

Abstract
Fluorogenic RNA aptamers are used to genetically encode fluorescent RNA and to construct RNA-based metabolite sensors. Unlike naturally occurring aptamers that efficiently fold and undergo metabolite-induced conformational changes, fluorogenic aptamers can exhibit poor folding, which limits their cellular fluorescence. To overcome this, we evolved a naturally occurring well-folded adenine riboswitch into a fluorogenic aptamer. We generated a library of ~1015 adenine aptamer-like RNAs in which the adenine-binding pocket was randomized for both size and sequence, and selected Squash, which binds and activates the fluorescence of GFP-like fluorophores. Squash exhibits markedly improved in-cell folding and highly efficient metabolite-dependent folding when fused to a S-adenosylmethionine (SAM)-binding aptamer. A Squash-based ratiometric sensor achieved quantitative SAM measurements, revealed cell-to-cell heterogeneity in SAM levels, and revealed metabolic origins of SAM. These studies show that the efficient folding of naturally occurring aptamers can be exploited to engineer well-folded cell-compatible fluorogenic aptamers and devices.
Graphical Abstract
INTRODUCTION
Genetically encoded metabolite sensors reveal dynamic changes in metabolite concentrations in single cells in real time. Most sensors comprise fluorescent proteins flanking a metabolite-binding domain1. Metabolite binding induces conformational changes that reposition the fluorescent proteins, thus altering the Fӧrster resonance energy transfer (FRET) between these proteins2. Metabolite sensors rely on ratiometric fluorescence, in which fluorescence is measured at two excitation/emission wavelengths, and the ratio is used to establish metabolite levels. Ratiometric probes are thus ‘self-calibrating,’ which allows them to produce signals that are independent of probe concentration. This overcomes cell-to-cell variability in sensor expression levels, and also resolves the problem of different fluorescence levels in thin versus thick parts of a cell3. The lack of proteins that undergo suitable metabolite-induced conformational changes limits the overall number of sensors available for researchers4.
In addition to protein-based sensors, sensors can be composed of RNA5,6. RNA-based sensors utilize a metabolite-binding RNA aptamer connected to a fluorogenic aptamer, such as Broccoli, Spinach, or Corn6–9. Metabolite binding allosterically induces folding of the fluorogenic RNA aptamer, allowing it to bind and activate the fluorescence of its otherwise non-fluorescent cognate fluorophore5,6. RNA-based sensors have mostly been used in bacteria where the RNA is stable and can thus accumulate to sufficient concentrations for fluorescence detection7,10. RNA-based sensors have recently been used in mammalian cells as a result of an expression system that allow RNA-based sensors to be expressed as highly stable circular RNA11. However, ratiometric sensors have not yet been developed for mammalian cells, which limit the usefulness of RNA-based sensors.
Here we describe RNA-based ratiometric sensors for metabolite imaging in live mammalian cells. These ratiometric sensor are composed of two fluorogenic aptamers, one of which is constitutively fluorescent and provides signal normalization, while the other produces fluorescence in proportion to S-adenosylmethionine (SAM) levels. We developed Squash, a fluorogenic aptamer that exhibits orange fluorescence, which is spectrally separated from the green fluorescence of Broccoli, which is used as the normalizer. Squash was developed using a novel approach to obtain highly folded fluorogenic aptamers, since poor RNA folding is a major factor that limits overall fluorescence. In this approach, we evolved a naturally occurring well-folded adenine RNA aptamer using systematic evolution of ligands by exponential enrichment (SELEX) to bind and activate the fluorescence of green fluorescent protein (GFP)-like fluorophores. Squash was fused to a SAM-binding aptamer to generate Squash-SAM sensors that produce orange fluorescence in proportion to SAM levels. Using the ratiometric SAM sensor, we test the distinct metabolic pathways that control SAM levels and uncover cell-state specific heterogeneity in intracellular SAM metabolism.
RESULTS
Design of an expanding and contracting RNA library for SELEX
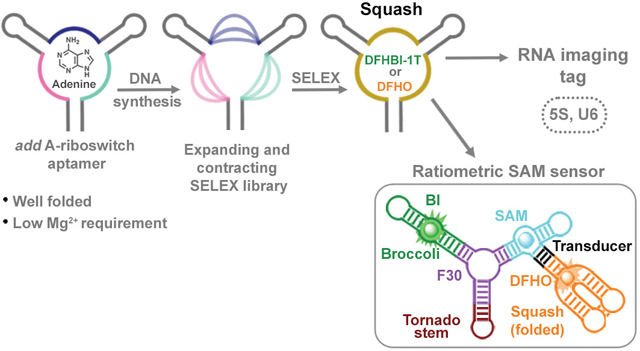
RNA aptamers that are selected in vitro often exhibit poor folding, unlike some naturally occurring riboswitch aptamers which can fold before mRNA transcription has been completed12. We reasoned that we could evolve the ligand-binding domain of a naturally occurring high-folding riboswitch to bind and activate a fluorogenic dye. We chose the adenine-binding aptamer in the add A-riboswitch from V. vulnificus13,14 (Fig. 1a,b) which folds in ~2 seconds15. This aptamer is unusual in that it folds in magnesium concentrations as low as 100 μM16. This may be useful since the mammalian cytoplasm typically contains lower magnesium levels17,18 (0.2 – 1.0 mM) than the bacterial cytoplasm, which can be in the millimolar range19.
Fig. 1 |. Evolution of the add A-riboswitch aptamer into fluorogenic aptamer Squash.

a, Strategy for evolution of the add A-riboswitch aptamer. Indicated are helices P1-P3, junctional strands (J1/2, J2/3, J3/1), kissing loop (gray lines), and adenine interactions with the aptamer (dotted line). The chemical structure of adenine and fluorophores are shown. Three regions (pink, blue and teal), which include J1/2, J2/3, and J3/1 were randomized in both size and sequence resulting in a sprouts/clips library for SELEX. Each nucleotide in the colored regions was replaced using a combination of random stochastic deletion (designated “N”) and random stochastic insertion (designated “n”). b, Three dimensional structure of the add A-riboswitch aptamer highlighting the adenine-binding pocket (PDB 1Y26). The mutagenized regions are shown in the same color as Fig. 1a. Adenine is shown in light green. c, Evolution of the add A-riboswitch aptamer into Squash, a DFHBI-1T-binding aptamer. The sprouts/clips library was selected for binding to DFHBI-conjugated agarose resulting in aptamer 9–1. Aptamer 9–1 was subjected to two consecutive rounds of directed evolution generating DE2–6. A pair of mutations was introduced in DE2–6 to strengthen the kissing loop interaction resulting in Squash. d, Fluorescence activation of DFHBI-1T by different aptamer intermediates that led to Squash. The values inside the bars indicate fold-activation of DFHBI-1T fluorescence by the corresponding aptamer. Data represent mean values ± s.d. for n=3 independent experiments. e, The Squash aptamer shows ligand-binding pocket expansion compared to the parental add A-riboswitch aptamer. Aptamers at different stages of Squash evolution are aligned. Constant sequences are indicated with gray dots. The sequences outside the randomized regions are shown in gray. Shown in red are U→C and U→G mutations that were introduced to enhance the kissing loop interaction.
GFP-like fluorophores are useful for fluorogenic aptamers since they exhibit low cytotoxicity, high cell permeability, minimal fluorescence when incubated in cells, and can be fluorescently activated by RNA aptamers20–24. We reasoned that we could evolve the ligand-binding domain of the adenine aptamer to bind and activate these dyes. However, the ligand-binding pocket may be too small to accommodate GFP-like fluorophores such as DFHBI-1T ((Z)-4-(3,5-difluoro-4-hydroxybenzylidene)-2-methyl-1-(2,2,2-trifluoroethyl)-1H-imidazol-5(4H)-one) and DFHO ((Z)-4-(3,5-difluoro-4-hydroxybenzylidene)-1-methyl-5-oxo-1H-imidazole-2-carbaldehyde oxime) (Fig. 1a). The nucleotides that comprise the ligand-binding pocket of an aptamer can be randomized to create RNA libraries that can be used in SELEX25,26 to discover new ligand-binding aptamers. These types of libraries were previously generated by either randomizing few residues27 or the entire binding pocket28 in purine riboswitches. However, these libraries only change the sequence, not the size, of the ligand-binding pocket.
To develop a SELEX library in which the ligand-binding pocket varies in both sequence and size, we modified the DNA synthesis protocol used to make random libraries29. In conventional oligonucleotide library synthesis, a phosphoramidite mixture representing all four nucleotides is prepared29. This mixture is used for site-specific incorporation of a random nucleotide at specified positions in each growing oligonucleotide strand29. To induce spontaneous shortening of the SELEX library, we reduced the coupling time from 25 s to 2 s. This reduces the coupling efficiency from ~100% to ~93% (Supplementary Table 1a). As a result, ~7% of the strands lose a nucleotide every time this mixture is used. We designated the loss of any nucleotide relative to the full-length sequence as a “clip”.
To create random additions of nucleotides, we created a new mixture containing all four phosphoramidites, but at 1% the normal concentration. This dilute mixture exhibits a ~5% coupling efficiency (Supplementary Table 1b), thus causing stochastic insertions at specific positions in the oligonucleotide. We designated the appearance of a stochastically added nucleotide as a “sprout” in the library.
Importantly, the “capping step” during oligonucleotide synthesis was omitted. The capping step comprises 5’-OH acetylation of any oligonucleotide that fails to couple to a phosphoramidite30. Removing this step is important since clips rely on stochastically inefficient coupling. Similarly, removing the capping step is important for sprouts, because coupling reactions typically do not occur when using the dilute phosphoramidite mixture for sprouts. Thus, removing the capping step is required for sprouts and clips.
The adenine aptamer ligand-binding pocket comprises three strands at the center of a three-way junction (Fig. 1a,b). We therefore created a sprouts and clips library in which the ligand-binding pocket was randomized for both sequence and size (Fig. 1a,b). Every nucleotide in the ligand-binding pocket was randomized sequentially using the standard phosphoramidite mixture with reduced coupling time, followed by the dilute phosphoramidite mixture (Fig. 1a). Thus, a 6 nt-long strand in the ligand-binding pocket can be transformed into as few as 0 and as many as 12 random nucleotides. Overall, the theoretically diversity of this sprouts/clips library is >7.3 × 1026 members, which contrasts with a diversity of only 1.7 × 1013 if standard randomization is used (Supplementary Figure 1a,b). In this library, the overall structure of the aptamer should remain, while the ligand-pocket will vary.
In vitro evolution and identification of Squash
To evolve the adenine aptamer into a fluorogenic aptamer, we selected for aptamers that bind to agarose-immobilized DFHBI20,21,23 (Fig. 1c, Supplementary Table 3). By the seventh round of SELEX, the pool exhibited fluorescence upon incubation with DFHBI-1T (Supplementary Figure 1c). At this point, the library was cloned into a bacterial expression vector, transformed into E. coli, and fluorescent aptamers were identified by sorting cells based on DFHBI-1T-induced fluorescence (Supplementary Figure 1d-f). After recovery of plasmids from the brightest cells, we found three library members which induced fluorescence activation of DFHBI-1T (Fig. 1d and Extended Data Fig. 1b). These aptamers also activated the fluorogenic dye DFHO, inducing an orange fluorescence (Extended Data Fig. 1a) suggesting that these aptamers could be used for different fluorescence applications depending on the fluorophore.
Notably, each aptamer contained an expanded ligand-binding pocket with sprouts in one or more of the randomized strands (Fig. 1e). The aptamer with the highest fluorescence activation (aptamer 9–1, ~521-fold fluorescence activation) was selected for optimization by directed evolution21,30 (Fig. 1d and Extended Data Fig. 1b). For directed evolution, we synthesized a DNA library of 9–1 aptamer mutants such that each nucleotide in the ligand-binding pocket has a controlled probability of being converted into one of the other three nucleotides21,31 (Supplementary Table 5c). We performed four rounds of SELEX using this library to enrich for binding to DFHBI-agarose (Supplementary Table 4a). After expression in E. coli and FACS (Supplementary Figure 2a,b), the brightest aptamer (designated DE1–2) exhibited ~790-fold fluorescence enhancement of DFHBI-1T (Fig. 1d). A subsequent round of directed evolution resulted in aptamer DE2–6, which showed ~949-fold activation of DFHBI-1T and 492-fold activation of DFHO (Fig. 1d, 2c and Supplementary Figure 2c-f).
Fig. 2 |. Squash activates the fluorescence of DFHBI-1T and DFHO without utilizing a G-quadruplex.

