-regulated (Figure 2i,j), indicating that cGAMP may induce apoptosis by an intrinsic apoptosis pathway. These results are consistent with other reports showing that the STING signaling pathway induces apoptosis of T cells and tumor cells.20,21 In addition, it has been reported that IFN-β can inhibit the proliferation and induce the apoptosis of pancreatic cancer cells.22,23 We proposed that cGAMP induces the apoptosis of pancreatic cancer cells both in an intrinsic apoptosis pathway by inducing the cGAMP-STING signal pathway, and through the production of IFN-β by pancreatic cancer cells upon cGAMP stimulation.
Furthermore, up-regulation of NKG2D ligands and chemokines was detected in pancreatic cancer cells following cGAMP stimulation (Figure 3c,d), which was consistent with a previous report that genomic damage of tumor cells could lead to activation of STING signaling, thereby up-regulating the expression of retinoic acid early transcript 1 (RAE1), a ligand of NKG2D.43 Activation of STING signaling may also trigger the production of chemokines such as CCL5 and CXCL10,44 which may initiate the recruitment of protective NK cells to the tumor microenvironment26 and enhance the sensitivity of pancreatic cancer cells to NK cell cytotoxicity. Given the important role of cGAMP in NK cell activation, it may become a novel adjuvant for NK cell therapy.
Chimeric antigen receptor (CAR), an artificially modified receptor protein, is composed of the extracellular domain of the single chain variable fragment (scFv) of the antibody, the hinge region, the transmembrane region, and an intracellular signal transduction domain.45 Compared to NK cells, CAR-modified NK cells can directly recognize and target specific antigens on the surface of tumor cells and exhibit enhanced cytotoxicity against tumor cells.46,47 In our previous work, we successfully established that CAR-NK-92 cells target MSLN, which were more effective in killing pancreatic cancer cells than NK-92 cells (Figure 5b,c). Because anti-MSLN CAR-NK-92 cells were derived from NK-92 cells, we did not repeat the cGAMP stimulation experiments in anti-MSLN CAR-NK-92 cells.
In our study, combination treatment with cGAMP and CAR-NK-92 cells displayed a robust anti-tumor effect in vitro and in vivo. We determined that NK cells first recognize and kill tumor cells nonspecifically through additional mechanisms besides that involving a CAR-specific mechanism. NK cells can kill tumor cells through direct cell cytotoxicity (release perforin and granzymes), mediated antibody-dependent cell-mediated cytotoxicity (ADCC), or the production of pro-inflammatory cytokines (IFN-γ, TNF-α).28,48 Furthermore, the STING agonist cGAMP promotes the antitumor effector functions of NK cells in two ways (Figure 6). First, cGAMP stimulation may lead to direct activation of NK cells and enhance its killing activity against tumor cells. Second, cGAMP stimulation may lead to indirect activation of NK cells through several mechanisms, including the induction of apoptosis of pancreatic cancer cells, up-regulation of IFN-β, NKG2D ligands, CCL5, and CXCL10 expression by pancreatic cancer cells, and by increasing the sensitivity of cancer cells to NK cell cytotoxicity. Taken together, these results suggest that a such a combination treatment may represent a novel immunotherapy strategy against pancreatic cancer.
Figure 6.