a, Secondary structure of Squash as predicted by mFOLD. Squash retained the main structural elements of add A-riboswitch aptamer including a 3-way junction and predicted kissing loop interactions (yellow residues, solid gray line). An additional basepair in the kissing loop between loop L2 and L3 (red residues, dotted gray line) improved fluorescence activation. b, Squash binds and activates the fluorescence of both DFHBI-1T and DFHO. Shown are the excitation (Ex) and emission (Em) spectra of Squash bound to DFHBI-1T and DFHO (structures shown in inset). Spectra were measured using 20 μM RNA and 2 μM of the indicated fluorogenic dye. c, Squash shows similar fluorescence activation of DFHBI-1T as Broccoli but much higher activation of DFHO than Corn. Fluorescence activation was measured by incubating 200 nM dye and 10 μM RNA. By using a large excess of RNA compared to the fluorophore, we ensured that the fluorescence observed is from 200 nM RNA-fluorophore complex. Data represent mean values ± s.d. for n=3 independent experiments. The values inside the bars indicate fold activation. AU, arbitrary units. d, The dissociation constant (Kd) between Squash and DFHO was measured by titration of 50 nM Squash with increasing concentration of DFHO, and then the data were fitted using a one-site saturation model. Data represent mean values ± s.d. for n=3 independent experiments. e, Squash-DFHO fluorescence was measured in buffers containing exclusively the indicated cations. Squash was fluorescent in K+-free buffers suggesting that it lacks a G-quadruplex. Broccoli, which contains a G-quadruplex, exhibited markedly reduced fluorescence in the absence of K+. Data represent mean values ± s.d. for n=3 independent experiments. f, Squash-DFHO shows >80% fluorescence activation at 0.1 mM MgCl2. Similar to the parental add A-riboswitch aptamer (see ref. 16), Squash-DFHO has a low magnesium requirement for fluorescence activation.. Data represent mean values ± s.d. for n=3 independent experiments.
In certain bacteria, adenine riboswitches contain an extra G•U basepair in the kissing loop interaction compared to the two-basepair kissing loop in V. vulnificus13. We therefore tested the effect of an additional basepair by replacing U residues in loops L2 and L3 with G and C, respectively. These mutations increased the fold activation of both DFHBI-1T and DFHO (949 fold to 1064 fold, and 492 fold to 550 fold, respectively) (Fig. 1d and Extended Data Fig. 1a,g). This improved aptamer binds DFHBI-1T and DFHO (Kd of 45 nM and 54 nM, respectively) with high quantum yield (0.71 and 0.60 for DFHBI-1T and DFHO, respectively) (Table 1). This aptamer also shows high brightness and high photostability in mammalian cells (Extended Data Figs. 2-4). The resulting aptamer was designated Squash, in keeping with previous nomenclature systems, and to reflect the multi-colored nature of Squash-fluorophore complexes (Fig. 2).
Table 1 |.
Comparison of photophysical and biochemical properties of Squash with previous fluorogenic aptamers
| Excitation maximum (nm) | Emission maximum (nm) | Extinction coefficient (M−1cm−1) | Fluorescence quantum yield | Brightnessc | Kd (nM) | % folded in vitro | |
|---|---|---|---|---|---|---|---|
| DFHBI-1T | 426 | 495 | 19,900a | 0.00098 | 0.12 | - | - |
| Broccoli/DFHBI-1T | 472 | 507 | 29,600 | 0.94 | 100 | 360 | 55 |
| Squash/DFHBI-1T | 452 | 503 | 24,200 | 0.71 | 62 | 45 | 78 |
| DFHO | 473 | 561 | 27,000b | 0.0006 | 0.22 | - | - |
| Corn/DFHO | 505 | 545 | 29,000 | 0.25 | 100 | 70 | 52 |
| Squash/DFHO | 495 | 562 | 24,600 | 0.60 | 204 | 54 | 80 |
All in vitro measurements for Squash reported here were performed in 40 mM HEPES-KOH (pH 7.4), 100 mM KCl and 0.5 mM MgCl2 buffer. Folding experiments for all the aptamers were also performed in the buffer mentioned above. The photophysical properties of Broccoli/DFHBI-1T26 and Corn/DFHO28 are taken from previous reports.
The extinction coefficient of DFHBI-1T and
DFHO are reported here at 452 nm and 495 nm respectively.
Squash activates fluorescence without a G-quadruplex
Squash maintains sequence similarity to the parental adenine aptamer except for expansion and mutation of its ligand-binding pocket (Fig. 1e and 2a). The kissing loop interactions were confirmed by mutation of the G30•C57 basepair to C30•C57, which markedly impaired Squash-induced fluorescence activation of DFHO (Extended Data Fig. 1g). A compensatory mutation (C30•G57) restored fluorescence. Although diverse fluorogenic aptamers bind fluorophores through a G-quadruplex32, Squash fluorescence is not dependent on potassium (Fig. 2e), suggesting a G-quadruplex-independent mechanism of fluorophore binding and activation. Similar to the parental add A-aptamer, Squash also has low Mg2+ requirement for folding (Fig. 2f).
We next wanted to identify fluorophores that would allow Squash to be imaged in cells expressing Broccoli. Broccoli can be imaged with BI, a DFHBI-1T derivative that binds and enhances the folding and brightness of Broccoli33. Importantly, BI binds Squash with much weaker affinity than Broccoli (Extended Data Fig. 2g). For Squash, we used DFHO, since Squash-DFHO shows a markedly distinct excitation/emission spectra (Ex=495 nm; Em=562 nm) than Broccoli-BI (Ex=470 nm; Em=505 nm) (Fig. 2b and Extended Data Fig. 1h). Additionally, DFHO binds very weakly to Broccoli (Kd ~ 11 μM) (Extended Data Fig. 2h). Thus, Broccoli-BI and Squash-DFHO can be imaged together as parts of a ratiometric sensor without substantial spectral interference.
Squash shows efficient folding in vitro and in cells
To determine if Squash exhibits improved folding, we used an in vitro folding assay34 where the first step is to determine the fluorescence of a fully folded Squash-fluorophore complex. To prepare this complex, 10 μM Squash is incubated with 0.2 μM DFHO. By having large excess of Squash, there is likely to be enough folded Squash to form 0.2 μM Squash-DFHO complex. Next, 0.2 μM Squash is incubated with excess (10 μM) DFHO. In this assay, the percent of folded Squash will determine the amount of Squash−DFHO complex that can form during the second step, up to a maximum of 0.2 μM (see Methods and ref.34 for more details). Using this approach, we found that ~80% of Squash is folded (Fig. 3a), compared to 60% for Spinach234 and 55% for Broccoli21.
Fig. 3 |. Squash shows efficient folding in vitro and in cells.

a, Squash does not require the F30 folding scaffold. In vitro folding of Broccoli (with DFHBI-1T, green bars) is substantially improved when placed in F30 scaffold. By contrast, Squash (with DFHO, orange bars) does not show increased folding when inserted into F30, suggesting that it already exhibits high folding. Data represent mean values ± s.d. for n=3 independent experiments. b, Squash-DFHO shows similar thermostabilty as Broccoli-BI, an aptamer-fluorophore pair evolved for improved binding and thermostability (see ref. 33). Melting temperature was measured using 1 μM RNA and 10 μM fluorophore. c, F30 enhances Broccoli but not Squash fluorescence in HEK293T cells. Left column, flow cytometry analysis of DFHBI-1T-treated cells expressing linear unscaffolded Broccoli or linear F30-Broccoli (expressed using U6+27 promoter). Right column, DFHO-treated cells expressing linear Squash or linear F30-Squash. Cells were analyzed in the green (ex. 488 nm, em. 525 ± 25 nm) or orange (ex. 488 nm, em. 570 ± 20 nm) channel for Broccoli and Squash fluorescence, respectively. An auxiliary far-red channel (ex. 635 nm; em. 780 ± 30) was used to measure cellular autofluorescence. F30 enhances the cellular fluorescence levels of Broccoli, but not Squash. d, HEK293T cells expressing either 5S-Broccoli or 5S-F30-Broccoli were incubated with 25 μM DFHBI-1T and imaged. The 5S-Broccoli appeared as faint dots while 5S-F30-Broccoli appeared as bright perinuclear puncta (see inset). Both 5S-Squash or 5S-F30-Squash appeared as bright perinuclear puncta (see inset). These data suggest that F30 is not needed to enhance Squash fluorescence. Scale bar, 20 μm.
We next asked if Squash folding increases when it is inserted into a “folding scaffold.” The F30 folding scaffold is based off of a naturally occurring three-way junction packaging RNA in the ϕ29 bacteriophage35. Aptamers are inserted via their helical stem regions into either arm of F30, which facilitates aptamer folding36. Although Broccoli folding increases after insertion into F30, Squash folding was unaffected by F30 (Fig. 3a), suggesting that Squash is already highly folded.
To further assess folding, we measured the thermal stability of Squash-DFHO. As a control we used Broccoli bound to BI, which markedly improves Broccoli thermal stability33. Squash-DFHO exhibited a similar Tm (50.5oC) as Broccoli-BI (52.5oC) (Fig. 3b). Thus, Squash’s thermostability is consistent with a highly folded structure.
We next asked if Squash is highly folded in HEK293T cells. As observed previously36, cells expressing F30-Broccoli showed higher fluorescence than Broccoli without F30 (Fig. 3c and Supplementary Figure 4a). However, cells expressing Squash and F30-Squash showed similar cellular fluorescence (Fig. 3c and Supplementary Figure 4). These results suggest that Squash folding is already so high that it cannot be further enhanced by F30.
Lastly, we imaged 5S rRNA in HEK293T cells. The 5S-Broccoli in DFHBI-1T-incubated cells appeared as faint dots using a 200 ms imaging time (Fig. 3d). This signal was markedly higher when 5S was tagged with F30-Broccoli (Fig. 3d). By contrast, 5S-Squash was readily detectable as a perinuclear punctate signal without using F30 (Fig. 3d). We confirmed that the tagged 5S constructs were expressed at similar levels (Supplementary Figure 5). Overall, these data suggest that Squash is well-folded in HEK293T cells.
Design of a Squash-based SAM-sensor
We next converted Squash into a sensor of SAM, a metabolite that influences cellular differentiation and cancer progression37. We previously created non-ratiometric RNA-based SAM sensors using Broccoli, Red Broccoli and Corn8,9,11,38. The SAM sensors comprise the SAM-binding aptamer portion of the SAM-III riboswitch39 fused to a fluorogenic aptamer via a transducer domain. The transducer domain is a thermodynamically unstable helix, which is stabilized upon SAM binding, thus allosterically inducing the folding of the fluorogenic aptamer8,11.
To identify an optimal transducer domain for the Squash-SAM sensor, we prepared a sprouts/clips DNA library by randomizing the transducer helix connecting the SAM aptamer and Squash (Fig. 4a and Supplementary Table 5h). We transcribed the library into RNA, and captured library members that bind to DFHO-agarose beads in the presence of 100 μM SAM (Supplementary Figure 6). After washing the beads with buffer containing 100 μM SAM, RNA was eluted by switching to a SAM-free buffer. After reverse transcription, the process was repeated for two more rounds, which resulted in an RNA pool that exhibited SAM-dependent fluorescence (Extended Data Fig. 5a).
Fig. 4 |. Development of a ratiometric S-adenosylmethionine (SAM) sensor from Squash.

a, Design of the Squash-SAM sensor transducer library. The SAM-III aptamer (teal) was fused to Squash (orange) through a transducer helix (black). The sensor was selected from a library of randomized transducer sequences generated using the sprouts/clips method. b, After three rounds of SELEX, two Squash-SAM sensors, 5–1 and 4–2, were tested for SAM-induced fluorescence. Squash-SAM sensor RNAs (1 μM) were incubated with 10 μM DFHO and 0 or 0.1 mM SAM for 1 h at 37 ˚C and then fluorescence was measured (ex. 495 nm; em. 562 nm). Data represent mean values ± s.d. for n=3 independent experiments. c-d, Kinetics of fluorescence activation (c) and deactivation (d) of Squash-SAM sensors. Sensor activation was induced by adding 0.1 mM SAM to 1 μM sensor RNAs and 10 μM DFHO at 37˚C. Deactivation was measured after removal of SAM by gel filtration. Comparison of the Squash-SAM sensors with Corn-SAM sensor (see ref. 8) showed markedly faster activation kinetics of both Squash-based sensors compared to the Corn-based sensor. e, Schematic of the Squash-based ratiometric SAM sensor. The ratiometric sensor comprises Broccoli in one arm of F30 and the Squash-SAM sensor in the other arm. SAM binding activates the orange fluorescence by promoting Squash folding and subsequent binding with DFHO. The constitutive green fluorescence of Broccoli-BI is used for normalization, thus eliminating the confounding effect of cell-to-cell variation in sensor expression level, and different fluorescence measurements in thick vs. thin parts of the cell. f, The orange to green (O/G) fluorescence ratio measured using the ratiometric sensor in individual HEK293T cells showed low cell-to-cell variation in SAM levels (also see next panel). Inhibition of SAM biosynthesis using cycloleucine showed marked reduction of SAM levels in all cells, as measured by the reduced O/G fluorescence ratio. The SAM levels were restored after withdrawal of cycloleucine. A control RNA with constitutive Squash fluorescence (see Extended Data Fig. 7b) did not show any notable change in the O/G ratio upon cycloleucine treatment. g, SAM trajectory plots of individual HEK293T cells upon cycloleucine treatment and withdrawal, measured by the ratiometric signal. Each trajectory plot (8 cells from three experiments) was generated based on the mean O/G fluorescence ratio (mean of O/G ratio of all the pixels comprising a cell) in single cells. The ratio was measured at 10 min time intervals. Scale bar, 20 μm.
We tested SAM-induced fluorescence of 48 individual library members (Extended Data Fig. 5b). Sensor 5–1 showed the highest fold increase in fluorescence (8.7-fold) upon incubation with SAM (Fig. 4b). Sensor 4–2 showed higher total fluorescence, but a lower fold activation due to a higher baseline signal (Fig. 4b). Sensor 5–1 is expected to give greater ability to detect different SAM levels, while Sensor 4–2 is expected to be useful if sensor RNA expression is low.
Both sensor 5–1 and 4–2 were highly selective for SAM over other related metabolites (Extended Data Fig. 5c,f). Both Squash-SAM sensors showed rapid fluorescence activation upon addition of 100 μM SAM, with 90% fluorescence achieved in 2 min (Sensor 5–1) and 6 min (Sensor 4–2), compared with 11 min for the Corn-based SAM sensors, with similar deactivation kinetics (Fig. 4c,d).
To use the Squash-SAM sensor in HEK293T cells, we expressed each sensor as a circular RNA using the Tornado system, which enables RNA-based sensors to be expressed at high levels in mammalian cells11 (Extended Data Fig. 6a). For both sensors, we observed a rapid decrease in fluorescence within 30 min of treatment with cycloleucine, an inhibitor of SAM biosynthesis8,9,11 (Extended Data Fig. 6). Upon removal of cycloleucine, the fluorescence returned over 60 min, indicating restoration of SAM levels. Overall, these data indicate that sensors 5–1 and 4–2 detect SAM in mammalian cells.
A ratiometric sensor for SAM imaging in live mammalian cells
An RNA-based sensor could become ratiometric by co-expressing a fluorescent protein for normalization. However, we found that RNA and protein expression were poorly correlated, even when expressed from the same plasmid (Extended Data Fig. 7a). Thus, we developed a single RNA that functions as a ratiometric sensor. This RNA contains the Squash-SAM sensor in one arm and Broccoli in other arm of the F30 three-way junction (Fig. 4e). As a control, expression of an RNA with Squash in one arm and Broccoli in the other arm of F30 showed high correlation and minimal spectral overlap between the orange and green fluorescence signals using both microscopic imaging and flow cytometry (Fig. 4f and Extended Data Fig. 7b-d).
Before using the sensor for ratiometric imaging, we first confirmed that the fluorophores used to detect Squash and Broccoli are selective for their cognate aptamer and do not affect the fluorescence of the other aptamer. To test this, we first measured Squash fluorescence in Squash-expressing mammalian cells incubated with 10 μM DFHO. Subsequent application of 10 μM BI did not affect Squash fluorescence levels (Extended Data Fig. 7e). Similarly, Broccoli fluorescence in BI-incubated cells expressing circular Broccoli was not affected by subsequent addition of 10 μM DFHO (Extended Data Fig. 7e). These data confirm that these fluorophores are selective for their cognate aptamers. We also verified that Broccoli and Squash do not exhibit FRET which would adversely affect fluorescence signals from the SAM sensor (Extended Data Fig. 7f). Finally, the Broccoli-BI pair is not affected by SAM or structurally related molecules making it an ideal normalizer (Extended Data Fig. 5i).
We next asked whether the ratiometric sensor detects endogenous SAM. The ratiometric signal was markedly reduced in HEK293T cells within 30 minutes of cycloleucine treatment (Fig. 4f,g). Notably, the overall time course and magnitude in the drop of fluorescence was relatively homogeneous across all imaged cells (Fig. 4f,g). When cycloleucine was removed, the ratiometric signal returned to basal levels within 90 min (Fig. 4f,g). Importantly, the effect of cycloleucine is not due to cytotoxicity because a control Squash RNA is not affected by this treatment (Fig. 4f).
Biochemical measurements of SAM levels at each time point after cycloleucine treatment correlate with the average ratiometric signal in cells (Extended Data Fig. 8a-d). Based on this correlation between cellular fluorescence and SAM levels, the average SAM concentration in HEK293T cells varies between 60 and 90 μM (Extended Data Fig. 8e-f). Taken together, the ratiometric Squash-SAM sensor enables quantitative detection of SAM levels in cells.
Cell-to-cell heterogeneity and metabolic origins of SAM in different cell types
Mouse embryonic stem cells (mES cells) contain enzymatic machinery to metabolize threonine to promote SAM biosynthesis40,41. Threonine is a precursor for 5-methyltetrahydrofolate, which is used to generate methionine from homocysteine42. However, bulk metabolic labeling studies using isotopically labeled threonine shows that only 5% of SAM is derived from threonine in mES cells41. Therefore, it remains unclear to what extent threonine is required to maintain methionine levels for SAM biosynthesis in mES cells.
To test this, we used mES cells cultured in media containing serum and leukemia inhibitory factor, as well as inhibitors of mitogen-activated protein kinase and glycogen synthase kinase-3β. mES cells cultured in this media (designated +2i), are highly homogenous with low propensity for differentiation43. In contrast, mES cells cultured without the two kinase inhibitors (−2i media) exhibit more cellular heterogeneity and varying tendencies to differentiate44,45.
For SAM measurements in mES cells, Squash-SAM sensor 4–2 was used since it produced more fluorescence than the Squash-SAM sensor 5–1 (Extended Data Fig. 9a). The intracellular SAM level in +2i media were homogeneous and varied between 40–70 μM (Fig. 5a,b and Extended Data Fig. 8f). This is similar to HEK293T cells which showed relatively homogeneous SAM levels at 60–90 μM (Fig. 4f and Extended Data Fig. 8f). However, mES cells cultured in −2i showed considerable cell-to-cell variability with SAM concentrations varying between 10–120 μM (Fig. 5a,c and Extended Data Fig. 8f).
Fig. 5 |. Metabolic origin of SAM in different cell types measured by the ratiometric sensor.