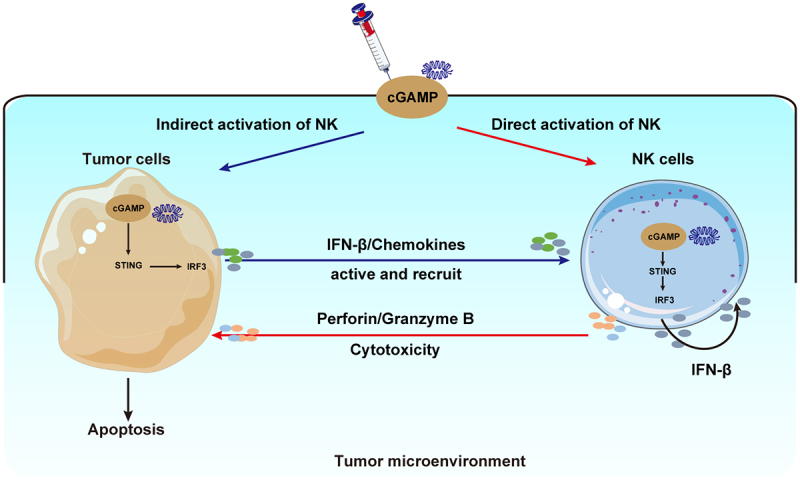
A schematic diagram showing the STING agonist-mediated enhancement of NK cell function. The STING agonist cGAMP promotes the anti-tumor effector functions of NK cells through two pathways. cGAMP directly activates STING signaling in NK cells, leading to the up-regulation of the expression of IFN-β, CD69, CD107a, perforin, granzyme B, and the cytokines IFN-γ, and TNF-α in cGAMP-treated NK cells, which ultimately enhanced the cytotoxicity of NK cells. Alternatively, cGAMP activates STING signaling in pancreatic cancer cells, up-regulates the expressions of NKG2D ligands, and induces the production of chemokines. Thus, cGAMP indirectly activates and recruits NK cells, and enhances the sensitivity of tumor cells to NK cell cytotoxicity.
In summary, this study offers the first evidence that cGAMP may not only directly increase the anti-tumor activity of NK cells but may also enhance the sensitivity of pancreatic cancer cells to NK cell cytotoxicity. Furthermore, our data suggest that the combinational treatment of cGAMP and CAR-NK cells may become a novel therapeutic strategy against pancreatic cancers in the clinic.
Supplementary Material
Funding Statement
This study is supported in part by the National Natural Science Foundation of China (91842305, 81771686), the Shandong Provincial Key Research and Development Program (Major Scientific and Technological Innovation Project) (Shanxi Provincial Key Research and Development Project 2019JZZY021013), and the Shandong Provincial Natural Science Foundation (Natural Science Foundation of Shandong Province ZR2020MH260), the National Health Commission of People’s Republic of China on the project entitled “Development of scientific system and service platform for cancer precision medicine” (2020-2025), and the operational funds from The First Affiliated Hospital of Xi’an Jiaotong University.
Abbreviations
- STING
Stimulator of interferon gene
- NK
Natural killer
- cGAMP
Cyclic GMP-AMP
- IRF3
Interferon regulatory factor 3
- IFN-β
Interferon β
- TNF-α
Tumor necrosis factor α
- CAR
Chimeric antigen receptor
- MSLN
Mesothelin
Disclosure statement
The authors declare no competing interests.
Author contributions
Yanyan Da designed and performed experiments, analyzed data, and wrote the manuscript; Yuxia Liu, Yuan Hu and Wenzeng Liu designed, performed experiments, and analyzed data; Junpeng Ma analyzed the data and improved the quality of Figures; Nan Lu provided guidance for the experiment design, analyzed and discussed the data, and wrote the manuscript; Chengsheng Zhang analyzed and discussed the data, and revised the manuscript; Cai Zhang conceived and supervised the study, designed experiments, and wrote the manuscript. All authors read and approved the final manuscript.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Connor AA, Gallinger S.. Pancreatic cancer evolution and heterogeneity: integrating omics and clinical data. Nat Rev Cancer. 2022;22(3):131–12. doi: 10.1038/s41568-021-00418-1. [DOI] [PubMed] [Google Scholar]