a, SAM levels in individual mES cells were determined using the O/G fluorescence ratio at 30 min intervals after withdrawing threonine, methionine or both. Threonine depletion did not lead to a reduction in SAM levels. The control constitutively fluorescent Squash was not affected by withdrawal of threonine and methionine. Scale bar, 20 μm. b, SAM trajectory plots of O/G ratio in individual mES cells cultured in +2i upon withdrawal of methionine or both threonine and methionine. Overall, low cell-to-cell variation was seen (n = 8 cells from three separate experiments). c, SAM trajectory plots of O/G ratio in individual mES cells cultured in −2i upon withdrawal of methionine or both threonine and methionine shows high cell-to-cell variability. The rate of SAM drop was slower than the rate in cells cultured in +2i (n = 8 cells from three separate experiments). d, Effect of methionine withdrawal on SAM level in HCT116 cancer cells. SAM levels showed high cell-to-cell variability at baseline. SAM levels were measured at 20 min time intervals after methionine withdrawal and after methionine (100 μM) addition. Cells indicated with a white arrow start with a very high level of SAM and show a slow drop in SAM levels over time, indicating low SAM utilization. Scale bar, 20 μm. e, Methionine withdrawal shows distinct metabolic subtypes based on SAM loss. Type I cells show high basal levels of SAM and a slow drop in SAM levels upon methionine depletion. Type II cells show intermediate levels of SAM and a rapid drop in SAM levels upon methionine depletion. Type III cells show low basal levels of SAM and a slow drop in SAM levels after methionine depletion. n = 5 cells for each type, from three separate experiments.
Upon switching mES cells to threonine-depleted media, we observed essentially no change in SAM levels in any mES cells examined over 3 h using either culturing condition (Fig. 5a). This suggests that threonine is not required to maintain SAM levels in mES cells.
Upon switching mES cells to methionine-free media, we observed a drop in SAM at a rate that depended on the culturing conditions (Fig. 5a-c). For +2i, the SAM levels dropped quickly within the first 30 min and then remained constant at a low level (Fig. 5a,b). The initial SAM concentration, as well as the rate of SAM depletion, was highly synchronous (Fig. 5a,b), suggesting that these cells were metabolically homogeneous.
In contrast, mES cells cultured in −2i showed considerable cell-to-cell metabolic heterogeneity upon methionine removal (Fig. 5a,c). mES cells that started with a low SAM concentration exhibited a minor drop in SAM levels. However, mES cells with higher SAM concentrations exhibited considerable heterogeneity in the rate of drop in SAM levels, although all cells showed a decline in SAM levels (Fig. 5c). These experiments indicate that mES cells cultured in −2i are metabolically distinct and that methionine derived from the media is the major source of SAM in mES cells under both culture conditions. Notably, in either +2i or −2i media, SAM levels were markedly depleted upon addition of cycloleucine (Extended Data Fig. 9b), supporting the idea that MAT2A is the major enzyme responsible for SAM biosynthesis in mES cells.
Although threonine depletion has minimal effects on SAM levels, threonine depletion may cause cells to switch to exogenous methionine to generate SAM. To test this, we switched mES cells to media lacking both methionine and threonine. Here, we observed a drop in SAM levels at a rate similar to when methionine alone was depleted (Fig. 5a-c). When individual cells were quantitatively examined, the overall trend was essentially identical in cells cultured in methionine-free media or both methionine- and threonine-free media, regardless of the culturing conditions (Fig. 5b,c). These results suggest that threonine does not substantially contribute to overall intracellular SAM levels in the conditions used here.
Notably, the effect of amino acid removal on SAM levels was not due to cytotoxicity, since SAM levels were restored by reintroducing the amino acids (Extended Data Fig. 9c,d).
We also examined whether SAM levels show heterogeneity in HCT116 colon cancer cells. Although both the 4–2 and 5–1 ratiometric SAM sensor produced readily detectable signals (Fig. 5d and Extended Data Fig. 10a), we used 5–1 since it exhibits a higher dynamic range. HCT-116 cells exhibited considerable cell-to-cell heterogeneity, with intracellular SAM levels ranging from 40–180 μM (Fig. 5d and Extended Data Fig. 8f). To understand the basis for these differences, we determined the SAM consumption rate by endogenous methyltransferases by depleting methionine and monitoring the rate of SAM loss. Using this approach, we found that HCT-116 cells exhibit three patterns of SAM consumption rates, which were related to the initial SAM concentration (Fig. 5d,e). For type I cells (initial SAM concentration 90–130 μM), SAM levels gradually dropped and substantial SAM levels were retained after 4 h (Fig. 5e). By contrast, type II cells (60–90 μM) showed rapid SAM consumption with 90% depleted within 40 min. Type III cells (40–60 μM), exhibited a more gradual drop in SAM levels over 4 h (Fig. 5e). These data suggest that individual HCT-116 cells exhibit distinct metabolic states.
Notably, addition of cycloleucine caused a slightly faster drop in SAM levels compared to methionine and threonine depletion in these cells (Extended Data Fig. 10c-e). The fact that methionine/threonine depletion was not as effective as cycloleucine at reducing SAM levels (Extended Data Fig. 10b-e) suggests that these cells may engage in threonine- and methionine-independent pathways for SAM biosynthesis37, albeit at a relatively low efficiency.
DISCUSSION
Here we describe a generalizable strategy for ratiometric imaging of metabolite levels using an RNA-based sensor. The ratiometric sensor is an RNA comprising Broccoli, which provides constitutive green fluorescence to normalize for sensor expression, and a SAM-regulated Squash aptamer, which produces orange fluorescence in proportion to SAM levels in cells. Broccoli and Squash bind different fluorophores, with minimal interference between their fluorescent emissions33. The sensor is expressed as a circular RNA, enabling expression levels that generate sufficient fluorescence signals for quantitative metabolite detection in diverse mammalian cells.
To create the ratiometric sensor, we generated Squash, a fluorogenic aptamer with high folding, and a corresponding increase in cellular fluorescence. RNA folding limits the fluorescence of fluorogenic aptamers in mammalian cells33,34,36. These aptamers likely fold poorly since they were created using fully randomized libraries that are selected only for their ability to bind to a fluorophore, rather than their ability to fold in the cytosol. These selected aptamers contrast with riboswitch aptamers which evolved to fold so efficiently that they can function as they are being transcribed12,46. We therefore evolved the naturally occurring add A-riboswitch aptamer into a fluorophore-binding fluorogenic aptamer. To do this, we used an RNA library comprising roughly 1015 library members in which randomization occurred exclusively in the ligand-binding pocket. The library members retained key structural features of the parental add A-riboswitch aptamer, including its kissing loop interaction and helical domains.
Squash was generated using a new approach for generating a randomized DNA library. Rather than simply randomizing the sequence of the nucleotides that comprise the ligand-binding pocket, we also randomized the size, thus allowing SELEX to sample larger and smaller ligand-binding pockets. Library members contain ligand-binding pockets that can theoretically range from 0 to 44 nucleotides in length, distributed across three junctional strands. To cause random increases in the size of the ligand-binding pocket, we added synthetic steps using phosphoramidites at low concentration, resulting in stochastic “sprouts” at defined positions in the DNA library. Random shortenings, which we term “clips,” were obtained by reducing the coupling time when using the standard phosphoramidite mixture. Using this approach, the ligand-binding pocket of Squash was expanded by 4 nucleotides relative to the parental aptamer. The sprouts/clips approach can be used to evolve any aptamer allowing it to expand or contract to accommodate ligands of different sizes.
Squash appears to have maintained the high folding efficiency of its parental aptamer based on its high folding in vitro and high fluorescence in cells. Riboswitch-derived aptamers have been used previously as templates for SELEX libraries based on the idea that their ligand-binding pockets may be readily evolved to bind different ligands28. Here we show that this approach also results in an efficiently folded fluorogenic aptamer, thus overcoming a major shortcoming of previous fluorogenic aptamers.
The Squash-SAM sensors show faster fluorescence induction upon addition of SAM. These faster kinetics may reflect Squash’s origin from a riboswitch, which normally undergoes adenine-dependent conformational changes46. Other natural aptamers may be suitable for developing fluorogenic aptamers, especially if their folding does not vary at different physiologic concentrations of magnesium. This helps to ensure that changes in the fluorescence signals reflect metabolite concentrations rather than changes in intracellular magnesium levels.
Ratiometric sensing has been previously demonstrated in E. coli using Broccoli and a dinitroaniline-binding aptamer, DNB47. However, this aptamer and its fluorophore, sulforhodamine B conjugated to dinitroaniline has only been used in bacterial cells47,48 possibly due to cellular background fluorescence. Thus, the approach described here provides a general strategy for ratiometric metabolite imaging in mammalian cells.
Intracellular SAM concentrations can vary in different disease contexts49. The Squash-SAM sensor could be adjusted to detect SAM at different concentrations by tuning the Kd of the sensor for SAM. This can be achieved either by changing the transducer sequence joining Squash and the SAM aptamer or by using a different SAM aptamer with suitable Kd.
Using the Squash-SAM sensor, we found that cells can exist in distinct metabolic states with respect to SAM metabolism. This effect is dependent on culturing conditions and cell type. These findings underscore the importance of interrogating metabolism at a single-cell level and illustrate the power of RNA-based ratiometric sensors to identify previously unanticipated heterogeneity in cellular metabolic networks.
METHODS
Reagents and equipment
Unless otherwise stated, all reagents were from Sigma-Aldrich except for cell culture reagents, which were from Invitrogen. These reagents were used without further purification. DFHBI-1T, DFHO and BI fluorophores were obtained from Lucerna Technologies (New York, NY) or were synthesized as described previously22,23,33. DNA libraries were ordered from the Keck Oligonucleotide Synthesis facility (Yale University) and primers were ordered from Integrated DNA Technologies (IDT). Absorbance spectra were recorded with a Thermo Scientific NanoDrop 2000 spectrophotometer with cuvette capability. ChemiDoc MP imager (Bio-Rad) was used to record bacterial colony fluorescence on agar plates as described previously21 and images were collected using Image Lab (ver. 5.2.1). Fluorescence was measured on FluoroMax-4 fluorimeter (Horiba Scientific) using FluorEssence (ver. 3.5). Fluorescence was also measured on Spectramax iD3 plate reader (Molecular Devices) using SoftMax Pro (ver. 7.1). FACS experiments were performed using BD FACSAria II instrument (BD Biosciences) and flow cytometry was performed using BD LSRFortessa (BD Biosciences). Sorting and flow cytometry data was collected using BD FACSDiva (ver. 8.0.1) software and analyzed using FlowJo (ver. 10.7.1). Fluorescence imaging experiments were performed using an Eclipse Ti-E microscope (Nikon) and images were collected using NIS Elements (ver. 3.22.15). Data were plotted using Sigmaplot (ver. 10.0) and GraphPad Prism (ver. 9.2.0).
Preparation of affinity matrix
DFHBI and DFHO affinity matrix were prepared as described before20,23. Amine-functionalized DFHBI or DFHO (50 mM in DMSO) was diluted to 5 mM in 100 mM HEPES-KOH buffer (pH 7.5). 2 mL of the fluorophore solution was then added to 1 mL of NHS-activated Sepharose (GE Life Sciences) beads, which had been washed with 2 × 1 mL of cold reaction buffer. The beads were then incubated with amine-functionalized fluorophore solution overnight at 4oC in the dark with gentle agitation. Then the beads were washed with the reaction buffer to remove the unreacted DFHBI or DFHO and incubated with 5 mL of 100 mM Tris.HCl (pH 8.0) for 2 h at room temperature to inactivate any remaining NHS-activated sites. After thorough washing with the reaction buffer and then with water, the beads were stored in 1:1 ethanol:0.1 M sodium acetate (pH 5.2) at 4˚C. The efficiency of the coupling was calculated by quantifying the amount of free DFHBI or DFHO in the flow-through using absorbance. Using this approach, we estimated that the Sepharose beads contain approximately 5 μmol of DFHBI or DFHO per mL of resin after the coupling reaction.
SELEX procedure
The single-stranded sprouts and clips random DNA library (Fig. 1a and Supplementary Table 5a) was purified using denaturing PAGE (8%, 7 M urea). See Supplementary Note for calculation of theoretical diversity of this library. ~1 nanomole of purified ssDNA library was amplified for five cycles in 10 mL final volume (divided in a 96 well plate) using Taq DNA polymerase with Thermopol buffer (New England Biolabs) to generate the dsDNA template for in vitro transcription.
~6 × 1014 different sequences of dsDNA template were transcribed in 250 μL T7 RNA polymerase transcription reaction using the AmpliScribe T7-Flash Transcription Kit (Lucigen). After treatment with DNase I (30 min at 37 °C), the RNA was purified using RNA Clean and Concentrator 100 columns (Zymo Research).
SELEX was performed essentially as described before20,21 using a single selection buffer for every round (40 mM HEPES-KOH (pH 7.4), 100 mM KCl and 5 mM MgCl2). First, library members capable of binding to the Sepharose beads were pre-cleared by incubation with “mock” beads consisting of aminohexyl-functionalized Sepharose. The mock beads (300 μL) were washed and then equilibrated in the selection buffer. ~2 nanomole of the library RNA was diluted in 500 μL of selection buffer containing 0.1 mg/mL of yeast transfer RNA (tRNA) and incubated with the mock beads for 30 min with gentle agitation. After collecting the flow-through, the mock beads were washed with 500 μL of selection buffer containing 0.1 mg/mL of yeast tRNA and the flow-through was collected again. The combined flow-through from both the steps was then incubated with pre-equilibrated DFHBI-functionalized beads (300 μL) for 30 mins with gentle agitation. Then the DFHBI-functionalized beads were washed with 3× 500 μL of selection buffer. Finally, RNAs that specifically bound to DFHBI were eluted with 1 mM DFHBI-1T.
The eluted RNA was ethanol precipitated using NaOAc (0.3 M final), glycoblue (0.1 mg/mL final) and 75% ethanol (final). Then the RNA was pelleted by centrifugation at 20000xg for 30 min after overnight incubation at −20oC. Then the RNA was reverse-transcribed using Superscript IV reverse transcriptase (Invitrogen) following manufacturer’s protocol in a total volume of 30 μL. The entire 30 μL reverse-transcription reaction was PCR-amplified in a total volume of 300 μL using standard Taq DNA polymerase conditions with Thermopol buffer (New England Biolabs). The PCR reaction was purified using QIAquick PCR Purification Kit (Qiagen) and used for in vitro transcription to generate the RNA pool for the next round. Selection pressure was exerted on the later rounds by more stringent washing (see Supplementary Table 3 for SELEX conditions). The presence of fluorescent RNA species in each round was assessed by mixing 20 μM RNA pool and 10 μM DFHBI-1T and measuring fluorescence emission of this solution on a fluorimeter in comparison with the fluorophore alone. After round 7, the RNA library were cloned into bacterial expression plasmid for FACS-based screening (described below).
Directed evolution of fluorogenic aptamers
The directed evolution using doped library was conducted as described in details previously21. In brief, these libraries were created in a way that each library member resembles the parental aptamer 9–1, except that there are on average eight mutations per sequence (Supplementary Table 5c). In order to obtain this library, every mutagenized position is chemically synthesized with a phosphoramidite mixture that contains primarily the original nucleotide at that position, but also contains a small fraction of the other three nucleotides21. After each selection, the in vitro fluorescence of individual library members were measured to select the candidate for the next round of directed evolution (Extended Data Fig. 1b-e and Supplementary Figure 3). During the directed evolution, selection pressure was exerted by lowering the magnesium concentration in selection buffer and increasing the temperature of the wash buffer (Supplementary Table 4a). A second directed evolution was performed using a doped library based on DE1–2 aptamer (Supplementary Table 5e) for both DFHBI and DFHO (Supplementary Table 4b). However, we obtained the same hits for both DFHBI- and DFHO- based directed evolution (Supplementary Table -5f). Finally the DE2–6 aptamer selected from the second directed evolution was further optimized by introducing rational structure-based mutations (Extended Data Fig. 1f,g)
Bacterial library generation and FACS sorting
The RNA pool after seven rounds of SELEX on the ‘sprouts and clips’ random library, or the RNA pool after four rounds of SELEX on the doped library, were reverse transcribed and PCR amplified. Then these PCR products were cloned into pET28c F30-Broccoli plasmid using BglII and NheI sites using T4 DNA ligase (New England Biolabs).
The resulting ligation mixtures were purified and electroporated into BL21 DE3 E. coli (Lucigen). These cells then were grown in LB media with kanamycin. Typical bacterial libraries contain 5–20×106 individual members. Next day the cells were diluted 1:10 in LB and induced for RNA expression with 1 mM IPTG for 3 h. Cells were preincubated with either 20 μM DFHBI-1T or 10 μM DFHO (in 1x PBS) and then sorted on a FACSAria II instrument. The sample holder of the sorter was maintained at 37oC to sort cells expressing thermostable aptamers. For DFHBI-1T-binding aptamers, cells were excited with the 488 nm laser and their emission was collected using 525±25 nm emission filter. To isolate yellow fluorescent aptamers, cells were excited with the 488 nm laser and emission was collected using 545±17.5 nm emission filter. Cells were sorted using arbitrary gates where roughly five hits are obtained in 10000 sorted cells. The hits from sorting were collected into 1 mL SOC media and recovered by shaking at 37oC for 1 h. Then the cells were plated on LB-agar supplemented with kanamycin and 10 μM DFHBI-1T or 5 μM DFHO.
The next day, the bacterial colonies were induced with 1 mM IPTG for 3h and imaged on a ChemiDoc MP imager using either green fluorescence (470 ± 15 nm ex; 532 ± 14 nm em), or yellow fluorescence (530 ± 14 nm ex; 605 ± 25 nm em). To normalize the colony size, autofluorescence signal from bacterial colonies were collected using the Cy5 channel (630 ± 15 nm ex; 697 ± 22.5 nm emission). Images were processed and normalized in ImageJ (NIH) to identify colonies expressing the brightest aptamers.
In vitro characterization of aptamers
After sequencing the plasmids from the brightest colonies, dsDNA templates (containing T7 promoter) were generated using PCR amplification. To generate truncation, deletion and point mutants, dsDNA templates containing T7 promoter were PCR amplified from the appropriate ssDNA sequences (IDT). PCR products were then purified with QIAquick PCR Purification Kit (Qiagen) and transcribed using AmpliScribe T7-Flash Transcription Kit (Lucigen). After treatment with DNase I (30 min at 37 °C), the RNA was purified using RNA Clean and Concentrator 25 columns (Zymo Research).
All RNAs used for in vitro studies (except the initial hits from SELEX and the DE2–6 mutants) were purified using 8% denaturing PAGE (7M urea) and eluted in 0.3 M NaOAc (pH 5.2) overnight. The RNA was ethanol precipitated and quantified using absorbance in a NanoDrop 2000 spectrometer. All in vitro RNA properties were measured in 40 mM HEPES-KOH (pH 7.4), 100 mM KCl, 0.5 mM MgCl2 buffer unless specified.
Absorption, excitation and emission spectra were measured using conditions where the RNA is in excess and the fluorophore is limiting to ensure that no free fluorophore contributes to the absorbance or fluorescence signal (Extended Data Fig. 2a-d). This approach also allows us to have a fixed concentration of RNA-fluorophore complex which is equal to the concentration of the fluorophore that was added34.
For potassium-dependence assay, separate buffers were prepared for K+, Na+ and Li+. HEPES solution was neutralized with KOH, NaOH or LiOH to generate a 1 M solution (pH 7.4). RNA (1 μM final) was diluted in a buffer containing 40 mM HEPES (pH 7.4) and 0.5 mM MgCl2. Then the salts (KCl, NaCl or LiCl) were added to a final concentration of 100 mM to the corresponding solutions. Finally, DFHBI-1T or DFHO was added to the RNA solution at a final concentration of 10 μM and incubated for 20 min. The fluorescence was measured on a FluoroMax-4 fluorimeter.
To measure magnesium dependence, 1 μM RNA was incubated with 10 μM DFHO in 40 mM HEPES-KOH (pH 7.4), 100 mM KCl buffer with different concentrations of MgCl2 for 20 min and then fluorescence emission was measured on a on a FluoroMax-4 fluorimeter.
To measure the thermostability RNA-fluorophore complexes, 1 μM of RNA was incubated with 10 μM fluorophore. Then the fluorescence values were recorded in 3°C increments from 19°C to 61°C, with 5-min incubation at each temperature to allow for equilibration.
All quantum yields were determined by comparing each RNA-fluorophore complex with Broccli-DFHBI-1T or Corn-DFHO. All measurements for quantum yield were taken in the presence of excess RNA (10 μM final) compared to the fluorophores (0.2 μM final) to avoid interference from unbound fluorophore. The integral of the emission spectra for each Squash-fluorophore complex was compared with the corresponding integrals for Broccoli or Corn to calculate the quantum yield.
Affinity measurements
Dissociation constants (Kd) for the RNA-fluorophore complexes were determined as described previously20,21. In brief, the RNA aptamer at a fixed concentration (50 nM) was titrated with increasing fluorophore concentration and the resulting increase in fluorescence was recorded. For each fluorophore concentration, a background signal for fluorophore only solution was also measured separately and subtracted from the measured RNA-fluorophore signal. Data was fitted to a single site saturation model using nonlinear regression analysis in Sigmaplot software.
In vitro folding of RNAs
Folding measurements were performed essentially as described before34. Briefly, the fluorescence intensities of two solutions were compared: one having excess fluorophore and limiting RNA, and the other with excess RNA and limiting fluorophore. As we described previously, this allows us to calculate the percent of the aptamer that is folded. For these experiments, we used 0.2 μM of fluorophore (or RNA) and 10 μM of RNA (or fluorophore). The signal from the limiting RNA condition was divided by the signal from the limiting fluorophore condition to determine the fraction folded.
Measurement of on and off rates for Squash-DFHO pair
To measure on and off rates of Squash binding to DFHO, we followed the same protocol that was reported previously to measure kinetic rates for Spinach-DFHBI50. Briefly, 50 nM RNA was mixed with different amounts of DFHO and binding kinetics was monitored on the fluorimeter as fluorescence signal increases. Each fluorescence signal trace was fitted with a monoexponential curve and the observed rate constant (kobs) was extracted. Then kobs values were plotted as a function of total RNA and fluorophore concentration. The resulting points were fitted with a line. This linear fit allowed extraction of the binding rate constant (kon) and the unbinding rate constant (koff) as the slope and intercept, respectively (Extended Data Fig. 2f). The koff value calculated for Squash-DFHO (0.014 ± 0.008 s−1) is very similar to that of Corn-DFHO (0.018 ± 0.002 s−1). However, the association rate constant (kon) for Squash-DFHO pair (162300 ± 8100 M−1 s−1) is seven times higher than that of Corn-DFHO pair (23000 ± 3000 M−1 s−1) which could be due to the highly folded nature of Squash aptamer.
SELEX for generating Squash-SAM sensors
The single stranded sprouts and clips transducer DNA library (Supplementary Table 5h) used for our sensor SELEX was synthesized by Keck Oligo Synthesis facility at Yale University. RNA library was generated as described previously for regular SELEX. The SELEX procedure was conducted to isolate RNAs which shows conditional folding of Squash upon binding SAM (Extended Data Fig. 6). Similar methods are also described in the literature51,52. The SELEX was performed using a single selection buffer for every round (40 mM HEPES-KOH (pH 7.4), 100 mM KCl and 0.5 mM MgCl2). During the first step, RNA species capable of binding to the Sepharose-DFHO beads without SAM (constitutively fluorescent) were removed by incubation with the beads. Approximately 0.5 nanomole of the library RNA was diluted in 300 μL of selection buffer containing 0.1 mg/mL of yeast tRNA and incubated with pre-equilibrated Sepharose-DFHO beads (50 μl) for 30 min with gentle agitation. After collecting the flow-through, the beads were washed with 300 μL of selection buffer containing 0.1 mg/mL of yeast tRNA and the flow-through was collected again. The combined flow-through from both the steps were mixed with SAM (0.1 mM final) and incubated for 10 min to induce folding of Squash. Then this solution was incubated with pre-equilibrated DFHO-functionalized beads (50 μL) for 30 mins with gentle agitation. Then the beads were washed gently with 3× 300 μL of selection buffer containing 0.1 mM SAM. Finally, sensor RNA was eluted with SAM free selection buffer (2 × 200 μL).
The eluted RNA was treated as previously to generate the RNA pool for the next round. Selection pressure was exerted on the later rounds by more stringent washing (5 × 200 μL for 2nd and 7 × 200 μL for 3rd round) with SAM containing buffer. To monitor the progress of SELEX, 2 μM of the RNA pool after each round of SELEX was mixed with 10 μM of DFHO and fluorescence was measured (ex = 495 nm, em = 562 nm) in the absence and presence of 0.1 mM SAM. After three rounds, RNA library members were cloned using TOPO cloning and 48 colonies were picked in random from two agar plates.
dsDNA templates from the selected bacterial colonies were PCR amplified from purified plasmid and the RNA was generated by in vitro transcription. For each library member, 1 μM of the RNA was mixed with 10 μM of DFHO in the absence and presence of 0.1 mM SAM and put into separate PCR tubes. The solutions were heated at 37˚C for 10 min and immediately imaged using a BioRad ChemiDoc MP imager (ex: 530 ± 14 nm, em = 605 ± 25 nm). Library members which showed substantial SAM dependent fluorescence enhancement were used for further characterization.
In vitro characterization of the sensors
For all in vitro measurements, the sensor RNAs (Supplementary Table 5i) were purified using 8% denaturing PAGE (7 M urea) as described above. To test the ability of each indicated transducer sequence to mediate SAM-induced fluorescence, 1 μM of the RNA was mixed with 10 μM of DFHO in the absence or presence of 0.1 mM SAM in a buffer containing 40 mM HEPES-KOH (pH 7.4), 100 mM KCl and 0.5 mM MgCl2. After 1 h incubation at 37˚C, fluorescence signal of each sample was measured at 37˚C using a Fluoromax-4 fluorimeter with 495 nm excitation and 562 nm emission. For both conditions, background signal (for 10 μM DFHO only) was deducted from the signal obtained for the sensor.
To measure the activation rate of the Squash-SAM sensors, a solution containing 1 μM sensor RNA and 10 μM DFHO (in buffer containing 40 mM HEPES-KOH (pH 7.4), 100 mM KCl and 0.5 mM MgCl2) was incubated at 37˚C for 30 min. Then SAM (0.1 mM final) was quickly added to the stirring RNA solution, and fluorescence was recorded over 20 min at 1 min intervals at 37˚C (495 nm ex; 562 nm em). The fluorescence signal was normalized to the intensity at 20 min (100) and intensity at 0 min (0).
To measure the deactivation rate of the sensors, 1 μM sensor RNA and 10 μM DFHO was incubated with 0.1 mM SAM (in buffer containing 40 mM HEPES-KOH (pH 7.4), 100 mM KCl and 0.5 mM MgCl2) for 1 h at 37˚C. Then this solution was buffer-exchanged with the same buffer used above (without SAM) using a Micro Bio-Spin Column with Bio-Gel P-30 beads (Bio-Rad) to remove SAM. DFHO (10 μM final) was added to the collected flow-through and the fluorescence emission was recorded as with the activation experiments. The fluorescence measurement was normalized to the intensity at 0 min (100) and the intensity at 20 min (0).
Dissociation constants (Kd) for the sensor complexes were determined as described previously8. For measurement of sensor-DFHO affinity, a solution containing the sensor RNA (100 nM final) and 0.1 mM SAM was titrated with increasing DFHO concentration and the resulting increase in fluorescence was recorded (Extended Data Fig. 5d,g). For each DFHO concentration, a background signal for DFHO only solution was also measured separately and subtracted from the signal measured for RNA-DFHO signal. For measuring Kd of the sensors to SAM, a solution containing the sensor RNA (1 μM final) and DFHO (10 μM final) was titrated with increasing concentration of SAM and the resulting increase in fluorescence was recorded (Extended Data Fig. 5e,h). The fluorescence was measured at 37˚C using Spectramax iD3 plate reader with 495 nm excitation and 562 nm emission. For each concentration of SAM measured, a background signal for no SAM sample was also measured separately and subtracted from the signal measured for RNA, DFHO and SAM together. In both cases, data was fitted to a single site saturation model using nonlinear regression analysis in Sigmaplot software.
Cloning of the plasmids for mammalian expression
U6 constructs were expressed from pAV-U6+27 plasmid, which expresses the 27 nt-leader sequence of the U6 small nucleolar RNA from the U6 promoter52. 5S constructs were expressed from pAV-5S plasmid, which expresses full length human 5S rRNA from its endogenous promoter. Different RNA constructs were fused to the 3ʼ end of either pAV-U6+27 or pAV-5S. The sequence encoding the constructs were amplified by PCR and then digested with XbaI and SalI and inserted into pAV-U6+27 or pAV-5S plasmids (digested with the same restriction enzymes) using T4 DNA ligase.
In the case of preparing pAV-Tornado aptamers and sensors, dsDNA templates containing the appropriate sequences were prepared with flanking NotI and SacII restriction sites. Then the appropriate sequences were inserted in a clonable Tornado expression cassette on a pAV-U6+27 vector through cloning using NotI and SacII restriction sites11. For the ratiometric sensors, the Squash-SAM sensor was inserted into one arm of an F30 scaffold and Broccoli aptamer was inserted into the other arm (Supplementary Table 5j). dsDNA sequence for the ratiometric sensors flanked by NotI and SacII restriction sites were generated using PCR and cloned into the pAV-Tornado plasmid as described above.
Cell culture conditions
All cell lines except mES cells were obtained directly from the American Type Culture Collection (ATCC). HEK293T (ATCC CRL- 3216) were cultured in DMEM media (Life Technologies, no. 11995–065) with 10% FBS, 100 U/mL penicillin and 100 μg/mL of streptomycin under standard tissue culture conditions (at 37˚C and 5% CO2). HCT116 cell s (ATCC CCL-247) were cultured RPMI 1640 media (Life Technologies, no. 11875–093) with 10% FBS, 100 U/mL penicillin and 100 μg/mL of streptomycin under standard tissue culture conditions. Cells were screened for mycoplasma contamination before passaging using Universal Mycoplasma Detection Kit (ATCC 30–1012K) according to ATCC recommendations.
Mouse embryonic stem cells (mES cells) were previously generated from C57BL/6 × 129S4/SvJae F1 male embryos53. After thawing the cells, they were cultured in gelatin-coated plates using proprietary Knockout DMEM formulation supplemented with 15% heat-inactivated FBS (Corning, 35–010-CV), 1x GlutaMAX (Life Technologies, no. 35050–061), 1 x Non-essential amino acids (Life Technologies no. 11140–050), β-ME (55 μM final), and 1000 units/mL of LIF (Millipore Sigma no. ESG1107). Because Knockout DMEM media is proprietary, it is not possible to generate customized media lacking certain ingredients. Therefore, after culturing the mES cells in Knockout DMEM for two passages, the mES cells were cultured in an experimental media53 containing a 1:1 mix of glutamine-free DMEM (Life Technologies no. 11960–069) and glutamine-free Neurobasal medium (Life Technologies no. 21103–049) supplemented with 10% heat-inactivated FBS (Corning, 35–010-CV), 1x GlutaMAX (Life Technologies, no. 35050–061), β-ME (55 μM final), and 1000 units/mL of LIF (Millipore Sigma no. ESG1107). This media described here is designated −2i media. To generate the +2i media, the −2i media was supplemented with a MEK inhibitor (PD0325901) and a GSK3β inhibitor (CHIR99021) with final concentration being 1 μM and 3 μM respectively. To dissociate the mES cells from plates during passages we used StemPro Accutase (Life Technologies, no. A1110501).
Media preparation for imaging
For imaging experiments media containing no phenol red were used to reduce background fluorescence. For imaging HEK293T cells, we used Flurobrite DMEM (Life Technologies, no. A1896701) supplemented with 10% FBS, 1x GlutaMAX, 100 U/mL penicillin, and 100 mg/mL streptomycin. For imaging in HCT116 cells, RPMI 1640 media lacking amino acids, glucose, and glutamine (MyBioSource, no. MBS653421) was used as starting point3. This media was supplemented with 1x Minimal Essential Media Non-essential Amino Acids (MEM NEAA), 5 mM glucose, 1x GlutaMAX, 10% dialyzed FBS (Gemini, no. 100–108), 100 U/mL penicillin, and 100 μg/mL streptomycin. Essential amino acids, except methionine and threonine, were added back at the same concentrations found in MEM amino acids solution to generate the methionine and threonine free media. Methionine (100 μM final) and threonine (400 μM final) were added to this media as required for the amino acid withdrawal experiments.
For mES cells, a custom version of the DMEM and Neurobasal media each lacking methionine, threonine and phenol red were prepared by the Media Preparation core at Sloan Kettering Institute. The −2i imaging media (without methionine and threonine) was prepared mixing these two media in 1:1 ratio and supplemented with 10 % dialyzed FBS (Gemini, no. 100–108), 1x GlutaMAX (Life Technologies, no. 35050–061), β-ME (55 μM final), and 1000 units/mL of LIF (Millipore Sigma no. ESG1107). Methionine (200 μM final) and threonine (800 μM final) were added to this media as required for the amino acid withdrawal experiments. To generate the +2i imaging media (without methionine and threonine), the −2i imaging media was supplemented with the MEK inhibitor (PD0325901) and the GSK3β inhibitor (CHIR99021) with final concentration being 1 μM and 3 μM respectively. Again methionine (200 μM final) and threonine (800 μM final) were added to this media as required for the amino acid depletion experiments.
Flow cytometry of mammalian cell
HEK293T cells were plated in 12-well plates and transfected with the appropriate RNA expressing plasmid next day using FuGENE HD (Promega) following manufacturer’s recommendation. After 36 h, the cells were washed with 1X PBS once, dissociated from the plate using TrypLE Express Enzyme (Life Technologies, no. 12604013), re-suspended in 4% FBS/1X PBS solution containing 10 μM DFHBI-1T or 5 μM DFHO as required, and kept on ice until analysis on the BD LSRFortessa instrument. Transfected cells were analyzed in green channels (ex=488 nm, em= 525 ± 25 nm) for DFHBI-1T and orange channel (ex=488 nm, em= 570 ± 20 nm) for DFHO. For Corn-DFHO, a yellow channel (ex 488 nm, em 545 ± 17.5 nm) was used due to blue shifted emission of Corn. An auxiliary far-red channel (ex=635 nm, em=780 ± 30 nm) was used to measures cellular auto-fluorescence. Processing and analysis of the data was performed in the FlowJo software.
Imaging aptamer-tagged RNAs and SAM sensors in HEK293T cells
Cell imaging for HEK293T cell was carried out as described previously23. HEK293T cells were plated on poly-D-lysine and mouse laminin-coated glass-bottom 24-well plates (MatTek). The next day, cells were transfected with the appropriate plasmid using FuGENE HD reagent (Promega). After 36 h, cells were washed with 1x PBS once and incubated with the imaging media described above containing appropriate fluorophore. After 30 min incubation in the incubator, Hoechst 33342 was added to a final concentration of 0.1 μg/mL.
The 24-well plate was transferred to a Tokai Hit stage top incubator (37 °C and 5% CO2) and live fluorescence images were taken with a Nikon Eclipse Ti-E microscope fitted with a CoolSnap HQ2 CCD camera (Photometrics) through a 40X air objective (NA 0.75) and analyzed with the ImageJ software to detect Broccoli (470 ± 20 nm ex; 495 nm dichroic; 525 ± 25 nm em), Corn (500 ± 12 nm ex; 520 nm dichroic; 542 ± 13.5 nm em), and Squash (500 ± 12 nm ex; 520 nm dichroic; 570 ± 20 nm em), and Hoechst (350 ± 25 nm ex; 400 nm dichroic; 460 ± 25 nm em).
Ratiometric Imaging of SAM in mammalian cells
For experiments in HEK293T cells, cell culture and transfection was performed as described above. HCT116 cells were plated on 24-well ibiTreat μ-Plate (ibidi GmbH, Germany) coated with poly-D-lysine and mouse laminin. For mES cells, cells were plated on 24-well ibiTreat μ-Plate coated with gelatin. For both of these cell lines, plasmids encoding RNA sensors were transfected using Lipofectamine Stem transfection reagent (Life Technologies, no. A1896701). Approximately 36 h after transfection, cells were washed with 1x PBS once and then incubated in the appropriate imaging media described above supplemented with required fluorophore(s). For imaging with the ratiometric sensors, 10 μM DFHO and 5 μM BI were used.
After placing the plates on a Tokai Hit stage top incubator (37 °C and 5% CO2), live fluorescence images were taken with a Nikon Eclipse Ti-E microscope fitted with a CoolSnap HQ2 CCD camera (Photometrics) through a 40X air objective (NA 0.75). For live cell imaging over long time period, the perfect focus system (PFS) was used to avoid focal drifts. For Squash-SAM sensors we used a modified YFP filter cube (500 ± 12 nm ex; 520 nm dichroic mirror; 570 ± 20 nm em). For the ratiometric sensors custom designed filter cubes were used to avoid bleed through of one channel into the other. For the green channel (Broccoli-BI), we used a filter cube with 460 ± 7 nm excitation filter, 473 nm dichroic mirror, and 500 ± 12 nm emission filter. For the orange channel (Squash-DFHO), we used a filter cube with 512 ± 12.5 nm excitation filter, 532 nm dichroic mirror, and 575 ± 29.5 nm emission filter. Hoechst-stained nuclei were imaged as described above. For ratiometric SAM imaging, we used 500 ms exposure time for the orange channel and 100 ms exposure time for the green channel.
Image analysis for generating SAM trajectories
Image analysis was performed in ImageJ (FIJI) ver. 1.53c. For the single-color SAM sensors, images analysis was performed as before8. Briefly, images obtained at each time points were background subtracted and the mean intensity of each cell at time 0 was defined as 100. The mean fluorescence intensity at any other time point is normalized to the value at time 0 and normalized mean fluorescence of each cell was plotted as a function of time.
For the ratiometric sensor, it is important to generate the orange to green (O/G) fluorescence ratio only for areas containing the cells. For each time points, images from both channels were background subtracted. Then the constitutively fluorescent green channel was used to create a binary mask to identify the cells. This mask was multiplied with the background subtracted images from each channel to generate masked images for each channel. Finally, images containing O/G fluorescence ratio was generated by dividing the masked orange channel image with masked green channel image. The ratio in the image is coded by pseudocolor.
Quantification of SAM using biochemical assay
SAM concentration in HEK293T cells were measured as described previously38 using a commercially available kit (Bridge-It S-adenosylmethionine (SAM) fluorescence assay kit, Mediomics, USA). For cycloleucine treatment experiments, SAM was extracted from HEK293T cells at each time points using 80% methanol as described previously38,55. After evaporating the liquid, the cell extract was dissolved in 25 μL of water. The SAM concentration was then determined according to the manufacturer’s protocol. Briefly, a standard curve was generated using standard solutions of SAM supplied with the kit. Then the concentration of SAM in the cellular extract was determined at each time point using the standard curve. Finally, the cellular concentration of SAM was determined with the assumption that cells are spherical with approximate diameter of 17 μm giving us a specific volume for each cell.
Statistics and Reproducibility
All data are expressed as means ± s.d. with the number of independent experiments (n) listed for each experiment. Statistical analyses were performed using Excel (Microsoft) and Prism (GraphPad). Experiments which show micrographs were repeated independently at least thrice and showed similar results. This applies to Fig. 3c, 4g, Extended Data Fig. 4a,c, 7d-f, 9a-d, 10a-d and Supplementary Figure 5a,b.
Data availability
All data supporting the findings of this study are available within the paper and its Supplementary Information files. They are also available from the corresponding author on reasonable request. The following plasmids generated in this study will be available through Addgene: pAV-U6+27-Squash (ID 177913), pAV-5S-Squash (ID 177914), pAV-U6+27-Tornado-Squash (ID 177915), pAV-U6+27-Tornado-Squash-SAM-sensor 4–2 (ID 177916), pAV-U6+27-Tornado-Squash-SAM-sensor 5–1 (ID 177917), pAV-U6+27-Tornado-F30-Squash-Broccoli (ID 177918), pAV-U6+27-Tornado-F30-Broccoli-Squash-SAM-sensor 4–2 (ID 177919), pAV-U6+27-Tornado-F30-Broccoli-Squash-SAM-sensor 5–1 (ID 177920).
Code availability
The custom code used to analyze the “sprouts and clips” library is deposited in Bitbucket. It can be accessed through the following link: https://bitbucket.org/jaffrey_lab_wcm/ncb_2021_dey_squash/src/master/
Extended Data
Extended Data Fig. 1. Characterization of the fluorescence properties of different intermediates during development of Squash.

a, Fluorescence activation of DFHO by Squash and its precursors. Fluorescence was measured with 200 nM dye and 10 μM RNA (ex: 495 nm, em: 562 nm, except for Corn; ex: 505 nm, em: 545 nm). Data represent mean values ± s.d. for n=3 independent experiments. Squash showed >2-fold activation of DFHO compared to Corn.
b, Fluorescence of initial hits after Round 7 of SELEX. In vitro transcribed RNAs from each hit (1 μM final) were mixed with DFHBI-1T (10 μM final) and the fluorescence was measured (ex: 452 nm, em: 503 nm, except for Broccoli; ex: 472 nm, em: 507 nm). Data represent mean values ± s.d. for n=3 independent experiments.
c,d, Fluorescence measurements of the hits with DFHBI-1T after first (c) and second (d) round of directed evolution, measured as in panel b. Data represent mean values ± s.d. for n=3 independent experiments.
e, Fluorescence measurements of hits using DFHO after the second round of directed evolution measured as in panel a. The fluorescence of the hits were normalized against Corn. Data represent mean values ± s.d. for n=3 independent experiments.
f, DE2–6 has one bulged nucleotide (U51) and one G•U pair based on mFold (Supplementary Figure 3d). Elimination of the bulge, the G•U pair, or both did not improve fluorescence of DE2–6. Fluorescence was measured as in a. Data represent mean values ± s.d. for n=3 independent experiments.
g, Mutation (G30C) of the kissing loop of DE2–6 resulted in loss of fluorescence which was recovered by a complementary mutation (C57G) in the opposite loop of the predicted kissing loop. Similar results were also observed for the other kissing loop basepair. Two mutations (U29C, U58G) created an extra G•C basepair in the kissing loop compared to add A-aptamer and resulted in ~20% increase in fluorescence of DE2–6. Further improvement of the kissing loop interaction cannot be achieved by further mutations (A28G + U59C in Squash). Data represent mean values ± s.d. for n=3 independent experiments.
h, Although Broccoli-BI and Squash-DFHO (20 μM RNA, 2 μM dye) excitation spectra overlap, their emission maxima show ~57 nm separation. Structure of BI is shown in the inset.
Extended Data Fig. 2. Photophysical and biophysical characterization of Squash.