- 2.Manrai M, Tilak T, Dawra S, Srivastava S, Singh A. Current and emerging therapeutic strategies in pancreatic cancer: challenges and opportunities. World J Gastroenterol. 2021;27(39):6572–6589. doi: 10.3748/wjg.v27.i39.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ettrich TJ, Seufferlein T. Systemic therapy for metastatic pancreatic cancer. Curr Treat Options Oncol. 2021;22(106). doi: 10.1007/s11864-021-00895-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Woo SR, Fuertes M, Corrales L, Spranger S, Furdyna M, Leung MK, Duggan R, Wang Y, Barber G, Fitzgerald K, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41(5):830–842. doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li A, Yi M, Qin S, Song Y, Chu Q, Wu K. Activating cGAS-STING pathway for the optimal effect of cancer immunotherapy. J Hematol Oncol. 2019;12(1):35. doi: 10.1186/s13045-019-0721-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu JX, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339(6121):826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwon J, Bakhoum SF. The cytosolic DNA-sensing cGAS-STING pathway in cancer. Cancer Discov. 2020;10(1):26–39. doi: 10.1158/2159-8290.CD-19-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang X, Bai XC, Chen ZJ. Structures and mechanisms in the cGAS-STING innate immunity pathway. Immunity. 2020;53(1):43–53. doi: 10.1016/j.immuni.2020.05.013. [DOI] [PubMed] [Google Scholar]
- 9.Ou L, Zhang A, Cheng Y, Chen Y. The cGAS-STING pathway: a promising immunotherapy target. Front Immunol. 2021;12:795048. doi: 10.3389/fimmu.2021.795048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang C, Mo F, Liu L, Luo M, Men K, Na F, Wang W, Yang H, Wei X. Oxidized mitochondrial DNA sensing by STING signaling promotes the antitumor effect of an irradiated immunogenic cancer cell vaccine. Cell Mol Immunol. 2021;18(9):2211–2223. doi: 10.1038/s41423-020-0456-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo Y, Jiang F, Kong L, Wu H, Zhang H, Chen X, Zhao J, Cai B, Li Y, Ma C, et al. OTUD5 promotes innate antiviral and antitumor immunity through deubiquitinating and stabilizing STING. Cell Mol Immunol. 2021;18(8):1945–1955. doi: 10.1038/s41423-020-00531-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, Mechette K, Leong JJ, Lauer P, Liu W, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7(283):283ra52. doi: 10.1126/scitranslmed.aaa4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flood BA, Higgs EF, Li SY, Luke JJ, Gajewski TF. STING pathway agonism as a cancer therapeutic. Immunol Rev. 2019;290(1):24–38. doi: 10.1111/imr.12765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nicolai CJ, Wolf N, Chang I-C, Kirn G, Marcus A, Ndubaku CO, McWhirter SM, Raulet DH. NK cells mediate clearance of CD8 + T cell–resistant tumors in response to STING agonists. Sci Immunol. 2020;5(45):eaaz2738. doi: 10.1126/sciimmunol.aaz2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, Raulet DH. Tumor-derived cGAMP triggers a STING-mediated interferon response in non-tumor cells to activate the NK cell response. Immunity. 2018;49(4):754–763. doi: 10.1016/j.immuni.2018.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461(7265):788–U740. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer. 2016;16(1):7–19. doi: 10.1038/nrc.2015.5. [DOI] [PubMed] [Google Scholar]
- 18.Tang CHA, Zundell JA, Ranatunga S, Lin C, Nefedova Y, Del Valle JR, Hu CCA. Agonist-mediated activation of STING induces apoptosis in malignant B cells. Cancer Res. 2016;76(8):2137–2152. doi: 10.1158/0008-5472.CAN-15-1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun F, Liu Z, Yang Z, Liu S, Guan W. The emerging role of STING-dependent signaling on cell death. Immunol Res. 2019;67(2–3):290–296. doi: 10.1007/s12026-019-09073-z. [DOI] [PubMed] [Google Scholar]