a, Absorbance spectra (50 μM RNA, 5 μM DFHBI-1T) of DFHBI-1T alone and in complex with Squash demonstrates a smaller red shift (Abs max: 450 nm) compared to Spinach (Abs max: 470 nm)20 and Broccoli (Abs max: 469 nm)21. Excess RNA was used to ensure binding of nearly all fluorophore.
b, DFHO shows a red-shifted absorbance spectrum upon binding Squash (50 μM RNA, 5 μM DFHO).
c, Both Squash and Broccoli shows similar emission maxima. Squash also showed higher fluorescence (10 μM RNA, 1 μM DFHBI-1T, ex. 452 nm for Squash and 472 nm for Broccoli) intensity with DFHBI-1T compared to DE2–6.
d, Fluorescence spectra (10 μM RNA, 1 μM DFHO, ex. 495 nm for Squash and 505 nm for Corn) showed a red-shifted Squash emission maxima (562 nm) compared to Corn (545 nm), consistent with a different mode of interaction between DFHO and Squash compared to Corn. Squash also showed more than two-fold higher fluorescence intensity with DFHO compared to Corn.
e, Kd was measured by titration of 50 nM RNA with DFHBI-1T and then fitting the data using a one-site saturation model. Data represent mean values ± s.d. for n=3 independent experiments.
f, Binding kinetics were performed as reported previously50. The fluorescence signal trace was fitted with a monoexponential curve to extract kobs. kobs was plotted as a function of total RNA (50 nM RNA) and fluorophore concentration. kon and koff were extracted as the slope and intercept, respectively. Squash koff (0.014 ± 0.008 s−1) is very similar to that of Corn (0.018 ± 0.002 s−1). kon for Squash-DFHO (162300 ± 8100 M−1 s−1) is seven-times higher than Corn-DFHO (23000 ± 3000 M−1 s−1) which could be due to the high folding of Squash.
g,h, Kd measurements for Broccoli and Squash binding to BI (g) and DFHO (h) were performed as in panel e, using 50 nM RNA and then fitting the data using a one-site saturation model. Data represent mean values ± s.d. for n=3 independent experiments.
Extended Data Fig. 3. Comparison of in cell photostability of Squash with other fluorogenic aptamers.

a, Comparison of in cell photostability of Squash-DFHBI-1T pair with Broccoli-BI. Compared to DFHBI-1T, BI shows improved photostability when bound to Broccoli33. We compared the photostability of Squash-DFHBI-1T with Broccoli-BI. Aptamers were expressed as circular RNAs11 and aptamer-expressing cells were incubated with dyes (10 μM). Continuous images (100 ms per frame) were taken for 10 s total while the cells were continuously illuminated. We used the highest available light power in the microscope since previously lower light levels were used and Broccoli-BI photobleaching was not readily detected33. Shown are every other frame starting from 0 ms to 1000 ms. Scale bar, 20 μm.
b, Comparison of in cell photostability of Squash-DFHO with Corn-DFHO. Corn aptamer exhibits high photostability with DFHO compared to Spinach or Broccoli with DFHBI-1T23. We compared the photostability of Squash-DFHO with tCorn (Corn in the tRNA scaffold)-DFHO. The images were taken in as in panel a except the exposure time was 500 ms per frame. Shown are frame between 0 and 2500 ms. Scale bar, 20 μm.
c, Quantification of in cell photostability for green emitting aptamers. Cellular mean fluorescence intensity was calculated and normalized to maximum intensity at 0 s. The normalized cellular fluorescence intensities were plotted against time. Both Broccoli-BI and Squash-DFHBI-1T showed a similar initial rate of drop in fluorescence, which reflects the rate of light-induced photoisomerization of the fluorophore to the low-fluorescence trans-isomer33. The higher plateau associated with Squash-DFHBI-1T likely reflects a higher on-rate for fluorophore binding since the plateau reflects the balance between light-induced isomerization/trans-fluorophore dissociation and cis-fluorophore rebinding33.
d, Quantification of in cell photostability for yellow-orange emitting aptamers. Assays were performed as in c. For comparison, we used mVenus, a fluorescent protein which has similar fluorescence emission properties as Corn-DFHO.
Extended Data Fig. 4. Squash exhibits higher cellular fluorescence than Corn when expressed as a circular RNA.

a, Comparison of in-cell fluorescence intensity of circular Squash or circular tCorn (tRNA-scaffolded Corn). HEK293T cells expressing 5S-control (no aptamer), Tornado-tCorn or Tornado-Squash plasmids were imaged using 10 μM DFHO and the same microscope settings. Cells expressing circular Squash exhibited much brighter cellular fluorescence than cells expressing circular tCorn. Circular Squash showed mostly nucleus-excluded signal. Scale bar, 20 μm.
b, Comparison of cellular brightness of Squash and Corn using flow cytometry of HEK293T cells prepared as in a. Cells were analyzed in the yellow (ex 488 nm, em 545 ± 17.5 nm) fluorescence channel. An auxiliary far-red channel (ex 635 nm; em 780 ± 30) was used to measure cellular auto-fluorescence. Cells expressing circular Squash exhibited substantially more fluorescence.
c, To determine if the higher fluorescence of circular Squash in a and b was due to higher expression, 10 μg total RNA was isolated from HEK293T cells and analyzed by gel staining with DFHO and SYBR Gold as described previously36. Bands corresponding to circular Squash and circular tCorn in SYBR Gold staining were identified by comparing them with the 5S-control lane and marked by white stars. Quantification of the band intensities indicates slightly higher expression of circular tCorn. Note, unlike Squash, two tCorn molecules are required to bind a single DFHO molecule. The higher fluorescence of circular Squash-expressing cells compared to tCorn is probably due to higher quantum yield and improved folding of Squash. Additionally, the DFHO fluorescence intensity of the band corresponding to circular Squash is much higher than that of the circular tCorn band, suggesting that Squash also folds better in the gel than Corn.
Extended Data Fig. 5. Development and characterization of Squash-SAM sensors.

a, Squash was fused with the SAM-binding SAM-III aptamer through a sprouts and clips randomized transducer (NnNnNnNn for each strand). 2 μM of the RNA pool after each SELEX round was mixed with 10 μM DFHO and fluorescence was measured in the absence and presence of 0.1 mM SAM at 37 ˚C.
b, For each library member, 1 μM of the in vitro transcribed RNA was incubated with DFHO in the absence or presence of 0.1 mM SAM (37 ˚C, 10 min) and put into separate tubes and imaged immediately (ex: 530 ± 14 nm, em = 605 ± 25 nm). Squash-SAM sensors 4–2 and 5–1 are highlighted.
c,f, Squash-SAM sensor 4–2 (c) and 5–1 (f) are activated by SAM and not related molecules. 1 μM RNA was mixed with 10 μM DFHO and indicated molecules. Data represent mean values ± s.d. for n=3 independent experiments.
d,g, Measurement of Kd of Squash-SAM sensor 4–2 (d) and 5–1 (g) with DFHO were measured as in Extended Data Fig. 2e. The Kds are slightly weaker than that of Squash with DFHO (see Fig. 2d). Data represent mean values ± s.d. for n=3 independent experiments.
e,h, Measurement of Kd of Squash-SAM sensor 4–2 (e) and 5–1 (h) with SAM. Sensor RNA (1 μM) was titrated with SAM in presence of 10 μM DFHO (37˚C) and the resulting data was fitted using one-site saturation model. The Kd for the SAM-III riboswitch is ~1 μM. Data represent mean values ± s.d. for n=3 independent experiments.
i, Broccoli-BI fluorescence (1 μM Broccoli, 10 μM BI) is not affected by SAM or related molecules making it an appropriate normalizer for ratiometric sensor. Data represent mean values ± s.d. for n=3 independent experiments.
Extended Data Fig. 6. Detection of intracellular SAM levels using Squash-based SAM sensors.

a, Detection of intracellular SAM levels using the Squash-SAM sensor 4–2 and 5–1. HEK293T cells were transfected with the Tornado plasmids expressing the sensors or the constitutively fluorescent Squash aptamer (no SAM dependent fluorescence enhancement) as circular RNAs. Cells expressing the circular SAM biosensors or circular Squash were incubated with 10 μM DFHO and imaged every 10 min after treatment with 30 mM cycloleucine (an inhibitor of the major SAM biosynthesis enzyme, MAT2A) for 90 min total. Following this, cycloleucine was removed by replacing the imaging media with cycloleucine-free media. Images were taken again at 10 min intervals for 90 min. Images are shown at specific intervals along with results for representative cells. Cells expressing the circular sensors showed drop in the cellular fluorescence during cycloleucine treatment and increase in cellular fluorescence during cycloleucine withdrawal. However, cell expressing circular Squash did not show any change in cellular fluorescence during this treatment. Scale bar, 20 μm
b, c, Quantification of live-cell SAM levels based on Squash-SAM sensors 4–2 (b) and 5–1 (c) respectively. Cellular mean fluorescence intensity was calculated for each sensor and normalized to maximum intensity at time point 0. The normalized values of mean cellular fluorescence intensity were plotted against time at 10 min intervals for six different cells. Each cell is represented using a different color. For each sensor, SAM decay was observed following addition of cycloleucine and SAM recovery is seen after withdrawal of cycloleucine. Interestingly for both the sensors, the SAM decay profile for each individual cell is very similar while the SAM recovery profiles are distinctly different.
Extended Data Fig. 7. Broccoli-BI and Squash-DFHO can be used as an orthogonal pair of fluorogenic aptamers for two color imaging.

a, Lack of correlation of eqFP670 and Squash fluorescence expressed from the same plasmid (U6+27 promoter for Squash and PGK for eqFP670). Many HEK293T cells have high Squash fluorescence and low eqFP670 fluorescence (white arrows). Some cells have correlated fluorescence (gray arrows). Flow cytometry also revealed poor correlation. Scale bar, 20 μm.
B, Schematic representation of the circular F30-Broccoli-Squash RNA used as a constitutive control for the ratiometric sensor (Fig. 4f). The RNA was generated by fusing Squash and Broccoli to each arm of an F30 scaffold.
C, High correlation between Squash and Broccoli fluorescence when expressed within a single RNA. The F30-Broccoli-Squash construct (panel b) was expressed as a circular RNA in HEK293T cells and imaged in both green and orange channels after incubation with 10 μM BI and 10 μM DFHO. Excellent correlation was observed for each cell (see Fig. 4f, bottom panel for O/G ratio). This was also confirmed by flow cytometry.
d, Circular Broccoli-expressing cells showed negligible bleedthrough in the orange channel and circular Squash-expressing cells showed no bleedthrough in the green channel. Circular F30-Broccoli-Squash shows bright signal in both channels.
e, To determine if DFHO can compete with BI’s ability to bind Broccoli, we imaged HEK293T cells expressing circular Broccoli (10 μM BI) and compared green fluorescence before and after addition of DFHO (10 μM, 1 hr). The green fluorescence signal was not substantially affected. Similarly, BI does not affect Squash-DFHO fluorescence when circular Squash is expressed in HEK293T cells.
f, To test the possibility of FRET in the ratiometric sensor, we expressed ratiometric Squash-SAM sensor 5–1 in HEK293T cells as a circle. After incubating the cells with 10 μM BI, we took an image in the green channel. Then we added DFHO (10 μM, 1 h incubation)). No decrease in the Broccoli-BI (possible donor) green fluorescence was seen upon formation of the Squash-DFHO pair (possible acceptor), consistent with lack of FRET.
Extended Data Fig. 8. Quantification of cellular SAM concentration using ratiometric sensors.

a, Standard curve measuring FRET signal (ex: 485 nm, em: 665 nm) in a biochemical assay to determine SAM concentration (see Methods). Data represent mean values ± s.d. for n=3 independent experiments.
b, The average cellular concentration of SAM was measured from 1.2 × 106 cells utilizing the standard curve in panel a. To calculate cellular volume, we assumed HEK293T cells are 17 μm-diameter spheres. We quantified the average SAM concentration at different time points during cycloleucine treatment and withdrawal. SAM concentrations were not affected by sensor expression. Data represent mean values ± s.d. for n=3 independent experiments.
c,d, Correlation between the ratiometric signal and biochemical SAM measurements for Squash-SAM sensors 5–1 (c) and 4–2 (d). To convert the sensor ratiometric signal into SAM concentration, we calculated the average ratiometric signal for HEK293T cells during cycloleucine treatment (n = 6 cells, from Fig. 4g). Using the biochemical assay we determined the average SAM concentration at the same time points, allowing extrapolation of intracellular SAM concentrations from the O/G ratio.
e, We measured the average O/G fluorescence ratio in 30 cells from three independent measurements for each indicated cell type and culture condition. HEK293T cells and mES cells cultured in +2i media showed a narrow distribution while HCT116 and mES cells cultured in −2i showed a wide range of O/G ratio. Sensors used for each cell type are indicated. Line indicates median, box shows the interquartile range, and whiskers are the minimum and maximum values.
f, The average O/G fluorescence ratio for each cell in panel e was converted into average cellular SAM concentration (based on panel c and d) and plotted for each condition. For HEK293T cells, the distribution of O/G fluorescence ratios was different for the two Squash-SAM sensors; however, when these O/G ratios are converted to SAM concentrations based on the specific O/G-to-SAM correlation for each sensor, the cellular SAM concentrations were similar regardless of the sensor. Line indicates median, box shows the interquartile range, and whiskers are the minimum and maximum values.
Extended Data Fig. 9. Effect of cycloleucine treatment and different amino acids depletion on SAM level in mESCs.

a, We expressed both Squash-SAM sensors 4–2 and 5–1 for ratiometric SAM imaging in mESCs. Unlike HEK293T cells, the SAM-dependent Squash signal (DFHO channel) was too dim for the 5–1 sensor. However, Squash-SAM sensor 4–2 gave much brighter cellular fluorescence signal in the DFHO channel. Thus, despite having lower dynamic range than Squash-SAM sensor 5–1, we used the Squash-SAM sensor 4–2 for ratiometric imaging of SAM in mESCs. Image acquisition time was 100 ms for BI channel and 500 ms for DFHO channel. Scale bar, 20 μm.
b, To test whether MAT2A is the major SAM biosynthetic enzyme in mESCs, we expressed Squash-SAM sensor 4–2 and treated the cells with cycloleucine (30 mM final), a MAT2A inhibitor. A rapid drop of cellular SAM levels was observed in 30 min for mESCs cultured in both +2i and −2i media. Note: mESCs cultured in −2i media exhibit a large flattened morphology, which accounts for their increased size relative to mESCs cultured in +2i. Scale bar, 20 μm.
c, d, The drop in SAM levels due to amino acid depletion in mESCs is reversible in cells cultured in either +2i (c) or −2i (d). In Fig. 5a, we showed that depletion of methionine leads to a drop in SAM in mESCs. To see if reintroduction of the amino acids in the media leads to rise in SAM level, we added the amino acids back to the media. Images are shown at indicated time points. For most mESCs cultured in both +2i and −2i conditions, we saw that the SAM levels go back to the pre-treatment condition indicating the reversible nature and lack of cytotoxicity of this process. Scale bar, 20 μm.
Extended Data Fig. 10. Effect of different amino acids depletion and cycloleucine treatment on SAM level in HCT116 cells.