- 20.Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, Radtke F, Ablasser A. Signalling strength determines proapoptotic functions of STING. Nat Commun. 2017;8(1):427. doi: 10.1038/s41467-017-00573-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang-Bishop L, Wehbe M, Shae D, James J, Hacker BC, Garland K, Chistov PP, Rafat M, Balko JM, Wilson JT, et al. Potent STING activation stimulates immunogenic cell death to enhance antitumor immunity in neuroblastoma. J Immunother Cancer. 2020;8(1):e000282. doi: 10.1136/jitc-2019-000282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vitale G, van Eijck CHJ, van Koetsveld Ing PM, Erdmann JI, Speel EJM, van der Wansem Ing K, Mooij DM, Colao A, Lombardi G, Croze E, et al. Type I interferons in the treatment of pancreatic cancer: mechanisms of action and role of related receptors. Ann Surg. 2007;246(2):259–268. doi: 10.1097/01.sla.0000261460.07110.f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Booy S, van Eijck CH, Dogan F, van Koetsveld PM, Hofland LJ. Influence of type-I Interferon receptor expression level on the response to type-I interferons in human pancreatic cancer cells. J Cell Mol Med. 2014;18(3):492–502. doi: 10.1111/jcmm.12200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smyth MJ, Swann J, Cretney E, Zerafa N, Yokoyama WM, Hayakawa Y. NKG2D function protects the host from tumor initiation. J Exp Med. 2005;202(5):583–588. doi: 10.1084/jem.20050994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu H, Wang S, Xin J, Wang J, Yao C, Zhang Z. Role of NKG2D and its ligands in cancer immunotherapy. Am J Cancer Res. 2019;9:2064–2078. [PMC free article] [PubMed] [Google Scholar]
- 26.Maghazachi AA. Role of chemokines in the biology of natural killer cells. Curr Top Microbiol Immunol. 2010;341:37–58. doi: 10.1007/82_2010_20. [DOI] [PubMed] [Google Scholar]
- 27.Marofi F, Rahman HS, Thangavelu L, Dorofeev A, Bayas-Morejón F, Shirafkan N, Shomali N, Chartrand MS, Jarahian M, Vahedi G, et al. Renaissance of armored immune effector cells, CAR-NK cells, brings the higher hope for successful cancer therapy. Stem Cell Res Ther. 2021;12(1):200. doi: 10.1186/s13287-021-02251-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu Y, Tian ZG, Zhang C. Chimeric antigen receptor (CAR)-transduced natural killer cells in tumor immunotherapy. Acta Pharmacol Sin. 2018;39(2):167–176. doi: 10.1038/aps.2017.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Siegler EL, Zhu YN, Wang P, Yang LL. Off-the-Shelf CAR-NK cells for cancer immunotherapy. Cell Stem Cell. 2018;23(2):160–161. doi: 10.1016/j.stem.2018.07.007. [DOI] [PubMed] [Google Scholar]
- 30.Zhang C, Hu Y, Shi C. Targeting natural killer cells for tumor immunotherapy. Front Immunol. 2020;11:60. doi: 10.3389/fimmu.2020.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu Y, Tian Z, Zhang C. Natural killer cell-based immunotherapy for cancer: advances and prospects. Engineering. 2019;5(1):106–114. doi: 10.1016/j.eng.2018.11.015. [DOI] [Google Scholar]
- 32.Argani P, Iacobuzio-Donahue C, Ryu B, Rosty C, Goggins M, Wilentz RE, Murugesan SR, Leach SD, Jaffee E, Yeo CJ, et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin Cancer Res. 2001;7(12):3862–3868. [PubMed] [Google Scholar]
- 33.Le K, Wang J, Zhang T, Chang H, Wang S, Zhu B. Overexpression of mesothelin in Pancreatic Ductal Adenocarcinoma (PDAC). Int J Med Sci. 2020;17(4):422–427. doi: 10.7150/ijms.39012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aldea M, Benitez JC, Chaput N, Besse B. New immunotherapy combinations enter the battlefield of malignant mesothelioma. Cancer Discov. 2021;11(11):2674–2676. doi: 10.1158/2159-8290.CD-21-1046. [DOI] [PubMed] [Google Scholar]