a, Both Squash-SAM sensors 4–2 and 5–1 produced signal that is substantially higher than the background in both the channels. We used 5–1 because it has higher dynamic range. Scale bar, 20 μm.
b, Threonine depletion does not change SAM levels in mESCs (see Fig. 5a). To test if this also holds in HCT116 cells, we expressed Squash-SAM sensor 5–1 in HCT116 cells and monitored SAM level after threonine depletion. Most cells did not show any notable change in SAM levels, consistent with a lack of functional threonine dehydrogenase enzyme in these cells.
c, Although threonine depletion does not change SAM levels (panel b), threonine could be important in methionine-depleted cells. To test this, we expressed Squash-SAM sensor 5–1 in HCT116 cells and monitored SAM levels for 240 min after depleting both threonine and methionine. Threonine depletion did not exacerbate the drop in SAM levels, suggesting that threonine does not have a major role in SAM levels in these cells.
d, Circular Squash-SAM sensor 5–1 expressed in HCT116 cells showed marked drop in SAM after cycloleucine (30 mM final) treatment, indicating MAT2A is required for SAM biosynthesis in these cells. A rapid drop in SAM levels was observed in 30 min for most cells. The cells indicated by white arrow (in c and d) start with a very high level of SAM and show slower drop in SAM level over time compared to other cells.
e, HCT116 cells have three different cell populations based on baseline SAM levels (see Fig. 5d and 5e). We quantified the average cellular ratio of O/G fluorescence for these cells at 15 min intervals after cycloleucine (30 mM final) treatment. All three populations showed slightly faster drop in SAM levels with cycloleucine treatment compared to depletion of methionine and threonine together.
Supplementary Material
{kind=link}
Acknowledgements
We thank members of the Jaffrey and Finley laboratories for helpful comments and suggestions. We thank S. Daniska and J. DeLuca from the Oligo Synthesis Facility, Keck Biotechnology Resource Lab at Yale University for their help in establishing the protocol for the sprout and clip library generation. We thank R. Rao for help with analysis of some mutant RNAs during initial developments of Squash. We also thank J. McCormick at the Flow Cytometry Core facility at Weill Cornell Medical College for help with FACS. This work was supported by NIH grants R01NS064516 and R35NS111631, the American Diabetes Association Pathway to Stop Diabetes Grant 1-18-VSN-02, the Weill Cornell Medicine Daedalus Fund for Innovation, and DoD ALSRP Research Grant AL1301111 to S.R.J, Starr Cancer Consortium grant I12-0051 to S.R.J. and L.W.S.F., NIH grants F32GM120987 and T32 CA062948 to A.O.O.-G.
Competing interests
S.R.J. is the co-founder of Lucerna Technologies and has equity in this company. Lucerna has licensed technology related to Spinach, Broccoli, and other RNA-fluorophore complexes. S.R.J. is a founder of Chimerna Therapeutics and has equity in this company.
REFERENCES
- 1.Sanford L. & Palmer A. Recent Advances in Development of Genetically Encoded Fluorescent Sensors. in Methods in Enzymology 589, 1–49 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Lindenburg L. & Merkx M. Engineering genetically encoded FRET sensors. Sensors 14, 11691–11713 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palmer AE, Qin Y, Park JG & McCombs JE Design and application of genetically encoded biosensors. Trends Biotechnol. 29, 144–152 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lechner H, Ferruz N. & Höcker B. Strategies for designing non-natural enzymes and binders. Curr. Opin. Chem. Biol 47, 67–76 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Paige JS, Nguyen-duc T, Song W. & Jaffrey SR Fluorescence Imaging of Cellular Metabolites with RNA. Science 335, 1194 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kellenberger CA, Wilson SC, Sales-Lee J. & Hammond MC RNA-based fluorescent biosensors for live cell imaging of second messengers cyclic di-GMP and cyclic AMP-GMP. J. Am. Chem. Soc 135, 4906–4909 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun Z, Nguyen T, McAuliffe K. & You M. Intracellular Imaging with Genetically Encoded RNA-based Molecular Sensors. Nanomaterials 9, 233 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim H. & Jaffrey SR A fluorogenic RNA-based sensor activated by metabolite-induced RNA dimerization. Cell Chem. Biol 26, 1725–1731 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li X. et al. Imaging Intracellular S-Adenosyl Methionine Dynamics in Live Mammalian Cells with a Genetically Encoded Red Fluorescent RNA-Based Sensor. J. Am. Chem. Soc 142, 14117–14124 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ortega AD et al. A synthetic RNA-based biosensor for fructose-1,6-bisphosphate that reports glycolytic flux. Cell Chem. Biol 10.1016/j.chembiol.2021.04.006 (2021). [DOI] [PubMed]
- 11.Litke JL & Jaffrey SR Highly efficient expression of circular RNA aptamers in cells using autocatalytic transcripts. Nat. Biotechnol 37, 667–675 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frieda KL & Block SM Direct Observation of Cotranscriptional Folding in an Adenine Riboswitch. Science 338, 397–401 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mandal M. & Breaker RR Adenine riboswitches and gene activation by disruption of a transcription terminator. Nat. Struct. Mol. Biol 11, 29–35 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Serganov A. et al. Structural basis for discriminative regulation of gene expression by adenine- and guanine-sensing mRNAs. Chem. Biol 11, 1729–1741 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dalgarno PA et al. Single-molecule chemical denaturation of riboswitches. Nucleic Acids Res. 41, 4253–4265 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lemay JF, Penedo JC, Tremblay R, Lilley DMJ & Lafontaine DA Folding of the Adenine Riboswitch. Chem. Biol 13, 857–868 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Grubbs RD Intracellular magnesium and magnesium buffering. BioMetals 15, 251–259 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Romani AMP Magnesium Homeostasis in Mammalian Cells. in Metallomics and the Cell (ed. Banci L) 69–118 (Springer, 2013). [Google Scholar]
- 19.Tyrrell J, Mcginnis JL, Weeks KM & Pielak GJ The Cellular Environment Stabilizes Adenine Riboswitch RNA Structure. Biochemistry 52, 8777–8785 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paige JS, Wu KY & Jaffrey SR RNA Mimics of Green Fluorescent Protein. Science 333, 642–646 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Filonov GS, Moon JD, Svensen N. & Jaffrey SR Broccoli: Rapid Selection of an RNA Mimic of Green Fluorescent Protein by Fluorescence-Based Selection and Directed Evolution. J. Am. Chem. Soc 136, 16299–16308 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song W, Strack RL, Svensen N. & Jaffrey SR Plug-and-Play Fluorophores Extend the Spectral Properties of Spinach. J. Am. Chem. Soc 136, 1198–1201 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song W. et al. Imaging RNA polymerase III transcription using a photostable RNA – fluorophore complex. Nat. Chem. Biol 13, 1187–1194 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steinmetzger C, Palanisamy N, Gore KR & Höbartner C. A Multicolor Large Stokes Shift Fluorogen-Activating RNA Aptamer with Cationic Chromophores. Chem. Eur. J 25, 1931–1935 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Tuerk C. & Gold L. Systematic Evolution of Ligands by Exponential Enrichment : RNA Ligands to Bacteriophage T4 DNA Polymerase. Science 249, 505–510 (1990). [DOI] [PubMed] [Google Scholar]
- 26.Ellington AD & Szostak JW In vitro selection of RNA molecules that bind specific ligands. Nature 346, 818–822 (1990). [DOI] [PubMed] [Google Scholar]
- 27.Dixon N. et al. Reengineering orthogonally selective riboswitches. Proc. Natl. Acad. Sci. U. S. A 107, 2830–2835 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Porter EB, Polaski JT, Morck MM & Batey RT Recurrent RNA motifs as scaffolds for genetically encodable small-molecule biosensors. Nat. Chem. Biol 13, 295–301 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hall B. et al. Design, Synthesis, and Amplification of DNA Pools for In Vitro Selection. Curr. Protoc. Mol. Biol 24, 24.2.1–24.2.27 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Bartel DP, Zapp ML, Green MR & Szostak JW HIV-1 Rev Regulation Involves Recognition of Non-Watson-Crick Base Pairs in Viral RNA. Cell 67, 529–536 (1991). [DOI] [PubMed] [Google Scholar]
- 31.Sunbul M. et al. Super-resolution RNA imaging using a rhodamine-binding aptamer with fast exchange kinetics. Nat. Biotechnol 39, 686–690 (2021). [DOI] [PubMed] [Google Scholar]
- 32.Truong L. & Ferré-D’Amaré AR From fluorescent proteins to fluorogenic RNAs: Tools for imaging cellular macromolecules. Protein Sci. 28, 1374–1386 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X, Kim H, Litke JL, Wu J. & Jaffrey SR Fluorophore-Promoted RNA Folding and Photostability Enables Imaging of Single Broccoli-Tagged mRNAs in Live Mammalian Cells. Angew. Chemie Int. Ed 59, 4511–4518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strack RL, Disney MD & Jaffrey SR A superfolding Spinach2 reveals the dynamic nature of trinucleotide repeat–containing RNA. Nat Methods 10, 1219–1224 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shu D, Khisamutdinov EF, Zhang L. & Guo P. Programmable folding of fusion RNA in vivo and in vitro driven by pRNA 3WJ motif of phi29 DNA packaging motor. Nucleic Acids Res. 42, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Filonov GS et al. In-Gel Imaging of RNA Processing Using Broccoli Reveals Optimal Aptamer Expression Strategies. Chem. Biol 22, 649–660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Su X, Wellen KE & Rabinowitz JD Metabolic control of methylation and acetylation. Curr. Opin. Chem. Biol 30, 52–60 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moon JD et al. Naturally occurring three-way junctions can be repurposed as genetically encoded RNA-based sensors. Cell Chem. Biol 28, 1–12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu C. et al. Crystal structures of the SAM-III/SMK riboswitch reveal the SAM-dependent translation inhibition mechanism. Nat. Struct. Mol. Biol 15, 1076–1083 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang J. et al. Dependence of Mouse Embryonic Stem Cells on Threonine Catabolism. Science 325, 435–439 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shyh-Chang N. et al. Influence of Threonine Metabolism on S-Adenosylmethionine and Histone Methylation. Science 339, 222–226 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanderson SM, Gao X, Dai Z. & Locasale JW Methionine metabolism in health and cancer: a nexus of diet and precision medicine. Nat. Rev. Cancer 19, 625–637 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Ying QL et al. The ground state of embryonic stem cell self-renewal. Nature 453, 519–523 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chambers I. et al. Nanog safeguards pluripotency and mediates germline development. Nature 450, 1230–1234 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Filipczyk A. et al. Biallelic expression of nanog protein in mouse embryonic stem cells. Cell Stem Cell 13, 12–13 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Lemay JF et al. Comparative study between transcriptionally- and translationally-acting adenine riboswitches reveals key differences in riboswitch regulatory mechanisms. PLoS Genet. 7, e1001278 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu R. et al. Genetically Encoded Ratiometric RNA-Based Sensors for Quantitative Imaging of Small Molecules in Living Cells. Angew. Chemie Int. Ed 58, 18271–18275 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sunbul M. & Jäschke A. SRB-2: a promiscuous rainbow aptamer for live-cell RNA imaging. Nucleic Acids Res. 46, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hao X. et al. Immunoassay of S-adenosylmethionine and S-adenosylhomocysteine: the methylation index as a biomarker for disease and health status. BMC Res. Notes 9, 1–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
METHODS REFERENCES
- 50.Han KY, Leslie BJ, Fei J, Zhang J. & Ha T. Understanding the photophysics of the Spinach-DFHBI RNA aptamer-fluorogen complex to improve live-cell RNA imaging. J. Am. Chem. Soc 135, 19033–19038 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Endoh T. & Sugimoto N. Selection of RNAs for Constructing ‘Lighting-UP’ Biomolecular Switches in Response to Specific Small Molecules. PLoS One 8, e60222 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Endoh T. & Sugimoto N. Signaling Aptamer Optimization through Selection Using RNA-Capturing Microsphere Particles. Anal. Chem 92, 7955–7963 (2020). [DOI] [PubMed] [Google Scholar]
- 53.Paul CP et al. Localized expression of small RNA inhibitors in human cells. Mol. Ther 7, 237–247 (2003). [DOI] [PubMed] [Google Scholar]
- 54.Carey BW, Finley LWS, Cross JR, Allis CD & Thompson CB Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mentch SJ et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab. 22, 861–873 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available within the paper and its Supplementary Information files. They are also available from the corresponding author on reasonable request. The following plasmids generated in this study will be available through Addgene: pAV-U6+27-Squash (ID 177913), pAV-5S-Squash (ID 177914), pAV-U6+27-Tornado-Squash (ID 177915), pAV-U6+27-Tornado-Squash-SAM-sensor 4–2 (ID 177916), pAV-U6+27-Tornado-Squash-SAM-sensor 5–1 (ID 177917), pAV-U6+27-Tornado-F30-Squash-Broccoli (ID 177918), pAV-U6+27-Tornado-F30-Broccoli-Squash-SAM-sensor 4–2 (ID 177919), pAV-U6+27-Tornado-F30-Broccoli-Squash-SAM-sensor 5–1 (ID 177920).