- 35.Wang Z, Li N, Feng K, Chen M, Zhang Y, Liu Y, Yang Q, Nie J, Tang N, Zhang X, et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell Mol Immunol. 2021;18(9):2188–2198. doi: 10.1038/s41423-021-00749-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Avula LR, Rudloff M, El-Behaedi S, Arons D, Albalawy R, Chen X, Zhang X, Alewine C. Mesothelin enhances tumor vascularity in newly forming pancreatic peritoneal metastases. Mol Cancer Res. 2020;18(2):229–239. doi: 10.1158/1541-7786.MCR-19-0688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takashima K, Takeda Y, Oshiumi H, Shime H, Okabe M, Ikawa M, Matsumoto M, Seya T. STING in tumor and host cells cooperatively work for NK cell-mediated tumor growth retardation. Biochem Bioph Res Co. 2016;478(4):1764–1771. doi: 10.1016/j.bbrc.2016.09.021. [DOI] [PubMed] [Google Scholar]
- 38.Basit A, Cho MG, Kim EY, Ewon D, Kang SJ, Lee JH. The cGAS/STING/TBK1/IRF3 innate immunity pathway maintains chromosomal stability through regulation of p21 levels. Exp Mol Med. 2020;52(4):643–657. doi: 10.1038/s12276-020-0416-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol. 2020;21(9):501–521. doi: 10.1038/s41580-020-0244-x. [DOI] [PubMed] [Google Scholar]
- 40.Klarquist J, Hennies CM, Lehn MA, Reboulet RA, Feau S, Janssen EM. STING-mediated DNA sensing promotes antitumor and autoimmune responses to dying cells. J Immunol. 2014;193(12):6124–6134. doi: 10.4049/jimmunol.1401869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhatelia K, Singh A, Tomar D, Singh K, Sripada L, Chagtoo M, Prajapati P, Singh R, Godbole MM, Singh R, et al. Antiviral signaling protein MITA acts as a tumor suppressor in breast cancer by regulating NF-kappa B induced cell death. Bba-Mol Basis Dis. 2014;1842(2):144–153. doi: 10.1016/j.bbadis.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 42.Murthy AMV, Robinson N, Kumar S. Crosstalk between cGAS-STING signaling and cell death. Cell Death Differ. 2020;27(11):2989–3003. doi: 10.1038/s41418-020-00624-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lam AR, Le Bert N, Ho SSW, Shen YJ, Tang MLF, Xiong GM, Croxford JL, Koo CX, Ishii KJ, Akira S, et al. RAE1 ligands for the NKG2D receptor are regulated by STING-dependent DNA sensor pathways in lymphoma. Cancer Res. 2014;74(8):2193–2203. doi: 10.1158/0008-5472.CAN-13-1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Su T, Zhang Y, Valerie K, Wang X-Y, Lin S, Zhu G. STING activation in cancer immunotherapy. Theranostics. 2019;9(25):7759–7771. doi: 10.7150/thno.37574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang C, Hu Y, Xiao WH, Tian ZG. Chimeric antigen receptor- and natural killer cell receptor-engineered innate killer cells in cancer immunotherapy. Cell Mol Immunol. 2021;18(9):2083–2100. doi: 10.1038/s41423-021-00732-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yilmaz A, Cui HW, Caligiuri MA, Yu JH. Chimeric antigen receptor-engineered natural killer cells for cancer immunotherapy. J Hematol Oncol. 2020;13(1):168. doi: 10.1186/s13045-020-00998-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daher M, Rezvani K. Outlook for new CAR-based therapies with a focus on CAR NK cells: what lies beyond CAR-engineered T cells in the race against cancer. Cancer Discov. 2021;11(1):45–58. doi: 10.1158/2159-8290.CD-20-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu SY, Fu T, Jiang YZ, Shao ZM. Natural killer cells in cancer biology and therapy. Mol Cancer. 2020;19(1):120. doi: 10.1186/s12943-020-01238-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.